

Abstract
In this paper, we have investigated the effects of the pore-forming toxin aerolysin, produced by Aeromonas hydrophila, on mammalian cells. Our data indicate that the protoxin binds to an 80-kD glycosyl-phosphatidylinositol (GPI)-anchored protein on BHK cells, and that the bound toxin is associated with specialized plasma membrane domains, described as detergent-insoluble microdomains, or cholesterol-glycolipid “rafts.” We show that the protoxin is then processed to its mature form by host cell proteases. We propose that the preferential association of the toxin with rafts, through binding to GPI-anchored proteins, is likely to increase the local toxin concentration and thereby promote oligomerization, a step that it is a prerequisite for channel formation. We show that channel formation does not lead to disruption of the plasma membrane but to the selective permeabilization to small ions such as potassium, which causes plasma membrane depolarization. Next we studied the consequences of channel formation on the organization and dynamics of intracellular membranes. Strikingly, we found that the toxin causes dramatic vacuolation of the ER, but does not affect other intracellular compartments. Concomitantly we find that the COPI coat is released from biosynthetic membranes and that biosynthetic transport of newly synthesized transmembrane G protein of vesicular stomatitis virus is inhibited. Our data indicate that binding of proaerolysin to GPI-anchored proteins and processing of the toxin lead to oligomerization and channel formation in the plasma membrane, which in turn causes selective disorganization of early biosynthetic membrane dynamics.
Many pathogenic bacteria secrete pore-forming toxins that are important determinants of the virulence of these organisms in mammals. Despite the importance of these membrane-damaging toxins in infectious processes, very little is known about the mechanisms by which they interact with nucleated mammalian cells.
Aerolysin is a pore-forming toxin secreted by the human pathogen Aeromonas hydrophila, a member of the Vibrinaceae family. The bacterium leads to a variety of infections ranging from gastroenteritis to deep wound infection and septicemia. Strong evidence implicates aerolysin as an important virulence factor produced by the bacterium (Donta and Haddow, 1978; Daily et al., 1981; Kaper et al., 1981; Janda et al., 1984). Using marker exchange mutagenesis, aerolysin was demonstrated to be required not only for the establishment but also for the subsequent maintenance of systemic infections associated with the bacterium (Chakraborty et al., 1987).
Since aerolysin was found to be hemolytic and able to form pores in lipid bilayer (Bernheimer et al., 1975; Howard and Buckley, 1982), most of the efforts have been focused on understanding the mechanisms leading to membrane insertion and pore formation using artificial membranes as well as erythrocytes as a model target cell (for reviews see Parker et al., 1996; Lesieur et al., 1997). The following sequence of events has emerged from these studies. The toxin is secreted by the bacterium as a dimeric inactive precursor (Howard and Buckley, 1982; van der Goot et al., 1993). Subsequent activation occurs by proteolytic removal of a COOH-terminal peptide (Howard and Buckley, 1985; van der Goot et al., 1992). The toxin then binds to the surface of the erythrocyte via a 47-kD, glycosyl-phosphatidylinositol (GPI)1-anchored glycoprotein that acts as a receptor (Gruber et al., 1994; Cowell et al., 1997). Because of the local increase in toxin concentration, aerolysin undergoes polymerization into a heptameric complex that inserts into the membrane and forms a water-filled channel (Howard and Buckley, 1982; Garland and Buckley, 1988; Wilmsen et al., 1990, 1992; Moniatte et al., 1996) thereby leading to osmotic lysis of the erythrocyte.
However, aerolysin effects on nucleated cells are likely to involve more complex mechanisms as suggested by the variety of pathological effects that this toxin has been shown to trigger. Purified aerolysin was shown to be lethal to mice (Janda et al., 1985; Chakraborty et al., 1987), enterotoxic in rabbit ileal segments (Asao et al., 1984), responsible, at sublethal doses, for the release of inflammatory mediators from granulocytes (Scheffer et al., 1988), chemotactic for human leukocytes (Jin et al., 1992), and cytotoxic to a variety of cell lines (Ljungh and Wadström, 1983). The mechanisms that mediate these various effects remain largely unknown.
In the present paper we have analyzed the effects of aerolysin on BHK cells. We show that proaerolysin binds to specific GPI-anchored receptors within cholesterol-glycolipid “rafts” and is subsequently processed to the mature toxin by host cell proteases. Intoxication leads to dramatic vacuolation in the cell cytoplasm. To our knowledge, such an intracellular effect has never been reported for any other pore-forming toxin. The aerolysin-induced vacuoles were shown to originate from the ER. In agreement with the fact that the early biosynthetic pathway is altered by the toxin, we have shown that transport of newly synthesized membrane proteins to the plasma membrane is inhibited. Our data thus indicate that interaction of proaerolysin with mammalian cells leads to a highly selective perturbation of the organization and dynamics of early biosynthetic membranes.
Materials and Methods
Cells and Materials
Monolayers of BHK cells were grown and maintained as described by Gruenberg et al. (1989) in Glasgow minimal essential medium (GMEM; Sigma Chemical Co., St. Louis), supplemented with 5% FCS, 2 mM l-glutamine under standard tissue culture conditions. For immunofluorescence experiments, cells were grown on 12 × 12-mm glass coverslips. Phosphatidylinositol-specific phospholipase C (PI-PLC) from Bacillus cereus, ionomycin, and A23187 were purchased from Boehringer Mannheim Corp. (Indianapolis, IN), Triton X-100 Ultra Pure from Pierce Chemical Co. (Rockford, IL), Triton X-114 from Merck (Darmstadt, Germany), BSA from Biomol (Hamburg, Germany). Triton X-114 was purified as described by Bordier (1981). Nocodazole, brefeldin A, trypsin/chymotrypsin inhibitor, and cycloheximide were from Sigma Chemical Co.; stock solutions (trypsin/chymotrypsin inhibitor in Hepes buffer saline, pH 7.4; cycloheximide: 10 mg/ml in water) were kept at −20°C. The LIVE/DEAD viability kit from Molecular Probes (Eugene, OR) was used according to the manufacturer's instructions.
Proaerolysin Purification and Iodination
Wild-type and variant proaerolysins were purified as described previously (Buckley, 1990). Concentrations were determined by measuring the OD at 280 nm, considering that a 1-mg/ml sample has an OD of 2.5 (van der Goot et al., 1994). Proaerolysin was labeled with 125I using Iodogen reagent (Pierce Chemical Co.) according to the manufacturer's recommendations. The radiolabeled toxin was separated from the free iodine by gel filtration on a PD10-G25 column (Pharmacia Biotech Sevrage, Uppsala, Sweden) equilibrated with PBS, pH 7.4. We consistently obtained a specific activity of ∼2 × 106 cpm/μg of proaerolysin. Radiolabeled proaerolysin ran as a single band on a SDS gel.
Proaerolysin Binding to BHK Cells
Confluent monolayers of BHK cells (1.5 × 107 cells per dish) were washed three times for 5 min with ice-cold PBS containing 1 mM CaCl2, 1 mM MgCl2, and 0.5% BSA (PBS2+-BSA), and 1 μg/ml of trypsin/chymotrypsin inhibitor. Cells were then incubated at 4°C with proaerolysin in incubation medium (IM) containing GMEM buffered with Hepes, pH 7.4, 1 μg/ml of trypsin/chymotrypsin inhibitor, and 0.5% BSA. In competition experiments, labeled and unlabeled proaerolysin were added simultaneously to the cells. The monolayer was then washed three times for 10 min with PBS2+-BSA at 4°C. The cells were scraped from the dish, collected by centrifugation at 1,500 rpm for 5 min, and then counted in a Packard auto-gamma scintillation spectrometer. Nonspecific binding was determined by incubating cells with radiolabeled proaerolysin in the presence of a 50-fold excess of unlabeled toxin.
Preparation of Triton X-100–Insoluble Membrane Fractions
The detergent extraction and flotation protocols were adapted from previously described procedures (Brown and Rose, 1992; Fra et al., 1994). Approximately 1.5 × 107 BHK cells were scraped, pelleted by centrifugation (5 min at 1,500 rpm), and then resuspended in 0.5 ml of buffer containing 25 mM Tris-HCl, 150 mM NaCl, 5 mM EDTA, 1% Triton X-100, as well as Complete, a cocktail of protease inhibitors (Boehringer Mannheim). The membranes were solubilized by rotary shaking at 4°C for 30 min. The sample was then adjusted to 40% sucrose in a SW40 tube (Beckman Instruments, Inc., Fullerton, CA), overlaid with 8.5 ml of 35% sucrose, topped up with 16% sucrose (in 10 mM Tris-HCl, pH 7.4), and then centrifuged for 18 h at 35,000 rpm at 4°C. The initial load (3 ml) as well as 10 fractions of 1 ml were collected.
PI-PLC Treatment
PI-PLC treatment was performed either on intact or on detergent-solubilized cells. In the first case, cells were incubated with 6 U/ml of PI-PLC in IM for 1 h at 37°C in the presence of 10 μg/ml cycloheximide. PI-PLC treatment of solubilized cells was performed as described by Lisanti et al. (1988). Briefly, BHK cells (3 × 107 cells) were extracted with 2 ml of TBS (10 mM Tris, pH 7.4, 150 mM NaCl, 1 mM EDTA) containing 1% (vol/ vol) Triton X-114 for 1 h at 4°C on a rotary shaker. The cell extract was clarified by centrifugation at 4°C (13,000 rpm for 10 min). The supernatant was subjected to temperature-induced phase separation as described by Bordier (1981) and centrifuged at room temperature for 5 min at 3,000 rpm. The aqueous phase was discarded and the detergent phase was reextracted with 10 volumes of TBS containing 0.06% Triton X-114. In between phase separations, the sample was clarified by centrifugation at 4°C (13,000 rpm for 10 min). The final detergent phase was diluted with 100 mM Tris, 50 mM NaCl, 1 mM EDTA, pH 7.4 to a final volume of 1 ml. The sample was then incubated with or without PI-PLC (6 U/ml) for 1 h at 37°C on an Eppendorf shaker. After incubation, an equivalent volume of TBS containing 2% Triton X-114 was added and the sample was subjected to phase separation. The aqueous phase was collected and reextracted three times by addition of 200 μl of 10% Triton X-114 in TBS without EDTA followed by phase separation.
Potassium Efflux Measurements
Confluent BHK monolayers were washed three times for 10 min with ice-cold PBS2+-BSA and 1 μg/ml of trypsin/chymotrypsin inhibitor. Monolayers were then incubated for 1 h at 4°C with proaerolysin in IM, washed three times for 10 min with PBS2+-BSA, and then further incubated at 37°C with IM for 15 min. Cells were subsequently washed with ice-cold potassium-free choline medium, pH 7.4, containing 129 mM choline-Cl, 0.8 mM MgCl2, 1.5 mM CaCl2, 5 mM citric acid, 5.6 mM glucose, 10 mM NH4Cl, 5 mM H3PO4, and then solubilized with 0.5% Triton X-100 in the same buffer for 20 min at 4°C. The potassium content of the cell lysates was determined by flame photometry using a PYE UNICAM SP9 atomic absorption spectrophotometer (Philips Electron Optics, Mahwah, NJ).
Membrane Potential Measurements
Trypsin-treated BHK cells were washed twice by centrifugation (900 rpm; 5 min) and resuspended in buffer containing 20 mM Hepes, pH 7.4, 143 mM NaCl, 5 mM KCl, 1 mM MgSO4, 1 mM CaCl2, 5.6 mM glucose, and 1 μg/ml trypsin/chymotrypsin inhibitor, to a final density of 1 × 107 cells/ml. 1 × 107 cells in 3 ml of the same buffer were placed in a thermostated quartz cuvette (37°C) and DiS-C3(5) (100 μM in DMSO) was added to a final concentration of 200 nM. Membrane incorporation of the dye was monitored using a fluorometer (excitation 625 nm; emission 670 nm; 10-nm slits). After reaching a steady state fluorescence (3–4 min after dye addition), proaerolysin was added. Maximal depolarization was obtained at the end of each experiment by adding 1 μg/ml (final concentration) of trypsin-activated aerolysin. Single fluorescent traces are expressed as the ratio I(t)/I max (i.e., the fluorescence intensity as a function of time over the fluorescence intensity corresponding to maximal depolarization).
Phase Contrast Microscopy of Living Cells
BHK cells were grown on glass-bottom microwell dishes (Mattek, Ashland, MA) at a density of 106 cells per dish. After the desired treatment, cells were visualized by phase contrast using an inverted microscope (Axiovert TV135; Carl Zeiss Inc., Thornwood, NY) equipped with a cooled slow-scan CCD camera (CH250 [1317 × 1035 pixels]; Photometrics Tucson, Arizona), controlled by a Power Macintosh 8100/100 (Apple Computer, Cupertino, CA). The IP Lab Spectrum 3.0 software (Signal Analytics Corp., Vienna, Virginia) was used for data acquisition.
