

Abstract
Bax, a member of the Bcl-2 protein family, accelerates apoptosis by an unknown mechanism. Bax has been recently reported to be an integral membrane protein associated with organelles or bound to organelles by Bcl-2 or a soluble protein found in the cytosol. To explore Bcl-2 family member localization in living cells, the green fluorescent protein (GFP) was fused to the NH2 termini of Bax, Bcl-2, and Bcl-XL. Confocal microscopy performed on living Cos-7 kidney epithelial cells and L929 fibroblasts revealed that GFP–Bcl-2 and GFP–Bcl-XL had a punctate distribution and colocalized with a mitochondrial marker, whereas GFP–Bax was found diffusely throughout the cytosol. Photobleaching analysis confirmed that GFP–Bax is a soluble protein, in contrast to organelle-bound GFP–Bcl-2. The diffuse localization of GFP–Bax did not change with coexpression of high levels of Bcl-2 or Bcl-XL. However, upon induction of apoptosis, GFP–Bax moved intracellularly to a punctate distribution that partially colocalized with mitochondria. Once initiated, this Bax movement was complete within 30 min, before cellular shrinkage or nuclear condensation. Removal of a COOH-terminal hydrophobic domain from GFP–Bax inhibited redistribution during apoptosis and inhibited the death-promoting activity of both Bax and GFP– Bax. These results demonstrate that in cells undergoing apoptosis, an early, dramatic change occurs in the intracellular localization of Bax, and this redistribution of soluble Bax to organelles appears important for Bax to promote cell death.
Programmed cell death is vital for both normal development and maintenance of many tissues in multicellular organisms. A form of programmed cell death, apoptosis, proceeds with morphological and biochemical characteristics that include blebbing of the cell membrane, a decrease in cell volume, nuclear condensation, and the intranucleosomal cleavage of DNA (37).
The family of Bcl-2–related proteins plays key roles in the regulation of apoptosis. Individual family members can function to either block or promote programmed cell death (38). Three extensively characterized mammalian family members are the antiapoptotic genes bcl-2 and bcl-XL, and the proapoptotic gene bax. In vivo studies suggest that a single protein product predominates for each gene: Bcl-2α, a 26-kD protein (36); Bcl-XL, a 27-kD protein (9); and Baxα, a 21-kD protein (27). (Bcl-2α and Baxα hereafter will be referred to as Bcl-2 and Bax.) Bcl-2, Bcl-XL, and Bax each contain a stretch of hydrophobic amino acids, ∼20 residues in length, at their COOH termini. There is little amino acid sequence conservation within these tails, but based on hydropathy plot analysis they are presumed to function in anchoring these proteins into organelle membranes (2, 25). Bcl-2 has been localized to the nuclear membrane, endoplasmic reticulum, and the outer mitochondrial membranes (3, 12, 19, 24). Removal of the COOH-terminal tail from Bcl-2 changes the subcellular localization to the cytosol, but it remains unclear what effect the removal of the tail has on the ability of Bcl-2 to inhibit apoptosis (1, 2, 13, 35). Bcl-XL also has been localized to the outer membrane of mitochondria (9). Although the localization of Bax has been less extensively investigated, Bax has been suggested to be targeted to organelle membranes (11), and in particular to mitochondria (39) by a COOH-terminal hydrophobic region. However, it was recently reported that in BHK cells, low level expression of Bax had a punctate, organelle localization whereas overexpressed Bax displayed a diffuse cytosolic distribution when visualized by immunocytochemistry (32). In that study, coexpression of high levels of Bcl-2 led to a punctate distribution of Bax, suggesting that Bax association with organelle membranes required Bcl-2. In another recent study, the endogenous Bax in murine thymocytes, splenocytes, and HL-60 human leukemia cells was found by subcellular fractionation to be predominantly soluble, localized separately from membrane-bound Bcl-2 (14, 15). During thymocyte apoptosis, a significant fraction of the Bax was found to redistribute from the cytosol to membranes. This latter finding raises the possibility of the regulation of Bax subcellular localization during apoptosis (14).
Previous investigations of Bax localization have relied upon cell fixation or fractionation. In this study, we examine Bax localization in living cells. The genes encoding human Bcl-2, Bcl-XL, and Bax were fused to that of the green fluorescent protein (GFP),1 and herein we report the localization by confocal microscopy of these fusion proteins in both healthy cells and cells undergoing apoptosis. The time course of changes in GFP–Bax localization relative to other cellular events occurring during apoptosis was characterized. Furthermore, the influence of Bcl-2 and Bcl-XL overexpression on Bax localization was examined. We have deleted the COOH-terminal hydrophobic tails from Bax, Bcl-2, and Bcl-XL to investigate the importance of these regions in the subcellular localization, and the ability of these proteins to influence cell death.
Materials and Methods
Except as noted, reagents were purchased from Sigma Chemical Co. (St. Louis, MO).
Generation of Expression Constructs
Full-length and COOH-terminal truncated forms (ΔCT) of human Bcl-2, Bcl-XL, and Bax were synthesized by PCR amplification and cloned into the pcDNA-3 mammalian expression vector (Invitrogen, Carlsbad, CA). A similiar strategy was used for GFP-fusion constructs, using the C3-EGFP plasmid (Clontech Laboratories, Inc., Palo Alto, CA). The cDNAs for human bcl-2, human bcl-XL, and human bax were gifts of C.M. Croee (Jefferson Cancer Center, Philadelphia, PA), C.B. Thompson (HHMI and University of Chicago, Chicago, IL), and S.J. Korsmeyer (HHMI and Washington University, St. Louis, MO), respectively. The bcl-2 ΔCT, bcl-XL ΔCT, and bax ΔCT forms constructed encode for proteins which have a deletion of 23, 25, and 21 amino acids at their COOH-terminal ends, respectively. For full-length constructs of each gene, oligonucleotides placing a restriction enzyme site immediately adjacent to the start codon (forward primers) or stop codon (reverse primers) were synthesized (GIBCO BRL, Gaithersburg, MD). For the truncated (ΔCT) forms, a stop codon was introduced by the reverse primers. PCR was performed using Pfu polymerase (Stratagene, La Jolla, CA) to minimize errors. PCR fragments were purified, digested with restriction enzymes, and ligated into the expression vectors. The bcl-XL, bcl-XLΔCT, bcl-2, and bcl-2 ΔCT genes were subcloned between the XhoI and XbaI sites in the polycloning regions of both pcDNA3 and C3-EGFP. The bax and bax ΔCT genes were subcloned between the HindIII and EcoRI sites in the polycloning regions of both pcDNA3 and C3-EGFP. All constructs were sequenced using the Sequenase 2 kit (United States Biochemical Corp., Cleveland, OH).
Cell Culture and Transient Transfection
L929 murine fibrosarcoma and Cos-7 green monkey renal epithelia cell lines (American Type Culture Collection, Rockville, MD) were grown in Minimum essential medium, Eagle's in Earle's balanced salt solution and DME, respectively, each supplemented with 2 mM glutamine, 1× nonessential amino acids, 2.5 mM sodium pyruvate, 100 U/ml penicillin, 100 μg/ ml streptomycin (all from Biofluids Inc., Rockville, MD), 50 μg/ml gentamicin (GIBCO BRL), and 10% heat-treated FCS. Cells were cultured at 37°C in 5% CO2 and then split twice weekly using 0.05% trypsin/0.02% versene (Biofluids Inc.). In preparation for transfection, cells were plated at a density of 2 × 105 cells per well in six-well tissue-culture plates (Becton Dickinson Labware, Bedford, MA). Either one (Cos-7) or two (L929) days later, cells were transiently transfected using the cationic lipid LipofectAmine (GIBCO BRL) as described by the manufacturer, using 2 μg of plasmid DNA and either 8 (L929) or 10 μl (Cos-7) of LipofectAmine per well. After 5 h in serum-free medium, an equal volume of medium containing 2× normal FCS was added. 1 d after transfection, the cells were washed once and placed in normal growth media. Transfected cells were then cultured for an additional 24 h, and visualized by microscopy. For cotransfection experiments (see Fig. 7), the above procedure was followed, except that a total of 4 μg of plasmid DNA was used per well: 1 of C3–EGFP–Bax and 3 of either pcDNA3–Bcl-2 or pcDNA3–Bcl-XL.
