

Abstract
Voltage-gated sodium channels (NaCh) are colocalized with isoforms of the membrane-skeletal protein ankyrinG at axon initial segments, nodes of Ranvier, and postsynaptic folds of the mammalian neuromuscular junction. The role of ankyrinG in directing NaCh localization to axon initial segments was evaluated by region-specific knockout of ankyrinG in the mouse cerebellum. Mutant mice exhibited a progressive ataxia beginning around postnatal day P16 and subsequent loss of Purkinje neurons. In mutant mouse cerebella, NaCh were absent from axon initial segments of granule cell neurons, and Purkinje cells showed deficiencies in their ability to initiate action potentials and support rapid, repetitive firing. Neurofascin, a member of the L1CAM family of ankyrin-binding cell adhesion molecules, also exhibited impaired localization to initial segments of Purkinje cell neurons. These results demonstrate that ankyrinG is essential for clustering NaCh and neurofascin at axon initial segments and is required for physiological levels of sodium channel activity.
Keywords: ankyrinG, sodium channel, neurofascin, clustering, action potential
Clustering of voltage-gated sodium channels (NaCh)1 at axon initial segments, nodes of Ranvier, and postsynaptic folds of the neuromuscular junction is vital for generating sufficient local current to overwhelm membrane capacitance and resistance, and to initiate and propagate a self-regenerating action potential. Understanding the mechanisms for formation and maintenance of these NaCh-enriched domains represents a challenge with important implications for the physiology of excitable membranes as well as general issues of cell polarity and membrane structure.
Initial clues regarding possible molecular neighbors of the NaCh that could determine membrane localization came from observations that the NaCh copurified with the membrane skeletal protein ankyrin and associated with ankyrin in in vitro assays (Srinivasan et al., 1988). Ankyrin subsequently was localized at axon initial segments and nodes of Ranvier (Kordeli et al., 1990; Kordeli and Bennett, 1991), sites where the existence of a high density of NaCh has been well documented (Catterall, 1981; Ellisman and Levinson, 1982; Wollner and Catterall, 1986). Ankyrin also was localized to the NaCh-enriched regions of the neuromuscular junction (Flucher and Daniels, 1989). The isoforms of ankyrin localized at nodes of Ranvier, and axon initial segments have been identified as 480- and 270-kD alternatively spliced variants of ankyrinG (Kordeli et al., 1995). AnkyrinG is present in the postsynaptic folds of the neuromuscular junction (Kordeli et al., 1998; Wood and Slater, 1998) and is associated with NaCh at early stages in morphogenesis of the node of Ranvier (Lambert et al., 1997). AnkyrinG also is associated with NaCh clusters in the dystrophic mouse in regions lacking myelination (Deerinck et al., 1997), as well as clusters of NaCh induced in cultured retinal ganglion neurons (Kaplan et al., 1997). Together, these results provide circumstantial evidence for a role of ankyrinG in the clustering of NaCh to specific membrane regions.
The ankyrins are a family of spectrin-binding proteins associated with the cytoplasmic surface of the plasma membrane in many cell types. Ankyrins associate via their membrane-binding domains with several ion channels in addition to the voltage-gated Na channel, as well as calcium release channels and the L1CAM family of cell adhesion molecules (Bennett et al., 1997). The nodal/initial segment ankyrinG isoforms are distinguished by a serine/ threonine-rich domain glycosylated with O-GlucNAc residues (Zhang and Bennett, 1996) and also contain an extended tail domain (Chan et al., 1993; Kordeli et al., 1995). The membrane-binding domain of ankyrins contains four subdomains, each composed of six ankyrin repeats (Michaely and Bennett, 1993). Biochemical studies (Michaely and Bennett, 1995a ,b) suggest that ankyrin is multivalent and could form lateral homo- and/or heterocomplexes between integral membrane proteins.
Isoforms of neurofascin and NrCAM, members of the L1CAM family of ankyrin-binding cell adhesion molecules (Hortsch, 1996), are colocalized with ankyrinG at the nodes of Ranvier in peripheral nerve and axon initial segments of Purkinje cell neurons (Davis et al., 1993, 1996). Clustering of neurofascin and NrCAM precedes redistribution of ankyrinG 480/270 kD and the NaCh during development of the node of Ranvier, which has prompted the hypothesis that these cell adhesion molecules may define the initial site for subsequent assembly of ankyrinG and the NaCh (Lambert et al., 1997).
This study evaluates the cellular and physiological consequences of the knockout of a cerebellar isoform of ankyrinG in mice. In this mutant mouse, we studied the targeting of neurofascin and NaCh and the firing properties of Purkinje cells in cerebellar slices. Immunofluorescence evidence suggests that NaCh are absent from axon initial segments of granule cells of mutant mice. In addition, Purkinje cells demonstrate severe deficits in action potential firing in response to somatic current injection, suggesting a reduced density or impaired localization of NaCh in mice lacking cerebellar ankyrinG. Neurofascin also exhibited impaired targeting to initial segments of Purkinje cell neurons. Together, these results provide direct evidence for an essential physiological role of ankyrinG in the normal function of NaCh as well as targeting of NaCh and a companion cell adhesion molecule to axon initial segments.
Materials and Methods
Targeted Disruption of Exon1b Locus
A 14-kb genomic DNA covering exon1b and upstream promoter sequence was isolated from a mouse 129SvEV genomic library (Stratagene, La Jolla, CA) and subcloned into pBS vector (Stratagene). A Neo gene flanked with SpeI and ApaI sites was inserted into the SmaI site. The resulting DNA was ligated with the tk gene and subcloned into pGEM11Z (Promega Corp., Madison, WI), linearized with NotI, and electroporated into an R1 ES cell line. Among 200 clones selected by G418 (200 μg/ml) and ganciclovir (2 μM), six positive clones were identified by Southern blot. They were expanded and injected into blastocysts. Male chimeras were bred to C57BL/6J to generate F1 offsprings. Heterozygous F1 mice were bred to generate the homozygous mutant mice. The exon1b-null (mutant) mice were confirmed by standard Northern, Southern, and Western blots. To reduce the variability of the mutant mouse phenotypes caused by different genetic backgrounds, the mutant mice and their littermates were kept at 50% C57Bl/6 and 50% Sv129 genetic background. All of the six founders from six independent ES cells gave rise to mutant mice with a similar phenotype.
Northern, Southern, and Western Blots
Northern blots were performed as described (Kordeli et al., 1995). The multiple tissue blot was purchased from CLONTECH Laboratories, Inc. (Palo Alto, CA). The blot shown in Fig. 2 was run according to the glyoxal/DMSO method outlined in Sambrook et al. (1989). The exon1b, exon1e, and spectrin-binding domain (common) probes were generated by PCR using the following primers: exon1e, sense, 5′GAATTCGGGCCGCTCGAC3′, antisense, 5′GAATTCTTTTTTCCCTTTCCTCTT3′; exon1b, sense, 5′TGAGATCTGGCCCCTGCT3′, antisense, 5′GACCGTTTGCGGTGTTT3′; common region probe, sense, 5′GTGAACCTGGTCTCAAGC3′, antisense, 5′GCATATCAGCTCTCTGTA3′. The blots were hybridized with the exon1b probe first and then stripped and rehybridized with exon1e probe. The blot was stripped again and hybridized with a probe common to all the isoforms. The results were obtained by 1-d exposure. After each strip, the blot was exposed for at least 2 d to confirm the complete removal of the previous signal. Tail DNA was isolated for standard Southern blot as described (Sambrook et al., 1989). The Western blot procedure was as described previously (Lambert et al., 1997).
Figure 2.

