

Abstract
The recent availability of full genomic sequence data for a large number of human influenza A (H3N2) virus isolates over many years provides us an opportunity to analyze human influenza virus evolution by considering all gene segments simultaneously. However, such analysis requires development of new computational models that can capture the complex evolutionary features over the entire genome. By analyzing nucleotide co-occurrence over the entire genome of human H3N2 viruses, we have developed a network model to describe H3N2 virus evolutionary patterns and dynamics. The network model effectively captures the evolutionary antigenic features of H3N2 virus at the whole-genome level and accurately describes the complex evolutionary patterns between individual gene segments. Our analyses show that the co-occurring nucleotide modules apparently underpin the dynamics of human H3N2 evolution and that amino acid substitutions corresponding to nucleotide co-changes cluster preferentially in known antigenic regions of the viral HA. Therefore, our study demonstrates that nucleotide co-occurrence networks represent a powerful method for tracking influenza A virus evolution and that cooperative genomic interaction is a major force underlying influenza virus evolution.
The rapid evolution of the influenza A virus poses a global challenge to public health. Recent events, such as induction of substantial morbidity and mortality by human H3N2 virus during the 2003–2004 influenza season (Bhat et al. 2005; Ghedin et al. 2005; Holmes et al. 2005) and the spread of highly pathogenic H5N1 influenza virus, have heightened concerns of potential pandemics. Thus, there is an urgent need for a better understanding of influenza virus evolution. Numerous full genomic influenza virus sequences are available in public archives, and analyses of these data have significantly enhanced our understanding of influenza evolution and its disease-causing mechanism (Chen et al. 2005; Fauci 2005; Ghedin et al. 2005; Holmes et al. 2005; Obenauer et al. 2006). However, opportunities remain to extract even more information from these valuable public archives in order to facilitate influenza prevention and control in the human populations.
The influenza A genome consists of eight gene segments that encode 11 proteins (Parrish and Kawaoka 2005). Five gene segments each encode a single protein: hemagglutinin (HA), neuraminidase (NA), nucleoprotein (NP), acidic polymerase (PA), and polymerase basic 2 (PB2). Three gene segments each encode two proteins: polymerase basic 1 (PB1) for PB1 and PB1-F2, NS for nonstructural proteins 1 and 2 (NS1 and NS2), and M for matrix proteins 1 and 2 (M1 and M2). Proteins NP, PA, PB1, and PB2 together mediate viral replication and transcription. The two surface glycoproteins, HA and NA, control viral entry into the cells and release from the infected cells and are the major antigenic targets of the host antibody responses. Pre-existing influenza-specific antibodies largely determine a host’s susceptibility to reinfection by related strains of virus.
Conventionally, analyses of influenza evolution have focused on individual viral genes, particularly HA, to understand and predict viral antigenic evolution (Bush et al. 1999; Ferguson et al. 2003; Fitch et al. 1991; Grenfell et al. 2004; Plotkin et al. 2002). These approaches effectively identify single mutations, as well as independent evolutionary behaviors of single genes. However, the evolutionary behavior of the virus often involves cooperative changes within and between genes. For example, mutations in the epitopes of influenza virus proteins that facilitate escape from the host immune response sometimes occur at the cost of viral fitness and thus require amino acid substitutions outside the epitope to restore optimal function (Rimmelzwaan et al. 2005). In addition, cooperative activities of both surface proteins, HA and NA, are critical for influenza virus infection and release (Wagner et al. 2002). Thus, important information about influenza evolutionary behavior is contained in the correlated changes between nucleotide positions both within genes and between genes.
Analysis of correlated mutations in human influenza viruses, however, can be obscured by its complex evolutionary events including co-circulation of distinct viral lineages and gene reassortment events that generate hybrid viruses from distinct ancestral viruses (Ghedin et al. 2005; Holmes et al. 2005). In this study, by considering co-occurrences or co-changes of human influenza genomic information as correlated changes in a loose sense, we have developed a computational approach that analyzes nucleotide co-occurrences across all genes to gain insight into evolution of influenza H3N2 viruses. We report how nucleotide co-occurrence networks are built and how they can be used to interpret evolutionary patterns of the influenza A viruses, including the significant changes recently observed for H3N2. We further demonstrate that co-occurring nucleotide modules, which are clustered preferentially in all five known antigenic epitopes of HA, likely underlie the dynamics of H3N2 evolution in humans. Thus, nucleotide co-occurrence networks are novel tools for tracking human influenza virus evolution.
Results
Construction of nucleotide co-occurrence networks
Influenza virus H3N2 first became widespread in humans during the 1968 “Hong Kong” flu and have been a major cause of influenza epidemics ever since. The recent availability of full genomic sequences for >1000 H3N2 isolates provides us an opportunity to examine how H3N2 viruses have evolved at the whole-genome level. To build nucleotide co-occurrence networks for human H3N2 viruses, we used the following five steps (Fig. 1A). In Step 1, genome sequences of 1032 H3N2 isolates from 1968 to 2006 were aligned. In Step 2, the eight gene segments were concatenated into a continuous sequence, similar to the approach reported by Ciccarelli et al. (2006). In Step 3, all nucleotide positions that were conserved in all isolates from all seasons were removed so that only regions of the concatenated genome that contained nucleotide variation (40% of all positions) were further considered. In Step 4, for each pair of nucleotide positions that remained after Step 3, the nucleotide pairs that exhibited perfect co-occurrence were tagged and connected (Fig. 1B). This resulted in a network for each of the 1032 viral isolates containing co-occurring nucleotide pairs over the entire concatenated genome. We termed this network the “nucleotide co-occurrence network,” in which nodes represent nucleotides at specific positions in the genome and edges between nodes represent co-occurring nucleotide pairs. In Step 5, we further clustered nucleotide pairs contained in the networks into co-occurring nucleotide modules, each of which represents a group of co-varying nucleotides that may reveal cooperative interactions underlying viral biological activity. Below, we illustrate how the network representation of complex viral genomic interactions can be used to understand and characterize human influenza H3N2 virus evolution.
Figure 1.

Overview of network construction. (A) Flow diagram showing the computational processes of analyzing H3N2 virus genomic co-occurrence network. (B) Mathematical framework of Step 4, construction of the viral genomic co-occurrence network. See Methods for details.
H3N2 nucleotide co-occurrence networks capture viral evolutionary patterns
To visualize how H3N2 viruses evolve over time, we examined the changes in network topology, also known as a connectivity map, by quantifying the average connectivity (K), or the average number of co-varying partners per nucleotide, for each of the 1032 nucleotide co-occurrence networks. The connectivity map shows that the human influenza H3N2 viruses evolve with large connectivity changes (Fig. 2A). For example, recent H3N2 viruses of Fujian strain that predominated in 2005 and 2006 underwent a significant connectivity change from those that predominated in 2003 and 2004 (Fig. 2A). A phylogenetic tree analysis of the recent strains revealed that the predominant strains in 2004 and 2005 belong to different branches of the phylogenetic tree (Fig. 2B, orange and blue). These results suggest that the large network topology changes between consecutive seasons, reflecting the replacement of epidemic strains.
Figure 2.