Antibodies and Transfection
The polyclonal antibodies against proaerolysin were raised in rabbits against purified toxin, and then affinity purified. Rabbit polyclonal antibodies against the mammalian KDEL receptor ERD2 (Griffiths et al., 1994), calnexin (Wada et al., 1991), caveolin-1 (Parton et al., 1994), and the mannose-6-phosphate receptor (Griffiths et al., 1990) were gifts from H-D. Söling (Zentrum Innere Medizin, Göttingen, Germany), A. Helenius (ETH, Zurich, Switzerland), R. Parton (University of Queensland, Brisbane, Australia), and B. Hoflack (Institut Pasteur, Lille, France). Mouse monoclonal antibodies against proaerolysin, the human transferrin receptor (Hopkins and Trowbridge, 1983), tubulin (Scherson et al., 1984), β-COP (maD [Pepperkok et al., 1993]; mannosidase II [Narula et al., 1992]; and the exoplasmic epitope of G protein of vesicular stomatitus virus (VSV-G) [17.2.21.4; Gruenberg and Howell, 1985]) were gifts from J.T. Buckley (University of Victoria, British Columbia, Canada), I. Throwbridge (The Salk Institute for Biological Studies, San Diego, CA), T. Kreis (University of Geneva, Geneva, Switzerland), B. Burke (University of Calgary, Calgary, Canada), and J. Gruenberg (University of Geneva). Monoclonal anti–caveolin-1 antibodies were purchased from Transduction Laboratories (Lexington, KT), polyclonal anti–placental alkaline phosphatase (PLAP) from DAKOPATTS (Copenhagen, Denmark). HRP-conjugated secondary antibodies were obtained from Amersham Corp. (Arlington Heights, IL). Fluorescent secondary antibodies were from Jackson ImmunoResearch Laboratories (West Grove, PA).
Human PLAP transfection experiments were performed as described by Graham and van der Eb (1973) using a PLAP expression construct driven by the Rous Sarcomavirus (RSV) promoter (pBC12/PLAPase), provided by D. Brown (State University of New York, Stony Brook, NY) (Brown et al., 1989). Plasmids were purified using columns (QUIAGEN Inc., Chatsworth, CA) according to the manufacturer's instruction. BHK cells from a confluent 60-cm2 tissue culture dish were diluted 1:80 and seeded on glass coverslips in a six-well tissue culture plate in view of immunofluorescence experiments. Per well, 3 μg of pBC12/PLAPase DNA were used. The expressing cells were analyzed by immunofluorescence after 48-h incubation at 37°C.
Immunofluorescence
Fixation and permeabilization of BHK cells were either performed by incubation for 20 min at room temperature in 3% paraformaldehyde/PBS followed by permeabilization of membranes with 0.1% Triton X-100 in PBS, or with 0.05% saponin in PBS for 4 min at room temperature, or by incubation for 4 min in methanol at −20°C. Unless specified otherwise, cells were fixed before the addition of antibodies. After three washes in PBS-0.5% BSA, fixed cells were reacted for 30 min at room temperature with either anti–KDEL receptor ERD2 (1:250; paraformaldehyde–saponin permeabilization), anti-calnexin (1:500; methanol permeabilization), anti–mannose-6-phosphate receptor (1:200; paraformaldehyde–Triton X-100 permeabilization), anti–caveolin-1 (1:200; methanol permeabilization), anti-transferrin receptor (1:400; methanol permeabilization), anti-tubulin (1:200; methanol permeabilization), anti–β-COP (1:100; methanol permeabilization), anti–mannosidase II (1:500; methanol permeabilization), or anti–VSV-G (1:400; paraformaldehyde–Triton X-100 permeabilization) antibody. Cells were then washed three times with PBS-0.5% BSA and incubated for 30 min at room temperature with FITC-conjugated, goat anti–rabbit or anti–mouse IgG (1:50), or with lissamine rhodamine sulfonyl chloride–conjugated, goat anti–rabbit or anti–mouse IgG (1:25). Immunofluorescence experiments were analyzed with the Zeiss inverted fluorescence microscope previously described, equipped with fluorescein, Cy3, and Cy5 filters.
To induce clustering of PLAP or receptor-bound proaerolysin, cells were incubated on ice with both primary antibodies for 1 h (polyclonal anti-PLAP, 1:35; monoclonal anti-proaerolysin, 1:20), and with both secondary antibodies for 1 h (FITC-conjugated, goat anti–rabbit, 1:50; lissamine rhodamine sulfonyl chloride–conjugated, goat anti–mouse IgG, 1:25) before fixation. Cells were then fixed with 3% paraformaldehyde/PBS for 20 min at room temperature.
Viral Infection
The protocol for infection with mutant tsO45 vesicular stomatitis virus (VSV) was adapted from previously described procedures (Kreis and Lodish, 1986; Pepperkok et al., 1993). Stock virus was diluted into PBS containing 1 mM CaCl2, 1 mM MgCl2, and 1% FCS, and was adsorbed to cells for 1 h at room temperature at a multiplicity of four. Unadsorbed virus was then washed away, and cells were further incubated in regular cell culture medium at 39.5°C for 3 h 10 min. The cells were then incubated with or without 0.38 nM proaerolysin in IM for 20 min at 39.5°C. Cycloheximide (10 μg/ml) was added to the cells before shifting them to 31°C. After 45 min at 31°C, cells were fixed in 3% paraformaldehyde/PBS for 5 min at 4°C, followed by 20 min at room temperature. When necessary, membranes were permeabilized with 0.1% Triton X-100 in PBS for 4 min at room temperature. Cells were then processed for immunofluorescence as described above. Fixed, and fixed and permeabilized cells were reacted with anti–VSV-G antibodies for 10 and 30 min, respectively.
Electron Microscopy
Cells were fixed with 2.5% glutaraldehyde in 100 mM cacodylate buffer, pH 7.35, for 1 h at room temperature. After six washes of 15 min with 100 mM cacodylate buffer, the cells were incubated with 1% osmium in water for 1 h at room temperature. The cells were then washed five times for 10 min in water before processing for Epon embedding (Gruenberg et al., 1989).
Western Blotting and Proaerolysin Overlays
The proaerolysin overlay procedure was adapted from a previously described GTP overlay protocol (Huber et al., 1994). Membrane fractions were run on a 15% SDS gel. The gel was then incubated in 50 mM Tris-HCl, pH 7.4, and 20% glycerol for 15 min. Proteins were then blotted onto a nitrocellulose membrane (Schleicher and Schuell, Inc., Keene, NH) for 5 h at 150 mA in the cold using a wet transfer chamber (Bio-Rad Laboratories, Hercules, CA). The transfer buffer contained 10 mM NaHCO3 and 3 mM Na2CO3, pH 9.8. Membranes were then subjected to Western blotting using the appropriate antibodies or submitted to proaerolysin overlay. For the overlay, the nitrocellulose membrane was incubated in a binding buffer containing 50 mM NaH2PO4, pH 7.5, and 0.3% Tween-20 for 20 min followed by a 2-h incubation in the presence of 1.4 nM radiolabeled or unlabeled proaerolysin diluted in the same buffer. The membrane was then washed six times for 5 min with binding buffer. Binding of proaerolysin was revealed either by autoradiography using BioMax films (Eastman Kodak, Rochester, NY) or using an antiproaerolysin antibody followed by HRP-labeled secondary antibody, which was then revealed using enhanced chemiluminescence (Amersham Corp.).
Analytical Techniques
SDS-PAGE was performed as described by Laemmli (1970). Protein concentration of BHK cellular fractions were determined with bicinchoninic acid (BCA; Pierce Chemical Co.).
Results
Evidence for an 80-kD Proaerolysin Receptor on BHK Cells
As a first step in the characterization of the interaction of proaerolysin with mammalian cells, we have analyzed the binding of 125I-labeled proaerolysin to BHK cells. All experiments were performed at 4°C to prevent possible internalization of the toxin. A detailed analysis of competition with unlabeled toxin showed that 125I-proaerolysin could bind to the cells, and that this binding was progressively inhibited at increasing concentrations of unlabeled toxin (Fig. 1, a); thus indicating that binding was specific. That binding was of high affinity is illustrated by the fact that 16 nM of unlabeled proaerolysin could inhibit binding of 125I-proaerolysin (4 nM) by 50%. Binding could also be inhibited by an excess of the hemolytically inactive G202C-I445C proaerolysin mutant (see below; van der Goot et al., 1994). Association of 125I-proaerolysin to BHK cells reached a plateau after 2 h (Fig. 1 b). Finally, binding was saturable, a maximum being reached at a 125I-proaerolysin concentration of about 50 nM (Fig. 1 c). Binding was essentially irreversible in the time range of our experiments since <10% of 125I-proaerolysin was released after 4 h of incubation in a toxin-free medium (not shown). These experiments show that 125I-proaerolysin binds to high affinity sites on BHK cells.
Figure 1.
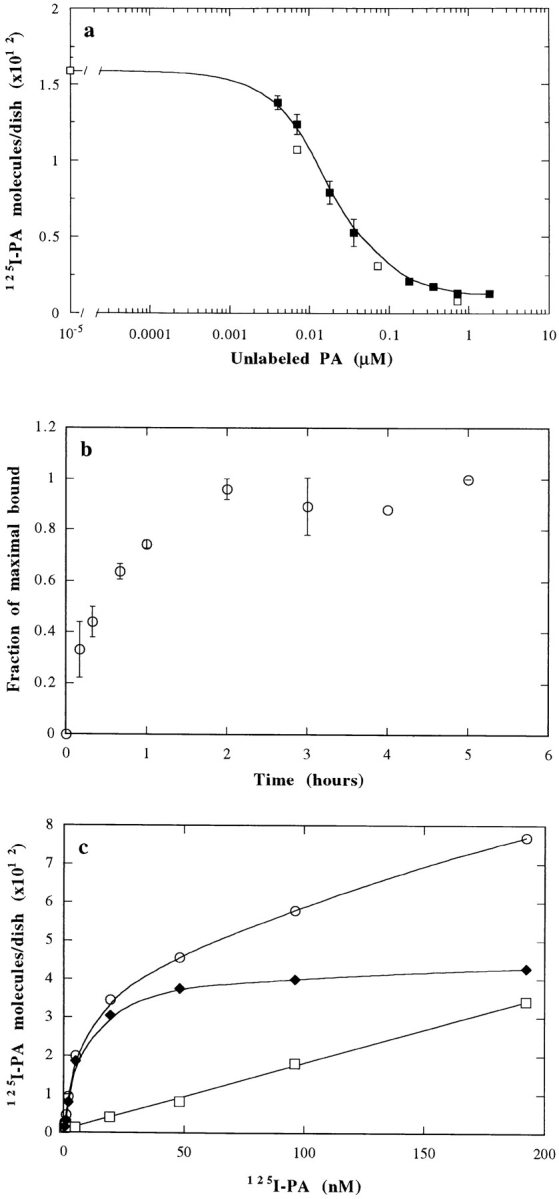
Binding of 125I-proaerolysin to BHK cells. (a) Inhibition of 125I-proaerolysin binding to BHK cells by unlabeled toxin. Cells were incubated for 1 h with 4 nM of 125I-proaerolysin plus the indicated amount of unlabeled wild-type toxin (▪). 50% inhibition occurred in the presence of 16 nM of unlabeled proaerolysin. Bars indicate the standard deviation (n = 3). Binding of wild-type 125I-proaerolysin could also be competed with an excess of unlabeled G202C-I445C mutant proaerolysin (□). (b) Kinetics of specific binding of 125I-proaerolysin to BHK cells at 4°C. Cells were incubated with 4 nM 125I-proaerolysin and washed at the indicated times. Specifically bound proaerolysin was determined by subtracting the values obtained in the presence of 50-fold excess of unlabeled toxin. Bars indicate the standard deviation (n = 3). (c) Concentration dependence of proaerolysin binding to BHK cells. Cells were incubated for 2.5 h at 4°C with 125I-proaerolysin in the absence (○) or in the presence of 50-fold excess unlabeled toxin (□). Specific binding (♦) was calculated by subtraction of the unspecific bound toxin (□) from the total bound toxin (○). The results represent the mean of two independent experiments. Maximal errors were <16%.
To identify proaerolysin-binding proteins, we have used a proaerolysin overlay assay: postnuclear supernatant (PNS) proteins were separated by SDS-PAGE, blotted onto a nitrocellulose membrane that was then incubated with proaerolysin. As shown in Fig. 2, proaerolysin bound predominantly to a protein with an apparent molecular weight of 80 kD, as well as to some lower molecular weight bands (26 and 28 kD), perhaps corresponding to degradation products. Indeed the intensity of the lower molecular weight bands varied from experiments to experiment (also see Fig. 4 b). As observed for binding of the toxin to living cells (see above), binding of 125I-proaerolysin to the proteins on the nitrocellulose membrane could be competed with an excess of unlabeled toxin. The 80-kD protein detected by this assay exhibited the biochemical properties of a membrane protein since it remained bound to a membrane fraction after higher or lower pH washes, or treatment with high salt concentrations (not shown).
Figure 2.

Proaerolysin binds to an 80-kD protein on BHK cells. Cells were treated or not with 2 nM proaerolysin for 1 h at 4°C, thoroughly washed, homogenized, and a PNS was prepared. 20 μg of PNS of toxin-treated cells (lane 1) and control cells (lane 2) were submitted to the proaerolysin overlay assay as described in Materials and Methods. The overlay was performed with unlabeled proaerolysin and revealed with anti-aerolysin antibodies. Arrowheads indicate proaerolysin and aerolysin (that were bound to the plasma membrane, only in lane 1) migrating at their expected molecular weights. Arrows indicate proaerolysin that bound to specific proteins on the nitrocellulose membrane during the overlay procedure.
Figure 4.

