

Abstract
Dissociated cerebellar granule cells maintained in medium containing 25 mM potassium undergo an apoptotic death when switched to medium with 5 mM potassium. Granule cells from mice in which Bax, a proapoptotic Bcl-2 family member, had been deleted, did not undergo apoptosis in 5 mM potassium, yet did undergo an excitotoxic cell death in response to stimulation with 30 or 100 μM NMDA. Within 2 h after switching to 5 mM K+, both wild-type and Bax-deficient granule cells decreased glucose uptake to <20% of control. Protein synthesis also decreased rapidly in both wild-type and Bax-deficient granule cells to 50% of control within 12 h after switching to 5 mM potassium. Both wild-type and Bax −/− neurons increased mRNA levels of c-jun, and caspase 3 (CPP32) and increased phosphorylation of the transactivation domain of c-Jun after K+ deprivation. Wild-type granule cells in 5 mM K+ increased cleavage of DEVD–aminomethylcoumarin (DEVD-AMC), a fluorogenic substrate for caspases 2, 3, and 7; in contrast, Bax-deficient granule cells did not cleave DEVD-AMC. These results place BAX downstream of metabolic changes, changes in mRNA levels, and increased phosphorylation of c-Jun, yet upstream of the activation of caspases and indicate that BAX is required for apoptotic, but not excitotoxic, cell death. In wild-type cells, Boc-Asp-FMK and ZVAD-FMK, general inhibitors of caspases, blocked cleavage of DEVD-AMC and blocked the increase in TdT-mediated dUTP nick end labeling (TUNEL) positivity. However, these inhibitors had only a marginal effect on preventing cell death, suggesting a caspase-independent death pathway downstream of BAX in cerebellar granule cells.
Extensive cell death is an important part of the development and ongoing maintenance of tissues in multicellular organisms (Glucksman, 1951; Clarke and Clarke, 1996). The demise of many cells is modulated by members of the BCL-2 family of proteins (Korsmeyer, 1996; White, 1996). BCL-2 was first recognized as a modulator of cell death in studies of follicular B cell lymphoma in which overexpression of BCL-2 promotes transformation by rendering cells more resistant to apoptosis. The antiapoptotic activity of BCL-2 has been borne out in numerous cell types in response to a variety of stimuli (for review see Korsmeyer, 1996; White, 1996).
Several homologues of BCL-2 have been isolated, including some members of this family that are antiapoptotic, like BCL-2, and others that are proapoptotic (for review see Korsmeyer, 1996; White, 1996). BCL-2 family members that inhibit apoptosis include CED-9, MCL-1, A1, and E1B-19K. Family members that promote apoptosis include BAX, BAK, and BAD. BCL-X, another BCL-2 homologue, has both an antiapoptotic, long-splice variant, BCL-XL, and a proapoptotic, short-splice variant, BCL-XS. While the essential biochemical function of BCL-2 family members remains under investigation, their activity is regulated, in part, by dimerization among the various family members (Korsmeyer, 1996; Oltvai et al., 1993). In this rheostat model, the ratio of proapoptotic versus antiapoptotic molecules determines the fate of a cell. Dimerization among family members is mediated by three highly conserved domains, BH1, BH2, and BH3. BH1 and BH2 appear critical for antiapoptotic members (BCL-2 and BCL-XL) to heterodimerize with the proapoptotic BCL-2 family member, BAX (Yin et al., 1994), while the BH3 domain of BAX is essential for its heterodimerization and activity (Chittenden et al., 1995) .
Studies of mice deficient in BCL-X or BAX have highlighted these two family members as particularly important for the nervous system. Apoptosis in developing sensory and central nervous system neurons is greatly increased in mice deficient in Bcl-X (Motoyama et al., 1995). Dissociated sympathetic neurons from Bax-deficient animals are remarkably resistant to trophic factor deprivation–induced death, and motor neurons from the facial nucleus do not degenerate in response to axotomy (Deckwerth et al., 1996). In contrast, isolated thymocytes from Bax-deficient mice are not resistant to apoptosis (Knudson et al., 1995). In this study, we determined whether BAX was critical for cerebellar granule cell apoptosis.
Dissociated cerebellar granule cells from early postnatal rats can be maintained in serum-containing medium by elevating extracellular potassium levels (25 mM) (Gallo et al., 1987), or by adding low concentrations of N-methyl-d-aspartic acid (NMDA)1 to the culture medium (Balazs et al., 1988). Both low concentrations of NMDA and depolarization are presumed to mimic endogenous excitatory activity (Burgoyne et al., 1993); the survival promotion is mediated by increases in intracellular calcium (Gallo et al., 1987). Overstimulation of glutamate receptors on granule cells leads to an excitotoxic death (Schramm et al., 1990; Dessi et al., 1993; Lafon-Cazal et al., 1993). Dissociated cerebellar granule cells develop characteristics of mature cerebellar granule cells in vivo including an extensive neuritic network, expression of excitatory amino acid receptors, and production and release of l-glutamate (Burgoyne et al., 1993). Removal of both potassium and serum from dissociated cerebellar granule cells triggers a cell death that is morphologically apoptotic, accompanied by DNA fragmentation, and dependent on macromolecular synthesis (D'Mello et al., 1993; Nardi et al., 1997). This apoptotic cell death presumably mimics the naturally occurring death of 20–30% of granule cells (Caddy and Biscoe, 1979), which is important for matching the number of granule cells with Purkinje cells, that occur during the third through fifth postnatal weeks (Wetts and Herrup, 1983; Williams and Herrup, 1988).
In the current study, we demonstrate that BAX is required for programmed cell death (PCD), but not excitotoxic cell death, of cerebellar granule cells. We used this homogeneous population of BAX-dependent neurons to assess when BAX was acting in the program that commits a cell to die. We found that while Bax-deficient granule cells did not die, they did progress through several early changes associated with neuronal PCD (Deckwerth and Johnson, 1993; Estus et al., 1994; Freeman et al., 1994; Miller and Johnson, 1996) including decreases in protein synthesis and glucose uptake, and an increase in the mRNA level of the AP-1 transcription factor c-jun. In contrast to these events that do occur in both Bax +/+ and Bax −/− cells, we found that Bax −/− lysates did not cleave Ac-DEVD-aminomethylcoumarin (DEVD-AMC), a fluorogenic substrate for caspases 2, 3, and 7 that was cleaved by lysates from wild-type cells undergoing apoptosis. These results identify a central nervous system cell type that requires BAX to undergo PCD and clearly place BAX downstream of changes in glucose uptake, protein synthesis, and certain mRNA levels and upstream of the activation of a DEVD-selective caspase. We further investigated the role of the caspases in PCD of granule cells by using peptide inhibitors of caspases. Although these compounds blocked cleavage of DEVD-AMC completely and blocked the increase in TdT-mediated dUTP nick end labeling (TUNEL)–positivity, they only had a marginal effect on cell survival, suggesting the presence of caspase-independent cell death effectors downstream of BAX.
MATERIALS AND METHODS
Breeding and Genotyping of Mice with Different Gene Dosages of Bax
Mice heterozygous for Bax (Knudson et al., 1995) were mated to yield F1 offspring with Bax −/−, Bax +/−, and wild-type genotypes. At postnatal day 4–5, tail DNA was prepared and screened by PCR as described (Deckwerth et al., 1996).
Cell Culture Media
All culture media were based on Basal Medium Eagle (Life Technologies, Grand Island, NY) containing 100 U/ml penicillin and 100 μg/ml streptomycin. The following additions were made: K5+S medium, 10% dialyzed FBS 10,000 mol wt cutoff (Sigma Chemical Co., St. Louis MO); K25+S medium, 10% dialyzed FBS, 20 mM KCl; K25−S medium, 20 mM KCl; K5−S medium, no additions. Dialyzed serum was used because adding fresh medium containing nondialyzed serum to cerebellar granule cells is toxic. This sensitivity to nondialyzed serum develops after several days in culture because of the glutamate in the serum (Schramm et al., 1990).
Neuronal Culture
This cell culture protocol is a modification of previous protocols (Levi et al., 1984) and is extensively detailed in Miller and Johnson (1996). The only difference was that cerebella from Bax +/+, Bax +/−, Bax −/− were treated as separate, parallel dissections. In brief, cerebella were dissected from postnatal day seven (P7) mice, sliced into 1-mm pieces, and incubated at 37°C for 15 min in 0.30 mg/ml trypsin (Worthington Biochemical Corp., Freehold, NJ). The tissue was then triturated in K25+S medium with 0.5 mg/ml trypsin inhibitor (Sigma Chemical Co.) by using a flame-polished Pasteur pipette. The resulting cell suspension was spun at 500 g for 6 min. The pellet was gently triturated in fresh K25+S medium and filtered through a nitex filter (size 3-20/14; Tetko Inc., Elmsford, NY). Trypan blue exclusion was used to determine the number of the living neurons before plating: 2–2.5 × 105 cells/cm2 in either four-well (Nunc, Naperville, IL) or 35-mm dishes (Corning Inc., Corning, NY). Before plating, dishes were coated with 0.1 mg/ml poly-l-lysine (P2636; Sigma Chemical Co.). The granule cells were kept at 35°C in a humidified incubator with 5% CO2/95% air for 7 d. Fresh K25+S medium was added after 4 d in vitro. To reduce the number of nonneuronal cells, aphidicolin (3.3 μg/ ml, Sigma Chemical Co.) was added to the medium 24–36 h after initial plating. The culture conditions do not support the survival of other neuronal cell types (Thangnipon et al., 1983; Kingsbury et al., 1985); the nonneuronal contamination was 1–2% (Miller and Johnson, 1996).
Treatment of Cultures
For studies of potassium deprivation, phosphatidylinositol-3 kinase (PI-3-K) inhibition, or inhibition of caspases, at 7 d in vitro, culture medium was replaced with either K5−S or K5+S medium after washing cells once with the respective medium. Control cultures were treated identically with K25+S medium. Special care was taken to assure that all media had been preincubated at 35°C, 5% CO2/95% air for 18–24 h. 30 μM LY 294002 (Biomol Research Laboratories, Inc., Plymouth, PA) was added to K25+S medium to inhibit PI-3-K. Boc-aspartyl(OMe)-fluoromethylketone (BAF) and Z-VAD-fluoromethylketone were obtained from Enzyme Systems Products (Dublin, CA).
