

Abstract
We prepared several monoclonal antibodies (mAbs) specific for the NH2- and COOH-terminal regions of the DNA helicase (WRN helicase) responsible for Werner's syndrome known as a premature aging disease. With these antibodies, we detected by immunoblot analysis the endogenous WRN helicase of a relative mass of 180 kD in several lines of cultured cells, but not in patient cells with a defined mutation. Immunocytochemical staining of proliferating fibroblasts and tumor cells showed that the major part of WRN helicase is in the nucleoplasm and not in the nucleolus. Similar experiments with a rat mAb specific to the mouse homologue of human WRN helicase yielded an identical conclusion. Although this nucleoplasmic staining was evident in cells in interphase, the condensed chromatin structure in metaphase was not stained by the same mAbs, suggesting that WRN helicases exist perhaps in a soluble form or bound to the unfolded chromatin structure. From quantitative immunoblot analysis, higher levels of WRN helicase were observed in all transformed cells and tumor cells examined than those of normal cells. The expression of WRN helicase was enhanced consistently in fibroblasts and B-lymphoblastoid cells by transformation with SV-40 and Epstein-Barr virus, respectively, suggesting that rapidly proliferating cells require a high copy numbers of WRN helicase.
Keywords: Werner's syndrome, RecQ DNA helicase, genetic instability, aging, nucleoplasmic localization
Werner's syndrome (WS)1 is an autosomal recessive disorder causing symptoms of premature aging accompanied by rare cancers. Patients with WS have a short stature, juvenile cataracts, atrophy of the skin, graying and loss of hair, diabetes mellitus, arteriosclerosis, osteoporosis, and neoplasia (Werner, 1904; Epstein et al., 1966; Martin, 1978; Goto et al., 1996). The early appearance of these symptoms generally associated with normal aging suggests that WS may represent an accelerated aging, with a 2–2.5-fold faster rate than normal aging. The life span of WS patients is 46 ± 11.6 yr due mainly to death caused by malignant tumor or cardiovascular infarctions (Goto, 1997). In vitro studies on the growth characteristics of fibroblast cells also suggest that WS may be related to normal aging: the life span of WS fibroblasts as expressed by population doubling levels (PDL) is much shorter than that of normal fibroblasts (Salk et al., 1985). We have already shown that the hypermutator phenotype, such as genetic instability, also occurs frequently in WS fibroblasts and lymphoblastoid B cells (B cells) transformed by the Epstein-Barr virus (EBV; Tahara et al., 1997).
The gene responsible for the WS (WRN) has been identified by positional cloning from the 8p11-12 region, and encodes a DNA helicase belonging to a RecQ DNA helicase family (Yu et al., 1996). Defective DNA metabolism is involved in a complex process of premature aging in WS patients, because DNA helicases are enzymes that unwind the energetically stable double-stranded structure of DNA to provide the single-stranded template for important cellular processes like replication, recombination and repair (Lohman, 1993). Using an expression system with insect cells and baculovirus, we and others previously demonstrated that the WRN helicase gene indeed encodes a functional DNA helicase that shows DNA-dependent ATPase and DNA unwinding activities (Gray et al., 1997; Suzuki et al., 1997). We also demonstrated that WRN helicase proteins tagged either with NH2-terminal FLAG peptide or enhanced green fluorescent protein (EGFP) localize exclusively in the nuclei of HeLa cells, consistent with the role of DNA helicase in nuclei (Matsumoto et al., 1997a; Suzuki et al., 1997).
Mutations occurring in WS patients have been extensively investigated by us and others with >100 patients. To date, ∼20 different types of mutations have been identified that are distributed in the entire coding regions of the WRN and exist in individual patients as either homozygous or compound heterozygous mutations (Matsumoto et al., 1997b; Oshima et al., 1997). More recently, we found that most of these mutations generate truncated helicase molecules lacking nuclear localization signals (NLS) in the COOH-proximal region, rendering all WRN products unable to be transported to the nucleus where the DNA helicase is supposed to function (Matsumoto et al., 1997a; Matsumoto et al., 1998a). Importantly, this finding clearly explains why WS patients show a set of similar clinical phenotypes no matter what type of mutation they carry.
Also, we found that both fibroblasts and EBV-transformed B cell lines from patients with homozygous mutation-4 or mutation-6, two of the most common mutation types in Japanese WS patients, have much less WRN mRNA than normal cells (Yamabe et al., 1997). This reduction in the WRN mRNA level suggests an augmented specific degradation of the WRN mRNA in patient cells, as was shown in other cases in which the nonsense codons affect RNA metabolism in vertebrate cells (Maquat, 1995). As regards the transcriptional regulation of WRN gene, we have recently characterized the promotor activity and found that the WRN gene expression is directed mainly by the SP1 transcriptional control system, like that of other housekeeping genes, but is further modulated by transcriptional factors, including Rb and p53 (Yamabe et al., 1998).
In this study, we established several mouse mAbs specific to WRN helicase, and their epitope regions were determined. With these defined antibodies, we investigated the subnuclear distribution of endogenous WRN helicases by immunocytochemical staining. In addition, the levels of endogenous WRN helicase expression were compared for the first time at the protein level for normal cells, transformed cells and for tumor cells. Strikingly, the expression level was highest for immortal EBV-transformed B cells, followed by high levels for mortal EBV-transformed B cells and a low level for untransformed B cells in the peripheral blood. Cellular WRN helicase levels determined by mAbs for other tumor cell lines are discussed in relation to the potential biological function of WRN helicase whose defects apparently result in chromosomal instability in WS patient cells.
Materials and Methods
Cell Lines
Skin fibroblasts were obtained from human biopsy samples and cultured by the method of Martin (1978); B cells were obtained by transforming peripheral blood leukocytes with EBV (Miller, 1990). In brief, peripheral blood leukocytes from normal individuals and WS patients were purified using a Lymphocyte Separation Medium and were transformed by EBV propagated in marmoset cell line B95-8 in the presence of 0.2 μg/ml Cyclosporin (Sandoz). Established cell strains were cultured in 25-cm2 bottles (Corning) using RPM11640 medium (Nissui Pharmaceutical Co. Ltd.) supplemented with 10% fetal calf serum (GIBCO BRL). The WI38 cell line originally from human embryonic lung fibroblasts and the WI38 line transformed by SV-40 were donated by Dr. Ide (Hiroshima University). Other cell lines were purchased from the American Tissue Culture Collection. All cell lines were maintained in RPMI 1640, McCoy's 5a medium or DME supplemented with 10% heat-inactivated FCS. The cell lines were cultured at 37°C in a humidified CO2 incubator.
Separation of Peripheral B-Lymphocytes
We used the panning method (Lewis and Kamin, 1980) to separate B-lymphocytes. In brief, leukocytes were collected from 20 ml peripheral blood by centrifuging with a lymphocyte separation medium (Organotechnika) following the instructions of the manufacturer and then suspended in PBS containing 5% FCS, and were added to a polystyrene culture flask whose surface had been coated with affinity-purified rabbit anti–human immunoglobulin antibodies (Organotechnika). Leukocytes were kept for 1 h at 20°C. After removing nonadherent cells by decantation and washing five times with PBS, the adherent B-lymphocytes were recovered from the surface of the culture bottle by leaving the cells 15 min at 20°C in PBS containing 4 mg/ml Lidocain-HCl and by subsequent pipetting. The peripheral blood B cells thus obtained were washed by PBS and were used for Western blot analysis.
