

Abstract
We have investigated the mechanism by which conventional kinesin is prevented from binding to microtubules (MTs) when not transporting cargo. Kinesin heavy chain (HC) was expressed in COS cells either alone or with kinesin light chain (LC). Immunofluorescence microscopy and MT cosedimentation experiments demonstrate that the binding of HC to MTs is inhibited by coexpression of LC. Association between the chains involves the LC NH2-terminal domain, including the heptad repeats, and requires a region of HC that includes the conserved region of the stalk domain and the NH2 terminus of the tail domain. Inhibition of MT binding requires in addition the COOH-terminal 64 amino acids of HC. Interaction between the tail and the motor domains of HC is supported by sedimentation experiments that indicate that kinesin is in a folded conformation. A pH shift from 7.2 to 6.8 releases inhibition of kinesin without changing its sedimentation behavior. Endogenous kinesin in COS cells also shows pH-sensitive inhibition of MT binding. Taken together, our results provide evidence that a function of LC is to keep kinesin in an inactive ground state by inducing an interaction between the tail and motor domains of HC; activation for cargo transport may be triggered by a small conformational change that releases the inhibition of the motor domain for MT binding.
Keywords: kinesin, microtubules, molecular motors
Conventional kinesin is a virtually ubiquitous molecular motor that moves cargo towards the plus ends of microtubules (MTs).1 Although kinesin has been implicated in the transport of various membrane-bound organelles and their positioning within the cell (reviewed in Goodson et al., 1997; Tanaka et al., 1998), its in vivo function is still controversial. It has been observed to be associated with various cellular membranes, but the majority is actually found as a soluble protein in the cytoplasm (Vale et al., 1985), neither associated with vesicles nor MTs. The existence of this sizable cytosolic pool is surprising in view of the fact that a kinesin motor attached to a bead or coverglass is able to walk processively along a MT (Howard et al., 1989; Block et al., 1990). One possibility is that there is a mechanism in cells that keeps kinesin in an inactive state (i.e., unable to bind to MTs) until it is needed to transport cargo (Hackney et al., 1992). However, there has been no direct evidence for inhibition of kinesin activity in vivo.
Kinesin is a tetramer composed of two heavy chains (110–130 kD) and two light chains (60–80 kD). Kinesin heavy chain (HC) consists of three structurally distinct domains, an NH2-terminal globular domain (head), an extended α-helical coiled-coil domain (stalk), and a globular COOH-terminal domain (tail). Motor activity is encoded by the NH2-terminal 350 amino acids of the globular head domains, which are responsible for MT binding and for MT-dependent ATPase activity. The stalk domains are involved in dimerization of the two HC polypeptides. Both the stalk and globular tail domains have been implicated in cargo binding (for reviews of kinesin domains see Bloom and Endow, 1994; Hackney, 1996; Vale and Fletterick, 1997; Block, 1998a ,b). A role for these COOH-terminal domains in cargo selection or regulation is also suggested by comparison of members of the kinesin superfamily; the proteins share extensive homology (35–45% identical) in their motor domains, but have little or no homology outside this domain. These divergent regions are thought to account for the independent targeting and/or regulation of particular kinesins (for reviews see Bloom and Endow, 1994; Moore and Endow, 1996; Hirokawa, 1996, 1998; Vale and Fletterick, 1997).
The function of the light chain (LC) of kinesin is not yet clear. LC contains two notable domains, a series of heptad repeats required for association with HC in vitro, and a series of tetratricopeptide repeat (TPR) protein–protein interaction motifs (Cyr et al., 1991; Gauger and Goldstein, 1993; Wedaman et al., 1993; Gindhart and Goldstein, 1996). Antibodies raised against LC bind to the globular tail domains of native kinesin (Hirokawa et al., 1989). The cloning of cDNAs for LC has shown that there are several alternatively spliced gene products that, when bound to HC, may target the kinesin tetramer to particular classes of organelles (Khodjakov et al., 1998; Liao and Gunderson, 1998). A role for LC in cargo binding is supported by studies in which antibodies specific for LC inhibit the binding of purified kinesin to a microsomal fraction or are able to displace kinesin already bound to membranes (Yu et al., 1992; Stenoien and Brady, 1997). However, other studies have suggested that LC may not be essential for cargo binding and that the regions required for this function are contained within the stalk and/or tail domains of HC (Skoufias et al., 1994; Bi et al., 1997).
While dimeric constructs of HC containing only the motor domains are fully capable of MT binding and motility, there are hints that native kinesin is inhibited (discussed in Hackney, 1996). Soluble kinesin purified from bovine brain has only a moderate ATPase activity upon interaction with MTs, but the ATPase activity is much higher after proteolytic removal of the COOH-terminal half of the molecule (Kuznetsov et al., 1989). Similarly, the in vitro expressed NH2-terminal motor domain of HC has a higher ATPase activity than the native protein (Gilbert and Johnson, 1993; Stewart et al., 1993; Huang and Hackney, 1994). Finally, binding of native kinesin to MTs is too weak in the presence of ATP to maintain the sustained contact required for processive motion (Vale et al., 1985; Hackney, 1996).
A potential mechanism for motor inhibition has been suggested by electron microscopy and sedimentation analysis of purified kinesin (Hirokawa et al., 1989; Hisanaga et al., 1989; Hackney et al., 1992). At physiological salt concentrations, purified kinesin is in a folded conformation in which the tail comes close to the motor domain, while, in high salt, kinesin is in an extended conformation. The existence of an inhibited ground state of kinesin may explain the large pool in cells that is not associated with MTs. However, an inhibition of kinesin's binding to MTs has not been directly demonstrated, a correlation of the folded conformation with the inhibited state has not been established, and the LC and HC domains responsible for the postulated inhibition have not been identified.
To investigate the regulation of kinesin under physiological conditions, we have developed an assay in which HC is expressed in COS cells, either alone or together with LC. Our results suggest that HC is inhibited from binding to MTs by its association with LC. This inhibition is dependent on the NH2-terminal domain of LC, including the heptad repeats, which interacts with a region of HC that includes the stalk domain and the NH2 terminus of the tail domain. In addition, inhibition of MT binding requires the COOH-terminal 64 amino acids of HC, which are not involved in the association with LC. We propose that LC induces a conformation in which the HC tail domain prevents the motor domain from binding to MTs. We also show that inhibition of MT binding can be released by shifting the pH from 7.2 to 6.8, suggesting at least one mechanism by which kinesin may be activated in vivo. Endogenous kinesin in COS cells also shows pH-sensitive inhibition of MT binding.
Materials and Methods
cDNAs for HC and LC and Construction of Mammalian Expression Vectors
A cDNA clone encoding the entire open reading frame (ORF) of the ubiquitous isoform of kinesin HC was isolated from a rat brain phage (λZAP) library (gift of J. Sullivan, Salk Institute, La Jolla, CA) by screening for the expression of kinesin-related proteins using a pan-kinesin peptide antibody (gift of T.J. Mitchison, UCSF, San Francisco, CA; Sawin et al., 1992). The library isolate provided HC cDNA cloned into Bluescript SK (Stratagene Inc., La Jolla, CA). A fragment of this plasmid encoding the entire ORF was subcloned into the mammalian expression vector pcDNA3-myc (gift of F. McKeon, Harvard Medical School, Boston, MA; Taylor and McKeon, 1997). The resultant plasmid, pcDNA3-myc-HC, contains an in-frame myc tag, 30 bases of the Bluescript SK plasmid (SalI– EcoRI), 60 bases of the 5′ untranslated region of HC and the HC ORF. The resulting 30 amino acid insertion does not affect the properties of HC, as this construct is able to bind to MTs in an ATP-dependent manner and interact with LC (see Results). Deletion mutants of HC were constructed by PCR such that stop codons were placed in the coding sequence of HC after amino acids 682, 810, and 891. The PCR products were subcloned back into pcDNA3-myc-HC.
The cDNAs encoding kinesin LCs were isolated by PCR from the same rat brain library. A PCR product corresponding to bp's 855–1477 of the published rat LC sequence (Cyr et al., 1991), which is found in all splice variants of LC, was obtained using the library as a template. This fragment was radiolabeled (T7 QuickPrime Kit; Pharmacia LKB Biotech Inc., Piscataway, NJ) and used to screen the library. Three full-length clones encoding different spliced variants of rat LC were isolated (LC-B, LC-C, and LC-F), each cloned into the EcoRI site of Bluescript SK (plasmids SK-LCs). LC-B is identical to the published sequences of rat LC-B (Cyr et al., 1991). LC-C is identical to the published sequence of rat LC-C, except for a short deletion of 8 amino acid residues (496–503) (Cyr et al., 1991); an identical deletion was noted in the hamster LC-C sequence (Khodjakov et al., 1998). The sequence of splice variant LC-F, though similar to LC-B and LC-C, is novel (the COOH-terminal 15 amino acids are DGGEEVSMSVEWNGA).
