

Abstract
Shiga toxin and other toxins of this family can escape the endocytic pathway and reach the Golgi apparatus. To synchronize endosome to Golgi transport, Shiga toxin B-fragment was internalized into HeLa cells at low temperatures. Under these conditions, the protein partitioned away from markers destined for the late endocytic pathway and colocalized extensively with cointernalized transferrin. Upon subsequent incubation at 37°C, ultrastructural studies on cryosections failed to detect B-fragment–specific label in multivesicular or multilamellar late endosomes, suggesting that the protein bypassed the late endocytic pathway on its way to the Golgi apparatus. This hypothesis was further supported by the rapid kinetics of B-fragment transport, as determined by quantitative confocal microscopy on living cells and by B-fragment sulfation analysis, and by the observation that actin- depolymerizing and pH-neutralizing drugs that modulate vesicular transport in the late endocytic pathway had no effect on B-fragment accumulation in the Golgi apparatus. B-fragment sorting at the level of early/recycling endosomes seemed to involve vesicular coats, since brefeldin A treatment led to B-fragment accumulation in transferrin receptor–containing membrane tubules, and since B-fragment colocalized with adaptor protein type 1 clathrin coat components on early/recycling endosomes. Thus, we hypothesize that Shiga toxin B-fragment is transported directly from early/recycling endosomes to the Golgi apparatus. This pathway may also be used by cellular proteins, as deduced from our finding that TGN38 colocalized with the B-fragment on its transport from the plasma membrane to the TGN.
Keywords: Shiga toxin, endosomes, Golgi, intracellular transport, clathrin
Bacterial and plant protein toxins use functions of higher eucaryotic cells for the transduction of their activity (O'Brien et al., 1992; Sandvig and van Deurs, 1996). For example, they rely on intracellular transport to reach their target molecules that often reside in the cytoplasm. Toxins such as diphtheria toxin are endocytosed and then cross membranes at the level of endosomes in a pH-dependent process. Other toxins such as Shiga toxin, verotoxins, Cholera toxin, and ricin do not depend on acidic pH in endosomes for intoxication of cells. Rather, these toxins are sensitive to treatments that affect the biosynthetic/secretory pathway suggesting that membrane transfer occurs at the level of the Golgi apparatus or the ER that these toxins have to reach by retrograde transport from the plasma membrane (Pelham et al., 1992). Especially toxins of the latter group are thus interesting models to study transport routes in animal cells that to date are still little explored.
Shiga toxin and the virtually identical VT-1 are produced by Shigella dysenteriae and by certain enterohemorrhagic strains of Escherichia coli, respectively (O'Brien et al., 1992; Sandvig and van Deurs, 1996; Johannes and Goud, 1998). These toxins have been identified as the etiologic agents of the hemolytic and uremic syndrome and of hemorrhagic colitis (O'Brien et al., 1992; Lingwood, 1996). They are composed of a catalytically active A-subunit and a receptor binding B-subunit. The A-subunit, a N-glycosidase that inhibits protein synthesis through the modification of 28S rRNA, depends upon a noncovalent interaction with the B-subunit for its binding to target cells and its intracellular transport. The B-subunit is a homopentamer of B-fragments that form a symmetrical ring-like structure in solution. The B-subunit binds with high affinity to the cellular toxin receptor, the glycolipid globotriaosylceramid (Lingwood, 1993). The toxin–receptor complex is then internalized, likely via clathrin-dependent mechanisms (Sandvig et al., 1989). In many cancer cell lines, internalized toxin or B-subunit is transported from endosomes via the TGN/Golgi apparatus to the ER (Sandvig et al., 1992, 1994; Johannes et al., 1997; Kim et al., 1998). The mechanisms of this retrograde transport remain elusive.
Notably, the precise site at which Shiga toxin leaves endosomes to target the TGN/Golgi apparatus is still unknown. The existence of two potential transport routes between endosomes and the TGN has been suggested because of the identification of proteins that are common to both compartments (for reviews see Gruenberg and Maxfield, 1995; Mellman, 1996; Mukherjee et al., 1997). A direct transport route from late endosomes (LE)1 to the TGN has been hypothesized for the cation-independent mannose 6-phosphate receptor (CI-MPR) and the MPR of 46 kD (MPR46), which both bind mannose 6-phosphate– tagged proteins at the level of the TGN and deliver these proteins to LE (von Figura, 1991; Munier-Lehmann et al., 1996). MPRs can also be found to a lesser extent in the plasma membrane and early endosomes (EE). Ligand dissociation occurs at low pH of LE, from where uncharged receptors are transported back to the TGN in a reaction regulated by the small GTP-binding protein Rab9 (Lombardi et al., 1993; Riederer et al., 1994). Based on the finding that small quantities of a marker protein for EE and recycling endosomes (RE), transferrin (Tf), can be detected in the TGN (Fishman and Fine, 1987; Stoorvogel et al., 1988), the existence of a direct transport route from EE/RE to the TGN has been hypothesized. Two other proteins that have been studied in some detail are TGN38 and the protease furin (Humphrey et al., 1993; Reaves et al., 1993; Chapman and Munro, 1994; Molloy et al., 1994; Ponnambalam et al., 1994). At steady-state, both are concentrated in the TGN. However, both proteins are also found in the plasma membrane and endosomes, and antibodies directed to these proteins are transported from the plasma membrane to the TGN. The itinerary of these proteins from endosomes to the TGN is still unknown.
In this study we have investigated the transport route taken by Shiga toxin B-fragment from EE to the Golgi apparatus. We found that the B-fragment accumulated with Tf in EE/RE when incubated with HeLa cells at 19.5°C. From this compartment, the B-fragment was rapidly transported to the TGN/Golgi apparatus, without apparent passage via LE. Our immunolocalization studies suggest that B-fragment sorting at the level of EE/RE involves adaptor protein type 1 (AP-1) clathrin coats. We thus hypothesize the existence of a direct transport route between EE/RE and the Golgi apparatus, which may also be used by cellular proteins, as judged from the finding that TGN38 colocalized with B-fragment during transport to the TGN.
Materials and Methods
Cells
HeLa cells were grown in DME containing 4.5 g/liter glucose (GIBCO BRL, Gaithersburg, MD) supplemented with 10% FCS (GIBCO BRL), 0.01% penicillin/streptomycin, 4 mM glutamine, and 5 mM pyruvate in a 5% CO2 incubator.
Reagents and Antibodies
Cy3, iodine, radioactive sulfate, and radiolabeled EGF (Amersham Life Science, Inc., Arlington Heights, IL), iodobeads, Tf-HRP, and FITC (Pierce Chemical Co., Rockford, IL), 5-([4,6-dichlorotriazin-2-yl]amino) fluorescein, ferro-Tf, CytoD, and Noc (Sigma, Saint-Quentin-Fallavier, France), Bafi (Fluka, Saint-Quentin-Fallavier, France), BFA (Epicentre Technologies Corp., Madison, WI), FITC-labeled Dex3, and tetramethylrhodamine-labeled EGF (Molecular Probes, Inc., Eugene, OR) were purchased from the indicated commercial sources. The anti–B-fragment mAb 13C4 was purified as described (Johannes et al., 1997). The anti-transferrin receptor mAb H68.4, the CTR433 mAb, the anti–rat TGN38 mAb 2-F7-1, the anti–γ-adaptin mAb 100/3, the anti-clathrin mAb X-22, the polyclonal anti–B-fragment antibody, the polyclonal anti–CI-MPR antibody, and the polyclonal anti-MPR46 antibody (MSC1, anti–cytoplasmic tail) were provided by I. Trowbridge (The Salk Institute, San Diego, CA), M. Bornens (Institut Curie, Paris, France), G. Banting (University of Bristol, Bristol, UK), E. Ungewickell (Max-Planck-Institut, Martinsried, Germany), F. Brotsky (University of California, San Francisco, CA), K. Niebuhr-Ebel (Institut Pasteur, Paris, France), B. Hoflack (Institut Pasteur, Lille, France), and K. von Figura (Universität Göttingen, Göttingen, Germany), respectively. The polyclonal anti-FITC antibody, the polyclonal anti-HRP antibody, and FITC- or Texas red–coupled secondary antibodies were purchased from Molecular Probes, Inc., Sigma, and Jackson ImmunoResearch Labs, Inc. (West Grove, PA), respectively.
Plasmids
To construct a plasmid expressing a mutant B-fragment carrying a tandem of sulfation sites at its COOH terminus (termed B-(Sulf)2), a previously described PCR-based strategy was adopted (Johannes et al., 1997). First, expression vector pSU108 was modified to introduce EcoRI and NotI restriction sites at the 3′ end of B-fragment cDNA (pB-EcoRI-NotI). PCR primers EN1 (5′-ACTAGCTCTGAAAAGCGGCCGCTAATGACTCAGAATAGCTC-3′) and EN2 (5′-CCGCTTTTCAGAGCTAGTGAATTCACGAAAAATAACTTCGC-3′) were used with plasmid-specific primers ShigaAtpE (5′-CACTACTACGTTTTAAC-3′) and Shiga-fd (5′-CGGCGCAACTATCGG-3′) to produce DNA fragments that, in a second PCR with primers ShigaAtpE and Shiga-fd, yielded a fragment that was cloned into the SphI and SalI restriction sites of pSU108. After polymerization cycles with sulfation site carrying oligodeoxynucleotides S1 (5′-TATGAATTCGAGGAACCTGAGTATGGAGAAGAGGAACCTGAGTATGG-3′) and S2 (5′-GCTACTTTTTTGCGGCCGCTTTCTCCATACTCAGGTTCCTCTTC-3′), the resulting fragment was cloned into the EcoRI and NotI restriction sites of pB-EcoRI-NotI. Sequences derived by PCR were verified by dideoxy-sequencing (Amersham Pharmacia Biotech, Uppsala, Sweden).
Purification and Labeling of Recombinant B-fragments and Tf
Purification of recombinant B-fragments, labeling with 5-([4,6-dichlorotriazin-2-yl]amino)fluorescein and radiolabeling with iodine were essentially done as described (Johannes et al., 1997). Labeling with Cy3 was done according to manufacturer's instructions (Amersham Life Science, Inc.). Tf was labeled with 5-([4,6-dichlorotriazin-2-yl]amino)fluorescein essentially as described for B-fragment (Johannes et al., 1997). In brief, 1 mg of ferro-Tf (Sigma) in 20 mM Hepes, pH 7.4, 150 mM NaCl, were added to 250 mM NaHCO3 and a 20-fold molar excess of 5-([4,6-dichlorotriazin-2-yl] amino)fluorescein (Sigma) and incubated by end-over-end rotation for 30 min at room temperature. 0.2 mM NH4Cl was added, and labeled protein was purified on gel filtration columns (Amersham Pharmacia Biotech).
Immunofluorescence Studies on Fixed Cells
For experiments on fixed cells, 0.75 × 105 HeLa cells on coverslips were placed on ice, fluorophore-labeled B-fragment was added at 1 μg/ml in Hepes-containing culture medium, and then incubated with cells for 30 min. The cells were then washed three times with ice-cold culture medium and shifted to either 19.5°C for 45–60 min in Hepes-containing culture medium or to 37°C in a CO2 incubator, as indicated in the figure legends. Marker proteins and antibodies were added at concentrations as follows: 10 μg/ml Tf, 1 μg/ml EGF, 1 mg/ml Dex3, polyclonal anti–CI-MPR at 1:500 dilution, anti-TGN38 (2-F7-1) at 2 μg/ml. Drugs were added at the concentrations and duration as indicated in the figure legends. After incubation, cells were washed three times with PBS containing 0.5 mM CaCl2 and 1 mM MgCl2, fixed with 3% PFA for 10 min, permeabilized with saponin, stained with the indicated primary and secondary antibodies, and then mounted, as described previously (Johannes et al., 1997). Slides were analyzed by confocal microscopy.
Confocal Microscopy
Confocal laser scanning microscopy was done on a TCS4D confocal microscope (Leica AG, Heerbrugg, Switzerland) based on a DM microscope interfaced with an Argon/Krypton laser. Simultaneous double fluorescence acquisitions were performed using the 488-nm and the 568-nm laser lines to excite FITC and Texas red dyes using 63× or 100× oil immersion Neofluar objectives (numerical aperture = 1.4). The fluorescence was selected with appropriate double fluorescence dichroic mirror and band pass filters and measured with blue-green–sensitive and red side–sensitive one photomultipliers.
Confocal Microscopy on Living Cells
HeLa cells were plated on 42-mm glass cover slides. Cy3-labeled B-fragment was bound to these cells and then internalized at 19.5°C, as described above. The cover slide was rapidly transferred to a POC-chamber (Bachofer Laboratoriumsgeräte, Reutlingen, Germany) on the stage at the confocal microscope, and culture medium at 37°C was added. Acquisitions during incubation at 37°C were done at frequencies as indicated in the figure legends. Care was taken to minimize light exposure as it was noticed that high image acquisition frequencies and high laser light intensities prevented B-fragment transport to the Golgi apparatus and caused the appearance of the protein in unidentified structures (not shown). Per time point, four optical slices were taken. Throughout an 80-min experiment, offset in z-direction was minimal. However, sometimes it was necessary to readjust the microscope. For quantification as shown in Fig. 3 B, the average fluorescent intensity in the Golgi area was divided by the average fluorescent intensity over the whole cell. To determine the fraction of Golgi-associated fluorescent material in percent after 80 min at 37°C, the ratio between total fluorescent contents in the Golgi area over whole cell total fluorescent contents was calculated. Background correction factors were determined from cells that did not have internalized B-fragment. All measurements were done using NIH image software (National Institutes of Health, Bethesda, MD).
Figure 3.




