

Abstract
During nuclear import, cytosolic transport factors move through the nuclear pore complex (NPC) to the nuclear compartment. Kap95p is required during import for docking the nuclear localization signal-receptor and ligand to the NPC. Recycling of this factor back to the cytoplasm is necessary for continued rounds of import; however, the mechanism for Kap95p recycling is unknown. We have determined that recycling of Kap95p requires a nuclear export signal (NES). A region containing the NES in Kap95p was sufficient to mediate active nuclear export in a microinjection assay. Moreover, the NES was necessary for function. Mutation of the NES in Kap95p resulted in a temperaturesensitive import mutant, and immunofluorescence microscopy experiments showed that the mutated Kap95p was not recycled but instead localized in the nucleus and at the nuclear envelope. Srp1p, the yeast nuclear localization signal-receptor, also accumulated in the nuclei of the arrested kap95 mutant cells. Wild-type and NES-mutated Kap95p both bound Gsp1p (the yeast Ran/TC4 homologue), Srp1p, and the FXFG repeat region of the nucleoporin Nup1p. In contrast, the NES mutation abolished Kap95p interaction with the GLFG repeat regions from the nucleoporins Nup116p and Nup100p. In vivo interaction was demonstrated by isolation of Kap95p from yeast nuclear lysates in either protein A–tagged Nup116p or protein A–tagged Nup100p complexes. The protein A–tagged Nup116p complex also specifically contained Gle2p. These results support a model in which a step in the recycling of Kap95p is mediated by interaction of an NES with GLFG regions. Analysis of genetic interactions suggests Nup116p has a primary role in Kap95p recycling, with Nup100p compensating in the absence of Nup116p. This finding highlights an important role for a subfamily of GLFG nucleoporins in nuclear export processes.
Communication between the nucleus and cytoplasm is mediated by large proteinaceous structures called nuclear pore complexes (NPCs).1 Cytosolic factors, as well as NPC proteins (nucleoporins), are required to mediate signal-dependent transport into the nucleus (for reviews see Moore and Blobel, 1994; Powers and Forbes, 1994; Melchior and Gerace, 1995; Simos and Hurt, 1995; Gorlich and Mattaj, 1996). After translation, a nuclear localization signal (NLS)–bearing protein binds in the cytoplasm to a heterodimeric complex comprised of a ∼60-kD NLS-receptor (importin α/karyopherin α/hSRP1/ Srp1p/Kap60p) (Adam and Gerace, 1991; Gorlich et al., 1994; Imamoto et al., 1995b ; Loeb et al., 1995; Moroianu et al., 1995a ; Weis et al., 1995) and a ∼95-kD subunit that acts as an adaptor for interaction with nucleoporins (importin β/karyopherin β/p97/Kap95p) (Adam and Adam, 1994; Chi et al., 1995; Enenkel et al., 1995; Gorlich et al., 1995a ; Imamoto et al., 1995a ; Radu et al., 1995a ,b). After docking at the cytoplasmic face of the NPC, translocation of the trimeric complex through the aqueous channel of the NPC may proceed by a succession of energy-dependent disassociation–reassociation steps with nucleoporins (Melchior and Gerace, 1995; Rexach and Blobel, 1995; Gorlich and Mattaj, 1996; Hu et al., 1996; Nehrbass and Blobel, 1996).
The energy required for movement through the NPC is due in part to GTP hydrolysis by the small G protein Ran/ TC4/Gsp1p (Melchior et al., 1993; Moore and Blobel, 1993; Melchior et al., 1995). The cumulative effect of the binding reactions is to propel the NLS protein–Srp1pKap95p trimer toward the nucleoplasm. At the nucleoplasmic face of the NPC, the import complex disassembles. Srp1p and the NLS-bearing protein are released into the nucleoplasm, but Kap95p appears to stay associated with the nuclear envelope (Gorlich et al., 1995b ; Moroianu et al., 1995b ). Continued rounds of import would require efficient return of these import factors to the cytoplasm.
The mechanism for recycling nuclear import factors is unknown. Return to the cytoplasm may be based on both diffusive and active export. Recent studies on proteins that shuttle continuously between the nucleus and cytoplasm, such as the HIV-1 Rev protein (Meyer and Malim, 1994) and the hnRNP A1 protein (Pinol-Roma and Dreyfuss, 1992; Michael et al., 1995), have led to the characterization of nuclear export signals (NESs). The NESs in Rev (Fischer et al., 1995; Wen et al., 1995), protein kinase inhibitor (PKI) (Wen et al., 1995), TFIIIA (Fridell et al., 1996b ), and yeast Gle1p (Murphy and Wente, 1996) are short stretches of hydrophobic residues that are sufficient for export. PKI mediates export of the bound protein kinase A catalytic subunit (Wen et al., 1995), whereas the other NES-bearing proteins are involved in RNA export. These NES motifs may serve both as signals for export and as adaptor regions for interaction with nucleoporins (Bogerd et al., 1995; Fritz et al., 1995; Gerace, 1995; Stutz et al., 1995; Fridell et al., 1996a ; Fritz and Green, 1996; Murphy and Wente, 1996).
Recent studies have shown that nuclear import factors can interact with some nucleoporins that contain regions of multiple FG (Phe, Gly), FXFG, or GLFG (Gly, Leu, Phe, Gly) peptide repeats (Iovine et al., 1995; Kraemer et al., 1995; Moroianu et al., 1995b ; Nehrbass and Blobel, 1995; Pascal and Gerace, 1995; Radu et al., 1995a ,b; Rexach and Blobel, 1995; Aitchison et al., 1996; Hu et al., 1996), suggesting a role for these particular nucleoporins in the docking and translocation steps of import. In a previous study, we examined the necessity for each of the individual GLFG tetrapeptide repeat regions in the yeast proteins Nup145p, Nup116p, Nup100p, Nup57p, and Nup49p (Iovine et al., 1995). In vitro ligand blot binding experiments show that the GLFG regions from Nup116p, Nup100p, and Nup145p all interact with Kap95p. Interestingly, only the GLFG region from Nup116p exhibits both loss-of-function and gain-of-function phenotypes in yeast cells. Deletion of the GLFG regions from the other four GLFG nucleoporins does not perturb cell viability (Wente et al., 1992; Wente and Blobel, 1994; Fabre et al., 1994; Grandi et al., 1995; Iovine et al., 1995), whereas internal deletion of the GLFG region from Nup116p results in temperaturesensitive cells with a diminished nuclear protein import capacity (Iovine et al., 1995). Moreover, overexpression of only the Nup116p–GLFG region (and not that from any of the other four) inhibits cell growth and nuclear transport (Iovine et al., 1995). Thus, if the in vivo import mechanism required a Kap95p–GLFG nucleoporin interaction, we predicted that the GLFG region of Nup116p was the most likely candidate for mediating this function. However, the discrete step of import that may require a Kap95p–Nup116p interaction has not been delineated.
In this report, we show that Kap95p can be isolated from yeast cells in complexes containing GLFG nucleoporins. We propose that this interaction is mediated by an NES motif in Kap95p. Mutation of the NES in Kap95p blocked continued rounds of nuclear import, resulted in the accumulation of the mutated protein at the nuclear envelope and in the nucleoplasm, and inhibited interaction with GLFG regions from Nup116p and Nup100p. These results suggest a model wherein Nup116p and Nup100p play a role in recycling Kap95p to the cytoplasm.
Materials and Methods
Strains and Plasmids
Yeast strains were grown in either YEP (1% yeast extract, 2% peptone) or synthetic minimal media plus appropriate amino acids supplemented with 2% of the indicated sugar (glucose, galactose, or raffinose). Yeast transformations were completed by the lithium acetate method (Ito et al., 1983), and general manipulations of yeast cells were conducted as described by Sherman et al. (1986). Bacterial strains were cultured in SOB media and transformed by standard methods (Sambrook et al., 1989). DH5α was used for all bacterial plasmids.
The sequence encoding five tandem IgG-binding domains was amplified by using the PCR and oligonucleotide primers (246 and 247) to flank the protein A fragment with unique PstI sites. The resulting fragment was ligated into pSW11 (Wente et al., 1992) partially digested with NsiI, yielding an in-frame fusion of protein A between amino acid residues 88 and 89 of Nup116p (pSW461).
Site-directed mutagenesis of KAP95 to kap95-L63A was completed as follows: pSW271 (Iovine et al., 1995) was used as the template for sequential PCRs. The first reaction used primers 259 and 279, with the 3′ 279 primer incorporating base pair changes for leucine to alanine (cta to gca) at nucleotides 2,830 and 2,831 of KAP95. The second reaction used primers 278 and 262, with the 5′ 278 primer incorporating the same two nucleotide changes. After mixing the products of these two reactions as a template, PCR was conducted with primers 259 (annealing at nucleotide 1,702) and 262 (at 4,357). The final product was digested with BamHI and ligated into pRS315 (pSW507). The AatII/NcoI fragment from pSW507 was ligated into the AatII/NcoI backbone of vector pSW503 to generate pSW509 harboring kap95-L63A. The site of the mutation was confirmed by DNA sequence analysis. A plasmid bearing the kap95-ΔNES allele (pSW696) with an in-frame deletion of the sequence encoding residues 55–65 was constructed in a similar manner with the following modifications: the first reaction used primers 259 and 281 (with deletion of 30 nucleotides of KAP95 sequence), the second reaction used primers 280 (with deletion of 30 nucleotides of KAP95 sequence) and 262. The AatII/NcoI fragment from the ΔNES product was ligated into the AatII/NcoI backbone of vector pSW503 to generate pSW510.
Nuclear Lysates and Coimmunoprecipitation
Nuclei were purified from yeast strain SWY960, SWY1381, or W303a/α cells as described by Rout and Kilmartin (1990). Lysates were prepared from 4 × 109 nuclei as described in Rout and Blobel (1993), and 400 μl of lysate was incubated with 50 μl of IgG-Sepharose (Pharmacia Fine Chemicals, Piscataway, NJ) for 1 h at room temperature. Control experiments used protein A–Sepharose (Pharmacia Fine Chemicals). The resin was washed (4 × 500 μl) with washing buffer (150 mM NaCl, 20 mM Tris, pH 6.5, 0.05% Tween-20) before elution with 0.1 M glycine, pH 2.8. The resin was also analyzed by adding SDS buffer directly to the washed and eluted beads. Samples from coimmunoprecipitation were separated by electrophoresis in SDS polyacrylamide gels and either transferred electrophoretically to nitrocellulose membranes or silver stained. Immunoblots were probed with an affinity-purified anti-Kap95p antibody at 1:100 (see below), affinity-purified anti-Gle2p antibody at 1:25, or affinity-purified anti-Gle1p antibody at 1:500 (gift of J. Watkins, Washington University, St. Louis, MO) as indicated. All dilutions were made in 10 mM Tris-HCl, pH 8.0, 150 mM NaCl, 0.05% Tween-20 (TBST)/2% milk. With washes in between, incubations with affinity-purified alkaline phosphatase–conjugated anti–rabbit IgG (Promega Corp., Madison, WI) (diluted 1:7,500) were conducted. Blots were developed via color visualization with nitro blue tetrazolium and 5-bromo-4-chloro-3-indolyl-1-phosphate (Promega Corp.).
For silver staining, the gels were washed first in 50% methanol for 30 min to overnight, and then in distilled water for 30 min to 2 h (four changes of water). Fresh stain was made by adding silver nitrate (0.2 g/ml water) dropwise to 4.2 ml of 0.36% NaOH and 280 μl NH4OH until saturated (final volume adjusted to 20 ml). The gel was stained for 15 min, washed with distilled water for 30 min (with four changes of water), and developed in 0.005% citric acid and 0.018% formaldehyde. Development was stopped by washing in 50% methanol.