Immunization and Preparation of Hybridomas
To prepare an immunogen, the whole WRN helicase molecule that contained NH2-terminal hexahistidine was produced in insect Sf21 cells with baculovirus expression systems as described by Suzuki et al. (1997). The expressed WRN helicase was purified to homogeneity by affinity column chromatography using Ni-chelating NTA agarose gel (Qiagen Inc.). In addition, a recombinant partial fragment containing NH2-terminal hexahistidine and the COOH-terminal half of WRN helicase consisting of amino acid residues 877–1,432 was produced in E. coli, and was used as an immunogen after purifying by affinity chromatography with NTA resin. Rat mAbs specific to the mouse homologue of human WRN helicase were made using the purified protein, containing NH2-terminal glutathione- S-transferase, and the COOH-terminal polypeptide fragment consisting of amino acid residues 273–513 of mouse WRN helicase, which were made in E. coli. BALB/c mice and Wistar rats were, respectively, immunized with these purified proteins in a complete Freund's adjuvant. Hybridoma clones that produced the antigen-specific antibodies were screened by ELISA and Western blot analysis.
Determination of the Epitope Regions in the WRN Helicase Molecule Recognized by mAbs
Various NH2- or COOH-terminal fragments of the WRN helicase protein that were NH2-terminally tagged with EGFP were produced in mouse melanoma cells B16F10 by transfecting a series of recombinant pEGFP-C3 expression plasmids (Clontech Laboratories Inc.) that contained the CMV promoter and the downstream DNA sequences coding for EGFP and fragments of WRN helicase. For example, the C49 and C59 polypeptides that contain NH2-terminal EGFP and COOH-terminal fragments of WRN helicase consisting of 49 aa and 59 aa, respectively, were generated by placing the cognate DNA fragments coding for these polypeptides in pEGFP-C3 plasmid downstream of the CMV promoter-EGFP sequence, and were transiently expressed in B16F10 cells by transfecting the constructed recombinant plasmid DNA (Matsumoto et al., 1997a). The N368 polypeptide containing the NH2-terminal 368 aa was similarly produced in B16F10 cells. Expression plasmids coding for other EGFP-conjugated proteins, N1304, N1162, N1046, C138, C128, C79, C69, and C19, were similarly constructed and were expressed in B16F10 cells. Cells expressing these polypeptides were fixed as described above in the immunocytochemistry section and were stained with the mAbs conjugated with fluorescent reagent Texas red. To define the epitope sites more precisely for mAbs to recognize the NH2-terminal region of WRN helicase, a recombinant partial fragment (C1201) containing NH2-terminal hexahistidine, but with no NH2-terminal 231 aa, was synthesized and was used for Western blot analysis after purification by Ni-NTA column chromatography.
Immunoblot Analysis
Cell lysates were prepared as described by Tada et al. (1996) from subconfluent cultures of cells. Approximately 107 cells were washed with ice-cold PBS, pelleted and disrupted in RIPA buffer containing 10 mM Tris-HCl buffer (pH 7.4), 1% NP-40, 0.1% sodium deoxycholate, 0.1% SDS, 150 mM NaCl, 1 mM ethylene diamine tetra acetate and a cocktail of protease inhibitors (PharMingen), and a portion of cell lysate containing 20 μg protein (derived from ∼1.5 × 105 fibroblast cells and 2.2 × 105 lymphoblastoid B cells) was electrophoresed on 7% SDS–polyacrylamide gels. A prestained SDS-PAGE standard protein marker (Bio-Rad Laboratories Inc.) was used to calibrate the molecular mass; it contained myosin (213 kD), β-galactosidase (119 kD), BSA (83 kD), and ovalbumin (47 kD). Proteins resolved in gel were electrophoretically transferred to a polyvinylidene difluoride membrane, Immobilon (Millipore). The transferred membrane was treated with TBS (20 mM Tris-HCl, 150 mM NaCl, pH 7.5) containing 5% (wt/vol) skim milk for 1 h. The membrane was then incubated overnight at 4°C with diluted antibodies to determine the specificity and affinity to WRN helicase of the antibodies. The membrane was washed and then was incubated with anti-mouse IgG-conjugated HRP (Dako). After washing again, the membrane was developed using ECL™ (Amersham Life Science, UK).
Immunocytochemistry by Indirect Immunofluorescence
Cells were grown to ∼80% confluency in 100-mm tissue culture dishes (Corning) and were stuck onto a silane-coated slide by cytospin. The spread cells were immediately fixed with 3.7% formaldehyde in PBS for 10 min and washed and permeabilized with PBS containing 0.05% Tween 20 (washing buffer). They were then treated with 3% (wt/vol) skim milk in PBS at room temperature for 2 h, treated with 0.1% Triton X-100 in PBS, and incubated with mouse anti-WRN helicase mAbs as the primary antibodies overnight at 4°C. The cells were washed again with the washing buffer three times for a total of 30 min, and they were further incubated with biotinylated goat anti–mouse IgG1 (Southern Biotechnology Associates Inc.) for 1 h at room temperature. The cells were washed again with the washing buffer and incubated with 5 μg/ml streptavidin FITC (PharMingen) for 30 min at room temperature.
Double immunocytostaining was made for WRN helicase and the nucleolus protein using mouse mAb 8H3 and the goat polyclonal antibody B23 (Santa Cruz) specific to nucleolar nucleophosmin/B23 protein as primary antibodies. These antibodies were mixed and incubated with the fixed HeLa cells. As secondary antibodies, the biotinylated donkey anti– mouse IgG and the Cy3 conjugated donkey anti–goat IgG (both from Chemicon International) were used to detect the primary antibodies that bound to WRN helicase and the nucleolus protein. The Cy3-conjugated IgG emitted red fluorescence, while the biotinylated anti–mouse IgG was stained green by streptavidin FITC by the same conditions described above. The nuclei were counterstained blue with DAPI. Fluorescent images were visualized with a Bionanoscope (Nikon Engineering) fitted with a 100× Nikon PlanApo oil immersion objective and two double-pass filter sets for fluorescein/DAPI and Texas red. An alternative procedure using acetone-methanol (1:1 vol/vol) was also carried out, which resulted in similar staining profiles (data not shown).