Figure 7.
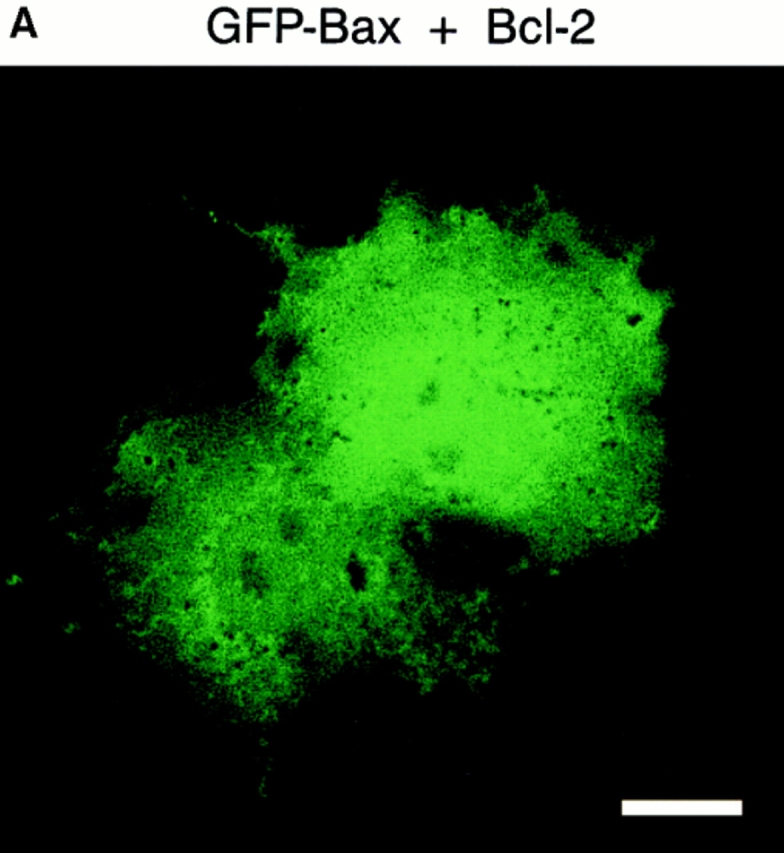

GFP–Bax distribution in Cos-7 cells is not affected by coexpression of Bcl-2. Cells were transiently transfected with a 3:1 ratio of plasmid containing Bcl-2 and GFP–Bax, and examined 48 h later by confocal microscopy. (A) In Cos-7 cells cotransfected with Bcl-2 and GFP– Bax, the GFP–Bax distributes throughout the cell in a pattern that is indistinguishable from that of cells expressing GFP–Bax alone. (B) The coexpression of GFP–Bax and human Bcl-2 in Cos-7 cells was monitored by Western blotting with α human Bax 2D2 and α human Bcl-2 8C8 monoclonal antibodies. Lanes a are the Cos-7 cell lysates from transfection with the pcDNA 3 vector alone. Lanes b are the cell lysates from the pcDNA 3–Bcl-2 transfection, and lanes c are from the cotransfection of C3–EGFP-Bax and pcDNA 3–Bcl-2. Arrows a, b, and c represent the GFP–Bax, its NH2-terminal translational variant, and endogenous simian Bax, respectively. Bar, 20 μm.
Generation of Anti–human Bcl-2 Monoclonal Antibody
A peptide corresponding to amino acids 41–54 of human Bcl-2 (GAAPAPGIFSSQPGC; Peptide Technologies Corp., Gaithersburg, MD) was conjugated to maleimide-activated keyhole limpet hemocyanin (KLH) (Pierce Chemical Co., Rockford, IL) through the cysteine residue according to the protocol provided by the manufacturer. Mice were immunized with the peptide–KLH conjugate and splenocytes from immunoreactive mice were fused by PEG 4,000 to murine NS-1 myeloma cells and selected with histone acetyltransferase (HAT) medium as previously described (15). The anti–human Bcl-2 antibody was designated αhBcl-2 8C8.
Cell Lysis and Western Blotting
Transfected cells from two wells of a six-well plate were washed once with PBS and then lysed in cold solubilization buffer (PBS, 25 μg/ml phenylmethylsulphonyl fluoride, 1 μg/ml leupeptin, and 1 μg/ml aprotinin) containing 1% Triton X-100. For Western blotting, 20 μl of each sample were separated by electrophoresis using 12% SDS-PAGE gels. Gels were electrotransferred onto Immobilon-P (Millipore Corp., Waters Chromatography, Milford, MA) membranes. The blots were blocked in PBS/0.05% Tween 20 containing 5% fetal bovine serum and incubated with the appropriate antibody: anti–human Bcl-2 8C8, anti-universal Bcl-XL 2H12 (15), or anti–human Bax 2D2 monoclonal antibodies (15). Primary antibody binding was detected by blotting with sheep anti–mouse F(ab') linked to horseradish peroxidase (Amersham Corp., Arlington Heights, IL), followed by band visualization using enhanced chemiluminescence as described by the manufacturer (Amersham Corp.).
Confocal Microscopy
Cells used for confocal microscopy were transfected as described above on 1.5 thickness glass coverslips pretreated with 5 μg/ml poly-l-lysine. After 24–48 h, cells were prepared for microscopy by incubation with 20 ng/ ml of a mitochondrion-specific dye (Mitotracker Red CMXRos; Molecular Probes Inc., Eugene, OR) and/or 100 ng/ml bis-benzamide for 30 min, as indicated in individual experiments. After pretreatment, cells were transferred to a sealed mounting chamber containing fresh medium; in indicated experiments, this medium was supplemented with 1–3 μM staurosporine (STS). Images were collected on a microscope (model LSM 410 with a 40× 1.2 NA Apochromat objective; Carl Zeiss, Thornwood, NY). The 488- and 568-nm lines of a krypton/argon laser were used for fluorescence excitation of GFP and Mitotracker Red CMXRos, respectively, and the 364-nm line of an argon laser was used for excitation of bis-benzamide dye. The temperature of the specimen was maintained between 35° and 37°C with an air stream incubator.
To investigate the mobility of the GFP-fusion proteins, the fluorescence within a region of a cell was photobleached by scanning the region with the 488-nm laser line at full power (no attenuation) and then the entire cell was scanned at 10-s intervals with low laser power (10% power, 0.3% attenuation) to monitor the recovery of fluorescence. The fraction of the fluorescent protein that was mobile was calculated as described (4) by comparing the fluorescence intensities in two regions of the cell, one inside the photobleach zone and the other elsewhere, before and 100 s after the photobleach (mobile fraction = ratio after 100-s recovery/ratio before photobleach). The time course of fluorescence recovery was determined by photobleaching a 16-μm strip within a flattened area of the cell and then monitoring the recovery of fluorescence within the strip (200 images at 0.46-s intervals).