For NMDA stimulation, cells were treated at 10 d in vitro as described in Lafon-Cazal et al. (1993). Culture medium was removed and placed at 35°C, 5% CO2/95% air. Cultures were washed twice for 15 min at 35°C in Mg2+-free Locke's solution (154 mM NaCl, 5.6 mM KCl, 3.6 mM NaHCO3, 2.7 mM CaCl2, 5.6 mM d-glucose, 5 mM Hepes, pH 7.4) and then incubated at 35°C for 30 min in Mg2+-free Locke's solution with 3 μM glycine (Sigma Chemical Co.) and 10, 30, or 100 μM NMDA (Sigma Chemical Co.), or 100 μM NMDA + 150 nM MK-801 (Research Biochemicals International, Natick, MA). Cells were washed twice for 15 min with Mg2+-free Locke's solution at 35°C. The original culture medium was replaced and the cells were incubated at 35°C, 5% CO2/95% air for 24 h after which cell viability was assessed.
Determination of Cell Viability
Cell viability was quantified from photomicrographs of representative fields of cells labeled with calcein AM (Molecular Probes Inc., Eugene, OR). Calcein AM is an acetoxymethyl ester fluorescein derivative, which is cleaved and trapped inside viable cells that have nonspecific esterase activity (Bozyczko et al., 1993). The photomicrographs were both taken and scored by an observer naive to the experimental condition and to the genotype of the animal from which the culture was derived. Six photomicrographs at 200 magnification were taken for each condition and the number of calcein AM–positive cells was counted. This method of quantifying cell viability is detailed and validated in Miller and Johnson (1996).
Metabolic Parameters
Experiments were performed in four-well dishes (Nunc) with ∼400,000 cells per well. After 7 d in vitro, culture medium was replaced with K5+S or K25+S medium (as described above). Detailed description of the following methods may be found in Deckwerth and Johnson (1993) and Miller and Johnson (1996).
Rate of Protein Synthesis.
Neuronal cultures were labeled for 1 h at 35°C with 10 μCi/ml l-[4,5-3H]leucine (159 Ci/mmol; Amersham Life Science, Inc., Arlington Heights, IL) in K25+S or K5+S medium containing 10 μM unlabeled l-leucine. Cultures were lysed, precipitated with 10% trichloroacetic acid (TCA), filtered, and counted in a liquid scintillation counter (Beckman Instrs., Fullerton, CA).
Rate of 2-deoxyglucose Uptake.
Neuronal cultures were labeled for 30 min at 35°C with 2.5 μCi/ml [1,2-3H]2-deoxy-d-glucose (30 Ci/mmol; ICN Biomedicals, Inc., Irvine,CA) in K25+S or K5+S medium containing 500 μM d-glucose. Cultures were washed three times, lysed, and added directly to liquid scintillation fluid and counted. Assays were linear with respect to time during the indicated measuring period (data not shown).
RT-PCR
Semiquantitative reverse transcription-PCR (RT-PCR) assays are based on those used for sympathetic neurons described in Freeman et al. (1994) and Estus et al. (1994), and extensively detailed by Estus (1997). Briefly, granule cells were switched to K5+S for the indicated times. Polyadenylated (poly-A) RNA was isolated from 400,000 cerebellar granule cells by using an oligo-dT-cellulose mRNA purification kit as directed by the manufacturer (QuickPrep Micro Kit; Pharmacia LKB Biotechnology Inc., Piscataway, NJ). Half of the poly-A RNA was converted to cDNA by reverse transcription (RT) with Moloney murine leukemia virus reverse transcriptase with random hexamers (16 μM) as primers. cDNA from ∼4,000 cells was used in a 50 μl PCR reaction. After amplification, the PCR products were separated by electrophoresis on 10% polyacrylamide gels, visualized by autoradiography of the dried gels, and quantified with a PhosphorImager (Molecular Dynamics, Inc., Sunnyvale, CA). Preliminary experiments with cerebellar granule cell cultures validated that the RT-PCR technique was linear with respect to the amount of input RNA used for RT and with respect to the amount of cDNA used for PCR within the ranges used in these experiments. No product was amplified when purified RNA was used as input for a PCR reaction. Results were repeated in at least two independent RNA preparations. The sequences of the PCR products were confirmed (Estus et al., 1994; Freeman et al., 1994; data not shown). Primer sequences for cyclophilin and cyclin D1 are reported in Freeman et al. (1994). Sequences for c-fos and c-jun are reported in Miller and Johnson (1996). Bax primer sequences are detailed in Greenlund et al. (1995). Primer sequences for CPP32 (EMBL/GenBank/DDBJ accession number D86352), ICH-1 (U13022), and GFAP (X02801) are as follows (5′ to 3′): CPP32 (−569) AGA GTA AGC ATA CAG GAA GTC GGC; CPP32(+351) GAT TCT AAG TCA TGG AGA TGA AGG; ICH1 (fwd) GGT TGA GAT GGC AAA CTG CT; ICH1 (rev) CCA GCA TCA CTC CC TCA CA; GFAP (+2092) CAA TGG AGT TGG AAG TTG TAG GC; and GFAP (−2245) GAT AGA CCT TCA CAA CTG AGA CG.
DEVD-AMC Cleavage Assay
DEVD-AMC cleavage was measured essentially as described (Armstrong et al., 1997). After 0, 4, 8, 12, 24, or 48 h in K5+S medium, 400,000 granule cells were washed once with PBS and lysed in 100 μl of buffer A (10 mM Hepes, pH 7.4, 42 mM KCl, 5 mM MgCl2, 1 mM DTT, 0.5% CHAPS, 1 mM PMSF, 1 μg/ml leupeptin). In a 96-well plate, 25 μl of lysate was combined with 75 μl of buffer B (25 mM Hepes, 1 mM EDTA, 0.1% CHAPS, 10% sucrose, 3 mM DTT, pH 7.5) containing 30 μM Ac-DEVD-AMC (Biomol Research Laboratories, Plymouth, PA) and incubated for 20 min at room temperature in the dark. Fluorescence was measured at excitation 360 nm and emission 460 nm in a fluorescent plate reader (Titertek Fluoroskan II; Flow Laboratories, Inc., McLean, VA). 20 min and 25 μl were both in the linear range of the assay with respect to time and amount of lysate added.
Although cultures were extensively washed before lysing cells, we were concerned that residual BAF or ZVAD from the culture medium might account for the block in DEVD-AMC cleavage in our enzyme assay. To test this possibility, we combined 8-h K+ deprivation lysates from BAF- or ZVAD-treated cultures with 8-h K+ deprivation lysates from untreated cultures and determined DEVD-AMC cleavage. We found that the 8-h lysates from the BAF- or ZVAD-treated cultures did not themselves contain any ability to inhibit the DEVD-AMC cleavage activity in our assay, demonstrating that the BAF and ZVAD in the culture medium had been adequately washed out before lysis (data not shown).
Transfections of Cerebellar Granule Cells
All constructs were under the cytomegalovirus (CMV) promoter. pGreen Lantern-1 (green fluorescent protein) was obtained from GIBCO BRL (Gaithersburg, MD). The BCL-2 construct is detailed in Greenlund et al. (1995a). The p35 construct was a generous gift from Dr. Lois K. Miller. pBluescript II was used as control DNA (Short et al., 1988). Granule cells were transfected essentially as described in Xia et al. (1995). In brief, after 5 d in vitro, cultures were switched to K25−S medium for 1 h after which DNA/CaCl2 precipitates were prepared as follows: An equal volume of solution containing 0.25 M CaCl2, 67 μg/ml pGreen Lantern-1 DNA, and 67 μg/ml of either Bax, BCL-2, or p35 DNA was added to 2× Hepes-buffered saline (274 mM NaCl, 10 mM KCl, 1.4 mM Na2HPO4-7H20, 15 mM dextrose, 42 mM Hepes-free acid, pH 7.07) and incubated in the dark at room temperature for 25 min. 30 μl of the CaCl2 precipitate (1 μg of DNA) was added dropwise to each well of a Nunc four-well plate containing ∼400,000 granule cells and incubated for 1 h at 37°C. Cells were washed twice with K25−S medium and returned to K25+S medium for 2 d after which the number of transfected cells was assessed by counting the number of pGreen Lantern-1 (fluorescent) cells (∼200) in a defined area for two separate wells of a Nunc four-well dish. Transfection efficiency was typically 0.2%. Cells were then switched to K5+S or K25+S medium, and the number of fluorescent cells was again assessed for the same defined area after 24 and 48 h and compared to the original number for a particular well. All cultures were counted by a naive observer. pGreen Lantern-1 positivity correlated well with a healthy, viable cell as assessed by phase-contrast microscopy, nuclear morphology, and exclusion of trypan blue. Preliminary experiments using both pGreen Lantern-1 and LacZ as markers of transfection demonstrated that all cells that were pGreen Lantern-1–positive were also LacZ-positive and vice versa. In addition, from the earliest time that pGreen Lantern-1 could reliably be detected (24 h), expression of the constructs was not toxic to the cells up to 9 d after transfection.
Phospho-Jun Staining
Neuronal cultures were washed once with PBS, pH 7.4, before a 30-min fixation with 4% paraformaldehyde in PBS at 4°C. Neuronal cultures were washed three times with TBS (100 mM Tris, 0.9% NaCl, pH 7.6), blocked for 30 min in TBS containing 5% goat serum (Sigma Chemical Co.) and 0.3% Triton X-100 at room temperature, and incubated overnight at 4°C with anti–phospho–c-jun antibody (Ser 63; New England Biolabs Inc., Beverly, MA) diluted 1:200 in TBS containing 1% goat serum and 0.3% Triton X-100. Cultures were washed three times with TBS then incubated overnight at 4°C in a 1:400 dilution of 1.5 mg/ml Cy3-donkey anti–rabbit antibody (Jackson Immunoresearch Laboratories, Inc., Westgrove, PA) in the same buffer as the primary antibody. Cultures were washed twice with TBS, stained with 1 μg/ml bisbenzimide (Hoechst 33258; Molecular Probes Inc., Eugene, OR) for 20 min to visualize nuclei, then washed two additional times with TBS. The phospho-jun antibody does not react with the nonphosphorylated form of c-Jun and does not appreciably cross-react with the phosphorylated form of JunD or JunB (New England Biolabs Inc.).
