

Abstract
The Arabidopsis DEMETER (DME) DNA glycosylase is required for the maternal allele expression of imprinted Polycomb group (MEDEA and FIS2) and transcription factor (FWA) genes in the endosperm. Expression of DME in the central cell, not in pollen or stamen, establishes gene imprinting by hypomethylating maternal alleles. However, little is known about other genes regulated by DME. To identify putative DME target genes, we generated CaMV:DME plants which ectopically express DME in pollen and stamens. Comparison of mRNA profiles revealed 94 genes induced by ectopic DME expression in both stamen and pollen. Gene ontology analysis identified three molecular functions enriched in the DME-inducible RNA list: DNA or RNA binding, kinase activity, and transcription factor activity. Semi-quantitative RT-PCR verified the candidate genes identified by GeneChip analysis. The putative target genes identified in this study will provide insights into the regulatory mechanism of DME DNA glycosylase and the functions of DNA demethylation.
Keywords: Gene imprinting, DNA demethylation, DNA glycosylase, GeneChip analysis
Introduction
Double fertilization is a unique characteristic of flowering plants. Fusion of an egg cell with a sperm cell gives rise to the diploid embryo and the second fusion of a diploid central cell with another sperm cell gives rise to the triploid endosperm. The endosperm is the functional homologue of the mammalian placenta and provides nutrients to the developing embryo.
Gene imprinting refers to the differential allele expression of paternal and maternal alleles depending on parental origin. Imprinting occurs in the placenta of mammals [1] and in the endosperm of plants [2, 3]. MEDEA (MEA) is a Polycomb group (PcG) gene imprinted in Arabidopsis [4, 5, 6]. The maternal MEA allele is expressed while the paternal allele is silenced [7]. There are two mechanisms regulating MEA imprinting. Demethylation by the DEMETER (DME) DNA glycosylase activates expression of the maternal MEA allele [8, 9] in the central cell of the female gametophyte. Silencing of the paternal MEA allele is not directly controlled by DNA methylation. Instead, PcG proteins, including maternally expressed MEA, repress expression of the paternal MEA allele in the endosperm [9]. Thus, MEA is a self-imprinted gene [9, 10, 11]. DME demethylation occurs by the base excision DNA repair process [9, 12]. DME first excises 5-methylcytosine by its glycosylase activity, and then the AP lyase activity of DME nicks the DNA strand. AP endonuclease cleavage generates a 3′ hydroxyl, DNA polymerase replaces the excised 5-methylcytosine with cytosine, and finally DNA ligase seals the nick. By excising 5-methylcytosine, DME prevents CpG hypermethylation of its target genes, thus activating their gene expression. DME is specifically expressed in the central cell of the female gametophyte before fertilization [8]. Its temporal and spatial expression is essential for the establishment of MEA imprinting and seed viability.
In mammals, there are approximately 100 imprinted genes with approximately half expressing the maternal allele and half expressing the paternal allele (http://www.geneimprint.com). In Arabidopsis, only four genes imprinted in the endosperm have been discovered [7, 13, 14, 15]. MEA, FWA and FIS2 are expressed maternally and are controlled by DME [8, 9, 13, 15]. PHERES1 (PHE1) is expressed paternally and PcG proteins including MEA, which is activated by DME, regulates its maternal silencing [14, 16]. Therefore, DME is a key regulator of genomic imprinting in flowering plants.
Considering the number of imprinted genes in animals, it is possible that there are more imprinted genes in Arabidopsis that are controlled by DME. To test this hypothesis, we used an RNA profiling approach to search for other genes regulated by DME. We generated transgenic plants that ectopically express DME under the control of the CaMV promoter in pollen and stamen [17]. Since the endogenous DME gene is not expressed in the wild type male reproductive organs (pollen and stamen), ectopic DME expression reveals the expression of novel target genes. We extracted poly (A)+ RNA from pollen and stamens harvested from CaMV:DME transgenic plants and wild type control plants. cRNAs were labeled and hybridized to Affymetrix Arabidopsis ATH1 GeneChip arrays. We compared and analyzed RNA profiles from both transgenic and wild type pollen and stamen and detected candidate DME-inducible genes, which were confirmed by semi-quantitative RT-PCR experiments. These DME-inducible genes shed light on the mechanism of DME-mediated demethylation and the role of gene imprinting in seed biology.