Cell Viability Assay
To assess the effects of the full-length and ΔCT forms of Bcl-2, Bcl-XL, and Bax, on cell viability, a cotransfection assay was performed in L929 or Cos-7 cells. Wells plated as described above were cotransfected with one vector construct containing a given gene in pcDNA3, and the second vector, EGFP (Clontech Laboratories Inc.), containing a gene for a red-shifted GFP. A 3:1 molar ratio of the former to the latter was used, with a total of 4 μg of DNA per well. Otherwise, cells were transfected as above. At 36–48 h after transfection, cells were washed twice, fresh culture medium was added, and the number of GFP-positive cells in a defined field was determined by examination with a fluorescence microscope. Apoptosis was then induced by the addition of medium containing STS at the concentrations indicated, and the number of GFP-positive cells within the same field was assessed at regular intervals over the next 24–42 h. To test the effect on bioactivity of adding GFP to the NH2 termini of the Bcl-2 family members, a similar assay was performed on cells transfected with 2 μg of the appropriate full-length or ΔCT gene fused to GFP inserted into C3-EGFP. For each construct, the assay was repeated three or four times. To normalize for variations in transfection efficiency, changes in viability are expressed as percentage decrease in the number of fluorescent cells within a given field over time.
Results
GFP–Bax Is a Cytosolic Protein in Healthy Cells Whereas GFP–Bcl-2 and the Majority of Bcl-XL Associate with Mitochondria
We examined the location of Bcl-2, Bcl-XL, and Bax in individual living cells by using GFP fusion constructs. Fusion constructs containing GFP have been successfully used to study the subcellular localization of a number of proteins (16, 26, 33). The gene encoding a high fluorescence, red-shifted GFP variant (5) was fused to the 5′ termini of cDNAs encoding human Bcl-2, Bcl-XL, or Bax. L929 murine fibrosarcoma and Cos-7 green monkey kidney epithelial cells were transfected with the fusion constructs encoding GFP–Bcl-2, GFP–Bcl-XL, and GFP–Bax.
The localization of these transiently expressed GFP– linked fusion proteins was visualized in living cells using confocal microscopy. In both Cos-7 (Fig. 1, a and b) and L929 cells (Fig. 2, a and b), GFP alone has a diffuse distribution, filling the entire cell as previously reported (28). GFP–Bcl-2 displays a punctate pattern of distribution, primarily in areas near the nucleus, in both Cos-7 (Fig. 1, e and f) and L929 (Fig. 2, e and f) cell lines. This distribution matches the localization determined previously for Bcl-2 by immunofluorescence (24, 41), and suggests association with an intracellular organelle or organelles. GFP–Bcl-XL displays a bright punctate distribution similar to that of GFP– Bcl-2 superimposed upon a fainter diffuse background (Figs. 1 and 2, i and j), suggesting both soluble and membrane-bound populations in individual cells. This is consistent with the dual localization of Bcl-XL in both the soluble and membrane pellets in cellular fractionation experiments (14, 15). In contrast to the localization of GFP–Bcl-2 and GFP–Bcl-XL, GFP–Bax has a diffuse distribution similar to that of GFP alone in both Cos-7 (Fig. 1, m and n) and L929 (Fig. 2, m and n) cell types.
Figure 1.
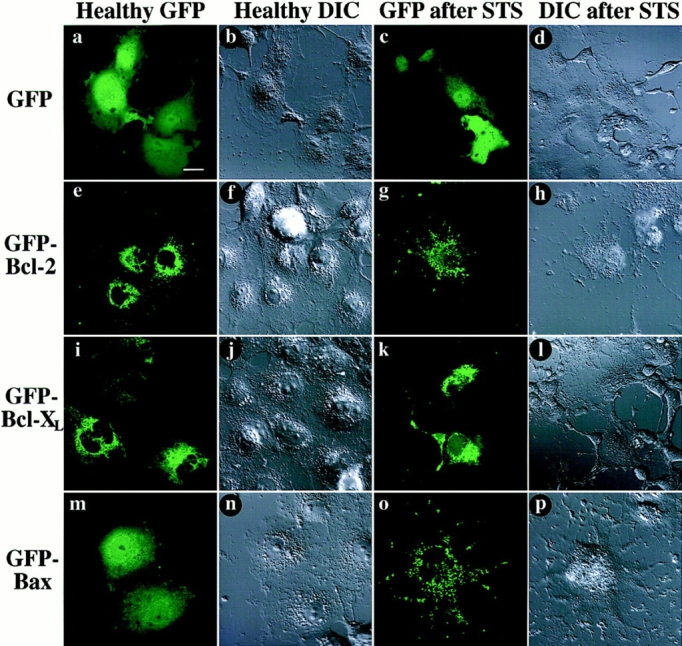
Distribution of GFP-fusion proteins expressed in living Cos-7 cells, before and after STS treatment. At 48 h after transfection, cells were examined by confocal microscopy. Each field was visualized by laser fluorescence to detect GFP (a, c, e, g, i, k, m, and o), and by DIC to illustrate cell morphology (b, d, f, h, j, l, n, and p). Native GFP (a and b) distributes throughout untreated cells, that display a healthy morphology. In contrast, GFP–Bcl-2 (e and f) displays a punctate distribution, mostly perinuclear. GFP–Bcl-XL (i and j) appears to distribute in a mainly punctate pattern similar to GFP–Bcl-2, but also has a faint diffuse fluorescence that extends throughout cells. GFP-Bax (m and n) distributes freely throughout cells, in a pattern indistinguishable from that of GFP alone. After 6 h of treatment with 1 μM STS, Cos-7 cells assume a morphology indicative of apoptosis, including loss of cell volume, retraction of processes, and blebbing of cell membrane. Despite these changes, native GFP (c and d) still distributes throughout the cell. GFP–Bcl-2 (g and h) retains a punctate distribution after STS treatment. GFP–Bcl-XL (k and l) also does not appear to change distribution with STS treatment, remaining mainly punctate with a background of diffuse fluorescence. However, GFP–Bax (o and p) undergoes a striking change in distribution, becoming entirely punctate after STS treatment. Bar, 20 μm.
Figure 2.

Distribution of GFP-fusion proteins expressed in living L929 cells, before and after STS treatment. At 48 h after transfection, cells were examined by confocal microscopy. Each field was visualized by laser fluorescence to detect GFP (a, c, e, g, i, k, m, and o), and by DIC to illustrate cell morphology (b, d, f, h, j, l, n, and p). Native GFP (a and b) distributes throughout healthy cells. GFP–Bcl-2 (e and f) displays a punctate perinuclear distribution. GFP–Bcl-XL (i and j) appears to have both punctate and diffuse fractions. GFP–Bax (m and n) distributes freely throughout cells. After 6 h of treatment with 1 μM STS, L929 cells undergo a loss of cell volume and retraction and blebbing of extended processes. Native GFP (c and d) is found throughout the cells after STS treatment, including blebs. GFP–Bcl-2 (g and h) retains a punctate distribution after STS treatment. After STS treatment, GFP–Bcl-XL (k and l) shows a largely punctate distribution with a background of diffuse fluorescence. As in Cos-7 cells, GFP–Bax (o and p) undergoes a change in distribution, becoming punctate after STS treament. Bar, 30 μm.
We examined the GFP fusion protein expression by Western blot analysis of transiently transfected Cos-7 cells 36 h after transfection. Immunoblotting of these lysates confirmed that GFP–Bcl-2 (Fig. 3, middle, arrow a), GFP– Bcl-XL (Fig. 3, bottom, arrow a), and GFP–Bax (Fig. 3, top, arrow a) are of appropriate size. Low molecular weight bands (lane c) representing native Bcl-2, Bcl-XL, and Bax were detected in all lysates. An additional, unexpected protein band (arrow b) of intermediate molecular weight was detected for each expression construct (Fig. 3). This intermediate molecular weight form was also found when COOH-terminal truncated forms of Bcl-2 (lane c), Bcl-XL (lane e), and Bax (lane a) fused to GFP were expressed and immunoblotted. When compared to full-length forms, all three COOH-terminal truncated proteins ran at the expected slightly lower molecular weights (Fig. 3). The COOH-terminal truncated constructs display slightly lower molecular weights for both the high molecular weight and intermediate molecular weight bands, indicating that the intermediate weight bands likely represent proteins shortened from the NH2-terminal, GFP end, of the fusion protein. Thus, the intermediate size bands may result from expression of transcripts initiated at an internal start codon within the GFP gene or proteolytic fragments lacking part of the GFP molecule, and thus they may or may not fluoresce.
