

Abstract
Introns are among the hallmarks of eukaryotic genes. Splicing of introns is directed by three main splicing signals: the 5′ splice site (5′ss), the branch site (BS), and the polypyrimdine tract/3′splice site (PPT-3′ss). To study the evolution of these splicing signals, we have conducted a systematic comparative analysis of these signals in over 1.2 million introns from 22 eukaryotes. Our analyses suggest that all these signals have dramatically evolved: The PPT is weak among most fungi, intermediate in plants and protozoans, and strongest in metazoans. Within metazoans it shows a gradual strengthening from Caenorhabditis elegans to human. The 5′ss and the BS were found to be degenerate among most organisms, but highly conserved among some fungi. A maximum parsimony-based algorithm for reconstructing ancestral position-specific scoring matrices suggested that the ancestral 5′ss and BS were degenerate, as in metazoans. To shed light on the evolutionary variation in splicing signals, we have analyzed the evolutionary changes in the factors that bind these signals. Our analysis reveals coevolution of splicing signals and their corresponding splicing factors: The strength of the PPT is correlated to changes in key residues in its corresponding splicing factor U2AF2; limited correlation was found between changes in the 5′ss and U1 snRNA that binds it; but not between the BS and U2 snRNA. Thus, although the basic ability of eukaryotes to splice introns has remained conserved throughout evolution, the splicing signals and their corresponding splicing factors have considerably evolved, uniquely shaping the splicing mechanisms of different organisms.
Splicing of pre-mRNA is a key step in eukaryotic gene expression, contributing to gene regulation, protein diversity, and phenotypic complexity. Introns are removed from the pre-mRNA by the spliceosome, which is composed of five snRNPs (small nuclear ribonucleoprotein) (U1, U2, U4, U5, and U6), each containing a small RNA bound by proteins. High-precision recognition of introns is required for correct splicing. This recognition is achieved by the binding of splicing factors to signals of varying specificity that are located both in the intron and its flanking exons (Hastings and Krainer 2001; Black 2003; Collins and Penny 2005). In vertebrates, three signals are known to direct splicing: The 5′ splice site (5′ss) at the 5′ end of the intron, the polypyrimdine tract/3′ splice site (PPT-3′ss) at the 3′ end of the intron, and a branch site (BS) upstream of the PPT-3′ss (Hastings and Krainer 2001; Black 2003). Spliceosome assembly is initiated by the binding of specific splicing factors to these signals: the U1 snRNP to the 5′ss, the protein SF1 to the BS, the U2 snRNP auxiliary factor U2AF large subunit (U2AF2; also known as U2AF65) to the PPT, and the U2AF small subunit (U2AF1; also known as U2AF35) to the 3′ss. In an ensuing reaction, U2 snRNP associates with the pre-mRNA through a base-pairing interaction between U2 snRNA (small nuclear RNA) and the BS; and subsequent recruitment of the U4/U5/U6 tri-snRNP leads to the formation of the mature spliceosome (Kent et al. 2005).
Comparing the splicing signals among available genomes is of great interest, because these signals are also known to be regulators of alternative splicing (Cartegni et al. 2002). Reconstructing the evolution of these signals is therefore important for understanding when and how alternative splicing evolved. We have previously suggested an evolutionary process for the appearance of alternative splicing in which ancestral splicing signals that supported constitutive splicing accumulated mutations. These mutations suboptimized the splicing signals, allowing them to be used in alternative splicing as well (Ast 2004). This is of special interest, because alternative splicing is believed to have contributed to the creation of phenotypic complexity among higher eukaryotes by increasing transcriptional and proteomic diversity within a given genome (Graveley 2001).
Nonetheless, only few studies attempting to characterize splicing signals among different organisms have been performed. A major drawback of these studies is that they are limited in their taxonomic sampling (e.g., Lim and Burge 2001; Bon et al. 2003; Kupfer et al. 2004; Abril et al. 2005; Sheth et al. 2006) and in terms of splicing signals investigated (e.g., Irimia et al. 2007). Moreover, since different methods have been applied for analysis of the various signals, it is difficult to integrate the results from different studies. In some cases, these studies have even yielded contradictory results. Such is the case, for example, regarding the PPT in Schizosaccharomyces pombe: Some studies maintain that this organism’s introns lack a PPT (Zhang and Marr 1994), while others maintain that its introns have a PPT (Kaufer and Potashkin 2000) and that it contributes to splicing (Romfo and Wise 1997).
Splicing signals are variable. In Saccharomyces cerevisiae, for example, the 5′ss is characterized by a set of highly conserved nucleotides at the 5′ end of the intron that serve as a binding platform for U1 snRNP during an early step of splicing. However, in humans these positions are considerably less conserved. Similarly, although the BS in S. cerevisiae is a highly conserved heptamer (“TACTAAC”) that binds the U2 snRNP, this signal is much more degenerate among vertebrates (Kaufer and Potashkin 2000; Izquierdo and Valcarcel 2006). The PPT shows even greater variability: In higher eukaryotes, the PPT adjacent to the 3′ss is a clear and essential splicing signal (Moore 2000; Reed 2000; Banerjee et al. 2004), whereas in some fungi its very existence is controversial.
What may underlie changes in splicing signals? Splicing signals serve as binding sites for splicing factors. Thus, changes in splicing signals may be linked with, or due to, corresponding changes in splicing factors. Different studies have examined various components of the spliceosome and found it to be highly conserved across evolution (Kaufer and Potashkin 2000; Anantharaman et al. 2002; Koonin et al. 2004; Collins and Penny 2005). However, no studies to date have attempted to correlate changes in the splicing signals, on the one hand, with the factors binding them, on the other. Shifts in terms of genome architecture and intron-exon structure may underlie changes in splicing signals as well. Intron lengths, for example, have changed dramatically across evolution (Aury et al. 2006). The number of introns per gene has also changed considerably, with introns being relatively scarce in fungi but much more abundant in vertebrates (Collins and Penny 2006). Splicing signals are influenced by such changes: In many organisms, for example, the strength of the splice sites correlates positively with intron length (Fields 1990; Kupfer et al. 2004; Weir and Rice 2004; Dewey et al. 2006).
The goal of this study was to characterize and analyze splicing signals and their corresponding splicing factors across the eukaryotic tree by employing a wide, systematic, comparative genomic approach to determine the extent to which changes in splicing signals can be attributed to complementary changes in splicing factors. We therefore compiled a data set of introns from 22 organisms, including organisms from each of the four major eukaryotic kingdoms: Plants, Protozoa, Fungi, and Metazoa. We adapted and developed a variety of algorithms for identifying and quantifying splicing signals in these introns. In parallel, we compiled data sets of the splicing factors that bind these signals during an early stage of spliceosome assembly. We found high variability in all splicing signals, often correlating with corresponding changes in the factors binding these signals. The most variable signal was the PPT: This signal is very weak among most fungi, intermediate in plants and protozoans, but gradually increasing in strength from invertebrates, to non-mammalian vertebrates, to vertebrates. This pattern correlated with changes in U2AF2, both in terms of domain conservation and in key residues that contact the PPT. Our results indicate that the three splicing signals underwent extensive changes during evolution, in parallel with considerable changes in terms of exon-intron architecture and domain structure of the splicing regulators. These changes were presumably shaped by the lifestyle of the organism, selective pressure on maintaining multi-intron genes, and the need to support alternative splicing.
Results
Database compilation and global overview of genomes
We compiled a database of over 1.2 million introns from 22 fully sequenced organisms, including one plant, two protozoans, 12 fungi, and seven metazoans, based on the NCBI databases and GenBank annotations. These annotations were previously shown to be reliable sources for global analysis of splicing patterns (Collins and Penny 2006). We chose to include a relatively large number of fungi in our data set because, as a monophyletic group, fungi comprise both unicellular and multicellular organisms, making them good candidates for studying the changes in intron-exon structure and in splicing signals at the transition stage from unicellular to multicellular organisms. For the sake of simplicity, fungi were divided into two groups: Hemiascomycetous fungi, including all fungi between S. cerevisiae and Yarrowia lipolytica, and non-hemiascomycetous fungi, including the fungi from Neurospora crassa to Cryptococcus neoformans. This classification was based on the grouping pattern in many of our results. Notably, whereas hemiascomycetous fungi form a monophyletic group, non-hemiascomycetous fungi were paraphyletic, containing euascomycetes, archiascomycetes, and basidiomycetes.
Considerable changes in terms of exon-intron architecture
In terms of exon-intron architecture, considerable differences were found between the organisms (Table 1). Metazoans and the plant Arabidopsis thaliana were rich in introns, as reflected by the percentage of genes with introns and by the measure of intron density; hemiascomycetous fungi and the protozoan Cryptosporidium parvum were extremely intron poor; non-hemiascomycetous fungi and the protozoan Dictyostelium discoideum formed an intermediate group. Variations were also observed with respect to intron length: Introns were found to be relatively short throughout eukaryotic evolution, with the exception of vertebrates. An opposite trend was observed with regard to exon length, where metazoans, but also the plant A. thaliana, had considerably shorter exons than most protozoans and fungi. These observations are all consistent with past findings (Ruskin et al. 1985; Zhang and Marr 1994; Deutsch and Long 1999; Bon et al. 2003; Aury et al. 2006). We could also confirm, using our large data set, that internal exons (those flanked by introns on both sides) tend to be considerably shorter than external exons (which lack an intron at one of their sides) (Chen et al. 2002). See Table 1 for further details.
Table 1.
General statistics pertaining to the genomes and intron-exon architecture of the 22 analyzed organisms

Genes are divided into three categories: Genes lacking introns (non–int genes), genes containing one intron (uni–int genes), and multi-intronic genes containing two or more introns (multi–int genes). All measures of length are medians. Intron density is the mean number of introns per spliced gene. All first or terminal exons were defined as external; all remaining introns were defined as internal.
Analysis of the 5′ splice site
Varying degrees of 5′ss conservation
Based on a preliminary analysis in which we found considerable nucleotide bias between position −4 and 8 (the fourth position upstream of the 5′ss and the eighth position downstream from the 5′ss, respectively), we defined this 12-nucleotide (nt) region as the 5′ss (see also Lim and Burge 2001; Carmel et al. 2004). The most striking observation regarding the 5′ss was its varying degrees of conservation (Fig. 1). In hemiascomycetous fungi and C. parvum, positions 1–6 were extremely highly conserved (the consensus nucleotide, defined as the most frequent nucleotide at each position, appears at a frequency close to 100%). Among all other organisms, the 5′ss signal was much more degenerate. On the other hand, the “G” at position −1 and the “A” at position −2 in the exonic portion of the 5′ss become increasingly conserved in metazoans with respect to fungi, consistent with results found in Ast (2004).
Figure 1.