The LC cDNAs were subcloned in two steps into the mammalian expression vector pcDNA3-HA (gift of F. McKeon; Taylor and McKeon, 1997). In the first step, an in-frame XhoI site was engineered at the NH2 termini of the LCs by PCR. The PCR product was subcloned into Bluescript SK to yield the plasmid SK-Xho-Nterm. Sequences encoding the COOH termini of the three LC isoforms were then subcloned into SK-Xho-Nterm. The resultant plasmids, SK-Xho-LCs, were used in the second step to transfer the LC coding sequences into the vector pcDNA3-HA. Each SK-Xho-LC plasmid was digested with XhoI and XbaI and ligated in-frame into pcDNA3-HA digested with the same enzymes. The final plasmids, pcDNA3-HA-LCs, consist of an in-frame hemagglutinin (HA) tag, 5 amino acids from the LC 5′-untranslated region (amino acids LPIGR), followed by the LC ORFs. Deletion mutants of LC-C were constructed by using PCR to engineer stop codons in the coding sequence of LC-C after amino acids 176, 237, 405, and 488. The PCR products were subcloned back into pcDNA3-HA-LC-C. To delete the heptad repeat region (pcDNA3-HA-LΔHR), an in-frame NotI site was constructed after amino acid 47 by PCR. The PCR product was cut with XhoI and NotI and subcloned back into pcDNA3-HA-LC-C cut with the same enzymes.
The sequences of all mutant constructs were confirmed by sequencing both strands of the resultant plasmids. Plasmid DNA was purified on a Qiagen column (QIAGEN Inc., Santa Clarita, CA).
Antibodies and Reagents
Mouse hybridoma cells producing monoclonal antibodies to the myc-tag (9E10) were purchased from American Type Culture Collection (ATCC, Rockville, MD). Mouse hybridoma cells producing monoclonal antibodies to the HA-tag (12CA5) were kindly provided by F. McKeon. Rabbit polyclonal antibodies to the myc- and HA-tags were purchased from Santa Cruz Biotechnology (Santa Cruz, CA). Mouse monoclonal antibodies to α-tubulin (DM 1A) were purchased from Sigma Chemical Co. (St. Louis, MO). Secondary antibodies for immunofluorescence microscopy (Oregon Green 488 and Rhodamine Red-X) were purchased from Molecular Probes (Eugene, OR). HK420 was kindly provided by R. Vale (UCSF, San Francisco, CA). All other reagents were from Sigma Chemical Co. unless otherwise indicated.
Antibodies to kinesin HC were raised in rabbits against the peptide khc13 (CKKLSGKLYLVDLAGSEKVSKTGAEG) coupled to keyhole limpet hemocyanin (Calbiochem Corp., San Diego, CA) via the NH2-terminal cysteine. Affinity purification was carried out with a SulfoLink (Pierce Chemical Co., Rockford, IL) column to which the peptide had been coupled. The acid-eluted antibodies were subsequently concentrated on a hydroxyapatite column.
Taxol was a gift of the Drug Synthesis & Chemistry Branch, Developmental Therapeutics Program, Division of Cancer Treatment, National Cancer Institute (Bethesda, MD). Taxol-MTs were prepared as described (Butner and Kirschner, 1991) except that taxol was added sequentially to 0.2, 2, and 20 μM to a stock solution of 5 mg/ml tubulin purified from bovine brain (Vallee, 1986a ) or purchased from Cytoskeleton (Denver, CO).
Cells, Transfection, and Immunofluorescence Microscopy
Hybridoma cells producing the 9E10 and 12CA5 monoclonal antibodies were grown in DMEM supplemented with 10% FBS (Hyclone, Logan, UT). After growing to confluency, the cells were pelleted and the supernatant was used undiluted for immunofluorescence microscopy, or diluted 1:5 for immunoblotting or 1:10 for immunoprecipitation.
African green monkey kidney cells (COS) were grown at 37°C in DMEM supplemented with 10% Fetal Clone III (Hyclone). Cells were plated on coverslips at least 6 h before transfection, which was carried out using Lipofectamine (Life Technologies/GIBCO BRL, Rockville, MD) as suggested by the manufacturer. 36–48 h after transfection, the cells were rinsed in PBS and fixed in 3% paraformaldehyde in PBS. The cells were permeabilized with 0.2% Triton X-100 in PBS, rinsed with 0.2% fish skin gelatin (FSG) in PBS, labeled with primary antibody in FSG/PBS for 1 h, rinsed three times with FSG/PBS, and then labeled with secondary antibody in FSG/PBS for 1 h. Coverslips were mounted in Mowiol (Calbiochem Corp.) and visualized with an Axiophot fluorescence microscope (Carl Zeiss Inc., Oberkochen, Germany). Images were taken with a CCD camera and stored in digital form using Northern Exposure software (Empix Imaging, Inc., Mississauga, Ontario, Canada). A strong immunofluorescent signal was detected with anti–myc or anti–HA in ∼20% of cells transfected with the corresponding construct. The fluorescent signal from the remainder of the cells was barely above that produced by the secondary antibody alone. For cells transfected with HC alone, 10–20% of the transfected cells expressed HC at high levels, 10–20% expressed low levels, and the rest expressed moderate levels based on visual analysis of fluorescence intensity. For double transfected cells, virtually every transfected cell expressed both chains at similar levels, and only rarely (1 in 10 coverslips) could cells transfected with only one chain be found.
Cell Lysis, Immunoprecipitations, MT Binding, and Sedimentation Assays
36–48 h after transfection, COS cells were trypsinized, pelleted, and lysed by resuspending in ice-cold Lysis buffer (LB; 25 mM Hepes/KOH, 115 mM potassium acetate, 5 mM sodium acetate, 5 mM MgCl2, 0.5 mM EGTA, 1% Triton X-100, 1 mM PMSF, 10 μg/ml leupeptin, 5 μg/ml chymostatin, 3 μg/ml elastatinal, 1 mg/ml pepstatin A, pH 7.2–7.4). For testing the effects of pH, LB was identical except that it was buffered with MES/KOH (pH 6.4), Pipes/KOH (pH 6.6, 6.8, 7.0, 7.2), or Hepes/KOH (pH 6.8, 7.0, 7.2, 7.4, 7.6). Insoluble material was removed by centrifugation of lysates in a TLA100.3 rotor at 50,000 rpm, 4°C for 30 min. Lysates were 1.5–3 mg/ml protein as determined by the BCA Protein Assay (Pierce Chemical Co., Rockford, IL) and were either used fresh or aliquots were frozen in liquid nitrogen and stored at −80°C. Lysates coexpressing HC+LC contained more HC in the soluble fraction than lysates expressing HC alone; thus, the former were diluted with lysates from untransfected COS cells such that similar amounts of expressed HC were subject to MT binding or immunoprecipitation. Immunoblotting with the anti–khc13 peptide antibody determined that transfected cells expressed ∼5–10-fold more HC than untransfected COS cells. Since only 20% of the cells were transfected, these cells likely express kinesin, on average, at levels 25–50-fold over the endogenous protein.
For immunoprecipitations, lysates were diluted with an equal volume of LB, and then hybridoma supernatant containing either the 9E10 or 12CA5 antibody was added. After incubating for 4–16 h at 4°C with rotation, Protein G Sepharose (Pharmacia LKB Biotech Inc.) was added and the incubation was continued for an additional 30–60 min. Beads were sedimented and washed three times with LB before resuspending in sample buffer. Proteins in the supernatant were precipitated with TCA and resuspended in sample buffer.
For MT binding assays, lysates were brought to room temperature and, unless otherwise indicated, taxol was added to 20 mM, AMP-PNP (5′-adenylylimidodiphosphate) or ATP to 2.5 mM, and taxol-stabilized MTs to 0.1 mg/ml. After incubating 30 min at room temperature with rotation, lysates were overlaid on 1/4 to 1/2 volume of 10% sucrose in LB + taxol. MTs were sedimented by centrifugation in an SW55 rotor for 30 min at 30,000 rpm, 22°C. The supernatant was carefully removed and the proteins were precipitated with TCA and resuspended in sample buffer. MT pellets were resuspended directly in sample buffer. Taxol-stabilized MTs are not affected by pH over a very wide range (Vallee, 1986b ) and immunoblotting with an antitubulin antibody confirmed that virtually all of the added taxol-stabilized MTs pellet, independent of the lysate pH values used here.
For sucrose gradient centrifugation, linear gradients were formed from 15, 13, 11, and 9% (wt/vol) layers of sucrose in LB by incubation for 4 h at 4°C. Lysates were loaded on top and the gradients centrifuged in a TLS55 rotor for 2.5 h at 55,000 rpm, 2°C. For sedimentation under high salt conditions, the lysates and sucrose solutions were brought to 0.5 M NaCl. Fractions collected from the top were diluted with sample buffer and analyzed by SDS-PAGE. The gradients were calibrated in parallel using protein standards with known sedimentation coefficients (malate dehydrogenase 4.3S, aldolase 7.7S, and catalase 11.3S).
Proteins were separated by SDS-PAGE, transferred to nitrocellulose, and immunoblotted after blocking with 5% nonfat dry milk in Tris-buffered saline + 0.1% (vol/vol) Tween 20. Recombinant proteins were detected by monoclonal or polyclonal antibodies to the myc- and HA-tags, followed by ECL (NEN Life Sciences, Boston, MA). Endogenous kinesin was detected with the affinity purified khc13 polyclonal antibody, detailed above. For quantitation, nitrocellulose exposed to primary antibodies were incubated with 125I goat anti–rabbit IgG secondary antibody (NEN Life Sciences) and the blots were exposed to a Phosphorimager screen and quantitated using a BAS1000 (both from Fuji Photo Film Co., Tokyo, Japan).