Determination of Subnuclear Distribution of WRN Helicase by Expression of the Full-Size cDNA in HeLa Cells
An expression plasmid was constructed with the full-size WRN cDNA and pEGFP-C3 plasmid (Clontech). The cloned full-size cDNA was placed downstream of the EGFP sequence in the pEGFP-C3 DNA, according to the manufacturer's instruction. It was transfected to HeLa cells and the WRN helicase NH2-terminally tagged with EGFP was generated as described before (Matsumoto et al., 1997a). HeLa cells were seeded onto a poly-l-lysine-coated cover glass (Iwaki Co.), cultured overnight and then transfected with 5 μg of expression plasmid DNA mixed with 10 μg lipofectin (GIBCO BRL). 24 h after transfection at 37°C, the cells were fixed with 4% paraformaldehyde. A cover glass was mounted with 0.2 μg/ml DAPI in antifade solution (Wako). The EGFP fusion proteins and nuclei were visualized by fluorescence microscopy with a Nikon Optiphot-2 microscope fitted with a 60× Nikon PlanApo oil immersion objective and a triple-pass filter set for fluorescein/DAPI and Texas red. Fluorescent images were collected using a high performance CCD camera and were processed by MacProbe (Perceptive Scientific Instruments).
Results
Preparation of mAbs Specific for WRN Helicase and Epitope Mapping
After screening multiple independent clones of hybridoma cells, we found five hybridoma cell lines generating the isotypes of mouse IgG1κ, referred to hereafter as 3D12, 4H12, 4D9, 4F8, and 8H3, that bind efficiently in immunoblot analysis to whole molecules of purified WRN helicase (Fig. 1). Their epitopes were determined by measuring the immunoreactivity of mAbs to parts of the WRN helicase generated by expression of the fragments of WRN cDNA in B16F10 mouse melanoma cells (Table I). The B16F10 cells express a very low level of endogenous murine WRN helicase protein (our unpublished data), and therefore do not disturb the detection of binding between mAbs and the recombinant derivatives of human WRN helicase expressed in the cells. The derivatives of human WRN helicase contained NH2-terminal EGFP and various lengths of fragments of WRN helicase (aa residues 1–368, 1–1,046, 1–1,162, 1–1,304, 1,294–1,432, 1,304–1,432, 1,353–1,432, 1,363–1,432, 1,373–1,432, 1,383–1,432, and 1,413–1,432). A convenient color reaction was used to assess the epitopes of each mAbs; the location of expressed fusion proteins were shown by green fluorescence, and positive immunoreactions between fusion proteins and mAbs were shown by a red color due to Texas red–conjugated avidin that binds to biotinylated anti–mouse IgG (Fig. 2). mAb 4H12 was found to react with COOH-terminal polypeptide C59 consisting of 59 aa residues but not to a shorter COOH-terminal polypeptide C49 consisting of 49 aa residues, indicating that the epitope of 4H12 is in the 10 aa residues that C49 lacks (Fig. 2 A). This 10-aa region contains the NLS required for WRN helicase to migrate from the cytoplasm to the nucleus (Matsumoto et al., 1997b). The mAbs 4D9, 4F8, and 8H3 could bind to the N368 polypeptide containing the NH2-terminal 368 aa residues of WRN helicase (Fig. 2 B shows the data obtained with mAb 8H3). The epitopes of 4D9, 4F8, and 8H3 were further narrowed by immunoblot analysis to a NH2-proximal region between amino acid residues 232 and 368 after examining their binding activities to a mutant WRN helicase (C1201) that lacked NH2-terminal aa residues 1–231 (Fig. 2 C). This region is downstream of the 3′-5′ exonuclease region (60–231 aa residues) predicted by Mushegian et al. (1997) from their homology search studies. The mAb 3D12 bound to recombinant WRN helicase proteins encompassing the helicase domain and the COOH terminus (877– 1,432 aa residues), suggests that its epitope is on the COOH-terminal side. Table I summarizes these results. Two mAb, 8H3 and 4H12, that recognize distinct epitopes on WRN helicase at NH2-terminal 232–368 aa and COOH-terminal 1,373–1,432 aa, respectively, were mainly used for the following studies.
Figure 1.

Immunoblot analysis of mAbs specific for WRN helicase. WRN helicase (0.74 μg) produced in insect cells by a baculovirus system and purified by Ni-chelate column chromatography (Suzuki et al., 1997) was used for immunoblot analysis to monitor the quality of mAbs. Lane 1, Coomassie brilliant blue stained WRN helicase; lanes 2 and 3, mAbs 3D12 and 4H12 raised against COOH-proximal polypeptide (aa residues 877–1,432), respectively; lane 4, mAb 4D9 raised against full-size WRN helicase; lane 5, mAb 4F8 raised against full-size WRN helicase; lane 6, mAb 8H3 raised against full-size WRN helicase. An arrow head shows the positions of full-size WRN helicase.
Table I.
Determination of Epitopes of mAbs Specific for WRN Helicase
| WRN proteins | Residues (aa) | mAbs | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 3D12 | 4H12 | 4D9 | 4F8 | 8H3 | ||||||||
| Western blotting | ||||||||||||
| Full | 1–1,432 | ○ | ○ | ○ | ○ | ○ | ||||||
| C1201 | 232–1,432 | − | ○ | ○ | ○ | ○ | ||||||
| C600 | 833–1,432 | ○ | ○ | × | × | × | ||||||
| Immunostaining | ||||||||||||
| Full | 1–1,432 | ○ | ○ | ○ | ○ | ○ | ||||||
| N1304 | 1–1,304 | × | × | − | ○ | − | ||||||
| N1162 | 1–1,162 | − | × | − | ○ | − | ||||||
| N1046 | 1–1,046 | × | × | ○ | ○ | ○ | ||||||
| N368 | 1–368 | × | − | ○ | ○ | ○ | ||||||
| C138 | 1,294–1,432 | × | − | − | − | − | ||||||
| C128 | 1,304–1,432 | × | ○ | − | × | − | ||||||
| C79 | 1,353–1,432 | × | ○ | − | × | − | ||||||
| C69 | 1,363–1,432 | × | ○ | − | − | − | ||||||
| C59 | 1,373–1,432 | × | ○ | − | − | − | ||||||
| C49 | 1,383–1,432 | × | × | − | − | − | ||||||
| C19 | 1,413–1,432 | × | × | − | × | − | ||||||
Results obtained by immunoblot analysis and immunostaining are summarized. The WRN proteins having various amino acid residues are shown in the two left columns; C and N stand for COOH and NH2 termini of intact WRN helicase. The open circles show positive immunoreactivity, while the crosses show no reactivity. The horizontal bars indicate that experiment was not done.
Figure 2.