Figure 3.

Expression of truncated and full-length GFP–Bax, GFP– Bcl-2, and GFP–Bcl-XL fusion proteins in COS cells. COOH-terminal truncated and full-length GFP fusion constructs of Bax (lanes a and b), Bcl-2 (lanes c and d) and Bcl-XL (lanes e and f) were transfected into COS cells. The expression of the fusion products was monitored by Western blotting of the cell lysates with α human Bax 2D2, α human Bcl-2 8C8, and α universal Bcl-XL 2H12 monoclonal antibodies for the detection of Bax, Bcl-2, and Bcl-XL, respectively. Arrows a are the expressed fusion products. Arrows b are likely the in-frame NH2-terminal translational variants. Arrows c represent the endogenous simian Bax, Bcl-2, or Bcl-XL that cross-react with the above specified antibodies.
To determine if the fusion of GFP to Bcl-2, Bcl-XL, and Bax affected their bioactivities, we compared the viability of either L929 or Cos-7 cells transfected with wild-type cDNA constructs to those transfected with GFP-fusion constructs after induction of apoptosis with STS. As shown in Fig. 4, the protective activities of GFP–Bcl-2 (Fig. 4 A) and GFP–Bcl-XL (Fig. 4 B) in L929 cells are comparable to those of the wild-type forms. Likewise, the activity of GFP–Bax appears to accelerate cell death of Cos-7 cells as effectively as does wild-type Bax (Fig. 4 C). Thus, adding GFP to the NH2-terminus of Bcl-2, Bcl-XL, or Bax does not block their abilities to influence apoptosis. However, the addition of a large protein to the NH2 terminus may alter the potency of Bcl-2 family members to regulate apoptosis or influence their intracellular localization.
Figure 4.
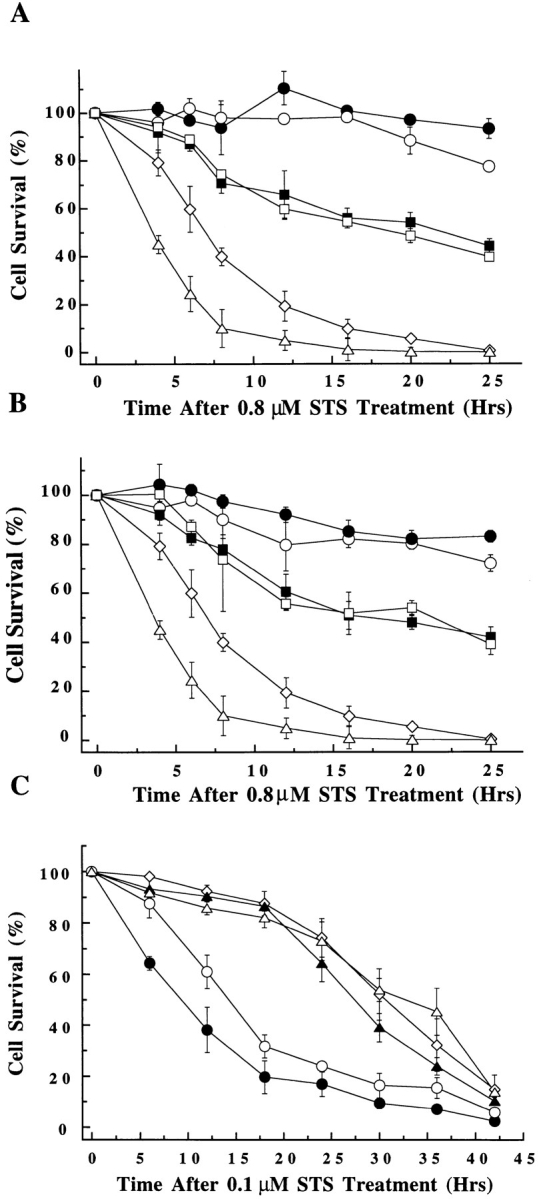
Effect on cell viability of transient expression of wild-type and COOH-terminal truncated forms of Bcl-2 family members, with and without GFP fused at the NH2-terminus. Cells were either singly transfected with the GFP fusion constructs or cotransfected with GFP and various Bcl-2 family members. At 48 h after transfection, cells were treated with STS. The number of GFP-positive cells was counted within a given field at regular intervals, starting at the time of STS addition (∼500 GFP-positive cells at time zero was typical for each experiment). Changes in cell viability are displayed as number of glowing cells within a field and expressed as a percentage of time zero value for that field. (A) Bcl-2 constructs expressed in L929 cells. (n = 4). pcDNA3 vector control (⋄), Bcl-2 (○), GFP–Bcl-2 (•), Bcl-2 ΔCT (□), GFP–Bcl-2 ΔCT (▪), and Bax (▵) (B) Bcl-XL constructs expressed in L929 cells. (n = 4). pcDNA3 vector control (⋄), Bcl-XL (○), GFP–Bcl-XL (•), Bcl-XL ΔCT (□), GFP–Bcl-XL ΔCT (▪), and Bax (▵) (n = 4). (C) Bax constructs expressed in Cos-7 cells. (n = 3). pcDNA3 vector control (⋄), Bax (○), GFP–Bax (•), Bax ΔCT (▵), GFP–Bax ΔCT (▴).
To investigate the identity of the organelle or organelles to which the GFP–Bcl-2 and the majority of the GFP–Bcl-XL localize, we treated transiently transfected Cos-7 cells with Mitotracker Red CMXRos. As shown in Fig. 5 (a–c), the pattern of GFP–Bcl-2 distribution coincides very closely with that of the mitochondrial marker, demonstrating that most of the GFP–Bcl-2 is found associated with mitochondria. The punctate fraction of the GFP–Bcl-XL also distributes to mitochondria (Fig. 5, d–f). These results match earlier findings localizing Bcl-2 and Bcl-XL to the membrane of mitochondria using other imaging modalities (19, 24). In constrast to GFP–Bcl-2 and GFP–Bcl-XL, GFP– Bax does not localize to mitochondria in healthy Cos-7 cells (Fig. 5, g–i).
Figure 5.

GFP–Bcl-2 and the punctate portion of GFP–Bcl-XL colocalize with mitochondria in Cos-7 cells. Transiently transfected Cos-7 cells were treated with 20 ng/ml Mitotracker Red CMXRos to stain mitochondria and then examined by laser fluorescence confocal microscopy. Each field was independently visualized with the appropriate wavelength for GFP (a, d, and g) and for Mitotracker Red CMXRos (b, e, and h) and then the two images were overlaid (c, f, and i). GFP–Bcl-2 localizes primarily to mitochondria (a–c). A majority GFP–Bcl-XL also localizes to mitochondria, although there is a faint, diffuse background which is not punctate (d–f). GFP–Bax does not localize to mitochondria in healthy cells (g–i). Bar, 20 μm.