Results
Immunolocalization of HC and LC Expressed in COS Cells
To study the regulation of kinesin in vivo, cDNAs encoding HC or LC were transfected into COS cells either alone or together. To aid in identification of the transiently expressed proteins, the myc epitope tag was engineered at the NH2 terminus of HC. An epitope tag from the influenza virus hemagglutinin (HA) protein was placed at the NH2 terminus of LC splice forms B, C, and F. Preliminary studies gave identical results for all three LC splice forms (data not shown); splice form C was used in the experiments described here.
Immunofluorescence microscopy of HC expressed alone showed diffuse staining throughout the cytoplasm. A filamentous pattern superimposed upon this diffuse staining was also visible in ∼5–10% of the transfected cells and only in those cells that expressed HC at high levels (Fig. 1, A and B). Double labeling with anti–myc and antitubulin antibodies indicated that the filamentous staining colocalized with MTs (Fig. 1, C and D). In addition, intense staining for HC was often seen at the tips of cellular processes, even when filamentous staining was barely detectable, suggesting that HC is able to move along MTs and accumulate at the plus ends (Fig. 1, C and D). These results confirm previous reports of HC association with MTs upon transient expression in CV-1 (Navone et al., 1992) and mouse L cells (Nakata and Hirokawa, 1995).
Figure 1.

Localization of HC expressed alone, LC expressed alone, and HC and LC expressed together. COS cells were transiently transfected with plasmids encoding HC alone (A–D), LC alone (E and F) or both HC and LC (G–J). The expressed proteins were localized by immunofluorescence microscopy. (A and B) HC was detected with an anti–myc monoclonal antibody followed by Rhodamine Red-X–labeled anti–mouse secondary antibody. (C and D) Cells were double labeled for HC and MTs. HC was detected with anti–myc polyclonal and Oregon green 488–labeled anti–rabbit antibodies, and tubulin was detected with antitubulin monoclonal and Rhodamine Red-X–labeled anti–mouse secondary antibodies. (E and F) LC was detected with anti–HA polyclonal and Oregon green 488–labeled anti–rabbit secondary antibodies. (G–J) Cells were double labeled for HC and LC. HC was detected with anti–myc monoclonal and Rhodamine Red-X–labeled anti–mouse antibodies, and LC was detected with anti–HA polyclonal and Oregon green 488– labeled anti–rabbit secondary antibodies. Bar, 10 μm.
Colocalization of HC with MTs was sensitive to ATP, suggesting that this interaction occurs via the HC motor domain. When COS cells expressing HC were permeabilized with digitonin or saponin before fixation, MT staining was observed in the presence of AMP-PNP but not ATP (data not shown). This is similar to the behavior of native kinesin, which binds tightly to MTs in the presence of AMP-PNP and is released by ATP (Vale et al., 1985). Therefore, an ATP-independent interaction of the HC tail domain with MTs (Navone et al., 1992) does not play a major role in these experiments.
Immunofluorescence microscopy of LC expressed alone showed diffuse staining throughout the cytoplasm at all levels of expression, as well as accumulation in the center of the cell (Fig. 1, E and F). In electron microscopy, the latter appeared as dense aggregates that labeled with anti– HA antibodies (data not shown). Even when expressed at very high levels, LC did not show accumulation at the tips of the cell or colocalization with MTs.
Immunofluorescence microscopy of COS cells coexpressing HC and LC showed an identical diffuse staining pattern for both proteins (Fig. 1, G–J). Most importantly, when coexpressed with LC, HC failed to colocalize with MTs or accumulate at the tips of processes, even when expressed at very high levels. Virtually every cell in the double transfections expressed both HC and LC, although very rarely, as depicted in Fig. 1, I and J, could cells expressing only one chain be detected. Note that in this case the cell on the left is expressing both HC and LC and shows the diffuse staining pattern, whereas the cell next to it, on the right, is expressing only HC and shows the characteristic MT staining pattern. These results suggest that HC and LC interact when coexpressed in COS cells and that this interaction inhibits the MT binding of HC. A similar lack of MT binding was observed for coexpressed HC and LC when cells were permeabilized with digitonin or saponin in the presence of either ATP or AMP-PNP before fixation (data not shown). An inhibitory effect of LC on the MT binding of HC was also observed in BHK cells (data not shown).
In Vitro Binding of Kinesin to Microtubules
To analyze the effect of LC on the MT binding of HC more directly, a MT cosedimentation assay was developed. COS cells transiently expressing HC with or without LC were lysed with Triton X-100 in a buffer mimicking intracellular conditions. Centrifugation yielded a soluble fraction (lysate) and a pellet containing the insoluble material. Immunoblotting experiments demonstrated that, when expressed alone, ∼30% of the HC remained in the lysate (experimental range 30–50%) (see Fig. 3 B, top, H). LC expressed alone was largely insoluble (see Fig. 3 B, top), in agreement with the observation of LC aggregates by electron microscopy. However, when HC and LC were coexpressed, essentially all of the HC and a significant percentage of LC remained in the lysate (see Fig. 3 B, bottom, H+L). The fraction of LC found in the pellet was variable, depending on the relative levels of expression of the two chains. These results are consistent with HC and LC interacting when coexpressed in COS cells while unassembled LC remains insoluble.
Figure 3.

Interaction of the LC deletion mutants with HC. (A) Deletion mutants of LC were constructed in which stop codons were engineered after amino acids 176, 237, 405, and 488. These constructs retain the heptad repeat region but contain deletions of some or all of the TPR motifs. The deletion mutant LΔHR is missing only the heptad repeat region. Amino acids numbers for the full length protein are shown below the schematic of LC. (B) COS cells expressing HC, LC, or the LC deletion mutants alone (H, L, L176, L237, L405, L488, LΔHR; top) or expressing HC together with LC or the LC deletion mutants (H+L, H+L176, H+L237, H+L405, H+L488, and H+LΔHR; bottom) were lysed with 1% Triton X-100. After centrifugation, the amount of expressed protein found insoluble in the pellet (P) and soluble in the lysate (S) was determined by immunoblotting with polyclonal antibodies to the myc- and HA-epitope tags. Molecular weight size standards (kilodaltons) are indicated on the left. (C) Coimmunoprecipitation of HC and the LC deletion mutants. Lysates were immunoprecipitated for HC using a monoclonal anti–myc antibody (left) or for LC using a monoclonal anti–HA antibody (right). Immunoprecipitates were immunoblotted to detect the expressed proteins using polyclonal antibodies to the epitope tags.
Next, taxol-stabilized MTs were added to the lysates from cells expressing either HC alone or both HC and LC. Incubations were carried out in the presence of AMP-PNP or ATP, the MTs were sedimented, and the amounts of HC and LC in the pellet were determined by immunoblotting. When expressed alone, the majority of HC cosedimented with MTs in the presence of AMP-PNP (Fig. 2 A, lane 5 vs. 7), but remained in the supernatant in the presence of ATP (lane 9 vs. 11). In contrast, most of the HC failed to bind to MTs when coexpressed with LC, even in the presence of AMP-PNP (Fig. 2 A, lane 6 vs. 8), although some binding was observed when very high concentrations of MTs were added (Fig. 2 B). These results confirm the conclusion drawn from the immunofluorescence experiments and show directly that the interaction of LC with HC reduces the ability of HC to bind microtubules.
Figure 2.

MT cosedimentation of HC expressed alone or together with LC. (A) Lysates from COS cells expressing HC alone (H) or together with LC (H+L) were compared under three conditions: no addition of MTs (left) or addition of taxol-stabilized MTs with either AMP-PNP (middle) or ATP (right). MTs and bound proteins were sedimented through a sucrose cushion and the MT pellets (P) and supernatants (S) were immunoblotted for the presence of the expressed HC and LC. (B) Quantitation of the amount of HC recovered in the MT pellet in the presence of AMP-PNP as a function of the amount of MTs added to the lysate. (•) Data from cells expressing HC alone, (▴) cells coexpressing HC and LC. Each data point represents the mean ± SD of at least four experiments.
Control experiments demonstrated that the presence of COS cell cytosol does not influence the interaction of the motor domain of HC with MTs. When increasing concentrations of MTs were added to COS cell lysates containing the bacterially expressed NH2-terminal 420 amino acids of human conventional kinesin heavy chain (HK420), half-maximal binding was obtained at ∼0.15 mM tubulin (data not shown). This value agrees with the apparent binding constant for MTs based on activation of the ATPase activity of purified dimeric kinesin constructs (Hackney, 1994, 1995; Rosenfeld et al., 1996; Jiang et al., 1997).