Evolving network connectivity of human influenza virus strains. (A) The average vertex connectivities (K) for each of the 1032 H3N2 nucleotide co-occurrence networks were calculated and plotted by flu season. Each marker represents one strain’s nucleotide co-occurrence network isolated from New York (circles), Memphis (asterisks), Hong Kong (squares), or Canterbury (crosses). The connectivity clusters for predominant strains (see Methods for detailed explanation of calculations) are colored according to their best-matched antigenic clusters in Smith et al’s analysis (Smith et al. 2004). The title denoted “Antigenicity” indicates the color-coded antigenic clusters in Smith’s work. (B) Phylogenetic tree derived from the concatenated viral genomes of H3N2 strains isolated between 1968 and 2006. Bootstrap values are shown for key nodes. The strains within a connectivity cluster in A are mapped to the phylogenetic tree and are shaded with the same color as the connectivity clusters. The clades are labeled using the season from which the majority of the strains were isolated. (C) Season-by-season analysis of whole-genome sequence evolution of 1032 strains. Genetic (nucleotide substitution) distances relative to A/Hong Kong/1/68 were calculated from the phylogenetic tree in B for each of the 1032 strains. The color and symbol representation are the same as in A. (D) Standardized vertex connectivities (K) for the simulated sampling of nucleotide co-occurrence networks are plotted by season. The blue line indicates the average K for all strains sampled from that season.
Distinct connection topologies were also observed between strains in the same season. For example, the connectivity map revealed that the recent H3N2 viruses started to emerge in 2003 (Fig. 2A, orange arrows), which have a big connectivity shift from the predominant strains in the same season. This result is consistent with the phylogenetic tree analysis, which shows that the minor strains in the 2003 flu season are on a different tree branch from those predominant strains (Fig. 2B; orange arrows indicate same strains as in Fig. 2A). Therefore, these findings suggest that the existence of significantly different topologies in the same season implies co-circulation of H3N2 strains from multiple phylogenetic lineages.
The connectivity map further reveals that connection topology of nucleotide co-occurrence networks evolves in clusters, while genetic evolution at the sequence level is relatively continuous (Fig. 2, cf. A and C; colors indicate network connectivity clusters for predominant H3N2 strains in consecutive seasons). Recently, Smith et al. (2004) systematically characterized the antigenic relationships between 273 H3N2 isolates sampled between 1968 and 2003 and showed that antigenic changes of H3N2 viruses are also clustered (Smith et al. 2004; the antigenic clusters are replotted in Fig. 2A). To determine if connectivity and antigenicity patterns of H3N2 strains are similar, we developed a simple clustering algorithm to cluster the predominant strains from each season based on their connectivities (see Methods). For ease of comparison, we considered connectivity clusters containing predominant strains from at least 2 yr from 1968 to 2003, which covers 26 seasons (Fig. 2A, color-coded connectivity clusters). Approximately 86% of all season pairs that shared a connectivity cluster also shared an antigenic cluster (P < 0.001), and only two of the 26 seasons in the connectivity clusters did not match their placements in the antigenic clusters (Fig. 2A, red arrows). The tight agreement between the connectivity and antigenic clusters suggests that changes in average network connectivity capture the antigenic drift of the H3N2 viruses. Therefore, it seems that nucleotide co-occurrence networks are a simple and efficient means of visualizing important characteristics of human influenza evolution.
We noted that the uneven distribution of H3N2 isolates with complete sequence data (>90% collected between 1993 and 2006) could potentially bias conclusions made for the entire period between 1968 and 2006. Therefore, we simulated an evenly distributed collection of viral isolates by randomly sampling only one viral isolate from each flu season from 1968 to 2006 and constructed nucleotide co-occurrence networks following the same procedure outlined in Figure 1. We calculated the connectivity pattern for these sampled viruses (corresponding to one dotted-line curve in Fig. 2D). By repeating this simulation many times, we were able to obtain connectivity patterns statistically for virus isolates that were evenly distributed among all seasons (Fig. 2D, bold blue line).
Overall, the pattern of changes in the network connectivity of the simulated networks is similar to that obtained with the 1032 H3N2 isolates (Fig. 2, cf. A and D). However, evolutionary patterns as indicated by the network connectivity changes became clearer. Namely, the evolution of human H3N2 viruses exhibited distinct features at different times (Fig. 2D). When the viruses first emerged in humans from 1968 to 1972, the connectivity of these early H3N2 viruses changed rapidly and significantly, indicating that viruses had to undergo dramatic and collective evolutionary changes for rapid adaptation to the new host. From 1972 to 2001, the connectivity was relatively steady and changed in clusters at a much slower pace, indicating relatively infrequent changes in the influenza activities. Interestingly, no severe diseases were associated with influenza viruses between 1979 and 1985 (Chakraverty et al. 1986). From 2001 to 2006, the connectivity increased steadily, with significant shifts during 2001–2002 and 2004–2005 (Fig. 2D, red arrows). The significant connectivity shift from 2004 to 2005 may reflect the combined effects of antigenic novelty and improved host adaptation on viral fitness of recent H3N2 viruses, late Fujian strains (Fig. 2A,D; Wolf et al. 2006). Taken together, these findings suggest that H3N2 whole-genome nucleotide co-occurrence networks capture the essential evolutionary characteristics of viruses.
H3N2 nucleotide co-occurrence networks reveal unique evolutionary patterns of individual gene segments
Nucleotide co-occurrence networks allow us to reveal the evolutionary pattern of H3N2 viruses at the whole-genome level. However, individual gene segments may evolve independently through gradual mutations or gene reassortment, exchange of a complete gene segment from one virus strain with another (Parrish and Kawaoka 2005). Therefore, it would be informative to follow the evolutionary trajectory of each viral gene segment separately. To track evolutionary patterns of individual gene segments, we analyzed changes in the nucleotide co-occurrence network connectivity of individual gene segments in each H3N2 isolate. To avoid biasing our observations, we continued to use the sampled distribution of H3N2 isolates and considered the period from 1998 to 2006, which has both most of the sequence data and the most interesting evolutionary changes of the viruses.
All individual gene segments have large connectivity shifts from 2004 to 2005, consistent with the whole-genome results discussed above. However, in some cases, patterns of connectivity changes are distinct for different gene segments (Fig. 3A,B). For example, the connectivity of NS changes significantly from 1999 to 2000, while HA has been changing significantly since 2003. MP evolves relatively slowly between 1997 and 2004 (Fig. 3A; significant changes highlighted in red). In contrast, the connectivity of NA, encoding a surface protein, changes with a pattern similar to gene segments encoding viral replication machinery, particularly NP, PA, and PB1, which all have significant network topology changes from 2001 to 2002 (Fig. 3B).
Figure 3.

Evolutionary patterns of individual influenza gene segments. (A,B) Average standardized connectivity K of H3N2 nucleotide co-occurrence networks for gene segments (A) HA, NA, MP, and NS; and (B) NA, NP, PA, PB1, and PB2 from 1998–2006. The largest changes in connectivity K in HA, NA, and NS before 2004 are labeled in red and indicated by arrows. (C,D) Phylogenetic trees of (C) HA and (D) NA nucleotide sequences from influenza A viruses sampled from 1997 to 2006. Bootstrap values are shown for the key nodes. The clades for strains during 2000–2006 were labeled using the season from which the majority of the strains were isolated.
One method for tracking the evolution of individual gene segments is to derive gene-segment phylogenetic trees and then analyze the major changes in their tree topologies, a method recently applied to influenza A gene segments by Holmes et al. (2005). When we compared our connectivity results with our own phylogenetic tree analyses for individual gene segments, the connectivity changes identified are consistent with transitions between two slender trunks of its phylogenetic tree (Fig. 3C,D; Supplemental Fig. S1). For example, the origins of the HA gene in 2004 do not come from strains in 2003, consistent with the connectivity shift in HA between 2003 and 2004 (Fig. 3A,C). In contrast, the NA gene shift in the phylogenetic structure occurred in 2001 and 2002 (Fig. 3D). Therefore, analysis of network connectivity relationships of individual gene segments can capture their evolutionary patterns, indicating that H3N2 nucleotide co-occurrence networks also present an effective and simple way to visualize evolutionary patterns of individual viral gene segments.