Materials and methods
Plant materials, transgenic plant isolation and growth condition
CaMV:DME-4 transgenic plants were generated as described [17] and wild type plants (Columbia-gl ecotype) were used in this experiment. Transgenic plants contained the NPTII gene, which confers resistance to kanamycin. Plants were grown in standard green house conditions (16 hrs light/8hrs dark).
Pollen collection and RNA extraction
Pollen grains and stamens were collected from stage 13 and 14 flowers and processed as described [17, 18]. Total RNAs were extracted using TRIzol (Invitrogen) as described [7]. Poly(A)+ RNA was selected from total RNA by using the Dynabeads mRNA Purification kit (Dynal Inc) according to procedures provided by the manufacturer.
Preparation of the probes, hybridization and scanning
Double-stranded cDNAs were generated using Superscript Choice system (Invitrogen) and were purified by phenol/chloroform extraction. Biotin-labeled cRNA probes were synthesized using BioArray High Yield RNA Transcript Labeling Kit (T7) (Enzo Life Sciences) and purified using RNeasy mini spin columns (Qiagen). Concentration of cRNA probe was determined using a UV-spectrophotometer and the size range of synthesized cRNA was determined by fractionating cRNAs on a 1.3 % agarose/formaldehyde gel. cRNAs were fragmented with fragmentation buffer (40 mM Tris-acetate, pH 8.1, 100 mM KOAc, 30 mM MgOAc) and cRNA size was determined on a 1.3 % agarose/formaldehyde gel. Fragmented biotin-labeled cRNAs were hybridized to the Arabidopsis ATH1 GeneChip Array (Affymetrix) for 16 hours according to manufacturer's protocol (Affymetrix). The arrays were stained with streptavidin phycoerythrin (SAPE, Molecular probes), goat IgG (Sigma) and anti-streptavidin biotinylated antibody. GeneChip arrays were scanned and analyzed using Affymetrix Microarray Analysis Suite (MAS) 5.0 (Santa Clara, CA). All probe sets on the array were scaled globally to a target intensity of 500 using MAS 5.0 default parameters.
RT-PCR analysis
Reverse transcription was done with Retro Transcript (Ambion) following instructions provided by the manufacturer. Primers used in these experiments are in the Supplementary Materials.
Results and Discussion
DME expression in CaMV:DME pollen
We previously generated transgenic plants bearing a transgene (CaMV:DME) with the 6.8 kb full-length DME cDNA ligated to the cauliflower mosaic virus promoter (CaMV) [17]. In multiple CaMV:DME transgenic lines, DME was ectopically expressed in stamen and pollen, and we detected expression of known DME target genes, MEA [17] and FWA (data not shown). This suggests that DME expression is both necessary and sufficient to activate target gene expression, and ectopic DME expression in stamen or pollen can induce DME target genes that are normally silenced in the male reproductive organs [17].
Transcriptional profiling of CaMV:DME and wild type
To gain a global view of the transcription network that is regulated by DME, we used Affymetrix GeneChip arrays to identify novel genes up-regulated by ectopic DME expression in CaMV:DME pollen and stamen compared to the wild type counterparts. Pollen grains were harvested from CaMV:DME and control wild type flowers at stages 13 (open flowers) and 14 (self-pollinating) [19]. We also collected stamens at the same stages from wild type and CaMV:DME plants. Stamens included filaments, and anthers containing mature and immature pollen grains. Poly(A)+ RNAs were extracted from the above four independent tissue samples, and their biotin–labeled cRNA probes were synthesized from double-stranded cDNAs generated from poly(A)+ RNAs. Biotinylated cRNA probes were hybridized to Affymetrix Arabidopsis ATH1 GeneChip arrays and data were analyzed using the GeneSpring program. Scatter plot analysis show that wild type and CaMV:DME RNA profiles from pollen are well correlated (r =0.99) (Fig. 1A). Similar results were seen when wild type and CaMV:DME stamen RNAs were compared (r =0.95) (Fig. 1B). The high correlation coefficients between wild type and CaMV:DME pollen and stamen indicate that ectopic expression of DME induces a small number of genes.
Fig. 1.