Generation of exon1b-specific ankyrinG knockout mouse by targeted recombination. (A) The restriction map of the ankyrinG genomic sequence covering exon1b, the target vector, and the expected mutant gene. (B) Southern blot analysis of genomic DNA identified mice corresponding to all three expected genotypes. The SpeI-digested DNA were hybridized with the BamHI-SpeI probe as indicated in A. The 14-kb band corresponds to the wild-type allele, while the 6-kb band corresponds to the mutant allele. (C) Northern blot analysis revealed the specific knockout of exon1b-containing transcripts in the homozygous (−/−) mutant mouse. The blot containing RNA isolated from homozygous (left lane) and wild-type (right lane) mouse brain was sequentially hybridized with probe to exon1b, exon1e, and the probe to the common region. The blot was stripped to remove the previous signal after each hybridization. The result indicated that the exon1b-containing ankyrinG transcripts were specifically knocked-out. The 15-kb band seen in Fig. 1 was further separated into three bands in this blot because the gel was run longer and the glyoxal/DMSO method was used. (D) Western blot analysis demonstrated the region-specific knockout of ankyrinG in exon1b-specific knockout mouse. The cerebellum and frontal brain homogenate from wild-type (+/+), heterozygous (+/−), and the homozygous mutant (−/−) was run on a 3.5–17% gradient gel and stained with Coomassie blue dye (left) to monitor the amount of loading. A duplicate gel was transferred to nitrocellulose membrane and probed with antibodies to the tail region of the ankyrinG, which detected the 480- and 270-kD isoforms (right). The results indicate a dramatic reduction of ankyrinG expression in cerebellum. The residual ankyrinG seen in cerebellum of mutant mice was from the axons in the white matter that originated from the other parts of brain or spinal cord.
In Situ Hybridization
Whole brains from control or mutant mice were frozen on a piece of luminal foil on powdered dry ice and then processed for cryosectioning. The sections (15 μm) were collected on poly-l-lysine–coated slides and air-dried. After fixation in ice-cold 4% paraformaldehyde, the sections were stored in 95% ETOH at 4°C. The sections were prehybridized in “minilist” solution (50% formamide, 4× SSC, 10% dextran sulfate) and then hybridized overnight with 100 μl of minilist solution containing 50 pg of 33P-labeled probes per section at 42°C. The probes were synthetic 45-mer oligomers: exon1e, 5′CCT TGG GTA CCT CTA ATC AGG CAT AGG GCA GCA TAG CCC TCG CAG3′; exon1b, 5′CTT TCC TTG AGA TCA GGG GAG TTG AGA CGA GAC AGA AGA TCA CCT3′. They were freshly labeled before use with a tailing kit (Boehringer Mannheim Corp., Indianapolis, IN). The sections were then washed with 1× SSC at 60°C, dried, and exposed to Kodak XAR-5 film (Rochester, NY).
Immunocytochemistry
The immunofluorescence labeling was performed according to published procedures (Lambert et al., 1997). Polyclonal anti–sodium channel, neurofascin, and NrCAM antibodies were described elsewhere (Davis et al., 1996; Lambert et al., 1997). A polyclonal anti–type II sodium channel from Upstate Biotechnology (Lake Placid, NY) was used with similar results (data not shown). In addition, the following sodium channel antibodies were used: a polyclonal antibody against loopII of sodium channel and a polyclonal antibody against the common epitope (TEEQKKYYNAMKKLGSKK) of type I and II NaCh generated in this laboratory, and two type I NaCh–specific antibodies, two type II NaCh–specific antibodies, and two type I/type II shared epitope (same sequence as above) antibodies purchased from Upstate Biotechnology and Alomone Labs (Jerusalem, Israel), including the antisera originally designated anti-SP19 (Gordon et al., 1988). Chicken anti–ankyrinG antibody is described elsewhere (Zhang and Bennett, 1996). Monoclonal anticalbindin antibody was purchased from Boehringer Mannheim Corp. Monoclonal antineurofilament antibody NN18 was from Sigma Chemical Co. (St. Louis, MO).
Electrophysiology
Parasagittal slices of cerebellar vermis were prepared from mice using standard techniques (Edwards, 1995). Mice (postnatal day [P]24–33) were anesthetized by inhalation of methoxyflurane and decapitated. The brain was immediately removed and immersed in ice-cold oxygenated artificial cerebrospinal fluid (O2-ACSF) containing (in mM) 125 NaCl, 2.5 KCl, 2 CaCl2, 1 MgCl2, 1.25 NaH2PO4, 26 NaHCO3, 25 glucose, pH 7.4. Using a vibratome, 300-μm parasagittal slices were cut and incubated for 1 h before experimentation in O2-ACSF at an elevated temperature (31–32°C), after which the bath was allowed to cool to room temperature (∼22– 23°C).
Whole-cell patch clamp recordings were performed using minor modifications of standard methods (Konnerth et al., 1990). A cerebellar slice was submerged in a recording chamber and held in place under a grid of nylon threads glued to a U-shaped platinum frame. The slice was perfused with room temperature O2-ACSF (with 10 μM bicuculline added) at 1–2 ml/min, and Purkinje cells were visually identified with a 40× ceramic-coated water immersion objective. Recordings in the current clamp mode were made using a patch pipette (1.6–2.6 MΩ) filled through the tip with (in mM) 140 KCl, 8 NaCl, 10 Hepes, 0.5 EGTA and backfilled with the addition of 4 MgATP, 0.3 TrisGTP. Using a Dagan 3900 amplifier equipped with a 3911A Whole Cell Expander, data were filtered at 5 kHz and acquired (3.3–50 kHz) using pCLAMP software (Axon Instruments, Inc., Foster City, CA).
Results
Cerebellum-specific Knockout of AnkyrinG in Mouse Brain
AnkyrinG cDNAs isolated from human brain (Kordeli et al., 1995) and mouse kidney (Peters et al., 1995) have distinct NH2-terminal amino acid sequences immediately preceding the ankyrin repeats (Fig. 1 A). A cDNA isolated from rat brain has an NH2-terminal sequence nearly identical to that of the isoform isolated from mouse kidney (Fig. 1 A). These considerations combined with genomic sequence data suggested the possibility of two classes of ankyrinG transcripts, each with distinct promoter elements (data not shown). Northern blot analysis confirmed that one exon (exon1b) is expressed only in mouse brain, while the other exon (exon1e) is expressed in brain as well as heart, skeletal muscle, and kidney (Fig. 1 B, left and middle). AnkyrinG transcripts from lung and some isoforms in heart and kidney did not hybridize with exon1b or exon1e, suggesting the existence of a third set of transcripts and possibly an additional promoter (Fig. 1 B, right).
Figure 1.