Nucleotide co-changes within and between H3N2 gene segments contribute independently to influenza evolution
To understand the evolutionary patterns of individual gene segments and their complex evolutionary relationships in a quantitative way, we introduced two additional metrics to describe the degree of co-changes in the nucleotide co-occurrence networks. In order to quantify the rate of co-changes within and between gene segments, we calculated R, the rate of a nucleotide losing or gaining a connection between two seasons. In order to quantify the extent of co-changes between gene segments from season to season, we calculated C, the ratio of R between two gene segments to R within each of the two gene segments. If C is >1, then the two gene segments are more likely to evolve together rather than separately.
When we plotted the rate of nucleotide connection changes (R) within and between all individual gene segments, we were able to explore network connection changes at a more detailed level (Fig. 4A). We have previously shown that the virus underwent large network connectivity changes from 2001 to 2002 and from 2004 to 2005 (Fig. 2A,D). Here we observe that co-changes both within gene segments (Fig. 4A, left side) and between gene segments (Fig. 4A, right side) contributed to these large connectivity changes.
Figure 4.

Quantification of correlated changes between H3N2 gene segments between 1998 and 2006. (A) Heat map of the rates (R) of season-to-season intragenic (left panel) and intergenic (right panel) connection changes for the simulated sampling of H3N2 strains from 1998 to 2006. Significant intragenic HA change is labeled with an arrow. See Methods for detailed calculations. (B) The extent of cooperative changes (C) between gene segments for the simulated sampling of H3N2 strains from 1998 to 2006. See Methods for detailed calculations.
In particular, we noted that HA uniquely underwent connectivity changes within itself from 2003 to 2004 (Fig. 4A, arrow), which indicates that the unique connectivity change of HA from 2003 to 2004 discussed above is mainly due to the co-changes within the gene. This independent evolution of HA indicates a gene reassortment event for HA, as has been suggested previously (Holmes et al. 2005). The highly similar evolutionary patterns among all gene segments from 2004 to 2005 may suggest a more complicated mechanism, such as multiple gene reassortments (Ghedin et al. 2005) or marked accumulated genetic drift (Parrish and Kawaoka 2005).
In contrast to HA, NA tends to have similar evolutionary patterns with other gene segments. Using the metric of cooperative evolution (C), we observed highly correlated changes between NA, NP, PA, PB1, and PB2 (Fig. 4B, C > 1). Proteins NP, PA, PB1, and PB2 encode the influenza virus replication machinery (Fodor et al. 2002), and therefore highly cooperative evolution between these genes is expected; however, the fact that NA also varies with this group of genes has not been noted before, indicating more complex interactions among viral genes during human influenza evolutions. By comparing nucleotide co-occurrence networks at the level of gene segments, we were able to characterize evolutionary relationships within and between influenza gene segments.
Co-occurring nucleotide modules identify detailed genomic changes that underlie human influenza dynamics
The nucleotide co-occurrence networks present a quantitative and accurate way to describe characteristics of human influenza evolution. We further sought to determine if these networks could reveal structural details about H3N2 evolution. We thus decomposed the viral networks into their 2324 individual co-occurring nucleotide (CN) modules (Supplemental Tables S1, S2). The distribution of a CN module among the isolates can be described as a vector of ones and zeros, indicating the presence or absence of this module in each of the H3N2 strains. Because this representation is similar to a gene phylogenetic profile, which denotes the distribution of genes across species (Slonim et al. 2006), we named it the strain-module profile.
To elucidate the dynamics of human influenza epidemics at the level of individual co-occurring nucleotides, we used the strain-module profiles to identify CN modules that are present in a majority of strains (≥50%) in a season and classified these as predominant modules. The predominant modules were further classified into conserved, transition, and transient modules based on their presence and relations over time from 1968 to 2006 (Supplemental Fig. S2). A conserved module is a predominant module that is present in every season from 1968 to 2006. The nucleotides present in the conserved modules may provide information about which genomic features are required for survival and predominance of epidemic strains. A transition module consists of two or more predominant modules that share at least one nucleotide position and alternate in different seasons. A transient module is a predominant module that shares no nucleotide positions with any other predominant modules and occurs only in some seasons. We clustered epidemic strains from 1998 to 2006 based on strain-module profiles for transition and transient modules only, and the strains cluster into four groups: 1998–1999, 2000–2001, 2002–2004, and 2005–2006. These clusters match the four phylogenetic groups derived from their genomic sequences (cf. Fig. 5A and Fig. 2B), suggesting that transition and transient CN modules alone contain enough information to capture the dynamics of strain evolution.
Figure 5.

Characteristics of co-occurring nucleotide modules. (A) Heat map of the distribution of transient and transition modules from 1998 to 2006 subject to hierarchical clustering based on the strain-module profile. Rows correspond to transient and transition modules, and columns correspond to the indicated season. If a transient module is present in a majority of strains in a season, it is blue; otherwise it is gray. Transition modules are labeled according to which of the two member modules is present in the majority of strains in a season (first member, blue; second member, red). (B) Module-based amino acid changes corresponding to the nucleotide changes in transient and transition modules from 1998 to 2006. Each row represents a single amino acid position in a viral protein (labeled on the right). Amino acids (single-letter abbreviations) are also color-coded, so that mutations can be seen as changes both in amino acid identity and color. See Supplemental Figure S3 for all module-based amino acid changes from 1968 to 2006.
By identifying amino acid substitutions corresponding to nucleotide changes within transient and transition modules, namely, predominant module-based amino acid substitutions (Supplemental Fig. S3), we hoped to gain functional insights into influenza virus evolution. When a colorgram of predominant module-based amino acid substitutions observed for human H3N2 virus from 1998 to 2006 was constructed, we observed that two transition modules, 225/1746 and 239/1522, underwent significant amino acid changes from 2004 to 2005 (Fig. 5B, arrows). The transition module 239/1522 consists of nine co-mutations involving nonstructural proteins and the viral RNA replication machinery. Interestingly, this module change has occurred recurrently, that is, the strains in 2001 had the same module structure as the strains in 2005. The transition module 225/1746 includes 10 co-substitutions from NA, M2, and subunits of viral RNA replication machinery. In contrast to the recurrent changes in module 239/1522, the changes in module 225/1746 in 2005 resulted in amino acids that are mostly in NA, have not been found in this module before, and therefore are more likely to contribute to the recent influenza epidemics. Thus, strain-module profiles can help distinguish detailed genomic changes contributing to viral activities.
Module-based amino acid substitutions determine the antigenic structures
We reasoned that if the genomic changes contained in CN-modules underlie the evolution of viral activities, the structural positions of these changes on major surface antigen, HA, should correspond to known antigenic positions (Wiley et al. 1981). Thus, for transient and transition modules with nucleotide changes that resulted in amino acid substitutions in HA, we mapped those substitutions onto the HA structure (Fleury et al. 2000). As shown in Supplemental Table S3 and Figure 6, these predominant module-based amino acid substitutions mostly occurred at the exposed side of the trimeric complex (∼86%) and are preferentially clustered in the five known antigenic epitopes of HA (∼72%, P < 0.001). In contrast, nonpredominant module-based amino acid changes were uniformly distributed across the entire HA protein (Fig. 6, yellow; Supplemental Table S3). These results suggest that predominant module-based amino acid substitutions may have been selected to interfere with antibody recognition by reshaping the antigenic structures of HA, whereas non-co-changes, or changes from CN modules that are present in only a low number of epidemic strains, are randomly distributed.