FRAP Indicates Bcl-2 Is Largely Immobile Whereas Bax Diffuses Freely
The similarity of the distribution of GFP–Bax to the distribution of GFP led us to investigate whether GFP–Bax is free to diffuse within the cytoplasm, as GFP has been shown to be (34). The mobilities of GFP–Bax and GFP were compared by photobleaching the fluorescence in a small region of the cell and then monitoring the recovery of fluorescence due to movement of unbleached molecules into the bleached zone. Both GFP and GFP–Bax moved rapidly into the bleached zone (Fig. 6). The time courses for fluorescence recovery of GFP and GFP–Bax were essentially identical (Fig. 6 B), and were consistent with diffusion kinetics. The proteins diffused too rapidly relative to the rates at which we could photobleach and monitor recovery to allow us to accurately determine diffusion constants.
Figure 6.


FRAP analysis of GFP, and GFP–Bax, and GFP–Bcl-2 in healthy and cells treated with STS. (A) Fluorescence recovery visualized in individual treated cells. In a healthy Cos-7 cell expressing GFP–Bax (a–d), fluorescence recovers rapidly in a photobleached area. After treatment with STS, a Cos-7 cell with punctate GFP–Bax (e–h) no longer recovers after photobleaching. In a healthy Cos-7 cell expressing GFP–Bcl-2 (i–l), recovery does not occur in the photobleached area. (B) Quantitative FRAP analysis to determine protein mobilities in cells expressing GFP alone, GFP–Bax, and GFP–Bcl-2. Fluorescence intensities during recovery after photobleach are plotted versus time. Data was collected at 0.46-s intervals during recovery. GFP and GFP–Bax rapidly recover with a time course consistent with diffusion. GFP–Bcl-2 shows minimal recovery, indicating that it is largely immobile. All intensity values were normalized to prebleach intensity (I = 100) and to the intensity immediately after photobleach (I = 0). Bars, 20 μm.
Photobleaching experiments also were performed on cells expressing GFP–Bcl-2 (Fig. 6). In contrast to GFP and GFP-Bax, GFP-Bcl-2 appeared to be largely immobile. The mobile fraction (percentage recovery) at 100 s after photobleaching (calculated as described in Materials and Methods) was 9 to 13% for GFP–Bcl-2 (n = 3) as compared with 45 to 85% for GFP–Bax (n = 4), and 82 ± 2% for GFP alone (34). The low mobility of GFP–Bcl-2 supports the conclusion that it resides in discrete organelles (mitochondria) rather than in a continuous membrane system such as the endoplasmic reticulum (where fluorescence recovers rapidly after photobleaching [4]).
Role of Bcl-2 in Docking Bax
Recent studies have proposed that Bax (32) and Bad (40) require Bcl-2 to associate with organelle membranes. To test the possibility that GFP–Bax localization to organelles is limited by the endogenous level of either Bcl-2 or Bcl-XL, vectors expressing either Bcl-2 or Bcl-XL were cotransfected with the GFP–Bax vector. As shown in Fig. 7 A, coexpression of Bcl-2 at high levels, verified by immunoblotting of cell lysates (Fig. 7 B), did not alter the cytoplasmic distribution of GFP–Bax. Similar results were obtained when Bcl-XL was coexpressed with GFP–Bax (data not shown). Thus, we find Bax is a soluble, cytosolic protein in healthy cells, which does not bind organelles even in the presence of elevated levels of Bcl-2 or Bcl-XL.
Bax Localization and Mobility Change during Apoptosis
Endogenous Bax and some of the endogeneous Bcl-XL migrate from the supernatant to the membrane pellets in cell fractionation experiments performed on thymocytes undergoing apoptosis (14). To investigate this phenomenon in living cells, Cos-7 cells were transiently transfected with native GFP, GFP–Bcl-2, GFP–Bcl-XL, and GFP–Bax. Cells expressing each construct were treated with 1 μM STS, and after 6 h both STS-treated and untreated cells were examined by confocal microscopy. STS-treated cells underwent cell shrinkage and blebbing of the cell membrane, a morphology indicative of apoptosis (Fig. 1, compare b, f, j, and n with d, h, l, and p). GFP–Bax markedly changed its intracellular distribution, relocating within cells during apoptosis from a diffuse (Fig. 1 m) to a punctate pattern (Fig. 1 o). This change was not observed with GFP alone (Fig. 1 c). No changes in the patterns of distribution of either GFP–Bcl-2 (Fig. 1 g) or GFP–Bcl-XL (Fig. 1 k) were observed in treated cells beyond those related to changes in cell size and shape. It is possible that redistribution of soluble GFP–Bcl-XL is reduced to some extent in this system due to the ability of overexpressed GFP–Bcl-XL to inhibit apoptosis (Fig. 4 B). Similiar experiments performed using another cell line, L929 fibroblasts, confirmed that GFP–Bax undergoes a shift from a diffuse to punctate distribution pattern during apoptosis whereas the localization patterns of GFP–Bcl-2 and GFP–Bcl-XL remain essentially unchanged (Fig. 2).
Mitochondrial staining of GFP–Bax–expressing Cos-7 cells undergoing apoptosis suggests that much of the GFP–Bax attaches to or enters mitochondria (Fig. 8, a–c). However, as there are also sites of Bax condensation that do not stain for mitochondria, it is possible that GFP–Bax is also localizing to other organelles upon induction of apoptosis or that mitochondria lose their Mitotracker Red CMXRos staining during apoptosis.
Figure 8.

GFP–Bax colocalizes with mitochondria in Cos-7 cells after STS treatment. Cos-7 cells transiently expressing GFP–Bax were treated with 1 μM STS to induce apoptosis and with 20 ng/ml Mitotracker Red CMXRos to stain for mitochondria and then examined after 4 h by laser fluorescence confocal microscopy. The field shown was independently visualized by laser fluorescence confocal microscopy at the appropriate wavelength for GFP (a) and for Mitotracker Red CMXRos (b), and the two images were then overlaid (c). Whereas a majority of the punctate GFP–Bax appears to localize to mitochondria, there are also areas where the GFP–Bax is punctate but which do not label with the mitochondrial dye (c, arrow). Bar, 20 μm.
The redistribution of Bax to organelles induced by STS treatment is associated with a reduction in Bax mobility (Fig. 6). FRAP experiments on cells in which GFP–Bax had a punctate (mitochondrial) localization indicated that the mobile fraction was 10 to 30% (n = 2), as compared with 45 to 85% in untreated cells. Thus, the punctate form of GFP–Bax is less mobile than the diffuse form of GFP–Bax consistent with the model that Bax redistributes from a cytoplasmic to membrane-bound localization during apoptosis.
Time Course of Bax Redistribution during Apoptosis
To relate the timing of the shift in GFP–Bax localization to other morphological events that occur during apoptosis, individual Cos-7 cells expressing GFP–Bax were followed by confocal microscopy over the course of STS-induced cell death. The change in GFP-Bax localization within individual cells occurred swiftly, over the course of ∼30 min after initiation (Fig. 9, c–e). Various cells in a given field may begin the process of GFP–Bax redistribution at different times, as seen in Fig. 9. At the time point when GFP–Bax begins redistribution, little alteration in the overall structure of the cell is evident when viewed by differential interference contrast (DIC) microscopy (Fig. 9 h), or by interference reflection microscopy that shows the cell– substrate interaction (data not shown). At 10–30 min after the initiation of Bax redistribution, cell shrinkage first became apparent by DIC imaging (Fig. 9, i and j, arrows).
Figure 9.

GFP–Bax redistribution occurs before cell shrinkage associated with apoptosis. A field containing three living Cos-7 cells expressing GFP–Bax was followed over time after addition of 1 μM STS. At each timepoint, the field was visualized by laser fluorescence to detect GFP–Bax (a–e and k–o), and by DIC to illustrate cell morphology (f–j and p–t). Time elapsed after addition of STS is indicated in the corner of each of the laser fluorescence panels. Redistribution of the GFP–Bax was first detectable for the two cells towards the top of the field at 1 h, 24 min after STS addition (c). Changes in cell shape first became apparent 12–24 min later, as evidenced by a retraction of cell outlines (arrows, i and j). Notice that at these timepoints, the lowermost cell has not yet initiated either GFP–Bax redistribution or cell shrinkage. GFP–Bax redistribution is first detectable for the lowermost cell at 2 h, 36 min (n). Bar, 20 μm.
