

Abstract
To infer the role of natural selection in shaping standing genetic diversity, it is necessary to assess the genomewide impact of demographic history on nucleotide diversity. In this study we analyzed sequence diversity of 16 nuclear loci in eight Pinus sylvestris populations. Populations were divided into four geographical groups on the basis of their current location and the geographical history of the region: northern Europe, central Europe, Spain, and Turkey. There were no among-group differences in the level of silent nucleotide diversity, which was ∼0.005/bp in all groups. There was some evidence that linkage disequilibrium extended further in northern Europe than in central Europe: the estimates of the population recombination rate parameter, ρ, were 0.0064 and 0.0294, respectively. The summary statistics of nucleotide diversity in central and northern European populations were compatible with an ancient bottleneck rather than the standard neutral model.
CHANGES in population size and other demographic events are part of the history of most natural populations and leave traces on the pattern of their genetic diversity. Often, demography can result in patterns similar to natural selection (e.g., Tajima 1989; Depaulis et al. 2003) and thus in spurious interpretations of genetic data. Early studies on nucleotide diversity were not able to take the demographic history into account, because in most cases the history of the populations was not known, and the number of loci was low. Studies on nucleotide diversity of multiple loci can help resolve the history, since the demographic events influence the whole genome, while selection affects only specific parts of the genome.
The amount of data on genomewide nucleotide diversity from different species has increased during the past few years. The human genome, as well as genomes of model species like Drosophila melanogaster and Arabidopsis thaliana, shows traces of past population size changes, mostly reflecting the recent colonizing of new areas (Akey et al. 2004; Nordborg et al. 2005; Ometto et al. 2005; Schmid et al. 2005). In crop plants, such as sunflower, maize, and alfalfa, bottlenecks caused by domestication have led to reduced genetic variation in their genomes (Liu and Burke 2006; Muller et al. 2006; Tenaillon et al. 2004). In the case of D. melanogaster, for example, simulations have shown that demographic models are able to explain the overall pattern of variation, and no selection needs to be incorporated (Haddrill et al. 2005).
Different populations of a species also have distinct histories, and even ancestral or refugial populations cannot be assumed to have a stable population history (Marth et al. 2004; Tenaillon et al. 2004; Voight et al. 2005; Schmid et al. 2006). Even when populations are not very isolated, their genetic polymorphism patterns may reflect differences in population history. For example, in humans, despite the rather low genetic differentiation between populations (Rosenberg et al. 2002), there are clear differences between demographic histories of Asians, Europeans, and Africans (Pluzhnikov et al. 2002; Marth et al. 2004; Voight et al. 2005).
Demography can leave various traces on sequence polymorphism data. Statistics describing the allele-frequency spectrum like Tajima's D or Fay and Wu's H may show distributions differing from neutral expectation as a result of an excess or a deficiency of rare or derived alleles (Tajima 1989; Fay and Wu 2000; Przeworski 2002; Depaulis et al. 2003). Not only allele frequencies, but also associations between different loci are affected by demography. Linkage disequilibrium (LD), the nonrandom association of alleles at different loci, increases during bottlenecks or as a result of admixture. Some demographic events may not affect the means of descriptive statistics, but increase their variance (Depaulis et al. 2003). This may be reflected, for instance, in heterogeneous values of nucleotide diversity or Tajima's D among loci, relative to neutral expectation (Depaulis et al. 2003). Generally, it is also expected that populations that have gone through bottlenecks during colonization or domestication have lost some nucleotide diversity (Nei et al. 1975). However, the power of any of these statistics to detect past events depends both on the severity and on the timing of the event (Depaulis et al. 2003). Voight et al. (2005) showed that combining multiple aspects of the data, namely the amount of polymorphism, characteristics of the allele-frequency spectrum, and the population recombination rate, into a new test statistic based on the P-values of the multiple summary statistics is a powerful method in searching for the correct demographic scenario.
So far most efforts to resolve the demographic histories of populations have been conducted on humans, model species and their relatives. One nonmodel plant species studied in this way is Norway spruce, where overall deviation from the standard neutral model (SNM) was detected (Heuertz et al. 2006). In this study we examine nucleotide variation at 16 nuclear loci of another conifer, Scots pine, Pinus sylvestris from the perspective of demographic history. P. sylvestris is an outcrossing coniferous tree with a large distribution in northern parts of Eurasia (Mirov 1967). Scots pine is locally adapted to various photoperiodic, temperature, and moisture conditions (Eiche 1966; Mirov 1967; Hurme et al. 1997). For understanding the genetic basis of local adaptation, it is important to distinguish between patterns of genetic diversity caused by population history and natural selection.
According to paleontological data, P. sylvestris or immediate ancestors existed in Europe already in the Tertiary period (2–65 MYA). During the existence of the species, the global climate has experienced several glacial and interglacial periods and the species has survived several large-scale changes in environment. On the basis of palynological data, the abundance of Pinus and other conifers has been oscillating during the last 100,000 years (Müller et al. 2003; Cheddadi et al. 2005), which probably reflects the environmental changes. Traditionally, it has been thought that most European forest trees have survived the glacial periods in refugial areas in Mediterranean peninsulas and colonized larger areas as the climate turned warmer. However, the history of boreal cold-tolerant tree species, such as P. sylvestris and Betula, might be different from that of more temperate species like Quercus and Tilia (Petit et al. 2003). Pollen and macrofossil records indicate that Pinus occurred in some parts of central Europe through glacial periods and probably did not totally disappear from there during the last glacial maximum (LGM) (25,000–18,000 YBP) (Müller et al. 2003; Willis and Van Andel 2004). The glacial stages may actually have given boreal trees a competitive advantage in some regions that are more suitable for other tree species at present.
Even though the genetic differentiation among the P. sylvestris populations studied here is known to be very low (Dvornyk et al. 2002; García-Gil et al. 2003), the populations can be divided into four groups on the basis of their present distribution, the history of glaciations, and the genetic data. Turkish and Spanish populations are isolated from the main part of the distribution and also harbor specific mitochondrial types, which suggests that they might have distinct histories and represent refugial parts of Scots pine distribution (Sinclair et al. 1999; Soranzo et al. 2000; Pyhäjärvi et al. 2007). Populations that are now found in the northern parts of Europe are considered a separate group, since they are known to have colonized their current locations during the last 10,000 years (Huntley and Birks 1983). Areas of the north group were covered by ice during the last glacial period (Svendsen et al. 1999), but the central group has possibly been at its current location over the last glacial period and has putatively been part of a large population covering central Europe during the LGM (Willis and Van Andel 2004).
In earlier studies on sequence diversity of P. sylvestris the detected amount of polymorphism was quite low and there was a very low level of linkage disequilibrium (Dvornyk et al. 2002; García-Gil et al. 2003). The level of nucleotide diversity is in contrast to the large census size and the high amount of allozyme diversity observed in earlier studies on conifers (Hamrick and Godt 1996). The same observation has also been made in studies on other conifers and a low mutation rate or population history has been suggested to explain the low levels of diversity (e.g., Brown et al. 2004; Heuertz et al. 2006). One goal of our study was to obtain nucleotide diversity data at the multilocus level from different P. sylvestris populations. We also wanted to ask whether there are differences in nucleotide variation between the geographic groups due to population history. Coalescent simulations were used to examine the effect of different demographic events on genomewide variation of P. sylvestris. Specific questions were: (1) Is the observed pattern of nucleotide diversity a result of stochastic processes in an equilibrium population (i.e., compatible with the standard neutral model)?, (2) In which time period did possible demographic events take place?, and (3) Does demographic history explain the low nucleotide diversity observed in P. sylvestris?