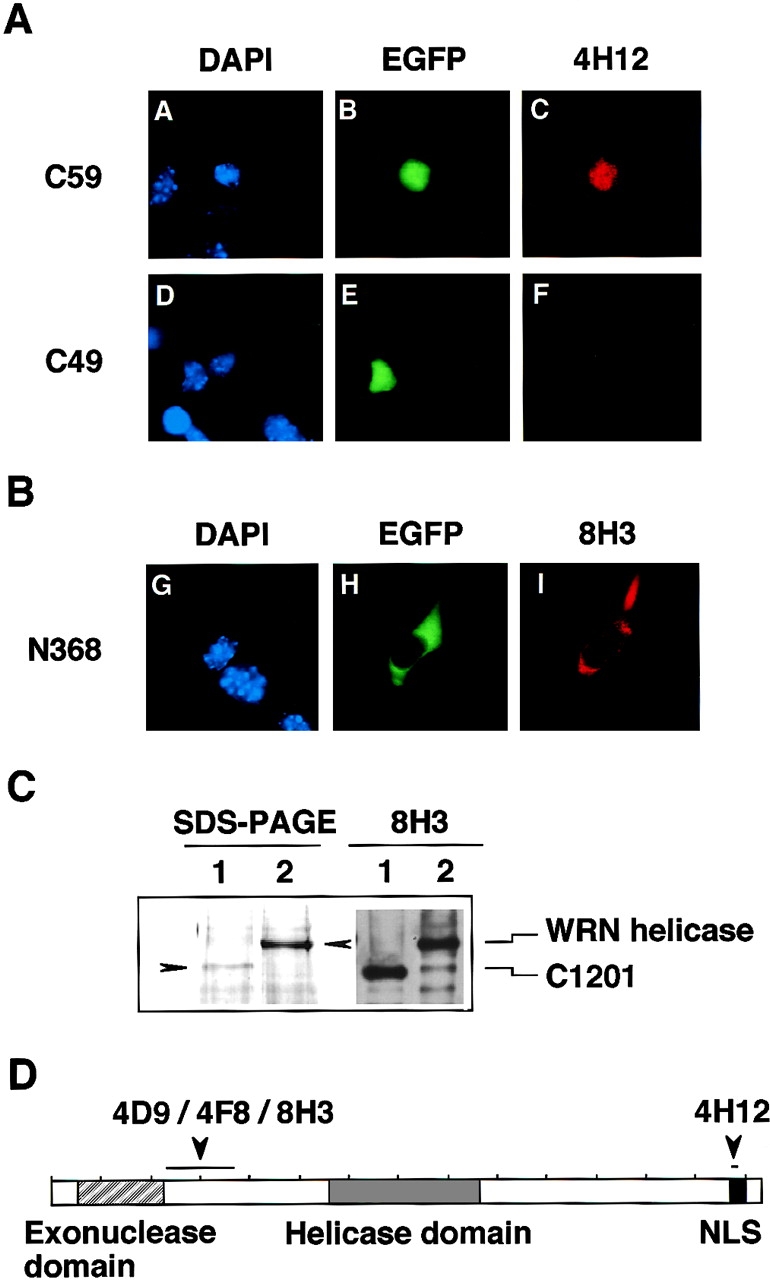
Determination of the epitopes of mAbs 4H12 and 8H3 that recognize COOH- and NH2-terminal region of WRN helicase. (A) The epitope of mAb 4H12 was determined using the parts of WRN helicase, C59 (COOH-terminal 59 aa) and C49 (COOH-terminal 49 aa), expressed as a fusion protein with EGFP in B16F10 mouse melanoma cells. The cells were fixed and tested for immunostaining by mAb 4H12, which was conjugated with fluorescent reagent Texas red (right). DAPI stains DNA and shows the location of the nucleus (left) and EGFP (middle) shows the locations of expressed EGFP fusion proteins. C59 contains the nuclear localization signal NKRRCF at aa residues 1,369–1,402 (Matsumoto et al., 1997a; Matsumoto et al., 1998a) and localizes in the nucleus, while C49 lacks the nuclear localization signal and does not migrate to the nucleus. (B) The epitope of mAb 8H3 was determined as for 4H12. The NH2-terminal fragment N368 (NH2-terminal 368 aa) was expressed in B16F10 cells as an EGFP-fused form and was tested for immunostaining with Texas red–conjugated mAb 8H3. The EGFP-N368 fusion protein does not have NLS and localizes in the cytoplasm of B16F10 cells. (C) Further epitope analysis of mAb 8H3 was made by Western blot analysis. Here, a full-size WRN helicase (lane 2) and a C1201-WRN helicase lacking NH2-terminal 231 aa synthesized by a baculovirus system (lane 1) were resolved by SDS-PAGE in 7% polyacrylamide gel and stained by Coomassie brilliant blue. They were blotted to filter and were tested for immunoreactivity with mAb 8H3 (lanes 3 and 4). (D) Schematic representation of WRN helicase molecule and the epitope sites for mAbs.
Detection of WRN Helicase Molecules in Fibroblasts and EBV-transformed B-Lymphoblastoid Cells from Normal Individuals
To detect endogenous WRN helicase in fibroblast cells from normals and WS patients, we first carried out an immunoblot analysis using mAb 4H12 that has the epitope in the COOH-terminal region of the WRN helicase. Fig. 3 A shows that mAb 4H12 detected WRN helicase with a molecular mass of 180 kD in the cell lysates prepared from normal individuals (lanes 3 and 4). As expected, no specific band to WRN helicase was seen in the patient cell lysates from homozygous gene mutation type 4/4 that yields a truncated protein (1–1,060 aa) lacking the epitope of 4H12 (lanes 1 and 2). To find out if this immunological detection extends to more easily obtainable EBV-transformed B cells, we used immunoblot analysis for B cells from two WS patients and two normals using mAb 8H3 that has an epitope in the NH2-terminal region (Fig. 3 B). Like fibroblast cells, the cell lysate of B cells from normal individuals showed a clear band of 180 kD WRN helicase (lanes 3 and 4). In contrast to a clear visualization of WRN helicase in the cell lysate prepared from the cells of normal individuals, we found no sign of immunoreactive WRN helicase-specific polypeptide in the cell lysates from WS patients carrying a homozygous mutation 4 (lanes 1 and 2). The amount of WRN helicase protein in 20 μg lysate protein of B cells was greater than that of fibroblast cells, as estimated from the intensities of the stained bands in immunoblot analysis.
Figure 3.


Immunoblot analysis for the amounts of WRN helicase in fibroblasts and B cells from WS patients and normal individuals. (A) Cell lysates containing 20 μg protein were prepared from fibroblast cells from patient (lanes 1 and 2) and normals (lanes 3 and 4), and were analyzed by Western blot analysis with mAb 4H12 that recognizes the COOH-terminal portion of WRN helicase. Lane 1, lysate from WS patient no. 15501 cells (PDL 14) containing homomutation type 4/4; lane 2, lysate from another WS patient no. 10801 cell (PDL 24) containing homomutation type 4/4; lane 3, lysate from normal no. TM36 cells (PDL 20); lane 4, lysate from normal primary cultured cell line WI38 (PDL 24). Arrow head indicates the position of 180-kD WRN helicase. Asterisks indicate the proteolytic degradation products of WRN helicase that also react with mAb 4H12. (B) Immunoblot analysis was made for EBV-transformed B cells from WS patients and normals as for A, but with mAb 8H3 that recognizes NH2-terminal 232–368-aa region. Lane 1, WS patient no. 2101 cell (PDL 53) containing homomutation type 4/4; lane 2, WS patient no. 10801 cells (PDL 36) containing homomutation type 4/4; lane 3, normal individual no. 0003 cells (PDL 53); lane 4, normal individual no. 0005 cells (PDL 18). Arrowhead indicates the position of 180-kD WRN helicase.
