

Abstract
The budding yeast lyt1 mutation causes cell lysis. We report here that lyt1 is an allele of cdc15, a gene which encodes a protein kinase that functions late in the cell cycle. Neither cdc15-1 nor cdc15-lyt1 strains are able to septate at 37°C, even though they may manage to rebud. Cells lyse after a shmoo-like projection appears at the distal pole of the daughter cell. Actin polarizes towards the distal pole but the septins remain at the mother–daughter neck. This morphogenetic response reflects entry into a new round of the cell cycle: the preference for polarization from the distal pole was lost in bud1 cdc15 double mutants; double cdc15-lyt1 cdc28-4 mutants, defective for START, did not develop apical projections and apical polarization was accompanied by DNA replication. The same phenomena were caused by mutations in the genes CDC14, DBF2, and TEM1, which are functionally related to CDC15. Apical polarization was delayed in cdc15 mutants as compared with budding in control cells and this delay was abolished in a septin mutant. Our results suggest that the delayed M/G1 transition in cdc15 mutants is due to a septin-dependent checkpoint that couples initiation of the cell cycle to the completion of cytokinesis.
Keywords: Saccharomyces cerevisiae, CDC15, septation, morphogenesis, checkpoint
The polarization of growth is the basis of cell morphogenesis in all eukaryotic cells, from unicellular microorganisms to specialized cells in mammalian tissues. Although it is still poorly understood at the molecular level, studies on the coordination of morphogenetic events along the cell division cycle in the budding yeast Saccharomyces cerevisiae are providing essential information in this regard. In recent years, the proteins and signals that promote the assembly of cytoskeletal structures to specific areas for the support of polarized secretion are slowly being unveiled (Cid et al., 1995; Roemer et al., 1996), as is the dependence of cytoskeletal dynamics on cell cycle signaling pathways (Lew and Reed, 1995). The cortical actin cytoskeleton assembles in the submembrane at growing areas and seems to be essential for the development of a functional cell wall (Drubin et al., 1993; Gabriel and Kopecká, 1995; Botstein et al., 1997). The location of the actin cytoskeleton in particular areas at different stages of cell cycle depends on signals elicited by the association and dissociation of the cell cycle-dependent protein kinase (CDK)1 Cdc28p with G1 or B cyclins. The nature of the signals that connect CDK-dependent phosphorylation to the activation of actin-anchorage sites at budding sites or at the septum area during cytokinesis is not understood, but Lew and Reed (1993) have shown that alterations in the timing of the Cdc28–G1 cyclin and Cdc28–B cyclin complexes lead to morphogenetic defects due to inaccurate cortical actin dynamics.
Cell wall deposition is probably supported by polarized secretion of β-glucan and chitin precursors to the growing regions, coupled with a local activation of the biosynthetic machinery. Besides actin, another cytoskeletal structure plays a crucial role in the development of cell shape along the mitotic cycle: a ring encircling the septum area that is essential for the localization of chitin to the mother–bud neck, an area of the cell wall very rich in this polymer, and the localization of the septum (De Marini et al., 1997). This ring comprises the septins Cdc3p, Cdc10p, Cdc11p, and Cdc12p, four homologous proteins that assemble in an interdependent fashion (Longtine et al., 1996). A dysfunction in any of these genes causes a cytokinetic defect and additional morphogenetic problems due to actin hyperpolarization (Adams and Pringle, 1984), reminiscent of those observed when the complex Cdc28p–G1 cyclin is hyperactivated or the Cdc28–B cyclin complex is inactivated (Lew and Reed, 1993). In sum, along the mitotic cycle actin locates to growing areas supporting polarized secretion while the septins remain attached to the bud neck, probably contributing to both the localization of chitin synthesis and to the arrangement of actin itself during bud emergence and septum formation. These hypotheses, however, await experimental confirmation.
Overexpression of B-cyclins, such as the one encoded by the CLB2 gene, causes an arrest of nuclear division in the anaphase stage (Surana et al., 1993) and, at morphogenetic level, the inability of cells to locate the actin cytoskeleton at the septum area (Lew and Reed, 1993). These observations indicate that B cyclin degradation, and hence inactivation of the CDK–B cyclin complex, is essential for the progression of late mitotic events, including septum formation. At least four other protein kinases are essential or important for the accomplishment of the M phase: Dbf2p, Dbf20p, Cdc15p, and Cdc5p (Johnston et al., 1990; Schweitzer and Philippsen, 1991; Kitada et al., 1993; Toyn et al., 1994), perhaps either directly or indirectly, by promoting B cyclin degradation (Surana et al., 1993; Toyn et al., 1996; Jaspersen et al., 1998). Conditional mutations in the genes encoding these kinases cause an M phase arrest at the restrictive temperature. Other genes which lead to this phenotype when mutated, and thus presumably also involved in the mechanisms responsible for the progression of mitosis are CDC14 and TEM1, coding respectively for a dual specific protein phosphatase (Wan et al., 1992; Taylor et al., 1997) and a putative small GTPase (Shirayama et al., 1994). Genetic interactions among these genes point to the existence of an interesting network (Kitada et al., 1993; Shirayama et al., 1996; Jaspersen et al., 1998), but so far the complexity of this network has not allowed workers to elucidate a pathway based on epistasis.
The study of autolytic mutants is a classic approach to address the cellular functions related to cell integrity, such as those that regulate cell wall dynamics and morphogenesis in response to cell cycle signals. In a screening for temperature-sensitive fragile mutants, Cabib and Durán (1975) isolated the lyt1 mutant. At the restrictive temperature of 37°C, strains bearing this mutation display cell lysis, defects in polarity and a cdc-like phenotype, as described by Molero et al. (1993). In this work we show that the lyt1 mutation is allelic to the CDC15 gene. In contrast to the clear cell cycle-arrest phenotype so far reported for cdc15 mutants, we offer evidence that cell lysis appears as a consequence of an abnormal morphogenetic response that takes place when late M phase–arrested cells commit a new round of the cell cycle in the absence of cytokinesis. Such a response involves an incorrect pattern of polarization of the actin and septin cytoskeletal structures. Based on genetic evidence, we also discuss the possibility that these events require the overcoming of a novel septin-dependent checkpoint that would prevent the cell from initiating a new round in the cell cycle in the absence of cytokinesis.
Materials and Methods
Strains, Media, and Culture Conditions
The S. cerevisiae strains used in this work are listed in Table I. DR1 strain was constructed by mating the RH210-3c strain to the 1784 strain, segregating the resulting diploid, and mating again two segregants of opposite mating type from this experiment that bore the cdc15 mutation. MY2, and MY3 diploid strains were constructed by mating the L2C24d and RH210-3c strains, but for MY2 and MY3 the parental L2C24d had been previously transformed with the pBS9 and the pLA10 plasmids, respectively, and diploid strains that maintained the plasmid were selected at 37°C. The strain DJY4 was constructed by mating the L2C24d strain to itself after expression of the HO gene included in the YCp50-pGAL/HO plasmid (provided by A. Aguilera, Seville University, Seville, Spain). Other strains were constructed by standard genetic means, as described briefly in Results. Unless specified, yeast genetic procedures and media were as described elsewhere (Ausubel et al., 1993). For general purposes, cells were grown in 250-ml flasks containing 50 ml or culture tubes containing 5 ml of YPD (2% wt/vol, glucose; 2%, wt/vol peptone; 1% yeast extract) or SD (1.7 g/liter Difco nitrogen base without amino acids; 0.5% wt/vol ammonium sulfate; 2% wt/vol glucose) plus the required mixture of amino acids for plasmid maintenance. For induction of the GAL1 promoter, cells were grown in media containing 2% raffinose instead of glucose and then transferred to media containing 1% raffinose plus 2% galactose when induction was required. Growth temperatures were 24°C for general purposes and 37°C for the expression of the lytic phenotype in Ts− strains. The strain of Escherichia coli used for cloning procedures was DH5α (Sambrook et al., 1989).
Table I.
S. cerevisiae Strains Used in This Work
| Strain | Genotype | Source | ||
|---|---|---|---|---|
| L2C24d | MATα cdc15-lyt1 ura3-52 | Molero et al., 1993 | ||
| JJY0-3c | MAT a cdc15-lyt1 ura3-52 leu2-3,112 trp1-1 his4 | This work | ||
| DU3 | MAT a/α cdc15-lyt1/cdc15-lyt1 ura3-52/ura3-52 | Molero et al., 1993 | ||
| RH210-3c | MAT a cdc15-1 ade2-1 his4 leu2-3,112 trp1-1 ura1 | C. Kuhre (Biozentrum, Basel, Switzerland) | ||
| MY1 | MAT a/MATα cdc15-1/cdc15-lyt1 | This work | ||
| MY2 | MAT a/MATα cdc15-1/cdc15-lyt1 + pBS9 | This work | ||
| MY3 | MAT a/MATα cdc15-1/cdc15-lyt1 + pLA10 | This work | ||
| L119-7d | MATα dbf2 ura3-52 trp11,2 ade1 | L. Johnston (National Institute for Medical Research, London, UK) | ||
| EO156 | MAT a tem1-3 ura3-52 leu2-3,112 his3 trp1-1 | A. Toh-e (Tokyo University, Tokyo, Japan) | ||
| RAY-3a | MAT a ura3-52 leu2-3,112 his3 trp1-1 | A. Toh-e (Tokyo University, Tokyo, Japan) | ||
| RH1779 | MAT a cdc14-1 ade2 his4 leu2 bar1 | C. Kuhre | ||
| JC223 | MATα bud1::URA3 leu2-3,112 ura3-52 | J. Chant (Harvard University, Cambridge, MA) | ||
| K1414 | MAT a cdc28-4 ura3-52 leu2-3 ade2-1 | C. Kuhre | ||
| VCY1 | MATα cdc10-11 ura3-52 leu2-3,112 trp1-1 his4 canR | Cid et al., 1998a | ||
| VCY242d | cdc10-11 cdc15-1 ura3-52 trp1 his3 leu2 | This work | ||
| DR1 | MAT a/α cdc15-1/cdc15-1 ade2/ade2 | This work | ||
| DJY4 | MAT a/α cdc15-lyt1/cdc15-lyt1 ura3-52/ura3-52 | This work | ||
| JJY1 | bud1::URA3 cdc15-1 leu2-3,112 ura3-52 | This work | ||
| JJY2 | bud1::URA3 cdc15-lyt1, ura3-52 | This work | ||
| JJY3 | cdc15-1 leu2-3,112 ura3-52 | This work | ||
| JJY4 | cdc15-lyt1 ura3-52 | This work | ||
| 1783 | MAT a leu2-3,112 trp1-1 ura3-52 his4 canR | D. Levin (Johns Hopkins University, Baltimore, MD) | ||
| 1784 | MATα leu2-3,112 trp1-1 ura3-52 his4 canR | D. Levin | ||
| YPH499 | MAT a ura3 leu2 his3 trp1 lys2 ade2 | H. Martín (Complutense University, Madrid, Spain) | ||
| TD28 | MAT a ura3-52 ino1-131 canR | H. Martín | ||
| 4965-3a | MAT a cdc16 ura3 leu2 his7 canR | L. Hartwell (University of Washington, Seattle, WA) | ||
| 9002 | MAT a cdc27 ade1 ade2 his7 lys2 tyr1 ura1 | L. Hartwell | ||
| 4086-23-2a | MAT a cdc23 ura3 leu2 his7 | L. Hartwell |
Analysis of Cell Lysis
For the expression of both lytic and morphogenetic phenotypes of Ts− mutants, yeast preinocula grown for 16 h at 24°C in YPD medium were used to inoculate two flasks containing fresh YPD medium that were immediately incubated at 24° and 37°C. Unless specified, the preinocula were at mid-log phase. Samples were taken at different intervals of time and the percentage of lysed cells was determined by staining with propidium iodide and subsequent fluorescence microscopy or FACS® analysis (see below).
DNA Manipulations
Except where specified, standard procedures were used for DNA manipulations (Sambrook et al., 1989). Restriction endonucleases and T4 DNA ligase were from Boehringer Mannheim (Indianapolis, IN). Yeast transformations were performed by the lithium acetate procedure (Ito et al., 1983). For DNA sequencing, fragments were subcloned in pBluescriptII KS and SK plasmids from Stratagene and single stranded DNA was sequenced by the Sanger technique (Sanger et al., 1977) using an ALF DNA sequencer from Pharmacia Biotech (Piscataway, NJ). The pJJ1 plasmid was constructed by cloning the 4-kb PvuII-PvuII fragment, containing the CDC15 gene from plasmid pBS9 (Schweitzer and Phillippsen, 1991), in the episomic vector YEp352 at the NruI site.