Sequence motifs of the 5′ss. The sequence motif of the 5′ss of each of the 22 organisms is displayed by using the PICTOGRAM program. The height of each letter is proportional to the frequency of the corresponding base at the given position, and bases are listed in descending order of frequency from top to bottom. The displayed sequence spans the exon-intron junction and includes the last four exonic nucleotides and the first eight intronic nucleotides (positions shown at the bottom). The arrow at the top marks the exon-intron junction. The organisms are displayed according to their phylogenetic grouping based on trees developed by Hedges (2002), Dujon (2006), and James et al. (2006). The eukaryotic kingdoms comprising these organisms, as well as phylogenetic subdivisions of these kingdoms, are plotted to the right of the sequence motifs. Sequence motifs of three major ancestral nodes, reconstructed by means of a maximum parsimony-based algorithm, are shown to the left of the tree. The top, middle, and bottom motifs represent the reconstructed sequences of the common ancestor of all metazoans, of all fungi and metazoans, and of all eukaryotes, respectively.
These trends were observed more clearly following quantification of the 5′ss signal by means of information content. Information content is a measure of sequence conservation, with high and low information content corresponding to high and low degrees of conservation, respectively (Hertz and Stormo 1999; Lim and Burge 2001). We divided the 5′ss region into exonic and intronic regions and calculated the information content in each of these regions (Fig. 2A). The total information content (exonic + intronic) was found to be high in the hemiascomycetous group (ranging from 9.3 to 11.7 bits) and in C. parvum (13.2 bits), while among all other organisms the information content was considerably lower (6.7–8.6). In addition, consistent with our above observations, we found the information content in the exonic portion to be relatively low in fungi and high in most metazoans, plants, and protozoans.
Figure 2.

(A) Information content (measured in bits) of the 5′ss sequence for each of the organisms. Information content is shown separately for the 4-nt exonic part and the 8-nt intronic part (marked in gray and black, respectively). Total information content is the sum of the two. (B) Multiple sequence alignments of the 5′ region of the U1 snRNA molecule in metazoans, protozoans, plant, and non–S. cerevisiae-like fungi. The scheme at the top shows a sample 5′ss containing the human consensus nucleotides, opposite the corresponding positions on U1 snRNA, and the location of the 5′ss positions relative to the exon/intron boundary is marked at the bottom. The binding site of the splicing factor U1–70K is marked as well. (C) Multiple sequence alignment of the 5′ region of U1 snRNA in S. cerevisiae-like fungi, which are characterized by a longer U1 snRNA sequence and by a different secondary structure (Kretzner et al. 1990). The 5′ss positions and the U1–70K binding site are marked as in B.
In view of the low conservation levels in the exonic part of the 5′ss in fungi, we next performed a functional analysis of these positions by assessing whether adherence to the consensus nucleotides in these positions anti-correlated with adherence to the consensus nucleotides in positions in the intronic part of the 5′ss. We found that, similar to the situation in metazoans, such anti-correlations between positions −1 and −2 and different exonic positions exist, which is indicative of the functional importance of these positions. Among many organisms, positions +7 and +8 were involved in significant correlations as well, highlighting the importance of these positions (see Supplemental Material and Supplemental Fig. S3).
Reconstruction of 5′ss among early eukaryotic ancestors
We next sought to understand whether the 5′ss of early eukaryotes was conserved, as we had proposed in the past (Ast 2004), or degenerate. For this purpose, we developed a maximum parsimony-based algorithm that receives a set of position-specific scoring matrices (PSSMs) and an evolutionary tree as input and reconstructs the most parsimonious ancestral PSSM at each node (see Methods). Examining the reconstruction of the ancestral 5′ss (Fig. 1), we found that it highly resembles the metazoan signal. This is true not only in terms of consensus nucleotides at most positions, but also in terms of conservation. The ancestral 5′ss contains 8.1 bits of information, on par with most organisms, but considerably less than among hemiascomycetous fungi. Also, the exonic part alone contained 1.3 bits, which is similar to metazoans and plants, but not to fungi. These results can be directly observed in Figure 1: The high conservation is mostly limited to one monophyletic group (hemiascomycetous fungi), whereas, among other organisms throughout the entire evolutionary tree, the 5′ss is degenerate.
Correlation between changes in U1 snRNA and in the 5′ss
Various fluctuations were observed when the consensus nucleotides for each of the 5′ss positions were compared among the different organisms. We were interested in assessing to what degree these fluctuations can be attributed to changes in the U1 snRNA sequence, which binds this signal. For this purpose, we compiled a data set of the U1 snRNA sequences in the various organisms. Using a battery of tools and algorithms (see Methods and Supplemental Material), we were able to identify the U1 snRNA sequence in 20 of the 22 species (Fig. 2B,C; Supplemental Fig. S1).
Many changes in the 5′ss cannot be correlated with changes in U1 snRNA. For example, the varying trends in conservation and in consensus nucleotides between position −1 and position +6 cannot be correlated with changes in U1 snRNA, as these seven positions remain unchanged throughout evolution. This underscores the importance of additional factors in determining the 5′ss. Nonetheless, various correlations between the 5′ss and U1 snRNA were found. These correlations involved predominantly positions −3 and +7, whose preferred nucleotide composition often correlated with the nucleotide binding this position in U1 snRNA (see Supplemental Material for complete analysis and discussion).
Analysis of the PPT-3′ss
PPT region
We analyzed the PPT and the 3′ss as two separate signals. The PPT analysis is presented in this section. The 3′ss analysis, involving the last four intronic positions and the first two exonic positions, is presented in Supplemental Material. The PPT is located between the BS and the 3′ss, in what we term the “PPT region.” The PPT region was defined as lying between two borders: The 3′ border was invariably set as position −5 (i.e., the fifth-to-last position within the intron), whereas the 5′ border of this region was set at the median distance between the termination of the BS and the 3′ss (see Results section “Analysis of Branch Site”). To globally assess whether PPTs exist in this region in various organisms, we first searched this region for bias in nucleotide composition.
The bias of each nucleotide at each position relative to the background frequency was defined as the difference between the position-specific and the background frequency of each nucleotide. Positional bias plots, visualizing statistically significant bias of each nucleotide at each position, are shown in Figure 3 for several metazoans and in Supplemental Figure S4 for all organisms. In metazoans, plants, and in one protozoan (D. discoideum), we observed a clear pyrimidine-rich signal near the 3′ end of introns. Among most fungi, there was an extremely low, albeit statistically significant, enrichment in pyrimidines toward the 3′ end of the intron. The three fungi S. cerevisiae, Kluyveromyces lactis, and Ustilago maydis were exceptions to the above observation; among them, a clearer, mostly T-rich enrichment was found near the 3′ end of the intron.
Figure 3.

PPT regions in metazoans. The PPT region was defined as the region between the BS and the 3′ss. For each organism, the median distance between the termination of the BS sequence and the 3′ss was determined, and positional bias plots of the region between this position and position −5 relative to the 3′ss are shown. Briefly, positional bias plots show the bias of each nucleotide at each position relative to the background frequency, by showing the difference (Δ) between the position-specific and the background frequency (see Methods). The y-axis presents the extent and nature of the bias, with positive and negative values denoting nucleotides that appear more, and less, than expected, respectively. Note: Adenosines, thymidines, cytosines, and guanines are visualized in green, yellow, red, and blue, respectively. Note: The bars at position −5 of C. elegans are truncated: The bias for T at this position is 0.54, while that of A is −0.27. See Supplemental Fig. S4 for results of other organisms.
Particularly interesting observations regarding the PPT region were made in metazoans, excluding Caenorhabditis elegans. In these organisms:
The enrichments in “T” and in “C” appear to be acting as two separate signals superimposed upon each other. The enrichment in “T” is more fixed in its pattern among the different organisms than is the “C” pattern and invariably peaks at position −10. The enrichment in “C”, when present, is at its lowest at position −10 but increases in strength in more downstream positions of the PPT. Moreover, the bias in “C” is not uniform across the phylogenetic tree. The bias is weakest in Drosophila melanogaster, stronger in zebrafish, stronger in chicken, and strongest in dog, mouse, and human. In addition, in chicken, dog, mouse, and human the bias in “C” is present in upstream positions of the PPT region, unlike D. melanogaster and zebrafish in which there is no bias for “C” in these positions. These results suggest a gradual shift in the PPT composition from invertebrates through vertebrates to mammals.
Using a measure of mean pyrimidine bias per position within the PPT region, we found a nonuniform bias across the phylogenetic tree: 0.143, 0.13, 0.152, 0.163, 0.164, and 0.167, in D. melanogaster, zebrafish, chicken, dog, mouse, and human, respectively. This suggests a gradual increase in the overall pyrimidine bias when moving from invertebrates through vertebrates to mammals.
Finally, a phenomenon highlighted by Figure 3 is the selection against “A” in vertebrates (zebrafish through human), in particular at the 3′ end of the PPT region: The enrichment in pyrimidines occurred mainly at the expense of “A” and much less so of “G”s.
Comparative analysis of the PPT
We next analyzed the PPT directly. For this purpose, we developed an algorithm that identifies stretches of pyrimidines and scores them based on their length and pyrimidine content. This is a good measure of PPT strength because longer, pyrimidine-rich PPTs are more efficient than shorter, purine-rich ones in directing splicing (Roscigno et al. 1993). We applied this algorithm to the last 50 nt of all introns and accepted a stretch as a putative PPT only if it ended within 10 nt of the 3′ss. This value was set because this is the maximal distance from the 3′ss in which PPTs were shown to be functional (Coolidge et al. 1997; Kol et al. 2005). Finally, a “PPT enrichment index” was calculated for each organism. This index was defined as the quotient of the mean PPT strength in introns of the particular organism divided by mean PPT strength in a randomized data set. The random data set corresponded to the original data set in terms of nucleotide composition and incorporated the BS and 3′ss signals in order to take into account the bias introduced by these two signals. Thus, the “PPT enrichment index” represents the fold-change in the mean PPT score in the original data set relative to the random one (see Methods).
Results of this analysis are plotted in Figure 4A and presented in Supplemental Table S2. Strikingly, among almost all organisms the PPT enrichment index was found to be >1 and to be statistically significant. This indicates a statistically significant bias for pyrimidine-rich stretches at the 3′ end of introns. However, organisms differed from each other dramatically in the extent of this bias: The strongest bias for PPTs was found among metazoans. In human, for example, an approximately ninefold increase was found in the mean PPT strength relative to the random data set. Most fungi, on the other hand, had very weak PPT enrichment indexes: In the case of non-hemiascomycetous fungi, the PPT enrichment indexes ranged from 1.14 to 1.57, whereas among hemiascomycetous fungi indexes were often even lower. Two exceptions were indexes for S. cerevisiae and K. lactis; both had much stronger PPTs. Among plants and protozoans, we found intermediate PPTs, with PPT enrichment indexes ranging from 1.8 to 2.3.
Figure 4.