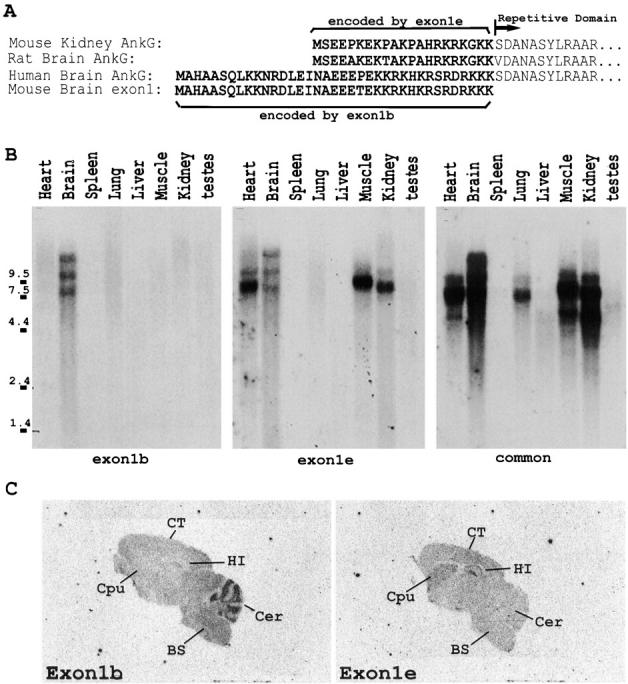
Expression of two classes of ankyrinG transcripts in mouse brain. (A) Mouse brain contains two different NH2-terminal amino acid sequences of ankyrinG. The predicted NH2-terminal amino acid sequences of ankyrinG from mouse kidney and rat and human brain cDNAs were aligned and compared. The amino acid sequence translated from the coding region of exon1 of ankyrinG genomic DNA was also included. (B) Northern blot analysis revealed that mouse brain contained two classes of ankyrinG transcripts that differed in the first exon. Isoform-specific probes to the brain-specific 5′UTR (exon1b) and the kidney isoform 5′UTR (exon1e) were PCR-amplified from mouse ankyrinG genomic DNA containing the first exon and from ankyrinG cDNA isolated from rat brain, respectively. The results indicated that exon1b-containing transcripts were only found in brain, while exon1e-containing transcripts were found in many tissues. (C) Tissue-specific expression of exon1b and exon1e in P20 mouse brain revealed by in situ hybridization. In situ hybridization with 33P-labeled oligonucleotides unique to exon1b (left) or exon1e (right) indicated that ankyrinG transcripts in frontal cortex (CT), hippocampus (HI), and caudate putamen (Cpu) contained exon1e, while exon1b shows the strongest signal detected in the cellular layers of the cerebellum (Cer). The signals were completely abolished when the sections were preincubated with 100-fold molar excess of the unlabeled probes (data not shown).
In situ hybridization using probes specific to exon1b and exon1e revealed selective expression of the two exons in adult mouse brain (Fig. 1 C). Exon1e-containing ankyrinG transcripts are expressed throughout the frontal brain with elevated levels in hippocampus, caudate putamen, and frontal cortex. Exon1b, in contrast, is most strongly expressed in cerebellum, but it is also detectable in the brain stem and thalamus at low levels. Developmental patterns of expression of these transcripts have not yet been examined.
The selective expression of exon1b in the cerebellum suggested a strategy for targeted gene disruption resulting in preferential loss of cerebellar ankyrinG, while maintaining ankyrinG expression in other tissues as well as other regions of the nervous system. We produced mutant mice with a neomycin gene incorporated in reverse orientation into the brain-specific exon1b by homologous recombination (Fig. 2 A). The success of the targeted disruption was confirmed by Southern blot (Fig. 2 B). The specific knockout of exon1b-containing ankyrinG transcripts in homozygous mutant mice was confirmed by Northern blot with probes specific for exon1b and exon1e (Fig. 2 C). No exon1b-containing transcripts were detected in the mutant mice, whereas the level of expression of exon1e-containing transcripts was unaffected (Fig. 2 C, middle). In situ hybridization performed on mutant mouse brain sections revealed no compensation of the exon1e-containing ankyrinG transcripts in cerebellum, while exon1b-containing ankyrinG transcripts were undetectable (data not shown). Immunoblots using an antibody to the tail domain of ankyrinG (anti–270/480 ankyrinG; Zhang and Bennett, 1996), which recognizes proteins derived from both classes of transcripts, revealed almost complete absence of ankyrinG protein in cerebellum of the mutant mouse (Fig. 2 D). Similar data were obtained using an antibody to the spectrin-binding domain of ankyrinG (data not shown). The mutant mouse forebrain exhibited only a moderate reduction of ankyrinG, suggesting that the alternative, exon1e-containing isoforms are normally expressed in forebrain.
The region-specific knockout of ankyrinG was further confirmed by immunofluorescence labeling (Fig. 3). AnkyrinG was almost completely missing in mutant mouse cerebellum (Fig. 3 A). Some ankyrinG immunoreactivity in the white matter of mutant mouse cerebellum was still detectable, suggesting that these fibers contain exon1e-ankyrinG. The expression of ankyrinG in hippocampal neurons of mutant mice was unaffected compared with the wild-type control mouse, indicating that these neurons also use exon1e-ankyrinG (Fig. 3, C and D).
Figure 3.
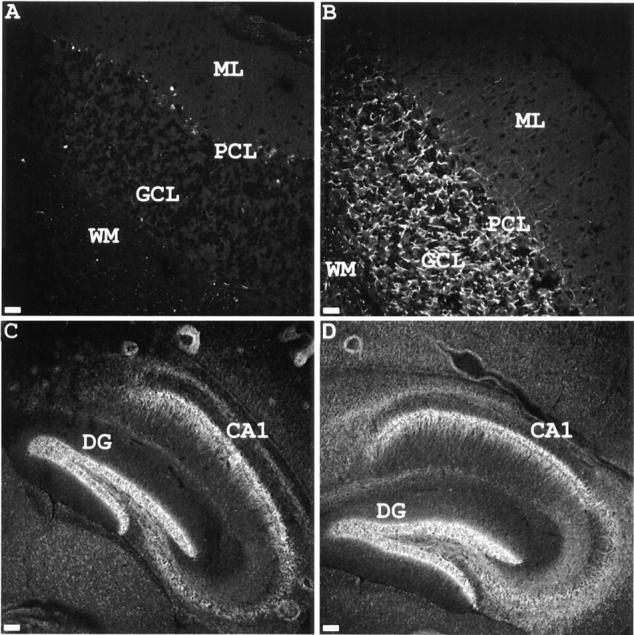
Regional loss of ankyrinG in exon1b knockout mouse. Cerebellar (A and B) and hippocampal (C and D) sections of exon1b knockout mouse (A and C) and control littermate (B and D) were stained with chicken antibodies to ankyrinG, followed by FITC-conjugated mouse anti–chicken secondary antibodies. The results indicated that ankyrinG is still expressed in hippocampus of the exon1b knockout mouse, while the expression in cerebellum is drastically reduced. DG, dentate gyrus; CA1, CA1 field of hippocampus; WM, white matter; GCL, granule cell layer; PCL, Purkinje cell layer; ML, molecular cell layer. Bars: (A and B) 20 μm; (C and D) 80 μm.
Mutant Mice Develop Progressive Ataxia
Mice with the exon1b-ankyrinG −/− genotype (hereafter referred to as mutant mice) were born in nearly Mendelian ratios (55/211) and had no obvious abnormality until P16–17. Mutant mice then developed characteristic cerebellar defects with symptoms of abnormal gait and tremor and reduced locomotion. When the mutant mice were prodded to walk, their hindlimbs were uncoordinated, and they fell frequently (Fig. 4). This phenotype had a variable time course of onset and severity but could generally easily be observed in mutant mice as young as P16. The ataxia became more severe with increasing age. The mutant mice are fertile but do not breed well. Some premature death of mutant mice occurred between 4 and 6 mo and in some cases was preceded by uncontrollable jumping and convulsions. Mice with the exon1b-ankyrinG +/− genotype (heterozygotes) were phenotypically normal and could not be distinguished from wild-type mice (+/+ for exon1b-ankyrinG).
Figure 4.

Photograph of the exon1b-ankyrinG knockout mouse and the control wild-type littermate. The moving wild-type littermate mouse (top) and the mutant mouse (bottom) were pulsed-flashed four times within 1/8 s, during which period of time the shutter of the camera was kept open. Their moving postures were captured and superimposed into one frame. The images demonstrate the tremor behavior of the mutant mouse.