Cloning of the CDC15 Gene
Strain L2C24d was transformed with a genomic library constructed by ligating 10–15-kb DNA fragments from S. cerevisiae strain AB320 into the BamHI site of the centromeric vector YCp50 (Rose et al., 1987). Approximately 7,000 transformants were screened, allowing the isolation of 14 clones that were able to grow at the restrictive temperature of 37°C. After plasmid extraction and retransformation, three of the plasmids, namely YCp50-T2, YCp50-T7, and YCp50-T12, were able to reproduce complementation of the lytic phenotype. Restriction analyses revealed that YCp50-T2 and YCp50-T12 were identical, so only YCp50-T2 and YCp50-T7 were further analyzed. YCp50-T7 contained an insert of ∼10 kb whereas the insert in YCp50-T2 seemed to be slightly shorter. A NruI-XbaI subclone of 5.5 kb from YCp50-T7 was still able to complement the lytic phenotype. Parallel restriction analyses of this subclone and the insert in YCp50-T7 led to the conclusion that they had a common XhoI-XhoI band of ∼1.1 kb that was essential for complementation. Insertion of this DNA fragment from the YCp50-T7 clone into a KS Bluescript vector and subsequent sequencing and comparison with databases using the FASTA program (Ebi, UK) revealed that it belonged to the open reading frame (ORF) of the CDC15 gene. To verify that the CDC15 gene was included in the YCp50-T2 and YCp50-T7 vectors, a 2.7-kb probe from the CDC15-containing plasmid pBS9 (Schweitzer and Philippsen, 1991) was hybridized in a Southern blot experiment against SalI/ClaI-digested YCp50-T2, YCp50-T7, and pBS9 (control) plasmids.
Rescue of the cdc15-lyt1 Allele and Mapping of the Mutation
Due to the length of the CDC15 ORF (2,922 bp), we devised a PCR strategy so that the gene could be amplified in three individual fragments for its analysis: (a) roughly the 3′ region, (b) an intermediate region, and (c) the 5′ region. Genomic DNA from strain L2C24d was used as template for the PCR reactions.
The 3′ region was amplified with the help of two oligonucleotides, one containing the HindIII site at position 1,930 of the ORF (5′-GTACTTCACCAAGCTTT-3′) and the other bearing an artificial HindIII site (5′-GGGAAGCTTAAGACTGTGCCACTGC-3′), which hybridized 300 bases downstream from the STOP codon. The 1.3-kb amplified DNA fragment was inserted in HindIII-cleaved vectors pBS9 (Schweitzer and Philippsen, 1991) and pJJ1 (this work) which are centromeric and episomal, respectively, substituting the equivalent fragment of the CDC15 gene. After checking the correct orientation, both plasmids containing the chimeric gene were transformed into strain L2C24d and verified for complementation. Both were able to complement a cdc15-lyt1 mutation as efficiently as the control pBS9 and pJJ1 plasmids, suggesting that the mutation was not included within the 3′ third of the gene.
The 5′ third of the gene was also amplified with two oligonucleotides (upper: 5′-GGAGGCTCGAGGAAGGA-3′; lower: 5′-CAGCGATGTTCTCGAGGG-3′) that included the XhoI recognition sites lying respectively in the promoter (56-bp upstream from the ATG) and at position 1,084 of the ORF. The PCR product was cloned in the pT7Blue vector and divided into two subfragments of 0.4 and 0.7 kb by XhoI-SacI cleavage. Subcloning of these fragments in the pBluescript SK vector allowed their sequencing. No differences were detected with respect to the previously reported CDC15 sequence (Schweitzer and Phillipsen, 1991).
To analyze the remaining region of the gene, a third PCR strategy was devised. The upper primer (5′-GGGAAGCTTTTCAGTGTTTGCT-3′) had a tail with a HindIII restriction site attached to the 5′-end and hybridized from base 1,065 of the ORF, whereas the lower primer (5′-GGGAAGCTTGTACTGTAAGGTAAC-3′) hybridized at position 1,950 and included the HindIII site at position 1,928. These primers were designed by flanking the target sequences of the oligonucleotides used in the above amplifications in order to include those regions within the new amplified fragment. The HindIII targets allowed the insertion of the PCR product in the pBluescript SK vector for subsequent sequencing, which revealed the presence of a single point mutation (see Results). To verify that this substitution indeed determined the cdc15-lyt1 mutation and was not a mistake introduced by the Taq-polymerase used during the amplification, we sequenced the products of three different PCR reactions and confirmed that they all carried the same substitution.
Staining Procedures
For nucleus staining, cells were fixed and permeated by 5 min of treatment at 4°C with 70% ethanol. They were then resuspended in 400 μl of 10 g/liter RNase A (Boehringer Mannheim) and incubated for 30 min at 37°C. Then, nuclei were stained by adding 0.005% propidium iodide and observed with a 100× objective in a Nikon Optiphot fluorescence microscope (Tokyo, Japan). For chitin staining, cells were treated with calcofluor white (Fluorescent Brightener 28; Sigma Chemical Co., St. Louis, MO) as previously described (Pringle, 1991). For actin staining, cells were fixed by adding 1 vol of solution of 8% p-formaldehyde to the culture media and kept at 4°C for 1 h. Cells were washed three times with buffer (0.1 M potassium dihydrogen phosphate, 0.01 M magnesium chloride, 0.01 M EGTA, pH 6.9), resuspended in 100 μl of the same buffer, and 50 μl of 1% Triton X-100 was added. After 2 min at room temperature, cells were washed several times with PBS buffer. They were then resuspended in 100 μl PBS and rhodamine-phalloidine (Sigma Chemical Co.) was added at a final concentration of 4 μg/ml. Cells were kept in the darkness for 1 h at room temperature and washed several times with PBS buffer. Finally, cells were resuspended in the mounting medium (0.01 M p-phenylenediamine in 1:9 PBS/glycerol) to avoid photobleaching. The fluorescence microscope was equipped with a HB-10101AF mercury fluorescent lamp from Nikon (Melville, NY). Photographs were taken with a Nikon FX-35A camera and the films used were Ilford 400 ASA (Mobberley, Cheshire, UK). Confocal microscopy was performed with an Olympus IMT-2 microscope (Tokyo, Japan) attached to a Bio-Rad MRC1000 confocal system (Hercules, CA).
Electron Microscopy
Cells were grown at 24°C and then switched to 37°C for 6 h to allow expression of the phenotype. For scanning electron microscopy, cells were prepared by filtering 1 ml of culture through a 0.2-mm Millipore filter (Waters Chromatography, Milford, MA) and fixing the cells with 5% (vol/ vol) glutaraldehyde in 0.2 M sodium cacodylate buffer, pH 7.2, for 60 min. After this fixation, the filters were washed with the same buffer and gradually dehydrated by passing them through graded ethanol concentrations (25, 50, 70, 90, and 100%, 10 min in each). Then the filters were preserved in acetone, dried in a critical point drier, and coated with gold and examined (Williams and Veldkamp, 1974).
For transmission electron microscopy, cells were prefixed in a solution of 2% p-formaldehyde and 1.5% glutaraldehyde in 0.005 M sodium cacodylate, pH 7.2. The sample cells were then washed several times with PBS buffer and fixed with 1% (wt/vol) potassium permanganate for 90 min at 4°C. Cells were then washed several times with water and gradually passed through acetone solutions of increasing concentrations ranging from 30 to 100%. After this dehydration process, they were embedded in resin following the procedure from the Embed 812 kit supplied by Electron Microscopy Sciences (Fort Washington, PA). Once solid, samples were cut on an ultramicrotome (Ultracut; Leica, St. Gallen, Switzerland), treated with lead citrate and uranyl acetate as described by Streiblová (1988), and scanned under a Zeiss 902 electron microscope (Carl Zeiss Inc., Thornwood, NY).
Cytometric Assays
FACS® determination of cell lysis was performed as described by De la Fuente et al. (1992). For the analysis of DNA contents, samples were prepared following the procedure described above for nucleus staining. 3,000 cells were analyzed per second on a FACScan® (Becton Dickinson, Mountain View, CA) cytometer on the FL3 log scale.
Bud Timing Assays
Cells were grown on YPD or SD plates for 16 h at 24°C. Mid-log phase cells were then collected and resuspended in 100 μl of liquid YPD. 1–2 μl of the resulting suspension were laid onto a thin square sheet of YPD solid medium. Thin sheets of medium were prepared by polymerizing molten medium between two sterile glass slides separated by 1 mm and, once solid, cutting the edges with a sterile scalpel to a square shape slightly smaller than the size of a coverslip. The sheet with the cells face up was settled between the slide and coverslip and sealed with vaseline. Bud development was followed on a Nikon Optiphot microscope under phase contrast with a 40× objective. The same field was followed for several hours and the appearance of new buds was recorded every 2–3 min. A thermostatted surface from Linkam Scientific (Waterfield, Surrey, UK) was used to maintain the desired temperature at 28° or 37°C. The mother budding cycle time was obtained by recording the interval of time since a given cell started a new bud until it started a second bud. The daughter budding cycle time was calculated by monitoring the time elapsed since a given cell started a new bud until the resulting daughter began budding again. Unless specified, the data presented for each strain or clone are averages from a total of about 30 cells studied by these means.
Results
The lyt1-1 Mutation Defines a Novel Temperature-sensitive cdc15 Allele
The lyt1 mutation had been previously isolated in a search for autolytic mutants (Cabib and Durán, 1975). To clone the structural gene, we screened the lyt1 strain L2C24d with a S. cerevisiae genomic library, isolating three clones able to grow at 37°C (Materials and Methods). As deduced from restriction, partial sequencing, and Southern analyses, all three clones bore the CDC15 gene and this gene alone was responsible for the complementation of the lyt1 phenotype. To obtain genetic evidence that lyt1 is allelic to cdc15 and to discard the possibility that the CDC15 gene might be an extragenic suppressor—even in a low number of copies—of the lyt1 mutation, we made a diploid strain by mating the RH210-3c, which carries the cdc15-1 mutation, with the L2C24d lyt1-1 mutant strain. We found that the resulting cdc15-1/lyt1-1 diploid strain (MY1) was temperature sensitive and displayed cell lysis, and concluded that the lyt1-1 mutation lies at the CDC15 locus. Thus, to avoid confusion due to overlapping nomenclatures, henceforth we shall refer to the lyt1-1 mutation as cdc15-lyt1.
Due to the length of the CDC15 ORF we adopted a strategy for the analysis of the cdc15-lyt1 allele in three subfragments of ∼1 kb each, based on individual substitutions in the CDC15 wild-type allele, complementation experiments and sequencing (Materials and Methods). Analysis of the DNA sequences revealed no differences with the wild-type CDC15 sequence published by Schweitzer and Phillipsen (1991) except for a G to A transition at position 1,229. This observation is consistent with the mechanism of action of ethylmethane sulfonate, the mutagen used by Cabib and Durán (1975) to obtain the cdc15-lyt1 mutant. A restriction map of the gene revealed that the substitution was part of an EcoRI cleavage site. Restriction analysis of PCR products cloned in pBluescript SK disclosed that the inserts amplified from the wild-type strain YPH499 had an EcoRI restriction site that was absent in inserts obtained from reactions in which the L2C24d had been used as template (Fig. 1). Moreover, sequencing of the PCR products from YPH499 genomic DNA showed that position 1,229 is occupied by a G in the wild-type allele, as in the original sequence obtained by Schweitzer and Philippsen (1991). We therefore conclude that the cdc15-lyt1 allele displays a unique point mutation that results in a substitution of the glycine at position 410 by glutamate.
Figure 1.
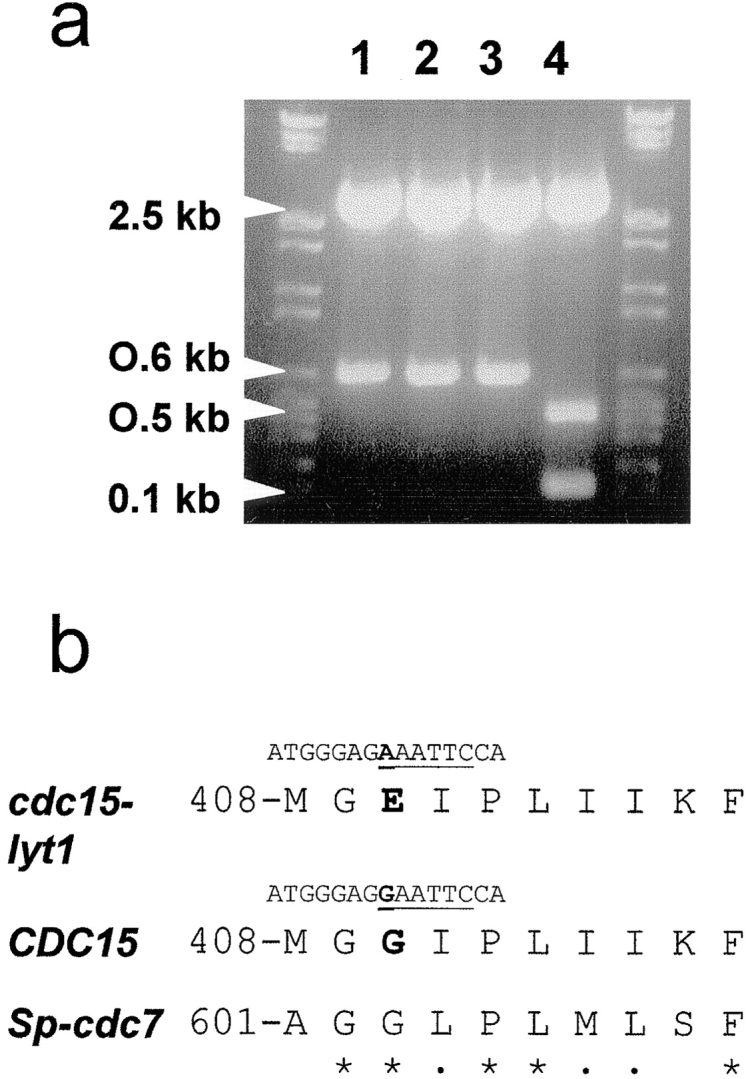
(a) EcoRI restriction analysis of PCR products from an inner fragment of the cdc15-lyt1 allele (lanes 1, 2, and 3 represent three different amplificates) and the CDC15 gene (lane 4), showing the disappearance of an EcoRI cleavage site in the mutant allele. (b) Sequence comparison of the CDC15-encoded peptide in the area where the lyt1 mutation maps from the mutant, the wild-type, and the putative homologue in the fission yeast S. pombe (Sp), cdc7. Underline, EcoRI site in the corresponding DNA sequence; boldface; residues affected by the mutation; asterisks, conserved residues; dots, structurally similar residues in the S. cerevisiae and S. pombe sequences.