TUNEL Staining
At 7 d in vitro, culture medium from cerebellar granule cells was replaced with either K5+S, K5+S plus 100 μM BAF, or fresh K25+S after washing the cells once with the appropriate medium. At 24 h after this treatment, cells were examined for DNA fragmentation using the In Situ Cell Death Detection Kit (Fluorescein; Boehringer Mannheim Biochemicals, Indianapolis, IN) according to the manufacturer's instructions. In brief, cells were fixed for 30 min in 4% paraformaldehyde/PBS at room temperature, washed once with PBS, and permeabilized in 0.1% Triton X-100, 0.1% sodium citrate for 20 min at 4°C. After two more washes with PBS, the cells were incubated with 50 μl of the TUNEL reagent for 1 h at 37°C in the dark. Cells were then washed twice with PBS, stained with 1 μg/ml Hoechst 33258 (Molecular Probes Inc.), and washed twice more with PBS. The percentage of TUNEL-positive cells was calculated as the number of TUNEL-positive cells divided by the number of Hoechst-stained cells. For each condition, a naive observer counted two to five fields, each containing over 200 bisbenzimide-stained neurons. Results represent mean ± range for two independent experiments.
RESULTS
Bax −/− Neurons are Resistant to Apoptosis
Dissociated cerebellar granule cells maintained in 25 mM K+ and serum undergo a PCD that is apoptotic if they are deprived of both potassium and serum (K5−S medium) (D'Mello et al., 1993). To determine whether BAX, a proapoptotic BCL-2 family member, was important in cerebellar granule cell apoptosis, we used cultures of dissociated cerebellar granule cells from P7 wild-type and Bax-deficient mice (Knudson et al., 1995). After 7 d in vitro, cultures of Bax +/+, and Bax −/− cerebellar granule cells were switched to medium containing low potassium and serum (K5+S) or maintained in high potassium and serum (K25+S). After 72 h, cultures were stained with calcein AM, which stains living cells. Phase-contrast images and the corresponding calcein AM photomicrographs are presented in Fig. 1. While virtually all of the Bax +/+ cells died by 72 h (compare Fig. 1, a and b with e and f ), granule cells from Bax −/− mice did not undergo apoptosis (compare Fig. 1, c and d with g and h). To determine the number of surviving cells, we counted the number of neurons on photomicrographs of cultures stained with calcein AM after 0, 12, 24, 48, or 72 h in K5+S medium (Fig. 2). All the granule cells from Bax −/− mice were protected from cell death (Fig. 2, open triangles) while more than 90% of the wild-type granule cells died by 72 h (Fig. 2, open circles). Granule cells from heterozygous animals died completely by 72 h (Fig. 2, open squares), though the time course of death was slightly slower. Nuclei from Bax −/− cultures deprived of K+ for 72 h were indistinguishable from control cultures maintained in K25+S medium (data not shown), while Bax +/+ and Bax +/− cultures displayed characteristic apoptotic nuclear changes 6 h after switching to K5+S medium (data not shown).
Figure 1.

Bax −/− cerebellar granule cells do not undergo apoptosis in response to K+ deprivation. Cerebellar granule cells from Bax +/+ (a, b, e, and f) and Bax −/− (c, d, g, and h) were maintained for 7 d in vitro and then switched to K25+S medium (a–d) or K5+S medium (e–h). Photomicrographs of phase contrast (a, c, e, and g) and the corresponding calcein AM–stained images (b, d, f, and h) were taken 72 h after the media were switched. Bar, 20 μm.
Figure 2.

Bax-deficient granule cells are fully protected from K+ or K+/serum deprivation-induced death. After 7 d in vitro, granule cells were switched to either K5+S (open circle, square, and triangle) or K5-S medium (closed circle, square, and triangle) for 12, 24, 48, or 72 h. Survival was assessed by counting neurons in photomicrographs of calcein AM-stained cultures and compared to cultures maintained in K25+S. Circles, squares, and triangles represent Bax +/+, Bax +/−, and Bax −/− cultures, respectively. Data represent mean ± range for two independent experiments.
In a more rigorous test of the ability of Bax −/− granule cells to resist apoptosis, we also determined the time course of death after both K+ and serum were removed (K5−S). Again, Bax −/− granule cells did not die while Bax +/+ and Bax +/− cells died completely by 72 h (Fig. 2); the death in Bax +/+ and Bax +/− was slightly faster when both serum and potassium were removed. Because removing K+ alone was an inherently simpler model in which only one source of trophic support was removed, this paradigm was used for all subsequent experiments.
We have previously shown that PI-3-K activity is increased by K+ depolarization (Miller et al., 1997). This increase appears critical to the survival of depolarized neurons as shown by the ability of two inhibitors of PI-3-K to produce death indistinguishable from that caused by removal of high K+ (Miller et al., 1997). Because BCL-2 family members have been recently implicated in signaling pathways (Gajewski and Thompson, 1996; Wang et al., 1996; Zha et al., 1996), we directly tested the ability of Bax −/− cells to survive in the absence of PI-3-K activity. Neurons maintained for 7 d in vitro were switched to K25+S with 30 μM LY 294002, an inhibitor of PI-3-K activity. Viability was assessed after 48 h by calcein AM staining (Fig. 3). Similar to previous results (Miller et al., 1997), LY 294002 blocked the survival-promoting activity of K+ in Bax +/+ cells. Results from cultures of animals heterozygous for Bax were similar to those from wild type. In contrast, neurons from Bax −/− cultures survived in the presence of LY 294002, implying that Bax deficiency blocked apoptosis downstream of PI-3-K.
Figure 3.

Inhibition of PI-3-K does not induce apoptosis in Bax −/− granule cells. After 7 d in vitro, granule cells were switched to K25+S medium in the presence of 30 μM LY 294002, an inhibitor of PI-3-K. Control cells were switched to K25+S medium. Survival was determined by counting neurons in photomicrographs of calcein AM–stained cultures 48 h after treatment. Data represent mean ± range for two independent experiments.
BAX −/− NEURONS ARE NOT PROTECTED FROM EXCITOTOXIC DEATH
Overexpression of BCL-2 is neuroprotective in some models of stroke and excitotoxic injury (Linnik, 1996). Therefore, we tested whether the absence of BAX would be protective in an NMDA model of excitotoxic death in cerebellar granule cells. After 10 d in vitro, cultures from Bax +/+ and Bax −/− were stimulated with 10, 30, or 100 μM NMDA for 30 min and viability was assessed 24 h later by calcein AM staining (Fig. 4). In contrast to K+ deprivation or treatment with an inhibitor of PI-3-K, the absence of Bax was not neuroprotective in this excitotoxic paradigm. The lack of death in cultures simultaneously exposed to 100 μM NMDA and 150 nM MK-801, a highly potent noncompetitive NMDA receptor antagonist (Wong et al., 1986), demonstrated that the death was specific to stimulation of NMDA receptors.
Figure 4.

Bax-deficient granule cells are not protected from NMDA-induced excitotoxic cell death. After 10 d in vitro, granule cells were stimulated with 0, 10, 30, and 100 μM NMDA, or 100 μM NMDA with 150 nM MK-801 for 30 min at 37°C. Survival was assessed after 24 h by counting neurons in photomicrographs of calcein AM–stained cultures. Data represent mean ± range for two independent experiments.
BAX Is Not Required for the Early Decrease in Metabolic Parameters
The experiments described above indicate that BAX is required for mediating apoptosis in cerebellar granule cells. We were able to use this granule cell culture model to gain insight into where BAX functions in the cell death pathway. One of the earliest changes in cerebellar granule cells undergoing PCD is a dramatic decrease in metabolic parameters such as glucose uptake and protein synthesis (Miller and Johnson, 1996; Nardi et al., 1997). We measured glucose uptake and protein synthesis in K+-deprived granule cells from Bax −/− animals to determine at what point Bax −/− cells are arrested in the cell death program and to provide an indication of the metabolic status of these cells. Cultures were switched to K5+S for 2, 6, 12, 24, or 48 h, and metabolic parameters were measured. The dramatic early fall in glucose uptake was seen in cells from Bax −/− animals; 2-deoxyglucose uptake fell to <20% of control within 2 h of K+ deprivation (Fig. 5 A). The early fall in protein synthesis, as measured by uptake of l-[4,5- 3H]leucine, was also observed in Bax −/− cultures (Fig. 5 B). However, Bax −/− cells did maintain a low basal level of protein synthesis even after 48 h of K+ deprivation. Therefore, BAX was not required for the early fall in metabolic parameters. Consistent with a decrease in metabolic parameters, granule cells from K+-deprived Bax −/− cultures, while alive, ceased to increase in diameter compared to those maintained in 25 mM potassium which continued to grow (Miller, T.M., personal observation). A similar relative decrease in soma diameter is seen in Bax −/− sympathetic neurons deprived of NGF (Deckwerth et al., 1996).
Figure 5.
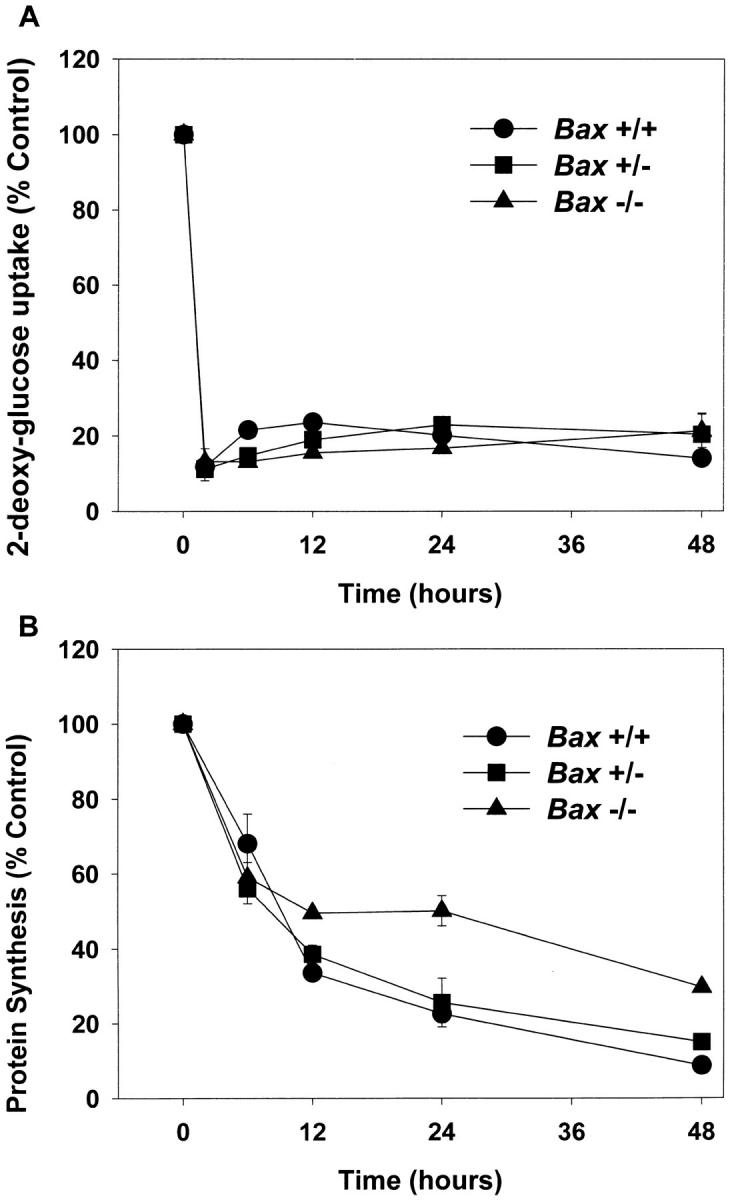
BAX deletion does not alter changes in metabolic parameters associated with PCD. (A) Glucose uptake was determined 2, 6, 12, 24, or 48 h after switching granule cell cultures to K5+S medium by measuring the incorporation of [1, 2-3H]2-deoxy-d-glucose and compared to control cultures that were switched to K25+S medium. (B) Protein synthesis was determined after 6, 12, 24, or 48 h by measuring the incorporation of l-[4, 5-3H]leucine and compared to control cultures that were switched to K25+S medium. Data represent mean ± range for two independent experiments.