Loss of Targeting of NaCh to Axon Initial Segments of Neurons in Mutant Mouse Cerebellum
At least three types of NaCh are expressed by neurons of mouse cerebellum (Westenbroek et al., 1989, 1992; De Miera et al., 1997). The type II NaCh is the major NaCh expressed by granule cells (De Miera et al., 1997), while NaCh6/Scn8a/CerIII (Burgess et al., 1995; Schaller et al., 1995; De Miera et al., 1997) is expressed in Purkinje cells and is likely one of the contributors to the generation of Purkinje cell action potentials (Raman et al., 1997). Type I NaCh are expressed in Purkinje neurons (Westenbroek et al., 1989; De Miera et al., 1997) but are not concentrated at axon initial segments (data not shown). Our NaCh antibodies were raised against peptides shared by type I and II NaCh (Lambert et al., 1997) and reacted with NaCh at nodes of Ranvier of peripheral nerve (Lambert et al., 1997) and axon initial segments of granule cells on tissue sections (Fig. 5 D). In addition, the molecular layer, which is composed of unmyelinated axons of granule cell neurons, was also labeled with this antibody, as previously reported (Westenbroek et al., 1989, 1992).
Figure 5.
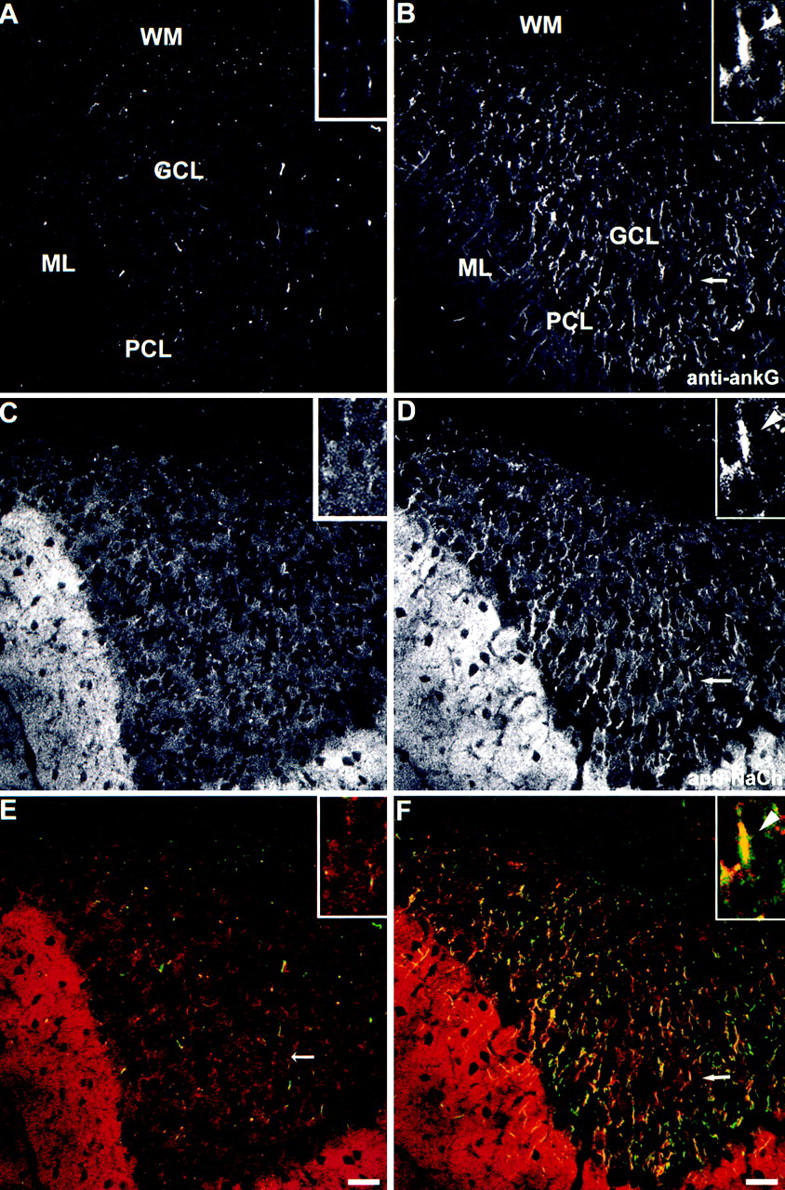
Loss of sodium channel clustering at initial segments of cerebellar granule cell axons of mutant mice. Cerebellar brain sections from mutant mouse (A, C, and E) and the wild-type control littermate (B, D, and F) were double-stained with antibodies to ankyrinG (A and B, green in E and F) and sodium channel (C and D, red in E and F). One typical example of colocalization of ankyrinG and NaCh at the initial segments of granule cells in wild-type mouse cerebellum (B, D, and F, arrows) was magnified and shown in the inset. The initial segment was marked with an arrowhead. Bars, 20 μm.
AnkyrinG is confined to initial segments of axons of granule cells (Fig. 3 B) as well as Purkinje neurons (see below), as previously reported (Kordeli et al., 1995). AnkyrinG at axon initial segments is clearly resolved in cultured granule cells (data not shown). This distribution pattern is also true in cerebellum sections (Fig. 5 B), where sodium channels colocalized with ankyrinG at the initial segments of granule cells (Fig. 5 F, see the enlarged view in the inset).
Immunofluorescence indicates that NaCh were not concentrated at initial segments of granule cells from mutant mice (Fig. 5 C), while NaCh were highly concentrated at granule cell initial segments and colocalized with ankyrinG in wild-type mice (Fig. 5 D). Mutant mouse cerebella retain strong labeling of NaCh in the molecular layer, where no ankyrinG is present, even in normal mice. Immunoblot data indicated the levels of NaCh protein detected by this antibody were the same in the mutant and control mouse cerebella of animals less than 40 d (data not shown). Although ankyrinG is absent from the molecular layer, ankyrinB is highly expressed in this region (Kunimoto et al., 1991), and this or some other protein such as syntrophin (Gee et al., 1998) could interact with NaCh in the unmyelinated granule cell axons. NaCh could not be consistently visualized in Purkinje cell initial segments with our antibody or with eight other antibodies (see Materials and Methods). These negative results suggest either that the NaCh did not react strongly with antibody because of problems with fixation or some other experimental parameter, or that axon initial segments of these neurons have a specialized NaCh isoform that is not recognized by available antibodies. However, the electrophysiological data presented below strongly support the conclusion that the density of NaCh in Purkinje neuron initial segments is dramatically reduced in the mutant mice.
Abnormal Physiology of Purkinje Cells of Mutant Mice
We studied the ability of Purkinje cells to fire action potentials upon injection of current at the soma in whole-cell patch-clamp recordings from cerebellar slices. Experiments were done blind to genotype, although the motor phenotype of each mouse was obvious. After analysis, the genotype of each animal was determined by Southern blot from tail-snip specimens. Wild-type (+/+) and heterozygote (+/−) mice could not be distinguished by cage behavior nor electrophysiological properties and were thus combined for data analysis as normal mice. Purkinje cells in cerebellar slices from mutant mice (−/−) were, however, impaired in their ability to fire action potentials. Three features were notable. Purkinje cells from mutant mice had a higher threshold to initiate action potentials, did not always fire an action potential in response to a short, 1-ms current injection, and showed a slower rate of maximal firing of multiple action potentials in response to long current injections. In Purkinje cells from normal mice, a single, “all-or-nothing” action potential (Fig 6 A, left) could always be elicited from a 1-ms pulse (10/10 cells from 7 animals). In contrast, most cells from mutant mice showed only passive membrane responses and were unable to initiate an action potential in response to the same magnitude 1-ms current injections (Fig. 6 B, left; 4/7 cells from 5 animals). In three cells from two mutant animals, an action potential could be initiated with a 1-ms current injection, but only when the stimulus intensity was increased compared with normal cells, indicating a higher threshold to initiate the action potential in mutant cells. Normal Purkinje cells always fired multiple action potentials in response to long (50–100 ms) current injections (Fig. 6 A, right). Although Purkinje cells in mutant mice could fire action potentials with long current injections (Fig. 6 B, right), the maximal firing rate was less than one half that of the normal cells (Fig. 6 C). The threshold to fire action potentials was also significantly higher in the Purkinje cells from mutant mice (Fig. 6 D). The action potentials in Purkinje cells from mutant mice were due to NaCh currents because they could be reversibly blocked by 1 μM TTX (data not shown). Purkinje cells from mutant and normal mice did not differ in their initial resting potential in whole-cell current clamp (−58 ± 3.0 mV for mutants, n = 7; −57 ± 2.9 mV for normals, n = 10) or resting input resistance (80 ± 16 MΩ for mutants, 125 ± 28 MΩ for normals).