Kinetics of B-fragment transport from EE/RE to the Golgi apparatus. (A) Confocal microscopy on living HeLa cells. Fluorophore-labeled B-fragment was internalized for 1 h into HeLa cells at 19.5°C, upon which the cells were transferred to the stage of a confocal microscope and incubated at 37°C. Digital images (four integration frames) were acquired at the indicated time points. Note that after 4 min, B-fragment was detected in peripheral structures, and then later concentrated in the perinuclear region. (B) Images as shown in A were quantified, and the fraction of average Golgi associated fluorescence over average total cell-associated fluorescence is represented in function of incubation time at 37°C. The means (± SE) of eight experiments are shown. The curve was fitted to f(x) = 1 + 2.97[1 − exp(−0.036×)], r = 0.9979. (C) Sulfation analysis. B-(Sulf)2 was internalized into HeLa cells at 19.5°C, and the cells were then shifted to 37°C. After 0, 15, 30, 60, and 90 min, radioactive sulfate was added for 15 min. Note that B-(Sulf)2 is at its peak concentration in the TGN during the 15–30 min interval. A representative of 2 experiments is shown. In each experiment, the data points were obtained in duplicate. (D) Cotransport of B-fragment and Tf in living cells. For the points 3 and 10 min at 37°C, fluorophore-coupled B-fragment and fluorophore-coupled Tf were internalized as described in A. For the point 30 min at 37°C, B-fragment alone was internalized continuously at 37°C, cells were then fixed and stained for the TfR. Note that the B-fragment concentrated in the Golgi area (large arrow at 10 min), while remaining cytoplasmic B-fragment–containing structures always were Tf (4 and 10 min) or TfR (30 min) positive (small arrows at 30 min). Single optical slices were obtained by confocal microscopy.
Recycling Experiment
FITC-coupled B-fragment was added to 0.75 × 105 HeLa cells and internalized at 19.5°C for 1 h, as described above. The cells were then put on ice, and in half of the wells anti-FITC antibody was added at 5–20 μg/ml. In pilot experiments on cell surface bound FITC-labeled B-fragment it was found that these concentrations allowed for >80% quenching of FITC fluorescence. When these concentrations of antibody were added to cells that had not been treated with FITC-coupled B-fragment, no internal staining could be detected showing that fluid phase internalization at this concentration was negligible (not shown). However, if cells had internalized FITC-coupled B-fragment, then also some anti-FITC antibody was transported inside the cells, presumably in interaction with FITC (not shown). After 30 min on ice in the presence of anti-FITC antibody, the cells were either fixed directly or shifted to 37°C for the indicated periods of time in the continued presence or absence of the anti-FITC antibody before fixation. The fluorescence per given cell number (30 cells per field) of mounted cells was then analyzed with an enhanced video camera (Princeton Instruments, Trenton, NJ). Exposure time was 1.5 s, and the average fluorescence intensity was determined (five fields per experiment). In the absence of anti-FITC antibody, the average fluorescence slightly decreased during incubation at 37°C, presumably resulting from a loss of some cell-associated, FITC-coupled B-fragment, an observation that is also made if cells that had bound radiolabeled B-fragment are shifted to 37°C (not shown). To determine quenching due to recycling, average fluorescence intensity at every time point after incubation at 37°C in the presence of anti-FITC antibody was corrected for loss of material (see above) and then divided by the average fluorescence intensity obtained after the 19.5°C incubation.
Immunoelectron Microscopy
For immunoelectron microscopy, B-fragment binding (1 μM) was performed on HeLa cells grown on 50-cm2 round tissue culture plates (107 cells), as described above. After washing, cells were incubated at 19.5°C in Hepes-containing culture medium supplemented in some experiments with BSA-gold (OD = 5; a gift from H.J. Geuze, University of Utrecht, Utrecht, The Netherlands) or Tf-HRP (Pierce Chemical Co.) (20 μg/ml), fixed directly or shifted to 37°C, as indicated in the figure legends. Fixation was done by adding to the medium an equivalent volume of 8% PFA in 0.2 M sodium phosphate buffer, pH 7.4, for 1 h, and further fixation was done with a fresh 4% PFA solution in 0.1 sodium phosphate buffer for 1 h more. Cryosectioning and immunogold labeling was done as described previously (Slot et al., 1991). In the case of mAbs, a rabbit anti–mouse linker antibody was used (DAKOPATTS AB, Älvsjö, Sweden). For triple labeling, cryosections were fixed with 1% glutaraldehyde between the first and second mAb.
Quantification on Cryosections
Evolution of colocalization between B-fragment and BSA-gold (see Fig. 5 D): membrane profiles that were labeled for BSA-gold (at least two gold particles) were identified by scanning over cryosections of cells that had internalized B-fragment and BSA-gold at 19.5°C, or that were shifted to 37°C for an additional 15 min. It was then determined whether these profiles were also labeled for B-fragment (100 membrane profiles were analyzed per condition).
Figure 5.



B-fragment is detected in compartments of the late endocytic pathway during transport to the Golgi apparatus. (A and B) HeLa cells were incubated with B-fragment and BSA-gold (5-nm gold particles) at 19.5°C. Cryosections were stained with anti–B-fragment antibody (10-nm gold particles). The arrow in B indicates a region of high BSA-gold concentration (bulk fluid phase) that is devoid of B-fragment. (C) Cells treated as in A and B were then shifted for 15 min to 37°C, fixed, and then cryosections were stained with anti–B-fragment antibody (15-nm gold particles) and anti–CI-MPR antibody (10-nm gold particles). Various endocytic structures are indicated by numbers: areas 1, BSA-gold–labeled EE; areas 2, CI-MPR- and BSA-gold–positive multivesicular LE; areas 3, CI-MPR–positive multilamellar LE. GA, Golgi apparatus. (D) Quantification of BSA-gold–positive structures containing or not B-fragment, after the 19.5°C incubation (lanes 1 and 2) or after an additional shift to 37°C for 15 min (lanes 3 and 4). (Lanes 1 and 3) Percentage of BSA-gold–containing structures that also contain B-fragment; (lanes 2 and 4), BSA-gold–containing structures without B-fragment. Bars, 100 nm.
To determine whether BSA-gold–positive bulk fluid phase elements (defined as 150–300-nm structures that contained ≥50 BSA-gold particles) were also positive for γ-adaptin, the former were identified on fields of observation that were labeled with ≥15 γ-adaptin–specific gold particles. 10 such fields with 15 bulk fluid phase elements were analyzed.
Determination of the fraction of marker protein–specific label in γ-adaptin–positive membrane profiles (Table I): all intracellular B-fragment (3,809 gold particles), Tf-HRP (2,170 gold particles), or BSA-gold–specific (8,245 gold particles) label on 298 fields of observation was analyzed for colocalization with γ-adaptin. Colocalization was scored as positive only if there were at least two γ-adaptin–specific gold particles on a membrane profile on which B-fragment, Tf-HRP, or BSA-gold–specific gold particles were localized in <100 nm. Depending on the marker protein, determinations were done on one to five independent labeling experiments.
Table I.
Localization of Marker Proteins in γ-Adaptin–positive Membrane Profiles
| Staining of γ-adaptin versus | Colocalization | |
|---|---|---|
| % | ||
| B-fragment | 9.7 (± 0.62; n = 5) | |
| Tf-HRP | 13.2 (± 3.1; n = 4) | |
| BSA-gold | 0.27 (n = 1) |
Quantification was done as described in Materials and Methods on cryosections of HeLa cells that had internalized BSA-gold and B-fragment or Tf-HRP and B-fragment for 1 h at 19.5°C. The percentage of marker protein–specific gold label in γ-adaptin–positive membrane profiles is given (± SE; n, the number of independent determinations on different labeling experiments).
To determine the distribution of B-fragment and Tf-HRP–specific immunogold label in γ-adaptin–positive membrane profiles (see Fig. 10 A), 204 fields of observation (a total number of 2,512 and 1,990 B-fragment and Tf-HRP–specific gold particles, respectively, were counted) with 278 γ-adaptin–positive membrane profiles (at least two γ-adaptin–specific gold labels) that contained 273 and 294 B-fragment and Tf-HRP–specific gold particles, respectively, were analyzed on triple-labeled cryosections (as shown for example in Fig. 9, F–I). For statistical evaluation, B-fragment or Tf-HRP–specific gold particles in γ-adaptin–positive membrane profiles were taken as denominators. The chi-square test showed a significative difference (P < 0.001) between the fraction of B-fragment–specific gold particles in only B-fragment and γ-adaptin–positive membrane profiles (180 out of 273), when compared with the fraction of Tf-HRP–specific gold particles in only Tf-HRP and γ-adaptin–positive membrane profiles (122 out of 294).
Figure 10.
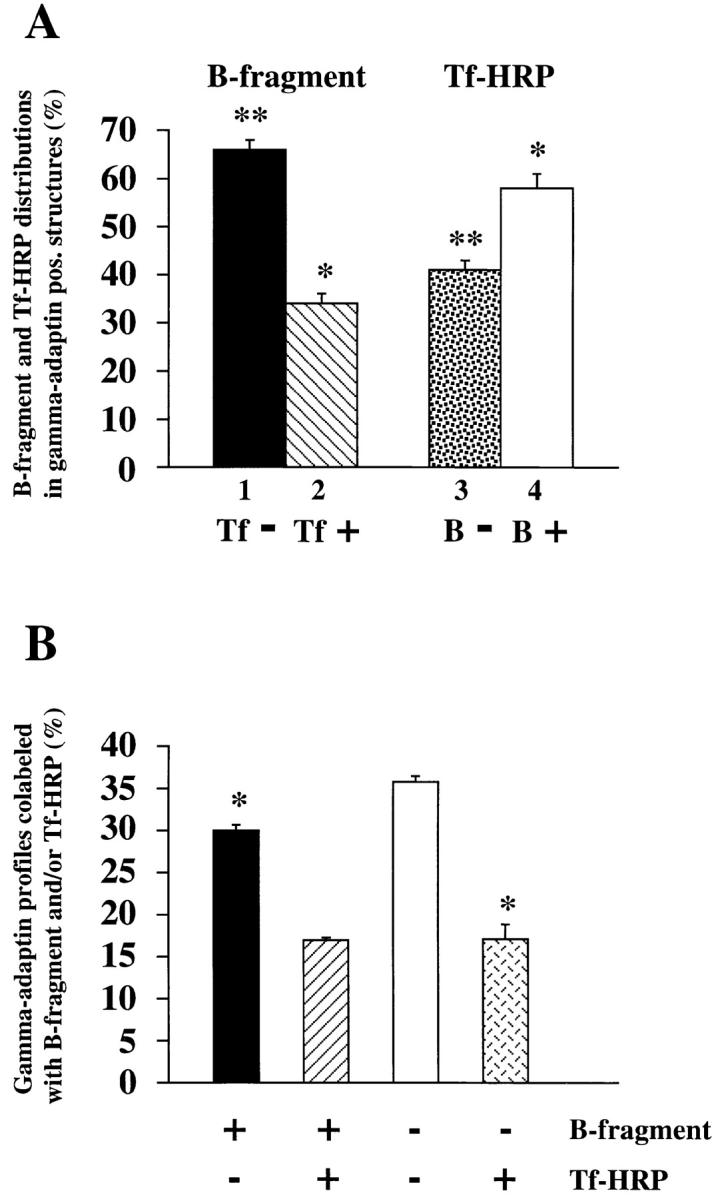
Quantification of γ-adaptin–positive membrane profiles. (A) Distribution of B-fragment and Tf-HRP–specific gold particles in γ-adaptin–positive membrane profiles. The columns represent the fraction of B-fragment (lanes 1 and 2) in γ-adaptin– positive structures that were labeled (lane 2) or not labeled (lane 1) for Tf-HRP, or the fraction of Tf-HRP (lanes 3 and 4) in γ-adaptin–positive structures that were labeled (lane 4) or not labeled (lane 3) for B-fragment. (B) Characterization of γ-adaptin–positive structures. Note that significantly more γ-adaptin– positive profiles were labeled for only B-fragment (left column), compared with such structures labeled for only Tf-HRP (right column). * and **, direct comparison shows that these conditions are significantly different (P < 0.01; see Materials and Methods).
Figure 9.
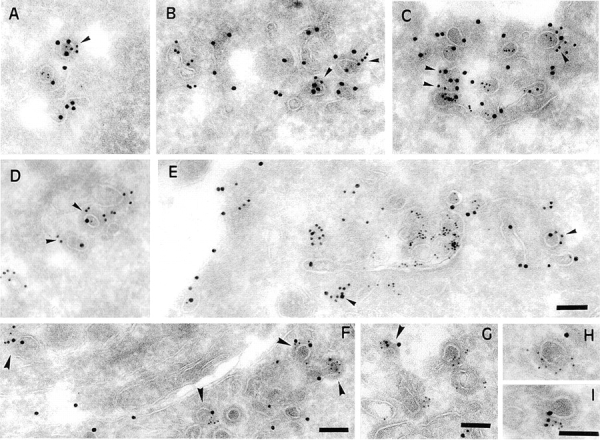
γ-Adaptin and clathrin colocalize with B-fragment and Tf-HRP at the ultrastructural level on coated membrane profiles of EE/RE. (A–E) HeLa cells were incubated for 1 h with B-fragment and BSA-gold (5-nm gold particles) at 19.5°C. Cryosections of these cells were labeled with anti–B-fragment antibody (15-nm gold particles) and anti–γ-adaptin antibody (10-nm gold particles in A–C) or anti-clathrin antibody (10-nm gold particles in D and E). In A–C, arrowheads indicate regions of colocalization between γ-adaptin and B-fragment. In D and E, arrowheads point out regions of colocalization between clathrin and B-fragment. (F-I) Serum-starved HeLa cells were incubated for 1 h with B-fragment and Tf-HRP at 19.5°C. Cryosections of these cells were labeled with anti–B-fragment antibody (15-nm gold particles), anti-HRP antibody (10-nm gold particles), and anti–γ-adaptin antibody (5-nm gold particles). Arrowheads in F and G point to double- or triple-labeled profiles. (H and I) Magnification of selected structures. Bars, 100 nm.
