

Abstract
Myoblast transplantation has been extensively studied as a gene complementation approach for genetic diseases such as Duchenne Muscular Dystrophy. This approach has been found capable of delivering dystrophin, the product missing in Duchenne Muscular Dystrophy muscle, and leading to an increase of strength in the dystrophic muscle. This approach, however, has been hindered by numerous limitations, including immunological problems, and low spread and poor survival of the injected myoblasts. We have investigated whether antiinflammatory treatment and use of different populations of skeletal muscle–derived cells may circumvent the poor survival of the injected myoblasts after implantation. We have observed that different populations of muscle-derived cells can be isolated from skeletal muscle based on their desmin immunoreactivity and differentiation capacity. Moreover, these cells acted differently when injected into muscle: 95% of the injected cells in some populations died within 48 h, while others richer in desmin-positive cells survived entirely. Since pure myoblasts obtained from isolated myofibers and myoblast cell lines also displayed a poor survival rate of the injected cells, we have concluded that the differential survival of the populations of muscle-derived cells is not only attributable to their content in desmin-positive cells. We have observed that the origin of the myogenic cells may influence their survival in the injected muscle. Finally, we have observed that myoblasts genetically engineered to express an inhibitor of the inflammatory cytokine, IL-1, can improve the survival rate of the injected myoblasts. Our results suggest that selection of specific muscle-derived cell populations or the control of inflammation can be used as an approach to improve cell survival after both myoblast transplantation and the myoblast-mediated ex vivo gene transfer approach.
Keywords: myoblast transplantation, inflammation, gene transfer, adenovirus, muscle derived stem cells
Duchenne Muscular Dystrophy (DMD)1 is an inherited muscle disease characterized by the absence of dystrophin in the membrane-associated cytoskeleton of muscle fibers (Hoffman et al., 1987; Arahata et al., 1989; Sugita et al., 1988; Zubryzcka-Gaarn et al., 1988). Dystrophin is associated with a large oligomeric complex of glycoproteins called dystrophin-associated proteins (DAPs) that provide linkage to the extracellular matrix (Ervasti and Campbell, 1991; Ibraghinov-Beskrovnaya et al., 1992; Matsumara et al., 1992; Matsumara and Campbell, 1994; Ozawa et al., 1995). The lack of dystrophin in dystrophic muscle, as well as the subsequent absence of all DAPs, disrupts the linkage between the subsarcolemmal cytoskeleton and the extracellular matrix (Matsumara et al., 1992; Matsumara and Campbell, 1994; Ozawa et al., 1995), resulting in muscle fiber necrosis and progressive muscle weakness (Watkins et al., 1988; Bonilla et al., 1988; Menke and Jokush, 1991). The mdx mouse, an animal model for DMD, also lacks dystrophin and DAPs in skeletal muscle fibers (Bulfield et al., 1984; Hoffman et al., 1987; Sincinsky et al., 1989).
Transplantation of normal myoblasts into dystrophin-deficient muscle can potentially create a reservoir of normal myoblasts capable of fusing with dystrophic muscle fibers and restoring dystrophin (Partridge, 1991). Previous experiments (Partridge et al., 1989; Alamedine et al., 1989; Morgan et al., 1990; Huard et al., 1991) showed that normal myoblasts fuse with dystrophic myoblasts to form hybrid myotubes expressing dystrophin, and transplantation of normal myoblasts resulted in dystrophin expression at the muscle fiber plasma membrane in the injected dystrophic muscle (mdx). The initial clinical trials of myoblast transfer to DMD patients demonstrated transient restoration of dystrophin-positive muscle fibers and an improvement of muscle strength in the injected DMD patients (Law et al., 1991; Huard et al., 1992a ; Huard et al., 1992b ; Karpati et al., 1993; Gussoni et al., 1992; Gussoni et al., 1997; Tremblay et al., 1993; Mendell et al., 1995). Although other limitations like the low spread and poor survival rate of the injected myoblasts may not be excluded (Huard et al., 1994c ; Beauchamps et al., 1994; Fan et al., 1996; Guerette et al., 1997; Tremblay et al., 1997; Merly et al., 1998), the failure of myoblast transfer in these clinical trials was at least partly related to immune rejection. This hypothesis was supported by the detection of serum antibodies against the injected cell antigens, such as dystrophin and dystrophin-associated proteins, in some DMD patients after transplantation (Huard et al., 1992b ). Moreover, prolonged efficacy of myoblast transplantation was observed in immunodeficient nude and SCID mice (Huard et al., 1994a ; Huard et al., 1994b ), as well as in mice adequately immunosuppressed using FK-506 (Kinoshita et al., 1994).