Unlike other point mutations in the CDC15 gene that lead to a temperature-sensitive phenotype (Shirayama et al., 1996), the mutation in the cdc15-lyt1 allele does not affect the conserved protein kinase domains. A search in the databases for domains similar to the sequence flanking the mutation in cdc15-lyt1 led to one only entry (Fig. 1 b): the cdc7 + gene from the fission yeast Schizosaccharomyces pombe (Frankhauser and Simanis, 1994). This gene encodes a putative protein kinase with a relatively high homology with the CDC15 gene in the protein kinase domain (46% identity and 66% structural similarity). Interestingly, these genes are quite divergent outside the kinase domains, the only exception being the region containing the cdc15-lyt1 mutation.
Cell Lysis Is Not Specific to the cdc15-lyt1 Allele
Molero et al. (1993) showed that at 37°C lyt1 strains arrest their cell cycle with a bud of the same size as that of the mother cell and eventually undergo cell lysis. All cdc15 ts mutant alleles described in the literature generate such doublets, which are characteristic of arrest in the late M phase (Culotti and Hartwell, 1971; Shirayama et al., 1996). Nevertheless, no cell lysis has been reported to occur in cdc15-arrested cells. In our hands, the cdc15-lyt1 mutant also showed an anaphase-arrest phenotype, but cell lysis was severe after 5 h at 37°C. Therefore, we wished to know whether the cell lysis phenotype was characteristic of the cdc15-lyt1 allele or whether other cdc15 alleles behaved similarly.
We tested whether a cdc15-1 mutant was also able to develop cell lysis by observing propidium iodide-treated cells from strain RH210-3c after incubation at the restrictive temperature. Cells clearly arrested in doublets but cell lysis was not as prominent in this strain as in a cdc15-lyt1 one (5% of lysed cells in the RH210-3c-cdc15-1 strain and more than 60% in the L2C24d-cdc15-lyt1- strain). Accordingly, either the specific cdc15-lyt1 allele conferred a lytic phenotype or the repercussion of any cdc15 mutation on cell integrity was strongly dependent on the genetic background. After mating the RH210-3c (cdc15-1) strain to the wild-type strain 1784 and subsequent study of the new cdc15-1 haploid segregants, we observed that 23 out of 29 of these clones displayed a very high degree of cell lysis at 37°C. The other six arrested as steady doublets and showed cell lysis below 10% after 5 h at 37°C. Moreover, segregants from 15 tetrads of the MY1 cdc15-1/cdc15-lyt1 diploid never gave rise to a 2:2 lysing/nonlysing progeny. By contrast, the severity of the lytic phenotype segregated in a rather randomized fashion, 4:0 lytic/nonlytic being the most frequent pattern. Therefore, cdc15-1 mutants develop lysis to the same extent as cdc15-lyt1 mutants in most genetic backgrounds. Interestingly, cell lysis seemed to parallel the expression of morphogenetic alterations in the doublet (see below). We decided to further study the nature of this abnormal response in order to gain insight into the ultimate cause of cell lysis.
Cell Lysis Is Preceded by the Formation of a Distal Projection in the Daughter Cell
An interesting observation was made when studying the lytic phenotype of cdc15 ts strains: lysed cells were seldom symmetrical doublets. 96.9% of lysed cells in a given population of the L2C24d cdc15-lyt1 strain showed an asymmetrical morphology after 5 h at 37°C: one of the cells in the doublet had developed a more or less conspicuous apical structure (Fig. 2 c, e, g, h–l, and s). This phenotype was precluded by introduction of the pBS9 plasmid, bearing the CDC15 gene (Fig. 2 a). To determine whether it was the mother, daughter, or either that was developing the apical projection, we grew a culture of the same strain at 24°C and then shifted some cells to a YPD microsheet and kept them under observation at 37°C in a thermostatted microscope. With this approach, we observed that the aberrant structure that seemed to precede cell lysis occurred in the daughter cell (Fig. 2, h–l). All 37 cells under these conditions behaved identically. Further support to the conclusion that it was essentially the daughter cell that developed the structure was provided by the staining of bud scars with calcofluor white. In 34 out of 35 scar-bearing doublets, the cell showing the apical shmoo-like structure never had chitin scars, suggesting that it was the newborn one. In turn, when present, scars were always situated on the nonaberrant cell of the doublet (data not shown).
Figure 2.
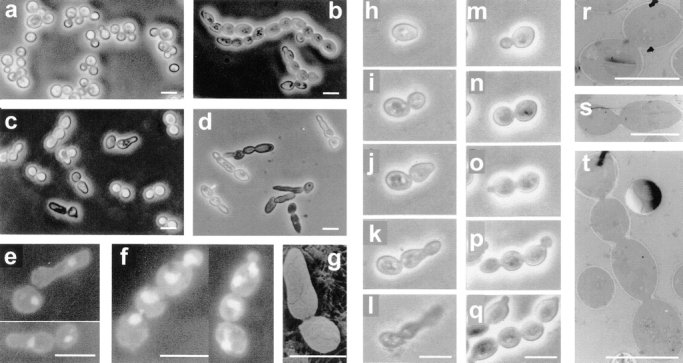
Microscopic observations by phase–contrast (a–d and h–q), fluorescence (e and f), scanning electron (g), and transmission electron (r–t) microscopy of haploid and diploid cdc15 mutant strains. (a) Strain L2C24d (cdc15-lyt1) transformed with the pBS9 plasmid bearing the CDC15 gene after 6 h of incubation at 37°C, thus displaying wild-type behavior. (b) Diploid MY1 strain (cdc15-1/cdc15-lyt1) under the same conditions showing abundant chained cells. (c) L2C24d (cdc15-lyt1) strain under the same restrictive conditions. (d) The same strain incubated in an osmotically stabilized medium (supplemented with 1 M sorbitol) under identical conditions, showing an exacerbated expression of its characteristic apical growth phenotype. (e) Nucleus staining in fixed RNase-treated cells from strain L2C24d (cdc15-lyt1) after 6 h of incubation at 37°C. (f) Cells from the diploid strain MY1 (cdc15-1/cdc15-lyt1) after the same treatment. (g) A characteristic asymmetric doublet from the L2C24d (cdc15-lyt1) strain incubated at the restrictive temperature for 6 h. (h–l) Series showing the development of a cell from the L2C24d (cdc15-lyt1) strain at 37°C. Pictures were taken at 0 h (h), 1 h (i), 2.5 h (j), 3.5 h (k), and 4.5 h (l). (m–q) Series showing the development of a cell from the MY1 (cdc15-1/cdc15-lyt1) strain in the same conditions. Pictures were taken at 0 (m), 0.5 h (n), 1.5 (o), 2.5 (p), and 3.5 h (q). (r and s) Section of cells from the L2C24d (cdc15-lyt1) strain after 6 h of incubation at 37°C, showing that septation has not been initiated. (t) A chain of cytokinetic-defective cells from the diploid MY1 (cdc15-1/ cdc15-lyt1) strain under the same conditions. Bars, 8 μm.
The same experiments were performed on the cdc15-lyt1/cdc15-1 MY1 strain. As in haploid strains, after 6 h at the restrictive temperature, most cells lysed and lysis was preceded by the development of a more or less conspicuous apical structure. Interestingly, linear chains of three, four, or more cells were frequently observed, a phenomenon not seen in the haploid mutants (Fig. 2, b, f, m–q, and t). cdc15-1/cdc15-1 and cdc15-lyt1/cdc15-lyt1 diploid strains also gave rise to a significant number of chains. To check that the occurrence of chained cells was indeed a consequence of ploidy and not due to the combined genetic backgrounds generated after mating, we constructed a diploid strain isogenic to the L2C24d cdc15-lyt1 strain (Materials and Methods). The resulting DJY4 cdc15-lyt1/ cdc15-lyt1 strain also generated a significant amount of chains in comparison to the isogenic haploid strain. Table II shows the frequency of chained cells, shmoo-like doublets, and plain doublets in the different cdc15 ts haploid and diploid strains. A sequential study of the formation of these chains is shown in Fig. 2, m–q. We stained the nuclei of fixed RNase-treated cells with propidium iodide and found that each cell of the chain had a nucleus (Fig. 2 f). Occasionally, cells at the end of the chain bore two nuclei, probably due to a discoordination between budding and nuclear division at advanced stages of phenotypic expression.
Table II.
Percentage of Doublets, Apical Projections, and Chains in Populations of cdc15 Mutants
| Strain | Culture medium |

|
|||||||
|---|---|---|---|---|---|---|---|---|---|
| % | % | % | |||||||
| L2C24d (cdc15-lyt1) | YPD | 19 (50 cells) | 79 (208 cells) | 2 (6 cells) | |||||
| YPD + sorbitol 1 M | 9 (44 cells) | 55 (260 cells) | 26 (171 cells) | ||||||
| RH210-3c (cdc15-1) | YPD | 80 (466 cells) | 18 (109 cells) | 2 (10 cells) | |||||
| YPD + sorbitol 1 M | 8 (26 cells) | 43 (133 cells) | 49 (152 cells) | ||||||
| MY1 (cdc15-1/cdc15-lyt1) | YPD | 27 (70 cells) | 45 (118 cells) | 28 (74 cells) | |||||
| YPD + sorbitol 1 M | 3 (8 cells) | 27 (86 cells) | 70 (224 cells) | ||||||
| DU3 (cdc15-lyt1/cdc15-lyt1) | YPD | 56 (217 cells) | 29 (112 cells) | 15 (56 cells) | |||||
| YPD + sorbitol 1 M | 12 (25 cells) | 60 (125 cells) | 28 (58 cells) | ||||||
| DR1 (cdc15-1/cdc15-1) | YPD | 89 (225 cells) | 11 (27 cells) | — (1 cell) | |||||
| YPD + sorbitol 1 M | 45 (102 cells) | 36 (81 cells) | 18 (41 cells) | ||||||
| DJY4 (cdc15-lyt1/cdc15-lyt1) | YPD | 31 (31 cells) | 31 (31 cells) | 38 (39 cells) | |||||
| YPD + sorbitol 1 M | 11 (9 cells) | 10 (8 cells) | 79 (63 cells) | ||||||
Percentage of cells showing a particular morphology (plain doublet, doublet with apical projection, or chained cells) in haploid and diploid cdc15-1 and cdc15-lyt1 mutants in different backgrounds in the absence or presence of 1 M sorbitol. In these experiments, cells were grown in 50 ml of YPD medium at 24°C overnight with or without sorbitol and then shifted to 37°C for 6 h.
Osmotic protection has been reported to delay or remedy lysis in some lytic mutants (Torres et al., 1991; Cid et al., 1998b ). We stabilized the culture media with 1 M sorbitol and studied the behavior of several cdc15 mutants at 37°C (Table II). 1 M sorbitol was unable to suppress the lytic effect of cdc15 ts mutations but seemed to stabilize the aberrant structures, delaying cell lysis to a slight extent and thus enhancing apical growth in the daughter cell, often leading to long tubular structures (Fig. 2 d). Also, in quantitative terms the proportion of cells able to develop these structures was much larger when sorbitol was present in the medium (Table II). Under these conditions, even the RH210-3c cdc15-1 strain, which in nonstabilized medium seldom develops apical structures, gave rise to a significant number of shmoo-like doublets. Additionally, in the presence of 1 M sorbitol, an increased number of haploid cells displayed a behavior similar to that described above for diploid strains, occasionally showing chained cells (Table II). To confirm that these effects were indeed due to the osmolarity of the medium and not to any other interference of the sorbitol molecule with cellular mechanisms, we repeated the experiments using 0.2 M NaCl as osmolyte, obtaining similar results (data not shown).
cdc15-lyt1 Mutants Do Not Commit Septation
The cell cycle arrest in cdc15 mutants has been described to occur in the anaphase and hence the segregated nuclei are still connected by the nuclear envelope and microtubules (Copeland and Snyder, 1993; Surana et al., 1993). We focused our interest on determining whether the septation process had been committed, especially in cells that developed the apical shmoo-like structure or gave rise to chains. To approach this issue, we grew cultures from the L2C24d (cdc15-lyt1) and MY1 (cdc15-1/cdc15-lyt1) strains at the permissive temperature and shifted them to 37°C for 6 h. Observation of these cells by transmission electron microscopy revealed that septation was never initiated (Fig. 2, r, s, and t). Indeed, septa were always absent, even in cells belonging to chains of three or four cellular units with separated nuclei. This observation implies that, at least in diploid and probably in osmotically stabilized haploid cdc15 mutants, a new round of the cell cycle can be started in the absence of cytokinesis. This offers a valuable new tool for study of the coordination of cycle-to-cycle transition in the budding yeast.