Figure 6.

Module-based H3N2 amino acid substitutions mapped onto the HA structure. Amino acid substitutions from 1968 to 2006 are mapped onto the (left panel) exposed and (right panel) buried surface of the HA monomer. Residues are colored by module-based amino acid changes (red), non-module-based changes (yellow), and intact residues (purple). The five antibody epitopes (Wiley et al. 1981) and the receptor-binding site (Skehel and Wiley 2000) are circled. The module-based amino acid substitutions carrying two or more residues in 1998–2006 are labeled.
We next determined to what extent the co-occurring nucleotide pairs captured in the predominant CN modules reflect functional correlations. Because there is little knowledge about functional correlations between these nucleotide pairs, we used the known antigenic features of HA to address this question. Numerous examples have shown that simultaneous two or more mutations at the five known epitope regions can help influenza virus evade host immunity more effectively than a single mutation (Wilson and Cox 1990; Jin et al. 2005). Thus, we regarded the amino acid co-substitutions as correlated changes when both mutations occur at the five known epitope regions, and used them to assess functional correlations of the CN modules. We only considered the nucleotide pairs in predominant CN modules whose changes are accompanied by pairs of amino acid substitutions on HA. Among the 77 pairs of co-occurring nucleotides that are accompanied by amino acid co-substitutions on HA during 1968–2006, 67 (87.0%, compared with 13.0% in other amino acid co-substitutions) pairs of amino acid co-substitutions occurred in the antigenic sites, indicating the ability of the nucleotide co-occurrence network to capture the correlated genomic changes during human influenza evolution. Furthermore, these CN modules involved co-substitution of amino acids located in distinct antigenic sites (Fig. 6A, left panel). Therefore, it appears that CN-module-based amino acid mutations can capture functional coordinations between different antigenic sites that are required for influenza virus to effectively evade the host immune response.
In addition, CN-module-based amino acid mutations may be required to maintain viral functionality. The two mutations W222R and G225D in a transition module, 3/1744 (Figs. 5B and 6, left panel) provided such an example. These two mutations were located in the receptor-binding region and coevolved in the predominant strains from 2003 to 2004. Multiple groups have shown experimentally that both the W222R and G225D single mutations reduce human H3N2 HA receptor-binding activity and viral replication efficiency (Skehel and Wiley 2000; Nakajima et al. 2005), indicating that the co-substitution of these sites maintains human H3N2 HA receptor-binding affinity. Thus, co-occurring nucleotide modules can also identify amino acid substitutions that result in functional changes in human influenza evolution.
Discussion
Analyses of viral sequence evolution, and influenza virus evolution in particular, have traditionally relied on phylogenetic techniques. In this report, we have developed a network model that can complement traditional methodologies by representing a virus as a complex genomic interaction network based on the co-occurring nucleotide pairs over the entire genome. The network representation of viral sequence provides new perspectives on the structure of their whole-genome evolution, which cannot be easily characterized by the traditional methodologies.
We have shown that large connectivity changes in the viral networks match both the antigenic changes and the transitions of side lineages in phylogenetic analyses. However, in many cases, the transitions of phylogenetic side lineages do not correspond to remarkable connectivity changes and antigenic shifts (Fig. 2, cf. B and A). For example, predominant strains from 1977 to 1985 split into several distant side lineages in the phylogenetic tree (Fig. 2B, green), but those isolates have very close connectivity (Fig. 2A, green) and similar antigenicity. This indicates that our network model has an advantage over the phylogenetic algorithms to detect such clusterwise antigenic evolutionary features.
Recently, Plotkin et al. (2002) used HA sequence clusters to describe the antigenic evolution of influenza A viruses. To determine how well the connectivity clusters correspond to the HA sequence clusters, we followed the same clustering method as developed by Plotkin et al. and clustered the HA sequences for the 1032 H3N2 isolates (see Methods). Overall, the connectivity clusters have a good correspondence (∼65.7%; 23 out of 35 seasons with data available from 1968 to 2006) with HA sequence clusters (Supplemental Fig. S4), but they match the antigenic clusters better than the HA sequence clusters. Moreover, by transforming viral genomic information into a network representation, we were able to study how influenza A evolves at multiple levels of complexity, from whole-genome, individual genes to specific amino acid residues.
We have demonstrated that the network connectivity for the entire viral genome provides a simple and effective way to associate a viral genomic sequence with its antigenic properties. The network model also provides a quantitative method to comparatively analyze the evolutionary patterns of individual genes and assess their coevolution. The distinct evolutionary patterns of individual gene segments for epidemic strains can be easily observed by visualizing the network connectivity of individual gene segments (Fig. 3A,B). Although different gene segments exhibit distinct evolutionary patterns, we observed extensive coevolution between gene segments, which may indicate their functional relatedness.
CN modules, the component clusters of the nucleotide co-occurrence networks, provide a means of organizing viral complex interactions of genomic information into units of potential biological activity. Two recent analyses of human H3N2 evolution suggest that epistatic or context-dependent interactions of amino acids in HA were largely associated with significant antigenic change that leads to an increase in viral fitness (Koelle et al. 2006; Wolf et al. 2006). Consistent with these analyses, CN modules also shed light on the organization of such context-dependent interactions of amino acid substitutions that underpin the evolution of human influenza. By mapping amino acids of predominant CN modules to their structural locations on the HA protein, we found that these module-based amino acids cluster in the known regions of HA antigenic structures, suggesting an evolutionary strategy used by influenza virus to reshape its antigenic structures. Therefore, the complex composition of gene segments involved in many CN modules may reveal even more complicated combinatorial effects of nucleotides/residues on human influenza evolution.
The key element of the network model construction requires identification of the correlated genomic information in terms of nucleotide or amino acid pairs. In this work, we regard the nucleotide co-occurrence as correlated changes in a loose sense. We found that many of these co-occurring nucleotide pairs are, indeed, correlated changes. In the case of 2003–2004 H3N2 isolates, our data suggest that a CN-module-based amino acid compensatory mutation maintains the receptor-binding specificity of the virus. In addition to compensating for functional losses, multiple mutations in antigenic structures are also more effective than a single mutation in enabling the virus to evade host immune response (Wilson and Cox 1990; Jin et al. 2005). We found that co-substitutions of amino acids in predominant CN modules mostly occur in the five known epitope regions on HA, indicating that the genomic co-changes captured in our network model are much more likely to be correlated mutations. However, we note that our simple method to compute co-occurring nucleotide pairs may introduce uncorrelated mutations due to gene reassortments or by chance. Correlated mutations of amino acids have been widely investigated and implicated in the evolution of protein stabilities, structures, and functions (Gobel et al. 1994; Pollock 2002; Choi et al. 2005; Chen and Lee 2006; Shapiro et al. 2006). Recently, numerous groups have also explored the correlated mutations in sequence evolution of some RNA viruses, including vesicular stomatitis virus (VSV), human immunodeficiency virus type 1 (HIV-1), and human influenza viruses (Rimmelzwaan et al. 2005; Shapiro et al. 2006; Shih et al. 2007). Accordingly, numerous factors have been explored to identify the correlated changes. In light of these works, a variety of ways should be considered to improve the capture of correlated genomic changes in the network construction. We have attempted some. One was to try different degrees of stringency of co-occurrence. Indeed, we obtained a higher coverage of co-occurring nucleotide pairs, but coverage of the corresponding amino acid pairs is only marginally improved (data not shown). In another analysis, based on the same method, we also constructed a network by identifying amino acid co-occurrence directly. The evolution of both networks for protein and genomic sequences observes exactly the same patterns, and the amino acid pairs captured in both networks are almost the same (data not shown). Future directions may also need to take into consideration the effect of gene reassortment and the structure of viral phylogenies.