Upregulation of the Expression of WRN Helicase by Transformation and Immortalization
The expression levels of the WRN gene product in fibroblasts derived from normal individuals were about one-tenth of those of EBV-transformed B-lymphoblastoid cells (Fig. 3). To gain more information about the levels of WRN helicase protein in various cells, we compared the levels of WRN helicase protein for the WI38 human diploid cell line derived from embryonic lung and the same WI38 cells transformed with SV-40. Comparative immunoblot analysis showed that the SV-40-transformed WI38 cells (WI38/SV) contained a higher level (∼10-fold) of WRN helicase protein than the original WI38 cells (Fig. 4 A, lanes 1 and 2). The level in WI38/SV cells was comparable to or slightly greater than that in HeLa cells (lane 3).
Figure 4.

Comparison of levels of WRN helicase expression in cells of normal, transformed and immortal states. Immunoblot analyses were made for 20 μg cell lysate proteins prepared from various cells, including fibroblasts and B cells, of normal, virus-transformed and immortal states. (A) Lane 1, normal WI38 cells; lane 2, WI38 cells transformed with SV-40; lane 3, HeLa cells. Arrowhead indicates the position of 180-kD WRN helicase, and the asterisks show the proteolytic degradation products of WRN helicase that also react with mAb 4H12. (B) Lanes 1 and 2, untransformed B cells from peripheral blood of normal individuals; lane 3, mortal EBV-transformed B cells from normal individual no. 0003 cells (PDL 53); lane 4, immortal B cells (PDL 269) developed from normal individual no. 0003 B cells; lane 5, mortal EBV-transformed B cells from normal individual no. 0005 cells (PDL 18); lane 6, immortal B cells (PDL 269) developed from no. 0005 B cells. (C) Comparison of expression levels of WRN helicase in tumor cell lines. Cell lysate proteins (20 μg) prepared from various human tumor cell lines were analyzed for WRN helicase by Western blot analysis with mAb 4H12. Lane 1, HeLa cells; lane 2, HT1080 cells (fibrosarcoma); lane 3, PA1 cells (ovarian teratocarcinoma); lane 4, Tera2 cells (embryonic carcinoma); lane 5, T24 cells (bladder carcinoma); lane 6, K562 cells (chronic myelogenous leukemia); lane 7, Molt4 cells (acute lymphoblastic leukemia); lane 8, A172 cells (glioblastoma).
The transformation of B cells by EBV can only extend the life span of most B cells up to a maximum 160 PDLs and does not necessarily make the cells immortal, as opposed to an often misunderstood concept that EBV infection makes B cells immortal (Tahara et al., 1997). However, a few populations of immortal B cells occur after a long period of culture of transformed B cells, similar to fibroblast cells transformed by SV-40. In our laboratory, three such immortalized B-lymphoblastoid cell lines were established from B cells transformed by EBV originally from normal individuals after a long period of culture, and the cells showed strong telomerase and infinitely proliferating activities (Kataoka et al., 1997). About twofold higher levels of WRN helicase were observed with these immortalized B cells (Fig. 4 B, lanes 4 and 6) than with their parental cells, which were mortal (lanes 3 and 5). In a separate comparative study, we examined the level of WRN helicase in untransformed B cells purified from peripheral blood cells, but no WRN helicase protein was detected (Fig. 4 B, lanes 1 and 2). Here, the order of WRN helicase expression was (a) immortal EBV-transformed B cells with telomerase activity, (b) mortal EBV-transformed B cells with no or very low telomerase activity, (c) untransformed B cells in peripheral blood. The data obtained with B cells predict that tumor cells may have higher levels of WRN helicase, as they are transformed and immortalized from original normal progenitor cells.
WRN Helicases in Tumor Cells
The expression levels of WRN helicase were compared in several human tumor cell lines by comparative immunoblot analysis with mAb 4H12 that recognizes the COOH-terminal epitope. All human tumor cell lines examined showed a level equivalent to that in HeLa cells or higher levels (Fig. 4 C). These cell lines were HT1080 (fibrosarcoma), PA1 (ovarian teratocarcinoma), Tera2 (embryonic carcinoma), T24 (bladder carcinoma), K562 (chronic myelogenous leukemia), Molt4 (acute lymphoblastic leukemia), and A172 (glioblastoma) cells, and are shown in lanes 2, 3, 4, 5, 6, 7 and 8, respectively (Fig. 4 C). The highest expression was found in K562 cells that showed much higher activity than that of HeLa cells (lane 6) followed by PA1, Tera2, and Molt4 cells (lanes 3, 4, and 7). The molecular size of WRN helicase was unaltered and remained at 180 kD for all tumor cells examined.
Immunocytochemistry Showing the Location of WRN Helicase in the Nucleoplasm
We used mAbs to determine the subnuclear location of WRN helicase. Previously, we showed that WRN helicases expressed in HeLa cells by transfected expression plasmids migrate exclusively to the nucleus (Matsumoto et al., 1997a; Suzuki et al., 1997). This nuclear migration was found to be dependent on the NLS encoded in the COOH-terminal region of WRN helicase (Matsumoto et al., 1998a). However, where in the nucleus WRN helicases reside, and whether the location changes upon cell cycling, remained unclear. To define the subnuclear location, we immunocytochemically stained the endogenous WRN helicase in skin fibroblasts from a normal individual (Fig. 5 I, A and B), skin fibroblasts from a WS patient (E and F), Tera2 cells (I and J), and WI38/SV cells (O and P). All the results showed that WRN helicase was mainly located in the nucleoplasm. In these cells the nucleolus regions were left unstained and were represented by black holes (Fig. 5 I, J and P). A higher magnification of the immunostaining of Tera2 cells by mAb 4F8 showed that the stained WRN helicase antibody exists as multiple nuclear dots (Fig. 6). Similar profiles were obtained by other mAbs 3D12, 8H3, and 4D9. To avoid the possibility that nucleolus proteins might be leaked by disruption of nucleoli during our immunocytostaining, as a reference, we stained the cells similarly with a different anti-human nucleolus antibody AE3 that has been broadly used to stain specifically the nucleolus. However, this was not the case, as shown in Fig. 5 I (C and D, G and H, and K and L for normal fibroblast, WS fibroblast and Tera2, respectively) in which the nucleoli of these cells were stained distinctly by the nucleolus-specific antibodies. When mouse colon carcinoma cells (colon 26) were stained with a rat monoclonal antibody specific to the mouse WRN helicase, again only the nucleoplasm was stained (Fig. 5 I, Q and R), indicating that the major part of WRN helicase is in the nucleoplasm and not in the nucleoli for both human and mouse cells.
Figure 5.