The NH2-terminal Domain of LC Interacts with HC and Is Required for Inhibition of MT Binding
Next, we identified the domain of LC required for inhibiting the MT binding of HC. To this end, we first determined the LC domain necessary for interaction with HC, and then analyzed whether the same domain is sufficient for inhibition of MT binding. Constructs of LC were made in which the heptad repeats or various numbers of the TPRs were deleted (Fig. 3 A). The proteins were expressed in COS cells alone or together with HC and lysates were analyzed by immunoblotting. In each case, a protein of the predicted size was detected (Fig. 3 B). LC constructs missing various numbers of TPR motifs (L176, L237, L405, and L488) aggregated to varying degrees when expressed alone or with HC and were thus found partially in the pellet after centrifugation (Fig. 3 B, top). Similar to full length LC, these LC constructs were able to shift the majority of HC to the soluble fraction (Fig. 3 B, bottom). Removal of the heptad repeat region (LΔHR) rendered LC insoluble even when coexpressed with HC, and this construct had no effect on the solubility of HC (Fig. 3 B). Taken together, these results suggest that the NH2-terminal region of LC, including the heptad repeat domain, is required for HC binding.
Coimmunoprecipitation experiments directly tested the interaction between the LC constructs and HC. When lysates were subjected to immunoprecipitation for HC using myc antibodies, LC and constructs retaining the heptad repeat domain, but lacking some or all of the TPR motifs were also precipitated (Fig. 3 C, left); small amounts that were not coprecipitated likely represent soluble, unassembled chains. When the same lysates were immunoprecipitated for LC using HA antibodies, essentially all the HC copelleted (Fig. 3 C, right). These results confirm that the NH2 terminus of LC, including the heptad repeat domain, is required for interaction with HC in vivo, in agreement with data obtained in vitro (Gauger and Goldstein, 1993). Because the construct in which the heptad repeat domain is deleted (LΔHR) is insoluble, we cannot exclude that LC contains an additional region that can interact with HC. However, we believe that LΔHR is insoluble because it is unable to interact with HC.
To determine whether the various LC constructs could inhibit MT binding of HC when they were coexpressed in COS cells, MT binding was first assessed by immunofluorescence microscopy. With the LC constructs lacking one or more TPR motifs, diffuse staining was seen for both the heavy and light chains (Fig. 4, A–H). A filamentous MT-like pattern and accumulation at the tips of processes, characteristic for HC alone, was never seen. However, with the construct lacking the heptad repeat domain (LΔHR), the characteristic MT-staining pattern was still detected for HC, whereas the LC mutant itself showed diffuse staining (Fig. 4, I and J). Similar to full length LC, the mutant LΔHR occasionally showed accumulations in the center of the cell that are likely related to its insolubility (data not shown). The nuclear staining seen for this construct was also variable. These results provide additional evidence that the NH2-terminal domain of LC, containing the heptad repeats, is responsible for the interaction with HC and suggest that this domain is also sufficient to inhibit the MT binding of HC.
Figure 4.

Localization of HC expressed with the LC deletion mutants. COS cells were transiently transfected with plasmids encoding the indicated proteins and the expressed proteins were localized by immunofluorescence microscopy. HC was detected with anti–myc monoclonal and Rhodamine Red-X–labeled anti–mouse secondary antibodies. The LC deletion mutants were detected with anti–HA polyclonal and Oregon green 488–labeled anti–rabbit secondary antibodies. For coexpression of HC with L488, note that the cell on the left expresses both proteins and shows diffuse staining, whereas the cell on the right expresses only HC and shows MT staining (G and H). Bar, 10 μm.
MT cosedimentation assays confirmed that the NH2-terminal domain of LC is required for inhibition of MT binding. Both intact LC and the TPR deletion constructs prevented HC from binding to MTs in the presence of AMP-PNP (Fig. 5). The expressed proteins did not pellet with MTs in the presence of ATP (data not shown). Since LC lacking the heptad repeats (LΔHR) is insoluble even in the presence of HC, only HC was detected in the supernatant after centrifugation and, as expected, bound to MTs in an ATP-sensitive manner (data not shown). Taken together, these results indicate that the NH2-terminal region of LC, including the heptad repeats, is required for both the interaction with HC and inhibition of the binding of HC to MTs.
Figure 5.

MT cosedimentation of HC expressed with the LC deletion mutants. Taxol-stabilized MTs and AMP-PNP were added to lysates from COS cells expressing HC alone (H), HC and LC (H+L), or HC and the LC deletion mutants (H+L176, H+L237, H+L405, and H+L488). MTs and bound proteins were sedimented through a sucrose cushion. The MT pellets (P) and supernatants (S) were immunoblotted to detect the expressed proteins using polyclonal antibodies to the myc- and HA-tags.
The COOH-terminal 64 Amino Acids of HC Are Required for Inhibition of MT Binding but Not for LC Interaction
A similar approach was used to determine the region of HC required for interaction with LC. Truncation mutants were made that either lacked the tail domain and the COOH-terminal portion of the stalk domain (H682), just the tail domain (H810), or the COOH-terminal half of the tail domain (H891) (Fig. 6 A). The proteins were expressed in COS cells alone or together with LC and lysates were analyzed by immunoblotting. In each case, a protein product of the predicted size was detected (Fig. 6 B). Whether expressed alone or together with LC, the expressed H682 and H810 were largely in the supernatant after centrifugation (Fig. 6 B, top). On the other hand, similar to full length HC, much of H891 was in the insoluble pellet when expressed alone, but was rendered soluble when coexpressed with LC. Nearly all of LC was insoluble when coexpressed with HC lacking the tail domain and some of the stalk (H682) (Fig. 6 B, bottom). In contrast, a significant portion of LC was in the supernatant when coexpressed with the longer HC constructs (H810 or H891). These data suggest that the COOH-terminal region of the HC stalk domain is required for the interaction with LC.
Figure 6.

Interaction of the HC deletion mutants with LC. (A) Deletion mutants of HC were constructed in which stop codons were engineered after amino acids 682, 810, and 891. These deletions remove both the tail domain and the conserved region of the stalk domain (H682), the entire tail domain (H810), or half of the tail domain (H891). (B) COS cells expressing HC or the HC deletion mutants alone (H, H682, H810, and H891; top) or together with LC (H+L, H682+L, H810+L, and H891+L; bottom) were lysed with 1% Triton X-100. After centrifugation, the amount of expressed protein found insoluble in the pellet (P) and soluble in the lysate (S) was determined by immunoblotting with polyclonal antibodies to the myc- and HA-epitope tags. Molecular weight size standards (kilodaltons) are indicated on the left. (C) Coimmunoprecipitation of the HC deletion mutants and LC. Lysates were immunoprecipitated for HC using a monoclonal anti–myc antibody (left) or for LC using a monoclonal anti–HA antibody (right). Immunoprecipitates were immunoblotted to detect the expressed proteins using polyclonal antibodies to the epitope tags.
Coimmunoprecipitation experiments tested directly which region of HC is required to interact with LC. When the lysates were subjected to immunoprecipitation for HC using myc antibodies, ∼50% of LC coprecipitated with H810, which lacks the entire HC tail, and ∼90% coprecipitated with H891, which lacks only the COOH-terminal 64 amino acids (Fig. 6 C, left). This suggests that the NH2-terminal, but not the COOH-terminal half of the HC tail domain is involved in LC binding. When LC was expressed with HC lacking both the tail domain and the COOH-terminal portion of the stalk domain (H682), the small amounts of LC in the soluble fraction could not be precipitated (Fig. 6 C, left). This suggests that the interaction of HC with LC also requires the COOH-terminal half of the HC stalk domain. Immunoprecipitation of LC from the same lysates using HA antibodies gave similar results. Thus, ∼50% of H810 and ∼90% of H891 were coprecipitated, whereas H682 was not precipitated (Fig. 6 C, right). Taken together with the effect of the HC constructs on the solubility of LC, these results suggest that the COOH-terminal portion of the HC stalk domain is required for the interaction with LC but that the NH2 terminus of the HC tail domain is also necessary for an optimal association.
To determine whether the same regions of HC that are required for interaction with LC are also involved in the inhibition of MT binding, the HC truncations were expressed alone or together with LC in COS cells. Lysates were subjected to MT cosedimentation assays. When expressed alone, essentially all of the HC truncations sedimented with MTs in the presence of AMP-PNP (Fig. 7, lanes 3, 5, and 7 vs. 11, 13, and 15). When coexpressed with LC, ∼90% of H682 and H810, and at least 50%, but usually much more (experimental range 50–95%), of H891 sedimented with MTs in the presence of AMP-PNP (Fig. 7, lanes 4, 6, and 8 vs. 12, 14, and 16; Fig. 8 C, lanes 11 and 12 vs. 15 and 16). In no case did the expressed HC deletions bind to MTs in the presence of ATP (data not shown). Taken together, the data indicate that although H891 is fully capable of interacting with LC (Fig. 6), this interaction is insufficient to inhibit its MT binding. We therefore conclude that the inhibition of MT binding requires not only an interaction of HC with LC, but also the COOH-terminal 64 amino acids of HC. Similar results were obtained when MT binding was assessed by immunofluorescence microscopy (data not shown).
Figure 7.

MT cosedimentation of the HC deletion mutants expressed alone or together with LC. Taxol-stabilized MTs and AMP-PNP were added to lysates from COS cells expressing HC (H) or the deletion mutants (H682, H810, and H891) alone or together with LC (H+L, H682+L, H810+L, and H891+L). MTs and bound proteins were sedimented through a sucrose cushion. The MT pellets (P) and supernatants (S) were immunoblotted to detect the expressed proteins using polyclonal antibodies to the myc- and HA-tags.
Figure 8.