Although recent effort has been focused on developing gene therapy approaches to deliver genes to skeletal muscle, it has also been hindered by some limitations. Intramuscular injection of naked or complexed DNA (liposomes) carrying reporter genes or dystrophin constructs has been relatively inefficient (Acsadi et al., 1991; Wolff et al., 1992; Katsumi et al., 1994). Retroviral vectors are capable of introducing truncated dystrophin to dystrophic myoblasts in vitro, but are incapable of efficiently transducing differentiated muscle cells such as myotubes and myofibers because they require dividing cells for integration and expression (Dunckley et al., 1992; Dunckley et al., 1993). While adenovirus (AV) appears to be a relatively good vector for skeletal muscle cells, potential limitations exist for the use of AV as a gene delivery vector to muscle, i.e., the differential transducibility of myofibers throughout muscle maturation, the immune rejection induced by first-generation adenoviral vectors, and the high titer of viral recombinants often required for successful muscle transduction (Quantin et al., 1992; Ragot et al., 1993; Vincent et al., 1993; Smith et al., 1993; Acsadi et al., 1994a ; Acsadi et al., 1994b; Acsadi et al., 1996; Yang et al., 1994; Engelhardt et al., 1994; Dai et al., 1995). Recent studies with mutant AV vectors in which all viral genes were removed suggest that these vectors may be able to circumvent the immunological problems related to viral antigens (Kochanek et al., 1996; Kumar-Singh and Chamberlain, 1996; Haecker et al., 1996; Clemens et al., 1996). Furthermore, the replication-defective Herpes Simplex virus type 1 (HSV-1) vector has been found capable of infecting and expressing a foreign reporter gene in muscle cells in vitro and in newborn myofibers in vivo. Limitations to the use of the first-generation HSV-1 vector for muscles include inefficient transduction of mature myofibers, cytotoxicity, and immune rejection (Huard et al., 1995; Huard et al., 1996; Huard et al., 1997). More recently, recombinant adeno-associated viral vectors have been used as gene delivery vehicles for skeletal muscle cells. Although a high efficiency of gene transfer occurs in mature muscle, and a long-term transgene expression of up to 18 mo has been observed (Kessler et al., 1996; Xiao et al., 1996; Reed Clark et al., 1997), the use of this viral vector remains limited in its application for DMD due to its restricted gene insert capacity (<5 kb).
Through the combination of myoblast transplantation and gene therapy, the ex vivo gene transfer approach has been investigated as a gene delivery approach to skeletal muscle. It has been shown that isogenic myoblasts transduced in vitro with adenovirus, retrovirus, and HSV-1 (Salvatori et al., 1993; Huard et al., 1994c ; Rando and Blau, 1994; Booth et al., 1997; van Deutekom et al., 1998a ; van Deutekom et al. 1998b ) are capable of delivering reporter genes to skeletal muscle. We have recently evaluated the feasibility of transferring full-length dystrophin into mdx mice through myoblast-mediated ex vivo gene transfer (Floyd et al., 1997; Floyd et al., 1998). We have infected myoblasts incapable of producing dystrophin with an adenoviral vector lacking all the viral genes, but carrying the expression of full-length dystrophin and β-galactosidase (Kochanek et al., 1996). The isogenic primary mdx myoblasts and immortalized mdx cell line, which were both transduced with the adenoviral vector, have been capable of expressing β-galactosidase and full-length dystrophin in vitro. Subsequently, these transduced myoblasts have been injected into dystrophic mdx muscle, and the injected cells have restored dystrophin and dystrophin-associated proteins (Floyd et al., 1997; Floyd et al., 1998).
The ex vivo procedure may possess some positive attributes as a gene transfer strategy to skeletal muscle: the approach can create a reservoir of myoblasts capable of regenerating and restoring dystrophin to dystrophic muscle. Furthermore, since one of the major barriers to the application of gene therapy to skeletal muscle is the inability of viral vectors to transduce mature myofibers efficiently, we have recently investigated the ex vivo technique as an approach to circumvent the maturation-dependent viral transduction of skeletal muscle. The efficiency in gene transfer of the same number of viral particles (adenovirus, retrovirus, and HSV-1) has been found superior when using the ex vivo approach rather than the direct gene transfer approach (Booth et al., 1997; Floyd et al., 1997; Floyd et al., 1998). The ex vivo approach could present an important alternative treatment, particularly for DMD patients over 10 yr of age whose myofibers become extremely inefficient in regeneration and may be refractory to direct viral transduction.
Both the ex vivo procedure and the myoblast transfer approach are limited by the poor survival of the injected myoblasts. The aim of this study is to identify the causes of the poor survival in order to eventually develop approaches to reduce early loss of injected myoblasts. We have investigated whether antiinflammatory treatment and different populations of skeletal muscle–derived cells can help to circumvent this major hurdle facing the application of cell and gene therapy to skeletal muscle. This study should aid in the development of strategies to achieve efficient gene delivery to skeletal muscle for inherited diseases and treatment of inadequate and deficient muscle healing after injuries.
Materials and Methods
Purification of Primary Myoblasts
Purification of the primary muscle–derived cells was performed using a previously described protocol (Rando and Blau, 1994). The forelimbs and the hindlimbs were removed from neonatal mdx mice, and the bone was dissected. The remaining muscle mass was minced into a coarse slurry using razor blades. Cells were enzymatically dissociated by adding 0.2% collagenase-type XI for 1 h at 37°C, dispase (grade II 240 ml) for 30 min, and 0.1% trypsin for 30 min. The muscle cell extract was preplated on collagen-coated flasks. We isolated different populations of muscle-derived cells based on the number of preplates performed on collagen-coated, flasks. Preplate (PP) 1 represented a population of muscle-derived cells that adhered in the first hour after isolation, PP2 in the next 2 h, PP3 in the next 18 h, and the subsequent preplates were obtained at 24-h intervals (PPs 4–6). The myogenic population in each flask was evaluated by desmin staining and on differentiation ability when cultured in a fusion medium. The proliferation medium was F10-Ham–supplemented with 20% FBS and 1% penicillin/streptomycin; the fusion medium was F10-Ham supplemented with 2% FBS and 1% antibiotic solution (penicillin/ streptomycin). All the culture medium supplies were purchased through Gibco Laboratories (Grand Island, NY). The different populations of cells were infected with β-galactosidase–expressing adenovirus. The adenovirus, an E1–E3 deleted recombinant adenovirus kindly obtained through Dr. I. Kovesdi (GeneVec Inc., Rockville, MD), had the β-galactosidase reporter gene under the control of the human cytomegalovirus promoter and followed by the SV40 t-intron and polyadenylation signal. The adenovirally transduced cells were transplanted into the hindlimb muscle (gastrocnemius and soleus) of mdx mice, and were assayed for their survival after implantation (see below).