BAX Is Not Required for Changes in mRNA Levels Associated with PCD
PCD of many cell types (Freeman et al., 1993), including cerebellar granule cells (D'Mello et al., 1993), is markedly attenuated by inhibitors of macromolecular synthesis, implying that transcription and translation of new gene products are important during cell death in these cells. Though most mRNA levels decline rapidly as cells die, we have previously identified increases in the mRNA levels of several genes during programmed cell death in granule cells and sympathetic neurons (Estus et al., 1994; Freeman et al., 1994; Miller and Johnson, 1996). The increase in the mRNA level of the AP-1 transcription factor c-Jun may be particularly important for cell death because microinjection of a dominant negative c-jun construct (Ham et al., 1995) or neutralizing antibodies directed against c-Jun (Estus et al., 1994) significantly delays PCD of sympathetic neurons.
Whether the increase in c-jun mRNA and presumably the action of c-jun is downstream or upstream of BAX is unknown. To address this issue and examine other message levels, cultures from Bax +/+ and Bax −/− animals were switched to K5+S for 1, 3, 6, 9, 12, 18, 24, 48, or 72 h and mRNA was isolated, reverse transcribed, and selectively amplified by PCR. mRNA levels of cyclophilin declined rapidly in wild-type granule cells (Fig. 6, A and B); actin and neuron-specific enolase were similar to cyclophilin (data not shown). mRNA levels of these same messages in Bax −/− cultures declined far less than in wild-type cultures reflecting the lack of death of Bax −/− cells (Fig. 6, A and C). Similar to results seen in rat cerebellar granule cells (Miller and Johnson, 1996), mRNA levels for c-jun increased approximately fourfold in K+-deprived wild-type granule cells (Fig. 6 B). c-jun mRNA also increased in K+-deprived, Bax-deficient granule cells (Fig. 6 C), demonstrating that increases in c-jun were upstream of Bax.
Figure 6.



mRNA levels of c-jun and CPP32 increase in both Bax +/+ and Bax −/− granule cells after K+ deprivation. Cultures were switched to K5+S and cDNA was prepared from granule cells after 1, 3, 6, 9, 12, 18, 24, 48, or 72 h. Control cultures were maintained in K25+S medium. cDNA from ∼4,000 cells was used in a 50-μl PCR reaction as described in Materials and Methods. Identical results were obtained in an independent neuronal preparation. (A) PCR products. (B) PhosphorImager quantitation of Bax +/+ PCR products. (C) PhosphorImager quantitation of Bax −/− PCR products. Time 0 was used as control level for B and C.
c-fos mRNA levels increase in dying sympathetic neurons and the Fos family of proteins may be important for cell death in sympathetic neurons (Estus et al., 1994). Examination of c-fos mRNA levels in K+-maintained granule cells was uninformative since K+ depolarization itself drives the expression of c-fos (Ghosh et al., 1994). cyclin D1, a cell cycle regulator that increases in dying sympathetic neurons (Freeman et al., 1994), remained relatively constant in both wild-type and Bax-deficient animals (Fig. 6 A). Since this message level did not decrease in the wild-type cultures even after 120 h of K+ deprivation (data not shown) when very few, if any, granule cells remained viable, we concluded that the majority of the cyclin D1 message level is in the few contaminating nonneuronal cells; therefore, we could not determine whether cyclin D1 increased in the neurons. Glial fibrillary acidic protein (GFAP) is expressed in the astrocytes, which are the main contaminating nonneuronal cell. GFAP, as expected, remained relatively constant as the neurons died. Bax mRNA levels did not increase in wild-type granule cells undergoing apoptosis and, as expected, no Bax mRNA was expressed in Bax −/− cells (Fig. 6 A).
mRNA levels of caspase 3 (CPP32) increased in both wild-type and Bax-deficient cultures deprived of K+ (Fig. 6). Caspase 3 has been clearly implicated in several cell death paradigms (for review see Henkart, 1996) and in developmental neuronal cell death (Kuida et al., 1996), although whether increases in mRNA levels of caspases have any significance for cell death is unclear. The mRNA level of at least one other caspase, caspase 2 (Ich-1), was not increased after K+ deprivation (data not shown).
c-Jun Is Phosphorylated on Ser 63 in Both Bax +/+ and Bax −/− Cultures
In addition to increases in mRNA levels, another indication of an increase in c-Jun activity is the phosphorylation of c-Jun on serines 63 and 73 of the transactivation domain (Binetruy et al., 1991; Smeal et al., 1991, 1992). Therefore, we determined the phosphorylation status of the c-Jun transactivation domain during PCD by immunostaining with an antibody that specifically recognizes the serine 63– phosphorylated form of c-Jun (Fig. 7). Cells maintained in K25+S medium (Fig. 7, a and e) had almost no staining. After 6 h of K+ deprivation, wild-type granule cells showed significant phosphorylation of c-Jun on serine 63 (Fig. 7 c). Similar to the parallel increases in c-jun mRNA levels in Bax +/+ and Bax −/− cultures, phospho-Jun staining also increased in Bax −/− granule cells deprived of K+ for 6 h (Fig. 7 g). Staining was not increased in the few nonneuronal cells in these cultures (data not shown). In nuclei counterstained with bisbenzimide (Fig. 7, b, d, f, and h), some K+-deprived, wild-type cells showed nuclear margination and chromatin condensation at 6 h (Fig. 7 d). The increase in c-Jun phosphorylation and c-jun mRNA in both wild-type and Bax −/− cultures indicates that BAX functions downstream of the increase in c-Jun activity that is associated with PCD.
Figure 7.

c-Jun phosphorylation increases in both Bax +/+ and Bax −/− granule cells after K+ deprivation. Bax +/+ (a–d) and Bax −/− (e–h) cultures were switched to K25+S (a, b, e, and f) or K5+S (c, d, g, and h) medium for 6 h and immunostained with an antibody that specifically recognizes the ser-63 phosphorylated form of c-Jun (a, c, e, and g) and stained with the nuclear dye bisbenzimide (b, d, f, and h). Bar, 5 μm.
BAX Is Required for Increases in Caspase Activity
Caspases have been implicated in several cell death models (for review see Henkart, 1996; Schwartz and Milligan, 1996). We examined whether caspases 2, 3, and 7 were activated after K+ deprivation. The activity of this subset of proteases can be measured by monitoring the cleavage of the fluorogenic substrate DEVD-AMC. In Bax +/+ and Bax +/− cells, DEVD-selective caspase activity increased to 18 times control levels by 8 h after K+ deprivation and then decreased as the cells died over the next 64 h. In stark contrast, granule cells from Bax −/− animals did not increase DEVD-specific caspase activity at any time after K+ deprivation (Fig. 8). These results indicate that BAX is required for activation of this subset of caspases in response to removal of potassium.
Figure 8.

BAX is required for increases in caspase activity. Cultures were switched to K5+S medium, lysed after 4, 8, 12, 24, 48, or 72 h, and cleavage of DEVD-AMC was determined. Control cultures were switched to K25+S medium and treated identically. Data represent mean ± SD for triplicate measurements from one experiment and are representative of two additional independent experiments.
Blocking Caspases Delays but Does Not Block Cell Death
Inhibiting caspases with peptide inhibitors blocks PCD in several models (Henkart, 1996; Schwartz and Milligan, 1996). For example, BAF or ZVAD-FMK, two inhibitors of caspases, have dramatic effects on saving sympathetic neurons from PCD induced by trophic factor deprivation (Deshmukh et al., 1996). We examined the effect of these compounds on K+ deprivation–induced cell death in granule cells. Neither compound was toxic to granule cells. After 3 d exposure to either 100 μM ZVAD or 100 μM BAF in K25+S medium, survival was 96 ± 1 and 103 ± 3%, respectively. In K5+S medium, while both BAF and ZVAD were able to delay PCD in granule cells at 12 h, neither afforded significant protection at later time points (Fig. 9 A). One possible explanation for the lack of protection was that BAF and ZVAD did not function as caspase inhibitors in these cells, perhaps because they did not effectively cross the plasma membrane. To rule out this possibility, we deprived cells of K+ in the presence of 100 μM BAF or 100 μM ZVAD and then measured caspase activity. Cells treated with either compound showed no increase in DEVD-AMC cleavage after K+ deprivation (Fig. 9 B), demonstrating that BAF and ZVAD did indeed penetrate granule cells and function as caspase inhibitors in these cells.
Figure 9.

Inhibitors of caspases do not block death, but do block DEVD-AMC cleavage. Cultures were switched to K5+S, K5+S plus 100 μM BAF, or K5+S plus 100 μM ZVAD-FMK. Control cultures were switched to K25+S medium. (A) After 12, 24, or 48 h neuronal survival was determined by calcein AM staining. (mean ± SD, N = 3 experiments) (B) After 8, 12, 18, 24, or 48 h cultures were lysed and assayed for DEVD-AMC cleavage. Fluorescence was measured after 20 min at room temperature (mean ± range, N = 2 experiments).