To determine the fraction of γ-adaptin–positive membrane profiles colabeled with B-fragment and/or Tf-HRP gold particles (see Fig. 10 B), 417 such profiles (at least two γ-adaptin–specific gold labels) were analyzed on three independent triple-labeling experiments (as shown for example in Fig. 9, F–I). Using the chi-squared test for paired series, profiles that were only labeled for B-fragment were found to be significantly more abundant than those labeled for only Tf-HRP (P < 0.01).
Retrograde Transport Assay and EGF Degradation
Iodinated B-Glyc-KDEL (5,000 cpm/ng) was bound to 105 HeLa cells as described above. After incubation at 19.5°C for 1 h and shift to 37°C for the indicated periods of time, cells were washed three times with PBS and lysed in SDS sample buffer. Samples were run on 10–20% polyacrylamide–SDS gradient gels, analyzed by autoradiography, and quantified with a PhosphorImager (Molecular Dynamics, Inc., Sunnyvale, CA) using the ImageQuant software. In each experiment, the percentage of glycosylated protein was determined.
For EGF degradation experiments, HeLa cells were incubated for 30 min in serum-free culture medium. Iodinated EGF (750 Ci/mmol; Amersham Life Science, Inc.) was then added in serum-free Hepes containing culture medium at 19.5°C for 1 h. Cells were washed, shifted to 37°C for the indicated periods, and then put on ice. Culture medium was taken off, the remaining cell-associated radioactivity and the amount of TCA-soluble material in the culture medium were determined. TCA-soluble counts were then expressed as percentage of total cell-associated radioactivity.
Sulfation Analysis
Analysis of sulfation on B-(Sulf)2 was essentially done as described (Johannes et al., 1997). In brief, B-(Sulf)2 was bound to sulfate-starved HeLa cells (105) on ice. After washing, the cells were incubated in sulfate-free, HBSS buffer (GIBCO BRL) at 19.5°C for 1 h, shifted to 37°C for the indicated times (0–90 min), and incubated for a final 15 min at 37°C in HBSS buffer containing 2.5 mCi/ml of [35S]sulfate (1,000 Ci/mmol; Amersham Life Science, Inc.). B-(Sulf)2 was immunoprecipitated from cell lysates obtained with RIPA buffer using the mAb 13C4 and protein A–Sepharose (Amersham Pharmacia Biotech). Washed immunoprecipitates were boiled in sample buffer and analyzed by PhosphorImager after separation by SDS-PAGE on Tris-Tricine gels (Schägger and von Jagow, 1987).
Results
Incubation at 19.5°C Induces B-fragment Accumulation in EE in HeLa Cells
We have previously shown that in HeLa cells, Shiga toxin B-fragment is transported from the plasma membrane via endosomes, the TGN and the Golgi apparatus to the ER (Johannes et al., 1997). To analyze transport from endosomes to the Golgi apparatus in further detail, we developed a low temperature incubation protocol that allowed the synchronization of this transport step. B-fragment was incubated with HeLa cells at various temperatures, and it was found that at 19.5°C, the protein accumulated efficiently in endosomes, while its transport to the Golgi apparatus was blocked (Fig. 1). These data are consistent with earlier studies that showed that the plant toxin ricin is not transferred to the Golgi apparatus during low temperature incubations, and that under these experimental conditions Shiga toxin is much less toxic to HeLa cells than at physiological temperatures (Sandvig et al., 1989).
Figure 1.
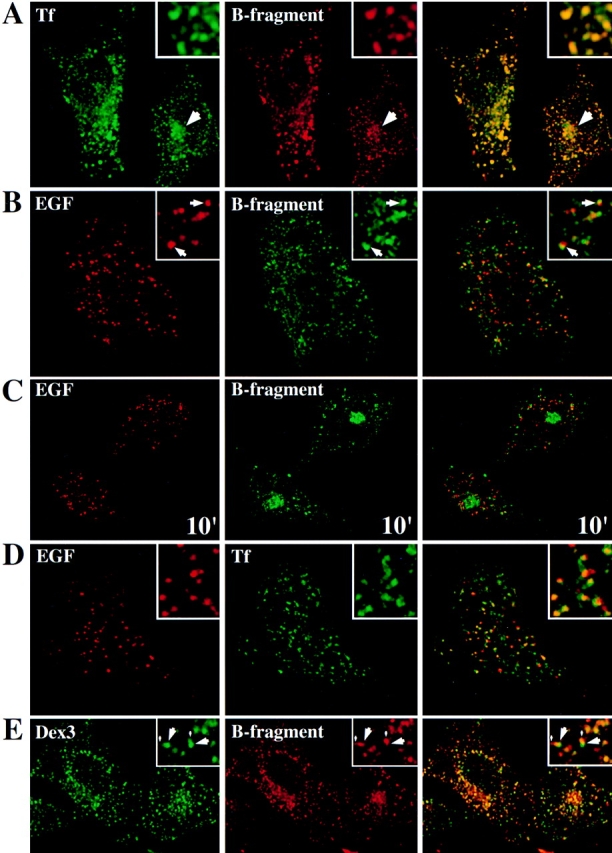
Study of B-fragment colocalization with established markers of the endocytic pathway during incubation at 19.5°C. The following proteins were incubated with HeLa cells at 19.5°C for 1 h: (A) Tf (green) and B-fragment (red), a large arrow indicates a region of perinuclear staining; (B) EGF (red) and B-fragment (green), arrows point out regions where B-fragment and EGF staining are juxtaposed; (D) TF (green) and EGF (red); (E) Dex3 (green) and B-fragment (red), note vesicular (large arrows) and tail-like (small arrows) Dex3 staining. For marker concentrations see Materials and Methods. Digital images (four integration frames) were acquired by confocal microscopy. The right panel represents the superposition of the red and green images. Insets show selected areas at higher magnification. In C, EGF (red) and B-fragment (green) were internalized at 19.5°C, as in B. The cells were then shifted to 37°C for 10 min before fixation. Note that B-fragment and EGF-specific labeling did basically not overlap.
We then compared B-fragment transport through the endocytic membrane system at 19.5°C to that of marker proteins with established pathways. When fluorophore-labeled B-fragment (Cy3-coupled, red) and ferro-Tf (fluorsceine-coupled, green), a marker protein of EE and RE, were incubated together with HeLa cells for 1 h at 19.5°C, both were found at the same intracellular sites (Fig. 1 A). As shown in the inserts of Fig. 1 A, the colocalization between both proteins was essentially complete. In some cells, B-fragment and Tf were found to label perinuclear structures in addition to the peripheral vesicular compartments (Fig. 1 A, arrows). These may represent perinuclear RE, an hypothesis that is consistent with the observation that when cells were shifted to 37°C, peripheral Tf-label was more rapidly lost than perinuclear Tf-label (not shown).
In addition to recycling receptors such as the transferrin receptor (TfR) back to the plasma membrane, EE also play an important role in providing access to LE and lysosomes. When B-fragment (green fluorescence) and lysosome-bound EGF (red fluorescence) were internalized at 19.5°C for 1 h, both gave staining patterns that were juxtaposed to each other, but which did not overlap (Fig. 1 B, insets, arrows), a distribution that was in marked contrast to the high degree of colocalization of B-fragment and Tf (see Fig. 1 A). As shown in Fig. 1 C, juxtaposition of B-fragment– (green fluorescence) and EGF-labeled (red fluorescence) structures was essentially lost when cells that had internalized B-fragment and EGF at 19.5°C were shifted for 10 min to 37°C. Under these conditions, EGF was transported to lysosomes and B-fragment to the Golgi apparatus (see also below). Since EGF and B-fragment colocalized at very early time points after internalization (not shown), these findings suggest that both proteins were partitioned away from each other at the level of sorting endosomes. Similar results, i.e., a separation between two marker proteins, were obtained when EGF (red fluorescence) was internalized together with Tf (green fluorescence) at 19.5°C (Fig. 1 D).
We next compared by confocal microscopy B-fragment (red fluorescence) transport to that of a fluid phase marker, FITC-coupled 3-kD dextran (Dex3, green fluorescence) (Fig. 1 E). After cointernalization of Dex3 and B-fragment at 19.5°C, Dex3-positive structures often had a comet-like appearance in that they were composed of strongly labeled bulk fluid phase elements (Fig. 1 E, insets, large arrows) with tail-like extensions (small arrows). Importantly, B-fragment containing structures were found in the same areas of the cell, but overlapped with the tail-like elements (Fig. 1 E, insets, small arrows). The situation was different for cells that had cointernalized Dex3 and EGF at 19.5°C, in the case of which EGF staining overlapped preferentially with the bulk-phase element of Dex3-containing structures (not shown).
These data suggest that the B-fragment accumulated at 19.5°C in EE/RE where the protein segregated from lysosome-bound, membrane-associated EGF and from the bulk of the fluid phase. The observation of B-fragment segregation from the bulk fluid phase was further confirmed by electron microscopical studies (see Fig. 5, A and B).
The B-fragment Passes within Minutes from Early/Recycling Endosomes into the Golgi Apparatus with a Fraction of Internalized Molecules Being Recycled to the Plasma Membrane
We next set out to follow B-fragment transport from EE/ RE to the Golgi apparatus. HeLa cells were incubated at 19.5°C with B-fragment, transferred for increasing times to 37°C, fixed, and then processed for ultracryomicrotomy followed by labeling with specific antibodies (Fig. 2). As described above, B-fragment (Fig. 2 A; 10-nm gold particles) accumulated at 19.5°C in vesicular and tubular structures (50–100 nm) that also contained the TfR (15-nm gold particles). The kinetics of B-fragment transport from EE/ RE to the Golgi apparatus was followed by warming the cells to 37°C. To identify membranes of the TGN, cryosections were also stained for MPR46, which at steady state in HeLa cells is mostly localized to the TGN, in contrast to the CI-MPR (not shown; see below). After 2-min incubation at 37°C (Fig. 2 B), some of the B-fragment (10-nm gold particles) accumulated already in membranes of the TGN in colocalization with MPR46 (15-nm gold particles) (Fig. 2 B), suggesting that the B-fragment entered the Golgi apparatus at the level of the TGN, in agreement with our previous results on sulfation site–carrying B-fragment mutants (Johannes et al., 1997). At 10 min (Fig. 2 C), B-fragment labeling (10-nm gold particles) at the Golgi apparatus increased significantly. As shown in Fig. 2 D, double-labeling experiments with B-fragment (15-nm gold particles) and MPR46 (10-nm gold particles) again identified some of the B-fragment–positive membranes as the TGN. After 30 min at 37°C (Fig. 2 E), all cisternae of the Golgi apparatus were strongly labeled with B-fragment (10-nm gold particles).
Figure 2.
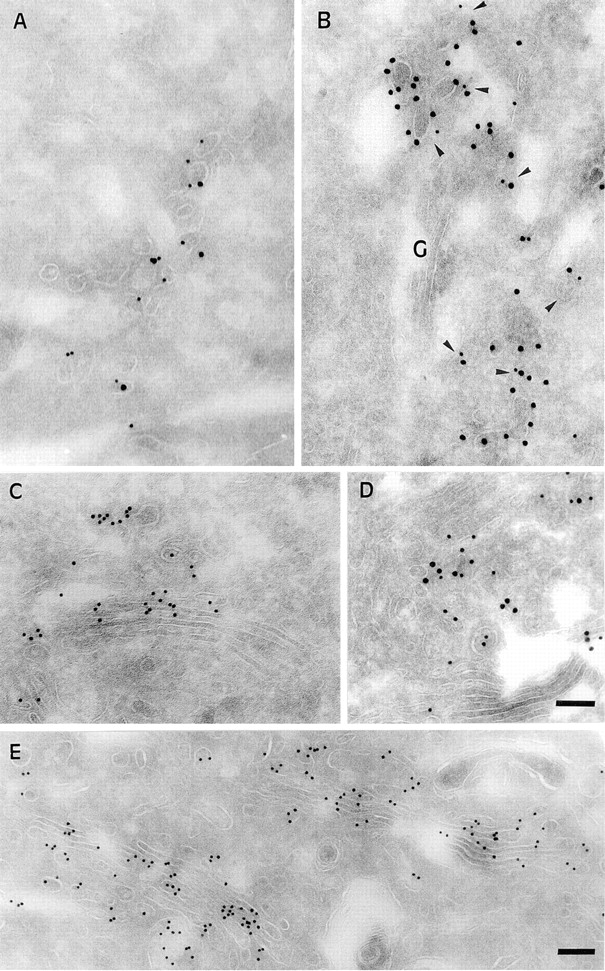
Kinetics of B-fragment transport from EE/RE to the Golgi apparatus revealed by immunoelectron microscopy in HeLa cells. (A) B-fragment was internalized for 1 h at 19.5°C, cells were fixed and prepared for cryosectionning as described under Materials and Methods. B-fragment (10-nm gold particles) was detected in tubular and vesicular elements that were also labeled for the TfR (15-nm gold particles). Cells that had internalized B-fragment at 19.5°C were then shifted for 2 min (B), 10 min (C–D), and 30 min (E) to 37°C. Cryosections were stained for B-fragment (10-nm gold particles in B, C, and E; 15-nm gold particles in D) and MPR46 (15-nm gold particles in B; 10-nm gold particles in D). Cryosections that were doubled stained for MPR46 (in B and D) showed that the B-fragment entered the Golgi apparatus via the TGN. Bars, 100 nm.
The fast appearance of B-fragment in the TGN and the cisternae of the Golgi apparatus was surprising since it seemed difficult to reconcile such a kinetics with transport via LE (Mukherjee et al., 1997; see below). The kinetics of B-fragment transport from EE/RE to the Golgi apparatus was therefore quantitatively studied following two independent protocols, first on living HeLa cells by confocal microscopy, and second by biochemical means using protein sulfation as a TGN-specific marker.