MATERIALS AND METHODS
Sampling:
P. sylvestris seeds were collected from eight natural populations throughout Europe: northern Finland (latitude 67°11′, longitude 24°03′), southern Finland (60°52′, 21°20′), Sweden (56°28′, 15°55′), Poland (50°41′, 20°05′), Austria (47°26′, 16°29′), France (48°51′, 07°52′), Turkey (39°27′, 30°18′), and Spain [37°22′, 02°50′ (W)] (Figure 1). Seeds were obtained from the Finnish Forest Research Institute. The sampling was designed so that different latitudes of the species distribution in Europe were covered. The sampled populations were situated on low land except for the Spanish (altitude 2050 m) and the Turkish (altitude 1600 m) populations. Five (haploid) megagametophytes from different trees per population were analyzed for each locus, so that the total sample size was 40 gametes. P. pinaster was used as an outgroup. P. pinaster sequences for phyo and dhy-like were kindly provided by M. T. Cervera from INIA, Madrid. Note that not all loci were analyzed from the same megagametophyte per tree. However, the sequence of one locus was always based on one megagametophyte per individual tree.
Figure 1.—

The distribution and sampled locations of P. sylvestris. The whole distribution of P. sylvestris in Eurasia is presented in the small map. In the larger map eight sampled European sites are indicated with open circles. The dashed line indicates the extent of the ice sheet during last glacial maximum according to Svendsen et al. (1999). FIN, northern Finland; FIS, southern Finland; SWE, Sweden; POL, Poland; AUS, Austria; FRA, France; TUR, Turkey; and SPA, Spain.
Primer design and molecular methods:
Total genomic DNA was isolated from the haploid megagametophyte tissue by the FastDNA kit (Q-BIO Gene) (1% polyvinylpolypyrrolidone was added to the lysis buffer) after germinating the seeds for a few days in moisturized petri dishes. The megagametophyte tissue was used to obtain haplotype information directly. Finnzymes DyNAzyme EXT polymerase was used to amplify most of the loci, but for six loci (adhc, hlh1, dhy-like, nir, lp2, and chs) Invitrogen (San Diego) Taq was used instead. Amplifications were performed using Mastercycler Gradient (Eppendorf, Madison, WI) with Taq and RoboCycler Gradient 96 (Stratagene, La Jolla, CA) with EXT. The typical amplification protocol for Taq was 4 min at 94° followed by 38 cycles of 45 sec at 94°, 1 min at 56°, 1 min 30 sec at 72°, and finally one cycle for 10 min at 72°; and for EXT it was 3 min at 94° followed by 38 cycles of 1 min at 94°, 1 min 30 sec at 56°, 2 min 30 sec at 72°, and finally one cycle for 20 min at 72°.
Primers for chcs, lp2, and nir were published in Plomion et al. (1999) and those for hlh1 in Komulainen et al. (2003). The rest of the 12 PCR-primer pairs were designed on the basis of available annotated conifer sequences and ESTs (mostly P. taeda and P. pinaster) in the NCBI GenBank. If only ESTs were available, putative intron positions were determined on the basis of A. thaliana homologs. In some cases the sequenced region was expanded using inverse PCR. Primers were designed in conserved regions, usually in exons with Oligo Primer Analysis Software 5.0. Long primers (22–25 nt) with high annealing temperature (at least 68°) were preferred to ensure amplifying the same gene family member. Additional sequencing primers were designed when PCR primers did not suffice to cover the whole amplified region in both directions. PCR and sequencing primers are reported in supplemental Table S1 at http://www.genetics.org/supplemental/. Primers designed for P. sylvestris amplified 14 loci from P. pinaster (supplemental Table S2).
PCR products were purified using the MinElute PCR purification kit (QIAGEN, Valencia, CA). Both strands were sequenced using the Big Dye Terminator kit v3.1 (Applied Biosystems, Foster City, CA). Multiscreen plates (Millipore, Bedford, MA) with Sephadex matrix (Sigma-Aldrich, St. Louis) were used for purification of the sequencing products. Sequencing was conducted with an ABI PRISM 377 and later with a 3730 DNA Analyzer (Applied Biosystems).
Data analyses:
Contigs were assembled with Sequencher 4.0.5 (Gene Codes, Ann Arbor, MI) and aligned with Clustal X (1.83) (Thompson et al. 1997). For editing and inspecting the alignments by eye GeneDoc (2.6.002) (Nicholas et al. 1997) was used. Intron positions were determined on the basis of ESTs, homologous genes in other plants, and a web-based gene identification tool GeneSeqer at PlantGDB (http://www.plantgdb.org/cgi-bin/PlantGDB/GeneSeqer/PlantGDBgs.cgi). For estimating the standard population genetics parameters (e.g., number of segregating sites, Tajima's D, and Fay and Wu's H) DNAsp 4.10.0 (Rozas et al. 2003) was used if not otherwise stated.
Populations were divided into four geographical groups: north (northern Finland, southern Finland, and Sweden), central (Austria, Poland, France), Spain, and Turkey. Population structure was studied locus by locus and also by averaging the pairwise FST's over all loci. FST statistics used for pairwise estimates of differentiation assume an infinite-sites model and are analogous to Weir and Cockerham's (1984) θ (Hudson et al. 1992b). For each locus, two statistics of spatial differentiation, based on the number of differences between haplotypes, Snn (Hudson 2000) and KST* (Hudson et al. 1992a), were calculated. Their statistical significance was evaluated by 1000 permutations, where samples were randomly partitioned into populations (Hudson 2000; Hudson et al. 1992a). Genetic clustering within the total data was analyzed with BAPS 4.0 as linked molecular data (Corander et al. 2003). Each segregating site was treated as a locus and all segregating sites of each gene were assigned to the same “linkage group” to account for dependences between segregating sites in the gene.
Exceptionally large or small FST values may signal the effect of natural selection, but detecting these outliers requires that the relationship between FST and heterozygosity is taken into account (Beaumont and Nichols 1996). FST estimates between the central and the northern group were analyzed with the program FDIST2 (http://www.rubic.rdg.ac.uk/∼mab). The distribution of FST values as a function of heterozygosity was characterized by coalescent simulations with 100 demes (maximum), two sampled populations, and an expected FST of 0.0095 which was the observed value. The observed values of FST for each locus were based on haplotypes, not single polymorphic sites, to avoid the issue of linkage between sites. Since sample sizes in Spain and Turkey were too limited, they were excluded from this analysis.
To obtain multilocus estimates for the population mutation parameter θ = 4Neμ per nucleotide site and to assess its random variation, a posterior distribution of θ close to the likelihood was computed using MCMC simulation under a Bayesian model. The model is based on results of coalescent theory regarding the number of segregating sites (Tavaré 1984). Details of the model and the simulation procedure are described in the appendix. Estimates are based on the number of segregating silent sites (noncoding and synonymous). The locus lp2 was excluded from this analysis, because of too many missing data. A gene with no variation (laccase) was included in the analysis.
To verify whether some of the loci had unexpectedly high or low amounts of nucleotide variation, 5000 coalescence simulations implemented in DNAsp were performed to infer a neutral expectation for the distribution of the level of polymorphism, given a common θ. For each locus, estimates of population recombination and θ were based on estimates for central European populations (θ = 0.005 and ρ = 0.02) and adjusted according to gene length and sample size in each case.
The HKA program (distributed by Jody Hey, http://lifesci.rutgers.edu/∼heylab) was used to examine the fit of the data to the neutral equilibrium model in a multilocus setting by using Tajima's D and the HKA statistic. The program creates new data sets by coalescent simulations based on parameters observed in the data and examines whether the observed values of diversity and divergence are compatible with the neutral model.