Further evidence that BAF was able to function inside cells was found in a TUNEL assay of K+-deprived granule cells. TUNEL serves as an in situ marker of DNA fragmentation (Gavrieli et al., 1992), one of the hallmarks of apoptosis. The number of TUNEL-positive cells was determined in cells switched to K25+S, K5+S, or K5+S with 100 μM BAF (Fig. 10, d–f, respectively). To facilitate visualization of the total number of cells, the nuclei were stained with bisbenzimide (Fig. 10, a–c). As expected, the percent of TUNEL-positive cells increased markedly after switching cells from K25+S to K5+S (Fig. 10, d and e) from 14 to 84% (Fig. 10 g). Although BAF did not block cell death (Fig. 9 A), BAF blocked the increase in TUNEL positivity associated with depriving granule cells of potassium (Fig. 10, f and g).
Figure 10.


Cell death in the presence of BAF is TUNEL-negative. Cultures were switched to K5+S (b and e) or K5+S plus 100 μM BAF (c and f). Control cultures were switched to K25+S medium (a and d). After 24 h, cultures were fixed, stained with the TUNEL reagent (d–f), and stained with bisbenzimide (a–c) as described in Materials and Methods. The percent TUNEL-positive cells is the number of TUNEL-positive cells divided by the number of bisbenzimide-stained cells (g). Data represent the mean ± range for two independent experiments.
A second method we used to inhibit caspase activity was expression of p35, a baculoviral protein. Recombinant p35 blocks the enzymatic activity of purified ICE, Ich-1, Ich-2, and CPP32 (caspases 1, 2, 4, and 3, respectively) (Bump et al., 1995; Xue and Horvitz, 1995). Expression of p35 delays PCD in NGF-deprived sympathetic neurons (Martinou et al., 1995) and a serum-deprived neuronal cell line (Rabizadeh et al., 1993), and also blocks developmental cell death in Caenorhabditis elegans (Sugimoto et al., 1994; Xue and Horvitz, 1995) and Drosophila (Hay et al., 1994). After 5 d in vitro, we transfected pGreen Lantern-1 in combination with constructs encoding p35, BCL-2, or the pBluescriptII plasmid into cerebellar granule cells (see Materials and Methods). At 7 d, the number of transfected cells was determined by counting pGreen Lantern-1–transfected (fluorescent) cells. Cultures were then switched to K5+S medium or maintained in K25+S medium. The number of surviving transfected cells was determined after 24 and 48 h and compared to the number of transfected cells before switching medium (Fig. 11). Although expression of BCL-2 significantly retarded cell death in K5+S medium, expression of p35 had no effect on cell death (Fig. 11). These data further suggest a caspase-independent cell death pathway downstream of BAX.
Figure 11.

p35 does not block cell death. After 5 d in vitro, granule cells were transfected, as described in Materials and Methods, with pGreen Lantern-1 and either pBluescriptII, a BCL-2 construct, or a p35 construct. At 7 d in vitro, the number of pGreen Lantern-1–positive (fluorescent) cells was determined before switching cells to K25+S or K5+S medium. The number of pGreen Lantern-1–positive cells was again determined after 24 and 48 h and compared to the original number of pGreen Lantern-1–positive cells before the medium was switched. Data represent mean ± SEM for four independent experiments. *indicates significance at P < 0.05 in a paired t test in which the data passed a test for normality.
DISCUSSION
In this study, we demonstrated that PCD in cerebellar granule cells induced by K+ deprivation, K+/serum deprivation, or inhibition of PI-3-K requires BAX. In contrast, BAX was not required for excitotoxic cell death in granule cells since we found that Bax −/− granule cells were not protected from NMDA-induced cell death. Because Bax −/− granule cells did not die in response to K+ deprivation, we were able to use this model to define where BAX is acting in the pathway leading to apoptosis. We analyzed several metabolic and genetic events associated with PCD in granule cells and found that Bax deficiency did not affect these early events. On the other hand, we found that BAX was required for the activation of caspases since Bax +/+, but not Bax −/− cells cleaved a fluorogenic DEVD substrate in response to K+ deprivation. Our data suggest that while likely to be involved in cell death, caspases may not be solely responsible for causing cell death in granule cells since blocking these proteases with peptide inhibitors delayed, but did not significantly block, the loss of cells. These studies of Bax-knockout cells are consistent with BAX or BAK induction studies in which cell death also proceeded in the presence of caspase inhibitors (Xiang et al., 1996; McCarthy et al., 1997). From these results and previous studies, we are now able to define a sequence of events associated with PCD in granule cells (Fig. 12).
Figure 12.

Diagrammatic representation of the events associated with programmed cell death in cerebellar granule cells.
Bax Deletion Does Not Appear to Result in Activation of Upstream Signal Transduction Pathways
Survival-promoting agents, such as IGF-I and K+, are likely to block the initial activation of the cell death program by activating intracellular signaling pathways. We have previously shown that both K+ and IGF-I activate two intracellular signaling pathways, PI-3-K and mitogen-activated protein (MAP) kinase in these cells (Miller et al., 1997). MAP kinase is not likely to be regulating survival directly since inhibition of this pathway does not lead to cell death in granule cells (Miller et al., 1997) or sympathetic neurons (Creedon et al., 1996; Virdee and Tolkovsky, 1996). In contrast, inhibition of PI-3-K in granule cells in the presence of IGF-I or K+ induces cell death that is indistinguishable from potassium deprivation, including a similar time course of death, dependence on macromolecular synthesis, and characteristic morphology of apoptotic nuclei (Miller et al., 1997). Further support for the importance of the PI-3-K pathway in granule cells is the survival-promoting effect of overexpressing AKT (Dudek et al., 1997), a downstream target of PI-3-K (Bos, 1995; Franke et al., 1995). The ability of Bax −/− granule cells to survive without PI-3-K activity and the fact that metabolic changes and changes in mRNA levels still occur in Bax −/− cells implies that BAX lies downstream of both.
BAX Is Not Required for Changes in Metabolic Parameters or mRNA Levels
The early, dramatic fall in metabolic parameters that we saw in Bax −/− cultures is part of the cell death program in cerebellar granule cells (Miller and Johnson, 1996; Nardi et al., 1997), sympathetic neurons (Deckwerth and Johnson, 1993), and thymocytes (Makman et al., 1966; Munck, 1968; Cidlowski, 1982). Thus, we have hypothesized that these events may represent an early trigger for PCD. The early decreases in glucose uptake and protein synthesis rate were not affected by the Bax genotype. This suggests that the actions of BAX are not related to early signal transduction pathways mediating neuronal survival by trophic signals. This conclusion is further supported by the inability of PI-3-K inhibitors to induce cell death in Bax −/− cells. It is also consistent with previous observations that overexpression of BCL-2, while greatly retarding sympathetic neuronal death after NGF deprivation, has no effect on the decrease in protein synthesis that presages that death (Greenlund et al., 1995b ).
The deletion of BAX also failed to block changes in mRNA levels, another step in the cell death program; c-jun mRNA increased in both Bax +/+ and Bax −/− cultures. Several reports have suggested that c-Jun may be important for cell death. Microinjection of neutralizing antibodies to c-Jun or a dominant negative c-jun construct delays the death of NGF-deprived sympathetic neurons (Estus et al., 1994; Ham et al., 1995). Dominant negative c-jun constructs also block cell death in human monocytic leukemia cells (U937) treated with ceramide (Verheij et al., 1996). An increase in c-Jun is evidently part of cell death in vivo. c-Jun increases in granule cells undergoing PCD in the weaver mouse (Gillardon et al., 1995) and in irradiated animals (Ferrer et al., 1996). c-Jun amino-terminal kinase (JNK) (Hibi et al., 1993) may be the kinase responsible for the increased phosphorylation on serine 63 of c-Jun in both Bax +/+ and Bax −/− cells (Fig. 7). Several reports have implicated this pathway during cell death (Xia et al., 1995; Chen et al., 1996; Latinis and Koretzky, 1996; Park et al., 1996; Verheij et al., 1996; Wilson et al., 1996). The increase in c-Jun both in vitro and in vivo, the fact that blocking c-Jun delays cell death, and increases in c-Jun phosphorylation during PCD imply that an increase in c-Jun is an important early occurrence during PCD. In PC12 cells, overexpression of Bcl-2 is able to block the increase in JNK activity associated with PCD, suggesting that the BCL-2 family functions upstream of c-Jun (Park et al., 1996). Our loss of function study with BAX is in apparent disagreement with these data. In cerebellar granule cells, the increase in c-jun message level and the phosphorylation of c-Jun are clearly upstream of BAX (Figs. 6 and 7).
Caspase Activity
We examined the role of caspases in PCD in granule cells by determining changes in mRNA levels of caspase 3 (CPP32) and increases in the activity of DEVD selective caspases. K+ deprivation increased caspase 3 mRNA levels in Bax +/+ and Bax −/− cultures. An increase in the message level of caspase 3 also occurs in response to focal ischemic injury (Asahi et al., 1997). Similarly, loss of expression of caspase 1 (ICE) in mammary epithelial cells correlates with the suppression of apoptosis by extracellular matrix (Boudreau et al., 1995). However, whether these changes in mRNA levels are important for cell death or lead to increases in activity of caspases is unclear.
We directly measured the activity of caspases 2, 3, and 7 and found that wild-type granule cells increased DEVD-specific substrate cleavage after K+deprivation (Fig. 8). Other groups have similarly reported that DEVD-AMC is cleaved in granule cells after K+/serum deprivation (Armstrong et al., 1996; Nath et al., 1996). The increase in the activity of DEVD-selective caspases is an event that clearly occurs downstream of BAX in cerebellar granule cells (Fig. 8) since Bax −/− cultures showed no increase in DEVD-AMC cleavage after K+ deprivation. Similarly, overexpression of BCL-2 prevents activation of caspases in Jurkat cells (Armstrong et al., 1996; Chinnaiyan et al., 1996), in GT1-7 neural cells (Srinivasan et al., 1996), and in PC12 cells (Stefanis et al., 1996).
Caspase Inhibitors Do Not Block Granule Cell Death
We were surprised by the failure of the baculoviral protein p35 or pharmacological caspase inhibitors to block PCD in cerebellar granule cells. Although inhibiting caspases does delay cell death in granule cells up to 24 h (Nath et al., 1996; Schulz et al., 1996; Armstrong et al., 1997), our data indicate that these compounds did not offer any substantial long-term protection (Fig. 9). This contrasts with sympathetic neurons in which PCD is prevented completely for at least 7 d by BAF (Deshmukh et al., 1996). These results suggest that granule cells have, in addition to a caspase-dependent pathway, a caspase-independent pathway downstream of BAX that insures cell death.