Distal Growth in cdc15 Mutants Is Supported by Actin but Not by the Septin Cytoskeleton
The above experiments had shown us that the mitotic arrest caused by cdc15 mutations occurs before the initiation of septation. During the last stages of the budding yeast cell cycle, actin patches localize to the bud–neck area to support polarized secretion for septum formation (Lew and Reed, 1993). We were interested in determining whether cdc15 mutants were able to localize the actin cytoskeleton to the septum area even though the morphogenetic response is inhibited or whether, as happens in a blockage of B-cyclin degradation (Lew and Reed, 1993), actin is not even polarized to the mother–bud neck.
We incubated the cdc15-lyt1 mutant strain L2C24d at 24°C overnight and then shifted it to the restrictive temperature. Samples were taken every 30 min, fixed, and then stained with rhodamine-conjugated phalloidine to visualize the actin cytoskeleton. An average of 120 nonlysed cells from each sample was analyzed for actin polarization under a fluorescence microscope. The dynamics of actin polarization are represented graphically in Fig. 3. As expected in a log phase culture, only 17% of the cells (single cells or doublets) displayed a randomly distributed pattern of actin deposition, whereas 76% of the population showed a polarization of actin towards the incipient, emerging or developing bud, and ∼7% of the cells concentrated most actin around the septum area. Total depolarization of the actin cytoskeleton was observed after 30 min at 37°C. This is probably not due to the expression of the cdc15 phenotype but rather would be the normal cellular response to the stress caused by a temperature shift (Pringle et al., 1989). After 1 h, the actin cytoskeleton had been repolarized, but the pattern of localization to the septum area was not detected. The same was observed subsequently, despite the progressive accumulation of cells in doublets, which invariably had a delocalized actin cytoskeleton, as corresponds to the expected anaphase arrest.
Figure 3.

Evolution of actin polarization in the L2C24d (cdc15-lyt1) strain at the restrictive temperature. Actin was stained with rhodamine-conjugated phalloidine. The plot represents the percentage of each cell type within the population (y axis) versus time of incubation at 37°C (x axis). Only live cells can be stained for the visualization of actin, so no lysed cells were included in this assay. (▴) Single unbudded cells with a random distribution of actin patches. (♦) Doublets with a depolarized actin cytoskeleton, as corresponds to anaphase-arrested cells. (▪) Doublets with a polarization of the actin cytoskeleton to the neck area for the promotion of septum morphogenesis. (•) Cells showing a pattern of actin polarization towards the incipient, emergent or developing bud. (✖) Doublets or asymmetric doublets that polarize the actin cytoskeleton towards the distal pole of one of the cells. Bars, 5 μm.
With these results, we concluded that at 37°C the actin cytoskeleton is never located at the mother–bud neck in cdc15 ts mutants. Thus, the absence of septation reported above could be a consequence of the failure of the morphogenetic apparatus to recognize the septum area. Remarkably, after 120 min at 37°C a growing population of cells appeared showing a strong polarization of actin towards the distal pole of one of the cells, which in most cases had started to develop the apical elongation described above. After 4 h, over 50% of the population had lysed and hence the study was not further continued in order to avoid collecting unrepresentative data from rare survivors.
Septins constitute another cytoskeletal structure assumed to play crucial roles in morphogenetic events along the cell cycle, such as the localization of septa (Longtine et al., 1996; Cid et al., 1998a ). Septins assemble at the bud neck as a ring before bud emergence and remain there until cytokinesis is complete. We studied the localization of the septin-based ring in cdc15-lyt1 mutants using a Cdc10p–green fluorescent protein (GFP) fusion. As shown in Fig. 4, a and b, septins remain as a double ring at the mother–daughter neck well after the apical structure has grown. Using confocal microscopy, we were able to simultaneously visualize rhodamine-stained actin and Cdc10p– GFP (Fig. 4, e–g). From this experiment we concluded that septins are never observed at the base of the distal actin-rich projections. Therefore, the aberrant shmoo-like structure characteristic of cdc15 mutants is supported by the actin but not by the septin cytoskeleton, which remains at the site of the aborted septation. It is likely that the lytic phenotype of cdc15 ts mutants would be due to a budding attempt that has been aborted because of the absence of essential morphogenetic components that rely on the presence of the septin ring. Consistent with this hypothesis, septins are present at the neck of the successful new buds that originate chains of cells in diploid cdc15/cdc15 mutants at 37°C, as shown in Fig. 4, b and c.
Figure 4.

Visualization of the septin ring by fluorescence (a–d) and confocal (e–g) microscopy of the L2C24d strain (cdc15-lyt1) transformed with the pLA10 plasmid bearing a CDC10–GFP fusion (Cid et al., 1998a ) (a, b, and e–g) and MY3 strain (cdc15-lyt1/cdc15-1 CDC10–GFP) (c and d). Cells were kept at 37°C for 4.5 h to allow expression of the characteristic morphogenetic phenotype. (a and b) Fluorescence microscopy shows that the septins remain at the mother–daughter neck in asymmetric doublets. (c and d) The formation of chains of cells in diploid cdc15/cdc15 mutants is supported by the formation of novel septin rings, but the first ring in the neck between the oldest cells persists. (e–g) Simultaneous staining with rhodamine-conjugated phalloidin allows the visualization of both the actin and septin cytoskeletons. Red, actin-rich areas; green, corresponds to the septin–GFP fusion. In e and g, the fluorescence image is overlapped to a phase contrast micrograph for better definition of cell shape. Bars, 5 μm.
Distal Growth in cdc15 Mutants Depends on Budding Pattern Signals and on the START Landmark of Cell Cycle
All the evidence obtained so far suggested that the morphogenetic response described could be due to the commitment of a new round of the cell cycle in the absence of cytokinesis. Based on cell polarity studies and assays on nuclear dynamics, we endeavored to clarify this point.
S. cerevisiae haploid strains always develop a new bud at the proximal pole; that is, next to the previous site of cytokinesis (for review see Roemer et al., 1997). The fact that when cytokinesis is absent the distal pole of the daughter cell is favored for subsequent morphogenetic events led us to wonder whether this phenomenon might depend on the signals for pole recognition, in which the small GTPase Bud1p/Rsr1p plays a major role (Chant and Herskowitz, 1991; for review see Chant, 1994; Park et al., 1997). To determine whether the choice of this abnormal site for morphogenesis depends on such cellular tags, we constructed double cdc15-1 bud1::URA3 and cdc15-lyt1 bud1::URA3 mutant strains by mating both the RH210-3c (cdc15-1) and JJY0-3c (cdc15-lyt1) strains to JC223 (bud1:: URA3) and subsequently studying the meiotic products. The resulting JJY1 and JJY2 strains showed a random budding pattern, due to the central role played by BUD1 in localization of the budding site (Chant, 1994). The polarity of the morphogenetic response described above was evaluated in these strains and compared with that of Ura− cdc15 segregants (JJY3 and JJY4) obtained in the same experiment (Table III). JJY1, JJY2, and other Ura+ cdc15 segregants analyzed that carried both cdc15-1 and bud1 mutations developed the abnormal projection described above in a random fashion; that is, often at sites other than the distal pole. By contrast, all the Ura− segregants, bearing only the cdc15 mutations, seldom failed to develop this structure precisely at the pole. Despite this, the morphogenetic response was still restricted to the daughter cell and determined cell lysis. We conclude from these experiments that the location of this abnormal morphogenetic response in cytokinesis-defective cdc15 mutants obeys the permanent polarity signals for budding.
Table III.
Polarity of the Projections in cdc15 Mutants
| Strain |

|
||||||
|---|---|---|---|---|---|---|---|
| % | % | % | |||||
| JJY1 (cdc15-1 bud1::URA3) | 27 (109 cells) | 44 (180 cells) | 29 (118 cells) | ||||
| JJY2 (cdc15-lyt1 bud1::URA3) | 24 (66 cells) | 64 (175 cells) | 12 (32 cells) | ||||
| JJY3 (cdc15-1) | 64 (110 cells) | 4 (6 cells) | 32 (56 cells) | ||||
| JJY4 (cdc15-lyt1) | 57 (85 cells) | 3 (4 cells) | 40 (59 cells) | ||||
Polarity of the projection formed by doublets expressing the cdc15-1 or cdc15-lyt1 mutations, as recorded after 6 h at 37°C. Percentages of doublets that develop a projection at the distal pole or at a random site, as well as the proportion of doublets that do not exhibit a conspicuous projection, are shown.
As can be seen in Fig. 2 e, the nucleus in cells with a distal projection is usually oriented towards this structure, suggesting that a new round of the cell cycle may be committed. If the apical projection is due to the overcoming of mitotic arrest and the entry of the cells in a new round of cell division, it must be dependent on the START landmark of cell cycle. cdc28-4 is a Ts− mutation that causes cell cycle arrest precisely at START (Reid and Hartwell, 1977) and therefore, if this hypothesis were true, one would expect that double cdc15 ts cdc28-4 mutants would be unable to polarize growth to the distal pole. A collection of these mutants was constructed by mating the K1414 (cdc28-4) strain to L2C24d (cdc15-lyt1). Double mutants were selected from tetrads that yielded 2:2 wild-type/Ts− segregation. When compared with single cdc15-lyt1 segregants from the same experiment, the START-defective cdc15 mutants did not give rise to apical projections at 37°C but remained either as large doublets or as large unbudded cells due to the G1 arrest. The data obtained on analyzing 198 cells from three different segregants carrying both mutations and 118 cells of a single cdc15 segregant held for 6 h at 37°C were as follows: 56.1% of the cdc28-4 cdc15-lyt1 population arrested as single large cells due to the START-specific arrest caused by the cdc28-4 mutation whereas no single cells of any size were observed in the single cdc15-lyt1 segregant; 42.1% of the population of the double mutants arrested as large-sized dumbbell-shaped doublets whereas this pattern was seen in 31.4% of the single cdc15 mutant population. More interestingly, only 1.7% of the double mutant population showed some polarization of growth at one end of the doublet, whereas 68.6% of the single cdc15-lyt1 mutants displayed polar growth. Therefore, the expression of cdc28-4 START-specific arrest was preventing haploid cdc15-lyt1 mutants from forming their characteristic apical projection.
To confirm that nuclei from cytokinetic-defective cells expressing the cdc15 phenotype were overcoming mitotic arrest, we decided to study the progression of the nuclear cell cycle in these cells. We performed quantitative DNA content assays by flow cytometry to determine whether the morphogenetic response was indeed accompanied by post-START events. As shown in Fig. 5, after incubation of the cells at the restrictive temperature in 1 M sorbitol, which retards lysis (as shown above), the peak corresponding to pre-S phase cells progressively decreased while the post-S phase peak increased due to the accumulation of anaphase-arrested cells, as expected. Interestingly, in longer exposures (times longer than 150 min) to the restrictive temperature a third peak was seen, indicating the presence of a new population bearing an amount of DNA equivalent to that of four nuclei. This last peak very likely represented cells that overcame the M phase arrest and started a new cell division, in agreement with the results reported above. When the same experiments were carried out on diploid cdc15/cdc15 strains, the graphics were difficult to interpret due to the appearance of subsequent diffuse peaks of higher fluorescence intensity, probably corresponding to chained cells (data not shown). In sum, these results suggest that a significant part of the population indeed overcomes the anaphase arrest at the restrictive temperature and initiates a new round of the cell cycle.
Figure 5.