Ligand Blot Overlays
Purified glutathione-S-transferase (GST)–Gsp1p (see purification of GST-Kap95p[1-77]) was loaded with GTP exactly as outlined in Chi et al. (1996). 6x-His-Kap95p and 6x-His-kap95pL63A were purified as described below (see Soluble Binding Assay), and 0.5 μg of each were separated via 7.0% SDS-PAGE and electrophoretically transferred to nitrocellulose membranes. The membranes were treated exactly as described in Iovine et al. (1995) except that they were overlaid with 0.5 mg of GST-Gsp1pGTP (in 5 ml total vol). Bound GST-Gsp1pGTP was detected using an affinitypurified anti–GST rabbit antibody at 1:2,000 (provided by J. Watkins), followed by enhanced chemiluminescence.
Soluble Binding Assay
GST-GLFG-Nup116p was purified as described below for GST-Kap95p(1-77). Bacterial cells expressing 6x-His-Kap95p were induced with 1 mM isopropylthio-B-d-galactoside, resuspended in sonication buffer (50 mM sodium phosphate, pH 8.0, 300 mM NaCl, 0.1 mM PMSF, 0.1 μM pepstatin A, 0.1 μM leupeptin, 0.1 mM benzamidine), and frozen at −70°C. After thawing, cells were sonicated with 60 1-s pulses (with 10 s of rest on ice after 10 pulses). The lysate was clarified by a 20,000 g spin for 20 min, and then incubated with 2 ml of packed Ni-NTA resin (Qiagen, Chatsworth, CA) for 1 h at 4°C. The resin was washed with 15 vol of sonication buffer and then with 20 vol of wash buffer (50 mM sodium phosphate, pH 6.0, 300 mM NaCl, 10% glycerol, 10 mM imidazole). The fusion protein was eluted with 20 ml of 0.15 M imidazole in wash buffer. The purified 6x-His-Kap95p was transferred into binding buffer (20 mM Hepes, pH 6.8, 150 mM potassium acetate, 2 mM magnesium acetate, 2 mM DTT, 0.1% Tween-20, 0.1% casaminoacids) using a Centricon 30 (Amicon Inc., Beverly, MA) or by dialysis. The soluble binding assay was conducted as described in Rexach and Blobel (1995).
β-Galactosidase Assays for Two-Hybrid Interactions
Yeast strain L40 (MATa, his3Δ200 trp1-901 leu2-3,112 ade2 LYS2::(lexAop)4-HIS3 URA3::(lexAop)8-LacZ) (gift of S. Hollenberg) was cotransformed with the various pCH436 and pCH358 constructs (generous gifts of C. Hardy (Washington University, St. Louis, MO); see above and Iovine et al., 1995; Hardy, 1996). Transformants were tested for β-galactosidase expression by scoring for blue color with the filter assay described in Breeden and Nasmyth (1985).
Preparation of GST-FITC Conjugates and Microinjection Assay
GST-Kap95-(1-77) and GST-kap95-(1-77)ΔNES fusion proteins were each purified from bacterial lysates generated as described (Iovine et al., 1995). The lysate (40 ml) was incubated with 1 ml of glutathione-agarose (Sigma Chemical Co., St. Louis, MO) for 1 h at 4°C. The resin was washed with MTPBS (150 mM NaCl, 20 mM sodium phosphate, pH 7.3, 0.01% NaN3, 10 mM EDTA, 0.1 mM DTT, 0.1 mM PMSF, 0.1 μM pepstatin A, 0.1 μM leupeptin, 0.1 mM benzamidine) until the OD280 reached baseline. The protein was eluted with 20 mM glutathione, 50 mM Tris, pH 9.0, 0.01% NaN3. 1 mg of protein was added to 1.5 ml of PBS, pH 7.2, and spun twice in a Centricon 30 concentrator. 0.4 mg of protein was labeled at a 50fold molar ratio using an FITC-FluorReporter protein labeling kit from Molecular Probes, Inc. (Eugene, OR).
COS-1 cells were grown to 80% confluence on 12-mm circular glass coverslips (Fisher Scientific Co., Pittsburgh, PA), and microinjected as described (Murphy and Wente, 1996). After microinjection, cells were incubated for 45 min at 37°C, and then fixed for 15 min with 3.0% formaldehyde in 10 mM sodium phosphate, pH 7.4, 150 mM NaCl, 2 mM magnesium chloride. The cells were washed once in buffer without formaldehyde, followed by mounting with 90% glycerol, 1 mg/ml p-phenylenediamine onto slides. Photographs were taken with ×40 objective on a microscope (Olympus Corp., Lake Success, NY) with T-MAX 400 film (Eastman Kodak Co., Rochester, NY).
Transport Assays and Immunofluorescence Microscopy
In situ hybridization for poly(A)+ RNA localization was conducted as previously described (Iovine et al., 1995). Nuclear import capacity was assessed by monitoring the localization of a GAL10-induced NLS–β-galactosidase fusion protein (expressed on pNLS-E1) (Underwood and Fried, 1990). Haploid strains SWY1312 and SWY1313 were each transformed with pNLS-E1 (URA3) and grown in SC-ura and 2% raffinose at 23°C overnight. The following morning, expression was induced by the addition of 2% galactose at the same time that the cells were shifted to 37°C. After 3 h at 37°C, the cells were processed for indirect immunofluorescence microscopy.
Early log phase or temperature-arrested yeast cells were prepared for immunofluorescence experiments as previously described (Wente et al., 1992). Cells were fixed for 2.5 min in 3.7% formaldehyde/10% methanol, and subsequently incubated with appropriate primary antibodies: the antiKap95p antibody (1:20) (see below), rabbit anti–importin α (Srp1p) antibody (1:1,500) (generous gift of D. Gorlich, University of Heidelberg, Germany), mouse mAb414 (1:1) (Davis and Blobel, 1986), or mouse monoclonal anti–β-galactosidase (1:100) (Boehringer Manneheim Biochemicals, Indianapolis, IN). After washing with 40 mM K2HPO4, 10 mM KH2PO4, 150 mM NaCl, 0.1% NaN3, 0.1% Tween 20, 2% nonfat dry milk (M buffer) alone, detection of bound antibody was accomplished by incubation with either affinity-purified Texas red–conjugated goat anti–rabbit IgG (Cappel Laboratories, Organon Teknika Corp., Durham, NC) at a 1: 200 dilution, or affinity purified FITC-conjugated goat anti–mouse IgG (1: 200) for 1 h at room temperature. The final washes in M buffer and then 1% BSA-PBS were followed by mounting with 90% glycerol, 1 mg/ml p-phenylenediamine, and 0.05 μg/ml 4′,6-diamidino-2-phenylindole DAPI at pH 8.0. Photographs were taken with the ×100 objective on an Olympus microscope with Kodak T-MAX 400 film.
For the confocal experiments, cells were treated identically except for two modifications. An FITC-conjugated donkey anti–rabbit IgG antibody (Cappel Laboratory) was used (1:200) in place of the Texas red anti–rabbit IgG for some samples, and propidium iodide (16 μg/ml) was incubated on slides for 15 min at room temperature before mounting with 90% glycerol plus 1 mg/ml p-phenylenediamine. Images were collected using the ×63 objective on the MRC 1024 confocal laser scanning microscope (Bio Rad Laboratories, Hercules, CA; LaserSharp software). Color was added to the images using NIH image, and the final figure was produced using Adobe Photoshop (Adobe Systems, Inc., Mountain View, CA).
Affinity Purification of Anti-Kap95p Antibody
GST-Kap95p (pSW327) (Iovine et al., 1995) was purified as described for the purification of GST-Kap95-(1-77). Purified protein was injected into rabbits to generate a polyclonal antiserum (Cocalico Biologicals, Inc., Reamstown, PA). The production bleeds were affinity purified against 6xHis-tagged Kap95p (dialyzed into PBS, pH 7.4) using the Affigel 15 system (Bio Rad Laboratories). After elution from the resin (0.1 M glycine, pH 2.8), the eluate was dialyzed against PBS, pH 7.4, and NaN3 was added to a final 0.05%. The affinity-purified antibody was titered against yeast lysates from W303 cells and recognizes a single band of ∼95 kD.
Results
Biochemical Copurification of Kap95p and Nup116p from Yeast Nuclei
To further characterize the physiological significance of a Kap95p–GLFG nucleoporin interaction, coimmunoprecipitation experiments were conducted from yeast cell fractions. These studies initially focused on Nup116p because its GLFG region is uniquely required for cell growth (Iovine et al., 1995). For these experiments, full-length Nup116p was tagged in its amino-terminal region with five tandem IgG-binding domains from Staphylococcus aureus (protein A) and expressed from the NUP116 promoter in an nup116 null strain. The protein A–Nup116p completely complemented the nup116 null phenotype, and it localized exclusively at NPCs as determined by indirect immunofluorescence microscopy (data not shown). Lysates of purified yeast nuclei containing protein A–Nup116p were incubated with IgG-Sepharose. Copurifying proteins were eluted with acidic buffer and analyzed by SDS-PAGE. Multiple polypeptides were detected by silver staining (Fig. 1, A and B). Immunoblotting with affinity-purified polyclonal anti-Kap95p antibodies revealed that the polypeptide of ∼95 kD corresponded to Kap95p (Fig. 1 A). The majority of the protein A–Nup116p remained associated with the IgG-Sepharose and was only eluted with SDS treatment (Fig. 1 C). Therefore, Kap95p was isolated from yeast cells in a complex containing protein A–Nup116p.
Figure 1.

Protein A–Nup116p interacts with Kap95p and Gle2p. (A and B) Nuclear lysates were prepared from the diploid strain SWY960 (protein A–Nup116p) and incubated with IgG-Sepharose. Lysate (L), unbound (U), and acid eluted (E) fractions were separated by electrophoresis in SDS polyacrylamide gels: 7.5% gels in A and 10.5% gels in B. The gels were either silver stained or transferred to nitrocellulose. Immunoblots were incubated with anti-Kap95p antibody (1:100) or anti-Gle2p antibody (1:25) as indicated. The dashes along the silver stain gel indicate the polypeptides that align with the corresponding Kap95p and Gle2p cross-reactive bands in the immunoblots (single stars). The double stars indicate the position of the protein A–Nup116. (C) A fraction of the protein A–Nup116p remains bound to the IgG-Sepharose. After elution with acidic buffer (yielding the samples in lanes E), IgG beads were treated with SDS buffer, and the bound fraction was analyzed on 7.5% SDS polyacrylamide gels. The immunoblot was developed with rabbit anti–IgG antibody (1:1,000): (lane 1) mock treated beads; (lane 2) beads incubated with protein A–Nup116p nuclear lysate. (Lane 3) A silver stain of the same fraction analyzed in lane 2. The double star indicates the position of the bound protein A–Nup116p (the upper band of the doublet when compared with untreated resin). (D) The Kap95p and Gle2p interactions with protein A–Nup116p are specific. Control experiments were conducted with nuclear lysates prepared from the diploid strain W303a/α (WT nuclei) incubated with IgG-Sepharose (left), or from SWY960 (protein A–Nup116 nuclei) incubated with protein A–Sepharose (right). Samples that eluted with acidic buffer from the IgG-Sepharose or that eluted with SDS buffer from the protein A–Sepharose were separated by electrophoresis in 7.5% (for Kap95p) or 10.5% (for Gle2p) SDS polyacrylamide gels and tested by immunoblotting. The respective positions expected for Kap95p and Gle2p bands are indicated by single stars. The bands by the plus sign (+) are due to the protein A from the resin. For all the panels, the sizes of the molecular mass markers are indicated at the right.
In separate studies we recently identified the nuclear export factors Gle1p and Gle2p in a synthetic lethal genetic screen with an nup100 mutation (Murphy et al., 1996; Murphy and Wente, 1996). Nup100p is a GLFG nucleoporin that possesses significant structural and functional overlap with Nup116p (Wente et al., 1992). Thus, we tested by immunoblotting whether either Gle1p (a 62-kD protein) or Gle2p (a 40-kD protein) was present in the protein A–Nup116p complex. Gle1p was not in the bound fraction (data not shown), but Gle2p was present and corresponded to the ∼41-kD band (Fig. 1 B). To confirm that the association of Kap95p and Gle2p with protein A–Nup116p was specific, binding of wild-type nuclear lysates to IgGSepharose and protein A–Nup116p nuclear lysates to protein A–Sepharose was analyzed. Kap95p and Gle2p showed no affinity for either resin (Fig. 1 D). Therefore, their cofractionation was dependent on the presence of Nup116p. However, whether Kap95p and Nup116p are directly interacting in this complex (or whether the complex reflects a single entity) will require further investigation.