MT cosedimentation of expressed recombinant HC+LC and endogenous kinesin is pH dependent. (A) Taxol-stabilized MTs and AMP-PNP were added to lysates from COS cells expressing HC (H) or HC and LC (H+L). Lysates were prepared in LB+Triton X-100 buffered with MES (pH 6.4), Pipes (pH 6.8), or Hepes (pH 7.2 and 7.6). MTs and bound proteins were sedimented through a sucrose cushion and the MT pellets and supernatants were immunoblotted with antibodies against myc- and HA-epitope tags to detect the expressed recombinant proteins. Only the immunoblots of the MT pellets are shown. (B) The pH dependence of MT binding of HC alone (circles) or HC and LC (triangles) was determined between pH 6.8 and 7.2 in LB + Triton X-100 buffered with Pipes (solid line) or Hepes (dashed line). Each data point represents the mean ± SD of at least five separate experiments. (C) Taxol-stabilized MTs and AMP-PNP were added to lysates, prepared at pH 6.8 or 7.2 as in A, from COS cells coexpressing the indicated recombinant proteins. MTs and bound proteins were sedimented through a sucrose cushion. The MT pellets (P) and supernatants (S) were immunoblotted to detect the expressed proteins using polyclonal antibodies to the myc- and HA-tags. (D) Sedimentation analysis of coexpressed HC and LC. COS cell lysates made at pH 6.8 (circles) and 7.2 (squares) were subjected to sedimentation on linear 9–15% sucrose gradients, both at physiological (closed symbols) and high salt (0.5 M NaCl; open symbols) concentrations. The expressed recombinant proteins were detected in the fractions by immunoblotting with polyclonal antibodies to the myc- and HA-tags. The mobility on the gradient of proteins with known S values is indicated. (E) Taxol-stabilized MTs and AMP-PNP were added to lysates, prepared at pH 6.8 or 7.2 as in A, from untransfected COS cells. MTs and bound proteins were sedimented through a sucrose cushion. The MT pellets (P) and supernatants (S) were immunoblotted to detect endogenous kinesin using a polyclonal antibody to kinesin HC.
Inhibition of MT Binding Is Dependent on pH
Our observation that kinesin in lysates is prevented from binding to MTs is consistent with the existence of a large pool of kinesin not associated with MTs in vivo, but is in apparent contradiction with the rationale for kinesin purification, which exploits its affinity for MTs in the presence of AMP-PNP (Vale et al., 1985). We considered whether pH differences could explain this discrepancy at least in part since kinesin is commonly purified at pH 6.6–6.8 (Kuznetsov and Gelfand, 1986; Hackney, 1988; Wagner et al., 1989, 1991), whereas our lysates were analyzed at pH 7.2–7.4.
COS cells expressing HC alone or together with LC were lysed in buffers ranging from pH 6.4 to 7.6. After removal of insoluble material, the lysates were subjected to MT cosedimentation assays. When expressed alone, HC bound to MTs in the presence of AMP-PNP at every pH, although less well at pH > 7.4 (Fig. 8, A and B). In contrast, when coexpressed with LC, HC bound well to MTs in the presence of AMP-PNP at pH 6.8 or lower, but, under the conditions of these assays, did not bind significantly at physiological pH. HC and LC remained associated with each other at all pH values. The effect of pH is not due to differences in salt concentration (data not shown). We conclude that kinesin is inhibited from MT binding at physiological pH, but not at slightly acidic pH.
The NH2-terminal region of LC, including the heptad repeat domain, behaved almost identically to the full-length LC (Fig. 8 C). At pH 6.8, ∼50% of HC and 20% of the truncated LC (L176) bound to MTs (Fig. 8 C, lane 2 vs. 6), the difference in the behavior of the chains being due to excess of L176. At pH 7.2, very little of either chain was associated with MTs (Fig. 8 C, lane 10 vs. 14). Thus, the NH2-terminal region, including the heptad repeats, is the only domain of LC required for both inhibition of MT binding at physiological pH and for release of inhibition at lowered pH. As expected, HC lacking the COOH-terminal 64 amino acids bound to MTs at both pH values, regardless of whether associated with intact LC or the truncated version (Fig. 8 C, lanes 3 and 4 vs. 7 and 8, and 11 and 12 vs. 15 and 16).
We next tested whether the pH-dependent release of inhibition of MT binding is accompanied by a conformational change in kinesin from a folded to an extended state (Hackney et al., 1992). Lysates made at pH 7.2 or 6.8 from cells coexpressing HC and LC were subjected to sucrose gradient centrifugation. At both pH values, the proteins sedimented at ∼9S (Fig. 8 D), corresponding to the folded conformation of kinesin in which the tail and motor domains are in proximity to each other (Hackney et al., 1992). Sedimentation in a high salt buffer (0.5 M NaCl) shifted the proteins to an extended 6S conformation at both pH 7.2 and 6.8 (Fig. 8 D), in agreement with the data obtained for purified kinesin (Hackney et al., 1992). These results indicate that under physiological conditions soluble kinesin is in a folded conformation and that activation at low pH is not due to gross conformational changes.
Endogenous Kinesin also Shows pH-dependent Inhibition of MT Binding
We were concerned that the results on the inhibition of MT binding of kinesin could be an artifact caused by the high level of overexpression of the protein in transiently transfected cells. To test whether endogenous kinesin in COS cells behaves identically, we raised a peptide-specific antibody against a region in the motor domain of rat HC. After affinity purification, the antibody recognized a major band of the expected size in immunoblots of lysates of nontransfected cells (data not shown). COS cell lysates were then subjected to the MT cosedimentation assay and the amounts of endogenous kinesin in the supernatant and MT pellet were determined by immunoblotting (Fig. 8 E). In the presence of AMP-PNP, kinesin bound to MTs at pH 6.8 (Fig. 8 E, lane 2 vs. 4), but not at pH 7.2 (lane 6 vs. 8). In the presence of ATP, no binding was observed at either pH (Fig. 8 E, lane 1 vs. 3 and 5 vs. 7). We conclude that endogenous kinesin also shows pH-sensitive inhibition of MT binding.
Discussion
We demonstrate here that kinesin, either present endogenously in COS cells or assembled by coexpression of HC and LC, is inhibited from binding MTs at physiological pH. These data thus provide an explanation for the large pool of soluble kinesin that is not associated with MTs in cells. Kinesin may be kept inactive when not transporting cargo to prevent its futile movement along MTs. This would not only be an economic use of the motor, but would also prevent the accumulation of kinesin at plus ends of MTs in the cell periphery.
Our results demonstrate that LC is required for inhibiting HC from binding MTs. In cell lysates, HC alone was able to bind in vitro to MTs in the presence of AMP-PNP, but not ATP, a characteristic property of the active motor. When expressed alone in intact cells, HC also appeared to interact with MTs and was apparently motile, as indicated by its accumulation at the periphery of the cells.
The NH2-terminal domain of LC, including the heptad repeats, was found to interact with HC in vivo, in agreement with previous in vitro data (Gauger and Goldstein, 1993). We believe that the heptad repeats themselves are responsible for this interaction, but a role for the 48 amino acids preceding this domain cannot be excluded. Our data also do not rule out an independent interaction of the COOH-terminal TPR domains with HC because this region alone proved to be insoluble when expressed in cells. However, the experiments of Gauger and Goldstein (1993) with in vitro translated, and apparently soluble, TPR domains argue against this possibility and suggest that the heptad repeat domain is sufficient for the interaction with HC. Our experiments additionally show that all LC fragments that interact with HC, minimally the NH2-terminal domain, also inhibit MT binding. Thus, much of the LC sequence, including the TPRs, is dispensable for both interaction with HC and for inhibition of MT binding.
The NH2-terminal domain of LC, including the heptad repeats, likely interacts both with a region of the HC stalk domain that is highly conserved across species and also with the NH2-terminal half of the HC tail domain. The COOH terminus of the HC stalk domain plus the tail domain is also sufficient to interact with LC in vitro (Gauger and Goldstein, 1993). Thus, the interaction between HC and LC appears to be mediated by relatively short regions (100–150 amino acids each) that contain sequences predicted to form α-helical coiled coils. Such an interaction would be extremely stable, consistent with the idea that, in vivo, the two chains always function together.
The interaction between LC and HC, although necessary, is not sufficient to inhibit the motor domain of HC from binding to MTs. Rather, the COOH-terminal 64 amino acids of HC are also required. These results suggest that LC stabilizes a conformation in which kinesin is folded back on itself, allowing the tail domain of HC to interact with the MT-binding motor domain. Indeed, the sedimentation coefficient of 9S determined for kinesin in cell lysates corresponds to the folded conformation of purified kinesin as determined by sedimentation and electron microscopy analysis (Hirokawa et al., 1989; Hisanaga et al., 1989; Hackney et al., 1992). Bending of the stalk domain of HC is likely due to helix-disrupting residues in the hinge region (Vale and Fletterick, 1997). Inhibition of motor activity by the HC tail is in agreement with previous reports that the motor domain alone has higher MT-stimulated ATPase activity than intact kinesin (Kuznetsov et al., 1989; Gilbert and Johnson, 1993; Stewart et al., 1993; Huang and Hackney, 1994; discussed in Hackney, 1996). The proposed role of LC is also consistent with previous reports that HC alone has a higher MT-stimulated ATPase activity than HC and LC together (Hackney et al., 1991). The agreement between our results and those with purified kinesin suggest that inhibition of MT binding is an intrinsic property of the motor protein. However, we cannot rule out the involvement of additional cytosolic proteins that were present in our lysates.