Cells expressing GFP–Bax were incubated with the nuclear stain bis-benzamide (Hoechst 33342), which labels DNA and is commonly used to detect the nuclear changes characteristic of apoptosis. At low concentrations, this stain can be used to study the location of DNA within living and even dividing cells without affecting cell viability (8). After bis-benzamide staining, cells were then treated with STS to induce apoptosis. As shown in Fig. 10, there is no evidence of nuclear changes when GFP-Bax initially begins redistribution. At ∼3 h after the initiation of the GFP–Bax transition, a more uneven pattern of bis-benzamide staining can be seen in the nuclei of cells (Fig. 10 e), suggestive of nuclear condensation. This gradually progresses to nuclear fragmentation, clearly apparent by 12 h after initiation of GFP–Bax redistribution (Fig. 10 f). Therefore, the shift in GFP–Bax distribution appears to be an early event in apoptosis, preceding several established morphological changes associated with apoptosis.
Figure 10.

GFP–Bax redistribution precedes nuclear fragmentation. A field containing three living Cos-7 cells expressing GFP–Bax was followed over time after addition of 1 μM STS, and 100 ng/ml of the nuclear stain bis-benzamide (a–e). In each panel, laser fluorescence confocal microscopy was used at the appropriate wavelength to visualize GFP (green) and bis-benzamide (blue). Time elapsed after STS addition is indicated in each panel. After 15 h (f) cells show fragmented nuclei associated with apoptosis. Bar, 20 μm.
COOH-terminal Hydrophobic Region Is Required for Bax Redistribution
The COOH-terminal hydrophobic region, spanning the last 20–24 amino acids of Bcl-2, Bcl-XL, and Bax is thought to act as an anchor localizing these proteins to the membranes of organelles. As our results suggest that Bax localizes to organelles only upon induction of apoptosis, we investigated the role of the COOH terminus in Bax trafficking. ΔCT constructs of GFP–Bcl-2 (Fig. 11, a and c), GFP-Bcl-XL (Fig. 11, e and g), and GFP–Bax (Fig. 11, i and k) all display a diffuse distribution in healthy cells, consistent with localization throughout the cytosol, when expressed in either Cos-7 or L929 cell lines. Induction of apoptosis causes no apparent redistribution of any of the ΔCT constructs in either cell line (Fig. 11). This indicates that the COOH terminus may play a pivotal role not only in anchoring Bcl-2 and Bcl-XL to organelles in healthy cells, but also in mediating the organelle association of Bax observed in cells undergoing apoptosis.
Figure 11.

Distribution of GFP-fusion proteins lacking COOH-terminal hydrophobic domains before and after STS treatment. Cos-7 and L929 cells were transiently transfected with the appropriate construct, and examined 48 h later by confocal microscopy. Each field was visualized by laser fluorescence to detect GFP. In both healthy Cos-7 cells (a) and L929 cells (c), ΔCT GFP– Bcl-2 was diffusely distributed throughout the cytosol. Likewise, in healthy Cos-7 cells (e) and L929 cells (g), ΔCT GFP–Bcl-XL was diffusely distributed throughout the cytosol. Finally, in both healthy Cos-7 cells (i) and L929 cells (k), ΔCT GFP– Bax was diffusely distributed throughout the cytosol. Cells expressing truncated GFP-fusion proteins were treated with 1 μM STS, and examined after 6 h. In both Cos-7 cells (b, f, and j) and L929 cells (d, h, and l), none of the truncated proteins redistributed after STS treatment. Bar, 20 μm.
Deletion of the COOH-terminal Tail Attenuates the Influence of Bcl-2, Bcl-XL, and Bax on Cell Viability
If Bax insertion into organelles is required for death promoting activity, then Bax ΔCT would be expected to have diminished potential to enhance apoptosis relative to wild-type Bax. The bioactivity of ΔCT constructs of Bax, Bcl-2, and Bcl-XL was compared to that of the full-length genes in a cell viability assay both with and without GFP fused to the NH2 terminus of each protein upon induction of apoptosis with STS. As illustrated in Fig. 4, A and B, for both Bcl-2 and Bcl-XL, deletion of the COOH-terminal hydrophobic domain resulted in a decrease in the apoptosis protection in L929 cells. For Bax, the deletion of the COOH-terminal domain also resulted in a sharp decrease in the ability of both wild-type and GFP-linked protein to accelerate apoptosis in both Cos-7 (Fig. 4 C) and L929 cells (data not shown). These results indicate that the COOH-terminal hydrophobic domain is important for Bax function, linking the ability of Bax to redistribute to organelles during apoptosis with its death-accelerating activity.
Discussion
Bax promotes programmed cell death in mammalian cells (18, 27). However, Bax expression per se, at least at physiological levels, is not lethal to cells. For example, sympathetic neurons express high levels of bax mRNA, yet will not undergo apoptosis unless deprived of growth factors (6). As initially characterized, even cells stably overexpressing Bax proliferate normally (27), and Bax only accelerates cell death after an external signal, such as interleukin-3 withdrawal. As protein levels of Bax do not change during apoptosis (14, 27), it seems plausible that a posttranslational activation of Bax occurs during apoptosis. Our findings indicate that Bax activity may be regulated at the level of intracellular localization, with Bax moving to the membranes of organelles after induction of apoptosis, but before nuclear fragmentation. Elimination of 21 amino acids from the COOH terminus prevents Bax redistribution and abrogates Bax apoptosis-promoting activity, correlating organelle binding with bioactivity.
In contrast to cytosolic GFP–Bax, GFP–Bcl-2 displays a punctate distribution in healthy cells that coincides very strongly with a mitochondrial marker. GFP–Bcl-XL demonstrates a mixed pattern, suggesting that although much of the protein is associated with mitochondria, a significant amount is found diffusely throughout the cytosol. In cell fractionation studies, this soluble Bcl-XL found in healthy cells redistributes to associate with the membrane during apoptosis (14). No change in distribution is observed for either GFP–Bcl-2 or GFP–Bcl-XL after the addition of STS. However, the levels of GFP–Bcl-XL in the transfection experiments are sufficient to delay or even block apoptosis (Fig. 4); thus, GFP–Bcl-XL may actually prevent alterations in the intracellular milieu that would lead to its redistribution. Further experiments will be necessary to explore the intracellular distribution of GFP–Bcl-XL during apoptosis, perhaps by using apoptosis pathways that are Bcl-XL insensitive.
For all three of the Bcl-2 family members examined here, the COOH-terminal deletion eliminates the ability of GFP-tagged proteins to associate with organelles. The dependency of Bax redistribution on the COOH-terminal hydrophobic domain suggests that a conformational change or other modification might occur in Bax during apoptosis, allowing the previously inaccessible or blocked hydrophobic tail to insert into membranes. It is striking that the translocation domain of diphtheria toxin, which has structural homology to Bcl-XL, displays a similar ability to change from a soluble to membrane-associated state. In the case of the diphtheria toxin translocation domain, this insertion is driven by a conformational change triggered by low pH (7, 30). It remains unclear how Bax insertion into membranes is controlled. One interesting candidate is phosphorylation: a regulatory mechanism involving phosphorylation has recently been described that influences both localization and activity of the apoptosis promoter Bad (40), and phosphorylation of Bcl-2 has been suggested to regulate its function (10, 17, 22).