Myoblasts Isolated from Single Viable Myofibers
Single muscle fibers were prepared from dissected extensor digitorum longus (EDL) muscle by enzymatic desegregation in 0.2% type 1 collagenase (Sigma Chemical Co., St. Louis, MO) as previously described (Rosenblatt et al., 1995; Feero et al., 1997). Isolated muscle fibers from 6-wk-old mice were used for this project. After isolating ∼200 myofibers per muscle, a minimum of 5–10 myofibers per well were plated on 6-well plates coated with 1 mg/ml Matrigel (Collaborative Biomedical Products, Bedford, MA) in 2 ml DMEM containing 10% horse serum, 1% chick embryo extract, 2% l-glutamine, and 1% penicillin/streptomycin (Gibco Laboratories). The plates were placed in a 37°C, 5% CO2 incubator for several days. The cells emerging from these myofibers were grown until confluence, assayed for desmin expression, transduced with adenovirus carrying LacZ reporter gene expression, and tested for myoblast survival after implantation following the protocol described below.
Immortalized Cell Line
We used an immortalized mdx cell line isolated from a transgenic animal carrying a thermolabile SV40 T antigen under the control of an inducible promoter (Morgan et al., 1994). The immortalized mdx cell line proliferated indefinitely under the permissive conditions of 33°C with gamma interferon (proliferation medium; DMEM + 20% FBS) and underwent normal differentiation at 37–39°C without gamma interferon (fusion medium; DMEM + 2% FBS). This myoblast cell line was assayed for desmin immunoreactivity and the ability to differentiate when cultivated in a fusion medium. Subsequently, these cells were transduced with adenovirus carrying LacZ reporter gene expression, and the survival of the injected myoblasts was analyzed as described below.
Myoblasts Engineered to Express Antiinflammatory Substance
The mdx myoblast cell line was used for engineering the myoblasts expressing antiinflammatory substance. The myoblasts were infected with a retroviral vector carrying the expression of interleukin-1 receptor antagonist protein (IL-1Ra) and a neomycin-resistance gene (Bandara et al., 1993). After infection, the myoblasts were selected using neomycin (1,000 μg/ml of medium) to obtain nearly 100% infected cells, because noninfected cells die when subjected to neomycin treatment. The selected myoblasts were first analyzed in vitro for their ability to express IL-1Ra (80 ng/ 1 × 106 cells 48 h after infection). The engineered myoblasts were found capable of differentiating into myotubes in vitro and forming myofibers after intramuscular transplantation in vivo. The modified myoblasts were subsequently infected with adenovirus carrying LacZ reporter gene expression and injected into mdx muscle. The early fate of the injected cells was monitored and compared with that of nonengineered cells using the protocol described below.
Immunohistochemistry for Desmin
The different muscle cell populations were fixed with methanol at −20°C for 1 min, followed by two rinses in PBS. The cell cultures were blocked with 10% horse serum for 1 h and incubated with the first antibody (1/200 monoclonal mouse anti-desmin; Sigma Chemical Co., St. Louis, MO) for 1 h. After three rinses in PBS, the sections were incubated with a second antibody, anti-mouse conjugated to Cy3 immunofluorescence (1/200; Sigma Chemical Co.).
Myoblast Transplantation
Different populations of muscle cells were used for these experiments. 0.5–1 × 106 cells were injected percutaneously into the midportion of the hindlimb muscle for each experimental group; the experimental groups that were compared together received the same amount of cells. Primary mdx muscle–derived cells, myoblasts isolated from single muscle fibers, immortalized mdx myoblasts, and IL-1Ra–expressing myoblast cultures, were transduced with an adenovirus carrying the LacZ reporter gene using a multiplicity of infection of 50. 48 h after transduction, the different groups of transduced myoblasts were harvested by trypsinization (0.1% trypsin), washed in HBSS, and intramuscularly injected with a Drummond syringe. At different time points after injection (0.5, 12, 24, 48, h, and 5 d), the animals were killed, and the injected muscles were assayed for LacZ expression (histochemistry and O-nitrophenyl-β-d-galactopyranoside [ONPG]). The β-galactosidase expression obtained from the injected muscles was compared with the transduced cell extract before transplantation (0 h after injection). Five to six C57 BL10J mdx/mdx mice (2 mo old) were used for each group. The experiment animals were kept in the Rangos Research Center Animal Facility of Children's Hospital of Pittsburgh. The policies and procedures of the animal laboratory were in accordance with those detailed in the guide for the Care and Use of Laboratory Animals published by the USA Department of Health and Human Services. Finally, the research protocols used for these experiments were approved by the Animal Research and Care Committee at Children's Hospital of Pittsburgh and the University of Pittsburgh (Protocol nos. 9–96 & 1–97).
LacZ Staining by Histochemical Technique
Cryostat sections of the injected and control muscles were stained for LacZ expression using the following technique: the muscles were fixed with 1.5% glutaraldehyde (Sigma Chemical Co.) for 1 min, and were rinsed twice in PBS and incubated in X-gal substrate (0.4 mg/ml 5-bromo-chloro-3-indolyl-β-d-galactoside [Boehringer-Mannheim, Indianapolis, IN], 1 mM MgCl2, 5 mM K4Fe(CN)6/5 mm K3Fe(CN)6 in PBS) overnight (37°C). After the LacZ histochemistry, the muscle sections were counterstained with hematoxylin/eosin and visualized by light microscopy (Optiphot microscope; Nikon, Inc., Melville, NY).