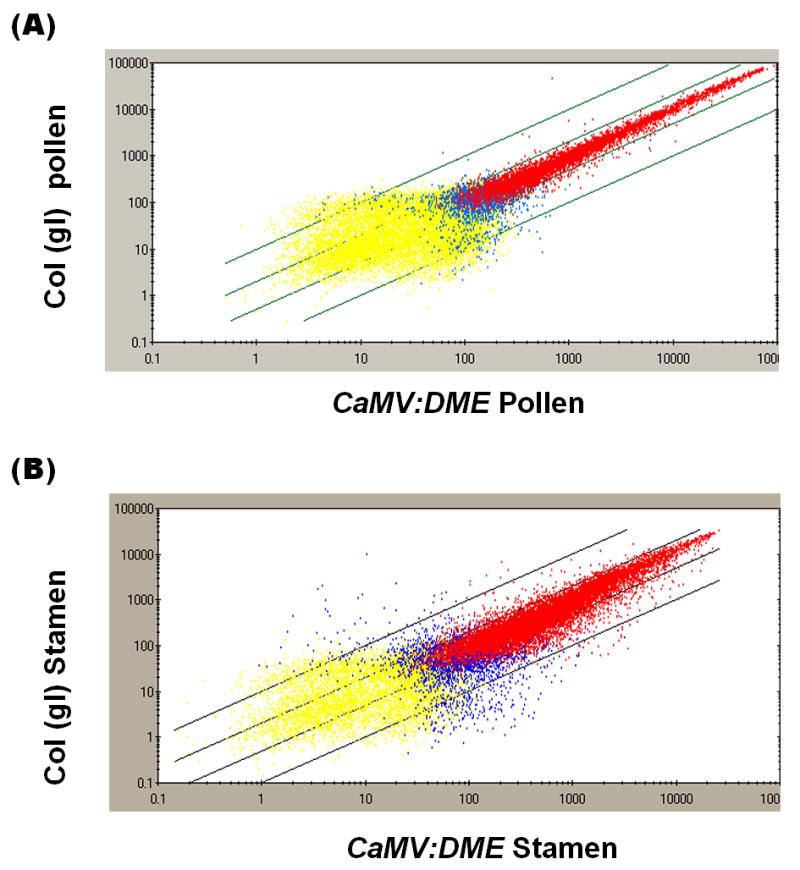
Scatter plots of wild type and CaMV:DME pollen and stamen. Scatter plots analysis of wild type and CaMV:DME pollen (A) and stamen (B) were generated using MAS 5.0 of all probe sets represented on the Affymetrix Arabidopsis ATH1 GeneChip array. A red dot indicates RNA with a detection call of present by MAS 5.0 in both wild type and CaMV:DME pollen or stamen. A yellow dot indicates RNA with a detection call of absent in both wild type and CaMV:DME pollen or stamen. Blue dots indicate different detection calls by MAS 5.0 for wild type and CaMV:DME pollen and stamen.
From the transcriptional profiling data, 6,392 and 7,281 RNAs were detected in wild type and CaMV:DME pollens, respectively (Fig. 2A). These numbers are in close agreement with RNA profiling data of pollen obtained by others [20, 21](Honys and Twell, 2004; Pina et al., 2005). 1,305 RNAs was detected in CaMV:DME pollen and not in wild type control pollen. Similarly, 13,095 and 14,450 RNAs were detected in wild type and CaMV:DME stamens, respectively (Fig. 2B). These numbers are in close agreement with RNA profiling data from stamens obtained by others [22]. 1,864 RNAs were detected in CaMV:DME but not in wild type stamens. We found 94 genes with DME-inducible expression in both CaMV:DME stamen and pollen when comparing CaMV:DME pollen- and stamen-specific RNAs (Fig. 2C). Thus, only a small number of DME-inducible genes were detected in both pollen and stamen.
Fig. 2.