To confirm a stable interaction was possible between Kap95p and Nup116p in vitro, the soluble binding assay described by Rexach and Blobel (1995) was conducted with bacterially expressed, purified proteins. The GLFG region of Nup116p was purified as a GST fusion and immobilized on glutathione-agarose beads. Purified 6x-His-tagged Kap95p was incubated with either the immobilized GST-Nup116pGLFG beads or immobilized GST alone. Bound and unbound fractions were analyzed by Coomassie blue staining of SDS polyacrylamide gels. Kap95p did not bind to GST alone (data not shown) (Rexach and Blobel, 1995), but binding of Kap95p was detected with the GST-Nup116pGLFG (Fig. 2). Therefore, this was a stable interaction and confirmed our previous studies detecting interaction between Nup116p and Kap95p with ligand blot overlay and two-hybrid assays. This specific in vitro interaction and the cofractionation of Kap95p and Nup116p from yeast cells reinforced our previous hypothesis proposing a discrete role for GLFG region–Kap95p binding during nuclear transport.
Figure 2.

The GLFG region of Nup116p stably interacts with Kap95p. Purified GST-GLFG-Nup116p (1.0 μg) was incubated with 10 μl of glutathione-agarose for 30 min at 4°C. After washing, 0.6 μg of purified 6x-His-Kap95p was added. The binding reaction was incubated for 45 min at room temperature. The unbound fraction and washes were combined and TCA precipitated. The bound proteins were eluted with SDS sample buffer and boiling for 5 min. Samples were separated by electrophoresis on an 8.0% SDS polyacrylamide gel and visualized by Coomassie blue stain. Both GST-GLFG-Nup116p and 6x-His+ Kap95p are found in the bound fraction. (Lane 1) “Mock” binding reaction (only buffer was added in the second binding reaction); (lane 2) 6x-His-Kap95p (0.6 μg); (lane 3) unbound sample; (lane 4) bound sample.
The Amino-terminal Region of Kap95p Has an NES
The goal of our continued studies was to determine when a Kap95p–Nup116p interaction might be required during nuclear transport. Previous studies showed that the first 130 amino acids of Kap95p are sufficient for a strong twohybrid interaction with the GLFG region of Nup116p (Iovine et al., 1995). We inspected this region of Kap95p amino acid sequence for motifs that may be facilitating binding to Nup116p–GLFG and observed a region from residues 55–65 with striking similarity to leucine-rich NES motifs (Fig. 3 A). Interestingly, NES motifs are necessary for the interaction of Rev and Gle1p with FG repeat–containing nucleoporins (Bogerd et al., 1995; Fritz et al., 1995; Stutz et al., 1995; Fritz and Green, 1996; Murphy and Wente, 1996).
Figure 3.

The NES region of Kap95p mediates nuclear export. (A) Alignment of a region of Kap95p with the NES sequences of HIV-1 Rev, PKI, TFIIIA, and Gle1p is shown. (B) GST-Kap95-(1-77) and GST-Kap95-(1-77)ΔNES fusion proteins were purified and conjugated to FITC. The fluorescent conjugates were individually coinjected into the nuclei of COS-1 cells with Texas red–HSA. After a 45min incubation at either 37°C or 4°C, the cells were fixed and examined by fluorescence microscopy. The Texas red– HSA remained nuclear localized (bottom row); however, the FITC-GST-Kap95-(1-77) moved to the cytoplasm (top, left) when incubated at 37°C. The FITC-GST-Kap95-(177) was confined to the nucleus at 4°C incubation (top, middle). The fusion lacking the NES motif (FITC-GSTKap95-(1-77)ΔNES) remained nuclear even at 37°C (top, right). Bar, 10 μm.
To test whether the NES-like motif in the amino-terminal region of Kap95p could mediate nuclear export, a series of microinjection experiments in COS-1 cells were conducted. The first 77 amino acids of Kap95p were linked in a carboxy-terminal fusion to GST (GST-Kap95-[1-77]). A similar construct that harbored an in-frame deletion spanning amino acids 55–65 of Kap95p was also made (GSTKap95-[1-77]ΔNES). The GST fusion proteins were expressed in bacteria, purified, and fluorescently labeled by conjugation to FITC. Each was coinjected with Texas red– labeled human serum albumin (HSA) into the nuclei of COS-1 cells (Fig. 3 B). After 45 min at 37°C, the Texas red–HSA remained within the nucleus in all experiments. In contrast, the FITC-GST-Kap95-(1-77) staining shifted to the cytoplasm, and the nuclear compartment staining was coincidentally reduced. The nuclear export of the FITC-GST-Kap95-(1-77) was dependent on incubation at 37°C because the staining was predominantly nuclear with incubation at 4°C (Fig. 3 B). These results suggested that the amino-terminal region of Kap95p was sufficient to mediate active nuclear export. In parallel experiments, microinjected GST-Kap95-(1-77)ΔNES remained localized in the nucleus after incubation at 37°C. Therefore, the NESlike motif was necessary for nuclear export.
The NES in Kap95p Is Required for Import Factor Recycling
To analyze whether the NES was necessary for Kap95p function, experiments in yeast cells were conducted. Mutagenesis studies of reported NESs have determined that the leucine residue at the position equivalent to 63 in Kap95p is important for function (Meyer and Malim, 1994; Wen et al., 1995; Fridell et al., 1996b ; Murphy and Wente, 1996). To examine whether this leucine in Kap95p was important, it was changed to alanine by site-directed mutagenesis (kap95-L63A). KAP95 is an essential gene (Iovine et al., 1995), but a haploid kap95 null strain harboring wild-type KAP95 on both a LEU2 CEN and a URA3 CEN plasmid is viable at all temperatures on media containing 5-fluoroorotic acid (5-FOA) (Fig. 4 A). To test whether the putative NES in Kap95p was necessary for cell viability, a plasmid shuffle assay was conducted with the kap95L63A mutated gene on the LEU2 CEN plasmid. If the mutated gene retains its essential function, the strain will grow in the absence of the wild-type KAP95 URA3 plasmid and form colonies on the 5-FOA media. While wildtype KAP95 complemented the kap95 null phenotype at all growth temperatures, the kap95-L63A allele did not grow at 37°C (Fig. 4 A). Therefore, changing the leucine residue at position 63 to alanine yielded a temperaturesensitive kap95 mutant.
Figure 4.
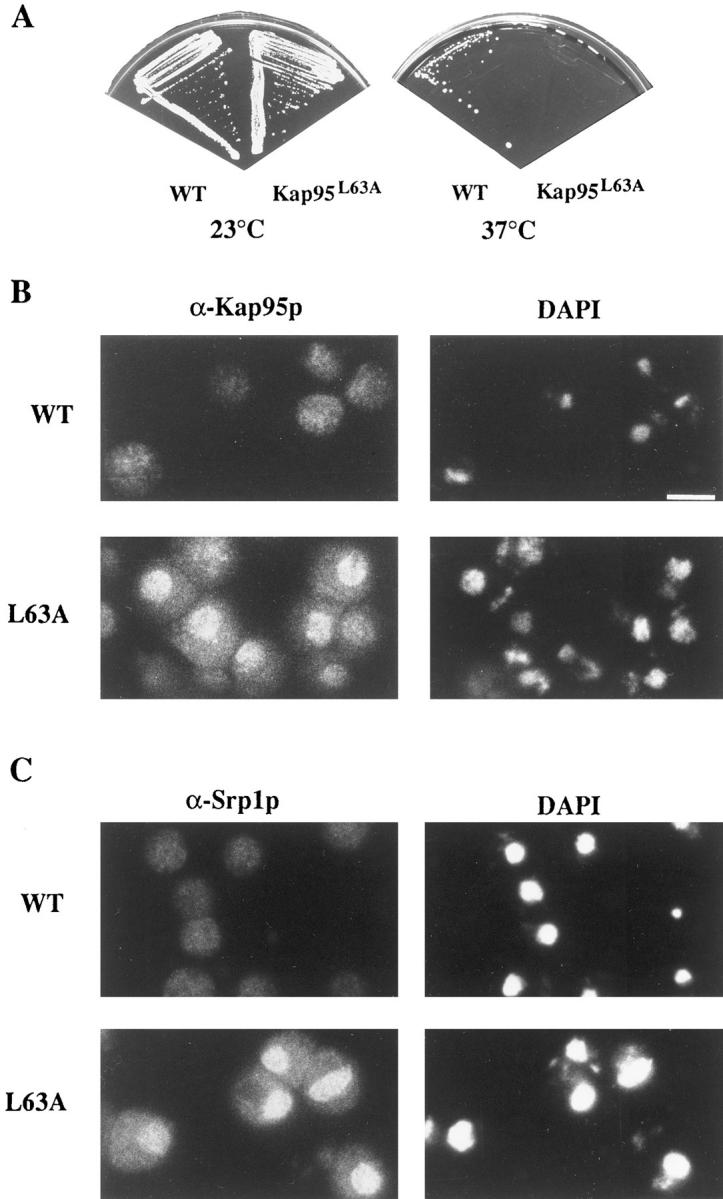
The NES in Kap95p is required for function. (A) Changing amino acid 63 in Kap95p from a leucine (L) to an alanine (A) causes a temperature-sensitive defect in haploid yeast cells. (B) kap95pL63A accumulates in the nucleus of arrested cells. Wild-type KAP95 (SWY1312) and mutant kap95-L63A (SWY1313) cells were grown at 23°C in YEPD before shifting to 37°C for 3 h. Cells grown at 37°C were fixed for 2.5 min and processed for immunofluorescence microscopy with anti-Kap95p antibodies. (C) The distribution of endogenous Srp1p is also perturbed in temperature-arrested kap95-L63A cells. Cells grown at 37°C were processed exactly as in B, except they were incubated with anti– importin α (Srp1p) antibody. Exposure and photograph printing time were identical for wild-type and mutant cells with a given antibody. Coincident staining of the nuclei with DAPI is shown. Bar, 5 μm.
The step of import that would likely require a Kap95p export event is the recycling process. If the role of the NES in Kap95p is to mediate export of itself back to the cytoplasm after a round of import, then Kap95p with the mutated NES should accumulate in the nucleus at the restrictive temperature. To test this hypothesis, immunofluorescence microscopy was used to examine the location of wild-type Kap95p and mutated kap95pL63A in cells grown at 23°C and shifted to 37°C. At 37°C, wild-type Kap95p was localized evenly throughout the cells (Fig. 4 B). In similarly treated kap95-L63A mutant cells arrested at 37°C, the staining pattern was strikingly different. A significant fraction of the mutated kap95pL63A protein was localized within the nucleus as well as some at the nuclear rim (Fig. 4 B). To confirm this localization, confocal microscopy was performed on arrested kap95-L63A cells that were double labeled with anti-Kap95p antibodies and either the anti-nucleoporin antibody mAb414 or propidium iodide for DNA. As shown in Fig. 5 A, the propidium iodide staining was intranuclear (red), the mAb414 staining was confined to the nuclear envelope (green), and there was also no detectable colocalization (yellow signal) between the two signals. Thus, intranuclear vs nuclear envelope localization was distinguishable by this method. In contrast, the propidium iodide and anti-Kap95p signals significantly colocalized as reflected by the yellow signal (Fig. 5 B). Some colocalization was also detected between the anti-Kap95p and mAb414 signals (Fig. 5 C). Therefore, at 37°C, kap95pL63A accumulated in the nucleus with a fraction also at the nuclear envelope. This suggested that lack of NES function inhibited Kap95p recycling.
Figure 5.