Figure 6.
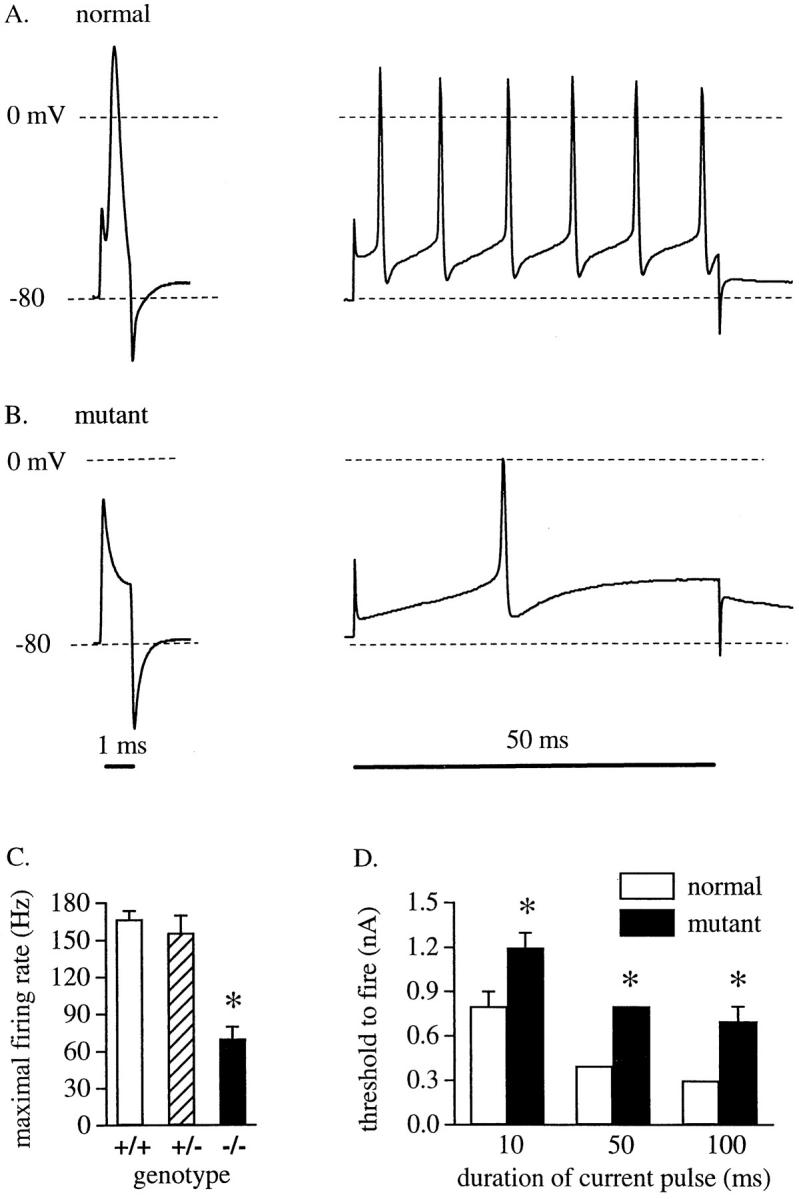
Deficits in action potential initiation and firing in mutant mice. Whole-cell current clamp recordings of Purkinje cells in cerebellar slices from (A) a normal mouse or (B) a mutant mouse with cerebellar knockout of ankyrinG. Cells were hyperpolarized to −80 mV and the membrane potential responses to 1-ms (left) or 50-ms (right) current injections at the soma were recorded. Dashed lines indicate −80 and 0 mV, as marked. (C) Comparison of the maximal action potential firing rate for 50– 100-ms current injections delivered to Purkinje cells from wild-type (+/+; n = 3), heterozygous (+/−; n = 7), and mutant (−/−; n = 7) mice. A series of depolarizing current injections from 0.1– 1.9 nA in 0.1-nA increments was delivered until the maximal firing rate was reached. (D) Comparison of the minimum current injection required to elicit the first action potential in Purkinje cells from normal (n = 10) and mutant (−/−; n = 7) mice for stimuli of 10, 50, or 100 ms duration. The normal group includes both wild-type and heterozygous mice since the physiological and behavioral phenotypes of the two groups were not different. Data (means ± SEM) were analyzed by a two-tailed t test. *P < 0.001.
Thus, although the molecular species of NaCh expressed by the mouse Purkinje cells cannot be demonstrated by immunocytochemistry (see above), the physiological data strongly support the idea that NaCh localization to the initial segment is impaired and/or NaCh density is reduced because of the knockout of a cerebellar isoform of ankyrinG. This is consistent with the immunofluorescence evidence for reduced localization of NaCh in cerebellar granule cell initial segments (Fig. 5) and neurofascin in Purkinje cell initial segments (Fig. 7, below).
Figure 7.
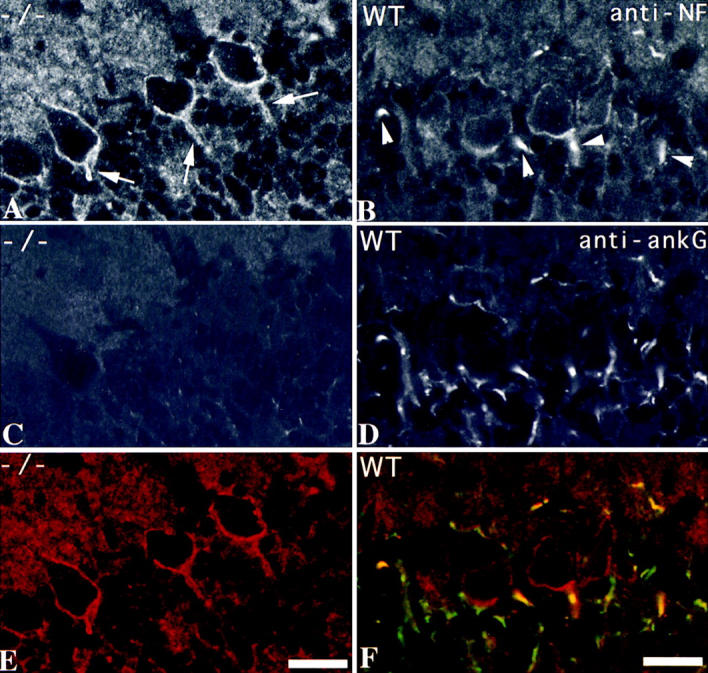
Redistribution of neurofascin in Purkinje cells of mutant mice. Sections of cerebellum from P40 mutant mouse (−/−; A, C, and E) and the wild-type control littermate (WT; B, D, and F) were stained with antibodies to ankyrinG (C and D, green in E and F) and neurofascin (A and B, red in E and F). Without ankyrinG, neurofascin was distributed uniformly at the plasma membrane of Purkinje cells (A, arrows). In normal mice, neurofascin was highly concentrated at the initial segments of Purkinje cells (B, arrowheads). Bar, 20 μm.