Comparison of the PPT strengths. (A) The PPT enrichment index indicates the extent of increase in PPT strength in the intron data set, relative to the PPT strength obtained in a corresponding, randomly permuted data set of equal nucleotide composition. Nonsignificant PPT enrichment indexes in C. glabrata, E. gossypi, and D. hansenii are plotted in white. (B) Percentage of introns in which PPTs were detected in metazoans. (C) Mean length of the detected PPTs in metazoans.
A further observation from Figure 4A is the gradual strengthening of the PPT along the metazoan lineages. In order to examine this strengthening further, we calculated the frequency of introns containing PPTs (Fig. 4B) and the mean length of the detected PPTs among metazoans (Fig. 4C). We found that across the metazoan lineages there was a gradual increase in the frequency of introns containing PPTs, as well as a gradual increase in PPT length. These analyses were all performed for the remaining organisms as well (see Supplemental Table S2 for full results). In the two organisms, Y. lipolytica and C. elegans, unique observations were made in the PPT analysis. In Y. lipolytica, the pyrimidine-rich signal is due to a C-rich stretch located upstream of the BS (see also Fig. 6D, below). Both the location and composition of this signal indicate that it may not function as a PPT. In C. elegans, the vast majority of PPTs were derived from the highly conserved “TTTTCAG” heptamer (see Supplemental Fig. S4). This conserved heptamer has been shown to interact with U2AF (Zorio and Blumenthal 1999) (see Discussion).
Figure 6.

Comparative analysis of the branch site. (A) Sample BS motifs, for selected organisms (see Supplemental Fig. S4 for all organisms). (B) Information content of the 7-nt BS, calculated as in Fig. 2. (C) Upstream extension of the BS signal in certain fungi. The region upstream of the BS in each intron was aligned, and positional bias plots of this region were formed. Results are shown for the seven positions immediately upstream of the BS in E. gossypii. Each of the significantly enriched (P < 0.05) nucleotides in this region is complementary to nucleotides in U2 snRNA directly downstream to the nucleotides that bind the BS sequence. The U2 snRNA nucleotides lie in a potential unpaired region, between two U2 snRNA stem-and-loop I and IIa structures (Ares 1986). The bold “A” nucleotide marks the BS nucleotide. (D) The 3′ intron end in Y. lipolytica. An alignment of the last 11 intronic positions is presented in the form of a pictogram. This alignment uses the 3′ss as anchor. The BS is clearly apparent, between positions −11 and −5. A C-rich signal upstream of the BS is presented in the form of positional bias plots.
PPT composition
Different analyses corroborated our findings regarding the compositional biases of the PPT. These analyses confirmed a general bias for “T” in the PPT, a gradual enrichment in the “C” content of the PPT when moving from nonvertebrates through vertebrates to mammals, and a general bias against “A.” In addition, these analyses confirmed that different nucleotide biases are present in the PPT in the positions preceding and following position −10. These analyses are fully presented and described in Supplemental Material.
Correlation between changes in the PPT and in factors binding it
We next set out to determine to what extent changes in the PPT were determined by corresponding changes in the splicing factors that bind the 3′ end of introns during early stages of splicing. Specifically, we focused on U2AF2 and U2AF1, which recognize the PPT and the 3′ss, respectively (Zamore and Green 1989; Kent et al. 2005), and on SF1, which binds the BS and facilitates the binding of U2AF2 to the adjacent PPT (Manceau et al. 2006). We moreover conducted an in silico functional analysis of the identified proteins: We concentrated on known functional residues in these proteins, including regions that are important for RNA binding, as well as residues that are important for interactions with other splicing factors, and predicted to what extent these residues have undergone changes compromising their functionality. A description of the functional domains in these proteins and the full analysis is described and presented in the Supplemental Material. Here we report the highlights of our findings.
For these three proteins, the 22 organisms can be divided into three groups: human-like (15 organisms), S. cerevisiae-like (four organisms), and human-reminiscent (three organisms). The human-like group comprises all sampled metazoans, plants, non-hemiascomycetous fungi, and the protozoan D. discoideum. The organisms in this group have functional homologs of all three proteins and we therefore concluded that in these organisms the 3′ intron end recognition is likely to take place as it does in human, with the U2AF heterodimer interacting with SF1 and with the PPT. This correlates with the fact that a PPT signal was observed in all these organisms. The S. cerevisiae-like group comprises S. cerevisiae, Candida glabrata, Eremothecium gossypii, and K. lactis. These organisms all lack functional copies of U2AF and instead contain homologs of MUD2, which is the analog of U2AF in S. cerevisiae. Therefore, 3′ss recognition in these organisms presumably follows the pattern of S. cerevisiae. The human-reminiscent group includes the two hemiascomycetes fungi Y. lipolytica and Debaryomyces hansenii and the protozoan C. parvum. In the two hemiascomycetes, fully functional SF1 and U2AF1 homologs were found. However, while the U2AF2 homolog has retained its ability of binding U2AF1 and SF1, it appears to have lost the ability to bind the PPT due to mutations in regions responsible for binding the PPT. We concluded that in this group the recognition of the 3′ss and BS is likely performed by U2AF1 and SF1, respectively, with U2AF2 functioning as a bridge between them. This is supported by the lack of PPT in these two organisms. In C. parvum, various mutations were found in all three proteins, suggestive of a 3′ss recognition mechanism considerably divergent from human.
We next focused on the three RNA recognition motifs (RRMs) of U2AF2, two of which (RRM1 and RRM2) bind the PPT and the third, called U2AF homology motif (UHM), mediates the interaction between SF1 and U2AF2. Comparing the RRMs of U2AF2 of the different species to the corresponding human RRM (Fig. 5A), we observed two phenomena. First, the RRM2 domain is more conserved in non-metazoans, with respect to human, than either RRM1 or UHM. This suggests that RRM2 may be the dominant domain in terms of PPT binding among non-metazoans. Second, we observed that among vertebrates there is almost 100% identity conservation in RRM1 and RRM2 with respect to human. This conservation gradually decreases from vertebrates to invertebrates, and the motifs are even less conserved in fungi than in invertebrates. This decreasing gradient correlates with the trend observed in the PPTs, which are weaker in invertebrates than in vertebrates and even weaker among most fungi.
Figure 5.

Comparative analysis of the RRM domains between U2AF2 homologs: (A) The conservation levels of the RRM domains of each organism are compared with their human counterpart. Xenopus tropicalis was used instead of chicken (see Methods). In B and C a conservation analysis of key residues in the RRM1 and RRM2 domains, respectively, is presented. These residues have been shown to be required for PPT binding in human (Sickmier et al. 2006). Residues labeled by “W” interact with the RNA through a water molecule, whereas residues labeled by “S” and “M” interact through the side-chain and main-chain, respectively. The dashed green boxes highlight the RNP1 and RNP2 motifs and the red boxes mark substitutions, with respect to metazoans, that change the biochemical properties in key residues that are responsible for RNA binding. In fungi, the following substitutions, with respect to human, can be observed: aromatic residues (F and Y) by nonaromatic ones (V, I, L, M, C, S) in the RNP1 and RNP2 regions of both RRMs. In the RRM1, polar to nonpolar substitutions: K to G at RNP1 and N to G in the RNP1 and RNP2; and polar basic (K) to nonpolar neutral (A) and polar acidic (E) at the C-terminal region (panel B). Similarly, in RRM2 of fungi we observed substitutions of a nonpolar residue (G) to polar ones (T and S) in the RNP2; polar to nonpolar substitutions: N to V, L, A near the RNP motifs, and Q to V at the C-terminal region; and basic to neutral (K to N, A) at the C-terminal region. In the RRM1 of A. thaliana there is a polar (N) to nonpolar (G) substitution in the RNP2 and a neutral (N) to basic (K) substitution in the RNP1. In the RRM1 of D. discoideum there is a substitution N to C, which reduces the polarity at this position. Finally, in the RRM2 of both species, there is a basic (K) to neutral (T) substitution in the C-terminal region.
While the above results suggest that the PPT coevolved with RRMs that bind it, the decreased conservation may also reflect increased phylogenetic distances. To assess the functional importance of the decreased conservation, we focused on specific, key residues in RRM1 and RRM2 that have previously been shown to be required for PPT binding in human (Sickmier et al. 2006). These included residues participating in main-chain, side-chain, and water-mediated interactions (Sickmier et al. 2006). The characteristics of these residues, in terms of polarity, charge, and aromaticity, are therefore important for U2AF2 binding to the PPT: A change in polarity will affect the water-mediated interactions, whereas any change in charge, polarity, or aromaticity is expected to affect the side-chain interactions. Among non-hemiascomycetous fungi, we identified many such changes, with respect to metazoans, in key residues both in RRM1 (Fig. 5B) and in RRM2 (Fig. 5C). These results suggest that the decrease in PPT strength among fungi, relative to metazoans, is linked to detrimental changes in key residues on U2AF2 required for PPT binding. This conclusion is strengthened by the fact that relatively fewer changes were observed in the RRMs of D. discoideum and A. thaliana, despite the fact that phylogenetically they are more distant from metazoans than non-hemiascomycetous fungi. This correlates with our findings pertaining to the PPTs, which are stronger in these two organisms than those in non-hemiascomycetous fungi (see Discussion).
Finally, we found that, in general, the UHM of the U2AF2 homologs has higher sequence similarity to the RRM domain of MUD2 than to the RRM1 and RRM2 domains (see Supplemental Fig. S17). In fact, the UHM from U2AF2 and the RRM from MUD2 present similar RNP motifs (see Supplemental Fig. S13). Moreover, the UHM in C. parvum represents an intermediate between the RRM of MUD2 and the UHM of U2AF2 in other organisms in terms of sequence conservation (see Supplemental Fig. S18). This provides further evidence for a common evolutionary history for U2AF2 and MUD2 (see also Abovich et al. 1994).
Analysis of branch site
To examine the BS signal, we developed a simple algorithm that extracts the putative BS motifs from the introns of the various organisms. The algorithm is based on the BS characteristics of two model yeasts, S. cerevisiae and S. pombe, as well as on general characteristics of the hemiascomycetous BS, as found by Bon et al. (2003) (see Methods). We validated our results by comparing our identified BSs with those identified by algorithms that have been used in the past (Kupfer et al. 2004; Kol et al. 2005) and against a set of biologically proven BSs in human (see Supplemental Material). The stringent requirements of the algorithm yielded satisfactory results, albeit at the price of (1) discarding a relatively large percentage of introns in which it cannot determine between two putative BSs, and (2) the branch site motifs yielded by the algorithm tending to be somewhat more conserved than they are (see Supplemental Material).
Branch site motifs and ancestral reconstruction
Branch motifs identified by the algorithm for representative organisms are shown in Figure 6A and for all organisms in Supplemental Figure S4. Similar to the results for the 5′ss analysis, the hemiascomycetous group stood out with respect to sequence conservation: The canonical, practically invariable “TACTAAC” BS motif was identified among all species in this group. Among the remaining species, the BS motif was reminiscent of this motif, but more variable.