Although the connectivity model can accurately describe the evolutionary patterns of human H3N2 viruses based on their networks of nucleotide co-occurrences, it fails to take into account highly correlated changes, particularly those with co-occurrence of any nucleotide pair at a pair of positions. To investigate whether the connectivity model can reflect all correlated changes in the networks of nucleotide co-occurrence, we introduced another metric, S, similar to R, to denote the rate of network changes by considering not only the changes in connectivity but also the changes when a pair of nucleotides of co-occurrence changes to another pair of nucleotides of co-occurrence. It was found that in the viral networks, the changing pattern of S correlates well with that of connectivity (Pearson correlation coefficient = 0.87) (also see Supplemental Fig. S5), indicating that the connectivity model can reflect the pattern of the entire correlated changes in the networks. Despite this, how to develop a novel network metric that can capture the dynamical nature of genomic correlation networks more accurately and comprehensively presents an important direction in our future work.
The influenza virus constantly mutates, making prediction of its evolutionary trajectory difficult (Smith 2006). As a promising step to overcome this difficulty, we present a method of constructing nucleotide co-occurrence networks to capture evolutionary information encoded in the viral genome. The nucleotide co-occurrence networks accurately capture influenza evolution from genomic sequence data, and thus would be valuable for influenza prevention and control, such as strain surveillance and vaccine strain selection. Our findings suggest that nucleotide co-occurrence networks, by modeling the complex co-changes at the nucleotide level, present an efficient means of studying influenza virus evolution.
Methods
Influenza virus sequences used
Sequence data of 1032 complete genomes of influenza A virus (H3N2) sampled from Hong Kong of China, Memphis, and New York of the United States, and Canterbury of New Zealand during 1968–2006 were downloaded from the Influenza Sequence Database (Fauci 2005) (see complete list in Supplemental Table S2).
Construction of viral nucleotide co-occurrence networks and co-occurring nucleotide modules
Step 1
Coding regions for each gene segment were compiled by performing multiple nucleotide sequence alignments separately on the eight individual influenza virus gene segments using ClustalW (Thompson et al. 1994). In cases of incomplete sequencing near the start or stop codons, nucleotides were added to the ends of the sequences based on the consensus sequence.
Step 2
A single alignment was generated by concatenating the coding regions of the eight viral gene segments, resulting in a whole-genome sequence of 13,115 nt.
Step 3
All conserved nucleotide positions within the 1032 H3N2 isolates (7869 positions) were removed, so that only variable positions (5246 positions) remained.
Step 4
For all possible nucleotide position pairs (i, j), we first eliminated the nucleotide pairs (xiyj) (where x, y ∈{A, T, G, C, –}) that occurred with a frequency of <5% in every season, then calculated the frequency of occurrence for all nucleotide pairs f(xiyj) and single nucleotides f(xi),f(yj), disregarding the eliminated nucleotide pairs. The extent of co-occurrence of two nucleotides at position (i, j) is denoted as
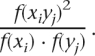 |
Finally, for each viral isolate, we defined a pair of its nucleotides at position (i, j) as perfectly co-occurring if the extent of their co-occurrence was 1. In this way, a nucleotide co-occurrence network for each H3N2 isolate was created by retaining the perfect co-occurring nucleotide pairs.
Step 5
We obtained co-occurring nucleotide modules by simply grouping all nucleotides with at least one connection. Thus, there are no connections between any two modules. In total, we obtained 2324 co-occurring nucleotide modules from the 1032 viral nucleotide co-occurrence networks, and each viral nucleotide co-occurrence network contained on average 280 co-occurring nucleotide modules.
Strain data simulation
To simulate the nucleotide co-occurrence networks for evenly distributed viral isolates from 1968 to 2006, we randomly sampled one viral strain from each season between 1968 and 2006 and constructed nucleotide co-occurrence networks for this sampled group of strains using the same procedure as we used for all 1032 strains (Fig. 1 and the method described above). The above simulation was repeated 1000 times, and all simulation results were plotted in Figure 2D. All computations in our study were performed using custom-written programs in Perl or Matlab on a Linux cluster of dual 2.8G Hz CPU machines with 2 GB of RAM, and simulation calculations were distributed to 20 CPUs at a cost of ∼13 h for each CPU.
Network property calculations
The basic measures for analysis, including network connectivity, clustering coefficients and path length, have been described previously (Watts and Strogatz 1998; Luscombe et al. 2004; Wuchty and Almaas 2005). The average vertex connectivity (K) of a nucleotide co-occurrence network is defined as
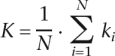 |
where N is the total number of nodes, and ki is the number of neighbors for node i. The average connectivity of all viral networks is computed as
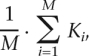 |
where M is the total number of H3N2 strains in the given season. We performed data standardization of K values derived from the simulated co-occurrence network to generate zero mean and unit variance.
The average per-season rate of connection changes R(m,n) when a network evolves from season m to season n (n > m) is computed as
 |
where i is the time in seasons, Xi is the number of edges changed when a network evolves from season i − 1 to season i, and Yi is the number of total edges of a network in season i. R(m,n) is the mean across 1000 simulated networks. For example, to produce Figure 4A, we considered edges between a pair of gene segments (i.e., HA:NA) or within a gene segment (i.e., HA:HA) as a subnetwork, and then computed its average R with one-season resolution (i.e., RHANA(98,99) denotes the rate of connection changes between gene segment HA and NA from 1998 to 1999).
To quantify the extent of cooperative evolution between gene segments a and b, we introduce a measure, Cab(m,n), defined as the ratio of average rate of intergene segment connection changes, Rab(m,n), to that of intragene segment connection change, Raa;bb(m,n). Therefore, Cab from 1998 to 2006 is computed as
Connectivity clusters and HA sequence clusters
To follow the evolution of network connectivities of predominant H3N2 strains over time, we developed a simple algorithm to cluster predominant strains in consecutive seasons. We ignored seasons with no samples (1970, 1979, 1989, and 1991), such that 1969 and 1971 are considered consecutive seasons. To represent the predominant strains in a season, we used a meta strain with a connectivity that is averaged over all strains in that season. Unfortunately, the number of H3N2 strains sampled in most of the seasons before 1993 is no more than five (Supplemental Table S2), which could cause the connectivity of the meta strain to deviate significantly from that of the actual predominant strains. Therefore, we use network connectivities for actual individual strains sampled before 1993 and only consider the predominant strains of a season as belonging to a cluster when it covers at least 50% of H3N2 strains from that season. All strains within a connectivity cluster are consecutive. Therefore, for a new strain to be placed in a cluster, we first require that the connectivity difference between the new strain and all other strains in the cluster be below a threshold (0.1 in our case); then we further require that the connectivity difference between the new strain and existing strains in the cluster from the same or adjacent seasons be below a more stringent threshold (0.03 in our case). A cluster containing predominant strains in consecutive seasons is obtained when at least 50% of H3N2 strains in any of these seasons are covered. The clustering runs iteratively until strains are reliably clustered. For a fair comparison with the antigenic clusters, the above two thresholds are chosen to ensure that the clustering generates the number of connectivity clusters close to that of antigenic clusters, 11, in the analysis of Smith and coworkers (Smith et al. 2004). Clusters that include the predominant strains from at least two seasons are plotted in Figure 2A.