Reduced Concentration of Neurofascin at Axon Initial Segments of Purkinje Neurons
Neurofascin and NrCAM colocalized with ankyrinG at initial segments of Purkinje neuron axons and the nodes of Ranvier of peripheral nerve (Davis et al., 1996; Lambert et al., 1997). The L1CAM family member(s) targeted to granule cell neuron initial segments has not been defined. In control mouse cerebellum, ankyrinG and neurofascin are colocalized at the initial segments of Purkinje cell axons (Fig. 7 F), with low levels of neurofascin also localized along the cell body plasma membrane, as previously reported (Davis et al., 1996). In the mutant mouse cerebellum, however, neurofascin was uniformly distributed at the plasma membrane of Purkinje neuron cell body and axon initial segment (Fig. 7 E). Some elevation of neurofascin was found in the molecular layer of ankyrinG-deficient mice. The same observations were made with an antibody to NrCAM (data not shown).
Neuronal Degeneration in the Mutant Mouse Brain
The cerebella of adult mutant mice (>5 mo old) is substantially smaller than those of control littermates, but the frontal brains were similar in size (data not shown). The molecular layer of the adult mutant mouse cerebellum was reduced in thickness, and the number of Purkinje cells was reduced by 60% based on staining with neurofilament antibody, hematoxylin and eosin, or toluidine blue (data not shown, n = 6). An antibody to calbindin, a Purkinje cell marker, revealed large gaps in the molecular layer of mutant mice where cell bodies and dendritic arbors of Purkinje cells were absent (Fig. 8 A).
Figure 8.

Neurodegeneration of Purkinje cells in mutant mice. Sections of cerebellum from a 5-mo-old mutant mouse (A) and the wild-type control littermate (B) were stained with antibodies to calbindin. The results show a dramatic reduction of Purkinje cells in the adult mutant mice. Bars, 50 μm.
Purkinje cells of mutant mice are likely to provide ineffective or inappropriate signaling to their target neurons because of an abnormal input from granule cells and from their own deficiencies in action potential initiation and firing rates. A potential consequence of abnormal signaling to the deep nuclei is that Purkinje cells would not receive sufficient neurotrophic factors by retrograde transport. These considerations may explain the progressive loss of Purkinje neurons in ankyrinG mutant mice. Interestingly, progressive, late-onset Purkinje cell loss was also found in jolting mice with a mutation in the Scn8a NaCh expressed in Purkinje cells (Boakes et al., 1984; Dick et al., 1986; Kohrman et al., 1996).
Discussion
This study demonstrates that ankyrinG is essential for normal targeting of NaCh to axon initial segments of granule cells and for normal firing properties in Purkinje cells. Targeted disruption of ankyrinG gene expression in the cerebellum results in progressive ataxia, abolishes targeting of voltage-gated NaCh to axon initial segments of granule cells, impairs action potential firing in Purkinje neurons, and results in progressive Purkinje neuron degeneration. Neurofascin and NrCAM, members of the L1CAM family of ankyrin-binding cell adhesion molecules, also exhibited reduced concentration at Purkinje cell axon initial segments in mutant mice. Although the molecular type of NaCh expressed by the Purkinje neurons could not be identified by immunocytochemistry, the physiological data (Fig. 6) strongly support the idea that NaCh localization to the initial segment is impaired and/or NaCh density is reduced because of the cerebellar knockout of ankyrinG. This is consistent with the immunocytochemical evidence for reduced targeting of NaCh in cerebellar granule cells (Fig. 5) and neurofascin in Purkinje cells (Fig 7).
The demonstration that ankyrinG is essential for clustering of sodium channels and neurofascin at the initial segments extends previous observations that ankyrinG coclusters with neurofascin and NaCh during morphogenesis of nodes of Ranvier (Davis et al., 1996; Lambert et al., 1997). The ankyrin membrane binding domain has distinct binding sites for the NaCh, located on subdomains 3 and 4 (Srinivasan et al., 1992), and for neurofascin, located on subdomains 2 and 3 as well as 3 and 4 (Michaely and Bennett, 1995b ). Therefore, ankyrinG could form heterocomplexes between NaCh and neurofascin in vivo by directly binding to both proteins. A similar interaction between an ion channel and cell adhesion molecule has been demonstrated at Drosophila neuromuscular junction, where PDZ-containing protein Discs-Large (Dlg) interacts with Shaker potassium channel and Fasciclin II directly (Zito et al., 1997). In dlg mutants, the localization of both Shaker channel and Fasciclin II to neuromuscular junction was altered (Tejedor et al., 1997; Zito et al., 1997), much like the altered localization of sodium channel and neurofascin in ankyrinG knockout mice. These observations suggest that heterocomplexes between ion channels and adhesion molecules mediated by cytoplasmic proteins may be a general feature of specialized cell domains. Possible physiological roles for such channel/adhesion molecule/ cytoplasmic protein complexes include directing extracellular interactions with adjacent cells to these sites, lateral organization of ion channel assemblies, and polarized delivery of newly synthesized ion channels to specialized sites. A potential benefit of heterocomplexes mediated by a cytoplasmic protein is that the interactions could be differentially regulated in response to cellular signals. For example, ankyrin association with neurofascin and other L1CAM family members is abolished by tyrosine phosphorylation of FIGQY residues in the cytoplasmic domain (Garver et al., 1997).
The role of ankyrinG in assembly of NaCh at nodes of Ranvier and the neuromuscular junction is not addressed in this study because of the selective disruption of ankyrinG in the cerebellum and the lack of antibodies recognizing NaCh of Purkinje neurons. However, similarities between initial segments and other sites of NaCh concentration support the prediction that ankyrinG is also required for restriction of NaCh at these domains. Nodes of Ranvier, the neuromuscular junction, and axon initial segments each have specialized features as well. Thus NaCh/ ankyrinG assemblies are likely to include additional proteins that perform specific functions adapted to each cell domain. Syntrophins, for example, associate with NaCh and are concentrated at the neuromuscular junction (Gee et al., 1998; Shultz et al., 1998). The β subunits of NaCh also are candidates to mediate important domain-specific interactions (Isom et al., 1995; Isom and Catterall, 1996).
The role of ankyrinG in molecular events leading to assembly of the specialized plasma membrane domain at axon initial segments remains an important issue for future investigation. One conclusion from this study is that signals for ankyrin-based targeting of NaCh to axon initial segments are likely to involve additional interacting proteins that have yet to be identified. Neurofascin and NrCAM have been proposed to direct assembly, first of ankyrin at nodes of Ranvier and other sites, followed by the localization of NaCh (Lambert et al., 1997). However, the finding that neurofascin is not distributed normally in the absence of ankyrinG (Fig. 7) suggests that ankyrinG is required for the concentration of neurofascin as well as the NaCh, at least at axon initial segments. It is of interest that restriction of 270-kD ankyrinG to axon initial segments of cultured dorsal root ganglion neurons requires multiple domains in addition to the membrane-binding domain (Zhang and Bennett, 1998). These findings imply the existence of unidentified ankyrin-binding protein(s) upstream in the pathway to formation of the initial segment specialized domain.
The finding that ankyrinG is required for the normal physiological function of NaCh is an example of a general principle of the critical importance of spatial organization of ion channels in cells of metazoan animals and the requirement for cytoplasmic proteins for proper cellular targeting. Other instances demonstrated in animal models include the role of rapsyn in the organization of acetylcholine receptors at the neuromuscular junction of mice (Gautam et al., 1995), and of the PDZ protein, Discs-Large (Dlg), in the organization of potassium channels and Fasciculin II in neuromuscular junctions of Drosophila (Tejedor et al., 1997; Zito et al., 1997). Apparently, evolution of mechanisms for spatial organization has proceeded through convergent pathways for different types of channels, with the selective advantage of rapid and precise response to stimuli. Ion channels with polarized localization in cells that associate with ankyrin include the Na/K ATPase (Nelson and Veshnock, 1987) and Na/Ca exchanger (Li et al., 1993). The Na/K ATPase requires ankyrinG119 for delivery to the plasma membrane in cultured cells (Devarajan et al., 1997) and may also require ankyrinG for targeting to basolateral domains of epithelial cells in tissues. Voltage-gated NaCh are members of a super-family that also includes voltage-gated channels for K+, and Ca2+ (Armstrong and Hille, 1998). It will be of interest to determine if ankyrin-binding and ankyrin-dependent cellular targeting are features shared by other members of the voltage-gated channel family. Deciphering the molecular code for cellular targeting of ion channels and most likely other signaling molecules is an issue connecting the fields of cell biology and physiology and is equivalent in functional significance to understanding primary and tertiary structures of these proteins.