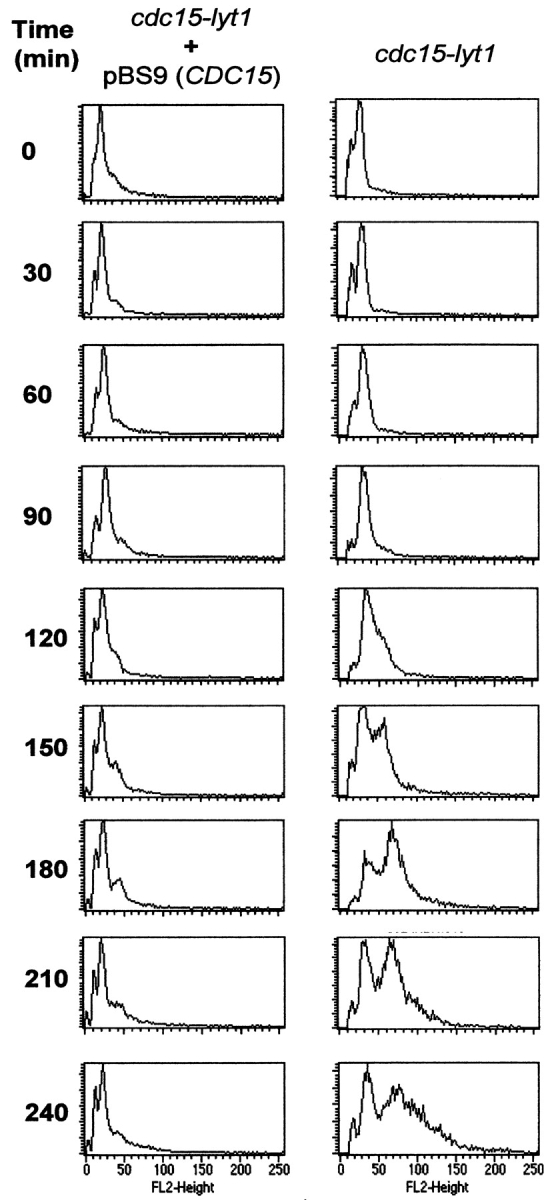
Flow cytometry graphics of RNase-treated propidium iodide-stained cells of the L2C24d strain. Cells were incubated at 37°C in osmotically stabilized media and samples were taken every 30 min. Peaks correspond to cell populations containing different amounts of DNA. The peak corresponding to a DNA content of 1 nucleus per cell (G1 peak) appears at ∼15 fluorescence units (see scale at the bottom), the G2/M peak (2 nuclei per unit) stands at ∼30 units, and cells displaying a DNA content equivalent to 4 nuclei appear at 60 U. Upon expression of the cdc15 phenotype (right), the G1 peak progressively disappears due to the accumulation of cells in anaphase. After 150 min, a new well-defined peak reveals a new population of cells with a larger amount of DNA. None of these phenomena occurred when the same strain was transformed with a CDC15-containing plasmid (left).
Mutants in Other Genes Controlling Late Mitosis Also Develop Apical Projections
There is genetic evidence that the products of the genes TEM1, CDC14, and DBF2 act in a common pathway with the Cdc15p protein kinase (Kitada et al., 1993, Shirayama et al., 1994, 1996). As in the case of CDC15, these genes are essential for viability and dysfunctions in them have been described to cause a late M phase arrest (Toyn and Johnston, 1994; Shirayama et al., 1994, 1996). By applying some of the same techniques as those described above for cdc15 mutants to strains EO156 (tem1-3), RH1779 (cdc14) and L119-7d (dbf2), we also detected a distal polarization pattern in these mutants (Fig. 6, a–c, Table IV). The data thus obtained are comparable to those obtained for cdc15-1 and cdc15-lyt1 in different backgrounds (Table II), suggesting that the eventual overcoming of mitotic arrest would not be restricted to cdc15 mutants, but that it would be common to other mutations in the pathway.
Figure 6.

(a–f) Morphological observations by phase contrast microscopy of mutants in genes involved in late mitotic events after 6 h of incubation at 37°C: (a) L119-7d (dbf2-1); (b) RH1779 (cdc14-1); (c) EO156 (tem1-3); (d) 4965-3a (cdc16); (e) 4086-23-2a (cdc23); (f) 9002 (cdc27). (g) L2C24d (cdc15-lyt1) strain transformed with the pGAL–CLB2 plasmid after 6 h of incubation at 37°C with galactose as a carbon source. (h) The same strain transformed with a pRS316 control plasmid under the same conditions. Arrows, distal projections. Bars, 5 μm.
Table IV.
Distal Polarization of Growth in Late Mitotic Mutants
| Strain |

|
||||||
|---|---|---|---|---|---|---|---|
| % | % | % | |||||
| EO156 (tem1-3) | 49 (38 cells) | 45 (35 cells) | 6 (5 cells) | ||||
| L119-7d (cdc14-1) | 67 (130 cells) | 29 (57 cells) | 4 (7 cells) | ||||
| RH1779 (dbf2) | 36 (54 cells) | 64 (95 cells) | 0 (0 cells) | ||||
| 4965-3a (cdc16) | 41 (124 cells) | 59 (179 cells) | 0 (0 cells) | ||||
| 4086-23-2a (cdc23) | 87 (268 cells) | 13 (41 cells) | 0 (0 cells) | ||||
| 9002 (cdc27) | 92 (131 cells) | 8 (11 cells) | 0 (0 cells) | ||||
| L2C24d + pGAL-CLB2 (cdc15-lyt1, CLB2 overexpression) | 82 (160 cells) | 14 (27 cells) | 4 (8 cells) | ||||
| L2C24d + pRS316 (cdc15-lyt1, control in galactose) | 41 (89 cells) | 43 (93 cells) | 16 (36 cells) | ||||
Percentages of cells showing a particular morphology (plain doublet, doublet with apical projection, or chained cells) in different mutants of genes genetically related to CDC15, APC mutants and cdc15-lyt1 mutants overexpressing the CLB2 gene under the GAL1 promoter. In all cases data were collected from samples incubated for 6 h at 37°C.
At this point, we tested mutants in the 20S proteosome/ anaphase-promoting complex (APC), which have been also reported to display an anaphase arrest due to the inability of the cell to remove B cyclins (Zachariae and Nasmyth, 1996). The rationale for this was to find out whether the formation of an apical projection in the doublet was a feature of mutants in the Cdc15p pathway or whether it could be ascribed to other key regulatory mechanism for late mitosis, such as B cyclin removal. Asymmetric doublets showing an apical projection were observed in cdc23 and a cdc17 strains (Fig. 6, e and f), although in a proportion much lower than that of the mutants in the Cdc15p pathway (Table IV). However, the 4956-3a strain, which bears a cdc16 mutation, gave rise to a very high proportion of asymmetric shmoo-like doublets (Table IV). Therefore, we concluded that the morphogenetic response described above is an effect of a cytokinetic failure, caused by a dysfunction either in the Cdc15p pathway or in the APC. These results could be better understood if cyclin degradation by the APC were a requisite for the functionality of the Cdc15p pathway or, as postulated by Jaspersen et al. (1998), a role of the Cdc15p pathway were the activation of the APC.
In view of these results, and since it is assumed that the overall control of exit from mitosis depends on the inactivation of the CDK-B cyclin complex, we wished to study the effect of B cyclin overexpression on the cdc15 distal polarization phenotype. We did this by using a GAL- CLB2 construction in a centromeric pRS316 plasmid, provided by M. Glotzer (Institute for Molecular Pathology, Vienna, Austria). Incubation of strains transformed with this vector in galactose-based media should lead to overproduction of CLB2. In the wild-type strain 1784, CLB2 overexpression caused an arrest of the cells in doublets but distal polarization in these doublets was rare (1–2% of the population, data not shown). We studied the behavior of L2C24d (cdc15-lyt1) GAL-CLB2 transformants in comparison with a control of the same strain bearing the pRS316 plasmid (Sikorski and Hieter, 1989). Although the distal projection phenotype is expressed with less severity in galactose, microscopic observations (Fig. 6, g and h) showed that CLB2 overexpression made the appearance of projections more rare, thus significantly reducing cell lysis. In fact, the proportion of cells displaying an apical projection in a L2C24d (cdc15-lyt1) population decreased threefold when CLB2 was overexpressed (Table IV). We conclude that the deregulation of B cyclin expression prevents the appearance of the distal polarization phenotype and hence cell lysis. These results are in accordance with the idea that the Cdc15p pathway modulates morphogenetic events for cytokinesis within a more general regulatory program controlled by the cyclic activation/inactivation of Cdc28p.
The Timing of Rebudding after M Phase Arrest in cdc15-lyt1 Mutants
We have shown above that certain mutants in late M phase–controling pathways are able to exit their characteristic cell cycle arrest. However, they are unable to form septa and the initiation of a new round of budding in the absence of septation causes an abnormal morphogenetic response. These results imply that the cell cycle arrest previously described for these mutants is not definitive but temporary and that it might be a consequence of the cytokinetic failure. On this basis, we were prompted to measure the efficiency of this arrest.
To elucidate whether a difference in the timing of bud emergence between wild-type cells and cdc15 mutants occurs at the restrictive temperature, we devised a simple “budding time” assay (Materials and Methods). In brief, at the restrictive temperature and using a thermostatted phase contrast microscope we measured the time elapsing since a single cell from the mutant strain L2C24d (cdc15-lyt1) starts to produce a bud until this bud starts to produce a second bud or a projection at its distal pole. As a control and under the same conditions we used the same strain transformed with the pBS9 centromeric plasmid, which contains the CDC15 wild-type gene, and measured the time between bud emergence in an original single cell and the emergence of the first bud of this newborn daughter. As shown in Table V, the average budding time thus obtained was ∼85 min for the control and 130 min for the mutant. Hence, the appearance of the apical bud in the cdc15-lyt1 mutant at the nonpermissive temperature was 45 min delayed with respect to the budding of the first daughter in the wild-type cell cycle. To confirm that this delay was not due to differences in growth efficiency based on the presence of the plasmid, we checked that there was no difference at all between the cycling time of the same clones at the permissive temperature of 28°C (Table V).
Table V.
Budding Delay in cdc15 and tem1 Mutants
| Strain | Average budding time at 37°C | Average budding time at 28°C | Delay at 37°C* | |||
|---|---|---|---|---|---|---|
| min | min | min | ||||
| L2C24d (cdc15-lyt1) | 131 ± 22 (32 cells) | 99 ± 6 (24 cells) | 45 | |||
| L2C24d + pBS9 (CDC15) | 86 ± 8 (23 cells) | 98 ± 8 (25 cells) | ||||
| EO156 (tem1-3) | 116 ± 5 (23 cells) | 96 ± 6 (21 cells) | 25 | |||
| RAY3-a (isogenic TEM1) | 91 ± 9 (10 cells) | 97 ± 8 (18 cells) |
Average budding time both at 37°C and 28°C in cells from cdc15-lyt and tem1-3 mutants and their respective complemented and isogenic wild type controls.
The delay is calculated by subtracting the budding time of the isogenic wild-type or complemented control from the budding time of the mutant.
We reproduced these assays in the EO156 strain bearing the tem1-3 mutation, using its isogenic wild-type strain RAY-3a as a control. In our hands, tem1-3 mutants were not as efficient as cdc15-1 and cdc15-lyt1 mutants in expressing the mitotic arrest phenotype since a few cells in the population continued to bud normally. Despite this, with these means we found a budding delay of 25 min (Table V).
The Overcoming of Mitotic Arrest Is Not Synchronous
All the data so far reported correspond to budding time assays taking the daughter cell as a reference. At this point we were prompted to determine to what extent the cytokinetic defect caused by the cdc15 mutation inhibited the ability of the mother cell to bud. As described above, haploid cdc15 strains lyse soon after the younger cell of the doublet has committed an abnormal morphogenetic response, offering no chance to measure the delay in bud emergence that might take place in the older cell, which under these circumstances remains unbudded. On the other hand, as seen in Fig. 2, b, m–q, and t, diploid cdc15/ cdc15 mutants, both mother and daughter cells, usually rebud successfully in the absence of cytokinesis at the restrictive temperature.
We performed timing assays using the MY1 diploid strain and a MY2 isogenic control. As shown in Table VI, the diploid was more efficient than the haploid strain tested above at overcoming the mitotic arrest. It took only 17 min more than the control for the daughter cell to start bud emergence, whereas the mother cell showed a delay of 37 min with respect to the transformed control. It is well known that during vegetative growth a wild-type S. cerevisiae strain divides in an asynchronous fashion in the sense that the mother cell starts a new round of cell division earlier than the daughter (Roemer et al., 1996). In fact, in the wild-type controls of our experiments daughters budded about 14 min later than mothers (Table VI), highlighting the delay detected in the cdc15/cdc15 mutant. From these results it may be concluded that although there is always a delay in bud emergence when cytokinesis has not been performed in the previous cycle, the daughter cell is able to overcome this arrest more readily than the mother cell.
Table VI.
Mother and Daughter Budding Delay in Diploid cdc15
| Mother cell | Daughter cell | Control mother cell | Control daughter cell | |||||
|---|---|---|---|---|---|---|---|---|
| min | min | min | min | |||||
| 37°C | 100 ± 5 (38 cells) | 84 ± 4 (38 cells) | 63 ± 5 (18 cells) | 68 ± 5 (18 cells) | ||||
| 28°C | 86 ± 9 (18 cells) | 99 ± 9 (18 cells) | 85 ± 7 (18 cells) | 98 ± 11 (18 cells) |
Average budding time at 37°C and 28°C for both mother and daughter cells in the diploid strain MY1 when expressing the cdc15 mutation (first and second column) and when this mutation is complemented with the pBS9 plasmid, which contains the CDC15 gene (MY2 strain) (third and fourth columns).