For the experiments on living cells, Cy3-labeled B-fragment was internalized into HeLa cells at 19.5°C, as described above. The cells were transferred to the stage of a confocal microscope and analyzed at 10–15-min intervals during incubation at 37°C. Data obtained in a typical experiment are shown in Fig. 3 A. At time point 4 min at 37°C (time necessary to adjust the microscope settings), B-fragment was found in vesicular cytoplasmic structures (Fig. 3 A) that corresponded to EE/RE since they were also labeled by cointernalized fluorescein-tagged Tf (Fig. 3 C). During further incubation at 37°C, accumulation in a juxtanuclear region became visible (Fig. 3 A), which at the end of the experiment was shown to be staining of the Golgi apparatus, as judged from colabeling with Golgi markers on fixed cells (not shown). It was also found that, using a double-fluorescence detection protocol on living cells, Cy3-tagged B-fragment accumulated in compartments that contained the green fluorescent protein–tagged Golgi marker Rab6 (not shown). Quantification of experiments as those of Fig. 3 A showed that the B-fragment moved with a t 1/2 of 19 min from EE/RE to the Golgi apparatus (Fig. 3 B). The plateau value of the Golgi over cell-ratio of 3.97 in Fig. 3 B corresponded to 32.1% (± 2.6%; n = 8) of total cell-associated B-fragment–specific labeling being concentrated in the Golgi area. It should be pointed out that at later time points, this value likely reflected a steady-state concentration of B-fragment in the Golgi apparatus as we have previously shown that in the absence of an ectopic ER retrieval signal, B-fragment distributed between the Golgi apparatus and the ER (Johannes et al., 1997).
Protein sulfation is a TGN-specific posttranslational modification that is catalyzed by sulfotransferase. We have previously shown that B-fragment with a COOH-terminal sulfation site and a KDEL signal can be sulfated (Johannes et al., 1997). Here we constructed a new mutant without retrieval signal and with a tandem of sulfation sites to increase sensitivity. This mutant, termed B-(Sulf)2, was internalized into HeLa cells for 1 h at 19.5°C, and the cells were then shifted to 37°C, as indicated in Fig. 3 C. At the end of the incubation periods, radioactive sulfate was added for 15 min to the cells to sample the amount of B-(Sulf)2 that was in the TGN at this time point. The peak value for sulfation on B-(Sulf)2 was obtained between 15 and 30 min (Fig. 3 C). During the 60–75-min interval, the amount of sulfated B-(Sulf)2 decreased to 25% of the peak value, and then remained stable when sampled after 90– 105 min (Fig. 3 C) or after 4 h (not shown). Likely, this residual material accessible to sulfation represents the steady state amount of B-(Sulf)2 in the TGN. In summary, the kinetics obtained by morphological and biochemical means were comparable, and both experiments confirmed the hypothesis of rapid B-fragment transport from EE/RE to the Golgi apparatus.
The possibility to follow transport of fluorophor-labeled proteins on living cells by confocal microscopy then allowed to compare the exit of B-fragment and Tf from EE/ RE. As shown in Fig. 3 D (left), 3 min after transfer to 37°C both proteins still largely colocalized to dispersed cytoplasmic structures. After 10 min at 37°C (Fig. 3 D, middle), B-fragment began to accumulate in the Golgi apparatus (large arrow; note that the plane of focus is not on the Golgi apparatus), while Tf staining had visibly diminished (the Tf image at 10 min was overexposed to compensate for loss resulting from recycling). However, it should be noted that the vesicular structures that remained B-fragment labeled also still contained Tf. After longer incubations at 37°C, Tf was recycled to the medium and the B-fragment regrouped in the Golgi apparatus (not shown). To label B-fragment–containing intermediate structures after longer periods of time, HeLa cells were incubated continuously for 30 min in the presence of Cy3-tagged B-fragment at 37°C, fixed, and then labeled for TfR (Fig. 3 D, right). In agreement with the data in vivo it was found that all B-fragment–labeled structures outside the Golgi apparatus were also positive for the TfR (Fig. 3 D, small arrows).
The presence of B-fragment in EE/RE in colocalization with Tf prompted us to test whether internalized B-fragment could be recycled to the plasma membrane, as described for other toxins (Sandvig and Olsnes, 1979; Nambiar and Wu, 1995; Alami et al., 1998). FITC-tagged B-fragment was internalized into HeLa cells at 19.5°C for 45 min. The cells were then put on ice and B-fragment that was still exposed at the cell surface was reacted with an anti-FITC antibody that quenched FITC fluorescence upon interaction with the fluorophore. The cells were subsequently transferred to 37°C in the continued presence of the anti-FITC antibody in the extracellular medium. It was found that 25% of FITC fluorescence that was emitted by intracellular B-fragments was quenched during a 30-min incubation at 37°C (Fig. 4). This value did not increase any more upon 50 additional min at 37°C in the presence of the anti-FITC antibody. 25% or 80% of total quenching was already obtained after 5 or 15 min at 37°C, respectively, indicating that B-fragment recycling to the plasma membrane was rapid. It can be concluded that only ∼25% of internalized B-fragment was recycled to the plasma membrane during an incubation period (80 min; Fig. 4) sufficiently long to have B-fragment at its peak concentration in the Golgi apparatus (Fig. 3). It should be added that following B-fragment internalization at 19.5°C and subsequent incubation at 37°C for 5–30 min, surface biotinylation studies failed to detect a significant increase in plasma membrane–associated B-fragment, consistent with the hypothesis of little B-fragment being recycled to the plasma membrane (not shown). The observation of limited recycling of B-fragment is also consistent with earlier studies that showed that plasma membrane associated Shiga toxin became rapidly insensitive to the neutralizing activity of anti-toxin antibody (Sandvig et al., 1989).
Figure 4.

A fraction of internalized B-fragment is recycled to the plasma membrane. FITC-labeled B-fragment was internalized into HeLa cells at 19.5°C, the cells were then incubated with anti-FITC antibody on ice for 30 min and shifted to 37°C in the continued presence of the anti-FITC antibody for the indicated periods. Fluorescence at each time point was determined as described in Materials and Methods and compared with the 0 time point to determine quenching due to newly recycled B-fragment. The means (± SE) for three independent experiments are shown.
B-fragment thus passed with a half time of 19 min from EE/RE to the Golgi apparatus with part of the molecules being recycled to the plasma membrane. The absence of B-fragment–positive structures outside the Golgi apparatus that would have been devoid of Tf or TfR suggested that the B-fragment did not accumulate outside EE/RE on its transport to the Golgi apparatus.
Upon Transport to the Golgi Apparatus, B-fragment Does Not Accumulate in Organelles of the Late Endocytic Pathway
To directly test whether B-fragment had access to compartments of the late endocytic pathway while moving from EE/RE to the Golgi apparatus, HeLa cells were incubated for 1 h with BSA coupled to gold particles (BSA-gold) and B-fragment at 19.5°C, and then fixed either directly (Fig. 5, A and B) or shifted for 15 min to 37°C before fixation (Fig. 5 C). The cells were then prepared for cryoultramicrotomy and cryosections were labeled with the indicated antibodies. Upon incubation at 19.5°C (Fig. 5, A and B), BSA-gold (5-nm gold particles) was found filling tubular and vesicular elements that also contained B-fragment (10-nm gold particles). In addition, strong BSA-gold accumulation was observed in large compartments (150– 300 nm) that were devoid of B-fragment (Fig. 5 B, arrow). At 19.5°C, B-fragment thus seemed to partition away from the bulk fluid phase, as described above (Fig. 1 E). We also noted that during low temperature incubation, no BSA-gold advanced to multivesicular or multilamellar late endosomes (not shown), demonstrating that transport to the late endocytic pathway was inhibited at 19.5°C (see also below).
To have quantitative data on the evolution of B-fragment staining with respect to that of lysosome-bound BSA-gold, we compared the percentage of B-fragment– positive, BSA-gold–containing structures after the 19.5°C incubation to the percentage of such structures observed after the additional 15 min shift to 37°C (Fig. 5 D). At 19.5°C, 76.9% of BSA-gold–positive endosomes also contained B-fragment, whereas after 15 min at 37°C (for example Fig. 5 C), only 16.7% of such double-labeled structures could be detected, showing that the high degree of colocalization between both markers was lost when the 19.5°C block was released (for quantification, see Materials and Methods). In Fig. 5 C we also show that the B-fragment (15-nm gold particles) had left EE after the 15-min incubation at 37°C, while these were still stained with BSA-gold (areas 1). The B-fragment had entered the Golgi apparatus (Fig. 5 C, GA, arrows), as described above. Importantly, the toxin subunit was neither detected in multivesicular LE that were already loaded with BSA-gold (5-nm gold particles) (areas 2), nor was it found multilamellar (mature) LE (areas 3). These structures were labeled with antibodies to the CI-MPR (10-nm gold particles), which, in contrast to MPR46, is mostly localized in LE in HeLa cells (not shown; see above).
Even after staining cryosections for only B-fragment in the absence of BSA-gold (experiments described in Fig. 2), structures with a characteristic morphology of multivesicular and multilamellar late endosomes were frequently observed. However, neither after the 19.5°C block nor during subsequent incubations (2, 10, 30 min) at 37°C, any B-fragment was detected in these structures (not shown). These direct observations and the quantification described above thus suggest that the B-fragment did not accumulate in compartments of the late endocytic pathway upon its transfer from EE/RE to the Golgi apparatus.
Drug Effects on B-fragment Transport
To test the influence of drugs with established effects on the endocytic pathway on B-fragment transport to the Golgi apparatus, cells that had internalized Cy3-labeled B-fragment at 19.5°C were incubated for 1 h at 37°C in presence of the vacuolar proton pump inhibitor bafilomycin A1 (Bafi), the actin-depolymerizing drug cytochalasin D (CytoD), or the fungal metabolite brefeldin A (BFA) (Fig. 6). Neither CytoD nor Bafi prevented B-fragment appearance in the Golgi apparatus, stained with the medial Golgi marker CTR433 (Fig. 6 A), consistent with recently published observations (Schapiro et al., 1998). The same results were obtained when the cells were pretreated for up to 2 h with these drugs (not shown). In the presence of BFA, which in addition to its profound effects on the biosynthetic/secretory pathway (for review see Klausner et al., 1992) has somewhat more subtle effects on the endocytic membrane system (Hunziker et al., 1991; Lippincott-Schwartz et al., 1991; Wood et al., 1991; Strous et al., 1993; for review see Hunziker et al., 1992), B-fragment accumulated in tubular elements that also contained the TfR (Fig. 6 B). The protein remained associated with these tubules even when the cells were incubated at 37°C for up to 4 h in presence of the drug (not shown). In the presence of the microtubule-destabilizing agent nocodazole (Noc), B-fragment was still transported to the dispersed cisternae of the Golgi apparatus where it colocalized with the Golgi marker CTR433 (Fig. 6 C, top row; see also Johannes et al., 1997) and with the TGN marker protein TGN38 (not shown). Interestingly, when Noc-treated cells were also exposed to BFA, CTR433-positive (Fig. 6 C, bottom row) Golgi compartments that persisted under these conditions (Lippincott-Schwartz et al., 1990) were not labeled by B-fragment any more, suggesting that transport between the BFA-induced, B-fragment-, and TfR-positive membrane system and the Golgi remnants had ceased.
Figure 6.

Drug effects on B-fragment transport to the Golgi apparatus. HeLa cells were incubated with fluorophore-labeled B-fragment for 1 h at 19.5°C, before being shifted to 37°C (A) in the absence (CTL) or presence of 1 μM Bafi or 1 μM CytoD, or (B) in the presence of 5 μg/ ml BFA. The cells were then fixed and labeled with CTR433 antibody (A) or anti-TfR antibody (B). (C) BFA inhibits B-fragment transport to dispersed Golgi cisternae in Noc-treated HeLa cells. HeLa cells were pretreated for 1 h with 10 μM Noc. The cells were then transferred on ice and incubated with fluorophore-labeled B-fragment for 30 min, washed, and then shifted for 30 min to 37°C in the absence (top row) or presence (bottom row) of 5 μg/ml BFA and in the continued presence of Noc. The cells were then fixed and stained for the Golgi marker CTR433. Note that in the absence of BFA, B-fragment associated with the dispersed cisternae of the Golgi apparatus (top), while in the presence of the drug, the CTR433-positive cisternae were devoid of B-fragment (bottom). Four optical slices were obtained by confocal microscopy.
To obtain quantitative data on drug effects on B-fragment transport, we used a previously constructed chimeric B-fragment in which the B-fragment was fused to a N-glycosylation site and the KDEL retrieval signal, yielding a protein termed B-Glyc-KDEL (Johannes et al., 1997). As wild-type B-fragment, B-Glyc-KDEL is transported from the plasma membrane to the ER, where the chimeric protein gets progressively glycosylated. The determination of the percentage of glycosylated, cell-associated B-Glyc-KDEL at any given time point after internalization represents thus a quantitative measure for the protein's transport to the ER. Fig. 7 A shows the results of glycosylation analysis on B-Glyc-KDEL, which was incubated with HeLa cells in the presence of drugs. Control cells (Fig. 7 A, closed circles) and cells that had been pretreated for 2 h with Bafi (open squares) or CytoD (open diamonds) and that had bound iodinated B-Glyc-KDEL on ice were shifted to 37°C in the continued presence of the drugs. BFA (Fig. 7 A, closed diamonds) was only added upon temperature shift to 37°C. Cell lysates taken after the indicated times at 37°C were analyzed by autoradiography. Bafi and CytoD had no effect on B-Glyc-KDEL glycosylation (Fig. 7 A). The same results were obtained when cells were incubated in the presence of the proton pump inhibitor concanamycin B, the weak base ammonium chloride or when Bafi treatment was combined with chloroquine treatment, the weak base chloroquine assuring a rapid dissipation of endosomal pH (not shown). In contrast, no glycosylation of B-Glyc-KDEL was detectable in the presence of BFA (Fig. 7 A, closed diamonds), even after incubations of ≤15 h (not shown).