The clustering of HA sequences for 1032 H3N2 isolates followed the method described by Plotkin et al. (2002). We tried a range of threshold distances and took the threshold distance (d = 4) that resulted in the best match between HA sequence clusters and the antigenic clusters by Smith et al. (2004). A comparison of the HA sequence clusters with connectivity clusters and antigenic groups by Smith et al. is shown in Supplemental Figure S4.
Phylogenetic analysis and genetic distance calculation
Phylogenetic trees of eight individual gene segments, the concatenated complete genome, and the strain-module profiles for 1032 H3N2 strains were inferred with the maximum likelihood (ML) method using PHYML (Guindon and Gascuel 2003). Such phylogenetic trees are also called ML-trees. The parameters use the default as provided by the software. To assess the reliability of key nodes on the phylogenetic trees, a bootstrap resampling analysis was also undertaken, which involved the inference of 1000 replicate ML-trees using neighbor-joining procedures. To estimate the genetic distance between two strains, we calculated the ML-tree distance between the two strains from the phylogenetic tree of the concatenated complete genome.
Classification of co-occurring nucleotide modules
The 2324 genomic modules were obtained using the method described above. A genomic module was defined as a predominant module if it occurred in at least 50% of the strains in at least one season during 1968–2006, and 417 out of 2324 modules were classified as predominant modules. Of the predominant modules, 153 were classified as conserved because they occurred predominantly in all seasons from 1968 to 2006. The remaining 264 nonconserved predominant modules were classified as transition modules if they shared at least two of the same positions occupied by different nucleotides. We obtained 114 transition modules. The remaining 39 modules were denoted as transient modules. For each module in transition and transient modules, we further identified the module-based amino acid substitutions corresponding to the nucleotide changes. Clustering analysis is done using hierarchical clustering algorithm implemented in the software EXPANDER (Shamir et al. 2005).
Acknowledgments
We are grateful to K. Miller-Jensen, J. Chen, S. Altman, A.E. Keating, C. Taylor, G. Grigoryan, P. Feng, and members of the Jiang lab for comments on the manuscript; and M.V. Olson, H. Tang, and G.G. Brownlee for stimulating discussions. This work was supported by the Bai Ren Project of Chinese Academy of Sciences to T.J., NSFC (Grant Nos: 30570428, 30599432), and Project “973” (Grant No. 2006CB911002). T.J. conceived and directed the project and analyzed the results. T.J., X.D., Z.W., A.W., L.S., and Y.C. performed computations, and T.J. and H.H. wrote the paper.
Footnotes
[Supplemental material is available online at www.genome.org.]
Article published online before print. Article and publication date are available at http://www.genome.org/cgi/doi/10.1101/gr.6969007
References
- Bhat N., Wright J.G., Broder K.R., Murray E.L., Greenberg M.E., Glover M.J., Likos A.M., Posey D.L., Klimov A., Lindstrom S.E., Wright J.G., Broder K.R., Murray E.L., Greenberg M.E., Glover M.J., Likos A.M., Posey D.L., Klimov A., Lindstrom S.E., Broder K.R., Murray E.L., Greenberg M.E., Glover M.J., Likos A.M., Posey D.L., Klimov A., Lindstrom S.E., Murray E.L., Greenberg M.E., Glover M.J., Likos A.M., Posey D.L., Klimov A., Lindstrom S.E., Greenberg M.E., Glover M.J., Likos A.M., Posey D.L., Klimov A., Lindstrom S.E., Glover M.J., Likos A.M., Posey D.L., Klimov A., Lindstrom S.E., Likos A.M., Posey D.L., Klimov A., Lindstrom S.E., Posey D.L., Klimov A., Lindstrom S.E., Klimov A., Lindstrom S.E., Lindstrom S.E., et al. Influenza-associated deaths among children in the United States, 2003-2004. N. Engl. J. Med. 2005;353:2559–2567. doi: 10.1056/NEJMoa051721. [DOI] [PubMed] [Google Scholar]
- Bush R.M., Bender C.A., Subbarao K., Cox N.J., Fitch W.M., Bender C.A., Subbarao K., Cox N.J., Fitch W.M., Subbarao K., Cox N.J., Fitch W.M., Cox N.J., Fitch W.M., Fitch W.M. Predicting the evolution of human influenza A. Science. 1999;286:1921–1925. doi: 10.1126/science.286.5446.1921. [DOI] [PubMed] [Google Scholar]
- Chakraverty P., Cunningham P., Shen G.Z., Pereira M.S., Cunningham P., Shen G.Z., Pereira M.S., Shen G.Z., Pereira M.S., Pereira M.S. Influenza in the United Kingdom 1982-85. J. Hyg. (Lond.) 1986;97:347–358. doi: 10.1017/s0022172400065438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen L., Lee C., Lee C. Distinguishing HIV-1 drug resistance, accessory, and viral fitness mutations using conditional selection pressure analysis of treated versus untreated patient samples. Biol. Direct. 2006;1:14. doi: 10.1186/1745-6150-1-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen H., Smith G.J., Zhang S.Y., Qin K., Wang J., Li K.S., Webster R.G., Peiris J.S., Guan Y., Smith G.J., Zhang S.Y., Qin K., Wang J., Li K.S., Webster R.G., Peiris J.S., Guan Y., Zhang S.Y., Qin K., Wang J., Li K.S., Webster R.G., Peiris J.S., Guan Y., Qin K., Wang J., Li K.S., Webster R.G., Peiris J.S., Guan Y., Wang J., Li K.S., Webster R.G., Peiris J.S., Guan Y., Li K.S., Webster R.G., Peiris J.S., Guan Y., Webster R.G., Peiris J.S., Guan Y., Peiris J.S., Guan Y., Guan Y. Avian flu: H5N1 virus outbreak in migratory waterfowl. Nature. 2005;436:191–192. doi: 10.1038/nature03974. [DOI] [PubMed] [Google Scholar]
- Choi S.S., Li W., Lahn B.T., Li W., Lahn B.T., Lahn B.T. Robust signals of coevolution of interacting residues in mammalian proteomes identified by phylogeny-aided structural analysis. Nat. Genet. 2005;37:1367–1371. doi: 10.1038/ng1685. [DOI] [PubMed] [Google Scholar]
- Ciccarelli F.D., Doerks T., von Mering C., Creevey C.J., Snel B., Bork P., Doerks T., von Mering C., Creevey C.J., Snel B., Bork P., von Mering C., Creevey C.J., Snel B., Bork P., Creevey C.J., Snel B., Bork P., Snel B., Bork P., Bork P. Toward automatic reconstruction of a highly resolved tree of life. Science. 2006;311:1283–1287. doi: 10.1126/science.1123061. [DOI] [PubMed] [Google Scholar]