Assays for β-galactosidase Activities
This technique was performed in order to achieve a better quantitation and comparison of the transgene expression level in the infected cells and the injected muscles (Sambrook et al., 1989). The injected muscles or cells were frozen in liquid nitrogen and homogenized in 0.25 M Tris-HCL (pH 7.8), and the homogenates were centrifuged at 3,500 g for 5 min. The muscle homogenate was disrupted by three cycles of freeze/thaw, and the supernatant was recolted by centrifugation (12,000 g/5 min at 4°C) and transferred to a microcentrifuge tube. 30 μl out of this extract was mixed with 66 μl of 4 mg/ml ONPG (O-nitrophenyl-β-d-galactopyranoside; Sigma Chemical Co.) dissolved in 0.1 M sodium phosphate (pH 7.5), 3 μl of 4.5 M β-mercaptoethanol dissolved in 0.1 M MgCl2, and 201 μl 0.1 M sodium phosphate. The mixture was then incubated at 37°C for 30 min. The reaction was stopped by adding 500 μl of 1 M Na2CO3, and the OD was read on a spectrophotometer at a wavelength of 420 nm. The level of β-galactosidase activity (U/sample) was extrapolated on a calibration curve that converted the OD at 420 nm to the concentration of β-galactosidase enzyme. We then compared the level of β-galactosidase enzyme in the transduced noninjected myoblasts with the amount obtained in the injected muscle 0.5, 12, 24, 48 h, and 5 d after injection.
Immunochemical Staining for Slow Myosin Heavy Chain (MyHC)
A monoclonal antibody specific for slow myosin heavy chain (MyHC) was used in this study. The antibody (M 8421; Sigma Chemical Co.) reacts with the slow MyHC of adult skeletal muscle. MyHC staining was performed using indirect immunoperoxidase techniques. In brief, 10-μm serial cryostat sections were collected on glass slides, fixed with cold acetone (−20°C) for 2 min., and blocked with 5% horse serum for 1 h. The sections were incubated overnight at room temperature in a humid chamber with primary antibodies diluted 1:500 in PBS, pH 7.4, containing 4% horse serum. The sections were subsequently rinsed three times in PBS and incubated with sheep anti-mouse antibodies conjugated with horseradish peroxidase (A7282; Sigma Chemical Co.) diluted 1:100 in PBS for 90 min. After three rinses in PBS, the peroxidase activity was then revealed by incubation with 1 mg/ ml diaminobenzidine in PBS containing 0.1% hydrogen peroxide (H2O2). Peroxide reaction was then revealed and stopped by repeated rinses in PBS. Sections were mounted in GelMount (Biomeda Corp., Foster City, CA) and observed under light microscopy (Nikon Optiphot microscope). Colocalization of the LacZ and slow myosin heavy chain expressing muscle fibers was then performed on serial muscle sections from tissue samples taken 2 and 5 d after transplantation.
Statistical Analysis
The average transduction level was computed at different time points for each group (n = 5) and compared over time by one-factor ANOVA (multicomparison type factorial) using statistical software (Stat View 512; Brain Power, Calabasas, CA).
Results
Isolation of Different Populations of Skeletal Muscle–derived Cells
We have observed that different populations of primary muscle–derived cells isolated from hindlimb muscle at different preplates contain a variable percentage of desmin positive cells. The different populations of cells consist of a mixture of muscle-derived cells, including myoblasts, fibroblasts, and adipocytes. We have found that populations of muscle-derived cells display different desmin immunoreactivity ranging from 7 to 78% after preplate (see Fig. 1 A). Furthermore, we have observed that the first preplate contained only 7% of desmin-positive cells, while the sequential preplates were enriched in their content of desmin-positive cells (2 = 14%; 3 = 25%; 4 = 72%; 5 = 75%; and 6 = 78%).
Figure 1.
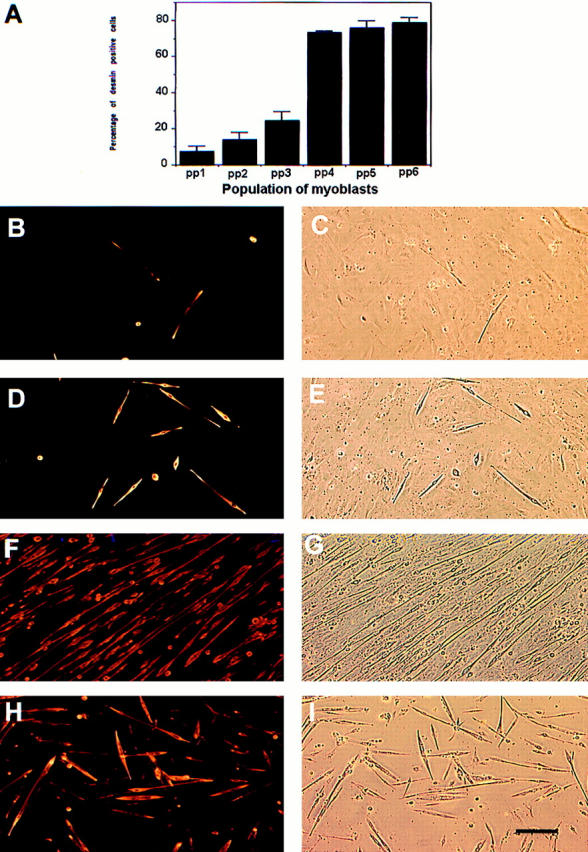
Characterization of the different populations of muscle-derived cells in vitro. The populations of muscle-derived cells after preplate (pp) displayed different desmin immunoreactivities ranging between 7 and 78% (A). The first preplate contained only 7% of desmin-positive cells, while the sequential preplates were enriched in myoblast content: pp2 = 14%, pp3 = 25%, pp4 = 72%, pp5 = 75%, and pp6 = 78% (A). Moreover, the ability of the muscle-derived cells to fuse into myotubes was found higher in pp5 (F and G) and pp6 (H and I) in comparison with PP1 (B and C) and PP3 (D and E). The desmin immunofluorescence is shown in B, D, F, and H, and the corresponding phase contrast field is shown in C, E, G, and I, respectively. Bar, 120 μm.