Abbreviations used in this paper
- NaCh
voltage-gated sodium channel(s)
- O2-ACSF
oxygenated artificial cerebrospinal fluid
- P
postnatal day
Footnotes
Cheryl Bock is gratefully acknowledged for culture and transfection of ES cells, as well as blastocyst injections. Paula Scotland is gratefully acknowledged for culture and identification of ES cells with homologous recombination.
This research was supported in part by a grant from the National Institutes of Health (V. Bennett), the McKnight Foundation (L.M. Boland) and the Edward J. Mallinckrodt, Jr. Foundation (S. Lambert).
Address all correspondence to Vann Bennett, Howard Hughes Medical Institute and Department of Cell Biology, Duke University Medical Center, Durham, NC 27710. Tel.: (919) 684-3105. Fax: (919) 684-3590. E-mail: vbennett@acpub.duke.edu
References
- Armstrong CM, Hille B .Voltage-gated ion channels and electrical excitability. Neuron. 1998;20:371–380. doi: 10.1016/s0896-6273(00)80981-2. [DOI] [PubMed] [Google Scholar]
- Bennett V, Lambert S, Davis JQ, Zhang X. Molecular architecture of the specialized axonal membrane at the node of Ranvier. Soc Gen Physiol Ser. 1997;52:107–120. [PubMed] [Google Scholar]
- Boakes RJ, Candy JM, Dick DJ, Harris JB, Pollard S, Thompson J. Abnormal electrophysiological and anatomical characteristics of cerebellar Purkinje cells in the mutant mouse jolting. J Physiol (Lond) 1984;346:27P. [Google Scholar]
- Burgess DL, Kohrman DC, Galt J, Plummer NW, Jones JM, Spear B, Meisler MH. Mutation of a new sodium channel gene, Scn8a, in the mouse mutant “motor endplate disease.” . Nat Genet. 1995;10:461–465. doi: 10.1038/ng0895-461. [DOI] [PubMed] [Google Scholar]
- Catterall WA. Localization of sodium channels in cultured neural cells. J Neurosci. 1981;1:777–783. doi: 10.1523/JNEUROSCI.01-07-00777.1981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan W, Kordeli E, Bennett V. 440-kD ankyrinB: structure of the major developmentally regulated domain and selective localization in unmyelinated axons. J Cell Biol. 1993;123:1463–1473. doi: 10.1083/jcb.123.6.1463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis JQ, McLaughlin T, Bennett V. Ankyrin-binding proteins related to nervous system cell adhesion molecules: candidates to provide transmembrane and intercellular connections in adult brain. J Cell Biol. 1993;121:121–133. doi: 10.1083/jcb.121.1.121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis JQ, Lambert S, Bennett V. Molecular composition of the node of Ranvier: identification of ankyrin-binding cell adhesion molecules neurofascin (mucin+/third FNIII domain−) and NrCAM at nodal axon segments. J Cell Biol. 1996;135:1355–1367. doi: 10.1083/jcb.135.5.1355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Miera EVS, Rudy B, Sugimori M, Llinas R. Molecular characterization of the sodium channel subunits expressed in mammalian cerebellar Purkinje cells. Proc Natl Acad Sci USA. 1997;94:7059–7064. doi: 10.1073/pnas.94.13.7059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deerinck TJ, Levinson SR, Bennett GV, Ellisman EH. Clustering of voltage-sensitive sodium channels on axons is independent of direct Schwann cell contact in the dystrophic mouse. J Neurosci. 1997;17:5080–5088. doi: 10.1523/JNEUROSCI.17-13-05080.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Devarajan P, Stabach PR, De Matteis MA, Morrow JS. Na,K-ATPase transport from endoplasmic reticulum to Golgi requires the Golgi spectrin-ankyrin G119 skeleton in Madin Darby canine kidney cells. Proc Natl Acad Sci USA. 1997;94:10711–10716. doi: 10.1073/pnas.94.20.10711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dick DJ, Boakes PJ, Candy JM, Harris JB, Cullen MJ. Cerebellar structure and function in the murine mutant “jolting.” . J Neurolog Sci. 1986;76:255–267. doi: 10.1016/0022-510x(86)90173-5. [DOI] [PubMed] [Google Scholar]
- Edwards, F.A. 1995. Patch clamp recording in brain slices. In Brain Slices in Basic and Clinical Research. A. Schurr and B.M. Rigor, editors. CRC Press, Boca Raton, FL. 99–116.
- Ellisman MH, Levinson SR. Immunocytochemical localization of sodium channel distributions in the excitable membranes of Electrophorus electricus. . Proc Natl Acad Sci USA. 1982;79:6707–6711. doi: 10.1073/pnas.79.21.6707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flucher BE, Daniels MP. Distribution of Na+channels and ankyrin in neuromuscular junctions is complementary to that of acetylcholine receptors and the 43 kD protein. Neuron. 1989;3:163–175. doi: 10.1016/0896-6273(89)90029-9. [DOI] [PubMed] [Google Scholar]
- Garver T, Ren Q, Tuvia S, Bennett V. Tyrosine phosphorylation at a site highly conserved in the L1 family of cell adhesion molecules abolishes ankyrin binding and increases lateral mobility of neurofascin. J Cell Biol. 1997;137:703–714. doi: 10.1083/jcb.137.3.703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gautam M, Noakes PG, Mudd J, Nichol M, Chu GC, Sanes JR, Merlie JP. Failure of postsynaptic specialization to develop at neuromuscular junctions of rapsyn-deficient mice. Nature. 1995;377:232–236. doi: 10.1038/377232a0. [DOI] [PubMed] [Google Scholar]
- Gee SH, Madhavan R, Levinson SR, Caldwell JH, Sealock R, Froehner SC. Interaction of muscle and brain sodium channels with multiple members of the syntrophin family of dystrophin-associated proteins. J Neurosci. 1998;18:128–137. doi: 10.1523/JNEUROSCI.18-01-00128.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gordon D, Merrick D, Wollner DA, Catterall WA. Biochemical properties of sodium channels in a wide range of excitable tissues studied with site-directed antibodies. Biochemistry. 1988;27:7032–7038. doi: 10.1021/bi00418a054. [DOI] [PubMed] [Google Scholar]
- Hortsch M. The L1 family of neural cell adhesion molecules: old proteins performing new tricks. Neuron. 1996;17:587–593. doi: 10.1016/s0896-6273(00)80192-0. [DOI] [PubMed] [Google Scholar]
- Isom LL, Ragsdale DS, De Jongh KS, Westenbroek RE, Reber BF, Scheuer T, Catterall WA. Structure and function of the β2 subunit of brain sodium channels, a transmembrane glycoprotein with a CAM motif. Cell. 1995;83:433–442. doi: 10.1016/0092-8674(95)90121-3. [DOI] [PubMed] [Google Scholar]
- Isom LL, Catterall WA. Na+ channel subunits and Ig domains. Nature. 1996;383:307–308. doi: 10.1038/383307b0. [DOI] [PubMed] [Google Scholar]
- Kaplan MR, Meyer-Franke A, Lambert S, Bennett V, Duncan ID, Levinson SR, Barres BA. Induction of sodium channel clustering by oligodendrocytes. Nature. 1997;386:724–728. doi: 10.1038/386724a0. [DOI] [PubMed] [Google Scholar]