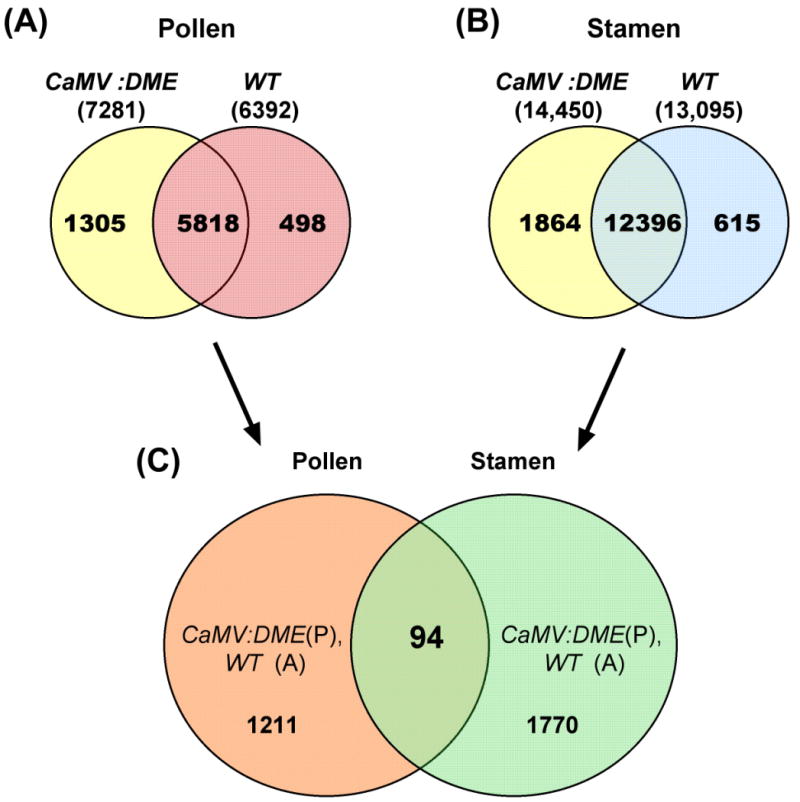
Analysis of RNA profiles. Venn diagrams of genes expressed in wild type and CaMV:DME pollen (A), and wild type and CaMV:DME stamen (B). 94 genes show DME-dependent expression in both CaMV:DME stamen and pollen (C).
These 94 genes were sorted into different functional categories (Supplementary Table 1). There are 4 genes related to cell growth and division, 5 genes to cell structure, 3 genes to disease and defense, 3 genes for energy, 2 genes for intracellular traffic, 9 genes for metabolism, 4 genes for post-transcription, 7 genes for protein destination and storage, 1 gene for protein synthesis, 3 genes for secondary metabolism, 10 genes for signal transduction, 14 genes for transcription, 4 genes for transporter, 2 genes for transposon and 23 genes unclassified (Supplementary Table 1).
We did not detect expression of MEA or FWA in the RNAs isolated from CaMV:DME pollen or stamen. It is possible that the level of MEA and FWA RNA was too low to be detected by the Arabidopsis whole genome Affymetrix probe array [23].
We also sorted the 94 genes into gene ontologies using the bulk retrieval tool at TAIR (http://www.arabidopsis.org/tools/bulk/go) (Fig. 3). The overall transcription profile is similar to the whole Arabidopsis genome profile [24] suggesting DME might regulate genes with a broad range of functional categories. Gene ontology analysis identified three molecular functions enriched in the DME-inducible RNA list compared to the whole Arabidopsis genome: DNA or RNA binding (11.1% versus 5.4%), kinase activity (8.3% versus 5.0%), and transcription factor activity (8.3% versus 5.0%). Although DME induces a wide range of genes, this enrichment suggests that target genes may share common regulatory pathways or be involved in similar functions such as DNA or RNA binding, kinase activity, and transcription factor activity.
Fig. 3.