Figure 7.
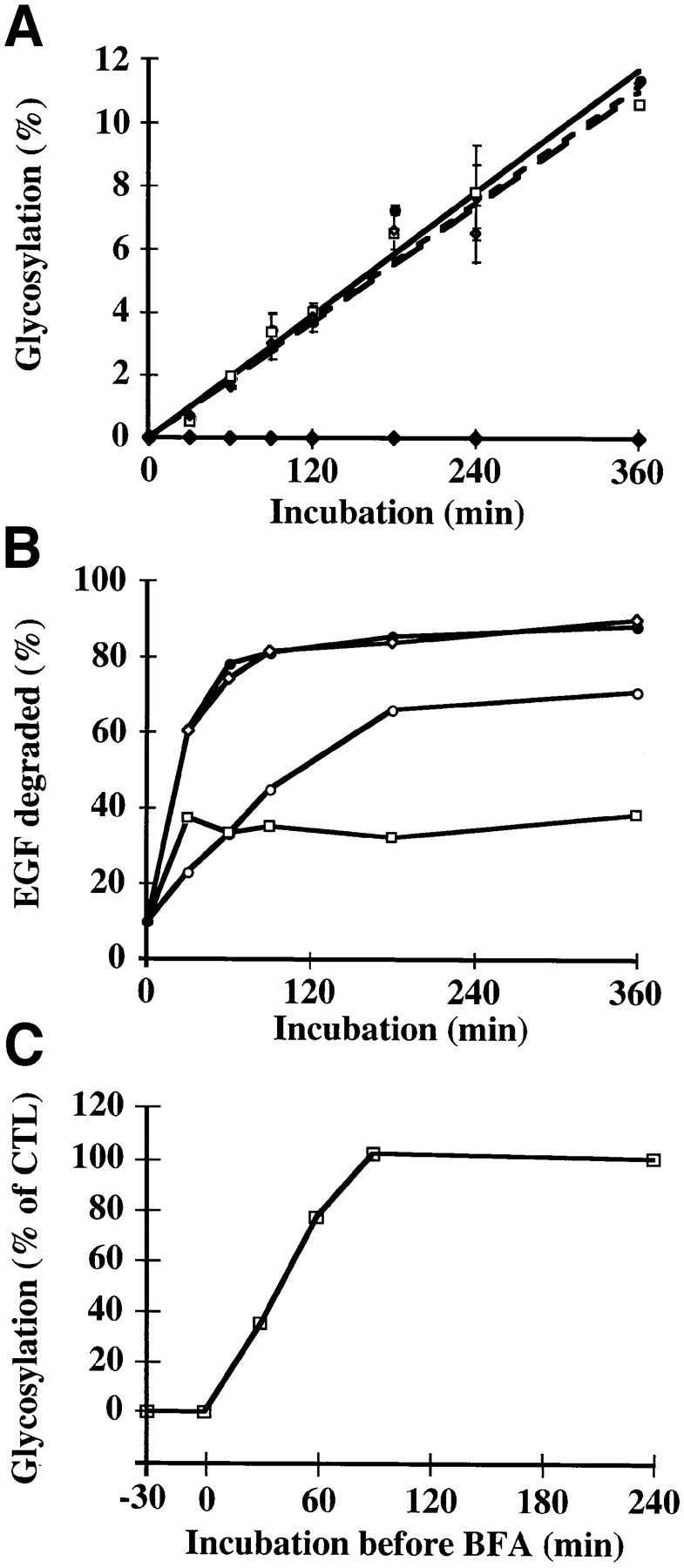
Quantification of drug effects on B-fragment transport to the biosynthetic/ secretory pathway. (A) HeLa cells were preincubated 2 h with 1 μM Bafi (open squares) and 1 μM CytoD (open diamonds). The cells were then transferred on ice and incubated with 50 nM iodinated B-Glyc-KDEL for 30 min. After washing, the cells were shifted to 37°C in the continued presence of the drugs. BFA (closed diamonds) was added only upon temperature shift to 37°C. Control cells (closed circles) were not treated with drugs. After the indicated times at 37°C, cells were lysed in sample buffer, and then lysates were analyzed on 10–20% polyacrylamide–SDS gradient gels, followed by autoradiography. The percentage of glycosylated B-Glyc-KDEL was expressed in function of incubation time. The means of three independent experiments (± SE) are shown. (B) Radiolabeled EGF was internalized into HeLa cells at 19.5°C. The cells were then shifted to 37°C in the absence (closed circles) or presence of 1 μM Bafi (open squares), 1 μM CytoD (open circles), or 5 μg/ml BFA (open diamonds). After the indicated times at 37°C, the number of TCA-soluble counts in the culture medium was determined as described in Materials and Methods. A representative of two experiments is shown. (C) Iodinated B-Glyc-KDEL was bound to HeLa cells, as described in A. The cells were then shifted to 37°C, and 5 μg/ml BFA was added after the indicated times (−30 min to 4 h, with respect to the beginning of B-Glyc-KDEL internalization). After 4 h at 37°C, cells were lysed and glycosylated B-Glyc-KDEL was determined as described in A. A representative of two experiments is shown.
In parallel, we analyzed the lysosomal degradation of EGF as a control for the efficiency of the drug treatments (Fig. 7 B). Iodinated EGF was internalized into HeLa cells at 19.5°C. To test whether the block at 19.5°C prevented access to lysosomes, cells were incubated for ≤8 h with radiolabled EGF at 19.5°C. The number of TCA-soluble counts remained stable under these conditions (not shown), indicating that EGF was not transferred to a degradative (late) compartment of the endocytic pathway. The cells that had internalized EGF at low temperatures were then shifted to 37°C. In the absence of the drugs (Fig. 7 B, closed circles), EGF degradation was complete after 90 min. As expected, Bafi (Fig. 7 B, open squares) almost completely inhibited degradation, as judged from the fact that the amount of extracellular TCA-soluble material derived from EGF remained stable. CytoD induced a delay in EGF degradation (Fig. 7 B, open circles), consistent with its proposed effect on transport to lysosomes (Durrbach et al., 1996). In contrast, despite its morphological effects (Fig. 6 B), BFA did not affect EGF degradation in lysosomes (Fig. 7 B, open diamonds), consistent with a study by Stoorvogel et al. (1993) that showed no significant inhibition by BFA of protein transport to lysosomes and lysosomal degradation.
B-fragment transport to the Golgi apparatus and to the ER thus seemed to be independent of the maintenance of an acidic environment in endosomes and independent of the actin cytoskeleton. In contrast, BFA induced the accumulation of the B-fragment in a tubular membrane compartment that contained the TfR, and inhibited B-Glyc-KDEL transport to the ER and B-fragment transport to Golgi remnants in Noc-treated cells.
In this context it should be mentioned that we have previously reported that in HeLa cells, radiolabeled B-fragment is not transformed into TCA soluble material (Johannes et al., 1997), suggesting that in these cells the protein is not transported to lysosomes, an observation which is consistent with the absence of colocalization with lysosomal marker proteins (Johannes et al., 1997), and with the absence of the protein from heavy fractions on Percoll gradients (not shown). Here we observed that during incubations of radiolabeled B-fragment with HeLa cells for ≤6 h in the presence of BFA, no TCA-precipitable counts appeared in the medium (not shown). Thus, even when its exit from the endosomal membrane system was blocked by BFA (see above), B-fragment did not “diffuse” to degradative (late) compartments of the endocytic pathway, despite that this transport route was still open for other proteins, e.g., EGF.
In another experiment, BFA was added at various times after the initiation of B-Glyc-KDEL internalization from the plasma membrane by temperature shift to 37°C (Fig. 7 C). It was found that when added after 90 min, the drug no longer had an effect on glycosylation. The t 1/2 of inhibition was 30 min (Fig. 7 C), in good agreement with the above described kinetics of B-fragment transport from EE/RE to the Golgi apparatus (see Fig. 3 B; note that in Fig. 7 C, transport is sampled from the plasma membrane). These data thus suggest a correlation between B-fragment transport from EE/RE to the Golgi apparatus and the passage of the BFA-sensitive transport step.
B-fragment Colocalizes with γ-Adaptin on Membranes of Early/Recycling Endosomes
The BFA-induced inhibition of B-fragment exit from TfR-positive membrane compartments suggested that ADP ribosylation factor–sensitive vesicular coats could play a role in this process (Donaldson et al., 1992; Helms and Rothman, 1992). We thus set out to study the distribution of vesicular coat proteins on endosomes. Different vesicular coats have been localized on endosomes, among the best studied are coatomer type I–like coats and clathrin coats (Louvard et al., 1983; Takei et al., 1995; Whitney et al., 1995; Aniento et al., 1996; Stoorvogel et al., 1996; Daro et al., 1997; Gu et al., 1997). We here made the observation that during a 1-h incubation at 19.5°C, a component of AP-1– type clathrin coats, γ-adaptin, was shifted from its localization at steady state in the TGN to B-fragment containing EE/RE (Fig. 8, A and B). If the cells were then incubated at 37°C for 10 min (not shown) or 30 min (Fig. 8, C and D), γ-adaptin reassociated with TGN membranes while the B-fragment accumulated in the TGN/Golgi apparatus.
Figure 8.
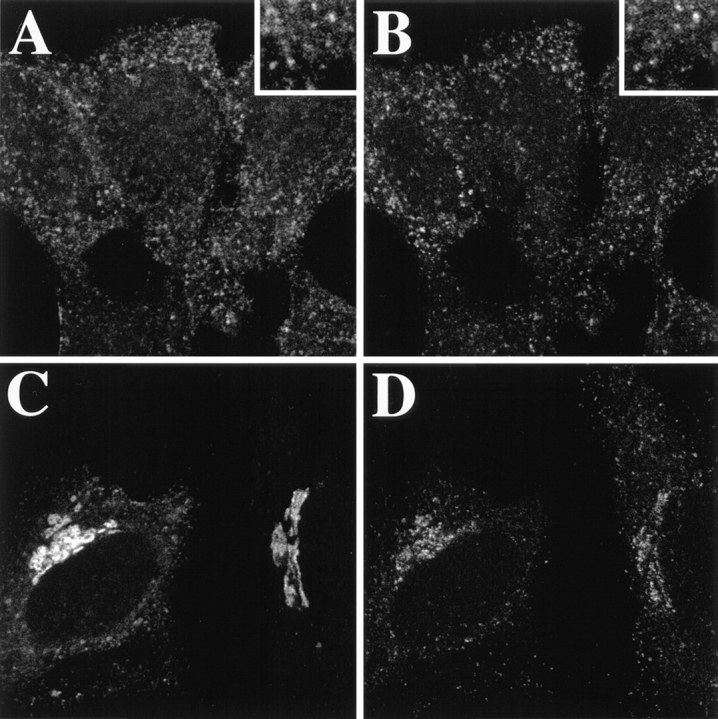
At 19.5°C, the γ-adaptin subunit of AP-1–type clathrin coats is relocalized from the TGN to EE/RE. Fluorophore-labeled B-fragment (A and C) was internalized for 1 h into HeLa cells at 19.5°C. The cells were then either fixed directly (A and B), or shifted for 30 min to 37°C (C and D) before fixation and staining for γ-adaptin (B and D). Note that at 19.5°C, γ-adaptin and B-fragment colocalized on EE/RE (A and B), and after shift to 37°C, γ-adaptin was relocalized to the TGN, in parallel with B-fragment accumulation in Golgi cisternae (C and D).
Cryosections of cells that had internalized B-fragment and BSA-gold at 19.5°C and that were then shifted for various times to 37°C, were labeled with anti–γ-adaptin and anti–B-fragment antibodies. At 19.5°C, B-fragment (15-nm gold particles) and γ-adaptin (10-nm gold particles) were found on the same coated membrane profiles (50–80 nm) (Fig. 9, A–C, arrowheads). The B-fragment and γ-adaptin– labeled structures sometimes were connected with endosomal membranes (Fig. 9, B and C), while in other instances seemingly free vesicles could be detected that may represent membrane buds that were cut perpendicularly (Fig. 9, A–C). In some cases, B-fragment–positive profiles were devoid of γ-adaptin, and vice versa (Fig. 9, B and C). In the same experiments it was noted that strongly BSA-gold–positive structures (bulk fluid phase) were devoid of γ-adaptin staining (not shown), suggesting that γ-adaptin localization on subdomains of EE/RE at 19.5°C was a specific event. Investigations using antibodies to clathrin (10-nm gold particles) combined with B-fragment (15-nm gold particles) labeling were also carried out on the experiment described above (Fig. 9, D and E). Again, membrane profiles stained with both antibodies (Fig. 9, D and E, arrows) were observed.