PI-PLC inhibits the binding of proaerolysin to BHK cells. Cells were incubated with or without 6 U/ml of PI-PLC for 1 h at 37°C in the presence of 10 μg/ml cycloheximide. The cells were then incubated in presence or absence of 2 nM proaerolysin for 1 h at 4°C, thoroughly washed, and homogenized. (a) The PNSs were probed for the presence of proaerolysin by Western blotting. (b) The PNSs were analyzed by proaerolysin overlay for the presence of proaerolysin binding proteins. Lane 1, control cells; lanes 2, proaerolysin-treated cells; lane 3, PI-PLC and proaerolysin-treated cells. Arrowheads indicate proaerolysin and aerolysin (that were bound to the plasma membrane) migrating at their expected molecular weights. Arrows indicate proaerolysin that bound to specific proteins on the nitrocellulose membrane.
Proaerolysin Binds to Triton X-100–insoluble Microdomains of the Plasma Membrane
Preliminary fractionation experiments indicated that proaerolysin, which had been bound to cells at 4°C, did not comigrate on sucrose gradients with the plasma membrane, thus suggesting that the toxin was associated to specific domains with physical properties distinct from those of the bulk of the plasma membrane. Some well-characterized membrane microdomains are the so-called Triton X-100–insoluble fractions, first described by Brown and Rose (1992), or cholesterol-glycolipid “rafts” (for review see Simons and Ikonen, 1997). We therefore investigated whether proaerolysin bound to BHK cells at 4°C could be recovered in Triton X-100–insoluble fractions. As shown in Fig. 3, proaerolysin was highly enriched in the floated Triton X-100–insoluble fractions (fractions 1 and 2). As expected, these fractions were very much enriched in the caveolae marker caveolin-1, but depleted in the transferrin receptor (Fig. 3). Interestingly the putative 80-kD proaerolysin receptor was also highly enriched in the floated fractions as shown by toxin overlay.
Figure 3.

Proaerolysin binds to detergent-insoluble microdomains of the plasma membrane of BHK cells. Cells treated with proaerolysin (4°C) were solubilized in 1% Triton X-100 and detergent-insoluble fractions were floated on a step sucrose gradient. The 10 fractions from the gradient (lanes 1 to 10; top to bottom of the gradient), the loading region (3 ml) and the starting homogenate were analyzed by Western Blot analysis for the presence of proaerolysin (PA), of caveolin-1 (Cav-1), and of the transferrin receptor (Trf-R). As is often the case, two forms of caveolin-1 could be observed. The nitrocellulose membrane was also subjected to the proaerolysin overlay assay, using radiolabeled toxin, to localize the 80-kD proaerolysin-binding protein (PA-R). 18 μg of protein was loaded in each lane except in lane 1, which contained 13 μg.
Proaerolysin Binds to BHK Cells Via GPI-anchored Proteins
Detergent-insoluble fractions have been shown to be enriched in GPI-anchored proteins (for reviews see Anderson, 1993; Parton, 1996). Therefore the observation that both proaerolysin and its putative receptor were recovered in Triton X-100–insoluble floated fractions prompted us to investigate whether the proaerolysin receptor on BHK cells was GPI anchored. As shown in Fig. 4 a, the toxin could no longer be visualized on Western blots of the PNS of cells that had been pretreated with PI-PLC, demonstrating that removal of GPI-anchored proteins dramatically inhibited binding. This observation is in agreement with recent work by Buckley and coworkers showing that the receptors for aerolysin on rat erythrocytes and T-lymphocytes are also GPI-anchored proteins (Cowell et al., 1997; Nelson et al., 1997). Also after PI-PLC treatment, the putative 80-kD proaerolysin receptor could no longer be detected by proaerolysin overlay (Fig. 4 b), consistent with our findings that this protein acts as a receptor for proaerolysin. However PI-PLC treatment did not destroy the binding capacity of the released receptor since the fraction of proteins that had been cleaved by PI-PLC still contained the binding activity on blots (not shown) as also observed by Nelson et al. (1997).
These experiments show that proaerolysin binds to one, or possibly several, GPI-anchored receptors, but that the interaction does not depend on the lipid moiety of the membrane-anchored receptor but on the protein and/or the sugar residues.
Proaerolysin Binds to Caveolae and Noncaveolar Microdomains
We then analyzed the distribution of the toxin at the cell surface by immunofluorescence. As shown in Fig. 5 a, the distribution of the toxin when bound to the cells at 4°C showed a rather homogeneous, slightly punctate cell surface staining. This pattern is very similar to the native distribution of GPI-anchored proteins such as the folate receptor or alkaline phosphatase (Mayor et al., 1994; for reviews see Parton, 1996; Maxfield and Mayor, 1997). Warming up the toxin-treated cells for 15 min at 37°C accentuated the punctate appearance of the proaerolysin distribution (Fig. 5 b) in agreement with the hypothesis that the toxin concentrates in microdomains. A more distinct punctate pattern could be obtained by forcing the toxin to cluster by antibody cross-linking. In these experiments, cells were incubated with the anti-proaerolysin monoclonal and the polyclonal secondary antibodies before fixation (see Materials and Methods). As observed for a variety of GPI-anchored proteins (Mayor et al., 1994; for review see Maxfield and Mayor, 1997), cross-linking with antibodies led to redistribution of the toxin into more punctate foci (Fig. 5 c). Using cells transiently overexpressing the placental GPI-anchored PLAP, we could show that the toxin coclustered to a very large extent with PLAP (Fig. 5, c and d). Upon antibody-induced cross-linking, endogenous alkaline phosphatase was found to cluster into caveolae (Parton et al., 1994). However it has been shown that overexpressed PLAP mostly clusters in smooth microdomains of the plasma membrane outside of caveolae (Harder, T., P. Scheiffele, P. Verkade, and K. Simons, submitted for publication). Finally, we performed double-labeling experiments of the antibody cross-linked toxin and caveolin-1. As shown in Fig. 5, e and f, some colocalization was evident although colocalization was not complete. These data therefore indicate that proaerolysin binds and tends to cluster into both caveolae and noncaveolar raft microdomains of the plasma membrane.
Figure 5.
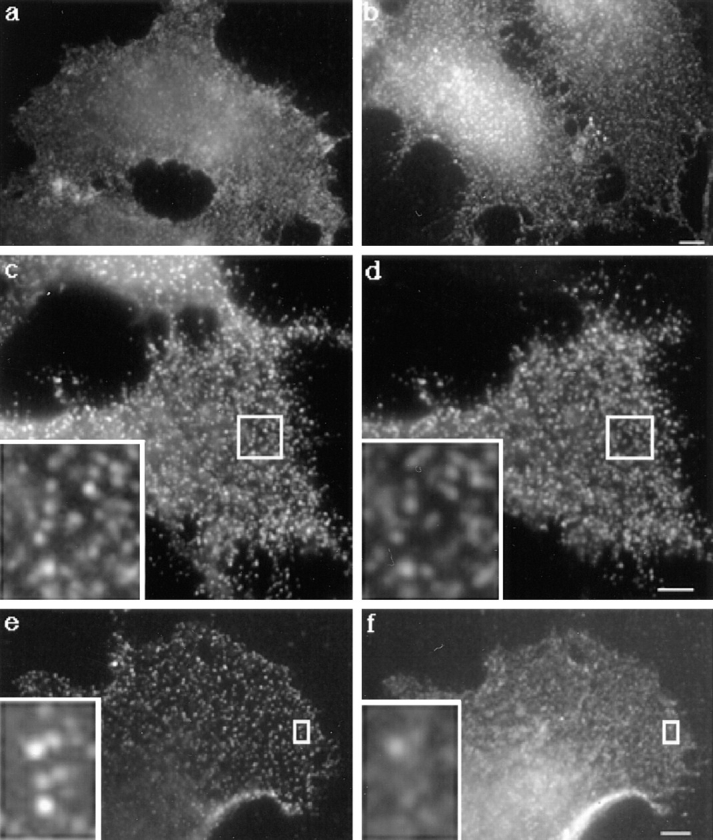
Distribution of proaerolysin at the plasma membrane of BHK cells. Cells were incubated with proaerolysin (2 nM) for 1 h at 4°C. (a) Cells were fixed with paraformaldehyde and further processed for immunofluorescence as described in Materials and Methods. (b) Cells were incubated for 15 min at 37°C, and then processed as in a. (c and d) BHK cells were transiently transfected with PLAP cDNA as described in Materials and Methods. Cells were kept at 4°C and incubated for 1 h with proaerolysin (2 nM), 1 h with anti-proaerolysin monoclonal and antialkaline phosphatase polyclonal antibodies, 1 h with the corresponding secondary antibodies. Cells were then fixed with paraformaldehyde. The same cell is shown stained for both proaerolysin (c) and PLAP (d). (e and f) BHK cells were kept at 4°C and incubated for 1 h with proaerolysin (2 nM), 1 h with anti-proaerolysin monoclonal antibody, 1 h with FITC-conjugated goat anti–rabbit, cells were then fixed and permeabilized with methanol (e). The cells were fixed with methanol and incubated 30 min at room temperature with anti–caveolin-1 antibody and for 30 min at room temperature with lissamine rhodamine sulfonyl chloride–conjugated goat anti–mouse IgG (f). Bars, 6.7 μm.
BHK Cells Express a Protease(s) That Activates Proaerolysin
To form channels in erythrocytes or artificial membranes, proaerolysin must be proteolytically activated. Since BHK cells exhibited a surprisingly high sensitivity to the proform of the toxin, we have investigated whether these cells express a converting enzyme. Cells were incubated with radiolabeled proaerolysin at 4°C, and then shifted to 37°C in a toxin-free medium. At various times, PNSs were prepared and the toxin was visualized by SDS-PAGE followed by autoradiography. As shown in Fig. 6, proaerolysin was gradually processed to a lower molecular weight form migrating slightly below trypsin-activated aerolysin. Concomitantly, a high molecular weight complex appeared corresponding to the aerolysin heptamer, which remained assembled in SDS after boiling even though the subunits were not covalently bound. We could exclude that cleavage was because of contaminating trypsin (e.g., because of low contamination remaining after passaging the cells) since the incubation medium used in the present work always contained 1 μg/ml trypsin/chymotrypsin inhibitor (see Materials and Methods). These results therefore demonstrate that BHK cells produce proteases able to process proaerolysin.
Figure 6.

Proaerolysin is processed by host cell proteases. BHK cells were incubated with 0.95 nM 125I-proaerolysin for 1 h at 4°C, thoroughly washed and incubated with a toxin-free medium at 37°C. After defined times, cells were homogenized, PNS was prepared and analyzed by SDS-PAGE (10% gel), followed by autoradiography. Lane a, proaerolysin marker; lane b, trypsin-activated aerolysin marker; all other lanes are labeled according to the incubation time at 37°C. 15 μg of total PNS proteins was loaded per lane. Note that the aerolysin heptamer although not covalent is not disassembled by boiling in SDS and can therefore be readily seen at the top of the gel.
It is of interest to note that both cleavage and oligomerization were very efficient since proaerolysin could no longer be detected after 25 min and since the nonheptameric form of aerolysin did not accumulate (Fig. 6). In contrast to what is observed in vitro, oligomerization does not appear to be a limiting step.
Proaerolysin Does Not Destroy the Integrity of the Plasma Membrane but Leads to Its Selective Permeabilization
We then investigated whether the toxin permeabilized the plasma membrane of BHK cells by measuring the intracellular potassium contents. As shown in Fig. 7 a, proaerolysin addition led to a decrease in intracellular potassium in a dose-dependent manner. No potassium efflux was observed when cells were kept at 4°C. As a control, we analyzed the effects of the G202C-I445C proaerolysin mutant, which has an engineered disulfide bridge that links the propeptide to the mature toxin (van der Goot et al., 1994). This mutant is unable to lyse erythrocytes presumably because it can no longer oligomerize. The intracellular potassium content of BHK cells was not affected by the mutant toxin, both in the proform and the mature form, even though it was able to bind to BHK cells, as shown by the fact that it could compete for binding with radiolabeled wild-type toxin (Fig. 1 a). We have also checked whether removal of GPI-anchored proteins from the cell surface would inhibit potassium efflux. PI-PLC treatment reduced the proaerolysin-induced potassium release by >75% (Fig. 7 b) again confirming that the proaerolysin receptor is GPI anchored. The remaining potassium efflux presumably reflects the fact that PI-PLC treatment was not complete.
Figure 7.

Effect of proaerolysin on the intracellular potassium concentration and the membrane potential of BHK cells. (a) Cells were incubated with different concentrations of proaerolysin for 1 h at 4°C, thoroughly washed and incubated with a toxin-free medium for 15 min at 37°C. Potassium contents were determined by flame photometry. Experiments were done in triplicate, and the standard deviation was calculated. (b) Cells were incubated with or without 6 U/ml of PI-PLC for 1 h at 37°C in the presence of 10 μg/ml cycloheximide, treated with or without 0.38 nM proaerolysin for 1 h at 4°C, thoroughly washed and incubated with a toxin-free medium for 15 min at 37°C. The potassium content was then determined by flame photometry. (c) Trypsinized-BHK cells were incubated with the membrane potential–sensitive dye DiS-C3(5) as described in Materials and Methods. The arrow indicates the time at which proaerolysin was added. Maximal depolarization was obtained at the end of each experiment by adding 1 μg/ml (final concentration) of trypsin-activated aerolysin (arrowheads). ♦, 100 ng/ml wild-type proaerolysin; □, 20 ng/ml wild-type proaerolysin; ○, 100 ng/ml G202C-I445C proaerolysin. The slight increase in fluorescence observed in the cells treated with the G202C-I445C mutant proaerolysin was not significant since a similar drift was observed in the absence of toxin.