- Fauci A.S. Race against time. Nature. 2005;435:423–424. doi: 10.1038/435423a. [DOI] [PubMed] [Google Scholar]
- Ferguson N.M., Galvani A.P., Bush R.M., Galvani A.P., Bush R.M., Bush R.M. Ecological and immunological determinants of influenza evolution. Nature. 2003;422:428–433. doi: 10.1038/nature01509. [DOI] [PubMed] [Google Scholar]
- Fitch W.M., Leiter J.M., Li X.Q., Palese P., Leiter J.M., Li X.Q., Palese P., Li X.Q., Palese P., Palese P. Positive Darwinian evolution in human influenza A viruses. Proc. Natl. Acad. Sci. 1991;88:4270–4274. doi: 10.1073/pnas.88.10.4270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fleury D., Daniels R.S., Skehel J.J., Knossow M., Bizebard T., Daniels R.S., Skehel J.J., Knossow M., Bizebard T., Skehel J.J., Knossow M., Bizebard T., Knossow M., Bizebard T., Bizebard T. Structural evidence for recognition of a single epitope by two distinct antibodies. Proteins. 2000;40:572–578. [PubMed] [Google Scholar]
- Fodor E., Brownlee G.G., Potter C.W., Brownlee G.G., Potter C.W., Potter C.W. Influenza virus replication. In: Potter C.W., editor. Perspectives in medical virology. Elsevier; Amsterdam, The Netherlands: 2002. pp. 1–29. [Google Scholar]
- Ghedin E., Sengamalay N.A., Shumway M., Zaborsky J., Feldblyum T., Subbu V., Spiro D.J., Sitz J., Koo H., Bolotov P., Sengamalay N.A., Shumway M., Zaborsky J., Feldblyum T., Subbu V., Spiro D.J., Sitz J., Koo H., Bolotov P., Shumway M., Zaborsky J., Feldblyum T., Subbu V., Spiro D.J., Sitz J., Koo H., Bolotov P., Zaborsky J., Feldblyum T., Subbu V., Spiro D.J., Sitz J., Koo H., Bolotov P., Feldblyum T., Subbu V., Spiro D.J., Sitz J., Koo H., Bolotov P., Subbu V., Spiro D.J., Sitz J., Koo H., Bolotov P., Spiro D.J., Sitz J., Koo H., Bolotov P., Sitz J., Koo H., Bolotov P., Koo H., Bolotov P., Bolotov P., et al. Large-scale sequencing of human influenza reveals the dynamic nature of viral genome evolution. Nature. 2005;437:1162–1166. doi: 10.1038/nature04239. [DOI] [PubMed] [Google Scholar]
- Gobel U., Sander C., Schneider R., Valencia A., Sander C., Schneider R., Valencia A., Schneider R., Valencia A., Valencia A. Correlated mutations and residue contacts in proteins. Proteins. 1994;18:309–317. doi: 10.1002/prot.340180402. [DOI] [PubMed] [Google Scholar]
- Grenfell B.T., Pybus O.G., Gog J.R., Wood J.L., Daly J.M., Mumford J.A., Holmes E.C., Pybus O.G., Gog J.R., Wood J.L., Daly J.M., Mumford J.A., Holmes E.C., Gog J.R., Wood J.L., Daly J.M., Mumford J.A., Holmes E.C., Wood J.L., Daly J.M., Mumford J.A., Holmes E.C., Daly J.M., Mumford J.A., Holmes E.C., Mumford J.A., Holmes E.C., Holmes E.C. Unifying the epidemiological and evolutionary dynamics of pathogens. Science. 2004;303:327–332. doi: 10.1126/science.1090727. [DOI] [PubMed] [Google Scholar]
- Guindon S., Gascuel O., Gascuel O. A simple, fast, and accurate algorithm to estimate large phylogenies by maximum likelihood. Syst. Biol. 2003;52:696–704. doi: 10.1080/10635150390235520. [DOI] [PubMed] [Google Scholar]
- Holmes E.C., Ghedin E., Miller N., Taylor J., Bao Y., St George K., Grenfell B.T., Salzberg S.L., Fraser C.M., Lipman D.J., Ghedin E., Miller N., Taylor J., Bao Y., St George K., Grenfell B.T., Salzberg S.L., Fraser C.M., Lipman D.J., Miller N., Taylor J., Bao Y., St George K., Grenfell B.T., Salzberg S.L., Fraser C.M., Lipman D.J., Taylor J., Bao Y., St George K., Grenfell B.T., Salzberg S.L., Fraser C.M., Lipman D.J., Bao Y., St George K., Grenfell B.T., Salzberg S.L., Fraser C.M., Lipman D.J., St George K., Grenfell B.T., Salzberg S.L., Fraser C.M., Lipman D.J., Grenfell B.T., Salzberg S.L., Fraser C.M., Lipman D.J., Salzberg S.L., Fraser C.M., Lipman D.J., Fraser C.M., Lipman D.J., Lipman D.J., et al. Whole-genome analysis of human influenza A virus reveals multiple persistent lineages and reassortment among recent H3N2 viruses. PLoS Biol. 2005;3:e300. doi: 10.1371/journal.pbio.0030300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin H., Zhou H., Liu H., Chan W., Adhikary L., Mahmood K., Lee M.S., Kemble G., Zhou H., Liu H., Chan W., Adhikary L., Mahmood K., Lee M.S., Kemble G., Liu H., Chan W., Adhikary L., Mahmood K., Lee M.S., Kemble G., Chan W., Adhikary L., Mahmood K., Lee M.S., Kemble G., Adhikary L., Mahmood K., Lee M.S., Kemble G., Mahmood K., Lee M.S., Kemble G., Lee M.S., Kemble G., Kemble G. Two residues in the hemagglutinin of A/Fujian/411/02-like influenza viruses are responsible for antigenic drift from A/Panama/2007/99. Virology. 2005;336:113–119. doi: 10.1016/j.virol.2005.03.010. [DOI] [PubMed] [Google Scholar]
- Koelle K., Cobey S., Grenfell B., Pascual M., Cobey S., Grenfell B., Pascual M., Grenfell B., Pascual M., Pascual M. Epochal evolution shapes the phylodynamics of interpandemic influenza A (H3N2) in humans. Science. 2006;314:1898–1903. doi: 10.1126/science.1132745. [DOI] [PubMed] [Google Scholar]
- Luscombe N.M., Babu M.M., Yu H., Snyder M., Teichmann S.A., Gerstein M., Babu M.M., Yu H., Snyder M., Teichmann S.A., Gerstein M., Yu H., Snyder M., Teichmann S.A., Gerstein M., Snyder M., Teichmann S.A., Gerstein M., Teichmann S.A., Gerstein M., Gerstein M. Genomic analysis of regulatory network dynamics reveals large topological changes. Nature. 2004;431:308–312. doi: 10.1038/nature02782. [DOI] [PubMed] [Google Scholar]
- Nakajima K., Nobusawa E., Nagy A., Nakajima S., Nobusawa E., Nagy A., Nakajima S., Nagy A., Nakajima S., Nakajima S. Accumulation of amino acid substitutions promotes irreversible structural changes in the hemagglutinin of human influenza AH3 virus during evolution. J. Virol. 2005;79:6472–6477. doi: 10.1128/JVI.79.10.6472-6477.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Obenauer J.C., Denson J., Mehta P.K., Su X., Mukatira S., Finkelstein D.B., Xu X., Wang J., Ma J., Fan Y., Denson J., Mehta P.K., Su X., Mukatira S., Finkelstein D.B., Xu X., Wang J., Ma J., Fan Y., Mehta P.K., Su X., Mukatira S., Finkelstein D.B., Xu X., Wang J., Ma J., Fan Y., Su X., Mukatira S., Finkelstein D.B., Xu X., Wang J., Ma J., Fan Y., Mukatira S., Finkelstein D.B., Xu X., Wang J., Ma J., Fan Y., Finkelstein D.B., Xu X., Wang J., Ma J., Fan Y., Xu X., Wang J., Ma J., Fan Y., Wang J., Ma J., Fan Y., Ma J., Fan Y., Fan Y., et al. Large-scale sequence analysis of avian influenza isolates. Science. 2006;311:1576–1580. doi: 10.1126/science.1121586. [DOI] [PubMed] [Google Scholar]