As described above, B-fragment colocalized extensively with Tf if incubated with HeLa cells at 19.5°C (see Fig. 1). We therefore investigated whether both molecules would also be found together in γ-adaptin/clathrin–coated membrane profiles. HeLa cells were incubated with B-fragment and Tf coupled to HRP (Tf-HRP) at 19.5°C for 1 h. Tf-HRP, which is transported as Tf (see for example Stoorvogel et al., 1996), was chosen since good anti-HRP antibodies exist. Cryosections from these cells were triple-labeled for B-fragment (15-nm gold particles), Tf-HRP (10-nm gold particles), and γ-adaptin (5-nm gold particles) (Fig. 9, F–I). Again, B-fragment was found in colocalization with γ-adaptin, and some of the γ-adaptin–positive membrane profiles were also labeled for Tf-HRP, while in other cases triple-labeled profiles were detected (Fig. 9, F–I, arrowheads). Quantification of double- and triple-labeled sections showed that 9.7% of total internalized B-fragment and 13.2% of total internalized Tf-HRP colocalized with γ-adaptin (Table I). Despite their comparable presence in γ-adaptin positive structures, B-fragment and Tf-HRP were found to have significantly different distributions in these structures (Fig. 10). Most B-fragment gold particles that colocalized with γ-adaptin were found in structures that were not labeled for Tf-HRP (66%), while in the case of Tf-HRP most gold particles that colocalized with γ-adaptin were found in triple labeled structures (58%) (Fig. 10 A). The same differences were observed when γ-adaptin– positive structures were analyzed for the presence or absence of B-fragment or Tf-HRP (Fig. 10 B). Significantly more γ-adaptin–positive structures were labeled for only B-fragment (30%), when compared with γ-adaptin–positive structures labeled for Tf-HRP only (17.1%).
In contrast to B-fragment and Tf-HRP, only a small percentage of internalized BSA-gold was present in γ-adaptin–positive structures (0.27%) (Table I), consistent with earlier studies which showed that fluid phase is basically excluded from transport to the Golgi apparatus (see Sandvig et al., 1994 and references therein), and that even at 37°C only a small fraction of fluid phase is recycled (Goud et al., 1984).
TGN38 and the B-fragment Colocalize during Transport to the Golgi Apparatus
To test whether a cellular protein could follow the same route as the B-fragment, B-fragment was incubated at 37°C with HeLa cells in the presence of antibodies directed either to the CI-MPR or to rat TGN38 (Fig. 11). The CI-MPR and TGN38 both cycle between endosomes and the TGN (Humphrey et al., 1993; Reaves et al., 1993; Chapman and Munro, 1994; Ponnambalam et al., 1994; Munier-Lehmann et al., 1996). The antibodies were added at concentrations at which fluid phase internalization was not detectable (see Materials and Methods). After increasing periods of time, the cells were washed, fixed, and then processed for immunofluorescence. At 10 min, colabeling was found for TGN38 and B-fragment in vesicular structures at the level of EE of rat TGN38-expressing HeLa cells (Fig. 11 A, top row). After 30 min (not shown) or 60 min at 37°C (Fig. 11 A, bottom row), both proteins arrived in the Golgi apparatus. They labeled overlapping structures with subtle differences that may have resulted from B-fragment transport into the cisternae of the Golgi apparatus, while TGN38 remained confined to the TGN. In accordance with the hypothesis that TGN38 and the B-fragment could both follow the same route from EE/RE to the Golgi apparatus it was also observed that after BFA treatment, both proteins were found in tubular elements (not shown).
Figure 11.
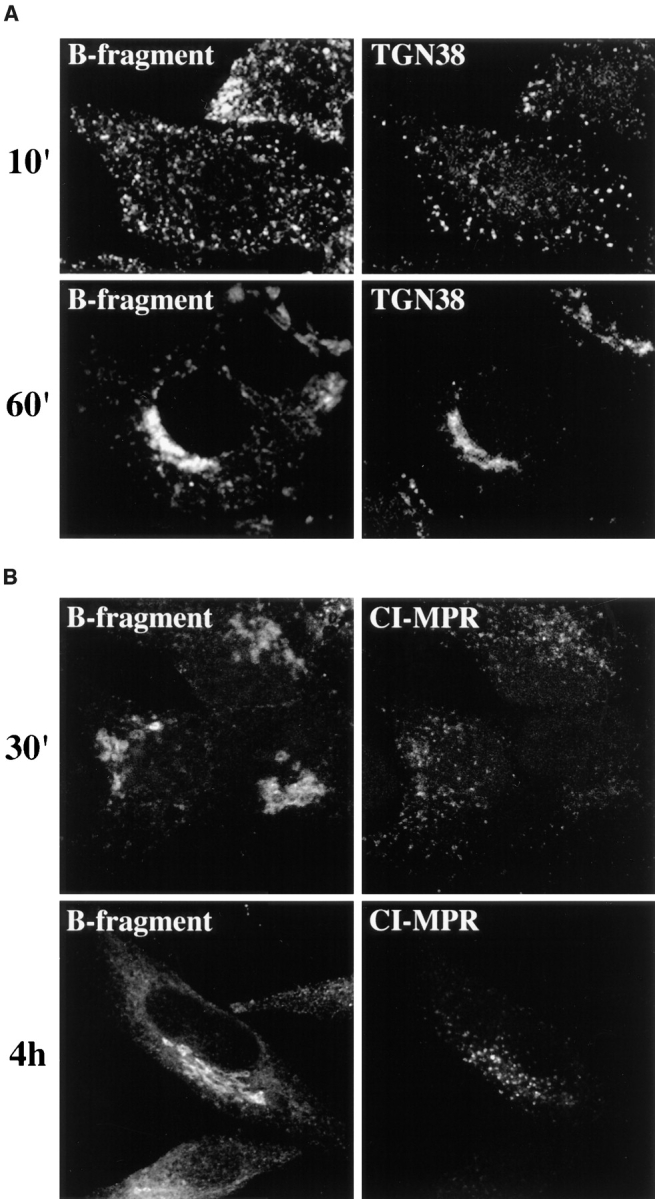
B-fragment and anti-TGN38, but not anti–CI-MPR, are found in the same structures. (A) Fluorophore-labeled B-fragment (left) and anti-TGN38 antibody (right) were bound to HeLa C7 cells on ice, upon which the cells were incubated at 37°C for 10 min (top) and 60 min (bottom) in the continued presence of both proteins. The cells were then fixed and stained for internalized anti-TGN38 antibody (right). (B) Fluorophore-labeled B-fragment (left) and anti– CI-MPR antibody (right) were bound to HeLa cells on ice, upon which the cells were incubated at 37°C for 30 min (top) and 4 h (bottom) in the continued presence of both proteins. The cells were then fixed and stained for internalized anti– CI-MPR antibody (right).
The internalized anti–CI-MPR antibody gave a distinctly different staining pattern than B-fragment. After 30 min (Fig. 11 B, top row) 1 h, 2 h (not shown), or 4 h (Fig. 11 B, bottom row) at 37°C, the antibody labeled vesicular elements that (especially after longer incubation) were concentrated in the perinuclear region consistent with LE or Golgi localization. At none of the analyzed time points, CI-MPR labeling and B-fragment–positive structures overlapped to a significant extend. These data thus suggest that B-fragment may be transported via the same pathway as TGN38 from EE/RE to the Golgi apparatus, while the CI-MPR may use a different transport route.
Discussion
In this study, we raised the question as to how Shiga toxin B-fragment is transported from the endocytic compartment to the Golgi apparatus. Our data suggest that B-fragment passes directly from EE/RE to the Golgi apparatus, circumventing the late endocytic pathway, and that in this basically unexplored transport route, B-fragment sorting at the level of EE/RE implicates AP-1–type clathrin coats.
Shiga Toxin B-fragment Transport from EE/RE to the Golgi Apparatus
To characterize the pathway followed by B-fragment between EE/RE and the Golgi apparatus, we took advantage of our finding that a temperature block at 19.5°C allowed a synchronization of B-fragment transport at the level of EE/RE. Using this temperature block protocol and subsequent shift to 37°C, the following observations were made that support the hypothesis of the existence of a direct transport pathway between EE/RE and the Golgi apparatus. First, at low temperature incubations, B-fragment was segregated from markers that were bound for LE and lysosomes, such as EGF, even if cells were incubated for prolonged periods. If the B-fragment was destined for the late endocytic pathway, one would have expected an accumulation at the entry site of this pathway, i.e., structures that were stained by EGF. Second, upon incubation at low temperatures, the fluid phase marker BSA-gold filled the membranes of EE, where it colocalized with cointernalized B-fragment. However, upon subsequent shift to 37°C, both markers efficiently separated when the bulk of BSA-gold was transferred to the late endocytic pathway, while the B-fragment was transported to the Golgi apparatus. Third, as judged from light microscopical studies, all cytoplasmic B-fragment containing structures also remained TfR positive, suggesting that on its transport to the Golgi apparatus, B-fragment did not accumulate outside EE. Electron microscopical observations further supported the idea of a direct transport route as B-fragment was not detected in multivesicular or multilamellar LE at any time point. Fourth, we show that after as little as 2 min at 37°C after the temperature block, B-fragment could be detected in the cisternae of the Golgi apparatus, which became strongly labeled after 10 min. This rapid kinetics is consistent with a direct passage from EE/RE to the Golgi apparatus. Fifth, the data presented in Fig. 3 B were fitted to an exponential raise curve with a time constant of 0.036 min−1, indicating the existence of a single rate-limiting step in EE/RE-to-Golgi transport. This observation is most easily explained if EE/RE derived transport intermediates fuse directly with the Golgi apparatus as acceptor compartment, without passing through other compartments. Sixth, drugs such as CytoD, ConB, Bafi, chloroquine, and ammonium chloride, with defined effects on the late endocytic pathway did not interfere with B-fragment transport to the Golgi apparatus and the ER. Particularly for the pH neutralizing drugs, an inhibition of the formation of LE-bound endosomal carrier vesicles (Clague et al., 1994; Aniento et al., 1996) and an inhibition of transport from LE to lysosomes (Benaroch et al., 1995; van Weert et al., 1995) has been described. Seventh, BFA was found to induce B-fragment accumulation in TfR-containing membrane tubules and also inhibited B-fragment transport to the ER. Strikingly, no B-fragment degradation (appearance of TCA-soluble material) became apparent, even after prolonged incubations at 37°C in the presence of BFA, whereas the transport route to LE/lysosomes was still open for other proteins (e.g., EGF), thus strongly suggesting that the B-fragment did not access late (degradative) compartments of the endocytic pathway. In addition, if LE were the exit point for B-fragment transport to the Golgi apparatus, one would have expected that in the presence of BFA, the protein would have accumulated in this compartment.
The existence of a direct transport route between EE/ RE and the TGN was first suggested in studies showing that small quantities of internalized Tf could be detected in the TGN (Fishman and Fine, 1987; Stoorvogel et al., 1988). Our hypothesis that protein toxins such as Shiga toxin may use this route is consistent with studies by Balch and colleagues (Simpson et al., 1995) who showed that another toxin, the plant toxin ricin, seemed to reach the Golgi apparatus even in cells that expressed a dominant-negative mutant of the small GTP-binding protein Rab9. Rab9 localizes to late endosomes and to the TGN and regulates transport of the CI-MPR, presumably from LE to the TGN (Lombardi et al., 1993; Riederer et al., 1994). One would thus have expected an effect of the dominant-negative mutant of Rab9 on ricin transport if this transport passed via LE.
The proposed B-fragment pathway from EE/RE to the Golgi apparatus may be shared by cellular proteins. We found that TGN38 colocalized with B-fragment during its transport to the TGN, and both, B-fragment and TGN38 distributions in cells were affected in the same way by BFA treatment. In addition, the kinetics of B-fragment and TGN38 transport to the Golgi apparatus were comparable. In agreement with these data, the group of F.R. Maxfield (Cornell University Medical College, New York) recently showed that an epitope-tagged version of TGN38 (Humphrey et al., 1993) is transported with a t 1/2 of 45 min from the plasma membrane to the TGN in CHO cells, with first TGN38-label appearing in the TGN after only 10 min (see Note Added in Proof). Such kinetics compares well with the t 1/2 of 19 min determined in this study for B-fragment transport from EE/RE to the TGN/Golgi apparatus, especially when it is considered that 80% of internalized TGN38 were recycled to the plasma membrane, thus inducing a delay with respect to Golgi transport. In contrast to TGN38, there was little colocalization between the CI-MPR and B-fragment in endosomes. These data suggest that the CI-MPR may follow a different transport route from endosomes to the Golgi apparatus, e.g., from LE. In this context it should also be added that the kinetics of B-fragment and CI-MPR transport to the TGN appears to be quite different. Whereas the t 1/2 for B-fragment is 19 min, resialylation in the TGN of CI-MPR that has been desialylated at the plasma membrane (thus sampling plasma membrane to TGN transport) occurs with a t 1/2 of 1–2 h (Jin et al., 1989).
The suggestion of a common pathway for both, B-fragment and TGN38 is in apparent contradiction with the fact that B-fragment transport to the Golgi apparatus (and the ER) is not affected by chemicals that raise endosomal and Golgi pH, while TGN38 has been reported to be retained in endosomes under similar conditions (Chapman and Munro, 1994; Reaves and Banting, 1994a ). It is possible that the transmembrane protein TGN38 relies on a sorting mechanism for Golgi transport whose activity is sensitive to luminal pH changes, while the glycolipid binding B-fragment would follow the flow of lipids which may be governed by different (pH insensitive) mechanisms, such as sorting into specialized environments in endosomes, as suggested for GPI-anchored proteins (Maxfield and Mayor, 1997). It should also be mentioned that on HeLa cells transfected with rat TGN38, we did not detect a major effect of Bafi and combined Bafi and chloroquine treatments on anti-TGN38 transport to the TGN (not shown). This divergence with the above mentioned studies (Chapman and Munro, 1994; Reaves and Banting, 1994a ) may be attributed to cell type differences.
Sorting of B-fragment into Golgi Transport Intermediates
Another important finding of this study is the observation that during incubation at 19.5°C, B-fragment and EGF were localized to juxtaposed structures suggesting that both proteins partitioned into separate compartments or into subdomains of the same compartment, i.e., sorting endosomes. In fact, it has been proposed that sorting at the level of sorting endosomes may occur through the formation of subdomains with high or low membrane-to-volume ratios (Geuze et al., 1983, 1987), the former being of tubular morphology and enriched in proteins and lipids destined for the recycling pathway, while the latter are of vesicular morphology and enriched in proteins and lipids destined for the late endosomal pathway (for review see Gruenberg and Maxfield, 1995; Mukherjee et al., 1997). Through such physical separation, enrichment of the respective cargo would be achieved. A prediction from this model is that a fluid phase marker should be mostly enriched in subdomains of sorting endosomes that contain proteins destined for late endosomes (bulk fluid phase domains). Using Dex3 and BSA-gold as fluid phase markers, we actually observed that the bulk of these markers segregated from B-fragment, but not from EGF.