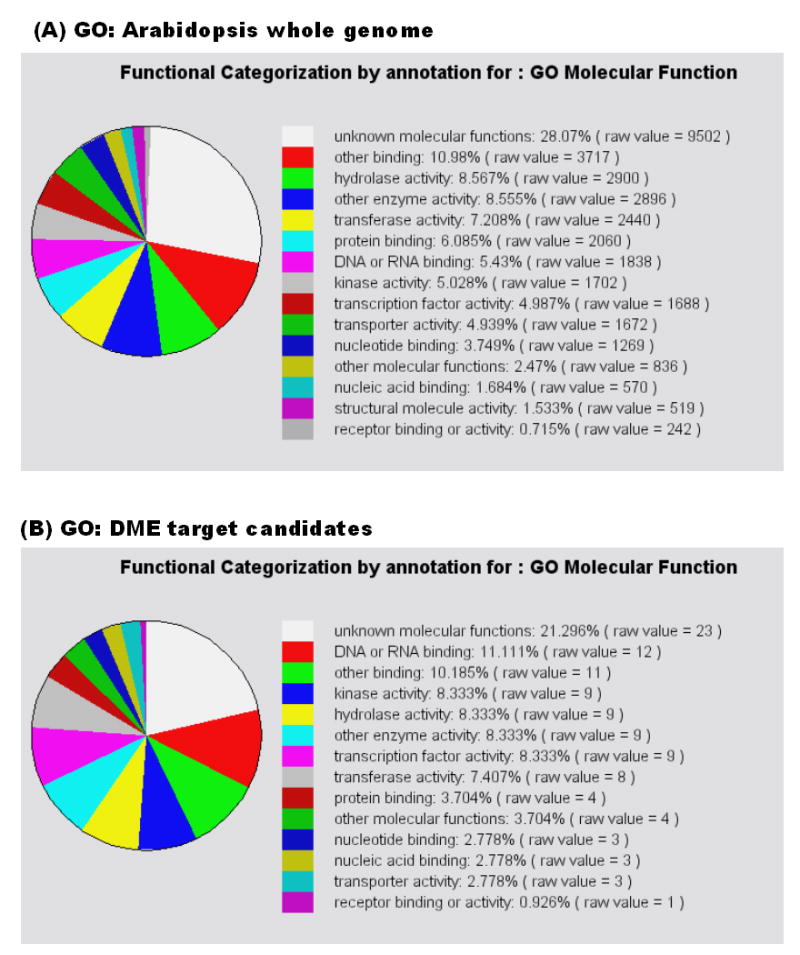
Functional categorization of wild type Arabidopsis whole genome (A) and genes induced by DME (B). 94 genes were sorted out to the functional categories classified in the databases of gene ontology at TAIR. DME-dependent downstream genes show similar distribution of molecular function with the Arabidopsis whole genome.
In Arabidopsis, a number of genes are demethylated by ROS1, DML2, and DML3 [25, 26]. ROS1 (REPRESSOR OF SILENCING 1) is a DME homolog that maintains the transcriptionally active states of a RD29A::LUCIFERASE (RD29A::LUC) reporter gene and the endogenous RD29A gene [12, 27, 28]. DML2 (DEMETER-LIKE 2) and DML3 (DEMETER-LIKE 3) are two other DME-like genes in Arabidopsis. Genes regulated by ROS1 have been identified by comparing RNA profiles from wild type and ros1 mutant plants [26]. Genes demethylated by ROS1, DML2, and DML3 have been revealed by comparing wild type and ros1, dml2 and dml3 triple mutant genome-tiling microarrays [25]. We found little overlap between our DME-target genes and these published results. Thus, although DME and DMLs have similar DNA demethylase activity [9, 12, 25], their putative target genes are very distinctive based on the data from microarray and genome-tiling analyses.
Semi-quantitative RT-PCR for the possible candidate genes
We chose six of the 94 genes that represent a wide range of biological processes for independent validation. Semi-quantitative RT-PCR was performed to measure the RNA level of putative DME-downstream genes from wild type and CaMV:DME pollen tissue. We found that RT-PCR results of these six genes were consistent with the microarray gene profiling results (Fig. 4). That is, expression was not detected in control wild type pollen, and was induced in CaMV:DME pollen. The list of representative candidate DME target genes includes: AT1G60930 which encodes a DEAD/DEAH box domain RecQ family DNA helicase involved in DNA replication, recombination, RNA metabolism, and gene silencing [29]; AT1G80960 which encodes a F-box-related protein with leucine-rich repeat domain; AT2G18050 that encodes a linker Histone H1-3, involved in DNA binding and chromatin structure that is induced by dehydration and ABA [30]; AT4G01780, encoding a XH/XS domain-containing protein; AT4G23800, a high mobility group (HMG) family protein and a non-histone component of chromatin that is involved in DNA binding inducing DNA-dependent transcription, replication and repair mechanism [31]; and AT5G18090, encoding a transcription factor B3 family protein. Taken together, these results suggest that DME, either directly or indirectly, regulates expression of the above genes in addition to MEA, FWA, and FIS2. Since their induction in expression is in response to ectopic expression of DME in tissues where DME is not normally active, they might be bona-fide target genes of DME.
Fig. 4.