We further investigated whether proaerolysin would lead to depolarization of the plasma membrane, as suggested by the potassium efflux measurements. For these experiments, trypsinized BHK cells were incubated with the fluorescent dye DiS-C3(5), a probe that has been extensively used to monitor changes in membrane potential (Papini et al., 1988). As shown Fig. 7 c, proaerolysin led to a decrease in membrane potential with dose-dependent kinetics. In contrast, the membrane potential was not affected by the G202C-I445C mutant toxin. We typically observed that the kinetics were biphasic, presumably reflecting sequential stages in channel formation or toxin action.
To investigate whether the overall integrity of the plasma membrane was maintained, toxin-treated cells were subjected to a viability assay (LIVE/DEAD assay; Molecular Probes). The principle of this assay is to incubate cells with calcein-AM, a membrane permeable reagent that can be cleaved by esterases in the living cells thereby yielding green fluorescence. Simultaneously, cells are incubated with ethidium homodimer–1, which only labels the nucleic acids of membrane-compromised cells because the dye is membrane impermeant, yet small enough to diffuse through the nuclear pore (red fluorescence). At various times, the number of cells, respectively, green or red were counted (Table I). Cells retained their plasma membrane integrity during several hours of toxin treatment since cellular esterases were not released and ethidium homodimer–1 was excluded (Table I; and Fig. 8 a). Similarly, propidium iodide and trypan blue were excluded from these cells.
Table I.
Viability of Proaerolysin-treated BHK Cells
| Time | Percent labeled with | |||
|---|---|---|---|---|
| Calcein | EthD-1 | |||
| (min) | ||||
| 0 | 100 | 0 | ||
| 75 | 100 | 0 | ||
| 120 | 88 | 12 | ||
| 195 | 77 | 20 | ||
| 240 | 60 | 38 | ||
BHK cells were incubated with 0.38 nM proaerolysin at 37°C for various times. Cells were then submitted to the viability assay (DEAD/LIVE from Molecular Probes). The percentage of cells positive for calcein or for ethidium dimer–1 (EthD-1) were counted. At least 150 cells were counted at each time point. Cells never stained with both markers. However after 3 h of toxin treatment, 2–3% of the cells did not stain for either of the markers.
Figure 8.
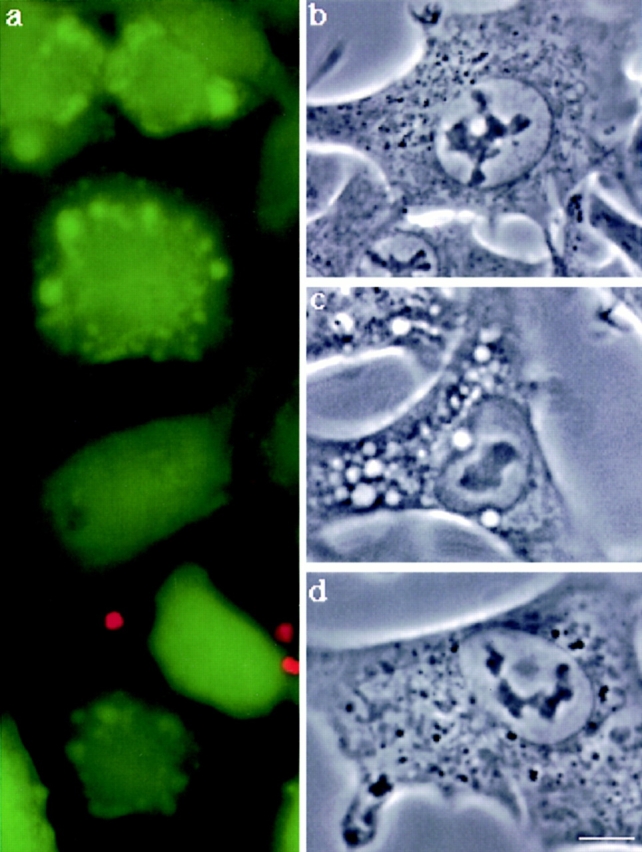
Effects of proaerolysin on the viability and morphology of BHK cells. (a) Cells were incubated with 0.38 nM proaerolysin at 37°C for 75 min, and then submitted to the DEAD/LIVE viability assay (see text). At this time point, all cells excluded ethidium dimer–1 and all retained cellular esterases as witnessed by the conversion of Calcein-AM to fluorescent calcein. Phase contrast image of control cells (b), cells incubated with 0.34 nM proaerolysin (c) for 1 h at 37°C. In d, cells were incubated with 6 U/ml of PI-PLC for 1 h at 37°C in the presence of 10 μg/ml cycloheximide, and then incubated with 0.38 nM proaerolysin for 1 h at 4°C, washed and incubated in a toxin-free medium for 1 h at 37°C. Bar, 10.5 μm.
Considering that the aerolysin channel has a diameter of 10 to 20 Å as determined by a variety of methods (Howard and Buckley, 1982; Wilmsen et al., 1990, 1992; Tschödrich-Rotter et al., 1996), it is somewhat surprising that ethidium homodimer–1, which has a molecular weight of 857 D, did not penetrate into the cells. One explanation would be that the channel formed by aerolysin in BHK cells is smaller than the one formed in erythrocytes or artificial membranes. However, far more likely, because of its structure ethidium homodimer–1 can not diffuse through the aerolysin channel. Similarly it has been shown that the channels formed by Staphylococcal α toxin, a toxin that shares many functional similarities with aerolysin (Lesieur et al., 1997), allows potassium efflux from human keratinocytes and T-lymphocytes but not influx of propidium iodide (Walev et al., 1993; Jonas et al., 1994).
The exclusion of ethidium homodimer–1 and the retention of cellular esterases in toxin treated cells clearly illustrate that proaerolysin does not affect plasma membrane integrity but has a selective effect on its permeability thereby lowering its membrane potential. We then investigated whether these permeability changes affected the subcellular organization of target cells.
Proaerolysin Induces Vacuolation of BHK Cells
To our surprise, we found that toxin-treated cells, while clearly viable as witnessed by the DEAD/LIVE assay (Fig. 8 a), contained a number of large vacuoles in their cytoplasm (Fig. 8 c) as well as ruffles and blebs at the plasma membrane. Vacuolation was not observed with the hemolytically inactive G202C-I445C aerolysin mutant nor with wild-type proaerolysin when the cells were kept at 4°C. Finally, vacuolation could be completely inhibited by treating the cells with PI-PLC before toxin addition (Fig. 8 d) further confirming that the proaerolysin receptor is GPI anchored. The ability of proaerolysin to induce vacuolation was not unique to BHK cells since we observed the same effects of the toxin on CHO and MDCK cells.
We then investigated whether the ability to induce vacuolation was a property common to all pore-forming toxins. BHK cells were treated with streptolysin O (SLO) (Bhakdi et al., 1996), which has been widely used to permeabilize cells. However at concentrations ranging from 25 to 1,000 ng/ml of SLO, we failed to observed the appearance of vacuoles in the cytoplasm even after several hours. We confirmed that SLO was indeed forming channels in BHK cells by measuring changes in membrane potential using the fluorescent dye DiS-C3(5). We found that 50 ng/ml of SLO led to depolarization of the plasma membrane with overall kinetics similar to those observed with proaerolysin in Fig. 7 c.
We then investigated whether massive calcium entry through the aerolysin channel was responsible for vacuolation by analyzing the effect of calcium ionophores. Neither ionomycin (2 and 10 μM) nor A23187 (2.5 and 10 μM) led to the appearance of vacuoles as monitored by phase contrast microscopy even after 6 h exposure to the ionophores (not shown). In certain cells however, a single, non-refringent vacuole could be observed near the nucleus as recently described by Subramanian and Meyer (1997). Since proaerolysin led to potassium efflux, we investigated whether its effects could be mimicked with the ionophore valinomycin. Similarly, no vacuolation was observed in the presence of valynomycin (2 μM).
These results indicate that proaerolysin induces vacuolation of a variety of cell lines and that this property is not common to all pore-forming toxins and ionophores.
Proaerolysin Leads to Vacuolation of the Endoplasmic Reticulum but Does Not Affect Later Stages of the Biosynthetic Pathway
To identify the origin of the proaerolysin-induced vacuoles, we investigated whether they contained the transferrin receptor, a well-established marker of early endosomes (Hopkins and Trowbridge, 1983; Trowbridge et al., 1993) or rab7, a small GTPase found on late endosomal compartments (Chavrier et al., 1990; Feng et al., 1995; Méresse et al., 1995). The transferrin receptor (see Fig. 10) and rab7 (not shown) were clearly absent from the vacuoles, indicating that these did not originate from compartments of the endocytic pathway.
Figure 10.
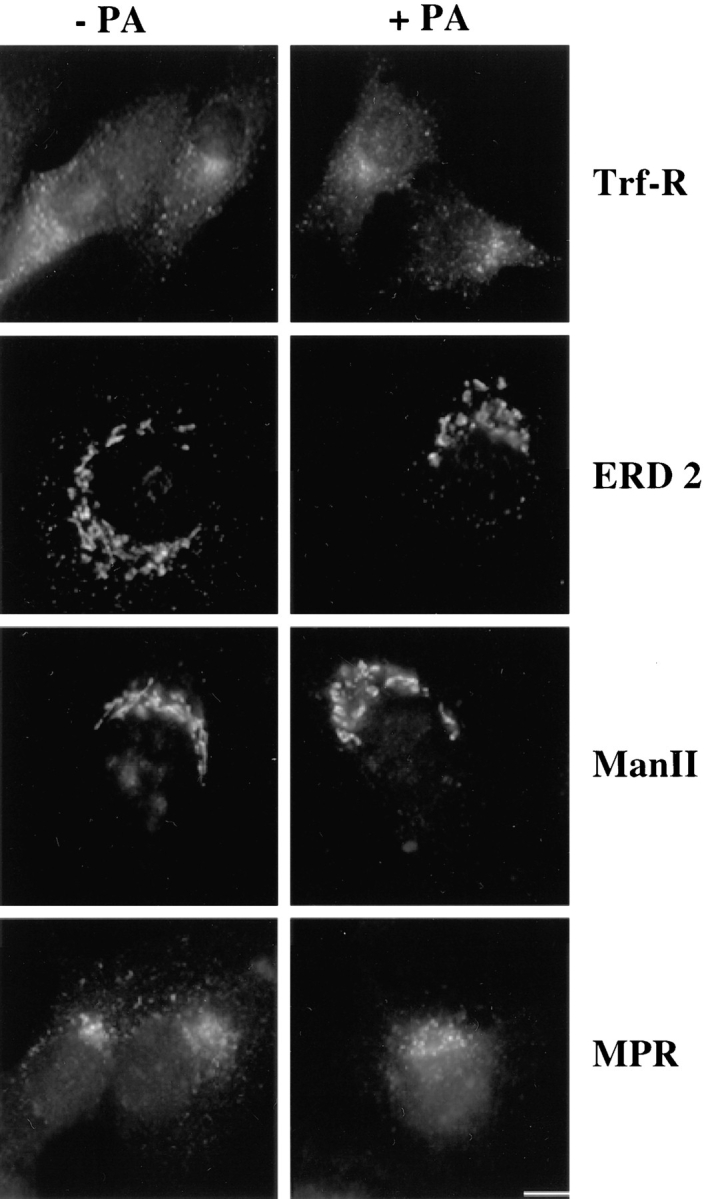
The distributions of the transferrin receptor (Trf-R), the KDEL receptor ERD2 (ERD2), mannosidase II (Man II), and the mannose-6-phosphate receptor (MPR) are not affected by proaerolysin in BHK cells. Cells were incubated in the presence or absence of 0.38 nM proaerolysin either for 50 min (MPR) or 1 h (Trf-R, ERD2, and Man II) at 37°C, and then processed for immunofluorescence as described in Materials and Methods. Bar: (Trf-R) 20 μm; (ERD2, Man II, and MPR) 13 μm.
We therefore investigated whether the toxin affected the biosynthetic pathway. The ER was analyzed using an antibody against calnexin, a transmembrane molecular chaperone involved in the folding and quality control of glycoproteins in the ER (Bergeron et al., 1994; Hammond and Helenius, 1995). As shown in Fig. 9, treatment of BHK cells with proaerolysin had a dramatic effect on the staining pattern of calnexin. After 20 min of proaerolysin treatment, the ER no longer showed its typical continuous reticulate appearance (Fig. 9 a), but appeared fragmented into small structures (Fig. 9 b). This initial ER fragmentation could be because of the increase of cytosolic calcium since a prolonged increase in the cytosolic calcium concentration has been shown to induce fragmentation, but not vacuolation, of the ER (Koch et al., 1988; Subramanian and Meyer, 1997). With time, the calnexin-positive vesicular structures increased in size. After 90 min, the large vesicular structures observed by phase contrast microscopy (Fig. 9 e) were clearly stained with the anti-calnexin antibody (Fig. 9 d).
Figure 9.

Effect of proaerolysin on the distribution of calnexin in BHK cells. Cells were incubated in the presence of 0.38 nM proaerolysin for various times at 37°C, immediately fixed with methanol, and then processed for immunofluorescence as described in Materials and Methods. Cells incubated without toxin (a) and with the toxin for 20 min (b), 60 min (c), and 90 min (d). e, phase contrast image of the cell shown in d. Bar: (a) 15 μm; (b) 17 μm; (c) 14 μm; (d and e) 12 μm.