Based on the degree of conservation in the BS (Fig. 6B), organisms can be divided into three groups corresponding to their phylogenies: Hemiascomycetous fungi had the largest degree of conservation with information content ranging between 7.9 and 12.7 bits; metazoans had the least information content, ranging between 5.0 and 5.7 bits; and non-hemiascomycetous yeasts formed an intermediate group, with information content ranging between 6.6 and 7.8 bits. Plants and protozoans most resembled metazoans, with information content ranging between 5.1 and 5.8 bits.
Based on the motifs of the various organisms, we reconstructed the ancestral BS motifs using the same maximum parsimony-based algorithm for reconstructing ancestral position-specific scoring matrices that was used for the reconstruction of the 5′ss. The ancestral BS at the root of the tree contained only 6.5 bits of information. This result is in agreement with the observation that highly conserved BSs were found only in one monophyletic group, the hemiascomycetous fungi, whereas among all other groups the BS was much more degenerate.
Correlation between changes in the BS and in U2 snRNA
To test whether changes in the BS motifs are correlated with changes in U2 snRNA, we compiled a data set of U2 snRNAs from the different organisms. Multiple sequence alignments of the regions binding the BS can be viewed in Supplemental Figure S2. The six positions of U2 snRNA that bind the BS were unchanged among all organisms. Therefore, variation in U2 snRNA cannot account for the variation in the BS motifs among the different organisms. However, we observed that for various species the bias around the BS signals extends beyond the BS heptamer, possibly reflecting an extended region of base-pairing with U2 snRNA. The hemiascomycetous fungi E. gossypii presented a clear example for such extended base-pairing (Fig. 6C). Similar results reflecting an extended branch site region were also found for N. crassa, Magnaporthe grisea, and Aspergillus fumigatus, and to a lesser extent in D. discoideum and D. hansenii.
Finally, we found that Y. lipolytica has a unique BS distance distribution. Although this organism has relatively long introns, the BSs were almost invariably found at the same location, immediately upstream of the 3′ss: Of the 709 introns in which BSs were identified in this organism, 560 (79%) are located at position −11 and 686 (97%) are located between positions −10 and −14. Thus, in Y. lipolytica, the BS and 3′ss form one consecutive stretch. This finding, along with the observation of the C-rich region upstream of the PPT (Fig. 6D) and lack of conservation of the U2AF2 RRMs, suggests that splicing in this organism has followed a unique path (see Discussion).
Discussion
In this study, the three major splicing signals were analyzed and compared across a wide array of eukaryotes. Our analyses have yielded several major findings. The first pertains to the high variability of the PPT signal. Although there is a certain bias for pyrimidines toward the 3′ end of the intron among all organisms, this signal is very weak among most fungi, stronger in plants and in protozoans, but by far strongest among metazoans. Moreover, among the latter group this signal appears to have evolved in terms of its nucleotide composition, length, and abundance. Second, we found various correlations between variations in splicing signals and in splicing factors: Such correlations were found between the 5′ss and U1 snRNA, as well as between the PPT-3′ss signal and the factors binding it. These findings highlight the importance of these splicing factors in determining splicing signals, but also underscore the importance of other factors in shaping them. Our final major finding pertains to the relatively low conservation of splicing signals at the root of the eukaryotic tree, based on a maximum parsimony reconstruction applied to the 5′ss and the BS.
Evolution of the PPT
The evolution of the PPT splicing signal and its functional importance in eukaryotes have been poorly addressed by previous studies. By using comparative genomics to analyze this signal, we were able to ask whether a PPT exists in an organism, to what extent, and how it compares with PPT signals found in vertebrates. Although a bias toward pyrimidines was detected just upstream of the 3′ss among most organisms, this bias was intermediate among plants and protozoans, very low, but existent, among most fungi, and very high among metazoans.
Particularly interesting trends were observed in the metazoan PPT. First, we found a gradual increase in PPT strength along the metazoan lineages. In C. elegans the detected PPTs are very short and stem from a highly conserved “TTTTCAG” heptamer at the 3′ intron end. The “TTTTC” sequence has been shown to cross-link with U2AF2 (Zorio and Blumenthal 1999) and to be critical for 3′ss recognition (Hollins et al. 2005), while the “AG” interacts with U2AF1 (Zorio and Blumenthal 1999). Therefore, this heptamer appears to be a very reduced version of the PPT combined with the 3′ss. As the phylogenetic distance from human decreases, the PPT gradually increases in strength, length, and abundance. This gradual increase was apparent predominantly in D. melanogaster, zebrafish, and chicken; among the three mammals, the PPT strength is similar. These results extend the results of a previous study, which examined the last 20 nt within introns and found them homogeneous within tetrapoda but noticeably distinct from those of other vertebrate and invertebrate taxa (Abril et al. 2005).
Using different approaches for analyzing the PPT, we consistently observed two different signals within the region just upstream of the 3′ss: A signal more biased in “T”, located toward the 5′ end of the PPT, and a more “C”-rich signal, located at the 3′ end of the PPT. Position −10, relative to the 3′ss, appears to serve as a key position, with the “T” signal peaking and the “C” signal falling at this position. What mechanism may underlie these two signals? One possibility is that they serve as binding regions for the RNA recognition motifs of U2AF2. It has previously been shown that RRM1 and RRM2 bind different regions along the PPT in vertebrates: RRM2 binds the more 5′ region of the PPT, whereas RRM1 binds the more 3′ region (Banerjee et al. 2003). Thus, the two signals may reflect the differential binding affinities of the two RRMs. In addition, the invariable “T” peak at position −10 may reflect function, as the maximal distance from the 3′ss in which PPTs were shown to be functional is 10 nt (Coolidge et al. 1997; Kol et al. 2005).
We further observed a gradual increase in the bias toward “C” and an increase in the length of the region with this bias from C. elegans to human: PPTs among lower metazoans are biased for “T”s with a limited “C” signal at the 3′ end of the PPT only, whereas among higher metazoans the “C” signal was more widespread throughout the entire PPT. Differential preferences of RRMs cannot explain these observations since the RRMs have remained highly conserved along these lineages. These findings may therefore reflect a gradual increase, or shift, in selective pressure exerted by factors other than U2AF2 that bind to the PPT. These factors may require a more “C”-rich PPT nucleotide composition. NOVA and PTB are examples of two such potential splicing factors. The consensus binding sequences of PTB are “UCUUC” and “CUCUCU” (Spellman and Smith 2006) and that of NOVA is “YCAY” (with Y representing pyrimidines) (Ule et al. 2006). Thus, the increase in “C” may reflect the growing importance of such factors in splicing regulation among higher metazoans.
Finally, with regard to the PPT composition, we noted two general phenomena: a preference for “T” over “C” and a preference for “G” over “A”. The preference for “T” over “C” is explained by past findings that “T”s and “C”s do not function equivalently within PPTs, with consecutive “T”s constituting the strongest PPT (Bouck et al. 1995; Coolidge et al. 1997). The bias against “A” may be due to a tendency to avoid the formation of a cryptic branch point (Ruskin et al. 1985; Kol et al. 2005). With “A” being the preferred branch point, there may be a selection against this nucleotide at positions following the true branch point.
Correlation between changes in the PPT and in U2AF2
We found that changes in the PPT signal correlate with changes in U2AF2. First, among all organisms in which the U2AF heterodimer was found to have retained its functionality, we found statistically significant PPTs. Second, the inability of U2AF2 to bind the PPT in D. hansenii and Y. lipolytica corresponds with the lack of PPT in these organisms. Third, the gradual decrease in the conservation of the RNA binding domains in U2AF2, with respect to human, correlates with the gradual decrease in strength of the PPT. Fourth, we found that various residues known to be critical for U2AF2 binding in metazoans are not conserved among fungi, but are conserved to a greater degree in D. discoideum and A. thaliana. This pattern corresponds to the pattern of PPT strengths among the various organisms, suggesting that these key residues are indeed related to the strength of the PPT.
However, the correlation between changes in U2AF2 and in the PPT is not perfect. Similar PPT enrichment indexes were found among the non-hemiascomycetous fungi and yet the pattern of substitutions in key residues in the RRMs of these fungi differed considerably. For example, the non-hemiascomycetous fungi C. neoformans and U. maydis exhibited fewer changes in the RRMs than other fungi, and yet share similar PPT strengths. In addition, in E. gossypii, C. glabrata, K. lactis, and S. cerevisiae, we would naively expect to find PPTs, as all contain S. cerevisiae-like factors. Yet, the first two organisms do not appear to have PPTs, although the latter two do.
Finally, we observed that RRM2 was the most conserved RRM among all organisms with U2AF2 homologs. This indicates that RRM2 may be the dominant RRM in terms of recognizing the PPT, in particular in non-metazoans. A factor that may have influenced the increased dominance of RRM2 is the fact that the size of the cross-linking site for RRM2 is much more variable than that of other RRMs (Banerjee et al. 2003).
Analysis and reconstruction of the 5′ss and BS
We previously suggested that the ancestral eukaryote was characterized by strong splice sites and little or no alternative splicing and that the weakening of the splicing signals brought forth the rise of alternative splicing in multicellular eukaryotes (Ast 2004). This hypothesis was based on the comparison of the human 5′ss to those of two yeasts. Based on a much broader taxonomic sampling, our current data suggest that the increased conservation of splicing signals is a phenomenon mostly limited to hemiascomycetic yeasts, whereas among most organisms the splicing signals are degenerate, probably reflecting the ancestral state. In this respect, our results agree with, and extend, recent results obtained by Irimia et al. (2007): After comparing the 5′ss of many eukaryotes, these authors suggest that the ancestral eukaryotic 5′ss was degenerate. Here, we have developed a maximum parsimony algorithm in order to quantify evolutionary shifts in splice site motifs. Our results validate the degeneracy of the ancestral 5′ss and suggest that the ancestral BS was degenerate as well. We note, however, that our analysis includes only one plant species. Increasing the taxonomic sampling of deep-branching organisms would increase the accuracy of the ancestral splicing motif reconstruction.
Splicing in the eukaryotic ancestor
Past studies concluded that the early, eukaryotic ancestors were relatively rich in introns and that their genes contained relatively high intron densities (Rogozin et al. 2003; Nguyen et al. 2005; Raible et al. 2005; Roy and Gilbert 2005a, b; Sverdlov et al. 2005; Carmel et al. 2007). This observation, combined with our finding that splicing signals are degenerate, may suggest that the genome of the ancestral eukaryote was more similar to the mammalian genome than previously anticipated and opens up the possibility that alternative splicing existed early in eukaryotic evolution. However, this conclusion must be tempered by a further observation emerging from this study, namely that there is no direct correlation between degenerate splicing signals and levels of alternative splicing. This can be demonstrated by the fact that various organisms with very little, or no, known alternative splicing in the form of exon skipping, such as S. pombe, N. crassa, M. grisea, and D. discoideum (Ast 2004), have splicing signals that are as degenerate as those of metazoans, such as human and mouse, with very high levels of alternative splicing (Kim et al. 2007).