Kinesin's inability to bind to MTs appears at first glance inconsistent with the rationale for its purification; i.e., nucleotide-dependent MT cosedimentation. The pH dependence of MT binding provides at least a partial explanation. Purification of kinesin is performed at the nonphysiological pH of 6.6–6.8 (Kuznetsov and Gelfand, 1986; Wagner et al., 1989, 1991; Hackney, 1991). At this pH, kinesin-MT binding is unregulated and efficient purification is predicted. With much lower affinity, kinesin also binds MTs at the physiologic pH 7.2–7.4 (Fig. 2 B), suggesting that kinesin could be purified at this pH if the concentrations of MTs and kinesin were sufficiently high. The reported purification of squid kinesin at neutral pH is consistent with this idea (Vale et al., 1985), although species differences cannot be excluded.
Our experiments with endogenous kinesin indicate that the inhibition of MT binding of kinesin at neutral pH is not an in vitro artifact produced by the high level of expression in transiently transfected cells. In addition, they confirm for COS cells that a large population of kinesin is soluble in the cytosol, nonassociated with MTs or membranes that were sedimented during the preparation of the cell lysates. The overexpression of kinesin in cells transiently transfected with both HC and LC did not have any obvious effect on the morphology of COS cells or on the distribution of markers for various organelles (data not shown). This is consistent with our view that kinesin is normally present in excess of demand, but is maintained in an inactive state.
With the exception of the TPR motifs of LC, all notable domains in both HC and LC can now be assigned a function in either motor activity or in the interaction between the two chains (Fig. 9 A). It therefore seems likely that the TPRs are involved in the one crucial motor function that thus far lacks a corresponding domain; namely, cargo binding. We consider it less likely that the COOH-terminal 10 amino acids of LC, which differ among the various splice forms, are responsible for cargo selection. The proposed role for the TPRs would be consistent with their high conservation across species (Gindhart and Goldstein, 1996), and the fact that TPRs are involved in protein–protein interactions (Lamb et al., 1995; Das et al., 1998). In support of this proposal, an antibody to LC inhibits the binding of purified chick brain kinesin to membranes in vitro (Yu et al., 1992) and an antibody to the third TPR of rat LC releases kinesin from a microsomal vesicle fraction (Stenoien and Brady, 1997). However, other studies have implicated the stalk and tail domains of HC in membrane binding. Purified sea urchin kinesin HC and a bacterially expressed HC stalk-tail domain, but not the stalk domain alone, were able to bind to membranes in vitro (Skoufias et al., 1994). Clearly, a definitive identification of the cargo binding domain has yet to be achieved and it is possible that both the TPR domain of LC and the tail domain of HC are involved in the interaction.
Figure 9.
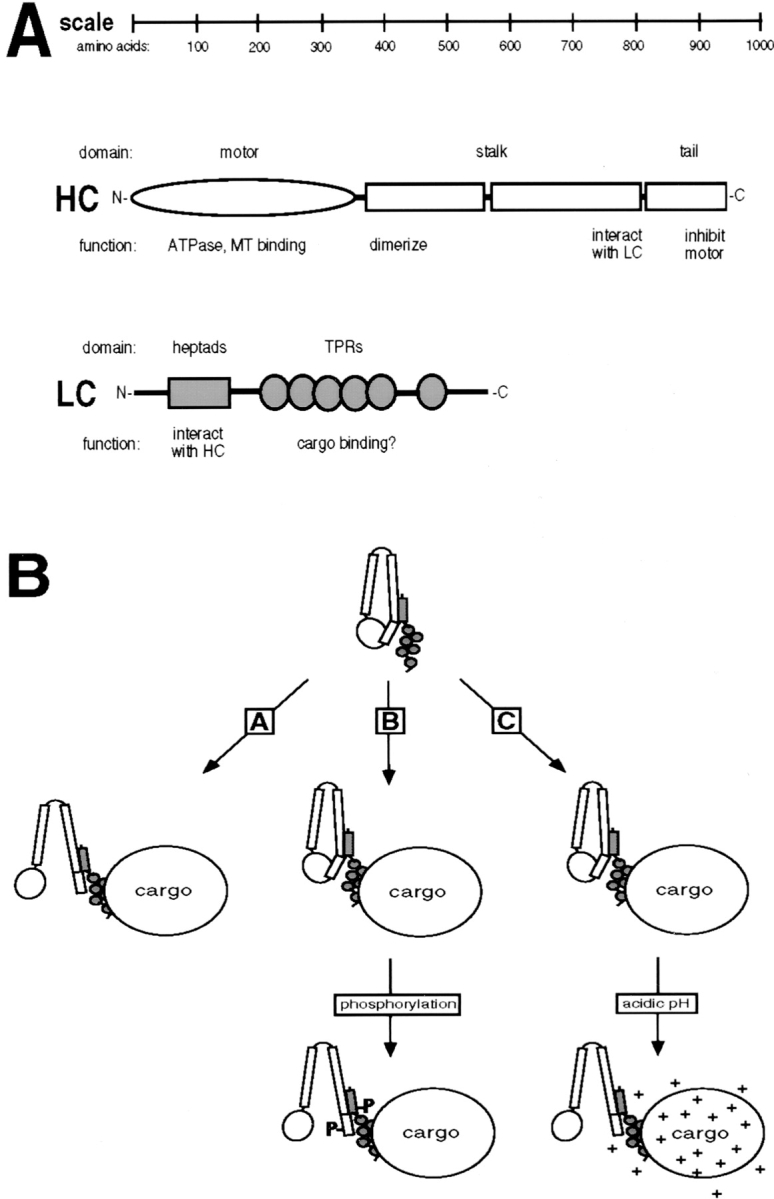
Functional domains of kinesin and models for the activation of kinesin in vivo. (A) Representation of the domains of kinesin HC and LC, drawn to scale, and the functions assigned to them. (B) Possible mechanisms to explain the activation of kinesin for MT binding in vivo. Only one HC and one LC of the kinesin tetramer are shown for simplicity. Soluble kinesin in the cell is in an inactive folded conformation such that the HC tail domain inhibits MT binding of the motor domain. Activation of kinesin, represented here by a hypothetical opening of the folded molecule, may be triggered: (A) by interaction of the motor with its cargo, (B) before or after (as drawn) cargo binding by posttranslational modification (e.g., phosphorylation) of either the HC or LC, or (C) after cargo binding by a localized shift of pH around the vesicle from neutral to slightly acidic.
How could kinesin be activated for cargo transport? Although previous experiments suggested that a transition from the folded into an extended conformation would correspond to an activation of the motor (Hackney et al., 1992), we find that activation by lowering pH is not accompanied by a transition to the extended conformation, suggesting that smaller conformational changes are sufficient. In vivo, activation need not necessarily be caused by a change in pH; the pH effect may cause a conformational change achieved in cells by other means. One possibility is that binding of kinesin to its cargo is the trigger for activation (Fig. 9 B, Scheme A). Consistent with this, a kinesin motor attached to a bead or a coverglass is able to walk processively along a MT (Howard et al., 1989; Block et al., 1990). Activation by nonspecific adsorption to these surfaces may mimic the effect of cargo binding in cells. Another possible means of activation may involve phosphorylation of kinesin. A kinase or phosphatase associated with the cargo may be responsible for activation of the motor (Fig. 9 B, Scheme B). Since our experiments were performed in whole cell lysates, it is in fact possible that the pH effect is due to an indirect effect on the phosphorylation status of kinesin. Both HC and LC can indeed be phosphorylated in vitro (Matthies et al., 1993; McIlvain et al., 1994; Lindesmith et al., 1997) and in vivo (Hollenbeck, 1993; Lee and Hollenbeck, 1995). However, while phosphorylation of LC modulates the MT gliding and ATPase activities of purified kinesin (Matthies et al., 1993; McIlvain et al., 1994; Lindesmith et al., 1997), the effect of phosphorylation on the membrane association of kinesin is still controversial (Sato-Yoshitake et al., 1992; Lee and Hollenbeck, 1995).
Yet another possibility is that pH is in fact the physiological trigger for activating kinesin. It is indeed striking that kinesin is activated for MT binding within a narrow pH range around the physiological pH. In addition, the NH2-terminal domain of LC, including the heptad repeat region, is involved in both inhibition of MT binding at physiological pH and release of inhibition at lowered pH, suggesting that acidification releases the inhibitory effect of LC and that this mode of activation is therefore physiologically relevant. Kinesin may initially be bound to cargo in an inactive form and be activated by a local shift of pH from neutral to slightly acidic (Fig. 9 B, Scheme C). Activation of plus-end–directed movement at acidic pH is supported by experiments in which lysosomes and late endosomes move from the perinuclear region out to the periphery upon exposure of the cells to acidic buffers (Heuser, 1989; Parton et al., 1991). In a physiological situation, global changes in cytosolic pH are unlikely, but a vesicle with low lumenal pH might, by proton leakage, create an acidic environment around itself, which could in turn activate vesicle-bound kinesin for MT binding. In support of such a hypothesis, inhibition of the vacuolar-type H+-ATPase by concanamycin blocks the plus-end–directed transport of Golgi enzymes into the peripheral ER (Cole et al., 1998). Although the pH gradient hypothesis would predict that acidic vesicles move towards the plus end of MTs, in cells the situation may be more complicated due to the presence of minus end motors and varying degrees of acidification and/or proton leakage among different kinds of vesicles. Further experiments are obviously required to test whether the pH gradient hypothesis or other mechanisms are responsible for activation of kinesin in vivo.