These cell populations consequently had a variable ability to differentiate into myotubes when cultivated into a fusion medium. The number of myotubes obtained in preplates 1 (Fig. 1, B and C) and 3 (Fig. 1, D and E) were much lower than in preplates 5 (Fig. 1, F and G) and 6 (Fig. 1, H and I). Although we observed that all preplates showed fusion of myoblasts into myotubes, the populations of muscle-derived cells with higher numbers of desmin-positive cells displayed a better ability to differentiate into myotubes.
We have obtained 97% of desmin-positive cells from a single myofiber isolated from EDL. In addition, the mdx myoblast cell line, isolated from transgenic mice carrying the SV40 T antigen, was nearly 100% desmin-positive. These cells were also capable of differentiating into myotubes, which demonstrates the high myogenicity index of those cell cultures (not shown).
Characterization of the Survival of the Different Populations of Muscle-derived Cells After Transplantation
We have observed that injection of the muscle-derived cells obtained after preplate 1 was rapidly lost 48 h after injection; only 17% of the transgene expression present on the injected cells before injection was measured in the injected muscle (Fig. 2). However, an improvement in the survival of the injected myoblasts was obtained with the subsequent preplates. The cells isolated at preplate 2 led to a 55% myoblast loss 48 h after injection, a 12% loss at preplate 3, and a 124% gain of the level of transgene expression present in the cells before transplantation preplate 6 (Fig. 2). These observations suggested that we isolated different populations of muscle-derived cells during preplating that displayed a better survival rate after transplantation.
Figure 2.
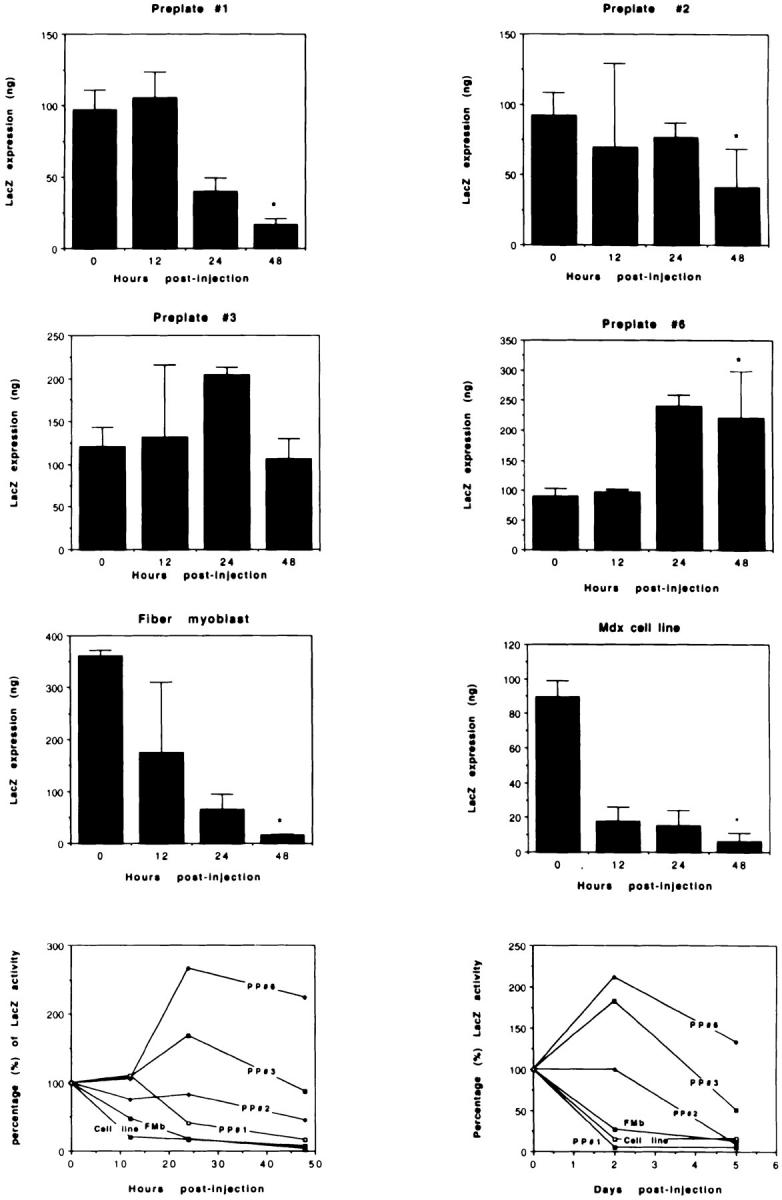
Characterization of the survival of different populations of muscle-derived cells after transplantation. Injection of the muscle-derived cells obtained after preplate 1 was rapidly lost 48 h after injection: only 17% of the transgene expression present on the injected myoblasts before injection was measured in the injected muscle. However, the cells isolated at preplate 2 led to 55% of the myoblast loss; preplate 3 led to a 12% loss, and preplate 6 gained 124% of the level of transgene expression present in the cells before transplantation. Surprisingly, a 96% loss of the pure population of myoblasts isolated from single myofibers was still observed 48 h after transplantation (fiber myoblast, FMb). Similarly, the immortalized myoblast cell line was rapidly lost after transplantation: 93% of the level of transgene expression present in the cell culture after implantation had disappeared 2 d after injection (Mdx cell line). Even though PP3 and PP6 displayed a better cell survival 2 d after injection, a decrease was still observed in the amount of LacZ reporter gene in the injected muscle 5 d after injection. However, the cells that displayed a better survival (PPs 3 and 6) remained with a higher level of gene transfer 5 d after injection. *P < 0.05 when compared with transduced noninjected myoblasts (0 h).