Confocal microscopy reveals that kap95pL63A accumulates both at the nuclear envelope and inside the nucleus. Mutant kap95L63A (SWY1313) cells were grown and processed exactly as in Fig. 4. Cells in A were labeled with the combination of propidium iodide (red; DNA stain) and mAb414 (green; nuclear envelope staining); cells in B were labeled with propidium iodide (red) and an antiKap95p antibody (green); cells in C were labeled with mAb414 (green) and an anti-Kap95p antibody (red). The combined images are shown in the third column, where yellow represents the overlap of red and green staining.
Studies by others have suggested that during the docking and translocation steps of import, Kap95p is associated with Srp1p. However, Kap95p and Srp1p apparently dissociate when the import complex reaches the nucleoplasmic face of the NPC (Gorlich et al., 1995b ; Moroianu et al., 1995b ). Based on these results, it has been proposed that Kap95p and Srp1p recycle to the cytoplasm by independent mechanisms (Gorlich and Mattaj, 1996). Indirect immunofluorescence microscopy was used to analyze the behavior of Srp1p in the kap95-L63A cells. Srp1p localization in KAP95 wild-type cells was diffuse and throughout the cells. However, Srp1p was markedly accumulated in the nuclei of temperature-arrested kap95-L63A cells (Fig. 4 C). Confocal microscopy experiments showed Srp1p was localized both inside the nucleus and at the nuclear envelope in a pattern similar to that shown for kap95pL63A (data not shown). Therefore, Srp1p recycling appeared inhibited when Kap95p NES function was blocked.
Nuclear Import Capacity Is Diminished when Kap95p Recycling Is Blocked
Since Kap95p is an essential import factor, mutations that perturb its function should coincidentally alter nuclear protein import capacity. In particular, if Kap95p was not recycled to the cytoplasm, the import capacity of the cell may become noticeably diminished with only limited (newly synthesized) Kap95p available in the cytoplasm. To examine the effect of the temperature-sensitive kap95L63A allele on import, the kap95-L63A strain was transformed with a plasmid expressing an NLS–β-galactosidase fusion protein under control of the inducible GAL10 promoter (Underwood and Fried, 1990). If import is diminished during growth at the nonpermissive temperature (37°C), the NLS–β-galactosidase will not accumulate in the nucleus. To induce reporter protein synthesis, cells grown in raffinose-containing media at 23°C were shifted to galactose-containing media and growth at 37°C. Localization of the fusion protein was determined by indirect immunofluorescence microscopy with anti–β-galactosidase antibodies. When grown at 37°C, the wild-type cells efficiently imported the NLS–β-galactosidase as reflected by the bright nuclear staining (Fig. 6 A). In contrast, the kap95-L63A mutant cells at 37°C showed diminished nuclear staining and the cytoplasmic signal was significantly enhanced (Fig. 6 A). Therefore, the NES-mutated protein was not capable of sustaining wild-type levels of nuclear import capacity at the nonpermissive temperature.
Figure 6.

Nuclear import capacity is diminished in arrested kap95-L63A cells. (A) To assay nuclear import, wild-type (SWY1312) and kap95-L63A (SWY1313) strains were transformed with the plasmid expressing NLS–β-galactosidase under GAL10 (pNLS-E1). Expression was induced by the addition of 2% galactose, and the cells were shifted to growth at 37°C. After 3 h at 37°C, the cells were fixed and processed for immunofluorescence microscopy. Localization of the reporter was determined using mAbs against β-galactosidase. (B) Export of poly(A)+ RNA was not inhibited. Wild-type and kap95-L63A cells grown in YEPD at 23°C were shifted to growth at 37°C and processed for in situ hybridization with a digoxigenin-oligo- (dT)30 probe. FITC-conjugated anti-digoxigenin antibodies were used to localize probe binding. Exposure and printing times are identical for wild-type and mutant cells in the given experiment. Coincident DAPI staining is shown. Bar, 5 μm.
To test whether the kap95-L63A phenotype was specific for import processes, poly(A)+ RNA export capacity was monitored. Wild-type and kap95-L63A cells were grown at 23°C and shifted to 37°C for 3 h, and samples were processed for in situ hybridization with a digoxigenin-labeled oligo(dT)30. The staining with an FITC-conjugated antidigoxigenin antibody was diffuse and cytoplasmic at 37°C in both wild-type and mutant cells (Fig. 6 C). These results suggested that the kap95-L63A cells were apparently competent for poly(A)+ RNA export over the same time frame that import levels were inhibited.
The NES Mutation Inhibits Interaction with Nup116p
Previous studies have reported interactions of Kap95p (and/or its vertebrate homologues) with GLFG nucleoporins (Iovine et al., 1995; Radu et al., 1995b ), FXFG/FG nucleoporins (Kraemer et al., 1995; Moroianu et al., 1995b ; Rexach and Blobel, 1995; Hu et al., 1996; Aitchison et al., 1996), Srp1p (Enenkel et al., 1995; Gorlich et al., 1995a ; Moroianu et al., 1995a ), and Gsp1p (Rexach and Blobel, 1995; Chi et al., 1996; Floer and Blobel, 1996; Gorlich et al., 1996; Lounsbury et al., 1996; Moroianu et al., 1996; Paschal et al., 1996). We predicted that only the interactions necessary for the recycling of Kap95p would be specifically perturbed by the NES mutation.
To examine whether the kap95-L63A mutation perturbed interactions with Srp1p, the FXFG region of Nup1p, and the GLFG region of Nup116p, the yeast two-hybrid assay was conducted (Fields and Song, 1989) (Fig. 7 A). As described in previous studies, wild-type Kap95p and Nup116p–GLFG specifically interact in the two-hybrid assay (Iovine et al., 1995). Fusions of full-length Srp1p and of the Nup1p–FXFG region to the activation domain of Gal4p (Gal4AD) were constructed. These were tested for specific interaction with a fusion of the LexA DNA binding domain (LexABD) to Kap95p. The relative level of transcriptional activation of a reporter lacZ gene with upstream LexA DNA binding sites was analyzed using a color filter assay for β-galactosidase activity. By this assay, Kap95p interacted with both Srp1p and the FXFG region of Nup1p. The Srp1p and Nup1p–FXFG Gal4AD fusions did not show any signal with control LexABD fusions (LexA–Orc2p). For testing the mutated kap95pL63A protein, LexABD–kap95pL63A was transformed into yeast strains harboring plasmids expressing the tester Gal4AD fusions. Compared with the activation levels with wild-type LexABD–Kap95p, Gal4AD–Srp1p and Gal4AD–FXFG interactions with the mutated LexABD–kap95pL63A were unchanged. However, β-galactosidase activity was not detected in the strain with the mutated LexABD–kap95pL63A and the Gal4AD-Nup116p-GLFG.
Figure 7.

The L63A mutation perturbs the Kap95p–GLFG nucleoporin two-hybrid interaction. (A) LexABD fusions of both wild-type Kap95p and mutant kap95pL63A were tested by a color β-galactosidase filter assay for interaction with GAD fusions of the GLFG region of Nup116p and Nup100p, the FXFG region of Nup1p, and Srp1p. Wild-type and mutant LexABD–Kap95p interacted with GAD-Nup1-FXFG and GAD– Srp1p to approximately the same level. Interaction of LexABD–kap95pL63A with GAD-Nup116p-GLFG or GAD-Nup100p-GLFG was not detected. LexABD–Orc2p is included as a specificity control, as well as Gal4AD alone. (B) Bacterially expressed GST–Gsp1p was purified and loaded with GTP. GST–Gsp1pGTP was used in a ligand blot overlay on purified 6x-His-Kap95p (0.5 μg) (lane 1) or 6x-His-kap95L63A (0.5 μg) (lane 2). This binding of GST–Gsp1pGTP was detected with an anti-GST antibody.
The two-hybrid assay could not be used to analyze Gsp1p binding to Kap95p because Gsp1p fused to either Gal4AD or the activation domain of VP16 did not show detectable β-galactosidase activity with wild-type LexA– Kap95p (data not shown). Therefore, Kap95p–Gsp1p interactions were analyzed by a blot overlay assay (Fig. 7 B). Wild-type 6x-His-tagged Kap95p and mutated 6x-Histagged kap95pL63A were bacterially expressed and purified. Equal amounts of each were separated by SDS-PAGE and transferred to nitrocellulose membranes. The blot was incubated with purified, GTP-loaded GST–Gsp1p, and subsequently probed with anti-GST antibodies to detect Gsp1p binding. The signal for GST–Gsp1pGTP binding was virtually identical for both samples. Therefore, the L63A change in Kap95p did not abolish Gsp1p binding.
Based on these results, the NES in the NH2-terminal region of Kap95p appeared critical for the Kap95p–Nup116p interaction. In a previous study (Iovine et al., 1995), we tested a panel of LexABD–Kap95p fusion proteins for interaction with the Gal4AD-Nup116p-GLFG region. The first 130 residues containing the NES were sufficient for interaction and had the highest activation levels in the two-hybrid assay. However, a low level of interaction was also detected with a fusion expressing an LexABD-Kap95pCOOH-terminal fusion lacking the NES region (LexABD– Kap95p[288-861]). Since the single point mutation in the NES alone significantly impaired interaction in the context of the full-length Kap95p protein, this suggests that the low levels of GLFG interaction detected with the LexABD–Kap95p(288-861) may not be important in the context of the full-length protein.
Nup100p May Facilitate Kap95p Recycling in the Absence of Nup116p
Taking into consideration that recycling was inhibited in kap95-L63A cells and that kap95pL63A did not bind Nup116p– GLFG, it is possible that lack of Nup116p–GLFG binding is at least partially responsible for impaired kap95pL63A recycling. However, Kap95p is an essential import factor (Iovine et al., 1995), but nup116 null cells are viable at 23°C (though growth is severely compromised compared with wild type) (Wente et al., 1992). Therefore, if Nup116p has a role in Kap95p recycling, some other factor must be compensating in its absence to maintain recycling at 23°C in nup116 null cells. Obvious candidates would include the other four yeast GLFG nucleoporins. It is unlikely that Nup49p and Nup57p play a role because both are isolated from yeast cells in a complex containing Nsp1p and Nic96p but lacking Kap95p (Grandi et al., 1993, 1995). Nup49p and Nup57p also do not bind Kap95p in respective ligand blot and soluble binding assays (Iovine et al., 1995; Rexach and Blobel, 1995). Nup100p and/or Nup145p are more likely candidates because both bind Kap95p in a ligand blot assay (Iovine et al., 1995), and their amino acid sequence similarities with Nup116p extend outside the GLFG regions (Wente et al., 1992; Wente and Blobel, 1994; Fabre et al., 1994). Moreover, redundant function of NUP100 and NUP145 with NUP116 has been previously suggested by the synthetic lethal phenotypes that result from combining the nup116 null allele with either an nup100 null or an nup145ΔN (amino-terminal deletion) allele (Wente and Blobel, 1994).
To specifically test for functional overlap between the GLFG regions of Nup116p, Nup100p, and Nup145p, strains were constructed that expressed mutated versions of each nucleoporin in which only the respective GLFG region was internally deleted. As shown by the plasmid shuffle assay in Fig. 8 B, haploid cells harboring both the nup116ΔGLFG and nup100ΔGLFG alleles were inviable at all growth temperatures. This demonstrated that the GLFG region of Nup145p was not sufficient for viability in the absence of those from Nup116p and Nup100p. However, the GLFG region from Nup116p was adequate for function because nup100ΔGLFG/nup145ΔGLFG cells were viable (Fig. 8 A). At 23°C, nup116ΔGLFG/nup145ΔGLFG cells were also viable (Fig. 8 B). Overall, these genetic interactions suggested that the GLFG region of Nup100p was necessary in the absence of Nup116p–GLFG function.
Figure 8.