The overall decay of LD with physical distance within genes was evaluated by nonlinear regression of r2 on distance between sites in base pairs (Hill and Robertson 1968). When the sample size is taken into account, the relationship
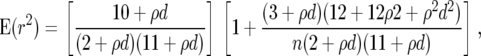 |
is expected, where n is the sample size, ρ = 4Nec between adjacent sites, d is a distance between sites in base pairs, and c is the recombination rate (Hill and Weir 1988). For each pair of parsimony-informative sites in a locus, the r2 estimate and distance were known. The nonlinear least-squares estimate of ρ was estimated by the nls function implemented in R version 2.2.0 (http://www.r-project.org/index.html). To test whether the least-squares ρ-estimates differ between the north and the central group, individuals were permutated among groups within each locus. The least-squares estimate and permutation of ρ were conducted only for those nine loci that had data from 15 individuals from both groups. Indel variation was not included. ρ was also estimated with a maximum-composite-likelihood method using the program maxhap (http://home.uchicago.edu/∼rhudson1/source/maxhap.html) (Hudson 2001). Information from two-site sample configurations from all loci was combined in the analysis. The program makenewh (http://home.uchicago.edu/∼rhudson1/source/twolocus/programs/makenewh.c) was used to generate the sampling distribution for a sample of 15 alleles. In this analysis, the indel variation was also included.
Coalescent simulations:
To assess the effect of demography on the central and the north group, we followed the approach of Haddrill et al. (2005) with some modifications. Spain and Turkey were not analyzed for demographic history, because of the low sample size (n = 5), which might have lead to misleading conclusions. The goal was not to extensively search for the best demographic history, but to examine whether the observed data were compatible with the SNM or with any alternatives from a grid of simple bottleneck scenarios. Coalescent simulations with various demographic scenarios were run with the software ms (Hudson 2002).
The simulations differed from those of Haddrill et al. (2005) in types of replications. We took into account the differences in sample size and length of the sequenced loci. The sequence length sets a limit to the maximum values of Tajima's D and it has a linear effect on the number of segregating sites (Tajima 1989) and on Fay and Wu's H. This is crucial because long sequences have many segregating sites. To achieve a comparable set of means and variances, this must be taken into account. The issue is especially important in these data, since the number of silent sites per gene in the observed data varied from 61.7 to 892.5 bp.
Simulations were conducted conditional on θ. For each simulation, the number of replications and the initial θ, θ0 must be given. θ0 = 4N0μ, where N0 is the effective population size at the start of the simulation, in other words, at present. Thus, θ0 is the expected value of θ in equilibrium, given the known mutation rate and the current very large effective size. In our modification, the value of θ0 is given as per site, not per locus. θ0 was adjusted so that the mean π of simulated data was similar to that observed (supplemental Table S2 at http://www.genetics.org/supplemental/). Note that θ0 may be different from the long-term θ estimated from the data, since the latter is dependent on the past population sizes that may vary through time, and the former is dependent only on N0, which is the population size at a single time point.
To mimic the observed data set of 16 loci as well as possible, 5000 replicates of the set of 16 loci were simulated with respective numbers of sites and sample sizes (Table 1, supplemental Table S2 at http://www.genetics.org/supplemental/). θ0 per site was kept constant across different loci, but θ0 per locus varied according to the number of silent sites in the gene. Also recombination rate (ρ) per adjacent sites was constant across all loci such that for each simulation, ρ/θ was at the observed level for the respective population. For recombination rate per gene, the total number of sites for each gene was also given.
TABLE 1.
Sequenced loci, their putative function, length of sequenced region, and sample size for each group of P. sylvestris
|
N
|
||||||
|---|---|---|---|---|---|---|
| Gene | Function | bp | North | Central | Spain | Turkey |
| dhy-like | Putative dehydrin | 441–449 | 15 | 15 | 5 | 5 |
| a3ip2 | ABI3-interacting protein 2 | 1037–1038 | 15 | 15 | 5 | 5 |
| chcs | Chalcone synthase | 332 | 15 | 15 | 5 | 5 |
| hprgp-like | Hydroxyproline-rich glycoprotein-like protein | 390 | 15 | 15 | 5 | 5 |
| gi | Putative gigantea | 389 | 15 | 15 | 5 | 5 |
| pal1 | Phenylalanine ammonia-lyase | 451 | 15 | 15 | 5 | 5 |
| hlh1 | Helix-loop-helix protein 1A (HLH1) | 752 | 13 | 15 | 4 | 5 |
| lp2 | S-adenosylmethionine synthetase | 1125 | 15 | 9 | 1 | 5 |
| nir | Nitrite reductase | 244 | 14 | 15 | 5 | 5 |
| scl | Putative SCARECROW gene regulator | 954 | 14 | 15 | 5 | 5 |
| phyo | Phytochrome O | 1083 | 13 | 13 | 3 | 4 |
| phyn | Phytochrome N | 2081 | 14 | 15 | 5 | 4 |
| phyp | Phytochrome P | 2594 | 15 | 15 | 5 | 5 |
| co | Constans | 631 | 15 | 14 | 3 | 3 |
| adhc | Alcohol dehydrogenase C | 579 | 15 | 15 | 5 | 5 |
| laccase | Laccase | 311 | 15 | 15 | 5 | 5 |
| Total | 13403 | |||||
The means and standard deviations were calculated for each summary statistic and for each set of 16 loci. The P-value for a certain summary statistic from the distribution generated under a demographic scenario was obtained by comparing the observed value to the distribution obtained from 5000 replicates (two-sided test).
Only observations from silent sites were compared to simulations, except for Fay and Wu's H statistics, where data from all sites were used. For each scenario, Fay and Wu H values were simulated with a different number of loci and number of sites, but with the same θ0 and ρ as for other statistics, since the observed data were also more restricted (supplemental Table S2 at http://www.genetics.org/supplemental/) compared to intraspecific data due to limited outgroup data.
The SNM and a grid of bottlenecks with four different timings of the end (thinking forward) of the bottleneck (from 0.001 to 1) and four different severities (from 0.001 to 0.5) were simulated. Duration of bottleneck was constant, 0.006, in all 16 bottleneck models. The detailed information on the simulated scenarios and the procedure can be found in supplemental methods at http://www.genetics.org/supplemental/. The main monitored summary statistics were average Tajima's D, average Fay and Wu 's H, and their standard deviations among loci. These were monitored because, in addition to being affected by selection, they are also affected by demographic history. Both are based on the difference between two θ-estimates and their expected value is zero under the standard coalescent, but they deviate from zero in some nonequilibrium situations. Fay and Wu's H depends on the difference between π and θH, the latter estimated based on the frequency of derived mutations. Average nucleotide diversity (π) per gene was monitored to adjust the variation to observed level and θ0 was used to infer the equilibrium level of variation.
RESULTS
Nucleotide diversity and allele-frequency spectrum:
A total of 13,289 bp were sequenced from 16 nuclear loci, excluding gaps and missing data. One locus, laccase was monomorphic, and the remaining 15 had a total of 153 polymorphic sites. All 16 indel variants occurred in three genes, adhc, a3ip2, and dhy-like, and were excluded from further analysis unless otherwise stated. One site in co had three variants and was treated as one segregating site in further analysis. The number of silent sites, the number of segregating sites (S), estimates of nucleotide diversity, Tajima's D, and Fay and Wu's H for each locus and group separately are presented in supplemental Table S2 at http://www.genetics.org/supplemental/.
A more detailed analysis of nucleotide variation was conducted for the four geographical groups separately. A plot of approximate posterior densities of multilocus θ for different groups is presented in Figure 2. The medians and credibility intervals of θ are presented in Table 2. All θ-estimates were between 0.0047 and 0.0057 with overlapping 95% confidence intervals, and there were no clear differences in the amount of nucleotide variation between different groups. In contrast, there was more heterogeneity in the amount of nucleotide variation among loci than is expected in the neutral equilibrium situation, given a common θ. Four loci had higher (dhy-like, chcs, hprgp-like, and adhc) and four had lower (hlh-1, scl, phyn, and phyp) θ-estimates than expected on the basis of neutral simulations. The highest variation in the estimate of the silent-site Watterson's (1975) θ (θw) among loci was in Turkey, from 0.0005 in scl to 0.0276 in adhc (if monomorphic laccase is ignored). The estimates of nucleotide diversity might be slightly biased upward since the most variable region of adhc was chosen for sequencing. For comparative purposes between species, the arithmetic mean of θw-estimates over all 16 loci and four groups (0.007) was used.