Our suggestion that at least two pathways exist downstream of BAX presumes that all potential caspases were effectively blocked in these studies. We demonstrated that DEVD-AMC cleavage was blocked completely in these cells, showing that the compounds used were able to penetrate the cells and function as caspase inhibitors. BAF, ZVAD-FMK (Armstrong et al., 1996; Cain et al., 1996; Deshmukh et al., 1996), and p35 (Bump et al., 1995; Xue and Horvitz, 1995) are nonspecific inhibitors with reasonable IC50s that block the activity of a wide spectrum of caspases. We cannot formally exclude the possibility that an atypical caspase might have been activated in granule cells in the presence of these inhibitors. Similarly, cell death in the presence of p35 might be accounted for by inadequate levels of protein expression.
The fact that cell death in the presence of caspase inhibitors was TUNEL-negative, while K+ deprivation induced was TUNEL-positive, provided further evidence for the ability of caspase inhibitors to function inside of cells. The nuclear morphology of dying BAF- and ZVAD-treated granule cells may also be an indication that these compounds blocked caspases. While BAF- and ZVAD-treated nuclei did condense, we found no evidence for margination or clumping of the chromatin typically observed in apoptotic cells, though these changes were apparent in untreated, K+-deprived cells (Miller, T.M., personal observation). The ability of caspase inhibitors to block lamin proteolysis (Lazebnik et al., 1995) may account for the lack of margination and chromatin clumping (Rao et al., 1996). In addition to demonstrating that these caspase inhibitors did function within cells, these data raise the possibility that the modestly slower, caspase-independent cell death in granule cells differs fundamentally from the cell death that normally occurs after K+ deprivation when caspases are unimpeded. Further studies of this caspase-independent, TUNEL-negative cell death may elucidate important differences between these two cell death pathways.
Evidence in a different system using an alternative approach has also recently suggested a BAX-dependent, protease-independent mechanism of cell death. BAX overexpression in Jurkat cells leads to an apoptotic-like death that is not blocked by caspase inhibitors, calpain inhibitors, serine protease inhibitors, granzyme B inhibitors, or proteasome inhibitors (Xiang et al., 1996). In these overexpressing Jurkat cells, as observed here, caspase inhibitors do block DNA fragmentation and cleavage of substrates for caspases; however, these cells still die. In Jurkat cells, a fall in mitochondrial membrane potential and an increase in ROS occurs (Xiang et al., 1996). These mitochondrial changes and the membrane localization of BAX suggest that a mitochondria-dependent cell death predominates in BAX gain of function experiments (Xiang et al., 1996). Data here in the experimental setting of BAX loss-of-function provide support for the possibility that a non–caspase-dependent, TUNEL-negative, cell death pathway resides downstream of BAX.
In summary (Fig. 12), K+ and IGF-I block the activation of the cell death program in granule cells. When these agents are removed and the program is activated, MAP kinase activity, PI-3-K activity, metabolic parameters, and most mRNA levels decrease, while some mRNA levels, e.g., c-jun, increase. Our data from the Bax −/− mice place BAX downstream of these changes. The PCD events subsequent to BAX include increased caspase activity and some other change(s) that promote DNA fragmentation, chromatin condensation, and cell death.
Bax −/− granule cell cultures offer an informative perspective on cell death because these cells clearly progress through part of the apoptotic program and then are completely halted. These cultures should be a valuable tool for further ordering the molecular pathways leading to cell death, for defining the role of the BCL-2 family in apoptosis, and, potentially, for identifying a caspase-independent cell-death pathway. Finally, Bax −/− animals bred to mice harboring various cerebellar mutations will determine whether BAX deletion can also rescue “diseased” granule cells in vivo. Assessing both the extent of neuroprotection and the recovery of cerebellar function may offer important insights into how modulation of the BCL-2 family may affect neurological disease.
Acknowledgments
We thank our colleagues at Washington University School of Medicine (St. Louis, MO) B. Klocke, G. Brown, and J. Harding for care of Bax-deficient mice, and R. Easton and P. Osborne for critically evaluating this manuscript.
Supported by National Institutes of Health Grants NS 24679 and AG12947 to E. Johnson, and by CA 49712 to S. Korsmeyer. K. Moulder is supported as a Lucille P. Markey predoctoral fellow, C. Knudson by a Pfizer Postdoctoral Fellowship, and M. Deshmukh by a Paralyzed Veterans of America Spinal Cord Research Foundation Grant 1786.
Abbreviations used in this paper
- BAF
Boc-aspartyl(OMe)-fluoromethylketone
- NMDA
N-methyl-d-aspartic acid
- PI-3-K
phosphatidylinositol-3 kinase
- PCD
programmed cell death
- TUNEL
TdT-mediated dUTP nick end labeling
Footnotes
Address all correspondence to Eugene M. Johnson, Jr., Washington University School of Medicine, Department of Molecular Biology and Pharmacology, 660 South Euclid Avenue, Box 8103, St. Louis, MO 63110. Tel.: (314) 362-3926. Fax: (314) 362-7058. E-mail: ejohnson@pharmdec.wustl.edu
REFERENCES
- Armstrong RC, Aja T, Xiang J, Gaur S, Krebs JF, Hoang K, Bai X, Korsmeyer SJ, Karanewsky DS, Fritz LC, Tomaselli KJ. Fas-induced activation of the cell death-related protease CPP32 is inhibited by Bcl-2 and by ICE family protease inhibitors. J Biol Chem. 1996;271:16850–16855. doi: 10.1074/jbc.271.28.16850. [DOI] [PubMed] [Google Scholar]
- Armstrong RC, Aja TJ, Hoang KD, Gaur S, Bai X, Alnemri ES, Litwack G, Karanewsky DS, Fritz LC, Tomaselli K. Activation of the CED3/ICE-related protease CPP32 in cerebellar granule neurons undergoing apoptosis but not necrosis. J Neurosci. 1997;17:553–562. doi: 10.1523/JNEUROSCI.17-02-00553.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asahi M, Hoshimaru M, Uemura Y, Tokime T, Kojima M, Ohtsuka T, Matsuura N, Aoki T, Shibahara K, Kikuchi H. Expression of interleukin-1β converting enzyme gene family and bcl-2 gene family in the rat brain following permanent occlusion of the middle cerebral artery. J Cerebr Blood Flow Metab. 1997;17:11–18. doi: 10.1097/00004647-199701000-00003. [DOI] [PubMed] [Google Scholar]
- Balazs R, Jorgensen OS, Hack N. N-Methyl-D-Aspartate promotes the survival of cerebellar granule cells in culture. Neuroscience. 1988;27:437–451. doi: 10.1016/0306-4522(88)90279-5. [DOI] [PubMed] [Google Scholar]
- Binetruy B, Smeal T, Karin M. HaRas augments c-Jun activity and stimulates phosphorylation of its activation domain. Nature (Lond) 1991;351:122–127. doi: 10.1038/351122a0. [DOI] [PubMed] [Google Scholar]
- Bos JL. A target for phosphoinositide 3-kinase: Akt/PKB. Trends Biochem Sci. 1995;20:441–442. doi: 10.1016/s0968-0004(00)89097-0. [DOI] [PubMed] [Google Scholar]
- Boudreau N, Sympson CJ, Werb Z, Bissell MJ. Suppression of ICE and apoptosis in mammary epithelial cell by extracellular matrix. Science (Wash DC) 1995;267:891–893. doi: 10.1126/science.7531366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bozyczko CD, McKenna BW, Connors TJ, Neff NT. A rapid fluorometric assay to measure neuronal survival in vitro. J Neurosci Methods. 1993;50:205–216. doi: 10.1016/0165-0270(93)90009-g. [DOI] [PubMed] [Google Scholar]
- Bump NJ, Hackett M, Hugunin M, Seshagiri S, Brady K, Chen P, Ferenz C, Franklin S, Ghayur T, Li P. Inhibition of ICE family proteases by baculovirus antiapoptotic protein p35. Science (Wash DC) 1995;269:1885–1888. doi: 10.1126/science.7569933. [DOI] [PubMed] [Google Scholar]
- Burgoyne RD, Graham ME, Cambray DM. Neurotrophic effects of NMDA receptor activation on developing cerebellar granule cells. J Neurocytol. 1993;22:689–695. doi: 10.1007/BF01181314. [DOI] [PubMed] [Google Scholar]
- Caddy KW, Biscoe TJ. Structural and quantitative studies on the normal C3H and Lurcher mutant mouse. Philos Trans R Soc Lond B Biol Sci. 1979;287:167–201. doi: 10.1098/rstb.1979.0055. [DOI] [PubMed] [Google Scholar]
- Cain K, Inayai-Hussain SH, Couet C, Cohen GM. A cleavage-site-directed inhibitor of interleukin-1β-converting enzyme-like proteases inhibits apoptosis in primary cultures of rat hepatocytes. Biochem J. 1996;314:27–32. doi: 10.1042/bj3140027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen YR, Meyer CF, Tan TH. Persistent activation of c-Jun N-terminal kinase 1 (JNK1) in gamma radiation-induced apoptosis. J Biol Chem. 1996;271:631–634. doi: 10.1074/jbc.271.2.631. [DOI] [PubMed] [Google Scholar]
- Chinnaiyan AM, Orth K, O'Rourke K, Duan HJ, Poirier GG, Dixit VM. Molecular ordering of the cell death pathway—bcl-2 and bcl-x(L) function upstream of the ced-3-like apoptotic proteases. J Biol Chem. 1996;271:4573–4576. doi: 10.1074/jbc.271.9.4573. [DOI] [PubMed] [Google Scholar]
- Chittenden T, Flemmington S, Houghton AB, Ebb RG, Gallo GJ, Elangovan B, Chinnadurai G, Lutz RJ. A conserved domain in Bak, distinct from BH1 and BH2, mediates cell death and protein binding functions. EMBO (Eur Mol Biol Organ) J. 1995;14:5589–5596. doi: 10.1002/j.1460-2075.1995.tb00246.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cidlowski JA. Glucocorticoids stimulate ribonucleic acid degradation in isolated rat thymic lymphocytes in vitro. Endocrinology. 1982;111:184–190. doi: 10.1210/endo-111-1-184. [DOI] [PubMed] [Google Scholar]
- Clarke PGH, Clarke S. Nineteenth-century research on naturally occurring cell death and related phenomenon. Anat Embryol. 1996;193:81–99. doi: 10.1007/BF00214700. [DOI] [PubMed] [Google Scholar]
- Creedon DJ, Johnson EM, Jr, Lawrence JC. Mitogen-activated protein kinase-independent pathways mediate the effects of nerve growth factor and cAMP on neuronal survival. J Biol Chem. 1996;271:20713–20718. doi: 10.1074/jbc.271.34.20713. [DOI] [PubMed] [Google Scholar]
- D'Mello SR, Galli C, Ciotti T, Calissano P. Induction of apoptosis in cerebellar granule neurons by low potassium: inhibition of death by insulin-like growth factor I and cAMP. Proc Natl Acad Sci USA. 1993;90:10989–10993. doi: 10.1073/pnas.90.23.10989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deckwerth TL, Elliott JL, Knudson CM, Johnson EM, Jr, Snider WD, Korsmeyer SJ. Bax is required for neuronal death after trophic factor deprivation and during development. Neuron. 1996;17:401–411. doi: 10.1016/s0896-6273(00)80173-7. [DOI] [PubMed] [Google Scholar]
- Deckwerth TL, Johnson EM., Jr Temporal analysis of events associated with programmed cell death (apoptosis) of sympathetic neurons deprived of nerve growth factor (NGF) J Cell Biol. 1993;123:1207–1222. doi: 10.1083/jcb.123.5.1207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deshmukh M, Vasilakos J, Deckwerth TL, Lampe PA, Shivers BD, Johnson EM., Jr Genetic and metabolic status of NGF-deprived sympathetic neurons saved by an inhibitor of ICE family proteases. J Cell Biol. 1996;135:1341–1354. doi: 10.1083/jcb.135.5.1341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dessi F, Charriaut-Marlangue C, Khrestchatisky M, Ben-Ari Y. Glutamate-induced neuronal death is not a programmed cell death in cerebellar culture. J Neurochem. 1993;60:1953–1955. doi: 10.1111/j.1471-4159.1993.tb13427.x. [DOI] [PubMed] [Google Scholar]
- Dudek H, Datta SR, Franke TF, Birnbaum MJ, Yao R, Cooper GM, Segal RA, Kaplan DR, Greenberg ME. Regulation of neuronal survival by the serine-threonine protein kinase Akt. Science (Wash DC) 1997;275:661–667. doi: 10.1126/science.275.5300.661. [DOI] [PubMed] [Google Scholar]
- Estus, S. 1997. Optimization and validation of RT-PCR as a tool to analyze apoptotic gene expression. In NeuroMethods. J. Poirier, editor. Humana Press, Totowa, NJ. 67–84.