Since the effect of proaerolysin on the ER was so striking, we investigated whether the toxin also had an effect on later stages of the biosynthetic pathway. We analyzed the distribution of the KDEL receptor ERD2, which localizes to the intermediate compartment between the ER and the Golgi as well as to the cis-Golgi (Lewis and Pelham, 1992; Griffiths et al., 1994; Tang et al., 1994), of mannosidase II, a marker of the cis-/medial-Golgi (Narula et al., 1992), and of the mannose-6-phosphate receptor, which in BHK cells (as well as in other cells) is primarily found in the TGN (Brown et al., 1986; Kobayashi et al., 1998). Surprisingly, the staining patterns of all three markers were not affected by toxin treatment even in cells that had dramatically vacuolated (Fig. 10). Similarly, the morphology of the Golgi complex studied in living cells by staining with C6-NBD-ceramide (Lipsky and Pagano, 1985) was not affected by the toxin (not shown).
These observations show that proaerolysin leads to fragmentation and vacuolation of the ER but does not affect the morphology of later compartments of the biosynthetic pathway nor that of the endocytic pathway.
Electron Microscopy of Proaerolysin-treated Cells
To confirm the above morphological observations, we analyzed proaerolysin-treated BHK cells by electron microscopy. As shown in Fig. 11, the cytoplasm contained large electron lucent vacuoles. These vacuoles were already apparent in some cells after 30 min, but were obvious in all cells after 60 min (Fig. 11). In many sections the vacuoles were clearly continuous with the nuclear envelope (Fig. 11, A and E) and had ribosomes on the cytoplasmic surface, confirming that the vacuoles are derived from the rough ER. There was clearly little effect of proaerolysin on the plasma membrane permeability to protein; the cytosol of the treated cells showed the same electron density as control cells. Effects of the toxin were highly selective since the cellular ultrastructures, in particular the Golgi, endosomes, and mitochondria, were well preserved (Fig. 11, A–E).
Figure 11.
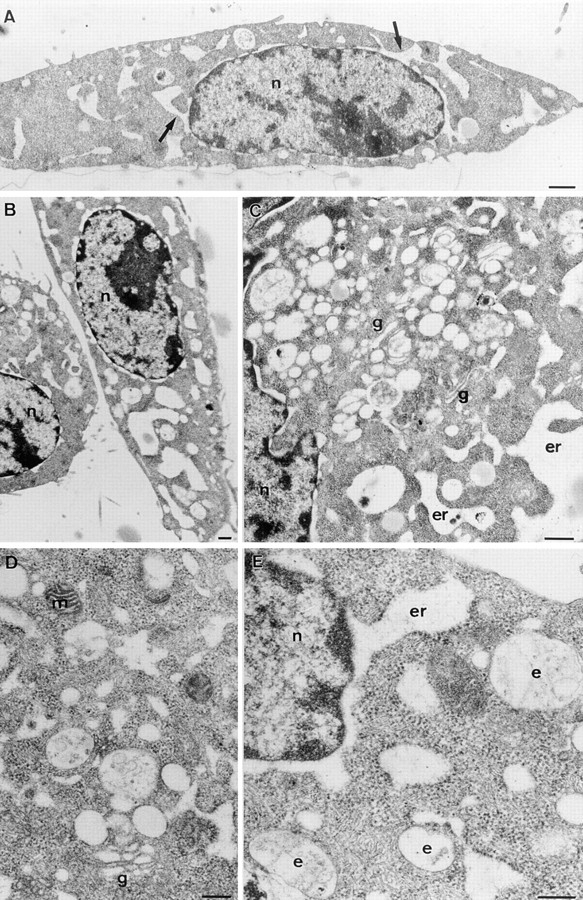
Ultrastructural analysis of aerolysin-treated BHK cells. Cells were incubated with 0.38 nM proaerolysin for 1 h at 37°C, fixed, and then processed for embedding in Epon and sectioning. A and B are low magnification overviews showing the typical morphology of the cells. Large electron lucent vacuoles are the striking characteristic of the treated cells. In many sections the vacuoles are clearly in continuity with the nuclear envelope (A, arrows; also see E). Together with the presence of membrane-associated ribosomes this identifies the vacuoles as part of the ER. Despite the drastic effects on the ER morphology the general ultrastructure of the cells is well preserved. Note that the cytoplasm is dense, mitochondria (m; e.g., D) are not swollen, and endosomes (e) show normal morphology (C–E). Golgi complexes are slightly swollen but still recognizable (g; C and D). n, nucleus. Bars: (A and B) 0.5 μm; (C–E): 0.25 μm.
Degradation of ε-COP and Brefeldin A Do Not Affect Aerolysin-induced Vacuolation
The observation that proaerolysin leads to vacuolation of the ER raised the possibility that a fraction of the toxin reaches by some mean the ER. To investigate whether the toxin enters the cells by vesicular transport, we have investigated its effect on the ldlF mutant CHO cell line (Guo et al., 1994, 1996). The ldlF cells have a temperature-sensitive mutation in the gene encoding for ε-COP, one of the components of the COPI coat involved in anterograde and retrograde biosynthetic membrane transport. At the restrictive temperature ε-COP is degraded, which leads to pleitropic membrane transport defects at early stages of the biosynthetic pathway and between early and late endosomes (Guo et al., 1994, 1996; Gu et al., 1997). In these experiments, cells were treated with the trypsin-activated toxin because ldlF cells lack, at the restrictive temperature, the cell surface proteases required to activate the protoxin (unpublished observations). As shown in Fig. 12, aerolysin led to vacuolation of ldlF cells both at the permissive (a) and the restrictive (b) temperature excluding the involvement of COPI-dependent membrane transport routes in toxin-mediated vacuolation.
Figure 12.
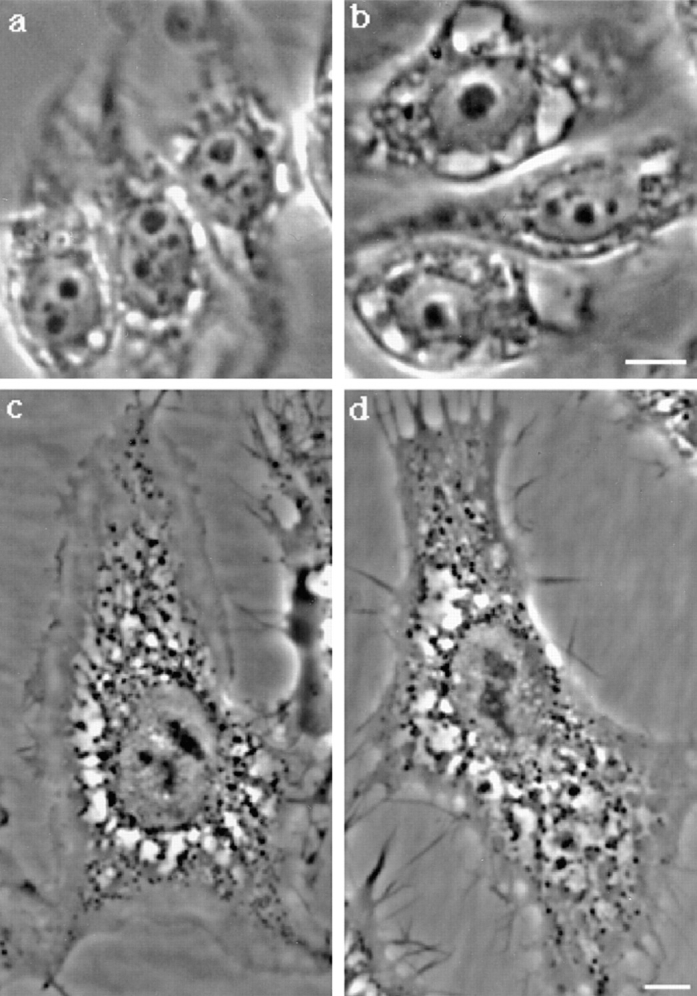
Degradation of ε-COP and BFA does not affect aerolysin-induced vacuolation. ldlF cells were incubated at 34° (a) and 40°C (b) for 6 h in culture medium. Cells were then shifted to 37°C and incubated for 60 min with 20 ng/ml proaerolysin. BHK cells were incubated with (d) or without (c) BFA for 45 min at 37°C before toxin addition. Images were taken on living cells 60 min after toxin addition. In d, BFA was also present during toxin treatment. Bar, 10.5 μm.
We then analyzed the effect of the fungal metabolite brefeldin A (BFA), which has been shown to block retrograde transport of toxins such as ricin and cholera toxin to the ER (Sandvig et al., 1991; Yoshida et al., 1991; Orlandi et al., 1993). As shown Fig. 12, c and d, BFA did not inhibit vacuolation, indicating that this process does not depend on retrograde transport processes. The drug was however effective under our conditions since it led to release of β-COP into the cytoplasm as witnessed by immunofluorescence (data not shown).
Having excluded that the toxin may be transported retrogradely to the ER, we considered the possibility that it might use a direct route from the plasma membrane to the ER (Smart et al., 1994). The simian virus 40 appears to use this route since this virus has been shown to associate to noncoated invaginations at the cell surface and then reaches the ER apparently via direct transport (Kartenbeck et al., 1989; Anderson et al., 1996; Stang et al., 1997). Preliminary observations (Parton et al., unpublished results) indicate that SV-40 infection is inhibited by BFA. Since we have seen no effect of this drug on proaerolysin induced vacuolation, it seems very unlikely that direct transport of the toxin from the plasma membrane to ER is required for vacuolation.
Taken together, these observations strongly indicate that vacuolation is not caused by membrane transport of the toxin to the ER.
Proaerolysin Affects the Dynamics of the Early Biosynthetic Pathway
Since the microtubule network has been shown to be required for assembly and maintenance of the ER (Lee et al., 1989; Terasaki et al., 1986), we investigated whether proaerolysin affected the cytoskeleton and thereby modified the ER organization. However, the microtubule network, visualized using anti-tubulin antibodies, was not perturbed even in cells that had dramatically vacuolated (Fig. 13, a and c).
Figure 13.
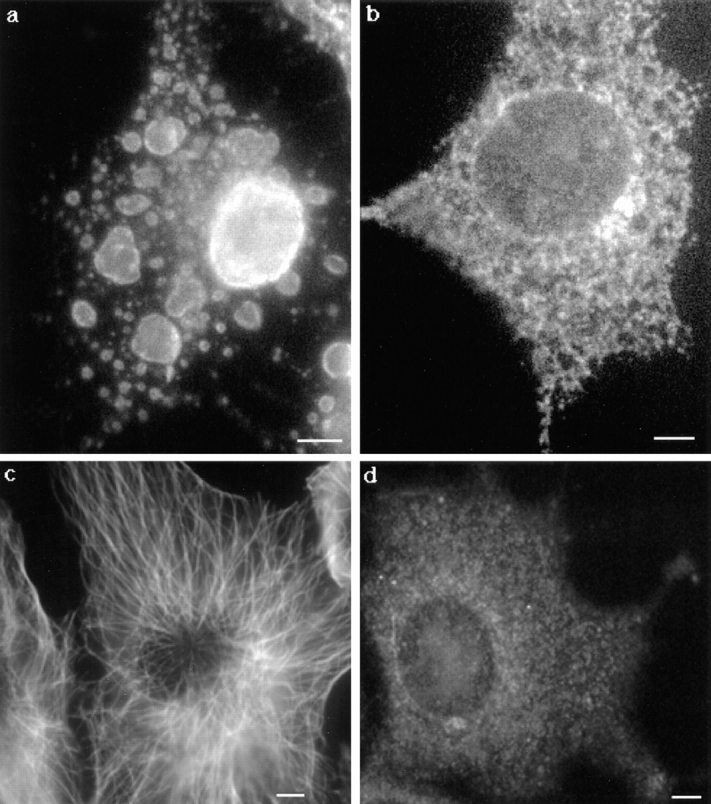
Proaerolysin-induced vacuolation is inhibited upon depolymerization of the microtubule network by nocodazole. Cells were treated with (b and d) or without (a and c) 10 μM of nocodazole for 1 h at 37°C before the addition of the toxin. The microtubule-depolymerizing drug remained present during the toxin treatment. After 60 min, cells were fixed with methanol and stained with anti-calnexin antibodies (a and b) or anti-tubulin antibodies. Bar, 6.7 μm
We then tried the reverse experiment, i.e., we tested whether nocodazole, which prevents polymerization of microtubules (Fig. 13 d) would affect vacuolation. Cells were treated with nocodazole for 1 h at 37°C followed by a 60-min incubation with proaerolysin. As shown in Fig. 13 b, there was no sign of ER vacuolation in nocodazole-treated cells. This observation indicates that the process requires the integrity of the microtubule network and that vacuolation is not because of mere swelling of the ER. The toxin therefore appears to interfere with ER membrane dynamics.
We then investigated whether the toxin would affect the distribution of β-COP, a component of the COPI coat that has been implicated in anterograde and retrograde transport between ER and Golgi (for review see Kreis et al., 1995). As shown in Fig. 14, the intensity of β-COP staining gradually decreased with time indicating that the COPI complex was being released into the cytoplasm. A similar, although more rapid release of β-COP has been observed when cells are treated with BFA or upon overexpression of a dominant-negative mutant of the small GTPase Arf1 (reviewed in Kreis et al., 1995). However, in contrast to what is observed with BFA or the Arf1 mutant, the release of COPI was not accompanied by a redistribution of Golgi markers to the ER (Fig. 10), demonstrating that Golgi markers can retain a Golgi localization even in the absence of bound COPI.