The Delay in Bud Emergence Caused by cdc15 Mutations Is Prevented in a cdc10 Background
With the above data, we concluded that the commitment of a new round of the cell cycle in cytokinesis-defective mutants involves nuclear entry in the S phase and actin-dependent morphogenetic events. However, at least in haploid backgrounds, septins fail to reassemble at the new bud site. Interestingly, mutations in any of the CDC3, CDC10, CDC11, and CDC12 septin-encoding genes lead to the development of elongated buds, a loss of cell polarity, and defective cytokinesis (for review see Longtine et al., 1996). To determine whether there was any relationship between the function of the CDC15 gene product and the function of septins at the time of cytokinesis, we constructed double cdc10-11 cdc15-1 strains by mating the VCY1 and RH210-3c strains and analyzing the meiotic products. Among the segregants, the cdc10 mutants were identified by their peculiar bud elongation phenotype at nonpermissive temperatures and the cdc15-1 mutation was followed by the red color of the colonies due to its genetic linkage to the ADE2 locus (Schweitzer and Phillipsen, 1991). The double mutants were viable and displayed the cdc10-characteristic phenotype of multinucleated cells with elongated buds at 37°C. However, these cells, unlike those of cdc10 single mutants, lysed before developing complex branched structures (data not shown). Peculiarly, these double mutants initiated several rounds of budding at 37°C before lysing, suggesting that a dysfunction in the septin ring somehow protects cdc15 mutant cells from lysis.
One of these segregants, VCY242d, was independently transformed with the YCP111-CDC10 plasmid, bearing the wild-type CDC10 gene, with the pBS9 plasmid, bearing the wild-type CDC15 gene, and with both plasmids simultaneously. The VCY242d strain and these transformants were subjected to further budding time experiments. At 37°C, we were able to measure the cycling time for the daughter when only the cdc15-1 mutation is expressed (VCY242d + YCP111-CDC10), when only the cdc10 mutation is expressed (VCY242d + pBS9), when both mutations are coexpressed (untransformed VCY242d) and when neither of the mutations is expressed in the same background (double transformant, used as control). The results of these experiments are shown in Table VII. Despite the severely altered morphology displayed by cdc10 mutants at 37°C, no delay in the budding cycle was observed in the untransformed double mutant. The VCY1 strain, bearing the cdc10-11 mutation, which displays a phenotype identical to the cdc10 deletion (Cid et al., 1998a ), was also studied in comparison with its isogenic wild type strain 1784. In this case, a delay of 8 min was recorded, which we do not consider significant taking into account that the buds developed by these mutants were much longer than normal buds. As expected, expression of the cdc15 mutation alone generated a 36-min delay as compared with the control. Surprisingly, in the double cdc10 cdc15 mutant only a 6-min delay was recorded, a figure comparable to the data on the expression of the cdc10 mutation alone. Controls at 28°C yielded no delay, suggesting that these observations derive from the phenotypic expression of the mutations and are not due to the idiosyncrasy of each transformant.
Table VII.
Rescue of the cdc15 Budding Delay in a cdc10 Background
| Strain | Average budding time at 37°C | Average budding time at 28°C | Delay at 37°C* | |||
|---|---|---|---|---|---|---|
| min | min | min | ||||
| VCY1 (cdc10-11) | 91 ± 9 (22 cells) | 104 ± 5 (20 cells) | 8 | |||
| 1784 (CDC10 isogenic to VCY1) | 83 ± 6 (11 cells) | 103 ± 6 (24 cells) | ||||
| VCY242d (cdc10-11 cdc15-1) | 84 ± 10 (25 cells) | 96 ± 6 (16 cells) | 7 | |||
| VCY242d + pBS9 (CDC15) | 81 ± 6 (27 cells) | 95 ± (14 cells) | 4 | |||
| VCY242d + YCp111-CDC10 | 119 ± 17 (40 cells) | 95 ± 6 (14 cells) | 42 | |||
| VCY242d + pBS9 (CDC15) + YCp111-CDC10 | 77 ± 6 (26 cells) | 95 ± 9 (16 cells) |
Average budding time both at 37°C and 28°C in cells from a cdc10-11 mutant and a double cdc15 cdc10 mutant and their respective isogenic wild-type or complemented controls.
The delay is calculated by subtracting the budding time of the isogenic wild-type or complemented control from the budding time of the mutant.
An experiment to demonstrate that rebudding runs parallel to a new S phase at nuclear level is shown in Fig. 7. FACS® analysis revealed that the evolution of the peak corresponding to 4n DNA content described above was delayed about 30 min in the transformant bearing a wild-type CDC10 with respect to the untransformed cdc10-11 cdc15-1 mutant. According to these results, the expression of a septin mutation prevents the temporal cell cycle arrest caused by the cytokinetic defects due to a loss of function in the CDC15 gene. A plausible explanation for this phenomenon is that the integrity of the septin ring would be essential for a putative cell cycle checkpoint (see Discussion), which would prevent the nucleus from starting a new round of the cell cycle unless cytokinesis has been committed.
Figure 7.

Flow cytometry plots of RNase-treated propidium iodide-stained cells of the VCY242d strain both untransformed (left) and transformed with a CDC10-bearing plasmid (right). Cells were incubated at 37°C in osmotically stabilized media. Samples were taken at the times indicated. Peaks that appear at 15, 30, and 60 arbitrary fluorescence units (see the scale at the bottom) correspond respectively to cells with DNA contents equivalent to 1, 2, and 4 nuclei. Comparison of the two series reveals an earlier (∼30 min) and larger amount of cells included in the third peak when both cdc10 and cdc15 mutations are coexpressed than when the cdc15 mutation is expressed alone.
Discussion
Evidence for a Role of the Cdc15p Pathway in Signaling for Cytokinesis
The connection between cell cycle regulation and morphogenesis is very poorly understood at the molecular level. Assuming that lysis in yeast cells can be a consequence of a failure in the pathways that coordinate morphogenetic responses, the study of lytic mutants may offer a valuable approach to this issue. The lyt1 mutant has been reported to display a pattern of cell division cycle arrest at the mitosis stage (Molero et al., 1993). Here we report that lyt1 in fact determines a new mutant allele of the CDC15 gene, which encodes a putative serine/threonine protein kinase (Schweitzer and Philippsen, 1991) essential for exit from the M phase in the S. cerevisiae cell cycle (Surana et al., 1993).
Some efforts have been made to introduce the product of the CDC15 gene into a signal transduction pathway. Shirayama et al. (1994) found that a high dose of CDC15 suppresses a mutation in the RAS-like gene TEM1 and proposed the existence of a hypothetical pathway that would be essential for the anaphase–telophase transition (Shirayama et al., 1994). Currently, the candidates for this pathway are the protein kinases Cdc15p, Cdc5p, Dbf2p, and Dbf20p, the protein phosphatase Cdc14p, the small GTPase Tem1p, and the SPO12 gene product (Toyn and Johnston, 1993; Kitada et al., 1993; Shirayama et al., 1994, 1996; Toyn et al., 1996; Jaspersen et al., 1998). Although there is no biochemical evidence of a pathway in which such components are sequentially integrated, these proteins must function as a regulatory network that controls important events for progression through the last stages of the cell cycle. The same events are dependent on B cyclin removal, but it remains uncertain whether the Cdc15p pathway (either directly or indirectly) promotes cyclin degradation by the APC or whether, conversely, it is cyclin depletion that activates the Cdc15p pathway. Based on the evidence that high Cdc28p kinase and low APC activity levels are observed in cdc15 mutants, Surana et al. (1993), Toyn et al. (1996), and Jaspersen et al. (1998) proposed that a role of Cdc15p might be to promote B cyclin removal. An alternative possibility would be that the pathways in which Cdc15p and functionally related proteins are involved have specific cellular targets for the promotion of telophase and/or cytokinesis themselves. In this case, the accumulation of B cyclins in these mutants would be due to negative feedback controls or checkpoint mechanisms.
Whatever the case, hints are gathering that the Cdc15p pathway may play a role in activation of the septum as a morphogenetic site, as it has been hypothesized for its putative counterpart in S. pombe, cdc7 (Sohrmann et al., 1998). The cdc15-lyt1 mutation maps in a domain different from the protein kinase consensus. Interestingly, the cdc7 and Cdc15p sequences are very similar in the region around Gly410, where the cdc15-lyt1 mutation maps (Fig. 1), but quite divergent in the rest of the sequence except for the protein kinase domains. The fission yeast cdc7 is essential for the formation of the septum but dispensable for nuclear division (Fankhauser and Simanis, 1994), strongly supporting the hypothesis of a direct involvement of the Cdc15p protein kinase in the commitment of cytokinesis. Moreover, a S. pombe homologue of Tem1p, namely spg1, has been reported to modulate the function of cdc7 in septum development (Schmidt et al., 1997). Sohrmann et al. (1998) have reported that cdc7 localizes to the spindle pole body at certain stages of mitosis, and that its localization depends on prior activation of the small GTPase spg1. Preliminary results on the localization of Cdc15p indicate that it mainly mimics that of cdc7 in the fission yeast (Jiménez, J., V.J. Cid, R. Cenamor, C. Nombela, and M. Sánchez, unpublished results), further supporting the notion that both kinases share a conserved function.
Recent evidence provided by Epp and Chant (1997), Lippincott and Li (1998), and Bi et al. (1998) has begun to unravel the molecular mechanisms that support cytokinesis. These authors report the existence of a contractile actin/myosin II ring-shaped structure at the time and site of septation. The correct organization of this structure depends on previous and accurate location of the earlier septin-based double ring and subsequent attachment to the neck of Myo1p and Iqg1/Cyk1p, respectively myosin II and an IQGAP-like protein. Lippincott and Li (1998) observed that the recruitment of both Iqg1/Cyk1p and actin to this ring depends on the function of the Cdc15p pathway. Despite this, Cdc15p may trigger additional events that are crucial for septation, since the contractile actin-myosin II ring is not essential for cytokinesis (Bi et al., 1998), whereas we show that CDC15 is strictly essential for the recognition of the septum area as a morphogenetic site.
A New Round of the Cell Cycle Can Be Started in the Absence of Cytokinesis
As seen in Figs. 2 and 6, cells that express mutations that arrest the cell cycle in anaphase thus failing to locate actin to the cytokinesis site, develop a distal projection. Pringle and Hartwell (1981) had already noticed the appearance of elongated cells on their early work on the characterization of certain cell cycle mutants. Several observations indicate that this abnormal morphogenetic response may correspond to a new round of the cell cycle: i.e., recognition of the distal pole depends on the basic polarity signaling pathways for budding led by Bud1p (Chant, 1994); START-defective cdc15 mutants do not develop such projections and, apparently correlative with this morphogenetic response, nuclei start DNA replication. Thus, assuming that the observed phenomena constitute the development of a new bud in cytokinesis-defective cells, we found that this new budding process was delayed by ∼40 min as compared with normal budding. A comparison of a normal cell cycle to the abnormal morphogenetic response in cdc15 mutants is offered in Fig. 8.
Figure 8.

Scheme of bud development and its estimated timing, as deduced from budding time assays, in a wild-type strain, haploid and diploid cdc15 mutants. Haploid cdc15 mutants fail to localize actin to the bud-daughter constriction at the end of mitosis and display an arrest in the transition from anaphase to telophase. The mother cell fails to accomplish any morphogenetic response in a haploid background, but the daughter cell polarizes growth with a delay of 45 min with respect to a wild-type daughter. Such polarization follows an incorrect distal pattern. This behavior leads to immediate cell lysis. In contrast, diploid cdc15/ cdc15 mutants are frequently able to develop a new bud at the distal pole after overcoming mitotic arrest and the mother cell also eventually starts a new round of the cell cycle.
From the morphogenetic point of view, the apical projection in cdc15 and functionally related mutants lacks a proper constriction at its base and tends to elongate, resembling a shmoo until cell lysis takes place. There is a strong polarization of actin towards this growing tip in cdc15 cell populations kept at 37°C for more than 2 h. The aberrant morphology may be due to a failure in the assembly of the septin ring at the new bud site since septin mutants also develop elongated buds (Hartwell, 1971). In this context, cell lysis in cdc15 mutants is probably a consequence of an actin-based morphogenetic response not sustained by the septin cytoskeleton. In diploid cdc15/cdc15 mutants, a new septin ring is assembled at the base of successful apical buds, leading to the development of nonlysing chains (Fig. 8). This suggests that in diploid strains the distal pole is more favored as a budding site than in haploid strains, because some determinants essential for the recruitment of septins may be present at that pole which are lacking in haploid strains (Zahner et al., 1996).
It is also worth noting that the development of either a distal projection or a new bud develops in an asynchronous fashion in mother and daughter cells even though they are not physically separated. In haploid backgrounds, the daughter cell develops a distal projection while the mother remains unchanged, although a 4n peak in FACS® analyses reveals that both nuclei do begin to replicate. In diploid backgrounds, which often escape immediate lysis, the mother cell does bud, although much later than the daughter. The fact that the daughter cell starts budding faster than the mother after the cytokinetic arrest contrasts with normal cellular behavior. In a typical cell cycle, the daughter cell must reach a critical size before starting a new cycle, whereas the mother can bud immediately after cytokinesis (Roemer et al., 1996).