In addition to the described partition mechanism, which may be especially important at the level of endosomes, it has been demonstrated that protein coats play a general role in sorting (Rothman and Wieland, 1996; Schekman and Orci, 1996). Different subunits of clathrin and coatomer protein coats have been detected on endosomes by morphological and biochemical means (Louvard et al., 1983; Takei et al., 1995; Whitney et al., 1995; Aniento et al., 1996; Stoorvogel et al., 1996; Daro et al., 1997; Gu et al., 1997). Here we have found that: (a) As shown for the TGN (Matlin and Simons, 1983; Brown et al., 1995), the temperature block at 19.5°C also prevented exit from EE/RE (B-fragment). (b) In the presence of BFA, B-fragment remained confined to TfR-containing membrane tubules. BFA is thought to affect the function of small GTP-binding proteins of the ADP ribosylation factor family (Donaldson et al., 1992; Helms and Rothman, 1992), and a role of ADP ribosylation factors in the formation of AP-1 vesicles has been suggested (Robinson and Kreis, 1992; Stamnes and Rothman, 1993; Traub et al., 1993; Le Borgne et al., 1996; Salamero et al., 1996; Le Borgne and Hoflack, 1997). In addition, AP-1 has been shown to bind to a Tyr-containing signal in the cytoplasmic tail of TGN38 (Reaves and Banting, 1994b ; Ohno et al., 1995). (c) During low temperature incubations (19.5°C), the γ-adaptin subunit of AP-1 was relocalized from the TGN to EE/RE where it colocalized at the light microscopical level with the B-fragment. (d) Our electron microscopical data show that γ-adaptin colocalized with the B-fragment in clathrin-coated membrane profiles on EE/RE. 47% of all γ-adaptin–positive structures were also labeled with B-fragment, and 9.7% of all internalized B-fragment was found in γ-adaptin–positive coated membrane profiles. This figure compares well with data on receptor concentration in coated pits at the plasma membrane, e.g., the Fc-receptor was found to localize at 8% in coated pits (Miettinen et al., 1992), and the TfR at ∼10% (Hansen et al., 1992, and references therein). Importantly, B-fragment accumulation in γ-adaptin–positive coated pits was much more pronounced than that of BSA-gold (33-fold), further suggesting that γ-adaptin–positive structures play a role in B-fragment sorting.
Tf was also found to colocalize with γ-adaptin in coated membrane profiles on endosomes. This observation is consistent with a recent study by Hopkins and colleagues who described the presence of the TfR in γ-adaptin–coated putative basolateral recycling intermediates in polarized cells (Futter et al., 1998). Since recycling continues at a reduced rate during low temperature incubations (Iacopetta and Morgan, 1983; van der Ende et al., 1989), B-fragment– and Tf-HRP–containing, γ-adaptin–positive membrane profiles observed at 19.5°C could correspond to recycling intermediates. However, our triple-labeling experiments further show that aside from γ-adaptin–positive structures that contain both, B-fragment and Tf-HRP, γ-adaptin– positive structures labeled for only B-fragment are significantly more abundant than γ-adaptin–positive structures labeled for Tf-HRP only. Altogether, these data directly implicate γ-adaptin in B-fragment sorting at the level of EE, and they suggest that in addition to a putative role in recycling, γ-adaptin may also participate in transport steps in which B-fragment rather than Tf is the predominant cargo molecule, i.e., transport from EE/RE to the Golgi apparatus. It may finally be noted that the B-fragment– and γ-adaptin–positive, but Tf-HRP–negative structures detected in this study by electron microscopy could correspond to the peripheral, γ-adaptin–positive, but TfR-negative membranes described by Futter et al. (1998) in immunofluorescence experiments.
Recently, Sandvig and colleagues (Llorente et al., 1998) demonstrated a role for dynamins in ricin transport from endosomes to the Golgi apparatus. Dynamins have been directly implicated through genetic, morphological, and biochemical evidence in the formation of clathrin-coated vesicles (for review see De Camilli et al., 1995; Warnock and Schmid, 1996), and a function for dynamins in transport of toxins from EE/RE to the Golgi apparatus would thus be consistent with the proposed role of clathrin coated vesicles in this pathway (this study).
The B-fragment represents an important tool to study an as yet basically unexplored transport route. Future studies will help to unravel in further detail the molecular mechanisms that underlie transport between EE/RE and the Golgi apparatus.
Acknowledgments
We thank R. Golsteyn, S. Amigorena, and P. Bénaroch for critical reading of the manuscript and D. Meur and D. Morineau for help with the photographic work.
This study was supported by grants from the European Union (ERB FMRX CT 96 0020) and Human Frontier Science Program to B. Goud, and from the Ligue Nationale Contre le Cancer to L. Johannes and C. Antony.
Abbreviations used in this paper
- AP-1
adaptor protein type 1
- Bafi
bafilomycin A
- BFA
brefeldin A
- BSA-gold
BSA coupled to gold particles
- CI-MPR
cation-independent mannose 6-phosphate receptor
- CytoD
cytochalasin D
- Dex3
dextran of 3 kD
- EE
early endosomes
- LE
late endosomes
- MPR46
mannose 6-phosphate receptor of 46 kD
- Noc
nocodazole
- RE
recycling endosomes
- Tf
transferrin
- TfR
transferrin receptor
Note Added in Proof
While this article was in press, the work by F.R. Maxfield and colleagues on TGN38 transport through the endocytic membrane system has been published (Ghosh, R.N., W.G. Mallet, T.T. Soe, T.E. McGraw, and F.R. Maxfield). 1998. J. Cell Biol. 142:923–936.
References
- Alami M, Taupiac M-P, Reggio H, Bienvenüe A, Beaumelle B. Involvement of ATP-dependent Pseudomonas exotoxin translocation from a late recycling compartment in lymphocyte intoxication procedure. Mol Biol Cell. 1998;9:387–402. doi: 10.1091/mbc.9.2.387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aniento F, Gu F, Parton RG, Gruenberg J. An endosomal beta COP is involved in the pH-dependent formation of transport vesicles destined for late endosomes. J Cell Biol. 1996;133:29–41. doi: 10.1083/jcb.133.1.29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benaroch P, Yilla M, Raposo G, Ito K, Miwa K, Geuze HJ, Ploegh HL. How MHC class II molecules reach the endocytic pathway. EMBO (Eur Mol Biol Organ) J. 1995;14:37–49. doi: 10.1002/j.1460-2075.1995.tb06973.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown WJ, DeWald DB, Emr SD, Plutner H, Balch WE. Role for phosphatidylinositol 3-kinase in the sorting and transport of newly synthesized lysosomal enzymes in mammalian cells. J Cell Biol. 1995;130:781–796. doi: 10.1083/jcb.130.4.781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chapman RE, Munro S. Retrieval of TGN proteins from the cell surface requires endosomal acidification. EMBO (Eur Mol Biol Organ) J. 1994;13:2305–2312. doi: 10.1002/j.1460-2075.1994.tb06514.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clague MJ, Urbe S, Aniento F, Gruenberg J. Vacuolar ATPase activity is required for endosomal carrier vesicle formation. J Biol Chem. 1994;269:21–24. [PubMed] [Google Scholar]
- Daro E, Sheff D, Gomez M, Kreis T, Mellman I. Inhibition of endosome function in CHO cells bearing a temperature-sensitive defect in the coatomer (COPI) component epsilon-COP. J Cell Biol. 1997;139:1747–1759. doi: 10.1083/jcb.139.7.1747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Camilli P, Takei K, McPherson PS. The function of dynamin in endocytosis. Curr Opin Neurobiol. 1995;5:559–565. doi: 10.1016/0959-4388(95)80059-x. [DOI] [PubMed] [Google Scholar]
- Donaldson JG, Finazzi D, Klausner RD. Brefeldin A inhibits Golgi membrane-catalysed exchange of guanine nucleotide onto ARF protein. Nature. 1992;360:350–352. doi: 10.1038/360350a0. [DOI] [PubMed] [Google Scholar]
- Durrbach A, Louvard D, Coudrier E. Actin filaments facilitate two steps of endocytosis. J Cell Sci. 1996;109:457–465. doi: 10.1242/jcs.109.2.457. [DOI] [PubMed] [Google Scholar]
- Fishman JB, Fine RE. A trans Golgi-derived exocytic coated vesicle can contain both newly synthesized cholinesterase and internalized transferrin. Cell. 1987;48:157–164. doi: 10.1016/0092-8674(87)90366-7. [DOI] [PubMed] [Google Scholar]
- Futter CE, Gibson A, Allchin EH, Maxwell S, Ruddock LJ, Odorizzi G, Domingo D, Trowbridge IS, Hopkins CR. In polarized MDCK cells basolateral vesicles arise from clathrin-gamma-adaptin-coated domains on endosomal tubules. J Cell Biol. 1998;141:611–623. doi: 10.1083/jcb.141.3.611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geuze HJ, Slot JW, Strous GJ, Lodish HF, Schwartz AL. Intracellular site of asialoglycoprotein receptor-ligand uncoupling: double-label immunoelectron microscopy during receptor-mediated endocytosis. Cell. 1983;32:277–287. doi: 10.1016/0092-8674(83)90518-4. [DOI] [PubMed] [Google Scholar]
- Geuze HJ, Slot JW, Schwartz AL. Membranes of sorting organelles display lateral heterogeneity in receptor distribution. J Cell Biol. 1987;104:1715–1723. doi: 10.1083/jcb.104.6.1715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goud B, Jouanne C, Antoine J-C. Reversible pinocytosis of horseradish peroxidase in lymphoid cells. Exp Cell Res. 1984;153:218–235. doi: 10.1016/0014-4827(84)90463-4. [DOI] [PubMed] [Google Scholar]
- Gruenberg J, Maxfield FR. Membrane transport in the endocytic pathway. Curr Opin Cell Biol. 1995;7:552–563. doi: 10.1016/0955-0674(95)80013-1. [DOI] [PubMed] [Google Scholar]
- Gu F, Aniento F, Parton RG, Gruenberg J. Functional dissection of COP-I subunits in the biogenesis of multivesicular endosomes. J Cell Biol. 1997;139:1183–1195. doi: 10.1083/jcb.139.5.1183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansen SH, Sandvig K, van Deurs B. Internalization efficiency of the transferrin receptor. Exp Cell Res. 1992;199:19–28. doi: 10.1016/0014-4827(92)90457-j. [DOI] [PubMed] [Google Scholar]
- Helms JB, Rothman JE. Inhibition by brefeldin A of a Golgi membrane enzyme that catalyses exchange of guanine nucleotide bound to ARF. Nature. 1992;360:352–354. doi: 10.1038/360352a0. [DOI] [PubMed] [Google Scholar]
- Humphrey JS, Peters PJ, Yuan LC, Bonifacino JS. Localization of TGN38 to the trans-Golgi network: Involvement of a cytoplasmic tyrosine-containing sequence. J Cell Biol. 1993;120:1123–1135. doi: 10.1083/jcb.120.5.1123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunziker W, Whitney JA, Mellman I. Selective inhibition of transcytosis by brefeldin A in MDCK cells. Cell. 1991;67:617–627. doi: 10.1016/0092-8674(91)90535-7. [DOI] [PubMed] [Google Scholar]
- Hunziker W, Whitney JA, Mellman I. Brefeldin A and the endocytic pathway. Possible implications for membrane traffic and sorting. FEBS (Fed Eur Biochem Soc) Lett. 1992;307:93–96. doi: 10.1016/0014-5793(92)80908-y. [DOI] [PubMed] [Google Scholar]
- Iacopetta BJ, Morgan EH. The kinetics of transferrin endocytosis and iron uptake from transferrin in rabbit reticulocytes. J Biol Chem. 1983;258:9108–9115. [PubMed] [Google Scholar]
- Jin M, Sahagian G, Snider MD. Transport of surface mannose 6-phosphate receptor to the Golgi complex in cultured human cells. J Biol Chem. 1989;264:7675–7680. [PubMed] [Google Scholar]
- Johannes L, Goud B. Surfing on a retrograde wave: how does Shiga toxin reach the endoplasmic reticulum? . Trends Cell Biol. 1998;8:158–162. doi: 10.1016/s0962-8924(97)01209-9. [DOI] [PubMed] [Google Scholar]
- Johannes L, Tenza D, Antony C, Goud B. Retrograde transport of KDEL-bearing B-fragment of Shiga toxin. J Biol Chem. 1997;272:19554–19561. doi: 10.1074/jbc.272.31.19554. [DOI] [PubMed] [Google Scholar]