- Parrish C.R., Kawaoka Y., Kawaoka Y. The origins of new pandemic viruses: The acquisition of new host ranges by canine parvovirus and influenza A viruses. Annu. Rev. Microbiol. 2005;59:553–586. doi: 10.1146/annurev.micro.59.030804.121059. [DOI] [PubMed] [Google Scholar]
- Plotkin J.B., Dushoff J., Levin S.A., Dushoff J., Levin S.A., Levin S.A. Hemagglutinin sequence clusters and the antigenic evolution of influenza A virus. Proc. Natl. Acad. Sci. 2002;99:6263–6268. doi: 10.1073/pnas.082110799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pollock D.D. Genomic biodiversity, phylogenetics and coevolution in proteins. Appl. Bioinformatics. 2002;1:81–92. [PMC free article] [PubMed] [Google Scholar]
- Rimmelzwaan G.F., Berkhoff E.G., Nieuwkoop N.J., Smith D.J., Fouchier R.A., Osterhaus A.D., Berkhoff E.G., Nieuwkoop N.J., Smith D.J., Fouchier R.A., Osterhaus A.D., Nieuwkoop N.J., Smith D.J., Fouchier R.A., Osterhaus A.D., Smith D.J., Fouchier R.A., Osterhaus A.D., Fouchier R.A., Osterhaus A.D., Osterhaus A.D. Full restoration of viral fitness by multiple compensatory co-mutations in the nucleoprotein of influenza A virus cytotoxic T-lymphocyte escape mutants. J. Gen. Virol. 2005;86:1801–1805. doi: 10.1099/vir.0.80867-0. [DOI] [PubMed] [Google Scholar]
- Shamir R., Maron-Katz A., Tanay A., Linhart C., Steinfeld I., Sharan R., Shiloh Y., Elkon R., Maron-Katz A., Tanay A., Linhart C., Steinfeld I., Sharan R., Shiloh Y., Elkon R., Tanay A., Linhart C., Steinfeld I., Sharan R., Shiloh Y., Elkon R., Linhart C., Steinfeld I., Sharan R., Shiloh Y., Elkon R., Steinfeld I., Sharan R., Shiloh Y., Elkon R., Sharan R., Shiloh Y., Elkon R., Shiloh Y., Elkon R., Elkon R. EXPANDER—An integrative program suite for microarray data analysis. BMC Bioinformatics. 2005;6:232. doi: 10.1186/1471-2105-6-232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shapiro B., Rambaut A., Pybus O.G., Holmes E.C., Rambaut A., Pybus O.G., Holmes E.C., Pybus O.G., Holmes E.C., Holmes E.C. A phylogenetic method for detecting positive epistasis in gene sequences and its application to RNA virus evolution. Mol. Biol. Evol. 2006;23:1724–1730. doi: 10.1093/molbev/msl037. [DOI] [PubMed] [Google Scholar]
- Shih A.C., Hsiao T.C., Ho M.S., Li W.H., Hsiao T.C., Ho M.S., Li W.H., Ho M.S., Li W.H., Li W.H. Simultaneous amino acid substitutions at antigenic sites drive influenza A hemagglutinin evolution. Proc. Natl. Acad. Sci. 2007;104:6283–6288. doi: 10.1073/pnas.0701396104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skehel J.J., Wiley D.C., Wiley D.C. Receptor binding and membrane fusion in virus entry: The influenza hemagglutinin. Annu. Rev. Biochem. 2000;69:531–569. doi: 10.1146/annurev.biochem.69.1.531. [DOI] [PubMed] [Google Scholar]
- Slonim N., Elemento O., Tavazoie S., Elemento O., Tavazoie S., Tavazoie S. Ab initio genotype–phenotype association reveals intrinsic modularity in genetic networks. Mol. Syst. Biol. 2006;2:2006.0005. doi: 10.1038/msb4100047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith D.J. Predictability and preparedness in influenza control. Science. 2006;312:392–394. doi: 10.1126/science.1122665. [DOI] [PubMed] [Google Scholar]
- Smith D.J., Lapedes A.S., de Jong J.C., Bestebroer T.M., Rimmelzwaan G.F., Osterhaus A.D., Fouchier R.A., Lapedes A.S., de Jong J.C., Bestebroer T.M., Rimmelzwaan G.F., Osterhaus A.D., Fouchier R.A., de Jong J.C., Bestebroer T.M., Rimmelzwaan G.F., Osterhaus A.D., Fouchier R.A., Bestebroer T.M., Rimmelzwaan G.F., Osterhaus A.D., Fouchier R.A., Rimmelzwaan G.F., Osterhaus A.D., Fouchier R.A., Osterhaus A.D., Fouchier R.A., Fouchier R.A. Mapping the antigenic and genetic evolution of influenza virus. Science. 2004;305:371–376. doi: 10.1126/science.1097211. [DOI] [PubMed] [Google Scholar]
- Thompson J.D., Higgins D.G., Gibson T.J., Higgins D.G., Gibson T.J., Gibson T.J. CLUSTAL W: Improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 1994;22:4673–4680. doi: 10.1093/nar/22.22.4673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagner R., Matrosovich M., Klenk H.D., Matrosovich M., Klenk H.D., Klenk H.D. Functional balance between haemagglutinin and neuraminidase in influenza virus infections. Rev. Med. Virol. 2002;12:159–166. doi: 10.1002/rmv.352. [DOI] [PubMed] [Google Scholar]
- Watts D.J., Strogatz S.H., Strogatz S.H. Collective dynamics of ‘small-world’ networks. Nature. 1998;393:440–442. doi: 10.1038/30918. [DOI] [PubMed] [Google Scholar]
- Wiley D.C., Wilson I.A., Skehel J.J., Wilson I.A., Skehel J.J., Skehel J.J. Structural identification of the antibody-binding sites of Hong Kong influenza haemagglutinin and their involvement in antigenic variation. Nature. 1981;289:373–378. doi: 10.1038/289373a0. [DOI] [PubMed] [Google Scholar]
- Wilson I.A., Cox N.J., Cox N.J. Structural basis of immune recognition of influenza virus hemagglutinin. Annu. Rev. Immunol. 1990;8:737–771. doi: 10.1146/annurev.iy.08.040190.003513. [DOI] [PubMed] [Google Scholar]
- Wolf Y.I., Viboud C., Holmes E.C., Koonin E.V., Lipman D.J., Viboud C., Holmes E.C., Koonin E.V., Lipman D.J., Holmes E.C., Koonin E.V., Lipman D.J., Koonin E.V., Lipman D.J., Lipman D.J. Long intervals of stasis punctuated by bursts of positive selection in the seasonal evolution of influenza A virus. Biol. Direct. 2006;1:34. doi: 10.1186/1745-6150-1-34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wuchty S., Almaas E., Almaas E. Evolutionary cores of domain co-occurrence networks. BMC Evol. Biol. 2005;5:24. doi: 10.1186/1471-2148-5-24. [DOI] [PMC free article] [PubMed] [Google Scholar]