Regardless of the consequences for cell polarity or the synchronicity of the phenomena derived from the cytokinetic failure, the most significant observations are that cdc15 (and other late mitotic mutants) eventually overcome their nuclear arrest in anaphase and that a new round of cell cycle can be successfully accomplished in the absence of cytokinesis. The dispensability of cytokinesis for bud emergence and DNA replication in a subsequent cycle was first noted by Hartwell (1971) studying septin mutants in his pioneer work on the S. cerevisiae cell cycle. Nevertheless, the cytokinetic failure in septin mutants is rather a consequence of the mislocalization of septa than a total inactivation of the morphogenetic machinery responsible for septation (Longtine et al., 1996; Cid et al., 1998a ).
Is There a Cytokinesis Checkpoint in the Budding Yeast?
Lew and Reed (1993) reported that the actin cytoskeleton is not polarized to the septum area to promote cytokinesis until the Cdc28p–Clb2p complex has become dissociated. Here we report that when anaphase arrest persists, a polar budding process is started in the absence of cytokinesis. It could be surmised that the inactivation of the Cdc28p–Clb complex by degradation of B cyclins would be sufficient for the promotion of both nuclear telophase events and cytokinesis and that the Cdc15p pathway would be essential for inactivating the CDK–cyclin complex at this stage. We shall refer to this as the classical hypothesis (Fig. 9 a). Following this interpretation, it is difficult to understand some of our results, such as the overcoming of anaphase arrest in the absence of cytokinesis in cdc15 and, especially, the absence of such an arrest in a double cdc15 cdc10 mutant. A plausible explanation is that there would be a late or slow cyclin degradation mechanism induced by a less efficient pathway, different from that of Cdc15p, and that such partial inactivation of Cdc28p would be sufficient to induce nuclear exit from mitosis but not to polarize actin to the mother–bud neck. Low cAMP levels rescue the cell cycle arrest caused by a cdc15 mutation (Spevak et al., 1993) and therefore alternative pathways for cyclin degradation could rely on intracellular cAMP levels. However, with our current knowledge it is still hard to explain how a failure in the septin ring could influence cAMP-dependent pathways. We studied the nature of the suppression of the cdc15-lyt1 phenotype by overexpressing the gene coding for S. cerevisiae phosphodiesterase, PDE1, and found that it rescued not only the anaphase arrest but also the septation defect (data not shown). Cdc15p might therefore activate morphogenesis in the septum via pathways involving an inactivation of the cAMP-dependent protein kinase.
Figure 9.
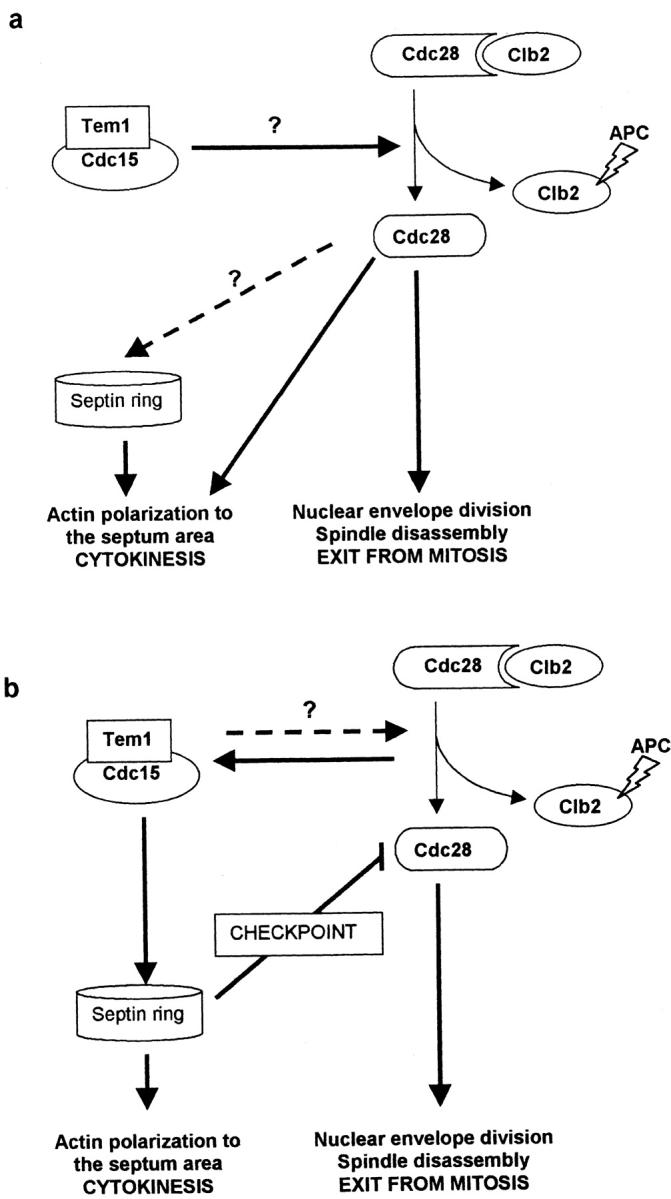
Hypotheses for the mechanisms involved in the coordination of morphogenetic events at the end of mitosis. Hypothesis (a) implies that the Cdc15p-mediated pathway is directly or indirectly involved in B cyclin degradation by the APC and that the consequent inactivation of the Cdc28p–B cyclin complex accounts for the polarization of the morphogenetic apparatus to the neck area for septum development. Hypothesis (b) argues that the Cdc15p pathway, similar to the cdc7 pathway in the fission yeast, would directly trigger the morphogenetic response for septum formation, perhaps activated by signals derived from Cdc28p–B cyclin inactivation or coordinated with it via feedback mechanisms (broken line). The arrest in anaphase observed in cdc15 and related mutants would be due to a cell cycle checkpoint that would inactivate Cdc28p until morphogenetic events at the neck are settled. The fact that a septin (cdc10) mutation prevents the delay in budding observed in a cdc15 mutant suggest that the components of such putative checkpoint rely on the integrity of the septin ring for their functionality.
Although we cannot discard the possibility that the Cdc15p pathway might play a direct role in cyclin degradation, we propose a second hypothesis that would involve the existence of a novel morphogenetic checkpoint (Fig. 9 b). According to the role of the homologous proteins in the fission yeast (Schmidt et al., 1997; Sohrmann et al., 1998) and to the results presented by Lippincott and Li (1998) together with our own, the Cdc15p pathway, directly or indirectly, could play an essential role in the localization of actin towards the neck area in order to accomplish septum formation. Therefore, as stated above, we could assume that Cdc15p, Tem1p, and functionally related proteins could be involved in signaling pathways for the activation of the neck as a morphogenetically active spot at a late mitotic stage. The activation of this pathway could be triggered by the inactivation of the Cdc28–B cyclin complex itself. How then can one explain why the Cdc15p pathway is also important, although transiently, for the nuclear exit from mitosis? A hypothetical checkpoint would inhibit the CDK activity, arresting the nuclear cycle in late anaphase until the septum area has become morphogenetically active. Such a mechanism would ensure the synchronicity of both cell-surface and nuclear events for successful completion of the cell cycle. In cdc15 or tem1 mutants the checkpoint would be activated, leading to a more or less efficient anaphase arrest. Here we report that such an arrest is effective for 30–45 min in the daughter cell and for even longer in the mother cell. Interestingly, a morphogenetic checkpoint dependent on cytoskeletal structures has already been characterized in S. cerevisiae (Lew and Reed, 1995; McMillan et al., 1998). Such checkpoint prevents DNA replication upon failures in the assembly of the cytoskeletal apparatus for bud emergence. Similar cytoskeleton-dependent checkpoint mechanisms could exist in other stages of cell cycle, such as cytokinesis.
From our results we conclude that the integrity of the septin ring is essential for the mitotic arrest observed in a cdc15 mutant. This might imply that some important component of the hypothetical cytokinesis checkpoint proposed above lies on the septin ring. In double cdc10 cdc15 mutants, the lack of a functional septin ring in previous stages of the cell cycle might cause some Cdc15p targets to be lacking or unavailable. In fact, a putative downstream target of the Cdc15p pathway, the Iqg1/Cyk1p IQGAP-like protein, fails to localize to the neck in septin-cdc12 mutants (Lippincott and Li, 1998).
Although so far molecular evidence is lacking, it is likely that the septin ring would translate signals dependent on Cdc28p–Clb2p kinase activity for depolarization of the actin cytoskeleton. Accordingly, mutations in the septins trigger an abnormal hyperpolarization of growth in G2 (Adams and Pringle, 1984), a morphogenetic response identical to that caused by defects in the activation of the Cdc28p–Clb2p complex (Lew and Reed, 1993). On this basis, it seems logical that septins could also translate the morphogenetic response elicited by inactivation of the Cdc28p–Clb2p complex at the end of mitosis to promote septation.
An alternative explanation to the budding delay in cytokinetic-defective mutants is that such a delay could be due to a sequestration of elements important for morphogenesis by the septin ring persisting at the neck. Two observations, however, detract from this hypothesis. First, the overcoming of the arrest is not a strictly morphogenetic phenomenon, but instead seems to be accompanied by nuclear post-START events. Second, diploid cdc15/cdc15 mutants successfully reassemble septins at the new bud site, but the delay still exists.
All the available evidence points to the notion that cytokinesis relies on the cooperative role of both the septin filaments for its localization and the Cdc15p pathway for its timely activation. This pathway, as in the case of the fission yeast, may transduce signals from the cell cycle universal modulator, the CDK–cyclin complex, to the neck area where septins are located. In this work we also analyze the consequences of a cytokinetic failure in the following cell cycle and conclude that, in these conditions, septin assembly to the sites of polarity is essential for cell viability during budding and that, when septins manage to localize to a new site, a new round of the cell cycle can be started in the absence of cytokinesis. This whole array of observations opens new perspectives in the study of the coordination of morphogenetic events along the mitotic cycle.
Acknowledgments
We thank A. Toh-e, J. Chant, C. Kuhne, and L. Hartwell for sending us strains, J.R. Pringle (University of North Carolina, Chapel Hill, NC), M.I. Castañón, A. Aguilera, and M. Glotzer for plasmids, M. Molina, A. Bueno, S. Moreno (both from University of Salamanca, Salamanca, Spain), and E. Herrero (Universitat de Lleida, Lleida, Spain) for helpful discussion, A. Álvarez and A. Vazquez for flow cytometry and confocal microscopy, and O. de Pablo for valuable help in solving photographic problems.
This work was supported by grants from the Dirección General de Investigación Científica y Técnica (CICYT) (MEC) to J. Jiménez (AP93-06566768) and V.J. Cid (PB91-919-01), and grants from CICYT (BIO95-0303) and Fondo de Investigaciones de la Seguridad Social (FISS) (97/ 0047-01).
Abbreviations used in this paper
- APC
anaphase-promoting complex
- CDK
cell cycle-dependent protein kinase
- GFP
green fluorescent protein
- ORF
open reading frame
Footnotes
J. Jimenez and V.J. Cid contributed equally to this work.
References
- Adams A, Pringle JR. Relationship of actin and tubulin distribution to bud growth in wild-type and morphogenetic mutant Saccharomyces cerevisiae. . J Cell Biol. 1984;98:934–945. doi: 10.1083/jcb.98.3.934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ausubel, F.M., R.E. Kingston, R. Brent, D.D. Moore, J.G. Seidman, J.A. Smith, and K. Struhl. 1993. Current Protocols in Molecular Biology. Greene Publishing Associates and Wiley Interscience, New York.
- Bi E, Maddox P, Lew DJ, Salmon ED, McMillan JN, Yeh E, Pringle JR. Involvement of an actomyosin contractile ring in Saccharomyces cerevisiaecytokinesis. J Cell Biol. 1998;142:1301–1312. doi: 10.1083/jcb.142.5.1301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Botstein, D., D. Amberg, J. Mulholland, T. Huffaker, A. Adams, D. Drubin, and T. Stearns. 1997. The yeast cytoskeleton. In The Molecular and Cellular Biology of the Yeast Saccharomyces cerevisiae. J.R. Pringle, J.R., Broach, and E.W. Jones, editors. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY. 1–90.