Surprisingly, the pure population of myoblasts obtained from the isolated myofibers (fiber myoblasts, FMb) displaying over 95% desmin immunoreactivity suffered a rapid loss after myoblast transplantation. In fact, a loss of 96% of the injected myoblasts was observed 48 h after transplantation (Fig. 2). Similarly, the mdx myoblast cell line (cell line) was rapidly lost after transplantation: 93% of the level of transgene expression present in the cell culture after implantation disappeared 2 d after injection. The high percentage of desmin-positive cells present in the muscle-derived cell population in preplate 6, therefore, was probably not the only factor explaining the improvement of cell survival after implantation.
Even though we have isolated populations of muscle-derived cells displaying a better survival after injection (PP3, PP6), all the cell populations decrease in reporter gene expression between days 2 and 5 after injection. The cells with the better survival rate consequently retain the better transgene expression at day 5.
Determination of the Ability of the Different Populations of Muscle-derived Cells to Achieve Gene Delivery in Skeletal Muscle In Vivo
All the myoblast populations after adenoviral transduction have been found to be capable of delivering β-galactosidase reporter gene to skeletal muscle 2 and 5 d after infection (Fig. 3). In fact, by using the same number of cells, we have observed that PP6, and, to a lesser extent, PP3, offer better gene transfer than the population of muscle-derived cells isolated at PP1 and PP2. The ability of the purified muscle cells (PP6 and PP3) to circumvent the poor survival of the injected cells may explain the better efficiency of gene transfer in the injected muscle. However, we have observed that PP6 displays a better ability to fuse with host myofibers (large diameter) when compared with muscle-derived cells isolated at earlier preplates. The myoblast cell line (cell line) and the highly pure myoblast culture isolated from myofibers (FMb) also display a reduction in gene transfer when compared with the muscle-derived cells isolated at preplate 6, suggesting that the ability of cells to bypass the poor survival after injection leads to an improvement of gene transfer to skeletal muscle.
Figure 3.
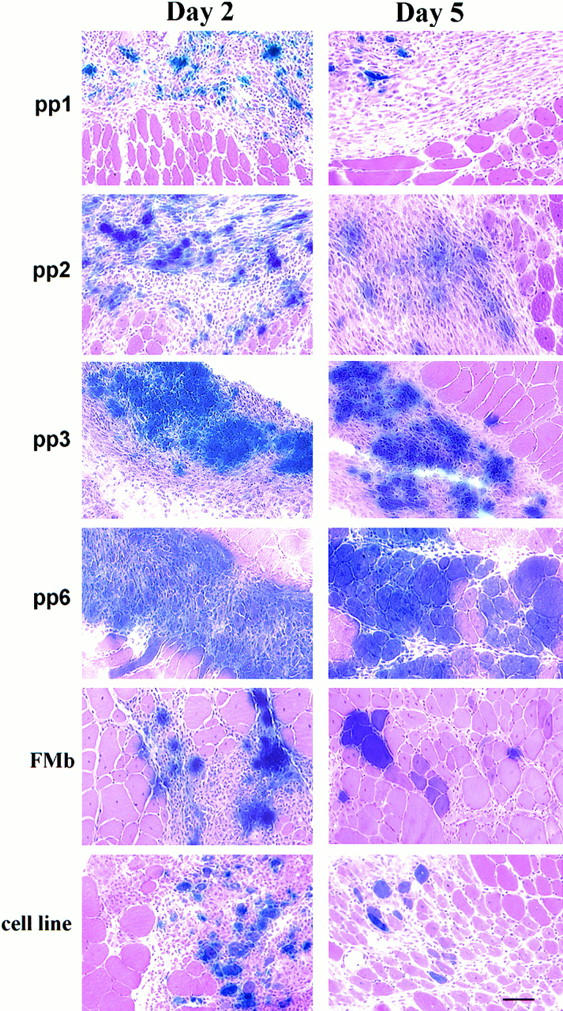
Determination of the ability of the different populations of muscle-derived cells to achieve gene transfer in muscle 2 and 5 d after injection. The population of purified primary myoblasts pp6 and, to a lesser extent, pp3, offered a better gene transfer than the population of muscle-derived cells isolated in pp1 and pp2. The myoblast cell line (cell line) and the highly pure myoblast culture isolated from fast single myofibers (FMb) also displayed a reduction in gene transfer when compared with the muscle-derived cells isolated in pp6. Bar, 50 μm.
The Influence of Muscle Fiber Type on the Survival of the Muscle-derived Cells
To determine whether the origin of the muscle cells influenced the cell survival after injection, we investigated the type of LacZ-positive myofibers (fast or slow twitch myofibers) formed in the injected gastrocnemius muscle (Fig. 4, E–H) by using anti-slow MyHC antibodies after FMb and PP6 cell transplantation. We evaluated whether a colocalization of myofibers expressing β-galactosidase and slow myosin heavy chains occurs in the injected muscle. After injection, the cells either fused together to form myotubes or with host myoblasts and muscle fibers to form mosaic myofibers. Serial sections revealed that transduced myoblasts obtained from single myofibers either fused together or with host myoblasts to form myotubes and immature myofibers in which no slow myosin heavy chain was detected (Fig. 4, A and B), or fused with host myofibers that showed an absence of expression of slow MyHC (Fig. 4, E and F). This result suggested that the myoblasts from single muscle fibers may preferentially fuse with host myofibers of the same phenotype. In contrast, the muscle-derived cells isolated at PP6 fused with both host myofibers that expressed (Fig. 4, C and D) or did not express slow myosin heavy chain (Fig. 4, G and H), suggesting that the muscle-derived cells at PP6 have the ability to fuse with fast and slow muscle fibers.
Figure 4.