Nup100p–GLFG may also mediate Kap95p recycling. (A) Synthetic lethal interactions between nup100ΔGLFG and nup 145Δ GLFG alleles. The heterozygous nup100ΔGLFG/NUP100 nup145ΔGLFG/NUP145 strain SWY585 was sporulated and dissected. The nup100ΔGLFG and nup145Δ GLFG alleles cosegregated and resulted in the viable haploid nup100ΔGLFG/nup145Δ GLFG strain SWY588. To demonstrate viability of cells, the double mutant haploid (SWY588), wild-type (W303), and respective single mutant parental strains (nup100ΔGLFG, SWY583; nup145Δ GLFG, SWY581) were grown on YPD at 30°C for 2 d. (B) Synthetic lethal interactions between nup116ΔGLFG, nup100Δ GLFG, and nup145ΔGLFG mutants. Strains containing the respective double ΔGLFG mutation combinations are shown on YPD and 5-FOA at 23°C. Strains harboring double deletions of the GLFG regions of NUP116, NUP100, and NUP145 were obtained by dissecting the appropriate diploid strains (see Table I). Confirmation of the nup116ΔGLFG chromosome segregation was determined by immunoblotting with an anti116 carboxy-terminal antibody (Iovine et al., 1995). On the YPD plate, the nup116Δ GLFG (SWY1407), nup100ΔGLFG/nup116ΔGLFG (SWY1406), and the nup145ΔGLFG/nup116ΔGLFG (SWY1429) cells also carry pSW131 (NUP116/URA3); the nup100ΔGLFG (SWY1401) cells also carry pSW132 (NUP100/URA3); and the nup145ΔGLFG (SWY656) cells also carry pSW190. (C) Protein A–Nup100p coimmunoprecipitates Kap95p from isolated nuclei. Nuclear lysates were prepared from SWY1381 and processed as described for Fig. 1. Immunoblots of the eluate fraction were incubated with either the anti-Kap95p antibody or an anti-IgG antibody. The latter antibody was used to detect the position of protein A–Nup100p (uppermost band) and any of its proteolytic fragments (asterisk).
If Nup100p mediates Kap95p recycling in Nup116p's absence, Nup100p should interact with Kap95p in vivo. Coimmunoprecipitation experiments were conducted with yeast cells expressing a protein A–Nup100p fusion protein as described for Fig. 1. Proteins copurifying with protein A–Nup100p were analyzed by immunoblotting with antiKap95p antibodies (Fig. 8 C). Kap95p was present in the fraction. To analyze whether the NES in Kap95p was required for interaction with Nup100p, the two-hybrid assay was conducted (Fig. 7 A). The Gal4AD-Nup100p-GLFG fusion interacted with wild-type LexABD–Kap95p, but β-galactosidase activity was not detected with LexABD– kap95pL63A. These results support the possibility that the GLFG region of Nup100p facilitates Kap95p recycling at 23°C in nup116 null cells.
Discussion
The mechanism for recycling nuclear import factors is poorly defined. We report here the first characterization of components necessary for recycling Kap95p, a factor required for docking the NLS-receptor and ligand to the NPC. First, a nuclear export signal in the amino-terminal region of Kap95p has been characterized. The NES region of Kap95p was necessary and sufficient to mediate nuclear export in COS-1 cells. Moreover, in yeast cells, recycling of the transport factor required a functional NES. A temperature-sensitive mutation in the NES of Kap95p resulted in the nuclear accumulation of the mutated kap95pL63A and an inhibition of nuclear import capacity. Secondly, the NES in Kap95p was also required for interaction with the GLFG regions of Nup116p and Nup100p. The wild-type Kap95p–Nup116p interaction was stable in vitro, and Kap95–Nup116p or Kap95p–Nup100p complexes were isolated from yeast cells. Therefore, we propose that the Kap95p–GLFG nucleoporin interaction has a specific role in recycling the import factor and defines the structural basis of a distinct step in the nuclear import cycle.
We have used two criteria for distinguishing whether a given protein is involved in Kap95p recycling. First, a fraction of Kap95p should be interacting with the protein in yeast cells (in vivo). Second, the interaction must be dependent on the NES in Kap95p. Our conclusion that Nup116p and/or Nup100p are required for Kap95p recycling is based on the observation that Kap95p was isolated from cells in complexes with protein A–tagged Nup116p or Nup100p. Moreover, the binding of Nup116p and Nup100p to Kap95p was significantly inhibited by a lossof-function point mutation in the NES. In contrast, although previous studies have documented in vivo interactions (Enenkel et al., 1995; Aitchison et al., 1996; Chi et al., 1996), Kap95p binding to either Nup1p–FXFG, Srp1p, or Gsp1p was not dependent on a wild-type NES. Thus, we predict that Nup1p–FXFG, Srp1p, and Gsp1p do not directly mediate Kap95p recycling.
It is very likely that Nup116p/Nup100p will not be the only factors required for recycling Kap95p. Thus far, we have not been able to isolate a viable haploid nup116Δ/ kap95-L63A strain (unpublished results). Experiments are currently underway to definitively test for this possible synthetic lethal relationship and to screen for novel genes that are lethal in combination with the kap95-L63A allele. Such genes may encode proteins that are also required for Kap95p recycling. In addition, the protein A–Nup116p complex may contain other cellular components of the nuclear export machinery. Besides Kap95p, Gle2p specifically copurified with protein A–Nup116p. Interestingly, in directed two hybrid assays, Gle2p interacts with Srp1p but not with Kap95p (Murphy et al., 1996). Temperature-arrested gle2 cells exhibit a poly(A)+ RNA defect (Murphy et al., 1996). Therefore, Gle2p may have multiple functions in export. In comparison, Gle1p did not cofractionate. Gle1p is an essential NES-containing protein required for the export of poly(A)+ RNA, and the NES region of Gle1p interacts with Rip1p and Nup100p (Murphy and Wente, 1996). Nup116p and Nup100p could represent parallel pathways for nuclear export that functionally overlap to some degree (Fig. 9). This is suggested by their biochemical similarities and by the synthetic lethality of nup116ΔGLFG/ nup100ΔGLFG cells and the viability of nup116ΔGLFG/ nup145ΔGLFG and nup100Δ GLFG/nup145ΔGLFG cells. Nup116p probably serves a primary role over Nup100p (thus the thicker arrow in Fig. 9) because the most severe growth perturbations are observed when it alone is deleted or overexpressed (Iovine et al., 1995).
Figure 9.

A model for the role of GLFG nucleoporins in nuclear export. The GLFG nucleoporins Nup100p and Nup116p are shown on the nuclear face of the nuclear envelope, whereas the FXFG nucleoporins are shown cytoplasmically faced. Factors that are reported to interact (biochemically and genetically) with the GLFG proteins are diagrammed, with the arrows indicating proposed export pathways. Molecules bound for export are shown as either containing an NES, or are bound to an NES adaptor. The right side of the diagram is based on the recent report of Gle1p by Murphy and Wente (1996). Gle1p may serve as an adaptor to mediate the export of poly(A)+ RNA via associations with the FG repeat protein Rip1p and the GLFG nucleoporin Nup100p. In this report an NES-dependent association of Kap95p with Nup116p and Nup100p is detailed. The NES in Kap95p is required for recycling the import factor. The basis for Srp1p recycling and Gle2p association in the protein A–Nup116p complex will require additional study.
It has been a formidable experimental challenge in the nuclear transport field to determine which step of transport requires particular nucleoporin interactions. Current models have suggested roles for repetitive motif-containing nucleoporins during cytoplasmic-docking and the subsequent inward movement of the NLS-ligand through the NPC to the nucleoplasm (Melchior and Gerace, 1995; Radu et al., 1995b ; Rexach and Blobel, 1995; Gorlich and Mattaj, 1996; Nehrbass and Blobel, 1996). These are largely based on a number of in vitro binding studies that have demonstrated interactions of Kap95p, or its vertebrate homologue, with nucleoporins that contain multiple repeats of “FXFG” peptide sequences separated by highly charged spacers, and with those that have degenerate “FG” repeats (Kraemer et al., 1995; Moroianu et al., 1995b ; Radu et al., 1995a ,b; Rexach and Blobel, 1995; Hu et al., 1996). However, the physiological significance of each of these interactions has not been clarified. The results from this in vivo and in vitro analysis of Kap95p–GLFG interactions suggest the following framework for the role of repeat-containing nucleoporins in import.
Our working model for the dynamics of Kap95p during the import cycle assumes that Kap95p has the capacity to interact with numerous different nucleoporins but each at discrete points in the transport pathway. This suggests that the type of repeat domain in a given nucleoporin will be specific for a given step of transport. The GLFG, FXFG, and FG regions would therefore not only be structurally distinct in terms of amino acid sequence, but also functionally distinct with a given repetitive region possibly dictating the direction of movement through the NPC. Such a scenario has also been suggested in a recent report by Hu et al. (1996). A schematic diagram is presented in Fig. 9, showing the GLFG nucleoporins Nup116p and Nup100p with pivotal roles in recycling and export functions. In contrast, interactions of Kap95p with the FXFG/FG nucleoporins are depicted in the steps required for docking on the cytoplasmic face of the NPC and movement from the cytoplasm to the nucleus. When Kap95p reaches the nucleoplasmic face of the NPC, interaction with GLFG regions may trigger the recycling of Kap95p from the nucleus to the cytoplasm after a round of import. In effect, the Kap95p interaction with Nup116p/Nup100p would serve two roles: the last step of inward movement, and the first step of recycling. The NES region in Kap95p could mediate the export of itself and perhaps associated proteins. Lack of NES function and GLFG nucleoporin binding may result in the disassociation of kap95pL63A from the nuclear envelope and inhibition of recycling to the cytoplasm.
Additional support for the model in Fig. 9 comes from several independent observations. The vertebrate homologue of Kap95p does not enter the nucleoplasm upon release of the import substrate, but instead stays associated with the nuclear envelope (Gorlich et al., 1995b ; Moroianu et al., 1995b ). The NES-like motif in the yeast Kap95p appears conserved in the amino acid sequence for its vertebrate homologue importin β/karyopherin β/p97. The amino acids spanning residues 52–61 of the human, rat, and bovine proteins are “VARVAAGLQI”, with the bold type face highlighting the hydrophobic residues typical of NESs (Chi et al., 1995; Gorlich et al., 1995a ; Radu et al., 1995a ). Finally, the only identified vertebrate GLFG nucleoporin is localized on the nucleoplasmic face of the NPC (Radu et al., 1995b ), and its depletion from Xenopus NPCs/nuclei limits nuclear import capacity in terms of nuclear growth and DNA replication (Powers et al., 1995).
One prediction of this model is a requirement for separate binding sites in Kap95p for the different nucleoporins or distinct conformations of Kap95p upon binding FXFG, FG, or GLFG repeat regions. Support for this interpretation comes from the observation that an FXFG region cannot functionally replace the GLFG region of Nup116p in vivo (Iovine et al., 1995). In addition, the reported in vitro interactions of different FG repeat regions with Kap95p or its vertebrate homologue have different requirements. High affinity binding of Kap95p to an FXFG region requires heterodimer formation with Srp1p (Rexach and Blobel, 1995). In contrast, the previously untested GLFG region of Nup116p (Fig. 2) exhibits stable binding to Kap95p alone. Finally, the amino- and carboxy-terminal FG repeat regions of rat p58 both bind vertebrate Kap95p alone, but only the carboxy-terminal region shows a stable interaction with a ligand/Srp1p/Kap95p complex (Hu et al., 1996).
Before this study, the mechanism of nuclear import could be biochemically divided into two distinct steps: NLS-dependent binding at the cytoplasmic face of the NPC, followed by energy-dependent transport through the pore (Newmeyer and Forbes, 1988; Richardson et al., 1988; Moore and Blobel, 1992). In vertebrate cell assays, import can be specifically blocked in vitro at each of these steps. N-ethylmaleimide treatment, or lack of the Srp1p or Kap95p homologues, inhibits the initial docking step (Adam et al., 1990; Newmeyer and Forbes, 1990; Adam and Gerace, 1991; Gorlich et al., 1995a ; Imamoto et al., 1995a ; Radu et al., 1995a ). Energy depletion or the absence of the small G protein Ran/TC4 results in an accumulation of docked import substrate at the nuclear envelope (Newmeyer and Forbes, 1988; Richardson et al., 1988; Moore and Blobel, 1992, 1993; Melchior et al., 1993). In this study, the accumulation of the import factor kap95pL63A in the nucleus potentially defines a third step in the import cycle. The kap95-L63A cells also exhibited a diminished nuclear import capacity at the nonpermissive temperature. We predict that this apparent import block is not due to the inability of kap95pL63A mutated protein to mediate the docking and translocation steps of import per se. Rather, the accumulation of kap95pL63A in the nucleus because of inhibited recycling may result in a limited cytoplasmic pool of the import factor and thus a corresponding decrease in the rate of import in the cells. Future experiments will be directed toward testing this hypothesis with in vitro import assays and recombinant proteins. Continued analysis of the arrest points for different transport mutants will also allow these models for import to be further tested.