Our findings imply that the eukaryotic ancestor resembled extant vertebrates in terms of splicing factors, as well. We found that the U2AF heterodimer and SF1 are conserved in most of the analyzed organisms. This indicates that these splicing factors existed in the early, eukaryotic ancestor. In support of this hypothesis, we also found that the RRM domain of MUD2 resembles the UHM domain, raising the possibility that MUD2 and U2AF2 have a common origin. The ancestral U2AF2 presumably had a domain organization similar to U2AF2 and has evolved differently along various lineages: In hemiascomycetous fungi, the UHM domain became dominant and other domains were lost, whereas among other organisms RRM2 became more dominant.
The residual similarities between RRM domains and the existence of possible pseudogenes for some of the splicing factors serve as further evidence that the changes in the splicing regulatory proteins have taken place gradually and have occurred independently in different organisms. In E. gossypii, for example, a U2AF1 homolog was found, but it appears to have lost its function. The splicing factors in the three organisms in the human-reminiscent group show similarity to, but a considerable functional divergence from, their human counterpart, serving as further proof for their gradual evolution. Thus, our results suggest that in terms of both splicing signals and splicing factors, the eukaryotic ancestor resembled extant vertebrates. This is in agreement with results of previous studies that have made similar conclusions both regarding the splicing signals (Irimia et al. 2007) and the spliceosome components (Kaufer and Potashkin 2000; Anantharaman et al. 2002; Koonin et al. 2004; Collins and Penny 2005).
Splicing in Y. lipolytica
Several unique features were observed in Y. lipolytica. Most prominently, we found that in this organism the BS and 3′ss form one combined sequence. It is noteworthy that a similar, 12-nt BS–3′ss juxtaposition was observed in the two deep-branching eukaryotes Trichomonas vaginalis and Giardia lamblia (Vanacova et al. 2005). With regard to these two organisms, it has been hypothesized that the juxtaposition of the BS and the 3′ss reflects a simplified spliceosomal assembly, combining the two steps of BS and 3′ss recognition (Vanacova et al. 2005). Our observation that the RRMs of U2AF2 are not conserved and apparently nonfunctional in Y. lipolytica sheds a somewhat different light on the BS–3′ss juxtaposition. Our findings suggest that U2AF2, which retained its capability to bind SF1 and U2AF1, may serve as a bridge between the two molecules, without binding the pre-mRNA. Alternatively, U2AF2 may have lost its functionality completely in Y. lipolytica.
Is U2AF2 loss, or modification of function, responsible for the BS–3′ss juxtapositions among other organisms as well? This analysis could not be carried out for G. lamblia as its genome sequence is not sufficiently complete to perform the homology search. However, an analysis of the splicing factors in T. vaginalis revealed a situation very similar to the one in Y. lipolytica. We were able to identify functional homologs for SF1 and U2AF1, but U2AF2 was found to be very divergent from the human protein. Additionally, the U2AF2 in T. vaginalis has only a UHM functional domain and no arginine-rich region, similar to the one we found in C. parvum. Thus, in this organism, too, the BS–3′ss juxtaposition appears to be coupled with modifications in the function of U2AF2, as in Y. lipolytica.
In Y. lipolytica we also found a clear, C-rich signal upstream of the BS. The composition, location, and lack of functional U2AF2 indicate that this signal is not a classic PPT. What may underlie this signal? Previous studies have indicated that initial binding of U2 snRNP to the BS region must be stabilized by an interaction with an anchoring site, located upstream of the BS (Gozani et al. 1996; Kramer 1996; Ast et al. 2001). Thus, this signal may serve as such an anchoring site.
Overall, our results indicate that the three major splicing signals have changed considerably throughout evolution, in parallel with shifts in exon-intron architecture and in concert with domain structure and key residues of splicing regulators. These components interact with and impact on each other and, though they are presumably determined by the lifestyle of organisms, they also, in turn, help determine an organism’s lifestyle by means of diversifying mechanisms such as alternative splicing.
Methods
Database assembly
The complete genomes of C. parvum (Build 1.1), D. discoideum (Build 2.1), S. cerevisiae (Build 2.1), C. glabrata (Build 1.1), K. lactis (Build 1.1), E. gossypii (Build 1.1), D. hansenii (Build 1.1), Y. lipolytica (Build 1.1), N. crassa (Build 1.1), M. grisea (Build 1.1), A. fumigatus (Build 1.1), S. pombe (Build 1.1), U. maydis (Build 1.1), and C. neoformans (Build 1.1) were downloaded from the National Center for Biotechnology Information website (http://www.ncbi.nlm.nih.gov/).
Gene, intron, and exon information was extracted from the annotated genomes using a BioPerl script. This script reads a sequence of annotated GenBank files and generates database records of exons and introns, along with statistical information pertaining to the exon-intron composition, the individual genes, and the entire genome. To allow analysis of the 5′ss and 3′ss, introns were extracted along with the last 15 nt of the upstream exon and the first 10 nt of the downstream exon.
Introns and exons for human (Homo sapiens, Build 35.4), mouse (Mus musculus, Build 34.1), dog (Canis familiaris, Build 2.1), chicken (Gallus gallus, Build 1.1), zebrafish (Danio rerio, release Zv4), C. elegans (release 2003), D. melanogaster (Build 4.1), and A. thaliana (release 2004) were extracted from the Exon-Intron Database (http://hsc.utoledo.edu/depts/bioinfo/database.html) (Saxonov et al. 2000), along with the upstream and downstream regions of the flanking exons. In cases of different alternatively spliced isoforms of the same gene, only the first annotated isoform was extracted; all other isoforms were excluded, in order to avoid redundancy.
Three filtrations were applied to the original data set: (1) All non-GT–AG introns were discarded (2) all introns <15 nt were discarded, and (3) U12 introns were discarded as well. U12 introns were identified based on conformance with position-specific scoring matrices of their 5′ss and BSs. A 20-position matrix representing the U12 5′ss and a 12-position matrix representing the BS were obtained from Levine and Durbin (2001). BSs were searched in the last 38 positions of the introns, as in Sheth et al. (2006). Any sequence whose log-odd score of both the 5′ss and the BS exceeded an empirically derived threshold of −43.54 and −19.73, respectively, was considered a potential U12 intron. This empirical threshold was designed to include 100% of the 5′ss and 99% of the BSs in the database of U12 introns composed by Levine and Durbin (2001). See Supplemental Table S1 for details on the number of introns filtrated at each step.
Position-specific scoring matrices (PSSMs) and scoring
Once a data set of 5′ss and BS was compiled for each organism, PSSMs containing the frequency of each nucleotide at each position were generated. Scores were assigned, both to the 5′ss and the BS of each intron, based on their adherence to their respective PSSMs. This score (S) is calculated as follows:
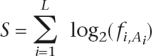 |
where A is the sequence to be scored, L is its length (12 for the 5′ss, and 7 for the BS) and fi,Ai is the PSSM frequency at position i of the ith nucleotide in sequence A.
Background frequencies
Intronic background frequencies were calculated by separately pooling all introns of each organism and calculating the relative frequency of each nucleotide. See Supplemental Table S1 for a list of the intronic background frequencies in each organism.
Information content
Information content (I) is a measure of sequence conservation, with high and low information content corresponding to high and low degrees of conservation, respectively. It is measured as follows:
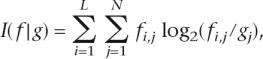 |
where L is the number of positions within the sequence, N the number of nucleotides (“A”, “C”, “G”, and “T”), fi,j is the observed frequency of nucleotide j at position i, and gj is the a priori probability of observing nucleotide j under the background distribution (Hertz and Stormo 1999; Lim and Burge 2001). In the calculation of the information content in the ancestral sequence reconstructions, a uniform background frequency of 25% was assumed for each nucleotide.
Pictograms
Graphical representations of PSSMs were composed by the PICTOGRAM application developed by Burge et al. (1999), downloaded from http://hollywood.mit.edu/burgelab/software. The height of each letter is proportional to the frequency of the corresponding base at the given position, and bases are listed in descending order of frequency from top to bottom.
Ancestral 5′ss and BS reconstruction
The ancestral reconstruction of the 5′ss and BS PSSMs was performed using the maximum parsimony paradigm. Specifically, a PSSM was assigned to each tree node so that the overall change in the PSSMs along the tree branches was minimized. We assume independence among PSSM positions, and in the following we formally describe the algorithm for reconstructing ancestral PSSM at a single position.
We first define a distance between two PSSMs at a specific position. Let f(X) be the frequency of nucleotide X (f(A) + f(C) + f(G) + f(T) = 1).
The distance between two PSSMs, x and y, at that position is defined as:
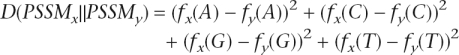 |
In this reconstruction problem, the PSSMs at the leaves of the tree are known, and the objective is to find the PSSMs at the internal nodes, such that the sum of distances, as defined above, along the tree branches is minimized. We first simplify the problem by considering only PSSMs in which each entry is a multiplication of 0.02 (e.g., a PSSM in which f(A) = 0.22, f(C) = 0.28, f(G) = 0.44, and f(T) = 0.06 is valid, but a PSSM in which f(A) = 0.23, f(C) = 0.27, f(G) = 0.44, and f(T) = 0.06 is not). This simplification reduces the number of PSSMs to a finite, computationally feasible number (there are 23,426 such valid PSSMs). Given this finite set of PSSMs and the cost matrix defined above, applying the classical Sankoff algorithm for parsimony reconstruction (Sankoff 1975) is straightforward. The algorithm is in linear time complexity with respect to the number of sequences and quadratic with respect to the alphabet size.
Positional bias plots
To provide graphical representation of bias in nucleotide composition at given positions with respect to expected background frequencies, we used positional bias plots. These plots visualize the bias of each nucleotide at each position relative to the background frequency, by showing the difference (Δ) between the position-specific and the background frequency. Only statistically significant biases (P < 0.01) are shown. Statistical significance was determined by performing χ2 tests between the observed frequency of each nucleotide at each position and the expected background frequency. The y-axis presents the extent of the bias: For example, assuming a 25% background frequency of “C”, a value of 0.1 for “C” at a given position indicates that at this position the frequency of “C” is 35%, while a value of −0.1 would indicate that “C” appears at a frequency of only 15% at that position.
BS detection
In the case of S. cerevisiae, the BS consensus sequence is a highly conserved heptamer composed of “TACTAAC”, but with large variability in its location, appearing as far as >100 nt upstream of the 3′ss (Pikielny et al. 1983; Langford et al. 1984; Fouser and Friesen 1987). Among other hemiascomycetous yeasts, the first two positions of the BS are less conserved (Bon et al. 2003). In the case of S. pombe, the BS sequence is more degenerate, based on different variations of the NNYTRAY sequence, where Y stands for pyrimidines, R for purines, and N for any nucleotide, but with a greater tendency to appear close to the 3′ss. Thus, to detect the BS, we implemented the following algorithm (“find_bs.pl”, available upon request):
Scan the 200 nt upstream of the 3′ss, and identify all heptamers conforming to any of the following BSs: NNYTRAY, NNCTYAC, NNRTAAC, and NNCTAAA. These sequences are based on findings by Bon et al. (2003), for hemiascomycetous yeasts, and by Sanger (http://www.sanger.ac.uk/Projects/S_pombe/intron.shtml), for S. pombe.