Figure 2.—

Posterior densities of multilocus θW (per base pair) for four geographical groups of P. sylvestris estimated from 15 loci (lp2 excluded). Ninety-five percent credibility intervals are reported in Table 2.
TABLE 2.
Descriptive statistics for nucleotide variation in four geographic groups of P. sylvestris
| Tajima's Da
|
|||||||||
|---|---|---|---|---|---|---|---|---|---|
| θb | C.I. (95%)c | ρd | ρe | ρd/θ | ρe/θ | Total | Silent | Fay and Wu's H | |
| North | 0.0056 | 0.0041–0.0075 | 0.0033 | 0.0064 | 0.60 | 1.14 | −0.63 (1.06)** | −0.53* | −0.34 |
| Central | 0.0050 | 0.0036–0.0067 | 0.0190 | 0.0294 | 3.80 | 5.88 | −0.32 (0.99) | −0.51* | −0.24 |
| Spain | 0.0047 | 0.0029–0.0074 | — | — | — | — | −0.16 (1.09) | 0.12 | −0.06 |
| Turkey | 0.0057 | 0.0037–0.0088 | — | — | — | — | −0.24 (0.88) | 0.21 | −0.09 |
Significance levels determined by coalescent simulations implemented in the HKA program: *0.01 < P < 0.05; **0.001 < P < 0.01.
Median of θ for silent sites.
95% credibility intervals for θ.
Least-squares estimate of ρ from nine loci, indels not included.
ρ estimate based on Hudson (2001), indels included.
Tajimas's D was calculated according to all and silent segregating sites. Tajima's D for all segregating sites tended to be negative in all groups, although the value was significantly negative only in the northern group (Table 2). Four of the Tajima's D values over all sites for individual loci (hprgb-like, phyn, co, and adhc) differed significantly from the neutral expectation in at least one population. When only silent sites were considered, significantly negative values were found in the northern and central groups, but slightly positive ones in Spain and Turkey. Average values of Fay and Wu's H statistics based on the all sites were negative in the northern and central groups and close to zero in Spain and Turkey.
Genetic clustering and population differentiation:
Analysis of genetic clustering with BAPS gave the best support for all 40 individuals belonging to the same genetic cluster. Average pairwise FST (Table 3) values between populations were also low, except those for the Spanish population, which was somewhat differentiated from the others. The KST* and Snn estimates of differentiation (Table 4) were also generally low, most KST*'s < 0.1. There were, however, four loci that were significantly differentiated among populations, gi, scl, phyp, and co. In these cases, there were usually some sites that were polymorphic in only one or two populations. These polymorphisms were never fixed in any population. Generally, the great majority of the polymorphisms were distributed equally among different populations.
TABLE 3.
Pairwise FST (Hudson et al. 1992b) values for four groups of P. sylvestris averaged over loci
| North | Central | Spain | |
|---|---|---|---|
| Central | 0.02 | — | |
| Spain | 0.09 | 0.14 | — |
| Turkey | −0.03 | 0.03 | 0.14 |
TABLE 4.
Genetic differentiation among four groups of P. sylvestris separately for 15 analyzed loci
| Gene | Snna | Kst*a |
|---|---|---|
| dhy-like | 0.306 | 0.006 |
| a3ip2 | 0.313 | −0.045 |
| chcs | 0.238 | 0.036 |
| hprgp-like | 0.325 | −0.005 |
| gi | 0.354* | 0.041 |
| pal1 | 0.269 | 0.056 |
| hlh1 | 0.327 | 0.012 |
| lp2 | 0.284 | −0.019 |
| nir | 0.311 | −0.015 |
| scl | 0.319 | 0.192** |
| phyo | 0.308 | −0.104 |
| phyn | 0.246 | −0.005 |
| phyp | 0.364* | 0.145** |
| co | 0.471 | 0.133* |
| adhc | 0.379 | −0.008 |
Probability obtained by the permutation test with 1000 replicates: *0.01 < P < 0.05; **0.001 < P < 0.01.
The observed loci fall in the neutral envelope of FST and heterozygosity when applying the method of Beaumont and Nichols (1996). Unexpectedly high FST values were not observed at any of the loci. At two loci, the FST was lower than average, just below the lower 0.95 limit, but the limit and the observations were negative.
Divergence between P. sylvestris and P. pinaster:
On the basis of 14 loci with outgroup sequence, synonymous divergence, KS between P. sylvestris and P. pinaster was 0.026. On the basis of the divergence and the mutation rate of 0.6 × 10−9/year (Savolainen and Wright 2004), the species have started to diverge ∼22 MYA. The only locus where KA was higher than KS was phyp (KA/KS = 35). The ratio was based on four nonsynonymous and one synonymous change. However, the multilocus HKA test detected no deviation from neutral expectations in divergence between P. sylvestris and P. pinaster.
Linkage disequilibrium and recombination:
The decay of intragenic linkage disequilibrium is presented in Figure 3. The recombination parameter ρ was estimated for the northern and the central European group, but not for Spain and Turkey because of the low sample size. The least-squares estimate of ρ for the northern group was 0.0033 and for the central group was 0.0190, which means that the expected r2 is 0.2 in the northern group at ∼1400 bp and in the central group at ∼250 bp. The composite-likelihood method produced somewhat higher ρ-estimates (Table 2), but both methods suggested that the central group had about five times as high as ρ compared to the northern one. However, the difference in least-squares estimate of ρ was not statistically significant (two-sided test, P = 0.12, 999 randomizations). There was no significant linkage disequilibrium between different genes according to either Fisher's exact or the chi-square test after Bonferroni correction. The relationship between recombination and mutation parameter estimates, ρ/θ (Table 2) varied depending on the method used for estimating ρ. Nevertheless, the ratio is smaller in the northern group in both cases.
Figure 3.—

Scatter plots describing the decay of linkage disequilibrium in northern and central European geographical groups of P. sylvestris. The squared correlation of allele frequencies r2 of two nucleotide sites is plotted against the distance between them. The data include all parsimony-informative sites pairs from nine loci.
Demographic history:
Despite the low differentiation between populations, the central and northern groups were analyzed separately for demographic history. The genetic clustering method is based on one aspect of the data (allele frequencies) and detailed aspects, such as the allele frequency spectrum might still differ among groups. The populations have clearly different colonization histories and we wanted to examine if they have left distinct marks on the populations' genomewide variation.
On the basis of Tajima's D neither group fitted well to simulation results of the SNM (P-value for northern, 0.015; and for central, 0.014). In addition, the standard deviation of nucleotide diversity among loci under the SNM did not fit to observed values of the central group (threshold P-value 0.05). Among the grids of bottleneck scenarios, only one bottleneck could not be rejected on the basis of any summary statistics monitored (Figure 4 and supplemental Table 3 at http://www.genetics.org/supplemental/). This bottleneck happened 0.1 × 4N0 generations ago and reduced the population to 1% of the present for 0.006 × 4N0 generations. Summing up, an ancient bottleneck in northern and central Europe fits the observed data better than the SNM.
Figure 4.—

P-values for Tajima's D and Fay and Wu's H under a grid 16 bottleneck models for northern and central groups of P. sylvestris. On the y-axis is the time of the end of a bottleneck in units of 4N0 generations. On the x-axis is the severity of a bottleneck in units of N0. The duration of bottlenecks was constant, 0.006.