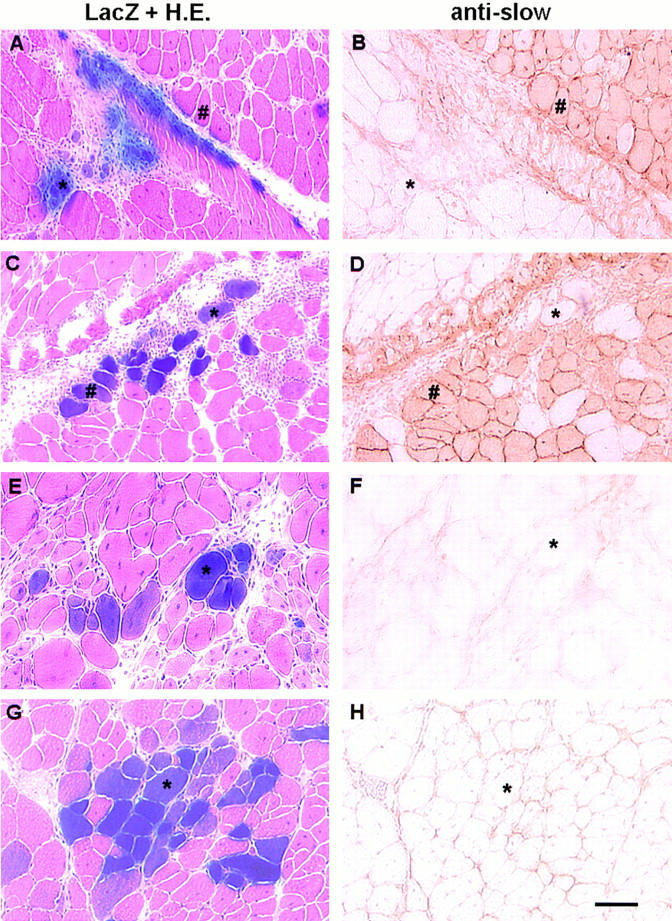
Expression of slow MyHC in LacZ-positive cells in soleus (slow) and gastrocnemius (fast) muscles 5 d after FMb (A, B, E, F ) and PP6 (C, D, G, H) implantation. Serial cryostat sections were used to reveal the colocalization of LacZ-expressing myofibers with the presence of myofibers expressing slow MyHC. The transduced myoblasts isolated from single myofibers (FMb) either fused together or with host myoblasts and formed immature myofibers in which no slow myosin heavy chain was detected (*, A and B), or fused with host myofibers that similarily did not express slow MyHC (*, E and F). This result suggested that the myoblasts from fast single fibers did not fuse with myofibers expressing slow MyHC (#, A and B) and may preferentially have fused with host muscle fibers of the same phenotype. In contrast, preplate 6 fused with host muscle fibers expressing (#, C and D) or not expressing slow myosin heavy chain (*, C, D, G, and H), suggesting that the muscle derived cells at PP6 had the ability to fuse with both fast- and slow-twitch muscle fibers. Bar, 50 μm.
Engineering of Muscle Cells that Display Antiinflammatory Action
In an attempt to investigate whether the cells capable of expressing antiinflammatory substances can improve the cell survival after injection, we genetically engineered myoblasts to express IL-1Ra, which is capable of competing with inflammatory cytokine IL-1 for binding to the IL-1 receptor, but does not induce IL-1 receptor signaling. We then compared the engineered myoblast survival with the nonengineered control cells (Fig. 5). The myoblast used for this experiment, the mdx cell line, displayed a drastic loss of the injected cells after injection (Fig. 2). Here, we again observed an 84% loss of the control myoblasts, nonengineered cells (Fig. 5 A) by 48 h after injection by the significant decrease in the amount of β-galactosidase expression in comparison to the noninjected transduced myoblast at 0 h. Moreover, a slight increase in the amount of reporter gene was detected at 120 h after injection, which remains significantly lower than that observed in control cells.
Figure 5.

Characterization of the ability of engineered myoblast expressing antiinflammatory substance to reduce the poor survival of the injected cells. The survival of the myoblasts engineered to express IL-1Ra was compared with the nonengineered control cells. The nonengineered cells were rapidly lost 48 h after injection (Control myoblast). In contrast, the cells engineered to express IL-1Ra significantly reduced the early loss of the injected cells (IL-1Ra– expressing myoblast): only 20% of the injected cells seemed to be lost 48 h after injection. However, a significant reduction in the amount of β-galactosidase expression was observed 24 h after injection in comparison to the noninjected myoblasts. We observed a high number of transduced myofibers that persisted between day 2 and day 5 after injection of the IL-1Ra expressing myoblasts (C and D). The absence of significant difference for both populations of cells at 0 and 0.5 h after injection suggested that the loss of myoblasts was minimal during injection. *P < 0.05 when compared with transduced noninjected myoblasts (0 h). Bar, 50 μm.
The cells engineered to express IL-1Ra, though, significantly reduced early loss of the injected cells 48 h after injection. Although we observed a major loss of 60% in the amount of β-galactosidase expressed in the noninjected cells (P < 0.05) 24 h after injection, the level of β-galactosidase expression detected 2 d after injection was not found to be significantly different than in the noninjected IL-1Ra–expressing myoblasts (Fig. 5 B). Moreover, the level of gene transfer did not change significantly between 2 and 5 d after injection, and still remained nondifferent to that observed in the noninjected myoblasts. (Fig. 5, B–D). This observation suggests that inflammation also plays a role in the poor survival rate of the injected cells; consequently, approaches capable of blocking the inflammation may aid in the development of strategies to achieve efficient myoblast transplantation.
Discussion
Several approaches have been investigated for efficient gene delivery to skeletal muscle, but all suffer from significant problems. Myoblast transplantation remains limited by the immune responses, poor survival rate of the injected myoblasts, and low spread of the injected cells that remain located in the vicinity of the injection site. Gene therapy in muscle has been hindered by the immunogenicity/cytotoxicity of the viral vectors and the differential transduction with viral vectors throughout the maturation of muscle fibers.