Figure 14.

Proaerolysin leads to the progressive release of β-COP into the cytosol. Cells were incubated in the presence of 0.38 nM proaerolysin for various times at 37°C, immediately fixed with methanol, and then processed for immunofluorescence as described in Materials and Methods. Cells incubated without toxin (a) and with the toxin for 20 min (b), 60 min (c), and 90 min (d). Bar, 16 μm.
Proaerolysin Inhibits Transport of Newly Synthesized VSV-G to the Plasma Membrane
Since proaerolysin induced fragmentation of the ER, affected its dynamics and led to the release of β-COP from membranes, we have investigated whether proaerolysin would affect transport to the plasma membrane of newly synthesized tsO45-G, a temperature-sensitive mutant of the transmembrane G protein of the VSV. BHK cells were infected with VSV tsO45 and incubated at the restrictive temperature (39.5°C) for 3.5 h to allow synthesis and accumulation of tsO45-G in the ER (Kreis, 1986; Kreis and Lodish, 1986). During the last 20 min of the incubation at 39.5°C, proaerolysin was added to cells. The cells were then shifted to 31°C for 45 min in the presence of cycloheximide to inhibit further protein synthesis, fixed, and then processed for immunofluorescence. As shown in Fig. 15, transport of VSV-G to the plasma membrane was dramatically inhibited. Then tsO45-G accumulated in the perinuclear region as seen after permeabilization of the fixed cells with Triton X-100 (Fig. 15). These data show that toxin action inhibits biosynthetic membrane transport of a transmembrane protein to the cell surface and suggests that the protein accumulates beyond the ER.
Figure 15.

Transport of newly synthesized VSV-G to the plasma membrane is inhibited in proaerolysin-treated cell. Cells were infected with tsO45-VSV for 3 h 10 min at 39.5°C in the absence of toxin. Cells were incubated an additional 20 min in IM with or without 0.38 nM proaerolysin. Cycloheximide was added to the cells before shifting them to 31°C for 45 min. Cells were then fixed with paraformaldehyde and either directly decorated with antibodies for plasma membrane staining or permeabilized with Triton X-100 (T-X100) to visualized intracellular staining. Processing for immunofluorescence was performed as described in Materials and Methods. Bar, 15 μm.
Discussion
The pore-forming toxin aerolysin is the major virulence factor secreted by Aeromonas hydrophila. In humans, this bacterium is able to induce effects as diverse as severe diarrhea, wound infection, septicemia, and meningitis, a disease spectrum that is reminiscent of certain Vibrio species. However the mechanism by which aerolysin leads to tissue damage remains largely unknown. In this study, we have analyzed the interaction of aerolysin with mammalian cells. Our data suggest the following sequence of events upon interaction of proaerolysin with BHK cells. The protoxin binds to GPI-anchored proteins at the surface of the target cells and clusters into Triton X-100–insoluble microdomains of the plasma membrane. A cellular protease then cleaves proaerolysin to its mature form, which can then oligomerize and insert into the plasma membrane to form channels. Channel formation allows passage of potassium, as well as presumably other small ions. This perturbation of the plasma membrane permeability in turn leads to selective fragmentation of the ER followed by its vacuolation. Also, transport of newly synthesized VSG-G protein from the ER to the plasma membrane is inhibited. The morphology of post-ER compartments in the biosynthetic pathway is however largely preserved. Similarly the morphology of compartments on the endocytic pathway is not affected.
Binding, Activation, Oligomerization, and Channel Formation
We showed that proaerolysin binds with high affinity to BHK cells via GPI-anchored proteins. Also treatment of cells with PI-PLC inhibited potassium efflux and protected them towards vacuolation. The most likely receptor candidate is a protein with an apparent molecular weight of 80 kD, which we are further characterizing. Two other receptors have been identified for proaerolysin: a 47-kD glycoprotein on rat erythrocytes (Gruber et al., 1994; Cowell et al., 1997) and Thy-1 on T-lymphocytes (Nelson et al., 1997). Interestingly, all three proaerolysin receptors are GPI anchored, and Buckley and co-workers have recently shown that the toxin appears to recognize both specific regions of the glycosyl anchor and some property of the polypeptide chain (Diep et al., 1998). Since toxins are the result of long-term coevolution between the pathogenic bacterium and the host, it is tempting to speculate that proaerolysin has evolved to bind to the type of protein that would most efficiently promote its channel formation. In that respect, a GPI-anchored protein seems an ideal receptor for a toxin that must first be proteolytically cleaved, and then oligomerize.
Several properties offered by the GPI anchor are likely to favor these processes because they increase the probability of collision between the protoxin and the protease as well as between aerolysin molecules. Firstly, the lipid anchor (versus a transmembrane segment) increases the lateral mobility of the receptor in the plane of the membrane (Zhang et al., 1991). Secondly, GPI-anchored proteins cluster transiently in sphingolipid-cholesterol rafts of the plasma membrane (Rothberg et al., 1992; Dupree et al., 1993), which as shown by the present immonufluorescence data leads to an increase in local toxin concentration (Fig. 5) that should favor oligomerization. Clustering might also favor the preceding activation step if the proteases were also present in the rafts. Finally, the GPI anchor might provide an increased spatial flexibility required for proper interaction between the protoxin and the host protease. Interestingly cleavage and polymerization of the cellular prion protein upon conversion to the scrapie isoform has also been suggested to occur within detergent-insoluble microdomains (Taraboulos et al., 1995; Vey et al., 1996; Naslavsky et al., 1997). Similarly the amyloid precursor protein has been localized to caveolae (Bouillot et al., 1996) therefore rafts might be a privileged site for processes such as cleavage and polymerization. In addition, it is interesting to note that aerolysin has evolved to interact selectively via a class of proteins that is only expressed on the apical surface of polarized epithelial cells (Lisanti et al., 1988, 1989, 1990). Since Aeromonas infection is increasingly associated with food-borne infections (Kirov, 1993), the toxin is most likely secreted inside the gut and must therefore interact with the apical surface of the epithelial cells.
After binding and maturation, the toxin led to potassium efflux and lowering of the membrane potential (Fig. 7). Despite its modified permeability, the plasma membrane maintained its overall integrity as ethidium homodimer–1 and propidium iodine were excluded from the cells, cellular esterases were retained and nucleotide-dependent mechanisms such as microtubule polymerization were not affected during several hours.
Selective Alterations in the Biosynthetic Pathway
The most striking effect of the toxin was the appearance of large vacuoles in the cytoplasm, an effect that had been previously seen on lung fibroblasts (Thelestam and Ljungh, 1981), as well as plasma membrane ruffling and blebbing. These morphological changes are reminiscent of those observed during apoptosis (Kerr et al., 1972). Other pore-forming toxins such as Staphylococcal α-toxin and listeriolysin from Listeria monocytogenes have been shown to trigger apoptosis as witnessed by degradation of genomic DNA (Jonas et al., 1994; Guzman et al., 1996). Our preliminary experiments however show that DNA was not degraded in proaerolysin-treated cells, also chromatin condensation was not observed (Fig. 11) and vacuolation could not be inhibited by overexpression of Bcl-2, a protein known to repress certain forms of apoptosis (Abrami, L., C. Borner, and F.G. van der Goot, unpublished observations) (Hueber et al., 1994; Gajewski and Thompson, 1996).
The proaerolysin-induced vacuoles also resemble those provoked by VacA from Helicobacter pylori, the bacterium involved in gastroduodenal ulcers (for reviews see Cover, 1996; Montecucco, 1996). However, whereas the VacA-induced vacuoles originate from late endosomes as shown by immunostaining for the vacuolar ATPase and for rab7 (Papini et al., 1996, 1997), proaerolysin-induced vacuoles originated from the ER as shown both by immunofluorescence and electron microscopy.
Although several steps in the mode of action of proaerolysin on mammalian cells remain to be elucidated, our data already enable us to conclude that the toxin must act at the cell surface and not at the site of vacuolation in the ER since inhibition of direct or indirect routes of retrograde transport to the ER does not prevent vacuolation. The observations that the G202C-I445C toxin mutant, which is unable to form channels, does not induce vacuolation illustrates that binding to the receptor is not sufficient and that perforation of the plasma membrane is necessary. Permeabilization per se of the plasma membrane however, might not be sufficient to induce vacuolation. Indeed SLO, which also permeabilizes the plasma membrane, does not lead to vacuole formation. Also Thelestham and Ljungh (1981) have observed that α-hemolysin, a second hemolysin secreted by Aeromonas hydrophila, also led to the formation of holes in the plasma membrane of lung fibroblasts but did not lead to vacuole formation. These observations suggest that the selectivity of the aerolysin-formed pore is important. Once the pore is formed, a highly speculative possibility is that a small fraction of the toxin is translocated through the plasma membrane into the cytoplasm, possibly through the aerolysin channel itself, and exerts an unknown enzymatic reaction. This latter mechanism would be similar to that of anthrax toxin, a toxin that is otherwise very different from aerolysin (for review see Leppla, 1991; Petosa and Liddington, 1996). However, the simplest explanation that is consistent with all our observations, is that selective permeabilization mediated by aerolysin is both necessary and sufficient.
It is less clear how this permeabilization results in the alteration of early biosynthetic membranes. It is tempting to speculate that pore formation triggers a cascade of events that selectively interfere with specific molecular machineries regulating membrane flow. Indeed, toxin action results in a microtubule-dependent vacuolation of the ER, indicating that this process requires ER membrane dynamics and suggesting that the homotypic ER fusion continues while fission is inhibited. In addition, we also observe that COPI coats are selectively released from the Golgi complex without affecting the overall Golgi organization. Both results may clearly contribute to explain why biosynthetic transport of VSV-G is inhibited. One of our future goals will be to further dissect the mechanisms of action of the toxin at the molecular level.
The present work describes the first detailed analysis of the effect of a pore-forming toxin on the morphology and intracellular dynamics of nucleated mammalian cells. Since aerolysin seems to trigger a variety of complex processes that affect the ER, new avenues of research aimed at understanding the mechanisms by which membrane-damaging proteins affect mammalian cells have been opened and can now be explored.
Abbreviations used in this paper
- BFA
brefeldin A
- GPI
glycosyl phosphatidylinositol
- IM
incubation medium
- PLAP
placental alkaline phosphatase
- PI-PLC
phosphatidylinositol-specific phospholipase C
- PNS
postnuclear supernatant
- ts045-G
VSV-G of VSV strain ts045
- SLO
streptolysin O
- VSV
vesicular stomatitis virus
- VSV-G
G protein of VSV
Footnotes
We are very grateful to J. Gruenberg, J.T. Buckley, T. Kobayashi, F. Perez, C. Lesieur, and M. Rojo for their helpful suggestions and critical reading of the manuscript. M-H. Beuchat is greatly acknowledged for her help in tissue culture. We are thankful to J.T. Buckley for providing us with the proaerolysin-producing strain, the purified proaerolysin mutant, and the anti-proaerolysin mAbs. We are also thankful to T. Kreis for giving us access to the fluorescence microscope and generous supply of antibodies. We thank H.D. Söling, A. Helenius, B. Hoflack, I. Trowbridge, and B. Burke for antibodies. We thank M. Krieger for providing us with the ldlF cell line; D. Brown for giving us PLAP DNA construct; and M. Kehoe for giving us purified SLO. We are very grateful to Thomas Harder for suggesting critical experiments and providing reagents.
L. Abrami was supported by the Roche Research Foundation and the Ciba-Geigy Jubileum Foundation. This work was supported by a grant from the Swiss National Science Foundation to G. van der Goot and a grant from the National Health and Medical Research Council of Australia to R.G. Parton.