- Estus S, Zaks WJ, Freeman RS, Gruda M, Bravo R, Johnson EM., Jr Altered gene expression in neurons during programmed cell death: identification of c-jun as necessary for neuronal apoptosis. J Cell Biol. 1994;127:1717–1727. doi: 10.1083/jcb.127.6.1717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrer I, Olive M, Ribera J, Planas AM. Naturally occurring (programmed) and radiation-induced apoptosis are associated with selective c-Jun expression in the developing rat brain. Eur J Neurosci. 1996;8:1286–1298. doi: 10.1111/j.1460-9568.1996.tb01297.x. [DOI] [PubMed] [Google Scholar]
- Franke TF, Yang SI, Chan TO, Datta K, Kazlauskas A, Morrison DK, Kaplan DR, Tsichlis PN. The protein kinase encoded by the Akt proto-oncogene is a target of the PDGF-activated phosphatidylinositol 3-kinase. Cell. 1995;81:727–736. doi: 10.1016/0092-8674(95)90534-0. [DOI] [PubMed] [Google Scholar]
- Freeman RS, Estus S, Horigome K, Johnson EM., Jr Cell death genes in invertebrates and (maybe) vertebrates. Curr Opin Neurobiol. 1993;3:25–31. doi: 10.1016/0959-4388(93)90031-s. [DOI] [PubMed] [Google Scholar]
- Freeman RS, Estus S, Johnson EM., Jr Analysis of cell cycle-related gene expression in postmitotic neurons: selective induction of cyclin D1 during programmed cell death. Neuron. 1994;12:343–355. doi: 10.1016/0896-6273(94)90276-3. [DOI] [PubMed] [Google Scholar]
- Gajewski TF, Thompson CB. Apoptosis meets signal transduction: elimination of a BAD influence. Cell. 1996;87:589–592. doi: 10.1016/s0092-8674(00)81377-x. [DOI] [PubMed] [Google Scholar]
- Gallo V, Kingsbury A, Balazs R, Jorgensen OS. The role of depolarization in the survival and differentiation of cerebellar granule cells in culture. J Neurosci. 1987;7:2203–2213. doi: 10.1523/JNEUROSCI.07-07-02203.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gavrieli Y, Sherman Y, Ben-Sasson S. Identification of programmed cell death in situ via specific labeling of nuclear DNA fragmentation. J Cell Biol. 1992;119:493–501. doi: 10.1083/jcb.119.3.493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghosh A, Ginty DD, Bading H, Greenberg ME. Calcium regulation of gene expression in neuronal cells. J Neurobiol. 1994;25:294–303. doi: 10.1002/neu.480250309. [DOI] [PubMed] [Google Scholar]
- Gillardon F, Baurle J, Grusser-Cornehls U, Zimmermann M. DNA fragmentation and the activation of c-Jun in the cerebellum of mutant mice (weaver, Purkinje cell degeneration) Neuroreport. 1995;6:1766–1768. doi: 10.1097/00001756-199509000-00014. [DOI] [PubMed] [Google Scholar]
- Glucksman A. Cell deaths in normal vertebrate ontogeny. Biol Rev Camb Philos Soc. 1951;26:59–86. doi: 10.1111/j.1469-185x.1951.tb00774.x. [DOI] [PubMed] [Google Scholar]
- Greenlund LJ, Deckwerth TL, Johnson EJ. Superoxide dismutase delays neuronal apoptosis: a role for reactive oxygen species in programmed neuronal death. Neuron. 1995a;14:303–315. doi: 10.1016/0896-6273(95)90287-2. [DOI] [PubMed] [Google Scholar]
- Greenlund LJ, Korsmeyer SJ, Johnson EM., Jr Role of BCL-2 in the survival and function of developing and mature sympathetic neurons. Neuron. 1995b;15:649–661. doi: 10.1016/0896-6273(95)90153-1. [DOI] [PubMed] [Google Scholar]
- Ham J, Babij C, Whitfield J, Pfarr CM, Lallemand D, Yaniv M, Rubin LL. A c-Jun dominant negative mutant protects sympathetic neurons against programmed cell death. Neuron. 1995;14:927–939. doi: 10.1016/0896-6273(95)90331-3. [DOI] [PubMed] [Google Scholar]
- Hay BA, Wolff T, Rubin GM. Expression of baculovirus P35 prevents cell death in Drosophila. Development (Camb) 1994;120:2121–2129. doi: 10.1242/dev.120.8.2121. [DOI] [PubMed] [Google Scholar]
- Henkart PA. ICE family proteases: mediators of all apoptotic cell death. Immunity. 1996;4:195–201. doi: 10.1016/s1074-7613(00)80428-8. [DOI] [PubMed] [Google Scholar]
- Hibi M, Lin A, Smeal T, Minden A, Karin M. Identification of an oncoprotein- and UV-responsive protein kinase that binds and potentiates the c-Jun activation domain. Genes Dev. 1993;7:2135–2148. doi: 10.1101/gad.7.11.2135. [DOI] [PubMed] [Google Scholar]
- Kingsbury AE, Gallo V, Woodhams PL, Balazs R. Survival, morphology and adhesion properties of cerebellar interneurones cultured in chemically defined and serum-supplemented medium. Dev Brain Res. 1985;17:17–25. doi: 10.1016/0165-3806(85)90128-2. [DOI] [PubMed] [Google Scholar]
- Knudson CM, Tung KSK, Tourtelotte WG, Brown GAJ, Korsmeyer SJ. Bax-deficient mice with lymphoid hyperplasia and male germ cell death. Science (Wash DC) 1995;270:96–99. doi: 10.1126/science.270.5233.96. [DOI] [PubMed] [Google Scholar]
- Korsmeyer SJ. Regulators of cell death. Trends Genet. 1996;11:101–105. doi: 10.1016/S0168-9525(00)89010-1. [DOI] [PubMed] [Google Scholar]
- Kuida K, Zheng TS, Na S, Kuan C-y, Yang D, Karasuyama H, Rakic P, Flavell R. Decreased apoptosis in the brain and premature lethality in CPP32-deficient mice. Nature (Lond) 1996;384:368–372. doi: 10.1038/384368a0. [DOI] [PubMed] [Google Scholar]
- Lafon-Cazal M, Pietri S, Culcasi M, Bockaert J. NMDA-dependent superoxide production and neurotoxicity. Nature (Lond) 1993;364:535–537. doi: 10.1038/364535a0. [DOI] [PubMed] [Google Scholar]
- Latinis KM, Koretzky GA. Fas ligation induces apoptosis and Jun kinase activation independently of CD45 and Lck in human T cells. Blood. 1996;87:871–875. [PubMed] [Google Scholar]
- Lazebnik YA, Takahashi R, Moir R, Goldman R, Poirier G, Earnshaw WC. Studies of the lamin proteinase reveal multiple parallel biochemical pathways during apoptotic execution. Proc Natl Acad Sci USA. 1995;92:9042–9046. doi: 10.1073/pnas.92.20.9042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levi G, Aloisi F, Ciotti MT, Gallo V. Autographic localization and depolarization-induced release of acidic amino acids in differentiating cerebellar granule cell cultures. Brain Res. 1984;290:77–86. doi: 10.1016/0006-8993(84)90737-6. [DOI] [PubMed] [Google Scholar]
- Linnik MD. Role of apoptosis in acute neurodegenerative disorders. Restor Neurol. 1996;9:219–225. doi: 10.3233/RNN-1996-9404. [DOI] [PubMed] [Google Scholar]
- Makman MH, Dvorkin B, White A. Alterations in protein and nucleic acid metabolism of thymocytes produced by adrenal steroids in vitro. J Biol Chem. 1966;241:1646–1648. [PubMed] [Google Scholar]
- Martinou I, Fernandez PA, Missotten M, White E, Allet B, Sadoul R, Martinou J-C. Viral proteins E1B19K and p35 protect sympathetic neurons from cell death induced by NGF deprivation. J Cell Biol. 1995;128:201–208. doi: 10.1083/jcb.128.1.201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCarthy NJ, Whyte MKB, Gilbert CS, Evan GI. Inhibition of Ced-3/ICE-related proteases does not prevent cell death induced by oncogenes, DNA damage, or the Bcl-2 homologue Bak. J Cell Biol. 1997;136:215–227. doi: 10.1083/jcb.136.1.215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller TM, Johnson EM., Jr Metabolic and genetic analysis of apoptosis in rat cerebellar granule cells. J Neurosci. 1996;16:7487–7495. doi: 10.1523/JNEUROSCI.16-23-07487.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller TM, Tansey MG, Johnson EM, Jr, Creedon DJ. Inhibition of phosphatidylinositol 3-kinase blocks depolarization- and insulin-like growth factor I-mediated survival of cerebellar granule cells. J Biol Chem. 1997;272:9847–9853. doi: 10.1074/jbc.272.15.9847. [DOI] [PubMed] [Google Scholar]
- Motoyama N, Wang FP, Roth KA, Sawa H, Nakayama K, Negishi I, Senju S, Zhang Q, Fujii S, Loh DY. Massive cell death of immature hematopoietic cells and neurons in Bcl-x-deficient mice. Science (Wash DC) 1995;267:1506–1510. doi: 10.1126/science.7878471. [DOI] [PubMed] [Google Scholar]