Our findings conflict with previous reports that suggest that Bax is constitutively localized to organelle membranes (for review see reference 29). One possible explanation for this discrepancy may be that in earlier work, Bax localization was examined in contexts where overexpression of Bax alone was sufficient to cause apoptosis, without the overt induction of apoptosis. In a study examining Bax localization in a transformed BRK cell line, Bax overexpression led directly to cell death (11). A recent paper on Bax localization to mitochondria (39) reported that the expression of fusion proteins containing full-length Bax was lethal to yeast cells, and removal of the COOH terminus abolished the ability of Bax to kill yeast cells and localize to yeast mitochondria. We have found that cationic lipid transfection procedures can themselves trigger apoptosis (Wolter, K.G., and X.-G. Xi, unpublished observations). Deletion of the COOH-terminal tail from GFP-Bax eliminated this increase in transfection-associated death. High levels of full-length GFP–Bax expression may, therefore, lead directly to the death of transfected cells, particularly if this expression closely follows the cellular stress of the transfection procedure. Perhaps some previous observations of organelle-located Bax were made on cells entering an apoptosis program. These reports and our own data concur in suggesting that Bax localization to organelles is a vital aspect of its death promoting ability.
However, our findings on the role of Bcl-2 in Bax localization clearly differ from those of Shibasaki and co-workers (32), who observed a redistribution of soluble Bax to a punctate pattern in BHK cells expressing high levels of Bcl-2. In our systems, we see no evidence for a localization of Bax to membranes driven by either Bcl-2 or Bcl-XL coexpression. This is consistent with earlier findings that Bax and Bcl-XL do not form heterodimers in healthy cells (15).
Recent reports indicate that Bcl-2 family members control the release of cytochrome C and apoptosis-inducing factor from mitochondria during apoptosis and contribute to cell death by caspase activation (20, 21). The overexpression of Bcl-2 has been reported to block the mitochondrial release of cytochrome C, and in doing so prevents apoptosis (38). These results are interesting, given the structural connection made between Bcl-XL and the diphtheria toxin translocation domain. This domain is capable of forming transmembrane ion channels, and is furthermore believed to multimerize to form pores that mediate the translocation of the catalytic domain of the diphtheria toxin across membranes. Bcl-XL and Bcl-2 have been reported to form transmembrane channels (23, 31). In this context, it is possible that upon Bax insertion into membranes, Bax may form channels or pores allowing for the release of factors such as cytochrome C from within the mitochondria to propagate the apoptotic pathway. This model does not preclude the interaction of Bax with Bcl-2 and Bcl-XL upon membrane insertion. In the future, it will be crucial to elucidate the trigger regulating Bax insertion into organelle membranes and the molecular consequences of membrane-bound Bax.
Acknowledgments
We would like to thank C. Croce, C. Thompson and S. Korsmeyer for the gifts of Bcl-2, Bcl-XL, and Bax genes, respectively. We thank R. Kocher, J. Castelli, X.-H. Liu, C. Ng, and P. Johnson for assistance, and L. Marrone (all from National Institutes of Health [NIH], Bethesda, MD), and K. Wood (Trevigen Inc., Gaithersburg, MD) for reading the manuscript.
Abbreviations used in this paper
- ΔCT
COOH-terminal truncation
- DIC
differential interference contrast
- GFP
green fluorescent protein
- STS
staurosporine
Footnotes
K.G. Wolter was supported by the Howard Hughes Medical Institute-National Institutes of Health Research Scholars Program.
Address all correspondence to Richard J. Youle, Bldg 10, Room 5D-37, National Institutes of Health, Bethesda, MD 20892. Tel.: (301) 496-6628. Fax: (301) 402-0380. E-mail: youle@helix.nih.gov
References
- 1.Alnemri ES, Robertson NM, Fernandes TF, Croce CM, Litwack G. Overexpressed full-length human BCL-2 extends the survival of baculovirus-infected Sf9 insect cells. Proc Natl Acad Sci USA. 1992;89:7295–7299. doi: 10.1073/pnas.89.16.7295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Borner C, Martinou I, Mattmann C, Irmler M, Schaerer E, Martinou J-C, Tschopp J. The protein bcl-2a does not require membrane attachment, but two conserved domains to suppress apoptosis. J Cell Biol. 1994;126:1059–1068. doi: 10.1083/jcb.126.4.1059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chen-Levy Z, Nourse J, Cleary ML. The bcl-2 candidate proto-oncogene product is a 24-kilodalton integral-membrane protein highly expressed in lymphoid cell lines and lymphomas carrying the t(14; 18) translocation. Mol Cell Biol. 1989;9:701–710. doi: 10.1128/mcb.9.2.701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cole NB, Smith CL, Sciaky N, Terasaki M, Edidin M, Lippincott-Schwartz J. Diffusional mobility of Golgi proteins in membranes of living cells. Science. 1996;273:797–801. doi: 10.1126/science.273.5276.797. [DOI] [PubMed] [Google Scholar]
- 5.Cormack BP, Valdivia RH, Falkow S. FACS-optimized mutants of the green fluorescent protein (GFP) Gene (Amst) 1996;173:33–38. doi: 10.1016/0378-1119(95)00685-0. [DOI] [PubMed] [Google Scholar]
- 6.Deckwerth TL, Elliott JL, Knudson CM, Johnson EM, Jr, Snider WD, Korsmeyer SJ. BAX is required for neuronal death after trophic factor deprivation and during development. Neuron. 1996;17:401–411. doi: 10.1016/s0896-6273(00)80173-7. [DOI] [PubMed] [Google Scholar]
- 7.Draper RK, Simon MI. The entry of diphtheria toxin into the mammalian cell cytoplasm: evidence for lysosomal involvement. J Cell Biol. 1980;87:849–854. doi: 10.1083/jcb.87.3.849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ellenberg J, Siggia ED, Moreira JE, Smith CL, Presley JF, Worman HJ, Lippincott-Schwartz J. Nuclear membrane dynamics and reassembly in living cells: targeting of an inner membrane protein in interphase and mitosis. J Cell Biol. 1997;138:1193–1206. doi: 10.1083/jcb.138.6.1193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gonzalez-Garcia M, Perez-Ballestero R, Ding L, Duan L, Boise LH, Thompson CB, Nunez G. Bcl-XL is the major bcl-x mRNA form expressed during murine development and its product localizes to mitochondria. Development (Camb) 1994;120:3033–3042. doi: 10.1242/dev.120.10.3033. [DOI] [PubMed] [Google Scholar]
- 10.Haldar S, Jena N, Croce CM. Inactivation of Bcl-2 by phosphorylation. Proc Natl Acad Sci USA. 1995;92:4507–4511. doi: 10.1073/pnas.92.10.4507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Han J, Sabbatini P, Perez D, Rao L, Modha D, White E. The E1B 19K protein blocks apoptosis by interacting with and inhibiting the p53-inducible and death-promoting Bax protein. Genes Dev. 1996;10:461–477. doi: 10.1101/gad.10.4.461. [DOI] [PubMed] [Google Scholar]