The observation that Srp1p also accumulated in the nucleus of arrested kap95-L63A cells suggests that Srp1p recycling was coincidentally inhibited. This may reflect a codependence of Kap95p and Srp1p recycling. Alternatively, a factor required for Srp1p recycling may not be efficiently imported. Srp1p recycling is sensitive to export processes, as reflected by its accumulation in the nuclei of nup120 mutant cells (Aitchison et al., 1995). In addition, the exchange of GDP for GTP on Gsp1p is required for Srp1p export (Koepp et al., 1996). Since Kap95p and Srp1p form a heterodimer during import (Chi et al., 1995; Enenkel et al., 1995; Gorlich et al., 1995a ; Imamoto et al., 1995c ; Moroianu et al., 1995a ; Radu et al., 1995a ), we tested whether the ∼65-kD band in the protein A–Nup116p complex was Srp1p. Immunoblotting confirmed that Srp1p was present; however, the interaction may not be specific because Srp1p also showed some affinity for protein A–Sepharose alone (our unpublished observations).
In conclusion, an essential role for an NES in recycling the import factor Kap95p has been demonstrated. This is the first description of a step required for import factor recycling and, by virtue of the NES in Kap95p, it is a striking illustration of the mutual dependence of import and export processes. The NES is also necessary for the interaction of Kap95p with the GLFG regions of Nup116p and Nup100p. In combination with other recent studies in our laboratory (Murphy et al., 1996; Murphy and Wente, 1996), this report also highlights specific roles in nuclear export for at least two members of the GLFG nucleoporin family (Nup100p and Nup116p).
Table I.
Yeast Strain Genotype and Construction
| Strain | Genotype | Derivation | ||
|---|---|---|---|---|
| W303 | Mata/Matα ade2-1/ade2-1 ura3-1/ura3-1 his3-11,15/his3-11,15 trp1-1/trp1-1 leu2-3,112/leu2-3,112 can1-100/can1-100 | |||
| W303α | Matα ade2-1 ura3-1 his3-11,15 trp1-1 leu2-3,112 can1-100 | |||
| SWY385 | Mata/Matα ade2-1/ade2-1 ura3-1/ura3-1 his3-11,15/his3-11,15 trpl-1/trp1-1 leu2-3,112/leu2-3,112 can1-100/can1-100 nup116::URA3 nup116ΔGLFG::TRP1/+ | Integrative transformation of SWY30 with pSW251(TRP1) | ||
| SWY386 | Mata ade2-1 ura3-1 his3-11,15 trp1-1 leu2-3,112 can1-100 nup116::URA3 nup116ΔGLFG::TRP1 | Segregant of tetrad from SWY385 | ||
| SWY387 | Matα ade2-1 ura3-1 his3-11,15 trp1-1 leu2-3,112 can1-100 nup116::URA3 nup116ΔGLFG::TRP1 | Segregant of tetrad from SWY385 | ||
| SWY480 | Mata/Matα ade2-1/ade2-1 ura3-1/ura3-1 his3-11,15/his3-11,15 trp1-1/trp1-1 leu2-3,112/leu2-3,112 can1-100/can1-100 nup100::TRP1 nup100ΔGLFG::LEU2/+ | Integrative transformation of SWY7 with pSW263(LEU2) | ||
| SWY545 | Mata/Matα ade2-1/ade2-1 ura3-1/ura3-1 his3-11,15/his3-11,15 trp1-1/trp1-1 leu2-3,112/leu2-3,112 can1-100/can1-100 nup145::LEU2 nup145ΔGLFG::URA3/+ | Integrative transformation of SWY203 with pSW280 (URA3) | ||
| SWY561 | Mata ade2-1 ura3-1 his3-11,15 trp1-1 leu2-3,112 can1-100 kap95::HIS3 pSW271(URA3) | Iovine et al., 1995 | ||
| SWY581 | Mata ade2-1 ura3-1his-11,15 trp1-1 leu2-3,112 can1-100 nup145::LEU2 nup145ΔGLFG::URA3 | Segregant of tretrad from SWY545 | ||
| SWY583 | Mata ade2-1ura3-1 his3-11,15 trp1-1 leu2-3,112 can1-100 nup100::TRP1 nup100ΔGLFG::LEU2 | Segregant of tetrad from SWY480 | ||
| SWY585 | Mata/Matα ade2-1/ade2-1 ura3-1/ura3-1 his3-11,15/his3-11,15 trp1-1/trp1-1 leu2-3,112/leu2-3,112 can1-100/can1-100 nup145::LEU2 nup145ΔGLFG::URA3/+ nup100::TRP1 nup100ΔGLFG:: LEU2/+ | Cross of SWY581 and SWY583 | ||
| SWY588 | Matα ade2-1 ura3-1 his3-11,15 tpr1-1 leu2-3,112 can1-100 nup145::LEU2 nup145ΔGLFG::URA3 nup100::TRP1 nup100ΔGLFG::LEU2 | Segregant of tetrad from SWY585 | ||
| SWY656 | Mata ade2-1 ura3-1 his3-11,15 trp1-1 leu2-3,112 can1-100 nup145::LEU2 pSW363 (HIS3) | Segregant of tetrad from SWY556 | ||
| SWY958 | Mata ade2-1 ura3-1 his3-11,15 trp1-1 leu2-3,112 can1-100 nup116::HIS3 pSW461(URA3) | Transformation of SWY27 with pSW461 (URA3) | ||
| SWY960 | Mata/Matα ade2-1/ade2-1 ura3-1/ura3-1 his3-11,15/his3-11,15 trp1-1/trp1-1 leu2-3,112/leu2-3,112 can1-100/can1-100 nup116::HIS3/nup116::HIS3 pSW461(URA3) | Cross of SWY958 and SWY29 carrying pRS314 (TRP1) | ||
| SWY1312 | Mata ade2-1 ura3-1 his3-11,15 trp1-1 leu2-3,112 can1-100 kap95::HIS3 pSW503(LEU2) | Transformation of SWY561 with pSW503(LEU2) and selected on 5-FOA | ||
| SWY1313 | Mata ade2-1 ura3-1 his3-11,15 trp1-1 leu2-3,112 can1-100 kap95::HIS3 pSW509(LEU2) | Transformation of SWY561 with pSW509(LEU2) and selected on 5-FOA | ||
| SWY1379 | Mata/Matα ade2-1/ade2-1 ura3-1/ura3-1 his3-11,15/his3-11,15 trp1-1/trp1-1 leu2-3,112/leu2-3,112 can1-100/can1-100 nup100::TRP1/nup100::HIS3 | Cross of SWY9 and SWY238 | ||
| SWY1381 | Mata/Matα ade2-1/ade2-1 ura3-1/ura3-1 his3-11,15/his3-11,15 trp1-1/trp1-1 leu2-3,112/leu2-3,112 can1-100/can1-100 nup100::TRP1/nup100::HIS3 ProteinA-NUP100::LEU2 | Integrative transformation of SWY1379 with pSW645 (LEU2) | ||
| SWY1400 | Mata/Matα ade2-1/ade2-1 ura3-1/ura3-1 his3-11,15/his3-11,15 trp1-1/trp1-1 leu2-3,112/leu2-3,112 can1-100/can1-100 nup116::URA3 nup116ΔGLFG::TRP1/+ nup100::TRP1 nup100ΔGLFG:: LEU2/+ | Cross of SWY387 and SWY583 | ||
| SWY1401 | Mata ade2-1 ura3-1 his3-11,15 trp1-1 leu2-3,112 can1-100 nup100::TRP1 nup100ΔGLFG::LEU2 pSW132(URA3) | Transformation of SWY583 with pSW132(URA3) | ||
| SWY1402 | Mata ade2-1 ura3-1 his3-11,15 trp1-1 leu2-3,112 can1-100 nup116ΔGLFG | 5-FOA selected SWY386 | ||
| SWY1404 | Mata/Matα ade2-1/ade2-1 ura3-1/ura3-1 his3-11,15/his3-11,15 trp1-1/trp1-1 leu2-3,112/leu2-3,112 can1-100/can1-100 nup116ΔGLFG/+ nup100::TRP1 nup100ΔGLFG::LEU2/+ | 5-FOA selected SWY1400 | ||
| SWY1405 | Mata/Matα ade2-1/ade2-1 ura3-1/ura3-1 his3-11,15/his3-11,15 trp1-1/trp1-1 leu2-3,112/leu2-3,112 can1-100/can1-100 nup116ΔGLFG/+ nup100::TRP1 nup100ΔGLFG::LEU2/+ pSW131(URA3) | Transformation of SWY1404 with pSW131(URA3) | ||
| SWY1406 | Matα ade2-1 ura3-1 his3-11,15 trp1-1 leu2-3,112 can1-100 nup116ΔGLFG nup100::TRP1 nup100ΔGLFG::LEU2 pSW131(URA3) | Segregant of tetrad from SWY1405 | ||
| SWY1407 | Mata ade2-1 ura3-1 his3-11,15 trp1-1 leu2-3,112 can1-100 nup116ΔGLFG pSW131(URA3) | Transformation of SWY1402 with pSW131(URA3) | ||
| SWY1428 | Mata/Matα ade2-1/ade2-1 ura3-1/ura3-1 his3-11,15/his3-11,15 trp1-1/trp1-1 leu2-3,112/leu2-3,112 can1-100/can1-100 nup116ΔGLFG/+ nup145::LEU2/+ pSW131 (URA3) pSW363 (HIS3) | Cross of SWY656 and SWY1407 | ||
| SWY1429 | Matα ade2-1 ura3-1 his3-11,15 trp1-1 leu2-3,112 can1-100 nup116ΔGLFG nup145::LEU2 pSW131 (URA3) pSW363 (HIS3) | Segregant of tetrad from SWY1428 | ||
| SWY1430 | Mata ade2-1 ura3-1 his3-11,15 trp1-1 leu2-3,112 can1-100 nup116ΔGLFG nup145::LEU2 pSW363 (HIS3) | Segregant of tetrad from SWY1428 after 5-FOA selection |
The following strains have been previously described: SWY7, SWY9 in Wente and Blobel, 1994; SWY27, SWY29 in Wente and Blobel, 1993; SWY30 in Wente et al., 1992; SWY491 and SWY561 in Iovine et al., 1995; SWY203, SWY556, and SWY656 in Emtage et al., 1997; SWY1017 in Murphy et al., 1996. All plasmids mentioned above are described in Table II.
Table II.