Score each heptamer according to its hamming-distance (the number of mismatches) from the optimal BS consensus “TAC TAAC”.
Discard all introns in which the best-scoring hit is not the most downstream hit.
Although step 3 discards a relatively large percentage of introns, it guarantees that those BSs that are detected have no “serious” competitors, thus reducing the false-positive rate. Supplemental Table S1 summarizes the percentage of introns in which BSs were identified.
Identification of PPT
An algorithm to find sequence segments, in which pyrimidines are statistically enriched, was developed. Not only consecutive stretches were considered, as we were also interested in identifying “gapped” stretches, i.e., stretches including non-pyrimidines. Each stretch was assigned a score quantifying its pyrimidine enrichment. The score was calculated as the χ2 test score with 1 degree of freedom, comparing the observed number of pyrimidines with the expected one, assuming a uniform nucleotide distribution (the inter-species nucleotide content heterogeneity was subsequently accounted for by using randomized data sets; see below). Having defined a score for each segment, the optimal segment was searched based on the following algorithm:
Identifying seeds of consecutive pyrimidines: The sequence is represented in binary terms, 1 and 0 representing pyrimidines and purines, respectively. Next, the algorithm compacts this sequence, representing it as the number of 1s appearing between each two 0s. Thus, if the initial stretch was TTTCTTTCTTTTTAATTTAC, then this will first be represented as 11111111111110011101, and in the next step as 13,0,3,1. The value in each position now reflects the length of a consecutive stretch of pyrimidines, and the distance between two positions reflects the number of gaps, or non-pyrimidines, between two such consecutive stretches. Each seed is scored as described above.
Combining adjacent seeds: The algorithm runs through all seeds and combines adjacent seeds if their combined score exceeds that of each of the individual seeds. “Adjacent seeds” were defined as all seeds distanced up to two positions from the original seed.
Purging redundant stretches: The algorithm identifies all stretches that are either fully included in other stretches or that fully include other identified pyrimidine-rich sequences. Only the highest scoring stretches are left.
Combining overlapping stretches: All stretches overlapping each other are identified, combined, and rescored based on their combined pyrimidine content.
As input, this algorithm receives a minimal score, serving as a threshold. This threshold score was set as the χ2 test score of six consecutive pyrimidines, as a minimal functional PPT has been shown to consist of 5–6 nt (Coolidge et al. 1997).
Random data set for the PPT analysis
To take into account the heterogeneity in nucleotide composition among the different organisms, we constructed a set of randomly permuted data sets of 50-nt intron ends. For each intron in each organism, a “random” 50-nt intron end was created by retaining a 3-nt 3′ss and a 7-nt BS (only if the BS was within the last 50 nt of the intron) and filling in the remaining 40 positions with 40 nt selected at random from within the intron. These 40 randomly selected nucleotides were obtained by first removing a 6-nt 5′ss, a 7-nt BS, and a 3-nt 3′ss from the intron sequence, randomly permuting the remaining positions, and selecting a 40-nt stretch from within the remaining sequence (or a shorter stretch, if the entire remaining intron sequence was <40 nt). We incorporated the BS and 3′ss signals into the randomized data sets, in order to take into account the bias introduced by these two signals. For organisms with only few introns (hemiascomycetous fungi and C. parvum), we created 30 random intron ends corresponding to each intron end, to obtain a representative, random data set.
Compilation of snRNA and protein data sets
We used a combination of BLASTN, the infernal package (Griffiths-Jones et al. 2005), and an algorithmic tool which we developed to identify the U1 snRNA and U2 snRNA homologs in the different organisms. For the identification of the SF1, U2AF2, and U2AF1 homologs, we used BLASTP (Altschul et al. 1990), Exonerate (Slater and Birney 2005), TBLASTN, and GeneWise (Birney et al. 2004). We used Pfam (http://pfam.sanger.ac.uk) and PROSITE (http://ca.expasy.org/prosite/) to confirm the existence of the characteristic domains in the three proteins. Multiple alignments of the entire protein sequences and of the domains were performed using T-COFFEE (Notredame et al. 2000). For a full description on the compilation of these data sets, view Supplemental Material.
Acknowledgments
We thank Yaron Racah for many insightful comments, critical observations, and stimulating suggestions. This work was supported by a grant from the Israel Science Foundation (1449/04 and 40/05), MOP Germany–Israel, GIF. E.E. is supported by the Catalan Institution of Research and Advanced Studies (ICREA) and by the grant BIO2005-01287 from the Spanish Ministry of Education and Culture. T.P. is supported by a Wolfson grant. S.S. and D.B. are fellows of the Edmond J. Safra Bioinformatics Program at Tel-Aviv University.
Footnotes
[Supplemental material is available online at www.genome.org.]
Article published online before print. Article and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.6818908
References
- Abovich N., Liao X.C., Rosbash M., Liao X.C., Rosbash M., Rosbash M. The yeast MUD2 protein: An interaction with PRP11 defines a bridge between commitment complexes and U2 snRNP addition. Genes & Dev. 1994;8:843–854. doi: 10.1101/gad.8.7.843. [DOI] [PubMed] [Google Scholar]
- Abril J.F., Castelo R., Guigo R., Castelo R., Guigo R., Guigo R. Comparison of splice sites in mammals and chicken. Genome Res. 2005;15:111–119. doi: 10.1101/gr.3108805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altschul S.F., Gish W., Miller W., Myers E.W., Lipman D.J., Gish W., Miller W., Myers E.W., Lipman D.J., Miller W., Myers E.W., Lipman D.J., Myers E.W., Lipman D.J., Lipman D.J. Basic local alignment search tool. J. Mol. Biol. 1990;215:403–410. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- Anantharaman V., Koonin E.V., Aravind L., Koonin E.V., Aravind L., Aravind L. Comparative genomics and evolution of proteins involved in RNA metabolism. Nucleic Acids Res. 2002;30:1427–1464. doi: 10.1093/nar/30.7.1427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ares M. U2 RNA from yeast is unexpectedly large and contains homology to vertebrate U4, U5, and U6 small nuclear RNAs. Cell. 1986;47:49–59. doi: 10.1016/0092-8674(86)90365-x. [DOI] [PubMed] [Google Scholar]
- Ast G. How did alternative splicing evolve? Nat. Rev. Genet. 2004;5:773–782. doi: 10.1038/nrg1451. [DOI] [PubMed] [Google Scholar]
- Ast G., Pavelitz T., Weiner A.M., Pavelitz T., Weiner A.M., Weiner A.M. Sequences upstream of the branch site are required to form helix II between U2 and U6 snRNA in a trans-splicing reaction. Nucleic Acids Res. 2001;29:1741–1749. doi: 10.1093/nar/29.8.1741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aury J.M., Jaillon O., Duret L., Noel B., Jubin C., Porcel B.M., Segurens B., Daubin V., Anthouard V., Aiach N., Jaillon O., Duret L., Noel B., Jubin C., Porcel B.M., Segurens B., Daubin V., Anthouard V., Aiach N., Duret L., Noel B., Jubin C., Porcel B.M., Segurens B., Daubin V., Anthouard V., Aiach N., Noel B., Jubin C., Porcel B.M., Segurens B., Daubin V., Anthouard V., Aiach N., Jubin C., Porcel B.M., Segurens B., Daubin V., Anthouard V., Aiach N., Porcel B.M., Segurens B., Daubin V., Anthouard V., Aiach N., Segurens B., Daubin V., Anthouard V., Aiach N., Daubin V., Anthouard V., Aiach N., Anthouard V., Aiach N., Aiach N., et al. Global trends of whole-genome duplications revealed by the ciliate Paramecium tetraurelia. Nature. 2006;444:171–178. doi: 10.1038/nature05230. [DOI] [PubMed] [Google Scholar]
- Banerjee H., Rahn A., Davis W., Singh R., Rahn A., Davis W., Singh R., Davis W., Singh R., Singh R. Sex lethal and U2 small nuclear ribonucleoprotein auxiliary factor (U2AF65) recognize polypyrimidine tracts using multiple modes of binding. RNA. 2003;9:88–99. doi: 10.1261/rna.2131603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banerjee H., Rahn A., Gawande B., Guth S., Valcarcel J., Singh R., Rahn A., Gawande B., Guth S., Valcarcel J., Singh R., Gawande B., Guth S., Valcarcel J., Singh R., Guth S., Valcarcel J., Singh R., Valcarcel J., Singh R., Singh R. The conserved RNA recognition motif 3 of U2 snRNA auxiliary factor (U2AF 65) is essential in vivo but dispensable for activity in vitro. RNA. 2004;10:240–253. doi: 10.1261/rna.5153204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Birney E., Clamp M., Durbin R., Clamp M., Durbin R., Durbin R. GeneWise and Genomewise. Genome Res. 2004;14:988–995. doi: 10.1101/gr.1865504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Black D.L. Mechanisms of alternative pre-messenger RNA splicing. Annu. Rev. Biochem. 2003;72:291–336. doi: 10.1146/annurev.biochem.72.121801.161720. [DOI] [PubMed] [Google Scholar]