- Kohrman DC, Smith MR, Goldin AL, Harris J, Meisler MH. A missense mutation in the sodium channel Scn8a is responsible for cerebellar ataxia in the mouse mutant jolting. J Neurosci. 1996;16:5993–5999. doi: 10.1523/JNEUROSCI.16-19-05993.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konnerth A, Llano I, Armstrong CM. Synaptic currents in currents in cerebellar Purkinje cells. Proc Natl Acad Sci USA. 1990;87:2662–2665. doi: 10.1073/pnas.87.7.2662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kordeli E, Bennett V. Distinct ankyrin isoforms at neuron cell bodies and nodes of Ranvier resolved using erythrocyte ankyrin-deficient mice. J Cell Biol. 1991;114:1243–1259. doi: 10.1083/jcb.114.6.1243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kordeli E, Davis J, Trapp B, Bennett V. An isoform of ankyrin is localized at nodes of Ranvier in myelinated axons of central and peripheral nerves. J Cell Biol. 1990;110:1341–1352. doi: 10.1083/jcb.110.4.1341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kordeli E, Lambert S, Bennett V. AnkyrinG. A new ankyrin gene with neural-specific isoforms localized at the axonal initial segment and node of Ranvier. J Biol Chem. 1995;270:2352–2359. doi: 10.1074/jbc.270.5.2352. [DOI] [PubMed] [Google Scholar]
- Kordeli E, Ludosky MA, Deprette C, Frappier T, Cartaud J. AnkyrinG is associated with the postsynaptic membrane and the sarcoplasmic reticulum in the skeletal muscle fiber. J Cell Sci. 1998;111:2197–2207. doi: 10.1242/jcs.111.15.2197. [DOI] [PubMed] [Google Scholar]
- Kunimoto M, Otto E, Bennett V. A new 440-kD isoform is the major ankyrin in neonatal rat brain. J Cell Biol. 1991;115:1319–1331. doi: 10.1083/jcb.115.5.1319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lambert S, Davis JQ, Bennett V. Morphogenesis of the node of Ranvier: co-clusters of ankyrin and ankyrin-binding integral proteins define early developmental intermediates. J Neurosci. 1997;17:7025–7036. doi: 10.1523/JNEUROSCI.17-18-07025.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Z, Burke EP, Frank JS, Bennett V, Philipson KD. The cardiac Na+-Ca2+exchanger binds to the cytoskeletal protein ankyrin. J Biol Chem. 1993;268:11489–11491. [PubMed] [Google Scholar]
- Michaely P, Bennett V. The membrane-binding domain of ankyrin contains four independently folded subdomains, each comprised of six ankyrin repeats. J Biol Chem. 1993;268:22703–22709. [PubMed] [Google Scholar]
- Michaely P, Bennett V. The ANK repeats of erythrocyte ankyrin form two distinct but cooperative binding sites for the erythrocyte anion exchanger. J Biol Chem. 1995a;270:22050–22057. doi: 10.1074/jbc.270.37.22050. [DOI] [PubMed] [Google Scholar]
- Michaely P, Bennett V. Mechanism for binding site diversity on ankyrin. Comparison of binding sites on ankyrin for neurofascin and the Cl−/HCO3−anion exchanger. J Biol Chem. 1995b;270:31298–31302. doi: 10.1074/jbc.270.52.31298. [DOI] [PubMed] [Google Scholar]
- Nelson WJ, Veshnock PJ. Ankyrin binding to (Na+ + K+)ATPase and implications for the organization of membrane domains in polarized cells. Nature. 1987;328:533–536. doi: 10.1038/328533a0. [DOI] [PubMed] [Google Scholar]
- Peters LL, John KM, Lu FM, Eicher EM, Higgins A, Yialamas M, Turtzo LC, Otsuka AJ, Lux SE. Ank3 (epithelial ankyrin), a widely distributed new member of the ankyrin gene family and the major ankyrin in kidney, is expressed in alternatively spliced forms, including forms that lack the repeat domain. J Cell Biol. 1995;130:313–330. doi: 10.1083/jcb.130.2.313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raman IM, Sprunger LK, Meisler MH, Bean BP. Altered subthreshold sodium currents and disrupted firing patterns in Purkinje neurons of Scn8a mutant mice. Neuron. 1997;19:881–891. doi: 10.1016/s0896-6273(00)80969-1. [DOI] [PubMed] [Google Scholar]
- Sambrook, J., E.F. Fritsch, and T. Maniatis. 1989. Molecular Cloning: A Laboratory Manual, 2nd edition. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY. 7.40–7.42.
- Schaller KL, Krzemien DM, Yarowsky PJ, Krueger B, Caldwell JH. A novel, abundant sodium channel expressed in neurons and glia. J Neurosci. 1995;15:3231–3242. doi: 10.1523/JNEUROSCI.15-05-03231.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schultz J, Hoffmuller U, Krause G, Ashurst J, Macias MJ, Schmieder P, Schneider-Mergener J, Oschkinat H. Specific interactions between the syntrophin PDZ domain and voltage-gated sodium channels. Nat Struct Biol. 1998;5:19–24. doi: 10.1038/nsb0198-19. [DOI] [PubMed] [Google Scholar]
- Srinivasan Y, Elmer L, Davis J, Bennett V, Angelides K. Ankyrin and spectrin associate with voltage-dependent sodium channels in brain. Nature. 1988;333:177–180. doi: 10.1038/333177a0. [DOI] [PubMed] [Google Scholar]
- Srinivasan Y, Lewallen M, Angelides K. Mapping the binding site on ankyrin for the voltage-dependent sodium channel from brain. J Biol Chem. 1992;267:7483–7489. [PubMed] [Google Scholar]
- Tejedor FJ, Bokhari A, Rogero O, Gorczyca M, Zhang J, Kim E, Sheng M, Budnik V. Essential role of dlg in synaptic clustering of Shaker K+channels in vivo. J Neurosci. 1997;17:152–159. doi: 10.1523/JNEUROSCI.17-01-00152.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westenbroek RE, Merrick DK, Catterall WA. Differential subcellular localization of the RI and RII Na+channel subtypes in central neurons. Neuron. 1989;3:695–704. doi: 10.1016/0896-6273(89)90238-9. [DOI] [PubMed] [Google Scholar]
- Westenbroek RE, Noebels JL, Catterall WA. Elevated expression of type II Na+channels in hypomyelinated axons of shiverer mouse brain. J Neurosci. 1992;12:2259–2267. doi: 10.1523/JNEUROSCI.12-06-02259.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wollner DA, Catterall WA. Localization of sodium channels in axon hillocks and initial segments of retinal ganglion cells. Proc Natl Acad Sci USA. 1986;83:8424–8428. doi: 10.1073/pnas.83.21.8424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wood SJ, Slater CR. β-Spectrin is colocalized with voltage-gated sodium channels and ankyrinG at the adult rat neuromuscular junction. J Cell Biol. 1998;140:675–684. doi: 10.1083/jcb.140.3.675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Bennett V. Identification of O-linked N-acetylglucosamine modification of ankyrinGisoforms targeted to nodes of Ranvier. J Biol Chem. 1996;271:31391–31398. doi: 10.1074/jbc.271.49.31391. [DOI] [PubMed] [Google Scholar]
- Zhang X, Bennett V. Restriction of 480/270-kD ankyrin-G to axon proximal segments requires multiple ankyrin-G–specific domains. J Cell Biol. 1998;142:1571–1581. doi: 10.1083/jcb.142.6.1571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zito K, Fetter RD, Goodman CS, Isacoff EY. Synaptic clustering of Fascilin II and Shaker: essential targeting sequences and role of Dlg. Neuron. 1997;19:1007–1016. doi: 10.1016/s0896-6273(00)80393-1. [DOI] [PubMed] [Google Scholar]