Address all correspondence to Gisou van der Goot, Department of Biochemistry, University of Geneva, 30 quai Ernest Ansermet, 1211 Geneva 4, Switzerland. Tel./Fax: (41) 022-702-6414. E-mail: Gisou.vandergoot@biochem.unige.ch
References
- Anderson HA, Chen Y, Norkin LC. Bound simian virus 40 translocates to caveolin-enriched membrane domains, and its entry is inhibited by drugs that selectively disrupt caveolae. Mol Biol Cell. 1996;7:1825–1834. doi: 10.1091/mbc.7.11.1825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson RG. Caveolae: where incoming and outgoing messengers meet. Proc Natl Acad Sci USA. 1993;90:10909–10913. doi: 10.1073/pnas.90.23.10909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asao T, Kinoshita Y, Kozaki S, Uemura T, Sakaguchi G. Purification and some properties of Aeromonas hydrophilahemolysin. Infect Immun. 1984;46:122–127. doi: 10.1128/iai.46.1.122-127.1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergeron JJ, Brenner MB, Thomas DY, Williams DB. Calnexin: a membrane-bound chaperone of the endoplasmic reticulum. Trends Biochem Sci. 1994;19:124–128. doi: 10.1016/0968-0004(94)90205-4. [DOI] [PubMed] [Google Scholar]
- Bernheimer A, Avigad L, Avigad G. Interactions between aerolysin, erythrocytes and erythrocyte membranes. Infect Immun. 1975;11:1312–1319. doi: 10.1128/iai.11.6.1312-1319.1975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhakdi S, Bayley H, Valeva A, Walev I, Walker B, Kehoe M, Palmer M. Staphylococcal α-toxin, streptolysin-O, and Escherichia colihemolysin: prototypes of pore-forming bacterial cytolysins. Arch Microbiol. 1996;165:73–79. doi: 10.1007/s002030050300. [DOI] [PubMed] [Google Scholar]
- Bordier C. Phase separation of integral membrane proteins in Triton X-114 solution. J Biol Chem. 1981;256:1604–1607. [PubMed] [Google Scholar]
- Bouillot C, Prochiantz A, Rougon G, Allinquant B. Axonal amyloid precursor protein expressed by neurons in vitro is present in a membrane fraction with caveolae-like properties. J Biol Chem. 1996;271:7640–7644. doi: 10.1074/jbc.271.13.7640. [DOI] [PubMed] [Google Scholar]
- Brown DA, Rose JK. Sorting of GPI-anchored proteins to glycolipid-enriched membrane subdomains during transport to the apical cell surface. Cell. 1992;68:533–544. doi: 10.1016/0092-8674(92)90189-j. [DOI] [PubMed] [Google Scholar]
- Brown DA, Crise B, Rose JK. Mechanism of membrane anchoring affects polarized expression of two proteins in MDCK cells. Science. 1989;245:1499–1501. doi: 10.1126/science.2571189. [DOI] [PubMed] [Google Scholar]
- Brown WJ, Goodhouse J, Farquhar MG. Mannose-6-phosphate receptors for lysosomal enzymes cycle between the Golgi complex and endosomes. J Cell Biol. 1986;103:1235–1247. doi: 10.1083/jcb.103.4.1235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buckley JT. Purification of cloned proaerolysin released by a low protease mutant of Aeromonas salmonicida. . Biochem Cell Biol. 1990;68:221–224. doi: 10.1139/o90-029. [DOI] [PubMed] [Google Scholar]
- Chakraborty T, Hihle B, Berghauer H, Goebel W. Marker exchange mutagenesis of the aerolysin determinant in Aeromonas hydrophila demonstrates the role of aerolysin in A. hydrophila-associated infections. Infect Immun. 1987;55:2274–2280. doi: 10.1128/iai.55.9.2274-2280.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chavrier P, Parton RG, Hauri HP, Simons K, Zerial M. Localization of low molecular weight GTP binding proteins to exocytic and endocytic compartments. Cell. 1990;62:317–329. doi: 10.1016/0092-8674(90)90369-p. [DOI] [PubMed] [Google Scholar]
- Cover TL. The vacuolating cytotoxin of Helicobacter pylori. . Mol Microbiol. 1996;20:241–246. doi: 10.1111/j.1365-2958.1996.tb02612.x. [DOI] [PubMed] [Google Scholar]
- Cowell S, Aschauer W, Gruber HJ, Nelson KL, Buckley JT. The erythrocyte receptor for the channel-forming toxin aerolysin is a novel glycosylphosphatidylinositol-anchored protein. Mol Microbiol. 1997;25:343–350. doi: 10.1046/j.1365-2958.1997.4691828.x. [DOI] [PubMed] [Google Scholar]
- Daily OP, Joseph SW, Coolbaugh JC, Walker RI, Merrell BR, Rollins DM, Seidler RJ, Colwell RR, Lissner CR. Association of Aeromonas sobriawith human infection. J Clin Microbiol. 1981;13:769–777. doi: 10.1128/jcm.13.4.769-777.1981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diep, D.B.N., K.L. Nelson, S.M. Raja, R.W. McMaster, and J.T. Buckley. 1998. Glycosylphosphatidylinositol anchors of membrane glycoproteins are binding determinants for the channel-forming toxin Aerolysin. In press. [DOI] [PubMed]
- Donta ST, Haddow AP. Cytotoxic activity of Aeromonas hydrophila. . Infect Immun. 1978;21:989–993. doi: 10.1128/iai.21.3.989-993.1978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dupree P, Parton RG, Raposo G, Kurzchalia TV, Simons K. Caveolae and sorting in the trans-Golgi network of epithelial cells. EMBO (Eur Mol Biol Organ) J. 1993;12:1597–1605. doi: 10.1002/j.1460-2075.1993.tb05804.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng Y, Press B, Wandinger-Ness A. Rab 7: an important regulator of late endocytic membrane traffic. J Cell Biol. 1995;131:1435–1452. doi: 10.1083/jcb.131.6.1435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fra AM, Williamson E, Simons K, Parton RG. Detergent-insoluble glycolipid microdomains in lymphocytes in the absence of caveolae. J Biol Chem. 1994;269:30745–30748. [PubMed] [Google Scholar]
- Gajewski TF, Thompson CB. Apoptosis meets signal transduction: elimination of a BAD influence. Cell. 1996;87:589–592. doi: 10.1016/s0092-8674(00)81377-x. [DOI] [PubMed] [Google Scholar]
- Garland WJ, Buckley JT. The cytolytic toxin aerolysin must aggregate to disrupt erythrocytes, and aggregation is stimulated by human glycophorin. Infect Immun. 1988;56:1249–1253. doi: 10.1128/iai.56.5.1249-1253.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graham FL, van der Eb AJ. A new technique for the assay of infectivity of adenovirus 5 DNA. Virology. 1973;52:456–467. doi: 10.1016/0042-6822(73)90341-3. [DOI] [PubMed] [Google Scholar]
- Griffiths G, Matteoni R, Back R, Hoflack B. Characterization of the cation-independent mannose 6-phosphate receptor- enriched prelysosomal compartment in NRK cells. J Cell Sci. 1990;95:441–461. doi: 10.1242/jcs.95.3.441. [DOI] [PubMed] [Google Scholar]
- Griffiths G, Ericsson M, Krijnse LJ, Nilsson T, Goud B, Soling HD, Tang BL, Wong SH, Hong W. Localization of the Lys, Asp, Glu, Leu tetrapeptide receptor to the Golgi complex and the intermediate compartment in mammalian cells. J Cell Biol. 1994;127:1557–1574. doi: 10.1083/jcb.127.6.1557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gruber HJ, Wilmsen HU, Cowell S, Schindler H, Buckley JT. Partial purification of the rat erythrocyte receptor for the channel-forming toxin aerolysin and reconstitution into planar lipid bilayers. Mol Microbiol. 1994;14:1093–1101. doi: 10.1111/j.1365-2958.1994.tb01341.x. [DOI] [PubMed] [Google Scholar]
- Gruenberg J, Howell KE. Immuno-isolation of vesicles using antigenic sites either located on the cytoplasmic or the exoplasmic domain of an implanted viral protein. A quantitative analysis. Eur J Cell Biol. 1985;38:312–321. [PubMed] [Google Scholar]
- Gruenberg J, Griffiths G, Howell KE. Characterization of the early endosome and putative endocytic carrier vesicles in vivo and with an assay of vesicle fusion in vitro. J Cell Biol. 1989;108:1301–1316. doi: 10.1083/jcb.108.4.1301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu F, Aniento F, Parton RG, Gruenberg J. Functional dissection of COP-I subunits in the biogenesis of multivesicular endosomes. J Cell Biol. 1997;139:1183–1195. doi: 10.1083/jcb.139.5.1183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo Q, Vasile E, Krieger M. Disruptions in Golgi structure and membrane traffic in a conditional lethal mammalian cell mutant are corrected by ε-COP. J Cell Biol. 1994;125:1213–1224. doi: 10.1083/jcb.125.6.1213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo Q, Penman M, Trigatti BL, Krieger M. A single point mutation in ε-COP results in temperature-sensitive, lethal defects in membrane transport in a Chinese hamster ovary cell mutant. J Biol Chem. 1996;271:11191–11196. doi: 10.1074/jbc.271.19.11191. [DOI] [PubMed] [Google Scholar]
- Guzman CA, Domann E, Rohde M, Bruder D, Darji A, Weiss S, Wehland J, Chakraborty T, Timmis KN. Apoptosis of mouse dendritic cells is triggered by listeriolysin, the major virulence determinant of Listeria monocytogenes. Mol Microbiol. 1996;20:119–126. doi: 10.1111/j.1365-2958.1996.tb02494.x. [DOI] [PubMed] [Google Scholar]
- Hammond C, Helenius A. Quality control in the secretory pathway. Curr Opin Cell Biol. 1995;7:523–529. doi: 10.1016/0955-0674(95)80009-3. [DOI] [PubMed] [Google Scholar]
- Hopkins CR, Trowbridge IS. Internalization and processing of transferrin and the transferrin receptor in human carcinoma A431 cells. J Cell Biol. 1983;97:508–521. doi: 10.1083/jcb.97.2.508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howard SP, Buckley JT. Membrane glycoprotein receptor and hole forming properties of a cytolytic protein toxin. Biochemistry. 1982;21:1662–1667. doi: 10.1021/bi00536a029. [DOI] [PubMed] [Google Scholar]
- Howard SP, Buckley JT. Activation of the hole forming toxin aerolysin by extracellular processing. J Bacteriol. 1985;163:336–340. doi: 10.1128/jb.163.1.336-340.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huber LA, Beug H, Simons K, Reichmann E. Two-dimensional gel mapping of small GTPases reveals transformation-specific changes during oncogenesis. Electrophoresis. 1994;15:469–473. doi: 10.1002/elps.1150150164. [DOI] [PubMed] [Google Scholar]
- Hueber AO, Raposo G, Pierres M, He HT. Thy-1 triggers mouse thymocyte apoptosis through a bcl-2-resistant mechanism. J Exp Med. 1994;179:785–796. doi: 10.1084/jem.179.3.785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janda JM, Bottone DJ, Sinner CV, Calcaterra D. Phenotype markers associated with gastrointestinal Aeromonas hydrophilaisolates from symptomatic children. J Clin Microbiol. 1984;17:588–591. doi: 10.1128/jcm.17.4.588-591.1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janda JM, Clark RB, Brenden R. Virulence of Aeromonas spp. as assessed through lethality studies. Current Microbiol. 1985;12:163–168. [Google Scholar]
- Jin G-F, Chopra AK, Houston CW. Stimulation of neutrophil leukocyte chemotaxis by a cloned cytolytic enterotoxin of Aeromonas hydrophila. . FEMS Microbiol Lett. 1992;98:285–290. doi: 10.1016/0378-1097(92)90170-s. [DOI] [PubMed] [Google Scholar]
- Jonas D, Walev I, Berger T, Liebetrau M, Palmer M, Bhakdi S. Novel path to apoptosis: small transmembrane pores created by staphylococcal α-toxin in T lymphocytes evoke internucleosomal DNA degradation. Infect Immun. 1994;62:1304–1312. doi: 10.1128/iai.62.4.1304-1312.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaper JB, Lockman H, Colwell RR. Aeromonas hydrophila: ecology and toxigenicity of isolates from an estuary. J Appl Bacteriol. 1981;50:359–377. doi: 10.1111/j.1365-2672.1981.tb00900.x. [DOI] [PubMed] [Google Scholar]
- Kartenbeck J, Stukenbrok H, Helenius A. Endocytosis of simian virus 40 into the endoplasmic reticulum. J Cell Biol. 1989;709:2721–2729. doi: 10.1083/jcb.109.6.2721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kerr JF, Wyllie AH, Currie AR. Apoptosis: a basic biological phenomenon with wide-ranging implications in tissue kinetics. Br J Cancer. 1972;26:239–257. doi: 10.1038/bjc.1972.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirov SM. The public health significance of Aeromonas spp. in foods. Int J Food Microbiol. 1993;20:179–198. doi: 10.1016/0168-1605(93)90164-c. [DOI] [PubMed] [Google Scholar]
- Kobayashi, T., E. Stang, K.S. Fang, P. de Moerloose, R.G. Parton, and J. Gruenberg. 1998. A lipid associated with the antiphospholipid syndrome regulates endosome structure/function. Nature. In press. [DOI] [PubMed]
- Koch, G.L., C. Booth, and F.B. Wooding. 1988. Dissociation and re-assembly of the endoplasmic reticulum in live cells. J. Cell Sci. 91:511–522. [DOI] [PubMed]
- Kreis TE. Microinjected antibodies against the cytoplasmic domain of vesicular stomatitis virus glycoprotein block its transport to the cell surface. EMBO (Eur Mol Biol Organ) J. 1986;5:931–941. doi: 10.1002/j.1460-2075.1986.tb04306.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kreis TE, Lodish HF. Oligomerization is essential for transport of vesicular stomatitis viral glycoprotein to the cell surface. Cell. 1986;46:929–937. doi: 10.1016/0092-8674(86)90075-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kreis TE, Lowe M, Pepperkok R. COPs regulating membrane traffic. Annu Rev Cell Dev Biol. 1995;11:677–706. doi: 10.1146/annurev.cb.11.110195.003333. [DOI] [PubMed] [Google Scholar]
- Laemmli UK. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature. 1970;227:680–685. doi: 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]
- Lee C, Ferguson M, Chen LB. Construction of the endoplasmic reticulum. J Cell Biol. 1989;109:2045–2055. doi: 10.1083/jcb.109.5.2045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leppla, S.H. 1991. The anthrax toxin complex. In Sourcebook of Bacterial Protein Toxins. J. & F. Alouf J.H., editors. Academic Press, New York. 277–302.