- Kim JH, Johannes L, Goud B, Antony C, Lingwood CA, Daneman R, Grinstein S. Nonivasive measurement of the pH of the endoplasmic reticulum at rest and during calcium release. Proc Natl Acad Sci USA. 1998;95:2997–3002. doi: 10.1073/pnas.95.6.2997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klausner RD, Donaldson JG, Lippincott-Schwartz J. Brefeldin A: Insights into the control of membrane traffic and organelle structure. J Cell Biol. 1992;116:1071–1080. doi: 10.1083/jcb.116.5.1071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Borgne R, Hoflack B. Mannose 6-phosphate receptors regulate the formation of clathrin-coated vesicles in the TGN. J Cell Biol. 1997;137:335–345. doi: 10.1083/jcb.137.2.335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Borgne R, Griffiths G, Hoflack B. Mannose 6-phosphate receptors and ADP-ribosylation factors cooperate for high affinity interaction of the AP-1 Golgi assembly proteins with membranes. J Biol Chem. 1996;271:2162–2170. doi: 10.1074/jbc.271.4.2162. [DOI] [PubMed] [Google Scholar]
- Lingwood CA. Verotoxins and their glycolipid receptors. Adv Lipid Res. 1993;25:189–211. [PubMed] [Google Scholar]
- Lingwood CA. Role of verotoxin receptors in pathogenesis. Trends Microbiol. 1996;4:147–153. doi: 10.1016/0966-842x(96)10017-2. [DOI] [PubMed] [Google Scholar]
- Lippincott-Schwartz J, Donaldson JG, Schweizer A, Berger EG, Hauri HP, Yuan LC, Klausner RD. Microtubule-dependent retrograde transport of proteins into the ER in the presence of brefeldin A suggests an ER recycling pathway. Cell. 1990;60:821–836. doi: 10.1016/0092-8674(90)90096-w. [DOI] [PubMed] [Google Scholar]
- Lippincott-Schwartz J, Yuan L, Tipper C, Amherdt M, Orci L, Klausner RD. Brefeldin A's effects on endosomes, lysosomes, and the TGN suggest a general mechanism for regulating organelle structure and membrane traffic. Cell. 1991;67:601–616. doi: 10.1016/0092-8674(91)90534-6. [DOI] [PubMed] [Google Scholar]
- Llorente A, Rapak A, Schmid SL, van Deurs B, Sandvig K. Expression of mutant dynamin inhibits toxicity and transport of endocytosed ricin to the Golgi apparatus. J Cell Biol. 1998;140:553–563. doi: 10.1083/jcb.140.3.553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lombardi D, Soldati T, Riederer MA, Goda Y, Zerial M, Pfeffer SR. Rab9 functions in transport between late endosomes and the trans Golgi network. EMBO (Eur Mol Biol Organ) J. 1993;12:677–682. doi: 10.1002/j.1460-2075.1993.tb05701.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Louvard D, Morris C, Warren G, Stanley K, Winkler F, Reggio H. A monoclonal antibody to the heavy chain of clathrin. EMBO (Eur Mol Biol Organ) J. 1983;2:1655–1664. doi: 10.1002/j.1460-2075.1983.tb01640.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matlin KS, Simons K. Reduced temperature prevents transfer of a membrane glycoprotein to the cell surface but does not prevent terminal glycosylation. Cell. 1983;34:233–243. doi: 10.1016/0092-8674(83)90154-x. [DOI] [PubMed] [Google Scholar]
- Maxfield FR, Mayor S. Cell surface dynamics of GPI-anchored proteins. Adv Exp Med Biol. 1997;419:355–364. doi: 10.1007/978-1-4419-8632-0_47. [DOI] [PubMed] [Google Scholar]
- Mellman I. Endocytosis and molecular sorting. Annu Rev Cell Dev Biol. 1996;12:575–625. doi: 10.1146/annurev.cellbio.12.1.575. [DOI] [PubMed] [Google Scholar]
- Miettinen HM, Matter K, Hunziker W, Rose JK, Mellman I. Fc receptor endocytosis is controlled by a cytoplasmic domain determinant that actively prevents coated pit localization. J Cell Biol. 1992;116:875–888. doi: 10.1083/jcb.116.4.875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molloy SS, Thomas L, VanSlyke JK, Stenberg PE, Thomas G. Intracellular trafficking and activation of the furin proprotein convertase: localization to the TGN and recycling from the cell surface. EMBO (Eur Mol Biol Organ) J. 1994;13:18–33. doi: 10.1002/j.1460-2075.1994.tb06231.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mukherjee S, Ghosh RN, Maxfield FR. Endocytosis. Physiol Rev. 1997;77:759–803. doi: 10.1152/physrev.1997.77.3.759. [DOI] [PubMed] [Google Scholar]
- Munier-Lehmann H, Mauxion F, Hoflack B. Function of the two mannose 6-phosphate receptors in lysosomal enzyme transport. Biochem Soc Trans. 1996;24:133–136. doi: 10.1042/bst0240133. [DOI] [PubMed] [Google Scholar]
- Nambiar MP, Wu HC. Ilimaquinone inhibits the cytotoxicities of ricin, diphtheria toxin, and other protein toxins in Vero cells. Exp Cell Res. 1995;219:671–678. doi: 10.1006/excr.1995.1278. [DOI] [PubMed] [Google Scholar]
- O'Brien AD, Tesh VL, Donohue-Rolfe A, Jackson MP, Olsnes S, Sandvig K, Lindberg AA, Keusch GT. Shiga toxin: biochemistry, genetics, mode of action, and role in pathogenesis. Curr Top Microbiol Immunol. 1992;180:65–94. doi: 10.1007/978-3-642-77238-2_4. [DOI] [PubMed] [Google Scholar]
- Ohno H, Stewart J, Fournier MC, Bosshart H, Rhee I, Miyatake S, Saito T, Gallusser A, Kirchhausen T, Bonifacino JS. Interaction of tyrosine-based sorting signals with clathrin-associated proteins. Science. 1995;269:1872–1875. doi: 10.1126/science.7569928. [DOI] [PubMed] [Google Scholar]
- Pelham HR, Roberts LM, Lord JM. Toxin entry: how reversible is the secretory pathway? . Trends Cell Biol. 1992;2:183–185. doi: 10.1016/0962-8924(92)90230-k. [DOI] [PubMed] [Google Scholar]
- Ponnambalam S, Rabouille C, Luzio JP, Nilsson T, Warren G. The TGN38 glycoprotein contains two non-overlapping signals that mediate localization to the trans-Golgi network. J Cell Biol. 1994;125:253–268. doi: 10.1083/jcb.125.2.253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reaves B, Banting G. Vacuolar ATPase inactivation blocks recycling to the trans-Golgi network from the plasma membrane. FEBS Lett. 1994a;345:61–66. doi: 10.1016/0014-5793(94)00437-4. [DOI] [PubMed] [Google Scholar]
- Reaves B, Banting G. Overexpression of TGN38/41 leads to mislocalisation of gamma-adaptin. FEBS (Fed Eur Biochem Soc) Lett. 1994b;351:448–456. doi: 10.1016/0014-5793(94)00813-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reaves B, Horn M, Banting G. TGN38/41 recycles between the cell surface and the TGN: brefeldin A affects its rate of return to the TGN. Mol Biol Cell. 1993;4:93–105. doi: 10.1091/mbc.4.1.93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riederer MA, Soldati T, Shapiro AD, Lin J, Pfeffer SR. Lysosome biogenesis requires Rab9 function and receptor recycling from endosomes to the trans-Golgi network. J Cell Biol. 1994;125:573–582. doi: 10.1083/jcb.125.3.573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson MS, Kreis TE. Recruitment of coat proteins onto Golgi membranes in intact and permeabilized cells: effects of brefeldin A and G protein activators. Cell. 1992;69:129–138. doi: 10.1016/0092-8674(92)90124-u. [DOI] [PubMed] [Google Scholar]
- Rothman JE, Wieland FT. Protein sorting by transport vesicles. Science. 1996;272:227–234. doi: 10.1126/science.272.5259.227. [DOI] [PubMed] [Google Scholar]
- Salamero J, Le Borgne R, Saudrais C, Goud B, Hoflack B. Expression of major histocompatibility complex class II molecules in HeLa cells promotes the recruitment of AP-1 Golgi-specific assembly proteins on Golgi membranes. J Biol Chem. 1996;271:30318–30321. doi: 10.1074/jbc.271.48.30318. [DOI] [PubMed] [Google Scholar]
- Sandvig K, Olsnes S. Effect of temperature on the uptake, excretion and degradation of abrin and ricin by HeLa cells. Exp Cell Res. 1979;121:15–25. doi: 10.1016/0014-4827(79)90439-7. [DOI] [PubMed] [Google Scholar]
- Sandvig K, van Deurs B. Endocytosis, intracellular transport, and cytotoxic action of Shiga toxin and ricin. Physiol Rev. 1996;76:949–966. doi: 10.1152/physrev.1996.76.4.949. [DOI] [PubMed] [Google Scholar]
- Sandvig K, Olsnes S, Brown JE, Petersen OW, van Deurs B. Endocytosis from coated pits of Shiga toxin: A glycolipid-binding protein from Shigella dysenteriae 1. J Cell Biol. 1989;108:1331–1343. doi: 10.1083/jcb.108.4.1331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sandvig K, Garred O, Prydz K, Kozlov JV, Hansen SH, van Deurs B. Retrograde transport of endocytosed Shiga toxin to the endoplasmic reticulum. Nature. 1992;358:510–512. doi: 10.1038/358510a0. [DOI] [PubMed] [Google Scholar]
- Sandvig K, Ryd M, Garred O, Schweda E, Holm PK, van Deurs B. Retrograde transport from the Golgi complex to the ER of both Shiga toxin and the nontoxic Shiga B-fragment is regulated by butyric acid and cAMP. J Cell Biol. 1994;126:53–64. doi: 10.1083/jcb.126.1.53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schagger H, von Jagow G. Tricine-sodium dodecyl sulfate-polyacrylamide gel electrophoresis for the separation of proteins in the range from 1 to 100 kDa. Anal Biochem. 1987;166:368–379. doi: 10.1016/0003-2697(87)90587-2. [DOI] [PubMed] [Google Scholar]
- Schapiro F, Lingwood CA, Furuya W, Grinstein S. pH-independent retrograde targeting of glycolipids to the Golgi complex. Am J Physiol Cell Physiol. 1998;43:319–332. doi: 10.1152/ajpcell.1998.274.2.C319. [DOI] [PubMed] [Google Scholar]
- Schekman R, Orci L. Coat proteins and vesicle budding. Science. 1996;271:1526–1533. doi: 10.1126/science.271.5255.1526. [DOI] [PubMed] [Google Scholar]
- Simpson JC, Dascher C, Roberts LM, Lord JM, Balch WE. Ricin cytotoxicity is sensitive to recycling between the endoplasmic reticulum and the Golgi complex. J Biol Chem. 1995;270:20078–20083. doi: 10.1074/jbc.270.34.20078. [DOI] [PubMed] [Google Scholar]
- Slot JW, Geuze HJ, Gigengack S, Lienhard GE, James DE. Immuno-localization of the insulin regulatable glucose transporter in brown adipose tissue of the rat. J Cell Biol. 1991;113:123–135. doi: 10.1083/jcb.113.1.123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stamnes MA, Rothman JE. The binding of AP-1 clathrin adaptor particles to Golgi membranes requires ADP-ribosylation factor, a small GTP-binding protein. Cell. 1993;73:999–1005. doi: 10.1016/0092-8674(93)90277-w. [DOI] [PubMed] [Google Scholar]
- Stoorvogel W, Geuze HJ, Griffith JM, Strous GJ. The pathways of endocytosed transferrin and secretory protein are connected in the trans-Golgi reticulum. J Cell Biol. 1988;106:1821–1829. doi: 10.1083/jcb.106.6.1821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stoorvogel W, Oorschot V, Geuze HJ. A novel class of clathrin-coated vesicles budding from endosomes. J Cell Biol. 1996;132:21–33. doi: 10.1083/jcb.132.1.21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strous GJ, van Kerkhof P, van Meer G, Rijnboutt S, Stoorvogel W. Differential effects of brefeldin A on transport of secretory and lysosomal proteins. J Biol Chem. 1993;268:2341–2347. [PubMed] [Google Scholar]
- Takei K, McPherson PS, Schmid SL, De Camilli P. Tubular membrane invaginations coated by dynamin rings are induced by GTP-gamma S in nerve terminals. Nature. 1995;374:186–190. doi: 10.1038/374186a0. [DOI] [PubMed] [Google Scholar]
- Traub LM, Ostrom JA, Kornfeld S. Biochemical dissection of AP-1 recruitment onto Golgi membranes. J Cell Biol. 1993;123:561–573. doi: 10.1083/jcb.123.3.561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Ende A, du Maine A, Schwartz AL, Strous GJ. Effect of ATP depletion and temperature on the transferrin-mediated uptake and release of iron by BeWo choriocarcinoma cells. Biochem J. 1989;259:685–692. doi: 10.1042/bj2590685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Deurs B, Petersen OW, Olsnes S, Sandvig K. Delivery of internalized ricin from endosomes to cisternal Golgi elements is a discontinuous, temperature-sensitive process. Exp Cell Res. 1987;171:137–152. doi: 10.1016/0014-4827(87)90257-6. [DOI] [PubMed] [Google Scholar]
- van Weert AW, Dunn KW, Gueze HJ, Maxfield FR, Stoorvogel W. Transport from late endosomes to lysosomes, but not sorting of integral membrane proteins in endosomes, depends on the vacuolar proton pump. J Cell Biol. 1995;130:821–834. doi: 10.1083/jcb.130.4.821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- von Figura K. Molecular recognition and targeting of lysosomal proteins. Curr Opin Cell Biol. 1991;3:642–646. doi: 10.1016/0955-0674(91)90035-w. [DOI] [PubMed] [Google Scholar]
- Warnock DE, Schmid SL. Dynamin GTPase, a force-generating molecular switch. Bioessays. 1996;18:885–893. doi: 10.1002/bies.950181107. [DOI] [PubMed] [Google Scholar]
- Whitney JA, Gomez M, Sheff D, Kreis TE, Mellman I. Cytoplasmic coat proteins involved in endosome function. Cell. 1995;83:703–713. doi: 10.1016/0092-8674(95)90183-3. [DOI] [PubMed] [Google Scholar]
- Wood SA, Park JE, Brown WJ. Brefeldin A causes a microtubule-mediated fusion of the trans-Golgi network and early endosomes. Cell. 1991;67:591–600. doi: 10.1016/0092-8674(91)90533-5. [DOI] [PubMed] [Google Scholar]