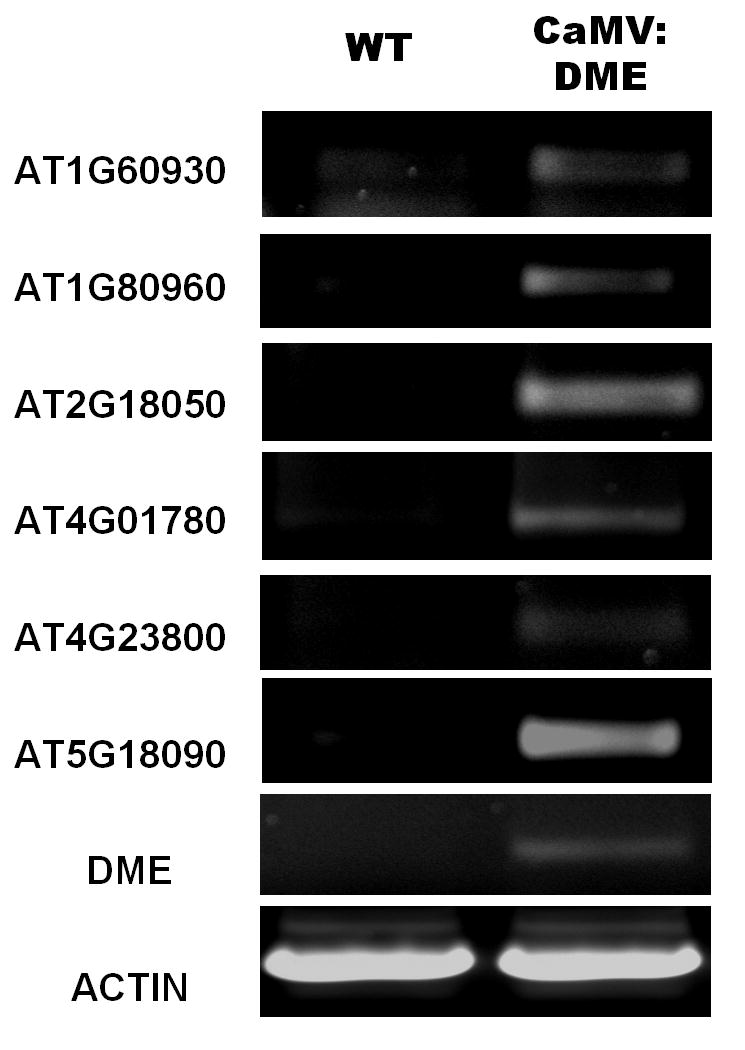
Semi-quantitative RT-PCR validation of putative DME target genes. Expression patterns of the possible DME-downstream genes were confirmed by semi-quantitative RT-PCR from independently isolated pollen tissue.
Whether DME changes the methylated status of each downstream gene, their biological function, and whether they are imprinted genes remains to be assessed. These future studies will provide a more global picture of gene imprinting and will help us to understand the mechanism whereby DME activates gene expression by DNA demethylation.
Supplementary Material
Acknowledgments
This work was supported by grants from BK21, Korea Research Foundation (KRF-2005-070-C00129) and BioGreen21 Program (20050401034633), Rural Development Administration, Republic of Korea to Y. Choi. This work was also supported by grants to R.L.F from NIH (GM069415) and the USDA (2005-02355).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Fowden AL, Sibley C, Reik W, Constancia M. Imprinted genes, placental development and fetal growth. Horm Res. 2006;65:50–58. doi: 10.1159/000091506. [DOI] [PubMed] [Google Scholar]
- 2.Gehring M, Choi Y, Fischer RL. Imprinting and seed development. Plant Cell. 2004;16 doi: 10.1105/tpc.017988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Scott RJ, Spielman M. Genomic imprinting in plants and mammals: how life history constrains convergence. Cytogenet Genome Res. 2006;113:53–67. doi: 10.1159/000090815. [DOI] [PubMed] [Google Scholar]
- 4.Grossniklaus U, Vielle-Calzada JP, Hoeppner MA, Gagliano WB. Maternal control of embryogenesis by MEDEA, a polycomb group gene in Arabidopsis. Science. 1998;280:446–50. doi: 10.1126/science.280.5362.446. [DOI] [PubMed] [Google Scholar]
- 5.Kiyosue T, Ohad N, Yadegari R, Hannon M, Dinneny J, Wells D, Katz A, Margossian L, Harada J, Goldberg RB, Fischer RL. Control of fertilization-independent endosperm development by the MEDEA polycomb gene in Arabidopsis. Proc Natl Acad Sci USA. 1999;96:4186–4191. doi: 10.1073/pnas.96.7.4186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Luo M, Bilodeau P, Koltunow A, Dennis ES, Peacock WJ, Chaudhury AM. Genes controlling fertilization-independent seed development in Arabidopsis thaliana. Proc Natl Acad Sci USA. 1999;96:296–301. doi: 10.1073/pnas.96.1.296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kinoshita T, Yadegari R, Harada JJ, Goldberg RB, Fischer RL. Imprinting of the MEDEA polycomb gene in the Arabidopsis endosperm. Plant Cell. 1999;11:1945–1952. doi: 10.1105/tpc.11.10.1945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Choi Y, Gehring M, Johnson L, Hannon M, Harada JJ, Goldberg RB, Jacobsen SE, Fischer RL. DEMETER, a DNA Glycosylase domain protein, is required for endosperm gene imprinting and seed viability in Arabidopsis. Cell. 2002;110:33–42. doi: 10.1016/s0092-8674(02)00807-3. [DOI] [PubMed] [Google Scholar]