- Lesieur C, Vécsey-Semjén B, Abrami L, Fivaz M, van der Goot FG. Membrane insertion: the strategy of toxins. Mol Membr Biol. 1997;14:45–64. doi: 10.3109/09687689709068435. [DOI] [PubMed] [Google Scholar]
- Lewis MJ, Pelham HR. Ligand-induced redistribution of a human KDEL receptor from the Golgi complex to the endoplasmic reticulum. Cell. 1992;68:353–364. doi: 10.1016/0092-8674(92)90476-s. [DOI] [PubMed] [Google Scholar]
- Lipsky NG, Pagano RE. A vital stain for the Golgi apparatus. Science. 1985;228:745–747. doi: 10.1126/science.2581316. [DOI] [PubMed] [Google Scholar]
- Lisanti MP, Sargiacomo M, Graeve L, Saltiel AR, Rodriguez BE. Polarized apical distribution of glycosyl-phosphatidylinositol-anchored proteins in a renal epithelial cell line. Proc Natl Acad Sci USA. 1988;85:9557–9561. doi: 10.1073/pnas.85.24.9557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lisanti MP, Caras IW, Davitz MA, Rodriguez BE. A glycophospholipid membrane anchor acts as an apical targeting signal in polarized epithelial cells. J Cell Biol. 1989;109:2145–2156. doi: 10.1083/jcb.109.5.2145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lisanti MP, Le BA, Saltiel AR, Rodriguez BE. Preferred apical distribution of glycosyl-phosphatidylinositol (GPI) anchored proteins: a highly conserved feature of the polarized epithelial cell phenotype. J Membr Biol. 1990;113:155–167. doi: 10.1007/BF01872889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ljungh A, Wadström T. Toxins of Vibrio parahaemolyticus and Aeromonas hydrophila. . J Toxicol Toxin Rev. 1983;1:257–307. [Google Scholar]
- Maxfield FR, Mayor S. Cell surface dynamics of GPI-anchored proteins. Adv Exp Med Biol. 1997;419:355–364. doi: 10.1007/978-1-4419-8632-0_47. [DOI] [PubMed] [Google Scholar]
- Mayor S, Rothberg KG, Maxfield FR. Sequestration of GPI- anchored proteins in caveolae triggered by cross-linking. Science. 1994;264:1948–1951. doi: 10.1126/science.7516582. [DOI] [PubMed] [Google Scholar]
- Méresse S, Gorvel J-P, Chavrier P. The rab7 GTPase resides on a vesicular compartement connected to lysosomes. J Cell Sci. 1995;108:3349–3358. doi: 10.1242/jcs.108.11.3349. [DOI] [PubMed] [Google Scholar]
- Moniatte M, van der Goot FG, Buckley JT, Pattus F, Van Dorsselaer A. Characterization of the heptameric pore-forming complex of the Aeromonastoxin aerolysin using MALDI-TOF mass spectrometry. FEBS (Fed Eur Biochem Soc) Lett. 1996;384:269–272. doi: 10.1016/0014-5793(96)00328-6. [DOI] [PubMed] [Google Scholar]
- Montecucco C. Bacterial protein toxins and cell vesicle trafficking. Experientia (Basel) 1996;52:1026–1032. doi: 10.1007/BF01952098. [DOI] [PubMed] [Google Scholar]
- Narula N, McMorrow I, Plopper G, Doherty J, Matlin KS, Burke B, Stow JL. Identification of a 200-kD, brefeldin-sensitive protein on Golgi membranes. J Cell Biol. 1992;117:27–38. doi: 10.1083/jcb.117.1.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naslavsky N, Stein R, Yanai A, Friedlander G, Taraboulos A. Characterization of detergent-insoluble complexes containing the cellular prion protein and its scrapie isoform. J Biol Chem. 1997;272:6324–6331. doi: 10.1074/jbc.272.10.6324. [DOI] [PubMed] [Google Scholar]
- Nelson KL, Raja SM, Buckley JT. The GPI-anchored surface glycoprotein Thy-1 is a receptor for the channel-forming toxin aerolysin. J Biol Chem. 1997;272:12170–12174. doi: 10.1074/jbc.272.18.12170. [DOI] [PubMed] [Google Scholar]
- Orlandi PA, Curran PK, Fishman PH. Brefeldin A blocks the response of cultured cells to cholera toxin. Implications for intracellular trafficking in toxin action. J Biol Chem. 1993;268:12010–12016. [PubMed] [Google Scholar]
- Papini E, Sandona D, Rappuoli R, Montecucco C. On the membrane translocation of diphtheria toxin: at low pH the toxin induces ion channels on cells. EMBO ( Eur Mol Biol Organ) J. 1988;7:3353–3359. doi: 10.1002/j.1460-2075.1988.tb03207.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papini, E., E. Gottardi, B. Satin, M. de Bernard, P. Massari, J. Telford, R. Rappuoli, S. B.Sato, and C. Montecucco. 1996. The vacuolar ATPase proton pump is present on intracellular vacuoles induced by Helicobacter pylori. J. Med. Microbiol. 45:84–89. [DOI] [PubMed]
- Papini E, Satin B, Bucci C, de Bernard M, Telford JL, Manetti R, Rappuoli R, Zerial M, Montecucco C. The small GTP binding protein rab7 is essential for cellular vacuolation induced by Helicobacter pylori cytotoxin. EMBO (Eur Mol Biol Organ) J. 1997;16:15–24. doi: 10.1093/emboj/16.1.15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parker MW, van der Goot FG, Buckley JT. Aerolysin-the ins and outs of a channel forming toxin. Mol Microbiol. 1996;19:205–212. doi: 10.1046/j.1365-2958.1996.355887.x. [DOI] [PubMed] [Google Scholar]
- Parton RG. Caveolae and Caveolins. Curr Opin Cell Biol. 1996;8:542–548. doi: 10.1016/s0955-0674(96)80033-0. [DOI] [PubMed] [Google Scholar]
- Parton RG, Joggerst B, Simons K. Regulated internalization of caveolae. J Cell Biol. 1994;1274:1199–1215. doi: 10.1083/jcb.127.5.1199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pepperkok R, Scheel J, Horstmann H, Hauri HP, Griffiths G, Kreis TE. β-COP is essential for biosynthetic membrane transport from the endoplasmic reticulum to the Golgi complex in vivo. Cell. 1993;74:71–82. doi: 10.1016/0092-8674(93)90295-2. [DOI] [PubMed] [Google Scholar]
- Petosa, C., and R.C. Liddington. 1996. The Anthrax toxin. In Protein Toxin Structure. M.W. Parker, editor. Springer Verlag. Heidelberg, Germany. 97–121.
- Rothberg KG, Heuser JE, Donzell WC, Ying YS, Glenney JR, Anderson RG. Caveolin, a protein component of caveolae membrane coats. Cell. 1992;68:673–682. doi: 10.1016/0092-8674(92)90143-z. [DOI] [PubMed] [Google Scholar]
- Sandvig K, Prydz K, Hansen S H, van Deurs B. Ricin transport in brefeldin A–treated cells: correlation between Golgi structure and toxic effect. J Cell Biol. 1991;115:971–981. doi: 10.1083/jcb.115.4.971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheffer J, Konig W, Braun V, Goebel W. Comparison of four hemolysin-producing organisms (Escherichia coli, Serratia marcescens, Aeromonas hydrophila, and Listeria monocytogenes)for release of inflammatory mediators from various cells. J Clin Microbiol. 1988;26:544–551. doi: 10.1128/jcm.26.3.544-551.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scherson T, Kreis TE, Schlessinger J, Littauer UZ, Borisy GG, Geiger B. Dynamic interactions of fluorescently labeled microtubule- associated proteins in living cells. J Cell Biol. 1984;99:425–434. doi: 10.1083/jcb.99.2.425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simons K, Ikonen E. Functiomal rafts in cell membranes. Nature. 1997;387:569–572. doi: 10.1038/42408. [DOI] [PubMed] [Google Scholar]
- Smart EJ, Ying YS, Conrad PA, Anderson RG. Caveolin moves from caveolae to the Golgi apparatus in response to cholesterol oxidation. J Cell Biol. 1994;1274:1185–1197. doi: 10.1083/jcb.127.5.1185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stang E, Kartenbeck J, Parton RG. Major histopatibility complex class I molecules mediate association of SV40 with caveolae. Mol Biol Cell. 1997;8:47–57. doi: 10.1091/mbc.8.1.47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subramanian K, Meyer T. Calcium-induced restructuring of nuclear envelope and endoplasmic reticulum calcium stores. Cell. 1997;89:963–971. doi: 10.1016/s0092-8674(00)80281-0. [DOI] [PubMed] [Google Scholar]
- Tang ZL, Scherer PE, Lisanti MP. The primary sequence of murine caveolin reveals a conserved consensus site for phosphorylation by protein kinase C. Gene. 1994;147:299–300. doi: 10.1016/0378-1119(94)90087-6. [DOI] [PubMed] [Google Scholar]
- Taraboulos A, Scott M, Semenov A, Avrahami D, Laszlo L, Prusiner SB, Avraham D. Cholesterol depletion and modification of COOH-terminal targeting sequence of the prion protein inhibit formation of the scrapie isoform. J Cell Biol. 1995;129:121–132. doi: 10.1083/jcb.129.1.121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terasaki M, Chen LB, Fujiwara K. Microtubules and the endoplasmic reticulum are highly interdependent structures. J Cell Biol. 1986;103:1557–1568. doi: 10.1083/jcb.103.4.1557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thelestam M, Ljungh A. Membrane-damaging and cytotoxic effects on human fibroblasts of α- and β-hemolysins from Aeromonas hydrophila. . Infect Immun. 1981;34:949–956. doi: 10.1128/iai.34.3.949-956.1981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trowbridge IS, Collawn JF, Hopkins CR. Signal-dependent membrane protein trafficking in the endocytic pathway. Annu Rev Cell Biol. 1993;9:129–161. doi: 10.1146/annurev.cb.09.110193.001021. [DOI] [PubMed] [Google Scholar]
- Tschödrich-Rotter M, Kubitscheck U, Ugochukwu G, Buckley J, Peters R. Optical single-channel analysis of the aerolysin pore in erythrocyte membranes. Biophys J. 1996;70:723–732. doi: 10.1016/S0006-3495(96)79612-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Goot FG, Lakey JH, Pattus F, Kay CM, Sorokine O, Van Dorsselaer A, Buckley T. Spectroscopic study of the activation and oligomerization of the channel-forming toxin aerolysin: identification of the site of proteolytic activation. Biochemistry. 1992;31:8566–8570. doi: 10.1021/bi00151a026. [DOI] [PubMed] [Google Scholar]
- van der Goot FG, Ausio J, Wong K, Pattus F, Buckley J. Dimerization stabilizes the pore-forming toxin aerolysin in solution. J Biol Chem. 1993;268:18272–18279. [PubMed] [Google Scholar]
- van der Goot FG, Hardie KR, Parker MW, Buckley JT. The C-terminal peptide produced upon proteolytic activation of the cytolytic toxin aerolysin is not involved in channel formation. J Biol Chem. 1994;269:30496–30501. [PubMed] [Google Scholar]
- Vey M, Pilkuhn S, Wille H, Nixon R, DeArmond SJ, Smart EJ, Anderson RG, Taraboulos A, Prusiner SB. Subcellular colocalization of the cellular and scrapie prion proteins in caveolae-like membranous domains. Proc Natl Acad Sci USA. 1996;93:14945–14949. doi: 10.1073/pnas.93.25.14945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wada I, Rindress D, Cameron PH, Ou WJ, Doherty JJ, Louvard D, Bell AW, Dignard D, Thomas DY, Bergeron JJ. SSR alpha and associated calnexin are major calcium binding proteins of the endoplasmic reticulum membrane. J Biol Chem. 1991;266:19599–19610. [PubMed] [Google Scholar]
- Walev I, Martin E, Jonas D, Mohamadzadeh M, Muller KW, Kunz L, Bhakdi S. Staphylococcal α-toxin kills human keratinocytes by permeabilizing the plasma membrane for monovalent ions. Infect Immun. 1993;61:4972–4979. doi: 10.1128/iai.61.12.4972-4979.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilmsen HU, Pattus F, Buckley JT. Aerolysin, a hemolysin from Aeromonas hydrophila, forms voltage-gated channels in planar bilayers. J Membr Biol. 1990;115:71–81. doi: 10.1007/BF01869107. [DOI] [PubMed] [Google Scholar]
- Wilmsen HU, Leonard KR, Tichelaar W, Buckley JT, Pattus F. The aerolysin membrane channel is formed by heptamerization of the monomer. EMBO (Eur Mol Biol Organ) J. 1992;11:2457–2463. doi: 10.1002/j.1460-2075.1992.tb05310.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshida T, Chen CC, Zhang MS, Wu HC. Disruption of the Golgi apparatus by brefeldin A inhibits the cytotoxicity of ricin, modeccin, and Pseudomonas toxin. Exp Cell Res. 1991;192:389–395. doi: 10.1016/0014-4827(91)90056-z. [DOI] [PubMed] [Google Scholar]
- Zhang F, Crise B, Su B, Hou Y, Rose JK, Bothwell A, Jacobson K. Lateral diffusion of membrane-spanning and glycosylphosphatidylinositol-linked proteins: Toward establishing rules governing the lateral mobility of membrane proteins. J Cell Biol. 1991;115:75–84. doi: 10.1083/jcb.115.1.75. [DOI] [PMC free article] [PubMed] [Google Scholar]