DISCUSSION
Patterns of nucleotide diversity among geographic groups of P.sylvestris:
Scots pine, as most other forest trees, is wind pollinated, which has a homogenizing effect on the distribution of genetic variation. On the basis of the overall FST values (0–0.14), genetic clustering, and amount of diversity, P. sylvestris populations form a genetically quite uniform group compared to many other plant species and even to Norway spruce (FST = 0–0.26 in a study with sampling equivalent to that in this study) (Heuertz et al. 2006).
The Spanish and Turkish populations were expected to be somewhat different from populations in central and northern Europe, because they are isolated from the main distribution and have a distinct mitochondrial composition suggesting differences in their phylogeographic history (Soranzo et al. 2000; Pyhäjärvi et al. 2007). The low differentiation of the Turkish population was surprising, because the population differs clearly from European populations with respect to the mitochondrial type. The Spanish population had slightly higher FST values compared to other groups, as also found by Dvornyk et al. (2002) for another Spanish population. Tajima's D was positive in both southern European populations as opposed to negative values found in central and northern populations (Table 2). Also, Fay and Wu's H is very close to zero in southern populations. Despite general uniformity of allele frequencies, there seem to be some differences between groups in terms of the detailed allele-frequency spectrum. However, part of the differences may result from differences in sample sizes among groups.
Intragenic linkage disequilibrium:
Generally, the rapid decay of LD is related to outcrossing breeding system and large effective population size. The LD of outcrossing species like P. taeda (Brown et al. 2004) and sunflower (Liu and Burke 2006) decays fast compared to that of selfing species such as rice (Garris et al. 2003) and A. thaliana (Nordborg et al. 2005), where LD extend over several kilobases. Scots pine ρ, as well as ρ/θ estimates, falls into the same magnitude as that of other outcrossers (Lynch 2006), but seems to be smaller in northern populations compared to central European populations.
In humans, the ancestral population in Africa has less LD compared to that of non-Africans, who have probably gone through more drastic population size changes (Frisse et al. 2001; Voight et al. 2005). The pattern of higher LD was also observed in P. taeda populations that have probably experienced bottlenecks, compared to populations from refugial areas in Florida (Brown et al. 2004; González-Martínez et al. 2006). It is even possible that the nucleotide diversity is not affected in recently colonized areas, but LD is higher compared to that in the ancestral population. For example, in a comparison between maize and teosinte, domestication has reduced ρ more than θ (Wright et al. 2005). The higher LD in the northern P. sylvestris might thus be a result of a more severe bottleneck, admixture, or a different outcrossing rate in northern populations (Nordborg 2000). However, the latter is unlikely, since the northern P. sylvestris is known to be highly outcrossing (Muona and Harju 1989). The distribution of mitochondrial haplotypes shows that admixture between lineages may have occurred in northern Europe (Pyhäjärvi et al. 2007). Admixture, however, should also result in elevated amounts of variation in the north, which is not observed. Simulations suggest that skewed allele-frequency distributions in P. sylvestris populations are a result of a bottleneck. A bottleneck might also be the reason behind the elevated levels of linkage disequilibrium in the north.
Timescale of demographic events affecting the genomic diversity of P.sylvestris:
Paleoecological data suggest that European Scots pine populations have experienced population size changes in the past. Indeed, the SNM did not fit well to the observed data from central and northern Europe. Rather, the observed nucleotide diversity patterns could result from an ancient bottleneck.
When in the geological time did the bottleneck take place? Events in the coalescent simulations are in the timescale of 4N0 generations. Therefore an estimate of N0 is required for calibrating simulations with geological time. One of the variables in the coalescent simulations was θ0 (4N0μ) per nucleotide site. With an estimate of mutation rate per generation, N0 can also be estimated.
The estimate of the mutation rate, μ in P. sylvestris is based on the divergence between P. sylvestris and Picea abies. The rate of synonymous substitution, d = μ2T, where d is the synonymous divergence between two species and T is the time since the species started to diverge, provides an estimates of the mutation rate (Graur and Li 2000). P. sylvestris and P. abies separated ∼140 MYA (Miller 1977; Savard et al. 1994). The level of divergence has been estimated to be 0.16 in (García-Gil et al. 2003), but we used the value 0.17 based on a broader set of EST-based data (Savolainen and Wright 2004). When generation time is assumed to be 20 years (Rohmeder 1972), the estimate for the per generation mutation rate becomes 12.1 × 10−9.
For example, in the bottleneck that fits best with data in the north, θ0 of 0.0115 was required to produce the data that were similar to those observed (supplemental Table 3 at http://www.genetics.org/supplemental/). Assuming mutation rate per generation 12.1 × 10−9 we get N0 of ∼238,000 individuals. That puts the bottleneck to ∼2 MYA. This calculation depends on the observed θ, the Picea–Pinus divergence, and many other unverified assumptions that are prone to errors. Therefore, the timing should be taken only as an example of a possible scenario. What it shows, however, is the timescale of population history that still could have an effect on the observed nucleotide variation in P. sylvestris.
Classical population genetic theory predicts that the amount of nucleotide variation in a colonizing population should decrease as a result of sequential bottlenecks (Nei et al. 1975). Traces of postglacial colonization (since 10,000 YBP) were expected to distinguish the northern group from others. However, the multilocus estimates of θ did not differ among the four groups of P. sylvestris studied here. Similar findings on uniformity of allele frequencies were made earlier with allozymes (Muona and Harju 1989).
The power to detect different demographic histories varies between different markers and species. Life-history traits, ecology, and population size are crucial determinants of how the history of the population is reflected in its genomewide variation. Probably the most recent colonization event to the north (scattering phase) is too recent for new mutations to have accumulated. Thus, it has not affected the pattern of nucleotide variation. Furthermore, the bottleneck effect during the colonization has probably not been severe enough to have reduced the amount of variation. Both central and northern groups are rather reflecting their common ancestor's (collecting phase) than their own distinct demographic histories (Wakeley and Aliacar 2001).
To infer demography and biogeography of conifers on a wider range of timescales, and to consider more recent events, mitochondrial and chloroplast DNA have been studied because they have a smaller population size and different migration rates compared to nuclear DNA (e.g., Soranzo et al. 2000; Robledo-Arnuncio et al. 2005; Anderson et al. 2006; Navascués et al. 2006). Due to their low mutation frequency, single-nucleotide polymorphisms are not the most suitable for detecting postglacial demographic events in long-lived trees such as P. sylvestris. However, the traces of postglacial colonization in nuclear markers can be found in other species. Annual plants, for example, have shorter generation time and populations usually are more fragmented compared to trees. Species with a shorter generation time accumulate mutations faster than species with a long generation time. In addition, a fragmented distribution probably results in severe bottlenecks during the colonization, which can be detected as reduced diversity (e.g., Van Rossum and Prentice 2004; Muller et al. 2007).
Low nucleotide diversity in P.sylvestris:
The multilocus θ-estimate, 0.005 in this study is close to 0.0056 estimated on the basis of pal1 and a small data set of 10 other loci of P. sylvestris in Dvornyk et al. (2002). The average nucleotide diversity, 0.007 is slightly higher than the multilocus θ-estimate. The value is similar to what has been found in other trees, but lower than that in plants in general (Savolainen and Pyhäjärvi 2007).
The advantage of the multilocus θ-estimate presented here, compared to the arithmetic mean, is that it does not put too much weight on the most variable loci, which generally results in an upward bias of the estimate. The method unrealistically assumes no intragenic recombination, which should not change the point estimate, but makes the credibility interval wider. Nonvariable loci were included in the estimate, which can also lower the genomic θ-estimate, since those loci might have been excluded from many studies on nucleotide diversity. In addition, surveys designed for SNP detection have sometimes concentrated on the more diverse loci.