- 9.Gehring M, Huh JH, Hsieh TF, Penterman J, Choi Y, Harada JJ, Goldberg RB, Fischer RL. DEMETER DNA glycosylase establishes MEDEA polycomb gene self-imprinting by allele-specific demethylation. Cell. 2006;124:495–506. doi: 10.1016/j.cell.2005.12.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Baroux C, Gagliardini V, Page DR, Grossniklaus U. Dynamic regulatory interactions of Polycomb group genes: MEDEA autoregulation is required for imprinted gene expression in Arabidopsis. Genes Dev. 2006;20:1081–6. doi: 10.1101/gad.378106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jullien PE, Katz A, Oliva M, Ohad N, Berger F. Polycomb group complexes self-regulate imprinting of the Polycomb group gene MEDEA in Arabidopsis. Curr Biol. 2006;16:486–92. doi: 10.1016/j.cub.2006.01.020. [DOI] [PubMed] [Google Scholar]
- 12.Morales-Ruiz T, Ortega-Galisteo AP, Ponferrada-Marin MI, Martinez-Macias RR, Ariza RR, Roldan-Arjona T. DEMETER and REPRESSOR OF SILENCING1 encode 5-methylcytosine DNA glycosylases. Proc Natl Acad Sci USA. 2006;103:6853–6858. doi: 10.1073/pnas.0601109103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kinoshita T, Miura A, Choi Y, Kinoshita Y, Cao X, Jacobsen SE, Fischer RL, Kakutani T. One-way control of FWA imprinting in Arabidopsis endosperm by DNA methylation. Science. 2004;303:521–523. doi: 10.1126/science.1089835. [DOI] [PubMed] [Google Scholar]
- 14.Kohler C, Page DR, Gagliardini V, Grossniklaus U. The Arabidopsis thaliana MEDEA Polycomb group protein controls expression of PHERES1 by parental imprinting. Nat Genet. 2005;37:28–30. doi: 10.1038/ng1495. [DOI] [PubMed] [Google Scholar]
- 15.Jullien PE, Kinoshita T, Ohad N, Berger F. Maintenance of DNA methylation during the Arabidopsis life cycle is essential for parental imprinting. Plant Cell. 2006;18:1360–72. doi: 10.1105/tpc.106.041178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Makarevich G, Leroy O, Akinci U, Schubert D, Clarenz O, Goodrich J, Grossniklaus U, Kohler C. Different Polycomb group complexes regulate common target genes in Arabidopsis. EMBO Rep. 2006;7:947–952. doi: 10.1038/sj.embor.7400760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Choi Y, Harada JJ, Goldberg RB, Fischer RL. An invariant aspartic acid in the DNA glycosylase domain of DEMETER is necessary for transcriptional activation of the imprinted MEDEA gene. Proc Natl Acad Sci USA. 2004;101:7481–6. doi: 10.1073/pnas.0402328101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Preuss D, Lemieux B, Yen G, Davis RW. A conditional sterile mutation eliminates surface components from Arabidopsis pollen and disrupts cell signaling during fertilization. Genes Dev. 1993;7:974–985. doi: 10.1101/gad.7.6.974. [DOI] [PubMed] [Google Scholar]
- 19.Bowman JL, Mansfield SG. Embryogenesis. In: Bowman J, editor. Arabidopsis: An atlas of morphology and development. Springer-Verlag; New York: 1994. pp. 351–361. [Google Scholar]