- 12.Hockenbery D, Nunez G, Milliman C, Schreiber RD, Korsmeyer SJ. Bcl-2 is an inner mitochondrial membrane protein that blocks programmed cell death. Nature (Lond) 1990;348:334–336. doi: 10.1038/348334a0. [DOI] [PubMed] [Google Scholar]
- 13.Hockenbery DM, Oltvai ZN, Yin X-M, Milliman CL, Korsmeyer SJ. Bcl-2 functions in an antioxidant pathway to prevent apoptosis. Cell. 1993;75:241–251. doi: 10.1016/0092-8674(93)80066-n. [DOI] [PubMed] [Google Scholar]
- 14.Hsu Y-T, Wolter KG, Youle RJ. Cytosol-to-membrane redistribution of Bax and Bcl-XLduring apoptosis. Proc Natl Acad Sci USA. 1997;94:3668–3672. doi: 10.1073/pnas.94.8.3668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hsu Y-T, Youle RJ. Nonionic detergents induce dimerization among members of the Bcl-2 family. J Biol Chem. 1997;272:13829–13834. doi: 10.1074/jbc.272.21.13829. [DOI] [PubMed] [Google Scholar]
- 16.Htun H, Barsony J, Renyi I, Gould DL, Hager GL. Visualization of glucocorticoid receptor translocation and intranuclear organization in living cells with a green fluorescent protein chimera. Proc Natl Acad Sci USA. 1996;93:4845–4850. doi: 10.1073/pnas.93.10.4845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ito T, Deng X, Carr B, May WS. Bcl-2 phosphorylation required for antiapoptosis function. J Biol Chem. 1997;272:11671–11673. doi: 10.1074/jbc.272.18.11671. [DOI] [PubMed] [Google Scholar]
- 18.Knudson CM, Tung KSK, Tourtellotte WG, Brown GAJ, Korsmeyer SJ. Bax-deficient mice with lymphoid hyperplasia and male germ cell death. Science. 1995;270:96–99. doi: 10.1126/science.270.5233.96. [DOI] [PubMed] [Google Scholar]
- 19.Krajewski S, Tanaka S, Takayama S, Schibler MJ, Fenton W, Reed JC. Investigation of the subcellular distribution of the bcl-2 oncoprotein: residence in the nuclear envelope, endoplasmic reticulum, and outer mitochondrial membranes. Cancer Res. 1993;53:4701–4714. [PubMed] [Google Scholar]
- 20.Kroemer G, Zamzami N, Susin SA. Mitochondrial control of apoptosis. Immunol Today. 1997;18:44–51. doi: 10.1016/s0167-5699(97)80014-x. [DOI] [PubMed] [Google Scholar]
- 21.Liu X, Kim CN, Yang J, Jemmerson R, Wang X. Induction of apoptotic program in cell-free extracts: requirement for dATP and cytochrome c. Cell. 1996;86:147–157. doi: 10.1016/s0092-8674(00)80085-9. [DOI] [PubMed] [Google Scholar]
- 22.May WS, Tyler PG, Ito T, Armstrong DK, Qatsha KA, Davidson NE. Interleukin-3 and Bryostatin-1 mediate hyperphosphorylation of Bcl2a in association with suppression of apoptosis. J Biol Chem. 1994;269:26865–26870. [PubMed] [Google Scholar]
- 23.Minn AJ, Velez P, Schendel SL, Liang H, Muchmore SW, Fesik SW, Fill M, Thompson CB. Bcl-x(L) forms an ion channel in synthetic lipid membranes. Nature. 1997;385:353–357. doi: 10.1038/385353a0. [DOI] [PubMed] [Google Scholar]
- 24.Monaghan P, Robertson D, Amos TA, Dyer MJ, Mason DY, Greaves MF. Ultrastructural localization of bcl-2 protein. J Histochem Cytochem. 1992;40:1819–1825. doi: 10.1177/40.12.1453000. [DOI] [PubMed] [Google Scholar]
- 25.Nguyen M, Millar DG, Yong VW, Korsmeyer SJ, Shore GC. Targeting of Bcl-2 to the mitochondrial outer membrane by a COOH-terminal signal anchor sequence. J Biol Chem. 1993;269:16521–16524. [PubMed] [Google Scholar]
- 26.Olson KR, McIntosh JR, Olmsted JB. Analysis of MAP 4 function in living cells using green fluorescent protein (GFP) chimeras. J Cell Biol. 1995;130:639–650. doi: 10.1083/jcb.130.3.639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Oltvai ZN, Milliman CL, Korsmeyer SJ. Bcl-2 heterodimerizes in vivo with a conserved homologue, Bax, that accelerates programmed cell death. Cell. 1993;74:609–619. doi: 10.1016/0092-8674(93)90509-o. [DOI] [PubMed] [Google Scholar]
- 28.Pines J. GFP in mammalian cells. Trends Genet. 1995;11:326–327. doi: 10.1016/s0168-9525(00)89092-7. [DOI] [PubMed] [Google Scholar]
- 29.Reed JC. Double identity for proteins of the Bcl-2 family. Nature. 1997;387:773–776. doi: 10.1038/42867. [DOI] [PubMed] [Google Scholar]
- 30.Sandvig K, Olsnes S. Diphtheria toxin entry is facilitated by low pH. J Cell Biol. 1980;87:828–832. doi: 10.1083/jcb.87.3.828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Schendel SL, Xie Z, Montal MO, Matsuyama S, Montal M, Reed JC. Channel formation by anitapoptotic protein Bcl-2. Proc Natl Acad Sci USA. 1997;94:5113–5118. doi: 10.1073/pnas.94.10.5113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Shibasaki F, Kondo E, Akagi T, McKeon F. Suppression of signalling through transcription factor NF-AT by interactions between calcineurin and Bcl-2. Nature. 1997;386:728–731. doi: 10.1038/386728a0. [DOI] [PubMed] [Google Scholar]
- 33.Stauber R, Gaitanaris GA, Pavlakis GN. Analysis of trafficking of Rev and transdominant Rev proteins in living cells using green fluorescent protein fusions: transdominant Rev blocks the export of Rev from the nucleus to the cytoplasm. Virology. 1995;213:439–449. doi: 10.1006/viro.1995.0016. [DOI] [PubMed] [Google Scholar]
- 34.Swaminathan R, Hoang CP, Verkman AS. Photobleaching recovery and anisotropy decay of green fluorescent protein GFP-S65T in solution and cells: cytoplasmic viscosity probed by green fluorescent protein translational and rotational diffusion. Biophys J. 1997;72:1900–1907. doi: 10.1016/S0006-3495(97)78835-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tanaka S, Saito K, Reed JC. Structure-function analysis of the Bcl-2 oncoprotein. J Biol Chem. 1993;268:10920–10926. [PubMed] [Google Scholar]
- 36.Tsujimoto Y, Ikegaki N, Croce CM. Characterization of the protein product of bcl-2, the gene involved in human follicular lymphoma. Oncogene. 1987;2:3–7. [PubMed] [Google Scholar]
- 37.Wyllie AH. Apoptosis and the regulation of cell numbers in normal and neoplastic tissues: an overview. Cancer Meta Rev. 1992;11:95–103. doi: 10.1007/BF00048057. [DOI] [PubMed] [Google Scholar]
- 38.Yang E, Korsmeyer SJ. Molecular thanatopsis: a discourse on the BcL-2 family and cell death. Blood. 1996;88:386–401. [PubMed] [Google Scholar]
- 39.Zha H, Fisk HA, Yaffe MP, Mahajan N, Herman B, Reed JC. Structure-function comparisons of the proapoptotic protein Bax in yeast and mammalian cells. Mol Cell Biol. 1996;16:6494–6508. doi: 10.1128/mcb.16.11.6494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zha J, Harada H, Yang E, Jockel J, Korsmeyer SJ. Serine phosphorylation of death agonist BAD in response to survival factor results in binding to 14-3-3 not BCL-X(L) Cell. 1996;87:619–628. doi: 10.1016/s0092-8674(00)81382-3. [DOI] [PubMed] [Google Scholar]
- 41.Zhu W, Cowie A, Wasfy GW, Penn LZ, Leber B, Andrews DW. Bcl-2 mutants with restricted subcellular location reveal spatially distinct pathways for apoptosis in different cell types. EMBO (Eur Mol Biol Organ) J. 1996;15:4130–4141. [PMC free article] [PubMed] [Google Scholar]