Low nucleotide variation could result either from low mutation rate or low effective population size, possibly reflecting past census size changes in history. However, a recurrent-hitchhiking model or genetic draft can result in independence between Ne and nucleotide diversity (Gillespie 2000). If this were the case, a negative genomewide Tajima's D as well as a positive Fay and Wu's H should be observed (Przeworski 2002; Haddrill et al. 2005). However, in Spain and Turkey Tajima's D was positive and in all groups Fay and Wu's H is negative. Further, given that LD seems to be so low, it would seem difficult to propose an overall effect of selective sweeps.
On the basis of divergence between P. sylvestris and P. abies the mutation rate is 12.1 × 10−9 per generation and seems to be at a comparable level with that of angiosperms. The estimate is of same magnitude as the mutation rate estimated for the whole genus Pinus by Willyard et al. (2007). Our estimate of the divergence time between P. pinaster and P. sylvestris, 22 MYA is also in agreement with their results concerning the divergence times of section Pinus.
As concluded by Heuertz et al. (2006) in the case of Norway spruce, the changes in population size, rather than recurrent hitchhiking or low mutation rate explain the observed low nucleotide variation in Scots pine. In our example, a bottleneck around 2 MYA could still have an effect that reduces the nucleotide diversity to half of the equilibrium value. This suggests that time required reaching the equilibrium and consequently the equilibrium level of θ would be very long, much longer than the timescale of most recent consecutive ice ages, for example. The timescale could explain the difference of nucleotide diversity level between trees and other plants that typically have shorter generation times. Trees are more prone to fluctuations in population size: More environmental changes happen in fewer generations due to long generation time and they practically never would have time to reach the equilibrium.
The effect of selection on the genome of P.sylvestris:
Even though only silent sites were considered for demography, they are in the genic regions and purifying selection might have reduced the nucleotide variation more than in intergenic regions. In addition, some of the chosen loci are part of the light-induced pathway in A. thaliana and might be connected to clinally varying traits. However, there are no strong imprints of selection at any specific loci and there was no clear reason for leaving any specific loci out of this study. The average negative Fay and Wu's H and Tajima's D are not caused merely by some individual loci (supplemental Table S2 at http://www.genetics.org/supplemental/), but the trend is present in different parts of the genome of P. sylvestris. The linkage disequilibrium decays very rapidly, which indicates that selective sweeps have not had a strong impact on the nucleotide variation.
Conclusion:
As concluded, e.g., in the review by Wright and Gaut (2005), the sampling of local populations is crucial for plant population genetics. In P. sylvestris the pattern of nucleotide diversity differs between geographic groups despite the apparent low genetic differentiation. Specieswide pooled samples might have concealed the important details about the nucleotide diversity, such as amount of linkage disequilibrium or sign of Tajima's D.
As presented in this study as well as earlier studies by, e.g., Haddrill et al. (2005) and Heuertz et al. (2006), a great deal of nucleotide variation in different species can be explained by demographic history. There are some exceptions; e.g., selective sweeps were found to describe the observed pattern of nucleotide variation in D. simulans better than purely demographic models (Quesada et al. 2006). We know from patterns of genetic variation of quantitative traits in P. sylvestris that some loci are probably under strong selection (Hurme et al. 1997). To find the loci that underlie these traits is a demanding task. Whatever the demographic history is that has shaped the genome of P. sylvestris, it seems to have increased the variances of summary statistics. Hence the standard neutrality tests may result in an excess of false positives. In human data, demographic history has been successfully taken into account while mapping positive selection (Voight et al. 2006). In nonmodel species like P. sylvestris, where the amount of data from putatively neutral loci, outside the coding area, is limited, genome scans looking for deviations from the genomewide pattern of nucleotide diversity might not be the ideal approach.
Acknowledgments
We thank Katri Kärkkäinen from the Finnish Forest Research Institute for providing seed material, Päivi Komulainen for her contribution to the data, Soile Finne for skillful technical assistance, Esa Läärä for contributing to the method for estimating multilocus θ, Martin Lascoux for discussions and comments on the manuscript, and two anonymous reviewers whose suggestions improved analysis. T.P. acknowledges the Finnish Graduate School in Population Genetics. This work was supported by the European Commission, project TREESNIPS (QLRT-2001-01973).
APPENDIX
The single-locus likelihood of θ under the infinite-sites model due to Watterson (1975) is the probability, with given θ, of observing Si segregating sites at locus i. Tavaré (1984) derived an explicit form of this probability,
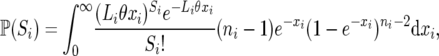 |
(A1) |
where ni is the number of sequences and Li the total number of sites at locus i. An alternative expression for this probability was obtained by substituting u = e−x in the integral of (A1):
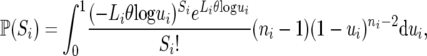 |
(A2) |
Thus, Si given ui is Poisson distributed with mean −Liθ log ui, where ui is beta distributed with mean 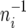 and variance
and variance 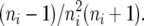 This dependency of the observed variable Si on the latent variable ui was exploited to construct the posterior distribution of θ in a Bayesian framework.
This dependency of the observed variable Si on the latent variable ui was exploited to construct the posterior distribution of θ in a Bayesian framework.
In the first stage of the model, the multilocus likelihood of θ is given by the product of the Poisson distributions of single loci, which are assumed independent. This likelihood depends on the nuisance parameters ui. In the second stage, these parameters are assigned priors given by the beta distributions mentioned above. In the third and final stage, a prior for the parameter of interest, θ, needs to be specified. Several families of distributions were considered: the uniform and gamma families as priors for θ directly as well as the Gauss family for log θ. To obtain a posterior distribution of θ close to its likelihood, only uninformative priors with large variances were considered. With variances large enough, the different families of distributions yielded virtually identical posteriors. The results presented here were obtained using a Gauss distribution with mean 0 and variance 106 as the prior for log θ.
The model was implemented using the BRugs package for the software R (http://www.r-project.org/), which embeds and provides a convenient interface to the OpenBugs software specialized in MCMC simulation of Bayesian models (http://mathstat.helsinki.fi/openbugs/). The convergence of the simulations was assessed using the Gelman–Rubin statistic, as modified by Brooks and Gelman (1998). The results presented here are based on simulations with 400,000 iterations after a burn-in phase of 5000 iterations. The posterior distributions were summarized in graphs of smoothed estimates of density functions. The medians of the simulated samples were calculated to serve as point estimates for θ. The 2.5% and the 97.5% sample quantiles of the simulation runs, respectively, were taken as the lower and upper limits of 95% credibility intervals.