- Cabib E, Durán A. Simple and sensitive procedure for screening yeast mutants that lyse at nonpermissive temperatures. J Bacteriol. 1975;124:1604–1606. doi: 10.1128/jb.124.3.1604-1606.1975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chant J. Cell polarity in yeast. Trends Genet. 1994;10:328–333. doi: 10.1016/0168-9525(94)90036-1. [DOI] [PubMed] [Google Scholar]
- Chant J, Herskowitz I. Genetic control of bud site selection in yeast by a set of gene products that constitute a morphogenetic pathway. Cell. 1991;65:1203–1212. doi: 10.1016/0092-8674(91)90015-q. [DOI] [PubMed] [Google Scholar]
- Cid, V.J., L. Adamiková, R. Cenamor, M. Molina, M. Sánchez, and C. Nombela. 1998a Cell integrity and morphogenesis in a budding yeast septin mutant. Microbiology. 142: In press. [DOI] [PubMed]
- Cid VJ, Cenamor R, Sánchez M, Nombela C. A mutation in the Rho1-GAP-encoding gene BEM2 of Saccharomyces cerevisiaeaffects morphogenesis and cell wall functionality. Microbiology. 1998b;144:25–36. doi: 10.1099/00221287-144-1-25. [DOI] [PubMed] [Google Scholar]
- Cid VJ, Durán A, del Rey F, Snyder MP, Nombela C, Sánchez M. Molecular basis of cell integrity and morphogenesis in Saccharomyces cerevisiae. . Microbiol Rev. 1995;59:345–386. doi: 10.1128/mr.59.3.345-386.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Copeland CS, Snyder M. Nuclear pore complex antigens delineate nuclear envelope dynamics in vegetative and conjugating Saccharomyces cerevisiae. . Yeast. 1993;9:235–249. doi: 10.1002/yea.320090304. [DOI] [PubMed] [Google Scholar]
- Culotti J, Hartwell LH. Genetic control of cell division cycle in yeast. III. Seven genes controlling nuclear division. Exp Cell Res. 1971;67:389–401. doi: 10.1016/0014-4827(71)90424-1. [DOI] [PubMed] [Google Scholar]
- De la Fuente J, Álvarez A, Nombela C, Sánchez M. Flow cytometric analysis of Saccharomyces cerevisiaeautolytic mutants and protoplasts. Yeast. 1992;8:39–45. doi: 10.1002/yea.320080104. [DOI] [PubMed] [Google Scholar]
- De Marini DJ, Adams AEM, Fares H, DeVirgilio C, Valle G, Chuang JS, Pringle JR. A septin-based hierarchy of proteins required for localized deposition of chitin in Saccharomyces cerevisiaecell wall. J Cell Biol. 1997;139:75–93. doi: 10.1083/jcb.139.1.75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drubin DG, Jones HD, Wertman KF. Actin structure and functions: roles in mitochondrial organization and morphogenesis in budding yeast and identification of the phalloidin binding site. Mol Cell Biol. 1993;4:1277–1294. doi: 10.1091/mbc.4.12.1277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Epp J.A., and J. Chant. 1997. An IQGAP-related protein controls actin-ring formation and cytokinesis in yeast. Curr. Biol. 7:921–929. [DOI] [PubMed]
- Fankhauser C, Simanis V. The cdc7protein kinase is a dosage-dependent regulator of septum formation in fission yeast. EMBO (Eur Mol Biol Organ) J. 1994;13:3011–3019. doi: 10.1002/j.1460-2075.1994.tb06600.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gabriel M, Kopecká M. Disruption of the actin cytoskeleton in budding yeast results in formation of an aberrant cell wall. Microbiol. 1995;141:891–899. doi: 10.1099/13500872-141-4-891. [DOI] [PubMed] [Google Scholar]
- Hartwell LH. Genetic control of cell division cycle in yeast. IV. Genes controlling bud emergence and cytokynesis. Exp Cell Res. 1971;69:265. doi: 10.1016/0014-4827(71)90223-0. [DOI] [PubMed] [Google Scholar]
- Ito H, Fukada Y, Murata K, Kimura A. Transformation of intact yeast with alkali cations. J Bacteriol. 1983;153:163–168. doi: 10.1128/jb.153.1.163-168.1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaspersen SL, Charles JF, Tinker-Kulberg RL, Morgan DO. A late mitotic regulatory network controlling cyclin destruction in Saccharomyces cerevisiae. . Mol Biol Cell. 1998;9:2803–2817. doi: 10.1091/mbc.9.10.2803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnston LH, Eberly SL, Chapman JW, Araki H, Sugino A. The product of the Saccharomyces cerevisiae cell cycle gene DBF2has homology with protein kinases and is periodically expressed in the cell cycle. Mol Cell Biol. 1990;10:1358–1366. doi: 10.1128/mcb.10.4.1358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitada K, Johnson AL, Johnston LH, Sugino A. A multicopy suppressor gene of the Saccharomyces cerevisiae G1 cell cycle mutant gene dbf4 encodes a protein kinase and is identified as CDC5. . Mol Cell Biol. 1993;13:445–457. doi: 10.1128/mcb.13.7.4445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lew DJ, Reed SI. Morphogenesis in the yeast cell cycle: regulation by cdc28 and cyclins. J Cell Biol. 1993;120:1305–1320. doi: 10.1083/jcb.120.6.1305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lew DJ, Reed SI. Cell cycle control of morphogenesis in the budding yeast. Curr Opin Genet Dev. 1995;5:17–23. doi: 10.1016/s0959-437x(95)90048-9. [DOI] [PubMed] [Google Scholar]
- Lippincott J, Li R. Sequential assembly of myosin II, an IQGAP-like protein, and filamentous actin to a ring structure involved in budding yeast cytokinesis. J Cell Biol. 1998;140:355–366. doi: 10.1083/jcb.140.2.355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Longtine SM, de Marini DJ, Valencik ML, Al-Awar OS, Fares H, De Virgilio C, Pringle JR. The septins: roles in cytokinesis and other processes. Curr Opin Cell Biol. 1996;8:106–119. doi: 10.1016/s0955-0674(96)80054-8. [DOI] [PubMed] [Google Scholar]
- McMillan JN, Sia RAL, Lew DJ. A morphogenesis checkpoint monitors the actin cytoskeleton in yeast. J Cell Biol. 1998;142:1487–1499. doi: 10.1083/jcb.142.6.1487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molero G, Yuste-Rojas M, Montesi A, Vázquez A, Nombela C, Sánchez M. A cdc-like autolytic Saccharomyces cerevisiae mutant altered in budding site selection is complemented by SPO12, a sporulation gene. J Bacteriol. 1993;172:6562–6570. doi: 10.1128/jb.175.20.6562-6570.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park HO, Erfei B, Pringle JR, Herskowitz I. Two active states of the Ras-related Bud1/Rsr1 protein bind to different effectors to determine yeast cell polarity. Proc Natl Acad Sci USA. 1997;94:4463–4468. doi: 10.1073/pnas.94.9.4463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pringle JR. Staining of bud scars and other cell wall chitin with calcofluor. Methods Enzymol. 1991;194:732–735. doi: 10.1016/0076-6879(91)94055-h. [DOI] [PubMed] [Google Scholar]
- Pringle, J.R., and L.H. Hartwell. 1981. The Saccharomyces cerevisiae cell cycle. In The Molecular and Cellular Biology of the Yeast Saccharomyces cerevisiae. J.N. Strathern, E.W. Jones and J.R. Broach, editors. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY. 97–142.
- Pringle JR, Preston RA, Adams AEM, Stearns T, Drubin DG, Haarer BK, Jones EW. Fluorescence microscopy methods for yeast. Methods Cell Biol. 1989;31:357–435. doi: 10.1016/s0091-679x(08)61620-9. [DOI] [PubMed] [Google Scholar]
- Reid B, Hartwell LH. Regulation of mating in the cell cycle of S. cerevisiae. . J Cell Biol. 1977;75:355–365. doi: 10.1083/jcb.75.2.355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roemer T, Vallier LG, Snyder M. Selection of polarized growth sites in yeast. Trends Cell Biol. 1996;6:434–441. doi: 10.1016/s0962-8924(96)10039-8. [DOI] [PubMed] [Google Scholar]
- Rose MD, Novick P, Thomas JH, Botstein D, Fink GR. A S. cerevisiaegenomic plasmid bank based on a centromere-containing shuttle vector. Gene. 1987;60:237–243. doi: 10.1016/0378-1119(87)90232-0. [DOI] [PubMed] [Google Scholar]
- Sambrook, J., E.F. Fritsch, and T. Maniatis. 1989. Molecular Cloning: A Laboratory Manual. Cold Spring Harbor Laboratory Press. Cold Spring Harbor, NY. 546 pp.
- Sanger F, Nicklen S, Coulson AR. DNA sequencing with chain-terminating inhibitors. Proc Natl Acad Sci USA. 1977;74:5463–5467. doi: 10.1073/pnas.74.12.5463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt S, Sohrmann M, Hofmann K, Woollard A, Simanis V. The Spg1p GTPase is an essential, dosage-dependent inducer of septum formation in Schizosaccharomyces pombe. . Genes Dev. 1997;11:1519–1534. doi: 10.1101/gad.11.12.1519. [DOI] [PubMed] [Google Scholar]
- Schweitzer B, Philippsen P. CDC15, an essential cell cycle gene in Saccharomyces cerevisiae, encodes a protein kinase domain. Yeast. 1991;7:265–273. doi: 10.1002/yea.320070308. [DOI] [PubMed] [Google Scholar]
- Shirayama M, Matsui Y, Toh-e A. The yeast TEM1gene, which encodes a GTP-binding protein, is involved in termination of M phase. Mol Cell Biol. 1994;14:7476–7482. doi: 10.1128/mcb.14.11.7476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shirayama M, Matsui Y, Toh-e A. Dominant mutant alleles of yeast protein kinase gene CDC15 suppress the lte1 defect in termination of M phase and genetically interact with CDC14. . Mol Gen Genet. 1996;251:176–185. doi: 10.1007/BF02172916. [DOI] [PubMed] [Google Scholar]
- Sikorski RS, Hieter P. A system of shuttle vectors and yeast host strains designed for efficient manipulation of DNA in Saccharomyces cerevisiae. . Genetics. 1989;122:19–27. doi: 10.1093/genetics/122.1.19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sohrmann M, Schmidt S, Hagan I, Simanis V. Asymmetric segregation on spindle poles of the Schizosaccharomyces pombeseptum-inducing protein kinase Cdc7p. Genes Dev. 1998;12:84–94. doi: 10.1101/gad.12.1.84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spevak W, Keiper BD, Stratowa C, Castañón MJ. Saccharomyces cerevisiae cdc15 mutants arrested at late stage in anaphase are rescued by Xenopus cDNAs encoding N-rasor a protein with β-transducin repeats. Mol Cell Biol. 1993;13:4953–4966. doi: 10.1128/mcb.13.8.4953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Streiblová, E. 1988. Cytological Methods in Yeast: A Practical Approach. I. Campbell and J.H. Duffus, editors. IRL Press, Oxford, UK. 9–49.
- Surana U, Amon A, Dowzer C, McGrew J, Byers B, Nasmyth K. Destruction of yeast CDC28/CLB mitotic kinase is not required for the metaphase to anaphase transition in budding yeast. EMBO (Eur Mol Biol Organ) J. 1993;12:1969–1978. doi: 10.1002/j.1460-2075.1993.tb05846.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor SG, Liu Y, Baskerville C, Charbonneau H. The activity of Cdc14p, an oligomeric dual specificity protein phosphatase from Saccharomyces cerevisiae, is required for cell cycle progression. J Biol Chem. 1997;272:24054–24063. doi: 10.1074/jbc.272.38.24054. [DOI] [PubMed] [Google Scholar]
- Torres L, Martín H, García-Saez MI, Arroyo J, Molina M, Sánchez M, Nombela C. A protein kinase gene complements the lytic phenotype of Saccharomyces cerevisiae lyt2mutants. Mol Microbiol. 1991;5:2845–2854. doi: 10.1111/j.1365-2958.1991.tb01993.x. [DOI] [PubMed] [Google Scholar]
- Toyn JH, Johnston LH. Spo12 is a limiting factor that interacts with the cell cycle protein kinases Dbf2 and Dbf20, which are involved in mitotic chromatic disjunction. Genetics. 1993;135:963–971. doi: 10.1093/genetics/135.4.963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toyn JH, Johnston LH. The Dbf2 and Dbf20 protein kinases of budding yeast are activated after the metaphase to anaphase cell cycle transition. EMBO (Eur Mol Biol Organ) J. 1994;13:1103–1113. doi: 10.1002/j.1460-2075.1994.tb06359.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toyn JH, Johnson AL, Donovan D, Toone WM, Johnston LH. The Swi5 transcription factor of Saccharomyces cerevisiaehas a role in exit from mitosis through induction of the cdk-inhibitor Sic1 in telophase. Genetics. 1996;145:85–96. doi: 10.1093/genetics/145.1.85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wan J, Xu H, Grunstein M. CDC14 of Saccharomyces cerevisiae. . J Biol Chem. 1992;267:11274–11280. [PubMed] [Google Scholar]
- Williams ST, Veldkamp CY. Preparation of fungi for scanning electron microscopy. Trans Br Mycol Soc. 1974;63:409–412. [Google Scholar]
- Zachariae W, Nasmyth K. TPR proteins required for anaphase progression mediate ubiquitination of mitotic B-type cyclins in yeast. Mol Biol Cell. 1996;7:791–801. doi: 10.1091/mbc.7.5.791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zahner JE, Harkins HA, Pringle JR. Genetic analysis of the bipolar pattern of bud site selection in the yeast Saccharomyces cerevisiae. . Mol Cell Biol. 1996;16:1857–1870. doi: 10.1128/mcb.16.4.1857. [DOI] [PMC free article] [PubMed] [Google Scholar]