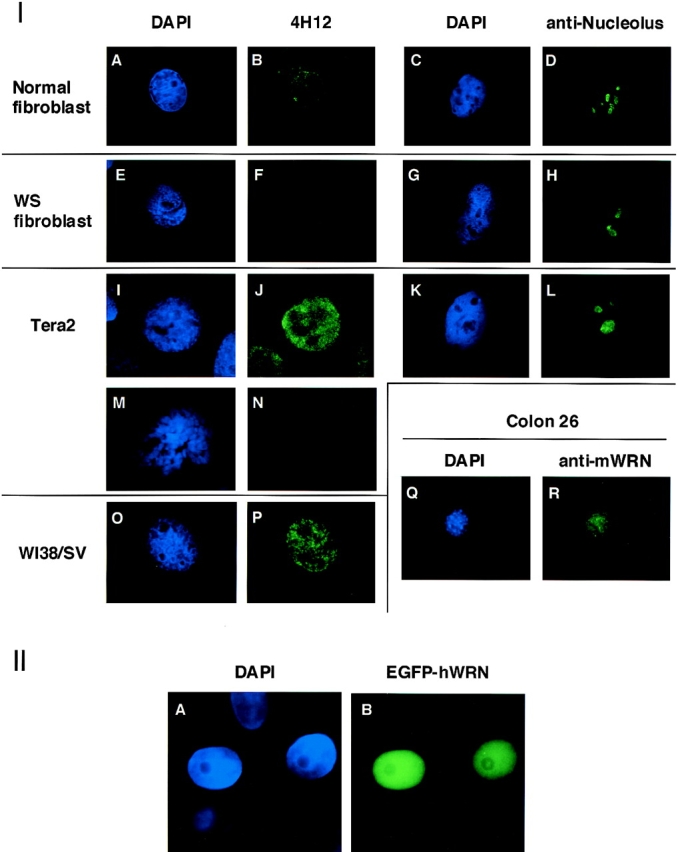
Determination of subnuclear distribution of WRN helicase by immunocytochemical staining and expression of a full-size WRN cDNA. (I) Subnuclear distribution of endogenous WRN helicase was examined by indirect immunocytochemical staining with mAb 4H12 for fibroblast cells from normal donors and WS patient no. 15501, and for Tera2 and WI38/SV cells. For controls, the nucleoli of normal fibroblast, WS patient no. 15501 and Tera2 cells were stained separately by mAb, anti human nucleolus clone AE3 (Leinco Technology Inc.), specific for human nucleolus. The nuclei of these cells were also stained by DAPI. (A and B) Fibroblast cells (interphase) from normal individual stained by DAPI and 4H12 antibody, respectively. (C and D) Fibroblast cells (interphase) from normal individual stained by DAPI and anti-nucleolus antibody, respectively. (E and F) Fibroblast cells (interphase) from WS patient no. 15501 containing homomutations 4/4 stained by DAPI and 4H12 antibody, respectively. (G and H) Fibroblast cells (interphase) from WS patient no. 15501 containing homomutations 4/4 stained by DAPI and anti-nucleolus antibody, respectively. (I and J) Tera2 cells (interphase) stained by DAPI and 4H12 antibody respectively. (K and L) Tera2 cells (interphase) stained by DAPI and anti-nucleolus antibody, respectively. (M and N) Tera2 cells chromatins from the metaphase stained by DAPI and 4H12 antibody, respectively. (O and P) WI38/SV cells (interphase) stained by DAPI and 4H12 antibody, respectively. (Q and R) Mouse colon 26 cells stained by DAPI and rat mAb to mouse WRN protein, respectively. (II) Subnuclear distribution in HeLa cells of WRN helicase synthesized as fusion protein with EGFP. (A) The DAPI stained HeLa cell nuclei. (B) The EGFP-human WRN helicase expressed in HeLa cells.
Figure 6.

A magnified profile of immunostained WRN helicase in the nucleoplasm. WRN helicases in Tera2 cells were immunostained by mAb 4F8 at the interphase of cell cycle. The green fluorescent dots demonstrate WRN helicase, and the black holes within the stained cell correspond to the nucleoli. Bar, 10 μm.
To confirm the nucleoplasmic localization of WRN helicase, we made a double immunostaining of growing HeLa cells using a mixture of two antibodies, mouse mAb 8H3 specific to WRN helicase and goat polyclonal antibody B23 specific to the nucleolar protein nucleophosmin/B23. The WRN helicase was simultaneously stained green using FITC and the nucleolus protein red using Cy3 (Fig. 7, 2 and 3). Under the conditions described in Materials and Methods, whole HeLa nuclei were stained blue by DAPI (which stains DNA), leaving several nucleolar regions unstained (Fig. 7, 1). The green WRN helicase signals spread in the entire nucleus consistent with a localization of WRN helicase mainly in nucleoplasm (Fig. 7, 2), while the red B23 signals were restricted in the nucleolar regions that were unstained by DAPI (Fig. 7, 3). The overlaid picture (Fig. 7, 4) showed that the nucleolar regions were distinctly stained with red/orange, and the surrounding nucleoplasm was stained with green, indicating nonoverlapping of WRN helicase and the nucleolar nucleophosmin/B23. These double immunocytostaining data confirmed our conclusion that the WRN helicase is mainly in the nucleoplasm of growing HeLa cells and not in the nucleolus.
Figure 7.

Double immunocytochemical staining of WRN helicase and the nucleolus protein of growing HeLa cells. Experimental conditions were described in Materials and Methods. (1) HeLa cell nuclei stained blue with DAPI. The unstained dark areas show the nucleoli. (2) WRN helicases (green) detected in nucleoplasm. (3) Nucleophosmin/B23 protein (red) detected in nucleoli. (4) Overlaid profile of stained WRN helicase (2) and nucleophosmin/B23 (3). The red/orange areas match with the nucleolus regions shown in panel 1.
Previously, we expressed the full-size WRN cDNA in HeLa cells to generate the WRN helicase NH2-terminally tagged with EGFP or FLAG. The expressed FLAG-WRN helicases localized in the entire nucleus of HeLa cells (Suzuki et al., 1997 [Fig. 2 therein]). In a similar experiment, the expressed EGFP-WRN helicases also localized in the nucleus (Matsumoto et al., 1997a), with a notable exclusion from small regions that presumably are nucleolus (Fig. 5 I, I). The results of these experiments are consistent with a conclusion of this paper that the WRN helicases mainly exist in the nucleoplasm.
To find out if WRN helicase exists in the condensed chromatin at metaphase, we analyzed the distribution of WRN helicase in Tera2 cells stained during their metaphase; however, we were unable to detect WRN helicase on the condensed chromatin (Fig. 5 I, M and N). These data indicate that WRN helicase is distributed in the nucleoplasm, not in the nucleoli or on the condensed chromatin at the metaphase.
Discussion
The gene, its product DNA helicase and various gene mutations responsible for Werner syndrome have been identified. Our previous findings that defective WRN helicase in patient cells are unable to migrate to the nucleus because of a lack of COOH-terminal NLS also explains why patients with WS show similar clinical phenotypes regardless of the type of gene mutation. Despite this fundamental knowledge of the pathogenesis of WS, several important issues concerning the endogenous protein level of WRN helicase in normal cells or cells of various states and its precise subnuclear location remained unknown. This report clarifies some of these issues.