- Munck A. Metabolic site and time course of cortisol action on glucose uptake, lactic acid output, and glucose 6–phosphate levels of rat thymus cells in vitro. J Biol Chem. 1968;243:1039–1042. [PubMed] [Google Scholar]
- Nardi N, Avidan G, Daily D, Zilkha-Falb R, Barzilai A. Biochemical and temporal analysis of events associated with apoptosis induced by lowering the extracellular potassium concentration in mouse cerebellar granule neurons. J Neurochem. 1997;68:750–759. doi: 10.1046/j.1471-4159.1997.68020750.x. [DOI] [PubMed] [Google Scholar]
- Nath R, Raser KJ, Stafford D, Hajimohammadreza I, Posner A, Allen H, Talanian RV, Yuen P, Gilbertsen RB, Wang KKW. Non-erythroid α-spectrin breakdown by calpain and interleukin 1β-converting enzyme-like protease(s) in apoptotic cells:contributory roles of both protease families in neuronal apoptosis. Biochem J. 1996;319:683–690. doi: 10.1042/bj3190683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oltvai ZN, Milliman CL, Korsmeyer SJ. Bcl-2 heterodimerizes in vivo with a conserved homolog, Bax, that accelerates programmed cell death. Cell. 1993;74:609–619. doi: 10.1016/0092-8674(93)90509-o. [DOI] [PubMed] [Google Scholar]
- Park DS, Stefanis L, Yan CYI, Farinelli SE, Greene LA. Ordering the cell death pathway. Differential effects of BCL2, an interleukin-1β-converting enzyme family protease inhibitor, and other survival agents on JNK activation in serum/nerve growth factor-deprived PC12 cells. J Biol Chem. 1996;271:21898–21905. doi: 10.1074/jbc.271.36.21898. [DOI] [PubMed] [Google Scholar]
- Rabizadeh S, LaCount DJ, Friesen PD, Bredesen DE. Expression of the baculovirus p35 gene inhibits mammalian neural cell death. J Neurochem. 1993;61:2318–2321. doi: 10.1111/j.1471-4159.1993.tb07477.x. [DOI] [PubMed] [Google Scholar]
- Rao L, Perez D, White E. Lamin proteolysis facilitates nuclear events during apoptosis. J Cell Biol. 1996;135:1441–1455. doi: 10.1083/jcb.135.6.1441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schramm M, Eimerl S, Costa E. Serum and depolarizing agents cause acute neurotoxicity in cultured cerebellar granule cells: role of the glutamate receptor responsive to N-methyl-D-aspartate. Proc Natl Acad Sci USA. 1990;87:1193–1197. doi: 10.1073/pnas.87.3.1193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schulz JB, Weller M, Klockgether T. Potassium deprivation-induced apoptosis of cerebellar granule neurons; a sequential requirement for new mRNA and protein synthesis, ICE-like protease activity, and reactive oxygen species. J Neurosci. 1996;16:4696–4706. doi: 10.1523/JNEUROSCI.16-15-04696.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwartz LM, Milligan CE. Cold thoughts of death—the role of ICE proteases in neuronal cell death. Trends Neurosci. 1996;19:555–562. doi: 10.1016/s0166-2236(96)10067-9. [DOI] [PubMed] [Google Scholar]
- Short JM, Fernandez JM, Sorge JA, Huse WD. Lambda ZAP: a bacteriophage Lambda expression vector with in vivo excision properties. Nucleic Acids Res. 1988;16:7583–7600. doi: 10.1093/nar/16.15.7583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smeal T, Binetruy B, Mercola D, Birrer M, Karin M. Oncogenic and transcriptional cooperation with HaRas requires phosphorylation of c-Jun on serines 63 and 73. Nature (Lond) 1991;354:494–496. doi: 10.1038/354494a0. [DOI] [PubMed] [Google Scholar]
- Smeal T, Binetruy B, Mercola D, Grover-Bardwick A, Heidecker G, Rapp UR, Karin M. Oncoprotein-mediated signalling cascade stimulates c-Jun activity by phosphorylation of serines 63 and 73. Mol Cell Biol. 1992;12:3507–3513. doi: 10.1128/mcb.12.8.3507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Srinivasan A, Foster LM, Testa MP, Ord T, Keane RW, Bredesen DE, Kayalar C. Bcl-2 expression in neural cells blocks activation of Ice/ced-3 family proteases during apoptosis. J Neurosci. 1996;16:5654–5660. doi: 10.1523/JNEUROSCI.16-18-05654.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stefanis L, Park DS, Yan CYI, Farinelli SE, Troy CM, Shelanski ML, Greene LA. Induction of CPP32-like activity in PC12 cells by withdrawal of trophic support—dissociation from apoptosis. J Biol Chem. 1996;271:30663–30671. doi: 10.1074/jbc.271.48.30663. [DOI] [PubMed] [Google Scholar]
- Sugimoto A, Friesen PD, Rothman JH. Baculovirus p35 prevents developmentally programmed cell death and rescues a ced-9 mutant in the nematode Caenorhabditis elegans. . EMBO (Eur Mol Biol Organ) J. 1994;13:2023–2028. doi: 10.1002/j.1460-2075.1994.tb06475.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thangnipon W, Kingsbury A, Webb M, Balazs R. Observations on rat cerebellar cells in vitro: influence of substratum, potassium concentration and relationship between neurones and astrocytes. Dev Brain Res. 1983;11:177–189. doi: 10.1016/0165-3806(83)90215-8. [DOI] [PubMed] [Google Scholar]
- Verheij M, Bose R, Lin XH, Yao B, Jarvis WD, Grant S, Birrer MJ, Szabo E, Zon LI, Kyriakis JM, et al. Requirement for ceramide-initiated SAPK/JNK signalling in stress-induced apoptosis. Nature (Lond) 1996;380:75–79. doi: 10.1038/380075a0. [DOI] [PubMed] [Google Scholar]
- Virdee K, Tolkovsky A. Inhibition of p42 and p44 mitogen-activated protein kinase activity by PD98059 does not suppress nerve growth factor-induced survival of sympathetic neurones. J Neurochem. 1996;67:1801–1805. doi: 10.1046/j.1471-4159.1996.67051801.x. [DOI] [PubMed] [Google Scholar]
- Wang H-G, Rapp UR, Reed JC. Bcl-2 targets the protein kinase Raf-1 to mitochondria. Cell. 1996;87:629–638. doi: 10.1016/s0092-8674(00)81383-5. [DOI] [PubMed] [Google Scholar]
- Wetts R, Herrup K. Direct correlation between Purkinje and granule cell number in the cerebella of lurcher chimeras and wild-type mice. Dev Brain Res. 1983;10:41–47. doi: 10.1016/0165-3806(83)90119-0. [DOI] [PubMed] [Google Scholar]
- White E. Life, death, and the pursuit of apoptosis. Genes Dev. 1996;10:1–15. doi: 10.1101/gad.10.1.1. [DOI] [PubMed] [Google Scholar]
- Williams RW, Herrup K. The control of neuron number. Annu Rev Neurosci. 1988;11:423–453. doi: 10.1146/annurev.ne.11.030188.002231. [DOI] [PubMed] [Google Scholar]
- Wilson DJ, Fortner KA, Lynch DH, Mattingly RR, Macara IG, Posada JA, Budd RC. JNK, but not MAPK, activation is associated with Fas-mediated apoptosis in human T cells. Eur J Immunol. 1996;26:989–994. doi: 10.1002/eji.1830260505. [DOI] [PubMed] [Google Scholar]
- Wong EHF, Kemp JA, Priestley T, Knight AR, Woodruff GN, Iverson LL. The anti-convulsant MK-801 is a potent NMDA antagonist. Proc Natl Acad Sci USA. 1986;83:7104–7108. doi: 10.1073/pnas.83.18.7104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia Z, Dickens M, Raingeaud J, Davis RJ, Greenberg ME. Opposing effects of ERK and JNK-p38 MAP kinases on apoptosis. Science (Wash DC) 1995;270:1326–1331. doi: 10.1126/science.270.5240.1326. [DOI] [PubMed] [Google Scholar]
- Xiang JL, Chao DT, Korsmeyer SJ. Bax-induced cell death may not require interleukin 1β–converting enzyme-like proteases. Proc Natl Acad Sci USA. 1996;93:14559–14563. doi: 10.1073/pnas.93.25.14559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xue D, Horvitz HR. Inhibition of the Caenorhabditis elegans cell-death protease CED-3 by a CED-3 cleavage site in baculovirus p35 protein. Nature (Lond) 1995;377:248–251. doi: 10.1038/377248a0. [DOI] [PubMed] [Google Scholar]
- Yin XM, Oltvai Z, Korsmeyer S. BH1 and BH2 domains of Bcl-2 are required for inhibition of apoptosis and heterodimerization with Bax. Nature (Lond) 1994;369:321–323. doi: 10.1038/369321a0. [DOI] [PubMed] [Google Scholar]
- Zha J, Harada H, Yang E, Jockel J, Korsmeyer SJ. Serine phosphorylation of death agonist BAD in response to survival factor results in binding to 14-3-3 not Bcl-XL. Cell. 1996;87:619–628. doi: 10.1016/s0092-8674(00)81382-3. [DOI] [PubMed] [Google Scholar]