Plasmid Construction
| Plasmid | Construction | Reference | ||
|---|---|---|---|---|
| pRS313 Backbone | ||||
| pSW363 | NUP145 locus lacking base pairs 2,191–2,850 (the GLFG region) | Emtage et al., 1997 | ||
| pRS315 Backbone | ||||
| pSW503 | Entire KAP95 locus in BamHI in pRS315 | This study | ||
| pSW509 | Entire kap95-L63A locus in BamHI in pRS315 | This study* | ||
| pSW696 | Entire kap95-ΔNES locus in BamHI in pRS315 | This study* | ||
| pRS316 Backbone | ||||
| pSW131 | Entire NUP116 locus in SalI/Xbal of pRS316 | Wente and Blobel, 1994 | ||
| pSW132 | Entire NUP100 locus is SalI/Xbal of pRS316 | Wente and Blobel, 1994 | ||
| pSW190 | Entire NUP145 locus in BamHI/SalI of pRS316 | Emtage et al., 1997 | ||
| pSW271 | Entire KAP95 locus in BamHI of pRS316 | Iovine et al., 1995 | ||
| pSW461 | Insertion of five tandem IgG-binding domains into NsiI site at base pair 990 of NUP116 locus | This study* | ||
| pRS304 Backbone | ||||
| pSW251 | NUP116 locus lacking base pairs 1,260–2,873 (the GLFG region) | This study | ||
| pRS305 Backbone | ||||
| pSW263 | NUP100 locus lacking base pair 890–2,657 (the GLFG region) | This study | ||
| pSW645 | Insertion of five tandem IgG-binding domains into NsiI site at base pair 1,253 of NUP100 locus | This study | ||
| pRS306 Backbone | ||||
| pSW280 | NUP145 locus lacking base pairs 2,191–2,850 (the GLFG region) | |||
| pCH436 Backbone | ||||
| pSW511 | kap95-L63A coding region in BamHI | This study | ||
| pCH358 Backbone | ||||
| pSW368 | SRP1 coding region in BamHI | Murphy et al., 1995 | ||
| pSW392 | Fragment from base pair 890–2,657 (GLFG domain) of NUP100 locus in BamHI | Murphy and Wente, 1996 | ||
| pSW608 | Fragment from base pair 1,294–2,446 of (FXFG domain) NUP1 locus in BamHI | This study | ||
| pGEX-3X Backbone | ||||
| pSW304 | Fragment from base pair 1,260–2,873 (GLFG domain) of NUP116 locus in BamHI | This study | ||
| pSW568 | Fragment from base pair 3,383–3,641 (encoding amino acids 1–77) of KAP95 locus in BamHI/EcoRI | This study | ||
| pSW570 | Fragment from base pair 3,383–3,641 (encoding amino acids 1–77) of kap95-ΔNES locus in BamHI/EcoRI | This study | ||
| pGEX-2T Backbone | ||||
| pPS826 | GSP1 coding region | Corbett et al., 1995 | ||
| pQE-32 Backbone | ||||
| pSW329 | KAP95 coding region in BamHI | This study | ||
| pSW571 | kap95-L63A coding region in BamHI | This study | ||
Vector backbone references for pRS313, pRS315, pRS316, pRS304, pRS305, and pRS306 (Sikorski and Hieter, 1989); pCH436 for LexABD fusion proteins, and pCH358 for GAL4AD fusion proteins (generous gifts of C. Hardy); pQE-32 (Qiagen); pGEX-3X (Pharmacia Fine Chemicals).
See Materials and Methods section for a more detailed description.
Acknowledgments
We thank D. Schafer, R. Murphy, J. Cooper, and R. Mecham for generous assistance with the microinjection experiments; M. Levy for Kap95p localization assistance; D. Gorlich for anti–importin α (Srp1p) antibodies; and C. Dingwall and P. Silver for providing Gsp1p plasmids. We are indebted to J. Watkins for excellent technical assistance, and J. Cooper, C. Hardy, D. Schnell, and members of the Wente laboratory for critical discussion and comments on the manuscript.
Footnotes
1. Abbreviations used in this paper: DAPI, 4′,6-diamidino-2-phenylindole; 5-FOA, 5-fluoroorotic acid; GST, glutathione-S-transferase; HSA, human serum albumin; NES, nuclear export signal; NLS, nuclear localization signal; NPC, nuclear pore complex; PKI, protein kinase inhibitor.
M.K. Iovine performed this work as a predoctoral trainee supported by National Institutes of Health (NIH) training grant 2T32GM07067-21. This work was supported by a grant from the NIH, GM51219-02, and a Junior Faculty Research Award from American Chemical Society to S.R. Wente.
Please address all correspondence to Susan R. Wente, Department of Cell Biology and Physiology, Box 8228, Washington University School of Medicine, 660 S. Euclid Avenue, St. Louis, MO 63110. Tel.: (314) 362-2713. Fax: (314) 362-7463. e-mail: swente@cell.bio.wustl.edu
References
- Adam EJ, Adam SA. Identification of cytosolic factors required for nuclear location sequence-mediated binding to the nuclear envelope. J Cell Biol. 1994;125:547–555. doi: 10.1083/jcb.125.3.547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adam SA, Gerace L. Cytosolic proteins that specifically bind nuclear location signals are receptors for nuclear import. Cell. 1991;66:837–847. doi: 10.1016/0092-8674(91)90431-w. [DOI] [PubMed] [Google Scholar]
- Adam SA, Marr RS, Gerace L. Nuclear protein import in permeabilized mammalian cells requires soluble cytoplasmic factors. J Cell Biol. 1990;111:807–816. doi: 10.1083/jcb.111.3.807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aitchison JD, Blobel G, Rout MP. Nup120p: a yeast nucleoporin required for NPC distribution and mRNA transport. J Cell Biol. 1995;131:1659–1675. doi: 10.1083/jcb.131.6.1659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aitchison JD, Blobel G, Rout MP. Kap104p: a karyopherin involved in the nuclear transport of messenger RNA binding proteins. Science (Wash DC) 1996;274:624–627. doi: 10.1126/science.274.5287.624. [DOI] [PubMed] [Google Scholar]
- Bogerd HP, Fridell RA, Madore S, Cullen BR. Identification of a novel cellular cofactor for the Rev/Rex class of retroviral regulatory proteins. Cell. 1995;82:485–494. doi: 10.1016/0092-8674(95)90437-9. [DOI] [PubMed] [Google Scholar]
- Breeden L, Nasmyth K. Regulation of the yeast HO gene. Cold Spring Harbor Symp Quant Biol. 1985;50:643–650. doi: 10.1101/sqb.1985.050.01.078. [DOI] [PubMed] [Google Scholar]
- Chi NC, Adam EJ, Adam SA. Sequence and characterization of cytoplasmic nuclear protein import factor p97. J Cell Biol. 1995;130:265–274. doi: 10.1083/jcb.130.2.265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chi NC, Adam EJ, Visser GD, Adam SA. RanBP1 stabilizes the interaction of Ran with p97 in nuclear protein import. J Cell Biol. 1996;135:559–569. doi: 10.1083/jcb.135.3.559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis LI, Blobel G. Identification and characterization of a nuclear pore complex protein. Cell. 1986;45:699–709. doi: 10.1016/0092-8674(86)90784-1. [DOI] [PubMed] [Google Scholar]
- Emtage, J.L.T., M. Bucci, J.L. Watkins, and S.R. Wente. 1997. Defining the essential functional regions of the nucleoporin Nup145p. J. Cell Sci. 110: In press. [DOI] [PubMed]
- Enenkel C, Blobel G, Rexach M. Identification of a yeast karyopherin heterodimer that targets import substrate to mammalian nuclear pore complexes. J Biol Chem. 1995;270:16499–16502. doi: 10.1074/jbc.270.28.16499. [DOI] [PubMed] [Google Scholar]
- Fabre E, Boelens WC, Wimmer E, Mattaj IW, Hurt EC. Nup145p is required for nuclear export of mRNA and binds homopolymeric RNA in vitro via a novel conserved motif. Cell. 1994;78:275–289. doi: 10.1016/0092-8674(94)90297-6. [DOI] [PubMed] [Google Scholar]
- Fields S, Song O. A novel genetic system to detect protein-protein interactions. Nature (Lond) 1989;340:245–246. doi: 10.1038/340245a0. [DOI] [PubMed] [Google Scholar]
- Fischer U, Huber J, Boelens WC, Mattaj IW, Luhrmann B. The HIV-1 Rev activation domain is a nuclear export signal that accesses an export pathway used by specific cellular RNAs. Cell. 1995;82:475–483. doi: 10.1016/0092-8674(95)90436-0. [DOI] [PubMed] [Google Scholar]
- Floer M, Blobel G. The nuclear transport factor karopherin β binds stoichiometrically to Ran-GTP and inhibits the Ran GTPase activating protein. J Biol Chem. 1996;271:5313–5316. doi: 10.1074/jbc.271.10.5313. [DOI] [PubMed] [Google Scholar]
- Fridell RA, Bogerd HP, Cullen BR. Nuclear export of late HIV-1 mRNAs occurs via a cellular protein export pathway. Proc Natl Acad Sci USA. 1996a;93:4421–4424. doi: 10.1073/pnas.93.9.4421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fridell RA, Fischer U, Luhrmann R, Meyer BE, Meinkoth JL, Malim MH, Cullen BR. Amphibian transcription factor IIIA proteins contain a sequence element functionally equivalent to the nuclear export signal of human immunodeficiency virus type 1 Rev. Proc Natl Acad Sci USA. 1996b;93:2936–2940. doi: 10.1073/pnas.93.7.2936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fritz CC, Green MR. HIV Rev uses a conserved cellular protein export pathway for the nucleocytoplasmic transport of viral RNAs. Curr Biol. 1996;6:848–854. doi: 10.1016/s0960-9822(02)00608-5. [DOI] [PubMed] [Google Scholar]
- Fritz CC, Zapp ML, Green MR. A human nucleoporin-like protein that specifically interacts with HIV Rev. Nature (Lond) 1995;376:530–533. doi: 10.1038/376530a0. [DOI] [PubMed] [Google Scholar]
- Gerace L. Nuclear export signals and the fast track to the cytoplasm. Cell. 1995;82:341–344. doi: 10.1016/0092-8674(95)90420-4. [DOI] [PubMed] [Google Scholar]
- Gorlich D, Mattaj IW. Nucleocytoplasmic transport. Science (Wash DC) 1996;271:1513–1518. doi: 10.1126/science.271.5255.1513. [DOI] [PubMed] [Google Scholar]
- Gorlich D, Prehn S, Laskey RA, Hartmann E. Isolation of a protein that is essential for the first step of nuclear protein import. Cell. 1994;79:767–778. doi: 10.1016/0092-8674(94)90067-1. [DOI] [PubMed] [Google Scholar]
- Gorlich D, Kostka S, Kraft R, Dingwall C, Laskey RA, Hartmann E, Prehn S. Two different subunits of importin cooperate to recognize nuclear localization signals and bind them to the nuclear envelope. Curr Biol. 1995a;5:383–392. doi: 10.1016/s0960-9822(95)00079-0. [DOI] [PubMed] [Google Scholar]
- Gorlich D, Vogel F, Mills AD, Hartmann E, Laskey RA. Distinct functions for the two importin subunits in nuclear protein import. Nature (Lond) 1995b;377:246–248. doi: 10.1038/377246a0. [DOI] [PubMed] [Google Scholar]
- Gorlich D, Pante N, Kutay U, Aebi U, Bischoff FR. Identification of different roles for RanGDP and RanGTP in nuclear protein import. EMBO (Eur Mol Biol Organ) J. 1996;15:5584–5594. [PMC free article] [PubMed] [Google Scholar]
- Grandi P, Doye V, Hurt EC. Purification of NSP1 reveals complex formation with ‘GLFG' nucleoporins and a novel nuclear pore protein NIC96. EMBO (Eur Mol Biol Organ) J. 1993;12:3061–3071. doi: 10.1002/j.1460-2075.1993.tb05975.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grandi P, Schlaich N, Tekotte H, Hurt EC. Functional interaction of Nic96p with a core nucleoporin complex consisting of Nsp1p, Nup49p, and a novel protein Nup57p. EMBO (Eur Mol Biol Organ) J. 1995;14:76–87. doi: 10.1002/j.1460-2075.1995.tb06977.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardy CFJ. OAF1, and essential ORC2 associated factor, plays a role in DNA replication. Mol Cell Biol. 1996;16:1832–1841. doi: 10.1128/mcb.16.4.1832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu T, Guan T, Gerace L. Molecular and functional characterization of the p62 complex, an assembly of nuclear pore complex glycoproteins. J Cell Biol. 1996;134:589–601. doi: 10.1083/jcb.134.3.589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imamoto N, Shimamoto T, Kose S, Takao T, Tachibana T, Matsubae M, Sekimoto T, Shimonishi Y, Yoneda Y. The nuclear pore-targeting complex binds to nuclear pores after association with a karophile. FEBS Lett. 1995a;368:415–419. doi: 10.1016/0014-5793(95)00699-a. [DOI] [PubMed] [Google Scholar]