References
- Akey, J. M., M. A. Eberle, M. J. Rieder, C. S. Carlson, M. D. Shriver et al., 2004. Population history and natural selection shape patterns of genetic variation in 132 genes. PLoS Biol. 2: 1591–1599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson, L. L., F. Hu, D. M. Nelson, R. J. Petit and K. N. Paige, 2006. Ice-age endurance: DNA evidence of a white spruce refugium in Alaska. Proc. Natl. Acad. Sci. USA 103: 12447–12450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beaumont, M. A., and R. A. Nichols, 1996. Evaluating loci for use in the genetic analysis of population structure. Proc. R. Soc. Lond. Ser. B 263: 1619–1626. [Google Scholar]
- Brooks, S., and A. Gelman, 1998. General methods for monitoring convergence of iterative simulations. J. Comput. Graph. Stat. 7: 434–455. [Google Scholar]
- Brown, G. R., G. P. Gill, R. J. Kuntz, C. H. Langley and D. B. Neale, 2004. Nucleotide diversity and linkage disequilibrium in loblolly pine. Proc. Natl. Acad. Sci. USA 101: 15255–15260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheddadi, R., J. L. de Beaulieu, J. Jouzel, V. Andrieu-Ponel, J. M. Laurent et al., 2005. Similarity of vegetation dynamics during interglacial periods. Proc. Natl. Acad. Sci. USA 102: 13939–13943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corander, J., P. Waldmann and M. J. Sillanpää, 2003. Bayesian analysis of genetic differentiation between populations. Genetics 163: 367–374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Depaulis, F., S. Mousset and M. Veuille, 2003. Power of neutrality test to detect bottlenecks and hitchhiking. J. Mol. Evol. 57: S190–S200. [DOI] [PubMed] [Google Scholar]
- Dvornyk, V., A. Sirviö, M. Mikkonen and O. Savolainen, 2002. Low nucleotide diversity at the pal1 locus in the widely distributed Pinus sylvestris. Mol. Biol. Evol. 19: 179–188. [DOI] [PubMed] [Google Scholar]
- Eiche, V., 1966. Cold damage and plant mortality in experimental provenance plantations with Scots pine in northern Sweden. Stud. For. Suec. 36: 1–218. [Google Scholar]
- Fay, J. C., and C. I. Wu, 2000. Hitchhiking under positive Darwinian selection. Genetics 155: 1405–1413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frisse, L., R. R. Hudson, A. Bartoszewicz, J. D. Wall, J. Donfack et al., 2001. Gene conversion and different population histories may explain the contrast between polymorphism and linkage disequilibrium levels. Am. J. Hum. Genet. 69: 831–843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- García-Gil, M. R., M. Mikkonen and O. Savolainen, 2003. Nucleotide diversity at two phytochrome loci along a latitudinal cline in Pinus sylvestris. Mol. Ecol. 12: 1195–1206. [DOI] [PubMed] [Google Scholar]
- Garris, A. J., S. R. McCouch and S. Kresovich, 2003. Population structure and its effect on haplotype diversity and linkage disequilibrium surrounding the xa5 locus of rice (Oryza sativa L.). Genetics 165: 759–769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gillespie, J. H., 2000. Genetic drift in an infinite population: the pseudohitchhiking model. Genetics 155: 909–919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- González-Martínez, S. C., E. Ersoz, G. R. Brown, N. C. Wheeler and D. B. Neale, 2006. DNA sequence variation and selection of tag single-nucleotide polymorphisms at candidate genes for drought-stress response in Pinus taeda L. Genetics 172: 1915–1926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graur, D., and W. H. Li, 2000. Fundamentals of Molecular Evolution. Sinauer Associates, Sunderland, MA.
- Haddrill, P. R., K. R. Thornton, B. Charlesworth and P. Andolfatto, 2005. Multilocus patterns of nucleotide variability and the demographic and selection history of Drosophila melanogaster populations. Genome Res. 15: 790–799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamrick, J. L., and J. W. Godt, 1996. Effects of life history traits on genetic diversity in plant species. Philos. Trans. R. Soc. Lond. B 351: 1291–1298. [Google Scholar]
- Heuertz, M., E. De Paoli, T. Källman, H. Larsson, I. Jurman et al., 2006. Multilocus patterns of nucleotide diversity, linkage disequilibrium and demographic history of Norway spruce Picea abies (L.) Karst. Genetics 174: 2095–2105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hill, W. G., and A. V. Robertson, 1968. Linkage disequilibrium in finite populations. Theor. Appl. Genet. 38: 226–231. [DOI] [PubMed] [Google Scholar]
- Hill, W. G., and B. S. Weir, 1988. Variances and covariances of squared linkage disequilibria in finite populations. Theor. Popul. Biol. 33: 54–78. [DOI] [PubMed] [Google Scholar]
- Hudson, R. R., 2000. A new statistic for detecting genetic differentiation. Genetics 155: 2011–2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hudson, R. R., 2001. Two-locus sampling distributions and their application. Genetics 159: 1805–1817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hudson, R. R., 2002. Generating samples under a Wright-Fisher neutral model of genetic variation. Bioinformatics 18: 337–338. [DOI] [PubMed] [Google Scholar]
- Hudson, R. R., D. D. Boos and N. L. Kaplan, 1992. a A statistical test for detecting geographic subdivision. Mol. Biol. Evol. 9: 138–151. [DOI] [PubMed] [Google Scholar]
- Hudson, R. R., M. Slatkin and W. P. Maddison, 1992. b Estimation of levels of gene flow from DNA-sequence data. Genetics 132: 583–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huntley, B., and H. J. B. Birks, 1983. An Atlas of Past and Present Pollen Maps for Europe 1–13 000 Years Ago. Cambridge University Press, Cambridge, UK.
- Hurme, P., T. Repo, O. Savolainen and T. Pääkkonen, 1997. Climatic adaptation of bud set and frost hardiness in Scots pine (Pinus sylvestris). Can. J. For. Res. 27: 716–723. [Google Scholar]
- Komulainen, P., G. R. Brown, M. Mikkonen, A. Karhu, M. R. García-Gil et al., 2003. Comparing EST-based genetic maps between Pinus sylvestris and Pinus taeda. Theor. Appl. Genet. 107: 667–678. [DOI] [PubMed] [Google Scholar]
- Liu, A., and J. M. Burke, 2006. Patterns of nucleotide diversity in wild and cultivated sunflower. Genetics 173: 321–330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lynch, M., 2006. The origins of eukaryotic gene structure. Mol. Biol. Evol. 23: 450–468. [DOI] [PubMed] [Google Scholar]
- Marth, G. T., E. Czabarka, J. Murvai and S. T. Sherry, 2004. The allele frequency spectrum in genome-wide human variation data reveals signals of differential demographic history in three large world populations. Genetics 166: 351–372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller, C. N., 1977. Mesozoic conifers. Bot. Rev. 43: 217–280. [Google Scholar]
- Mirov, N., 1967. The Genus Pinus. Ronald Press, New York.
- Muller, M. H., C. Poncet, J. M. Prosperi, S. Santoni and J. Ronfort, 2006. Domestication history in the Medicago sativa species complex: inferences from nuclear sequence polymorphism. Mol. Ecol. 15: 1589–1602. [DOI] [PubMed] [Google Scholar]
- Muller, M. H., J. Leppälä and O. Savolainen, 2007. Genome wide effects of post-glacial colonization in Arabidopsis lyrata. Heredity (in press). [DOI] [PubMed]
- Muona, O., and A. Harju, 1989. Effective population sizes, genetic variability, and mating system in natural stands and seed orchards of Pinus sylvestris. Silvae Genet. 38: 221–228. [Google Scholar]
- Müller, U. C., J. Pross and E. Bibus, 2003. Vegetation response to rapid climate change in Central Europe during the past 140,000 yr based on evidence from the Furamoos pollen record. Quat. Res. 59: 235–245. [Google Scholar]
- Navascués, M., Z. Vaxevanidou, S. C. González-Martínez, J. Climent, L. Gil et al., 2006. Chloroplast microsatellites reveal colonization and metapopulation dynamics in the Canary Island pine. Mol. Ecol. 15: 2691–2698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nei, M., T. Maruyama and R. Chakraborty, 1975. The bottleneck effect and genetic variability in populations. Evolution 29: 1–10. [DOI] [PubMed] [Google Scholar]
- Nicholas, K. B., H. B. J. Nicholas and D. W. I. Deerfield, 1997. GeneDoc: analysis and visualization of genetic variation. EMBNEW. NEWS 4: 14. [Google Scholar]
- Nordborg, M., 2000. Linkage disequilibrium, gene trees and selfing: an ancestral recombination graph with partial self-fertilization. Genetics 154: 923–929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nordborg, M., T. T. Hu, Y. Ishino, J. Jhaveri, C. Toomajian et al., 2005. The pattern of polymorphism in Arabidopsis thaliana. PLoS Biol. 3: 1289–1299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ometto, L., S. Glinka, D. De Lorenzo and W. Stephan, 2005. Inferring the effects of demography and selection on Drosophila melanogaster populations from a chromosome-wide scan of DNA variation. Mol. Biol. Evol. 22: 2119–2130. [DOI] [PubMed] [Google Scholar]
- Petit, R. J., I. Aguinagalde, J. L. de Beaulieu, C. Bittkau, S. Brewer et al., 2003. Glacial refugia: hotspots but not melting pots of genetic diversity. Science 300: 1563–1565. [DOI] [PubMed] [Google Scholar]
- Plomion, C., P. Hurme, J. M. Frigerio, M. Ridolfi, D. Pot et al., 1999. Developing SSCP markers in two Pinus species. Mol. Breed. 5: 21–31. [Google Scholar]
- Pluzhnikov, A., A. Di Rienzo and R. R. Hudson, 2002. Inferences about human demography based on multilocus analyses of noncoding sequences. Genetics 161: 1209–1218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Przeworski, M., 2002. The signature of positive selection at randomly chosen loci. Genetics 160: 1179–1189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pyhäjärvi, T., M. J. Salmela and O. Savolainen, 2007. Colonization routes of Pinus sylvestris inferred from distribution of mitochondrial DNA variation. Tree Genet. Genomes (in press).