Diagnosis of Werner Syndrome by Immunoblot Analysis
In this study, two types of mAb recognizing the NH2- or COOH-terminal region of WRN helicase were established. We used these antibodies in Western blot analysis to make the immunological diagnosis of WS possible by analyzing the lysates of cells from patients and normal individuals. The WS patients with mutation type-4, the most frequent WRN mutation in the world, can be diagnosed immunologically by the absence of WRN helicase protein in EBV-transformed B cell lysates (Fig. 3). Recently, we developed two types of genetic diagnosis systems, mutant allele specific amplification and oligonucleotide ligation assay that use the nucleotide sequence information of mutated genes and PCR (Matsumoto et al., 1998b). However, because such a genetic diagnosis is made from only known mutated gene sequences and their specific primer settings, they can not be used for WS with unknown mutations. To detect truncated gene products or the absence of gene products, the immunological method using mAbs with a refined epitope, as is shown for WS in this report, will lead the way to the clinical field in combination with a genetic diagnosis. Most WRN mutations result in the generation of truncated proteins, and therefore, the absence of a 180-kD full-size WRN helicase molecule in EBV-transformed B cells from patients will indeed indicate WS. Previously, we reported that the levels of mutant WRN mRNA in WS patient cells are significantly lower (average 16%) than those of intact mRNA from normal individuals (Yamabe et al., 1997), and we anticipated a low level of truncated WRN gene products in WS patient cells. The results obtained from patient cell lysates by immunoblot analysis were beyond this anticipation, because the patient cells (with homozygous mutation 4) showed almost no detectable truncated WRN helicase molecules with predicted size of 120 kD. Our more extended studies showed that cells from WS patients carrying homozygous mutations 6, 7, and 9 also contained no mutant proteins in the predicted truncated forms of 40, 130, and 70 kD, respectively (data not shown). We speculate that the truncated WRN helicase molecules may be sensitive to cellular proteases, and may be removed from WS patient cells by proteolytic digestion. A slightly larger molecular mass (180 kD) of endogenous WRN helicase than the predicted 162 kD was observed for all cells used in this study. Whether this larger molecular mass is due to phosphorylation and/or other post translational protein modifications should be investigated.
WRN Helicase Gene Expression Is Upregulated in Rapidly Proliferating Cells
Our findings that the copy number of WRN helicase is markedly augmented in EBV-transformed B cells or SV-40-transformed fibroblasts apparently provide an important foresight into the role of WRN helicase in these cells that have acquired a rapidly proliferating phenotype. Today, whether the transformation by viruses fortuitously stimulates the transcription and translation of WRN, or whether the cells that proliferate fast require a high level of WRN helicase and thus such cells with a large copy number of WRN helicases are selected, is not clear. The latter hypothesis is plausible because all the tumor cell lines that are free from virus infection and proliferate rapidly showed high levels of WRN expression (Fig. 4 C). In this context, we recently found the transcription of WRN helicase gene is regulated by several SP1 and a RCE elements, characteristic of a housekeeping gene, but is further modulated by tumor suppressor Rb and p53 proteins: upregulation by Rb and downregulation by p53 (Yamabe et al., 1998). The high levels of WRN helicase in tumor cells observed in this study may be due to the combined effects of the increased Rb and the inactivation of p53 by mutation. Perhaps, greater numbers of WRN helicase may be needed for some rapidly growing cells to suppress the genomic instability resulting from the illegitimate recombinations incurred by the continual DNA replications. Supporting this hypothesis, we recently showed that WRN gene can suppress the increased homologous and illegitimate recombination of Saccharomyces cerevisiae mutant cells that lack the Sgs1 DNA helicase belonging to the RecQ helicase family (Yamagata et al., 1998).
WRN Helicases Are Distributed as Nuclear Dots in the Nucleoplasm and Are Not in the Condensed Chromatin
Our immunocytochemical studies clearly showed that WRN helicases are distributed in the nucleoplasm during the interphase, and are not in the condensed chromatin in the metaphase (Fig. 5 I, M and N). Marciniak et al. (1998), using polyclonal antibodies, showed that WRN helicase localizes in the nucleolus of WI38 cells, whereas the mouse WRN helicase is distributed in the nucleoplasm. Their data on the location of human WRN helicase, therefore, contradict the findings of this study. This discrepancy does not seem to stem from the methods used for fixing cells because we obtained unambiguous nucleoplasmic staining for WI38 and WI38/SV cells after fixation with the acetone-methanol method that Marciniak used. The conditions of the cultured cells, e.g., growing or nongrowing, or the extent of cell confluence, may rather affect the mode of nuclear localization. These important issues remain to be clarified by the specific and refined antibodies characterized in this study. In this context, the fact should be mentioned that the commercially available polyclonal antibody (sc-1956; Santa Cruz) produced a similar result as our mAbs.
Our findings described in this paper are also dissimilar to previous observations obtained with yeast sgs1 helicase, a homologue of WRN helicase in S. cerevisiae, in which sgs1 exists in the nucleolus when the cells are young and migrates to the nucleoplasm as the cells senesce and the nucleoli fragment (Sinclair et al., 1997). Rather, the profiles of immunostaining of WRN helicase shown in this report resemble those of p53 and RF-A (replication factor A) that bind to DNA exclusively in the nucleoplasm (Din et al., 1990; Wilcock and Lane, 1991). Further studies are needed to determine if yeast sgs1 has the same biological role as human WRN helicases in DNA metabolism. In addition, most importantly, the kind of DNA event(s) where WRN helicase fails to participate in patient cells remains to be clarified.
Acknowledgments
We thank Ms. Chie Itoh, Kumiko Fujita, Masako Okada, and Mr. Megumu Shio for their technical assistance. We also thank Dr. Aaron J. Shatkin at the CABM for valuable discussions.
This work was supported by the Drug Organization supervised by the Ministry of Health and Welfare of Japan.
Abbreviations used in this paper
- aa
amino acids
- EBV
Epstein-Barr virus
- EGFP
enhanced green fluorescent protein
- NLS
nuclear localization signals
- PDL
population doubling levels
- WS
Werner's syndrome
Footnotes
Address correspondence to Yasuhiro Furuichi, AGENE Research Institute, 200 Kajiwara, Kamakura, Kanagawa, 247 Japan. Fax: 81 467 48 6595. E-mail: furuichi@agene.co.jp
References
- Din S, Brill SJ, Fairman MP, Stillman B. Cell-cycle-regulated phosphorylation of DNA replication factor A from human and yeast cells. Genes Dev. 1990;4:968–977. doi: 10.1101/gad.4.6.968. [DOI] [PubMed] [Google Scholar]
- Epstein CJ, Martin GM, Schultz AL, Motulsky AG. Werner's syndrome. A review of its symptomatology, natural history, pathologic features, genetics and relationship to the natural aging process. Medicine. 1966;45:177–221. doi: 10.1097/00005792-196605000-00001. [DOI] [PubMed] [Google Scholar]
- Goto M. Hierarchical deterioration of body systems in Werner's syndrome: implication for normal aging. Mech Ageing and Dev. 1997;98:239–254. doi: 10.1016/s0047-6374(97)00111-5. [DOI] [PubMed] [Google Scholar]
- Goto M, Miller RW, Ishikawa Y, Sugano H. Excess of rare cancers in Werner's syndrome (adult progeria) Cancer Epidemiol Biomark Prev. 1996;5:239–246. [PubMed] [Google Scholar]
- Gray MD, Shen JC, Kamath-Loeb AS, Blank A, Sopher BL, Martin GM, Oshima J, Loeb LA. The Werner syndrome protein is a DNA helicase. Nat Genet. 1997;17:100–103. doi: 10.1038/ng0997-100. [DOI] [PubMed] [Google Scholar]
- Lewis, G.K., and R. Kamin. 1980. Separation of T and B cells using plastic surfaces coated with anti-immunoglobulin antibodies (“panning”). In Selected Methods in Cellular Immunol. B.B. Mishell and S.M. Shiigi, editors. W.H. Freeman and Company, New York. pp. 227–234.