- Bon E., Casaregola S., Blandin G., Llorente B., Neuvéglise C., Munsterkotter M., Guldener U., Mewes H.W., Van Helden J., Dujon B., Casaregola S., Blandin G., Llorente B., Neuvéglise C., Munsterkotter M., Guldener U., Mewes H.W., Van Helden J., Dujon B., Blandin G., Llorente B., Neuvéglise C., Munsterkotter M., Guldener U., Mewes H.W., Van Helden J., Dujon B., Llorente B., Neuvéglise C., Munsterkotter M., Guldener U., Mewes H.W., Van Helden J., Dujon B., Neuvéglise C., Munsterkotter M., Guldener U., Mewes H.W., Van Helden J., Dujon B., Munsterkotter M., Guldener U., Mewes H.W., Van Helden J., Dujon B., Guldener U., Mewes H.W., Van Helden J., Dujon B., Mewes H.W., Van Helden J., Dujon B., Van Helden J., Dujon B., Dujon B., et al. Molecular evolution of eukaryotic genomes: Hemiascomycetous yeast spliceosomal introns. Nucleic Acids Res. 2003;31:1121–1135. doi: 10.1093/nar/gkg213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouck J., Fu X.D., Skalka A.M., Katz R.A., Fu X.D., Skalka A.M., Katz R.A., Skalka A.M., Katz R.A., Katz R.A. Genetic selection for balanced retroviral splicing: Novel regulation involving the second step can be mediated by transitions in the polypyrimidine tract. Mol. Cell. Biol. 1995;15:2663–2671. doi: 10.1128/mcb.15.5.2663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burge C.B., Tuschl T., Sharp P.A., Tuschl T., Sharp P.A., Sharp P.A.1999. Splicing of precursors to mRNAs by the spliceosomes. In The RNA world II (eds. R.F. Gesteland et al.), 525–560. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY [Google Scholar]
- Carmel I., Tal S., Vig I., Ast G., Tal S., Vig I., Ast G., Vig I., Ast G., Ast G. Comparative analysis detects dependencies among the 5′ splice-site positions. RNA. 2004;10:828–840. doi: 10.1261/rna.5196404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carmel L., Wolf Y.I., Rogozin I.B., Koonin E.V., Wolf Y.I., Rogozin I.B., Koonin E.V., Rogozin I.B., Koonin E.V., Koonin E.V. Three distinct modes of intron dynamics in the evolution of eukaryotes. Genome Res. 2007 doi: 10.1101/gr.6438607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cartegni L., Chew S.L., Krainer A.R., Chew S.L., Krainer A.R., Krainer A.R. Listening to silence and understanding nonsense: Exonic mutations that affect splicing. Nat. Rev. Genet. 2002;3:285–298. doi: 10.1038/nrg775. [DOI] [PubMed] [Google Scholar]
- Chen C., Gentles A.J., Jurka J., Karlin S., Gentles A.J., Jurka J., Karlin S., Jurka J., Karlin S., Karlin S. Genes, pseudogenes, and Alu sequence organization across human chromosomes 21 and 22. Proc. Natl. Acad. Sci. 2002;99:2930–2935. doi: 10.1073/pnas.052692099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collins L., Penny D., Penny D. Complex spliceosomal organization ancestral to extant eukaryotes. Mol. Biol. Evol. 2005;22:1053–1066. doi: 10.1093/molbev/msi091. [DOI] [PubMed] [Google Scholar]
- Collins L., Penny D., Penny D. Proceedings of the SMBE Tri-National Young Investigators' Workshop 2005. Investigating the intron recognition mechanism in eukaryotes. Mol. Biol. Evol. 2006;23:901–910. doi: 10.1093/molbev/msj084. [DOI] [PubMed] [Google Scholar]
- Coolidge C.J., Seely R.J., Patton J.G., Seely R.J., Patton J.G., Patton J.G. Functional analysis of the polypyrimidine tract in pre-mRNA splicing. Nucleic Acids Res. 1997;25:888–896. doi: 10.1093/nar/25.4.888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deutsch M., Long M., Long M. Intron-exon structures of eukaryotic model organisms. Nucleic Acids Res. 1999;27:3219–3228. doi: 10.1093/nar/27.15.3219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dewey C.N., Rogozin I.B., Koonin E.V., Rogozin I.B., Koonin E.V., Koonin E.V. Compensatory relationship between splice sites and exonic splicing signals depending on the length of vertebrate introns. BMC Genomics. 2006;7:311. doi: 10.1186/1471-2164-7-311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dujon B. Yeasts illustrate the molecular mechanisms of eukaryotic genome evolution. Trends Genet. 2006;22:375–387. doi: 10.1016/j.tig.2006.05.007. [DOI] [PubMed] [Google Scholar]
- Fields C. Information content of Caenorhabditis elegans splice site sequences varies with intron length. Nucleic Acids Res. 1990;18:1509–1512. doi: 10.1093/nar/18.6.1499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fouser L.A., Friesen J.D., Friesen J.D. Effects on mRNA splicing of mutations in the 3′ region of the Saccharomyces cerevisiae actin intron. Mol. Cell. Biol. 1987;7:225–230. doi: 10.1128/mcb.7.1.225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gozani O., Feld R., Reed R., Feld R., Reed R., Reed R. Evidence that sequence-independent binding of highly conserved U2 snRNP proteins upstream of the branch site is required for assembly of spliceosomal complex A. Genes & Dev. 1996;10:233–243. doi: 10.1101/gad.10.2.233. [DOI] [PubMed] [Google Scholar]
- Graveley B.R. Alternative splicing: Increasing diversity in the proteomic world. Trends Genet. 2001;17:100–107. doi: 10.1016/s0168-9525(00)02176-4. [DOI] [PubMed] [Google Scholar]
- Griffiths-Jones S., Moxon S., Marshall M., Khanna A., Eddy S.R., Bateman A., Moxon S., Marshall M., Khanna A., Eddy S.R., Bateman A., Marshall M., Khanna A., Eddy S.R., Bateman A., Khanna A., Eddy S.R., Bateman A., Eddy S.R., Bateman A., Bateman A. Rfam: Annotating non-coding RNAs in complete genomes. Nucleic Acids Res. 2005;33:D121–D124. doi: 10.1093/nar/gki081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hastings M.L., Krainer A.R., Krainer A.R. Pre-mRNA splicing in the new millennium. Curr. Opin. Cell Biol. 2001;13:302–309. doi: 10.1016/s0955-0674(00)00212-x. [DOI] [PubMed] [Google Scholar]
- Hedges S.B. The origin and evolution of model organisms. Nat. Rev. Genet. 2002;3:838–849. doi: 10.1038/nrg929. [DOI] [PubMed] [Google Scholar]
- Hertz G.Z., Stormo G.D., Stormo G.D. Identifying DNA and protein patterns with statistically significant alignments of multiple sequences. Bioinformatics. 1999;15:563–577. doi: 10.1093/bioinformatics/15.7.563. [DOI] [PubMed] [Google Scholar]
- Hollins C., Zorio D.A., MacMorris M., Blumenthal T., Zorio D.A., MacMorris M., Blumenthal T., MacMorris M., Blumenthal T., Blumenthal T. U2AF binding selects for the high conservation of the C. elegans 3′ splice site. RNA. 2005;11:248–253. doi: 10.1261/rna.7221605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irimia M., Penny D., Roy S.W., Penny D., Roy S.W., Roy S.W. Coevolution of genomic intron number and splice sites. Trends Genet. 2007;23:321–325. doi: 10.1016/j.tig.2007.03.015. [DOI] [PubMed] [Google Scholar]
- Izquierdo J.M., Valcarcel J., Valcarcel J. A simple principle to explain the evolution of pre-mRNA splicing. Genes & Dev. 2006;20:1679–1684. doi: 10.1101/gad.1449106. [DOI] [PubMed] [Google Scholar]
- James T.Y., Kauff F., Schoch C.L., Matheny P.B., Hofstetter V., Cox C.J., Celio G., Gueidan C., Fraker E., Miadlikowska J., Kauff F., Schoch C.L., Matheny P.B., Hofstetter V., Cox C.J., Celio G., Gueidan C., Fraker E., Miadlikowska J., Schoch C.L., Matheny P.B., Hofstetter V., Cox C.J., Celio G., Gueidan C., Fraker E., Miadlikowska J., Matheny P.B., Hofstetter V., Cox C.J., Celio G., Gueidan C., Fraker E., Miadlikowska J., Hofstetter V., Cox C.J., Celio G., Gueidan C., Fraker E., Miadlikowska J., Cox C.J., Celio G., Gueidan C., Fraker E., Miadlikowska J., Celio G., Gueidan C., Fraker E., Miadlikowska J., Gueidan C., Fraker E., Miadlikowska J., Fraker E., Miadlikowska J., Miadlikowska J., et al. Reconstructing the early evolution of Fungi using a six-gene phylogeny. Nature. 2006;443:818–822. doi: 10.1038/nature05110. [DOI] [PubMed] [Google Scholar]
- Kaufer N.F., Potashkin J., Potashkin J. Analysis of the splicing machinery in fission yeast: A comparison with budding yeast and mammals. Nucleic Acids Res. 2000;28:3003–3010. doi: 10.1093/nar/28.16.3003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kent O.A., Ritchie D.B., Macmillan A.M., Ritchie D.B., Macmillan A.M., Macmillan A.M. Characterization of a U2AF-independent commitment complex (E′) in the mammalian spliceosome assembly pathway. Mol. Cell. Biol. 2005;25:233–240. doi: 10.1128/MCB.25.1.233-240.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim E., Magen A., Ast G., Magen A., Ast G., Ast G. Different levels of alternative splicing among eukaryotes. Nucleic Acids Res. 2007;35:125–131. doi: 10.1093/nar/gkl924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kol G., Lev-Maor G., Ast G., Lev-Maor G., Ast G., Ast G. Human-mouse comparative analysis reveals that branch-site plasticity contributes to splicing regulation. Hum. Mol. Genet. 2005;14:1559–1568. doi: 10.1093/hmg/ddi164. [DOI] [PubMed] [Google Scholar]
- Koonin E.V., Fedorova N.D., Jackson J.D., Jacobs A.R., Krylov D.M., Makarova K.S., Mazumder R., Mekhedov S.L., Nikolskaya A.N., Rao B.S., Fedorova N.D., Jackson J.D., Jacobs A.R., Krylov D.M., Makarova K.S., Mazumder R., Mekhedov S.L., Nikolskaya A.N., Rao B.S., Jackson J.D., Jacobs A.R., Krylov D.M., Makarova K.S., Mazumder R., Mekhedov S.L., Nikolskaya A.N., Rao B.S., Jacobs A.R., Krylov D.M., Makarova K.S., Mazumder R., Mekhedov S.L., Nikolskaya A.N., Rao B.S., Krylov D.M., Makarova K.S., Mazumder R., Mekhedov S.L., Nikolskaya A.N., Rao B.S., Makarova K.S., Mazumder R., Mekhedov S.L., Nikolskaya A.N., Rao B.S., Mazumder R., Mekhedov S.L., Nikolskaya A.N., Rao B.S., Mekhedov S.L., Nikolskaya A.N., Rao B.S., Nikolskaya A.N., Rao B.S., Rao B.S., et al. A comprehensive evolutionary classification of proteins encoded in complete eukaryotic genomes. Genome Biol. 2004;5:R7. doi: 10.1186/gb-2004-5-2-r7. http://genomebiology.com/2004/5/2/R7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kramer A. The structure and function of proteins involved in mammalian pre-mRNA splicing. Annu. Rev. Biochem. 1996;65:367–409. doi: 10.1146/annurev.bi.65.070196.002055. [DOI] [PubMed] [Google Scholar]