The proposed mechanism by which kinesin is kept in an inactive ground state may be valid for other motors as well. Many myosins are inhibited for actin interaction in a light chain-dependent manner by sequences downstream of their motor domains (Cross et al., 1988; Mooseker and Cheney, 1995). In some cases, release of inhibition can occur by phosphorylation (Tan et al., 1992). In others, changes in the Ca2+ concentration affect the light chain calmodulin and lead to motor activation (Wolenski, 1995). Particularly similar to the regulation of kinesin by pH is the case of Myr3, a myosin I, in which Ca2+ acts as a negative regulator by affecting allosteric interactions between the tail and motor domains (Stöffler and Bähler, 1998). Thus, local changes in ion concentrations may be a general mechanism controlling the activity of molecular motors in vivo.
Acknowledgments
We thank L. Sutherland for cloning of cDNAs, Dr. R. Vale for providing the construct HK420, Dr. F. McKeon for providing vectors, and Drs. V. Mursean, C. Shamu, A. Hudson, and A. Desai for critical reading of the manuscript.
Abbreviations used in this paper
- HA
hemagglutinin
- HC
heavy chain
- LB
Lysis buffer
- LC
light chain
- MT
microtubule
- ORF
open reading frame
- TPR
tetratricopeptide repeat
Footnotes
K.J. Verhey was supported by a National Institutes of Health (NIH) postdoctoral fellowship. T.A. Rapoport is a Howard Hughes Medical Institute investigator. This work was supported in part by NIH grants to B.J. Schnapp.
Address all correspondence to Bruce J. Schnapp, Department of Cell Biology, Harvard Medical School, 240 Longwood Ave., Boston, MA 02115. Tel.: (617) 432-3818. Fax: (617) 432-1144. E-mail: schnapp@warren.med.harvard.edu
References
- Bi GQ, Morris R, Liao G, Alderton JM, Scholey JM, Steinhardt RA. Kinesin- and myosin-driven steps of vesicle recruitment for Ca2+-regulated exocytosis. J Cell Biol. 1997;138:999–1008. doi: 10.1083/jcb.138.5.999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Block SM. Kinesin: what gives? . Cell. 1998a;93:5–8. doi: 10.1016/s0092-8674(00)81138-1. [DOI] [PubMed] [Google Scholar]
- Block SM. Leading the procession: new insights into kinesin motors. J Cell Biol. 1998b;140:1281–1284. doi: 10.1083/jcb.140.6.1281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Block SM, Goldstein LS, Schnapp BJ. Bead movement by single kinesin molecules studied with optical tweezers. Nature. 1990;348:348–352. doi: 10.1038/348348a0. [DOI] [PubMed] [Google Scholar]
- Bloom GS, Endow SA. Motor proteins 1: kinesins. Protein Profile. 1994;1:1059–1116. [PubMed] [Google Scholar]
- Butner K, Kirschner M. Tau protein binds to microtubules through a flexible array of distributed weak sites. J Cell Biol. 1991;115:717–730. doi: 10.1083/jcb.115.3.717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cole NB, Ellenberg J, Song J, DiEuliis D, Lippincott-Schwartz J. Retrograde transport of Golgi-localized proteins to the ER. J Cell Biol. 1998;140:1–15. doi: 10.1083/jcb.140.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cross RA, Jackson AP, Citi S, Kendrick-Jones J, Bagshaw CR. Active site trapping of nucleotide by smooth and non-muscle myosins. J Mol Biol. 1988;203:273–281. doi: 10.1016/0022-2836(88)90100-3. [DOI] [PubMed] [Google Scholar]
- Cyr JL, Pfister KK, Bloom GS, Slaughter CA, Brady ST. Molecular genetics of kinesin light chains: generation of isoforms by alternative splicing. Proc Natl Acad Sci USA. 1991;88:10114–10118. doi: 10.1073/pnas.88.22.10114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Das AK, Cohen PTW, Barford D. The structure of the tetratricopeptide repeats of protein phosphatase 5: implications for TPR-mediated protein–protein interactions. EMBO (Eur Mol Biol Organ) J. 1998;17:1192–1199. doi: 10.1093/emboj/17.5.1192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gauger AK, Goldstein LS. The Drosophilakinesin light chain. Primary structure and interaction with kinesin heavy chain. J Biol Chem. 1993;268:13657–13666. [PubMed] [Google Scholar]
- Gilbert SP, Johnson KA. Expression, purification, and characterization of the Drosophila kinesin motor domain produced in Escherichia coli. . Biochemistry. 1993;32:4677–4684. doi: 10.1021/bi00068a028. [DOI] [PubMed] [Google Scholar]
- Gindhart JG, Jr, Goldstein LS. Tetratricopeptide repeats are present in the kinesin light chain. Trends Biochem Sci. 1996;21:52–53. [PubMed] [Google Scholar]
- Goodson HV, Valetti C, Kreis TE. Motors and membrane traffic. Curr Opin Cell Biol. 1997;9:18–28. doi: 10.1016/s0955-0674(97)80147-0. [DOI] [PubMed] [Google Scholar]
- Hackney DD. Kinesin ATPase: rate-limiting ADP release. Proc Natl Acad Sci USA. 1988;85:6314–6318. doi: 10.1073/pnas.85.17.6314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hackney DD. Isolation of kinesin using initial batch ion exchange. Methods Enzymol. 1991;196:175–181. doi: 10.1016/0076-6879(91)96017-l. [DOI] [PubMed] [Google Scholar]
- Hackney DD. The rate-limiting step in microtubule-stimulated ATP hydrolysis by dimeric kinesin head domains occurs while bound to the microtubule. J Biol Chem. 1994;269:16508–16511. [PubMed] [Google Scholar]
- Hackney DD. Highly processive microtubule-stimulated ATP hydrolysis by dimeric kinesin head domains. Nature. 1995;377:448–450. doi: 10.1038/377448a0. [DOI] [PubMed] [Google Scholar]
- Hackney DD. The kinetic cycles of myosin, kinesin, and dynein. Annu Rev Physiol. 1996;58:731–750. doi: 10.1146/annurev.ph.58.030196.003503. [DOI] [PubMed] [Google Scholar]
- Hackney DD, Levitt JD, Suhan J. Kinesin undergoes a 9 S to 6 S conformational transition. J Biol Chem. 1992;267:8696–8701. [PubMed] [Google Scholar]
- Hackney DD, Levitt JD, Wagner DD. Characterization of alpha 2 beta 2 and alpha 2 forms of kinesin. Biochem Biophys Res Commun. 1991;174:810–815. doi: 10.1016/0006-291x(91)91490-4. [DOI] [PubMed] [Google Scholar]
- Heuser J. Changes in lysosome shape and distribution correlated with changes in cytoplasmic pH. J Cell Biol. 1989;108:855–864. doi: 10.1083/jcb.108.3.855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirokawa N. The molecular mechanism of organelle transport along microtubules: the identification and characterization of KIFs (kinesin superfamily proteins) Cell Struct Funct. 1996;21:357–367. doi: 10.1247/csf.21.357. [DOI] [PubMed] [Google Scholar]
- Hirokawa N. Kinesin and dynein superfamily proteins and the mechanism of organelle transport. Science. 1998;279:519–526. doi: 10.1126/science.279.5350.519. [DOI] [PubMed] [Google Scholar]
- Hirokawa N, Pfister KK, Yorifuji H, Wagner MC, Brady ST, Bloom GS. Submolecular domains of bovine brain kinesin identified by electron microscopy and monoclonal antibody decoration. Cell. 1989;56:867–878. doi: 10.1016/0092-8674(89)90691-0. [DOI] [PubMed] [Google Scholar]
- Hisanaga S, Murofushi H, Okuhara K, Sato R, Masuda Y, Sakai H, Hirokawa N. The molecular structure of adrenal medulla kinesin. Cell Motil Cytoskelet. 1989;12:264–272. doi: 10.1002/cm.970120407. [DOI] [PubMed] [Google Scholar]
- Hollenbeck PJ. Phosphorylation of neuronal kinesin heavy and light chains in vivo. J Neurochem. 1993;60:2265–2275. doi: 10.1111/j.1471-4159.1993.tb03513.x. [DOI] [PubMed] [Google Scholar]
- Howard J, Hudspeth AJ, Vale RD. Movement of microtubules by single kinesin molecules. Nature. 1989;342:154–158. doi: 10.1038/342154a0. [DOI] [PubMed] [Google Scholar]
- Huang TG, Hackney DD. Drosophila kinesin minimal motor domain expressed in Escherichia coli. Purification and kinetic characterization. J Biol Chem. 1994;269:16493–16501. [PubMed] [Google Scholar]