- 20.Honys D, Twell D. Transcriptome analysis of haploid male gametophyte development in Arabidopsis. Genome Biol. 2004;5:R85. doi: 10.1186/gb-2004-5-11-r85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Pina C, Pinto F, Feijo JA, Becker JD. Gene family analysis of the Arabidopsis pollen transcriptome reveals biological implications for cell growth, division control, and gene expression regulation. Plant Physiol. 2005;138:744–756. doi: 10.1104/pp.104.057935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mandaokar A, Thines B, Byongchul S, Lange BM, Choi G, Koo YJ, Yoo YJ, Choi YD, Choi G, Browse J. Transcriptional regulators of stamen development in Arabidopsis identified by transcriptional profiling. Plant J. 2006;46:984–1008. doi: 10.1111/j.1365-313X.2006.02756.x. [DOI] [PubMed] [Google Scholar]
- 23.Redman JC, Haas BJ, Tanimoto G, Town CD. Development and evaluation of an Arabidopsis whole genome Affymetrix probe array. Plant J. 2004;38:545–561. doi: 10.1111/j.1365-313X.2004.02061.x. [DOI] [PubMed] [Google Scholar]
- 24.Berardini TZ, Mundodi S, Reiser R, Huala E, Garcia-Hernandez M, Zhang P, Mueller LM, Yoon J, Doyle A, Lander G, Moseyko N, Yoo D, Xu I, Zoeckler B, Montoya M, Miller N, Weems D, Rhee SY. Functional annotation of the Arabidopsis genome using controlled vocabularies. Plant Physiol. 2004;135:1–11. doi: 10.1104/pp.104.040071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Penterman J, Zilberman D, Huh JH, Ballinger T, Henikoff S, Fischer RL. DNA demethylation in the Arabidopsis genome. Proc Natl Acad Sci U S A. 2007;104:6752–6757. doi: 10.1073/pnas.0701861104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhu J, Kapoor A, Sridhar VV, Agius F, Zhu JK. The DNA glycosylase/lyase ROS1 functions in pruning DNA methylation patterns in Arabidopsis. Curr Biol. 2007;17:54–59. doi: 10.1016/j.cub.2006.10.059. [DOI] [PubMed] [Google Scholar]
- 27.Gong Z, Morales-Ruiz T, Ariza RR, Roldan-Arjona T, David L, Zhu JJ. ROS1, a repressor of transcriptional gene silencing in Arabidopsis, encodes a DNA Glycosylase/Lyase. Cell. 2002;111:803–814. doi: 10.1016/s0092-8674(02)01133-9. [DOI] [PubMed] [Google Scholar]
- 28.Agius F, Kapoor A, Zhu JK. Role of the Arabidopsis DNA glycosylase/lyase ROS1 in active DNA demethylation. Proc Natl Acad Sci U S A. 2006;103:11796–11801. doi: 10.1073/pnas.0603563103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hartung F, Plchova H, Puchta H. Molecular characterisation of RecQ homologues in Arabidopsis thaliana. Nucl Acid Res. 2000;28:4275–4282. doi: 10.1093/nar/28.21.4275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ascenzi R, Gantt JS. Molecular genetic analysis of the drought-inducible linker histone variant in Arabiidopsis thaliana. Plant Mol Biol. 1999;41:159–169. doi: 10.1023/a:1006302330879. [DOI] [PubMed] [Google Scholar]
- 31.Riechmann JL, Heard J, Martin G, Reuber L, Jiang C, Keddie J, Adam L, Pineda O, Ratcliffe OJ, Samaha RR, Creelman R, Pilgrim M, Broun P, Zhang JZ, Ghandehari D, Sherman BK, Yu G. Arabidopsis transcription factors: genome-wide comparative analysis among eukaryotes. Science. 2000;290:2105–2110. doi: 10.1126/science.290.5499.2105. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.