- Quesada, H., S. Ramos-Onsins, J. Rozas and M. Aguadé, 2006. Positive selection versus demography: evolutionary inferences based on an unusual haplotype structure in Drosophila simulans. Mol. Biol. Evol. 23: 1643–1647. [DOI] [PubMed] [Google Scholar]
- Robledo-Arnuncio, J. J., C. Collada, R. Alía and L. Gil, 2005. Genetic structure of montane isolates of Pinus sylvestris L. in a Mediterranean refugial area. J. Biogeogr. 32: 595–605. [Google Scholar]
- Rohmeder, E., 1972. Das Saatgut in der Forstwirtschaft. Verlag Paul Parey, Hamburg, Germany/Berlin.
- Rosenberg, N. A., J. K. Pritchard, J. L. Weber, H. M. Cann, K. K. Kidd et al., 2002. Genetic structure of human populations. Science 298: 2381–2385. [DOI] [PubMed] [Google Scholar]
- Rozas, J., J. C. Sánchez-DelBarrio, X. Messeguer and R. Rozas, 2003. DnaSP, DNA polymorphism analyses by the coalescent and other methods. Bioinformatics 19: 2496–2497. [DOI] [PubMed] [Google Scholar]
- Savard, L., P. Li, S. H. Strauss, M. W. Chase, M. Michaud et al., 1994. Chloroplast and nuclear gene-sequences indicate late Pennsylvanian time for the last common ancestor of extant seed plants. Proc. Natl. Acad. Sci. USA 91: 5163–5167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Savolainen, O., and T. Pyhäjärvi, 2007. Genomic diversity in forest trees. Curr. Opin. Plant Biol. 10: 162–167. [DOI] [PubMed] [Google Scholar]
- Savolainen, O., and M. Wright, 2004. Estimating divergence rates of conifers based on EST sequences conifer EST sequences, p. 7 in Population, Evolutionary and Ecological Genomics of Forest Trees. IUFRO Sections Population Genetics and Genomics, September 13–17, 2004, Pacific Grove, CA.
- Schmid, K. J., S. Ramos-Onsins, H. Ringys-Beckstein, B. Weisshaar and T. Mitchell-Olds, 2005. A multilocus sequence survey in Arabidopsis thaliana reveals a genome-wide departure from a neutral model of DNA sequence polymorphism. Genetics 169: 1601–1615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmid, K., O. Torjek, R. Meyer, H. Schmuths, M. H. Hoffmann et al., 2006. Evidence for a large-scale population structure of Arabidopsis thaliana from genome-wide single nucleotide polymorphism markers. Theor. Appl. Genet. 112: 1104–1114. [DOI] [PubMed] [Google Scholar]
- Sinclair, W. T., J. D. Morman and R. A. Ennos, 1999. The postglacial history of Scots pine (Pinus sylvestris L.) in western Europe: evidence from mitochondrial DNA variation. Mol. Ecol. 8: 83–88. [Google Scholar]
- Soranzo, N., R. Alía, J. Provan and W. Powell, 2000. Patterns of variation at a mitochondrial sequence-tagged-site locus provide new insights into the postglacial history of European Pinus sylvestris populations. Mol. Ecol. 9: 1205–1211. [DOI] [PubMed] [Google Scholar]
- Svendsen, J. I., V. I. Astakhov, D. Y. Bolshiyanov, I. Demidov, J. A. Dowdeswell et al., 1999. Maximum extent of the Eurasian ice sheets in the Barents and Kara Sea region during the Weichselian. Boreas 28: 234–242. [Google Scholar]
- Tajima, F., 1989. Statistical method for testing the neutral mutation hypothesis by DNA polymorphism. Genetics 123: 585–595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tavaré, S., 1984. Line-of-descent and genealogical processes, and their applications in population genetics models. Theor. Popul. Biol. 26: 119–164. [DOI] [PubMed] [Google Scholar]
- Tenaillon, M. I., J. U'Ren, O. Tenaillon and B. S. Gaut, 2004. Selection versus demography: a multilocus investigation of the domestication process in maize. Mol. Biol. Evol. 21: 1214–1225. [DOI] [PubMed] [Google Scholar]
- Thompson, J. D., T. J. Gibson, F. Plewniak, F. Jeanmougin and D. G. Higgins, 1997. The ClustalX windows interface: flexible strategies for multiple sequence alignment aided by quality analysis tools. Nucleic Acids Res. 25: 4876–4882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Rossum, F., and H. C. Prentice, 2004. Structure of allozyme variation in Nordic Silene nutans (Caryophyllaceae): population size, geographical position and immigration history. Biol. J. Linn. Soc. 81: 357–371. [Google Scholar]
- Wakeley, J., and N. Aliacar, 2001. Gene genealogies in a metapopulation. Genetics 159: 893–905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watterson, G., 1975. On the number of segregating sites in genetical models without recombination. Theor. Popul. Biol. 7: 256–276. [DOI] [PubMed] [Google Scholar]
- Weir, B. S., and C. C. Cockerham, 1984. Estimating F-statistics for the analysis of population structure. Evolution 38: 1358–1370. [DOI] [PubMed] [Google Scholar]
- Willis, K. J., and T. H. van Andel, 2004. Trees or no trees? The environments of central and eastern Europe during the last glaciation. Quat. Sci. Rev. 23: 2369–2387. [Google Scholar]
- Willyard, A., J. Syring, D. S. Gernandt, A. Liston and R. Cronn, 2007. Fossil calibration of molecular divergence infers a moderate mutation rate and recent radiations for Pinus. Mol. Biol. Evol. 24: 90–101. [DOI] [PubMed] [Google Scholar]
- Voight, B. F., A. M. Adams, L. A. Frisse, Y. D. Qian, R. R. Hudson et al., 2005. Interrogating multiple aspects of variation in a full resequencing data set to infer human population size changes. Proc. Natl. Acad. Sci. USA 102: 18508–18513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voight, B. F., S. Kudaravalli, X. Wen and J. K. Pritchard, 2006. A map of recent positive selection in the human genome. PLoS Biol. 4: 446–458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wright, S. I., and B. S. Gaut, 2005. Molecular population genetics and the search for adaptive evolution in plants. Mol. Biol. Evol. 22: 506–519. [DOI] [PubMed] [Google Scholar]
- Wright, S. I., I. V. Bi, S. G. Schroeder, M. Yamasaki, J. F. Doebley et al., 2005. The effects of artificial selection of the maize genome. Science 308: 1310–1314. [DOI] [PubMed] [Google Scholar]