- Jiang W, Stock MF, Li X, Hackney DD. Influence of the kinesin neck domain on dimerization and ATPase kinetics. J Biol Chem. 1997;272:7626–7632. doi: 10.1074/jbc.272.12.7626. [DOI] [PubMed] [Google Scholar]
- Khodjakov A, Lizunova EM, Minin AA, Koonce MP, Gyoeva FK. A specific light chain of kinesin associates with mitochondria in cultured cells. Mol Biol Cell. 1998;9:333–343. doi: 10.1091/mbc.9.2.333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuznetsov SA, Gelfand VI. Bovine brain kinesin is a microtubule-activated ATPase. Proc Natl Acad Sci USA. 1986;83:8530–8534. doi: 10.1073/pnas.83.22.8530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuznetsov SA, Vaisberg YA, Rothwell SW, Murphy DB, Gelfand VI. Isolation of a 45-kDa fragment from the kinesin heavy chain with enhanced ATPase and microtubule-binding activities. J Biol Chem. 1989;264:589–595. [PubMed] [Google Scholar]
- Lamb JR, Tugendreich S, Hieter P. Tetratricopeptide repeat interactions: to TPR or not to TPR? . Trends Biochem Sci. 1995;20:257–259. doi: 10.1016/s0968-0004(00)89037-4. [DOI] [PubMed] [Google Scholar]
- Lee KD, Hollenbeck PJ. Phosphorylation of kinesin in vivo correlates with organelle association and neurite outgrowth. J Biol Chem. 1995;270:5600–5605. doi: 10.1074/jbc.270.10.5600. [DOI] [PubMed] [Google Scholar]
- Liao G, Gunderson GG. Kinesin is a candidate for cross-bridging microtubules and intermediate filaments. J Biol Chem. 1998;273:9797–9803. doi: 10.1074/jbc.273.16.9797. [DOI] [PubMed] [Google Scholar]
- Lindesmith L, McIlvain JM, Jr, Argon Y, Sheetz MP. Phosphotransferases associated with the regulation of kinesin motor activity. J Biol Chem. 1997;272:22929–22933. doi: 10.1074/jbc.272.36.22929. [DOI] [PubMed] [Google Scholar]
- Matthies HJ, Miller RJ, Palfrey HC. Calmodulin binding to and cAMP-dependent phosphorylation of kinesin light chains modulate kinesin ATPase activity. J Biol Chem. 1993;268:11176–11187. [PubMed] [Google Scholar]
- McIlvain JM, Jr, Burkhardt JK, Hamm-Alvarez S, Argon Y, Sheetz MP. Regulation of kinesin activity by phosphorylation of kinesin-associated proteins. J Biol Chem. 1994;269:19176–19182. [PubMed] [Google Scholar]
- Moore JD, Endow SA. Kinesin proteins: a phylum of motors for microtubule-based motility. Bioessays. 1996;18:207–219. doi: 10.1002/bies.950180308. [DOI] [PubMed] [Google Scholar]
- Mooseker MS, Cheney RE. Unconventional myosins. Annu Rev Cell Dev Biol. 1995;11:633–675. doi: 10.1146/annurev.cb.11.110195.003221. [DOI] [PubMed] [Google Scholar]
- Nakata T, Hirokawa N. Point mutation of adenosine triphosphate-binding motif generated rigor kinesin that selectively blocks anterograde lysosome membrane transport. J Cell Biol. 1995;131:1039–1053. doi: 10.1083/jcb.131.4.1039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Navone F, Niclas J, Hom-Booher N, Sparks L, Bernstein HD, McCaffrey G, Vale RD. Cloning and expression of a human kinesin heavy chain gene: interaction of the COOH-terminal domain with cytoplasmic microtubules in transfected CV-1 cells. J Cell Biol. 1992;117:1263–1275. doi: 10.1083/jcb.117.6.1263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parton RG, Dotti CG, Bacallao R, Kurtz I, Simons K, Prydz K. pH-induced microtubule-dependent redistribution of late endosomes in neuronal and epithelial cells. J Cell Biol. 1991;113:261–274. doi: 10.1083/jcb.113.2.261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenfeld SS, Rener B, Correia JJ, Mayo MS, Cheung HC. Equilibrium studies of kinesin-nucleotide intermediates. J Biol Chem. 1996;271:9473–9482. doi: 10.1074/jbc.271.16.9473. [DOI] [PubMed] [Google Scholar]
- Sato-Yoshitake R, Yorifuji H, Inagaki M, Hirokawa N. The phosphorylation of kinesin regulates its binding to synaptic vesicles. J Biol Chem. 1992;267:23930–23936. [PubMed] [Google Scholar]
- Sawin KE, Mitchison TJ, Wordeman LG. Evidence for kinesin-related proteins in the mitotic apparatus using peptide antibodies. J Cell Sci. 1992;101:303–313. doi: 10.1242/jcs.101.2.303. [DOI] [PubMed] [Google Scholar]
- Skoufias DA, Cole DG, Wedaman KP, Scholey JM. The carboxyl-terminal domain of kinesin heavy chain is important for membrane binding. J Biol Chem. 1994;269:1477–1485. [PubMed] [Google Scholar]
- Stenoien DL, Brady ST. Immunochemical analysis of kinesin light chain function. Mol Biol Cell. 1997;8:675–689. doi: 10.1091/mbc.8.4.675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stewart RJ, Thaler JP, Goldstein LS. Direction of microtubule movement is an intrinsic property of the motor domains of kinesin heavy chain and Drosophilancd protein. Proc Natl Acad Sci USA. 1993;90:5209–5213. doi: 10.1073/pnas.90.11.5209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stöffler H-E, Bähler M. The ATPase activity of Myr3, a rat myosin I, is allosterically inhibited by its own tail domain and by Ca2+binding to its light chain calmodulin. J Biol Chem. 1998;273:14605–14611. doi: 10.1074/jbc.273.23.14605. [DOI] [PubMed] [Google Scholar]
- Tan JL, Shoshana R, Spudich JA. Control of nonmuscle myosins by phosphorylation. Annu Rev Biochem. 1992;61:721–759. doi: 10.1146/annurev.bi.61.070192.003445. [DOI] [PubMed] [Google Scholar]
- Tanaka Y, Kanai Y, Okada Y, Nonaka S, Takeda S, Harada A, Hirokawa N. Targeted disruption of mouse conventional kinesin heavy chain, Kif5b, results in abnormal perinuclear clustering of mitochondria. Cell. 1998;93:1147–1158. doi: 10.1016/s0092-8674(00)81459-2. [DOI] [PubMed] [Google Scholar]
- Taylor SS, McKeon F. Kinetochore localization of murine Bub1 is required for normal mitotic timing and checkpoint response to spindle damage. Cell. 1997;89:727–735. doi: 10.1016/s0092-8674(00)80255-x. [DOI] [PubMed] [Google Scholar]
- Vale RD, Fletterick RJ. The design plan of kinesin motors. Annu Rev Cell Dev Biol. 1997;13:745–777. doi: 10.1146/annurev.cellbio.13.1.745. [DOI] [PubMed] [Google Scholar]
- Vale RD, Reese TS, Sheetz MP. Identification of a novel force-generating protein, kinesin, involved in microtubule-based motility. Cell. 1985;42:39–50. doi: 10.1016/s0092-8674(85)80099-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vallee RB. Reversible assembly purification of microtubules without assembly-promoting agents and further purification of tubulin, microtubule-associated proteins, and MAP fragments. Methods Enzymol. 1986a;134:89–104. doi: 10.1016/0076-6879(86)34078-3. [DOI] [PubMed] [Google Scholar]
- Vallee RB. Purification of brain microtubules and microtubule-associated protein 1 using taxol. Methods Enzymol. 1986b;134:104–115. doi: 10.1016/0076-6879(86)34079-5. [DOI] [PubMed] [Google Scholar]
- Wagner MC, Pfister KK, Bloom GS, Brady ST. Copurification of kinesin polypeptides with microtubule-stimulated Mg-ATPase activity and kinestic analysis of enzymatic properties. Cell Motil Cytoskelet. 1989;12:195–215. doi: 10.1002/cm.970120403. [DOI] [PubMed] [Google Scholar]
- Wagner MC, Pfister KK, Brady ST, Bloom GS. Purification of kinesin from bovine brain and assay of microtubule-stimulated ATPase activity. Methods Enzymol. 1991;196:157–175. doi: 10.1016/0076-6879(91)96016-k. [DOI] [PubMed] [Google Scholar]
- Wedaman KP, Knight AE, Kendrick-Jones J, Scholey JM. Sequences of sea urchin kinesin light chain isoforms. J Mol Biol. 1993;231:155–158. doi: 10.1006/jmbi.1993.1267. [DOI] [PubMed] [Google Scholar]
- Wolenski JS. Regulation of calmodulin-binding myosins. Trends Cell Biol. 1995;5:310–316. doi: 10.1016/s0962-8924(00)89053-4. [DOI] [PubMed] [Google Scholar]
- Yu H, Toyoshima I, Steuer ER, Sheetz MP. Kinesin and cytoplasmic dynein binding to brain microsomes. J Biol Chem. 1992;267:20457–20464. [PubMed] [Google Scholar]