- Imamoto N, Shimamoto T, Takeo T, Tachibana T, Kose S, Matsubae M, Sekimoto T, Shimonishi Y, Yoneda Y. In vivo evidence for involvement of a 58 kDa component of the nuclear-pore targeting complex in nuclear protein import. EMBO (Eur Mol Biol Organ) J. 1995b;14:3617–3626. doi: 10.1002/j.1460-2075.1995.tb00031.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imamoto N, Tachibana T, Matsubae M, Yoneda Y. A karoyphilic protein forms a stable complex with cytoplasmic components prior to nuclear pore binding. J Biol Chem. 1995c;270:8559–8565. doi: 10.1074/jbc.270.15.8559. [DOI] [PubMed] [Google Scholar]
- Iovine MK, Watkins JL, Wente SR. The GLFG repetitive region of the nucleoporin Nup116p interacts with Kap95p, an essential yeast nuclear import factor. J Cell Biol. 1995;131:1699–1713. doi: 10.1083/jcb.131.6.1699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito H, Fukuda Y, Murata K, Kimura A. Transformation of intact yeast cells treated with alkali cations. J Bacteriol. 1983;153:163–168. doi: 10.1128/jb.153.1.163-168.1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koepp DM, Wong DH, Corbett AH, Silver PA. Dynamic interactions of the nuclear import receptor and its interactions with transport factors. J Cell Biol. 1996;133:1163–1176. doi: 10.1083/jcb.133.6.1163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kraemer DM, Strambio de Castilla C, Blobel G, Rout MP. The essential yeast nucleoporin NUP159 is located on the cytoplasmic side of the nuclear pore complex and serves in karyopherin-mediated binding of transport substrate. J Biol Chem. 1995;270:19017–19021. doi: 10.1074/jbc.270.32.19017. [DOI] [PubMed] [Google Scholar]
- Loeb JD, Schlenstedt G, Pellman D, Kornitzer D, Silver PA, Fink GR. The yeast nuclear import receptor is required for mitosis. Proc Natl Acad Sci USA. 1995;92:7647–7651. doi: 10.1073/pnas.92.17.7647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lounsbury KM, Richards SA, Perlungher RR, Macara IG. Ran binding domains promote the interaction of Ran with p97/β-karopherin, linking the docking and translocation steps of nuclear import. J Biol Chem. 1996;271:2357–2360. doi: 10.1074/jbc.271.5.2357. [DOI] [PubMed] [Google Scholar]
- Melchior F, Gerace L. Mechanisms of nuclear protein import. Curr Opin Cell Biol. 1995;7:310–318. doi: 10.1016/0955-0674(95)80084-0. [DOI] [PubMed] [Google Scholar]
- Melchior F, Paschal B, Evans J, Gerace L. Inhibition of nuclear protein import by nonhydrolyzable analogues of GTP and identification of the small GTPase Ran/TC4 as an essential transport factor. J Cell Biol. 1993;123:1649–1659. doi: 10.1083/jcb.123.6.1649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melchior F, Guan T, Yokoyama N, Nishimoto T, Gerace L. GTP hydrolysis by Ran occurs at the nuclear pore complex in an early step of protein import. J Cell Biol. 1995;131:571–581. doi: 10.1083/jcb.131.3.571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer BE, Malim MH. The HIV-1 Rev trans-activator shuttles between the nucleus and the cytoplasm. Genes & Dev. 1994;8:1538–1547. doi: 10.1101/gad.8.13.1538. [DOI] [PubMed] [Google Scholar]
- Michael WM, Choi M, Dreyfuss G. A nuclear export signal in hnRNP A1: a signal-mediated, temperature-dependent nuclear protein export pathway. Cell. 1995;83:415–422. doi: 10.1016/0092-8674(95)90119-1. [DOI] [PubMed] [Google Scholar]
- Moore MS, Blobel G. The two steps of nuclear import, targeting to the nuclear envelope and translocation through the nuclear pore, require different cytosolic factors. Cell. 1992;69:939–950. doi: 10.1016/0092-8674(92)90613-h. [DOI] [PubMed] [Google Scholar]
- Moore MS, Blobel G. The GTP-binding protein Ran/TC4 is required for protein import into the nucleus. Nature (Lond) 1993;365:661–663. doi: 10.1038/365661a0. [DOI] [PubMed] [Google Scholar]
- Moore MS, Blobel G. A G protein involved in nucleocytoplasmic transport: the role of Ran. Trends Biochem Sci. 1994;19:211–216. doi: 10.1016/0968-0004(94)90024-8. [DOI] [PubMed] [Google Scholar]
- Moroianu J, Blobel G, Radu A. Previously identified protein of uncertain function is karyopherin alpha and together with karyopherin beta docks import substrate at nuclear pore complexes. Proc Natl Acad Sci USA. 1995a;92:2008–2011. doi: 10.1073/pnas.92.6.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moroianu J, Hijikata M, Blobel G, Radu A. Mammalian karyopherin alpha 1 beta and alpha 2 beta heterodimers: alpha 1 or alpha 2 subunit binds nuclear localization signal and beta subunit interacts with peptide repeat-containing nucleoporins. Proc Natl Acad Sci USA. 1995b;92:6532–6536. doi: 10.1073/pnas.92.14.6532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moroianu J, Blobel G, Radu A. Nuclear protein import: Ran-GTP dissociates the karopherin αβ heterodimer by displacing α from an overlapping binding site on β. Proc Natl Acad Sci USA. 1996;93:7059–7062. doi: 10.1073/pnas.93.14.7059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy R, Wente S. An RNA export mediator with an essential nuclear export signal. Nature (Lond) 1996;383:357–360. doi: 10.1038/383357a0. [DOI] [PubMed] [Google Scholar]
- Murphy R, Watkins JL, Wente SR. GLE2, a S. cerevisiae homologue of the S. pombe export factor RAE1, is required for nuclear pore complex structure and function. Mol Biol Cell. 1996;7:1921–1937. doi: 10.1091/mbc.7.12.1921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nehrbass U, Blobel G. Role of the nuclear transport factor p10 in nuclear import. Science (Wash DC) 1996;272:120–122. doi: 10.1126/science.272.5258.120. [DOI] [PubMed] [Google Scholar]
- Newmeyer DD, Forbes DJ. Nuclear import can be separated into distinct steps: in vitro nuclear pore binding and translocation. Cell. 1988;52:641–653. doi: 10.1016/0092-8674(88)90402-3. [DOI] [PubMed] [Google Scholar]
- Newmeyer DD, Forbes DJ. An N-ethylmaleimide–sensitive cytosolic factor necessary for nuclear protein import: requirement in signal- mediated binding to the nuclear pore. J Cell Biol. 1990;110:547–557. doi: 10.1083/jcb.110.3.547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paschal BM, Gerace L. Identification of NTF2, a cytosolic factor for nuclear import that interacts with nuclear pore complex protein p62. J Cell Biol. 1995;129:925–937. doi: 10.1083/jcb.129.4.925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paschal BM, Delphin C, Gerace L. Nucleotide-specific interaction of Ran/TC4 with nuclear transport factors NTF2 and p97. Proc Natl Acad Sci USA. 1996;93:7679–7683. doi: 10.1073/pnas.93.15.7679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinol-Roma S, Dreyfuss G. Shuttling of pre-mRNA binding proteins between nucleus and cytoplasm. Nature (Lond) 1992;355:730–732. doi: 10.1038/355730a0. [DOI] [PubMed] [Google Scholar]
- Powers MA, Forbes DJ. Cytosolic factors in nuclear transport: what's importin? . Cell. 1994;79:931–934. doi: 10.1016/0092-8674(94)90024-8. [DOI] [PubMed] [Google Scholar]
- Powers MA, Macaulay C, Masiarz FR, Forbes DJ. Reconstituted nuclei depleted of a vertebrate GLFG nuclear pore protein, p97, import but are defective in nuclear growth and replication. J Cell Biol. 1995;128:721–736. doi: 10.1083/jcb.128.5.721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Radu A, Blobel G, Moore MS. Identification of a protein complex that is required for nuclear protein import and mediates docking of import substrate to distinct nucleoporins. Proc Natl Acad Sci USA. 1995a;92:1769–1773. doi: 10.1073/pnas.92.5.1769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Radu A, Moore MS, Blobel G. The peptide repeat domain of nucleoporin Nup98 functions as a docking site in transport across the nuclear pore complex. Cell. 1995b;81:215–222. doi: 10.1016/0092-8674(95)90331-3. [DOI] [PubMed] [Google Scholar]
- Rexach M, Blobel G. Protein import into nuclei: association and dissociation reactions involving transport substrate, transport factors, and nucleoporins. Cell. 1995;83:683–692. doi: 10.1016/0092-8674(95)90181-7. [DOI] [PubMed] [Google Scholar]
- Richardson WD, Mills AD, Dilworth SM, Laskey RA, Dingwall C. Nuclear protein migration involves two steps: rapid binding at the nuclear envelope followed by slower translocation through nuclear pores. Cell. 1988;52:655–664. doi: 10.1016/0092-8674(88)90403-5. [DOI] [PubMed] [Google Scholar]
- Rout MP, Blobel G. Isolation of the yeast nuclear pore complex. J Cell Biol. 1993;123:771–783. doi: 10.1083/jcb.123.4.771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rout MP, Kilmartin JV. Components of the yeast spindle and spindle pole body. J Cell Biol. 1990;111:1913–1927. doi: 10.1083/jcb.111.5.1913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sambrook, J., E.F. Fritsch, and T. Maniatis. 1989. Molecular Cloning: A Laboratory Manual. Second edition. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY. 545 pp.
- Sherman, F., G.R. Fink, and J.B. Hicks. 1986. Methods in Yeast Genetics. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY.
- Sikorski RS, Hieter P. A system of shuttle vectors and yeast host strains designed for efficient manipulation of DNA in Saccharomyces cerevisiae. . Genetics. 1989;122:19–27. doi: 10.1093/genetics/122.1.19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simos G, Hurt EC. Nucleocytoplasmic transport: factors and mechanisms. FEBS Lett. 1995;369:107–112. doi: 10.1016/0014-5793(95)00674-x. [DOI] [PubMed] [Google Scholar]
- Stutz F, Neville M, Rosbash M. Identification of a novel nuclear pore-associated protein as a functional target of the HIV-1 Rev protein in yeast. Cell. 1995;82:495–506. doi: 10.1016/0092-8674(95)90438-7. [DOI] [PubMed] [Google Scholar]
- Underwood MR, Fried HM. Characterization of nuclear localizing sequences derived from yeast ribosomal protein L29. EMBO (Eur Mol Biol Organ) J. 1990;9:91–99. doi: 10.1002/j.1460-2075.1990.tb08084.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weis K, Mattaj IW, Lamond AI. Identification of hSRP1 alpha as a functional receptor for nuclear localization sequences. Science (Wash DC) 1995;268:1049–1053. doi: 10.1126/science.7754385. [DOI] [PubMed] [Google Scholar]
- Wen W, Meinkoth JL, Tsien RY, Taylor SS. Identification of a signal for rapid export of proteins from the nucleus. Cell. 1995;82:463–473. doi: 10.1016/0092-8674(95)90435-2. [DOI] [PubMed] [Google Scholar]
- Wente SR, Blobel G. A temperature-sensitive NUP116null mutant forms a nuclear envelope seal over the yeast nuclear pore complex thereby blocking nucleocytoplasmic traffic. J Cell Biol. 1993;123:275–284. doi: 10.1083/jcb.123.2.275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wente SR, Blobel G. NUP145encodes a novel yeast glycine-leucine-phenylalanine–glycine (GLFG) nucleoporin required for nuclear envelope structure. J Cell Biol. 1994;125:955–969. doi: 10.1083/jcb.125.5.955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wente SR, Rout MP, Blobel G. A new family of yeast nuclear pore complex proteins. J Cell Biol. 1992;119:705–723. doi: 10.1083/jcb.119.4.705. [DOI] [PMC free article] [PubMed] [Google Scholar]