- Lohman TM. Helicase-catalyzed DNA unwinding. J Biol Chem. 1993;268:2269–2272. [PubMed] [Google Scholar]
- Marcianiak RA, Lombard DB, Johnson FB, Guarente L. Nuclear localization of the Werner syndrome protein in human cells. Proc Natl Acad Sci USA. 1998;95:6887–6892. doi: 10.1073/pnas.95.12.6887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maquat LE. When cells stop making sense: effects of nonsense codons on RNA metabolism in vertebrate cells. RNA. 1995;1:453–465. [PMC free article] [PubMed] [Google Scholar]
- Martin GM. Genetic syndromes in man with potential relevance to the pathobiology of aging. Birth Defects Orig Artic Ser. 1978;14:5–39. [PubMed] [Google Scholar]
- Matsumoto T, Shimamoto A, Goto M, Furuichi Y. Impaired nuclear localization of defective DNA helicases in Werner's syndrome. Nat Genet. 1997a;16:335–336. doi: 10.1038/ng0897-335. [DOI] [PubMed] [Google Scholar]
- Matsumoto T, Imamura O, Yamabe Y, Kuromitsu J, Tokutake Y, Shimamoto A, Suzuki N, Satoh M, Kitao S, Ichikawa K, et al. Mutation and haplotype analyses of the Werner's syndrome gene based on its genomic structure: genetic epidemiology in the Japanese population. Hum Genet. 1997b;100:123–130. doi: 10.1007/s004390050477. [DOI] [PubMed] [Google Scholar]
- Matsumoto T, Imamura O, Goto M, Furuichi Y. Characterization of the nuclear localization signal in the DNA helicase involved in Werner's syndrome. Int J Mol Med. 1998a;1:71–76. doi: 10.3892/ijmm.1.1.71. [DOI] [PubMed] [Google Scholar]
- Matsumoto T, Tsuchihashi Z, Ito C, Fujita K, Goto M, Furuichi Y. Genetic diagnosis of Werner's syndrome, a premature aging disease, by mutant allele specific amplification (MASA) and oligomer ligation assay (OLA) J Anti-Aging Med. 1998b;1:131–140. [Google Scholar]
- Miller, G. 1990. Epstein-Barr virus biology, pathogenesis and medical aspects. In Virology. B.N. Fields and K.M. Knipe, editors. Raven Press, New York. pp. 1921–1958.
- Mushegian AR, Bassett DE, Boguski MS, Bork P, Koonin EV. Positionally cloned human disease genes: patterns of evolutionary conservation and functional motifs. Proc Natl Acad Sci USA. 1997;94:5831–5836. doi: 10.1073/pnas.94.11.5831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oshima J, Yu C-E, Piussan C, Klein G, Jabkowski J, Balci S, Miki T, Nakura J, Ogihara T, Ells J, et al. Heterozygous and compound heterozygous mutations at the Werner syndrome locus. Hum Mol Genet. 1996;5:1909–1913. doi: 10.1093/hmg/5.12.1909. [DOI] [PubMed] [Google Scholar]
- Salk D, Bryant E, Hoehn H, Johnston P, Martin GM. Growth characteristics of Werner syndrome cells in vitro. Adv Exp Med Biol. 1985;190:305–311. doi: 10.1007/978-1-4684-7853-2_14. [DOI] [PubMed] [Google Scholar]
- Sinclair DA, Mills K, Guarente L. Accelerated aging and nucleolar fragmentation in yeast sgs1 mutants. Science. 1997;277:1313–1316. doi: 10.1126/science.277.5330.1313. [DOI] [PubMed] [Google Scholar]
- Suzuki N, Shimamoto A, Imamura O, Kuromitsu J, Kitao S, Goto M, Furuichi Y. DNA helicase activity in Werner's syndrome gene product synthesized in a baculovirus system. Nucleic Acids Res. 1997;25:2973–2978. doi: 10.1093/nar/25.15.2973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tada S, Yanagisawa J, Sonoyama T, Miyajima A, Seki M, Ui M, Enomoto T. Characterization of the properties of a human homologue of Escherichia coliRecQ from Xeroderma Pigmentosum group C and from HeLa cells. Cell Struct Funct. 1996;21:123–132. doi: 10.1247/csf.21.123. [DOI] [PubMed] [Google Scholar]
- Tahara H, Tokutake Y, Maeda S, Kataoka H, Watanabe T, Satoh M, Matsumoto T, Sugawara M, Ide T, Goto M, et al. Abnormal telomere dynamics of B-lymphoblastoid cell strains from Werner's syndrome patients transformed by Epstein-Barr virus. Oncogene. 1997;15:1911–1920. doi: 10.1038/sj.onc.1201377. [DOI] [PubMed] [Google Scholar]
- Werner, O. 1904. On Cataract in Conjunction with Scleroderma. Ph.D. dissertation, Kiel University. Schmidt and Klauning Press, Kiel, Germany.
- Wilcock D, Lane DP. Localization of p53, retinoblastoma and host replication proteins at sites of viral replication in herpes-infected cells. Nature. 1991;349:429–431. doi: 10.1038/349429a0. [DOI] [PubMed] [Google Scholar]
- Yamabe Y, Sugimoto M, Satoh M, Suzuki N, Sugawara M, Goto M, Furuichi Y. Down-regulation of the defective transcripts of the Werner's syndrome gene in the cells of patients. Biochem Biophys Res Commun. 1997;236:151–154. doi: 10.1006/bbrc.1997.6919. [DOI] [PubMed] [Google Scholar]
- Yamabe Y, Shimamoto A, Goto M, Yokota J, Sugawara M, Furuichi Y. Sp1-mediated transcription of the Werner helicase gene is modulated by Rb and p53. Mol Cell Biol. 1998;18:6191–6200. doi: 10.1128/mcb.18.11.6191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamagata K, Kato J, Shimamoto A, Goto M, Furuichi Y, Ikeda H. Bloom's and Werner's syndrome genes suppress hyper recombination in yeast sgs1 mutant: implication for genomic instability in human diseases. Proc Natl Acad Sci USA. 1998;95:8733–8738. doi: 10.1073/pnas.95.15.8733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu C-E, Oshima J, Fu Y-H, Wijsman EM, Hisama F, Alisch R, Matthews S, Nakura J, Miki T, Ouais S, Martin GM, Mulligan J, Schellenberg GD. Positional cloning of the Werner's syndrome gene. Science. 1996;272:258–262. doi: 10.1126/science.272.5259.258. [DOI] [PubMed] [Google Scholar]