- Kretzner L., Krol A., Rosbash M., Krol A., Rosbash M., Rosbash M. Saccharomyces cerevisiae U1 small nuclear RNA secondary structure contains both universal and yeast-specific domains. Proc. Natl. Acad. Sci. 1990;87:851–855. doi: 10.1073/pnas.87.2.851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kupfer D.M., Drabenstot S.D., Buchanan K.L., Lai H., Zhu H., Dyer D.W., Roe B.A., Murphy J.W., Drabenstot S.D., Buchanan K.L., Lai H., Zhu H., Dyer D.W., Roe B.A., Murphy J.W., Buchanan K.L., Lai H., Zhu H., Dyer D.W., Roe B.A., Murphy J.W., Lai H., Zhu H., Dyer D.W., Roe B.A., Murphy J.W., Zhu H., Dyer D.W., Roe B.A., Murphy J.W., Dyer D.W., Roe B.A., Murphy J.W., Roe B.A., Murphy J.W., Murphy J.W. Introns and splicing elements of five diverse fungi. Eukaryot. Cell. 2004;3:1088–1100. doi: 10.1128/EC.3.5.1088-1100.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langford C.J., Klinz F.J., Donath C., Gallwitz D., Klinz F.J., Donath C., Gallwitz D., Donath C., Gallwitz D., Gallwitz D. Point mutations identify the conserved, intron-contained TACTAAC box as an essential splicing signal sequence in yeast. Cell. 1984;36:645–653. doi: 10.1016/0092-8674(84)90344-1. [DOI] [PubMed] [Google Scholar]
- Levine A., Durbin R., Durbin R. A computational scan for U12-dependent introns in the human genome sequence. Nucleic Acids Res. 2001;29:4006–4013. doi: 10.1093/nar/29.19.4006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim L.P., Burge C.B., Burge C.B. A computational analysis of sequence features involved in recognition of short introns. Proc. Natl. Acad. Sci. 2001;98:11193–11198. doi: 10.1073/pnas.201407298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manceau V., Swenson M., Le Caer J.P., Sobel A., Kielkopf C.L., Maucuer A., Swenson M., Le Caer J.P., Sobel A., Kielkopf C.L., Maucuer A., Le Caer J.P., Sobel A., Kielkopf C.L., Maucuer A., Sobel A., Kielkopf C.L., Maucuer A., Kielkopf C.L., Maucuer A., Maucuer A. Major phosphorylation of SF1 on adjacent Ser-Pro motifs enhances interaction with U2AF65. FEBS J. 2006;273:577–587. doi: 10.1111/j.1742-4658.2005.05091.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore M.J. Intron recognition comes of AGe. Nat. Struct. Biol. 2000;7:14–16. doi: 10.1038/71207. [DOI] [PubMed] [Google Scholar]
- Nguyen H.D., Yoshihama M., Kenmochi N., Yoshihama M., Kenmochi N., Kenmochi N. New maximum likelihood estimators for eukaryotic intron evolution. PLoS Comput. Biol. 2005;1:e79. doi: 10.1371/journal.pcbi.0010079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Notredame C., Higgins D.G., Heringa J., Higgins D.G., Heringa J., Heringa J. T-Coffee: A novel method for fast and accurate multiple sequence alignment. J. Mol. Biol. 2000;302:205–217. doi: 10.1006/jmbi.2000.4042. [DOI] [PubMed] [Google Scholar]
- Pikielny C.W., Teem J.L., Rosbash M., Teem J.L., Rosbash M., Rosbash M. Evidence for the biochemical role of an internal sequence in yeast nuclear mRNA introns: Implications for U1 RNA and metazoan mRNA splicing. Cell. 1983;34:395–403. doi: 10.1016/0092-8674(83)90373-2. [DOI] [PubMed] [Google Scholar]
- Raible F., Tessmar-Raible K., Osoegawa K., Wincker P., Jubin C., Balavoine G., Ferrier D., Benes V., de Jong P., Weissenbach J., Tessmar-Raible K., Osoegawa K., Wincker P., Jubin C., Balavoine G., Ferrier D., Benes V., de Jong P., Weissenbach J., Osoegawa K., Wincker P., Jubin C., Balavoine G., Ferrier D., Benes V., de Jong P., Weissenbach J., Wincker P., Jubin C., Balavoine G., Ferrier D., Benes V., de Jong P., Weissenbach J., Jubin C., Balavoine G., Ferrier D., Benes V., de Jong P., Weissenbach J., Balavoine G., Ferrier D., Benes V., de Jong P., Weissenbach J., Ferrier D., Benes V., de Jong P., Weissenbach J., Benes V., de Jong P., Weissenbach J., de Jong P., Weissenbach J., Weissenbach J., et al. Vertebrate-type intron-rich genes in the marine annelid Platynereis dumerilii. Science. 2005;310:1325–1326. doi: 10.1126/science.1119089. [DOI] [PubMed] [Google Scholar]
- Reed R. Mechanisms of fidelity in pre-mRNA splicing. Curr. Opin. Cell Biol. 2000;12:340–345. doi: 10.1016/s0955-0674(00)00097-1. [DOI] [PubMed] [Google Scholar]
- Rogozin I.B., Wolf Y.I., Sorokin A.V., Mirkin B.G., Koonin E.V., Wolf Y.I., Sorokin A.V., Mirkin B.G., Koonin E.V., Sorokin A.V., Mirkin B.G., Koonin E.V., Mirkin B.G., Koonin E.V., Koonin E.V. Remarkable interkingdom conservation of intron positions and massive, lineage-specific intron loss and gain in eukaryotic evolution. Curr. Biol. 2003;13:1512–1517. doi: 10.1016/s0960-9822(03)00558-x. [DOI] [PubMed] [Google Scholar]
- Romfo C.M., Wise J.A., Wise J.A. Both the polypyrimidine tract and the 3′ splice site function prior to the first step of splicing in fission yeast. Nucleic Acids Res. 1997;25:4658–4665. doi: 10.1093/nar/25.22.4658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roscigno R.F., Weiner M., Garcia-Blanco M.A., Weiner M., Garcia-Blanco M.A., Garcia-Blanco M.A. A mutational analysis of the polypyrimidine tract of introns. Effects of sequence differences in pyrimidine tracts on splicing. J. Biol. Chem. 1993;268:11222–11229. [PubMed] [Google Scholar]
- Roy S.W., Gilbert W., Gilbert W. Complex early genes. Proc. Natl. Acad. Sci. 2005a;102:1986–1991. doi: 10.1073/pnas.0408355101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roy S.W., Gilbert W., Gilbert W. Rates of intron loss and gain: Implications for early eukaryotic evolution. Proc. Natl. Acad. Sci. 2005b;102:5773–5778. doi: 10.1073/pnas.0500383102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruskin B., Greene J.M., Green M.R., Greene J.M., Green M.R., Green M.R. Cryptic branch point activation allows accurate in vitro splicing of human beta-globin intron mutants. Cell. 1985;41:833–844. doi: 10.1016/s0092-8674(85)80064-7. [DOI] [PubMed] [Google Scholar]
- Sankoff D. Minimal mutation trees of sequences. SIAM J. Appl. Math. 1975;28:35–42. [Google Scholar]
- Saxonov S., Daizadeh I., Fedorov A., Gilbert W., Daizadeh I., Fedorov A., Gilbert W., Fedorov A., Gilbert W., Gilbert W. EID: The Exon-Intron Database—An exhaustive database of protein-coding intron-containing genes. Nucleic Acids Res. 2000;28:185–190. doi: 10.1093/nar/28.1.185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheth N., Roca X., Hastings M.L., Roeder T., Krainer A.R., Sachidanandam R., Roca X., Hastings M.L., Roeder T., Krainer A.R., Sachidanandam R., Hastings M.L., Roeder T., Krainer A.R., Sachidanandam R., Roeder T., Krainer A.R., Sachidanandam R., Krainer A.R., Sachidanandam R., Sachidanandam R. Comprehensive splice-site analysis using comparative genomics. Nucleic Acids Res. 2006;34:3955–3967. doi: 10.1093/nar/gkl556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sickmier E.A., Frato K.E., Shen H., Paranawithana S.R., Green M.R., Kielkopf C.L., Frato K.E., Shen H., Paranawithana S.R., Green M.R., Kielkopf C.L., Shen H., Paranawithana S.R., Green M.R., Kielkopf C.L., Paranawithana S.R., Green M.R., Kielkopf C.L., Green M.R., Kielkopf C.L., Kielkopf C.L. Structural basis for polypyrimidine tract recognition by the essential pre-mRNA splicing factor U2AF65. Mol. Cell. 2006;23:49–59. doi: 10.1016/j.molcel.2006.05.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slater G.S., Birney E., Birney E. Automated generation of heuristics for biological sequence comparison. BMC Bioinformatics. 2005;6:31. doi: 10.1186/1471-2105-6-31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spellman R., Smith C.W., Smith C.W. Novel modes of splicing repression by PTB. Trends Biochem. Sci. 2006;31:73–76. doi: 10.1016/j.tibs.2005.12.003. [DOI] [PubMed] [Google Scholar]
- Sverdlov A.V., Rogozin I.B., Babenko V.N., Koonin E.V., Rogozin I.B., Babenko V.N., Koonin E.V., Babenko V.N., Koonin E.V., Koonin E.V. Conservation versus parallel gains in intron evolution. Nucleic Acids Res. 2005;33:1741–1748. doi: 10.1093/nar/gki316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ule J., Stefani G., Mele A., Ruggiu M., Wang X., Taneri B., Gaasterland T., Blencowe B.J., Darnell R.B., Stefani G., Mele A., Ruggiu M., Wang X., Taneri B., Gaasterland T., Blencowe B.J., Darnell R.B., Mele A., Ruggiu M., Wang X., Taneri B., Gaasterland T., Blencowe B.J., Darnell R.B., Ruggiu M., Wang X., Taneri B., Gaasterland T., Blencowe B.J., Darnell R.B., Wang X., Taneri B., Gaasterland T., Blencowe B.J., Darnell R.B., Taneri B., Gaasterland T., Blencowe B.J., Darnell R.B., Gaasterland T., Blencowe B.J., Darnell R.B., Blencowe B.J., Darnell R.B., Darnell R.B. An RNA map predicting Nova-dependent splicing regulation. Nature. 2006;444:580–586. doi: 10.1038/nature05304. [DOI] [PubMed] [Google Scholar]
- Vanacova S., Yan W., Carlton J.M., Johnson P.J., Yan W., Carlton J.M., Johnson P.J., Carlton J.M., Johnson P.J., Johnson P.J. Spliceosomal introns in the deep-branching eukaryote Trichomonas vaginalis. Proc. Natl. Acad. Sci. 2005;102:4430–4435. doi: 10.1073/pnas.0407500102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weir M., Rice M., Rice M. Ordered partitioning reveals extended splice-site consensus information. Genome Res. 2004;14:67–78. doi: 10.1101/gr.1715204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zamore P.D., Green M.R., Green M.R. Identification, purification, and biochemical characterization of U2 small nuclear ribonucleoprotein auxiliary factor. Proc. Natl. Acad. Sci. 1989;86:9243–9247. doi: 10.1073/pnas.86.23.9243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang M.Q., Marr T.G., Marr T.G. Fission yeast gene structure and recognition. Nucleic Acids Res. 1994;22:1750–1759. doi: 10.1093/nar/22.9.1750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zorio D.A., Blumenthal T., Blumenthal T. Both subunits of U2AF recognize the 3′ splice site in Caenorhabditis elegans. Nature. 1999;402:835–838. doi: 10.1038/45597. [DOI] [PubMed] [Google Scholar]