While the construction of viral vectors capable of delivering and expressing the full-length dystrophin into dystrophin-deficient muscle cells with perhaps little cytotoxicity and immunogenicity is being achieved, the incapacity of viral transduction in mature muscle fibers still remains. Even though multiple strategies, including induction of muscle regeneration and fenestration of the basal lamina, are under investigation to improve viral transduction in mature muscle (van Deutekom et al., 1998a ; van Deutekom et al., 1998b ), we have recently observed that the ex vivo gene transfer approach can help to improve viral entry and transduction in mature myofibers.
The ex vivo procedure was originally used to verify the success of myoblast transplantation without immunological problems. This method was performed using adenovirus, retrovirus, and herpes simplex virus carrying the expression of reporter genes (β-galactosidase or luciferase). It showed that transduced myoblasts (isogenic myoblasts) fused and reintroduced the reporter genes into the injected muscle (Salvatori et al., 1993; Huard et al., 1994c ; Rando and Blau, 1994; Booth et al., 1997; Floyd et al., 1997; Floyd et al., 1998; van Deutekom et al., 1998a ; van Deutekom et al., 1998b ). Recently we observed that the ex vivo approach could be used as an alternative to improve the efficiency of viral vectors in mature muscle. In fact, the ex vivo gene transfer approach of adenovirus, retrovirus, and herpes simplex virus resulted in a higher efficiency of gene delivery than that observed by direct injection of the same number of viral particles (Booth et al., 1997; Floyd et al., 1997; Floyd et al., 1998; van Deutekom et al., 1998a ; van Deutekom et al., 1998b ). The ability of these viral vectors to transduce myoblasts efficiently make the ex vivo approach an important alternative for viral gene delivery to skeletal muscle.
The well-documented poor survival of the injected myoblasts, however, limits the efficiency of myoblast-mediated ex vivo gene transfer to mature muscle. It has been observed that many of these injected myoblasts were rapidly lost within 48 h after transplantation (Huard et al., 1994c ; Beauchamps et al., 1994; Fan et al., 1996). Even though the causes of the poor survival rate of the injected myoblast after implantation remain unclear, the accelerated death of the injected myoblasts has been shown to be partly related to the inflammation (Guerette et al., 1997; Tremblay et al., 1997; Merly et al., 1998).
The present investigation has been performed in order to characterize whether inflammation is the only factor involved in the poor survival of the injected myoblast. Advances in cellular and molecular biology have identified IL-1Ra and soluble receptors for tumor necrosis factor-α as promising proteins to inhibit the inflammation and the progression of arthritis (Doherty, 1995; Bandara et al., 1993). The extremely wide range of biological activities of IL-1Ra may improve cell survival by blocking the action of an inflammatory cytokine (IL-1). We have then engineered myoblasts capable of expressing antiinflammatory substances such as IL-1Ra, and tested the ability of these engineered cells to prevent the rapid loss of the injected cells.
We have observed that engineered myoblasts expressing IL-1Ra allow for a better survival rate of the injected myoblasts 48 h after injection (Fig. 5). The nonengineered myoblast cell line has displayed poor survival of the injected cells, but the same cell line expressing IL-1Ra has significantly improved the survival rate of the injected cells 48 h after injection. We have observed a major improvement in the survival of the injected cells 48 h after injection by a local expression of an antiinflammatory substance, even if the amount of the β-galactosidase reporter gene decreased at 24 h in comparison to the 0 h. The result suggests that IL-1Ra–expressing myoblasts have probably fused between 24 and 48 h after injection, and have led to an increase of the expression of β-galactosidase reporter gene. However, we have observed a slight decrease in the amount of β-galactosidase reporter gene expression at 5 d after injection, which may be related to the immune rejection problem (see below).
Inflammation does not likely stand as the only factor involved in the rapid loss of the injected myoblasts after implantation. In fact, an improvement in the survival rate of the injected myoblasts has been obtained through the use of antiinflammatory drugs (anti-LFA-1), but a reduced loss of the injected myoblasts still remains (Guerette et al., 1997; Tremblay et al., 1997). Similarly, in this study, a substantial reduction in the loss of the injected myoblasts has been achieved with the IL-1Ra–expressing cells, but ∼20% loss is still observed 48 h after injection.
We have then evaluated whether the use of different populations of muscle-derived cells can help to circumvent the poor survival of the injected myoblasts after transplantation. To evaluate this hypothesis, mdx myoblast cell lines, pure myoblasts isolated from single myofibers, and different populations of primary muscle–derived cells have been injected into adult mdx muscle. The myoblasts have been adenovirally transduced, and the early fate of the injected cells after injection has been evaluated at different time points after injection (0.5, 12, 24, 48 h, and 5 d). We have observed that populations of primary muscle– derived cell cultures isolated from hindlimb muscle displayed a differential ability to express desmin and differentiate into myotubes, as well as a completely different cell survival after injection. The same number of muscle cells derived from preplate 1 vs. preplate 6 has resulted, respectively, in 83% loss and 124% gain in the transgene expression by 2 d in comparison to the noninjected transduced cells at 0 h. This observation suggests that the muscle-derived cells PP6 have totally overcome the rapid loss of the injected cells without the requirement of blocking the inflammation.
The content of desmin-positive cells could be involved with the ability of the purified muscle cells (preplate 6) to display a better survival rate than the cell population containing fewer desmin-positive cells. Even though this can stand as a likely explanation, especially between PP1 and PP6, the poor survival of pure cultures of myoblasts isolated from single fibers and the myoblast cell line, which both exhibited a nearly 100% desmin immunoreactivity, shows that the number of desmin-positive cells is not the only factor involved in the poor survival of the injected cells.
We have attempted to characterize whether the source of muscle-derived cells may have a primordial role in the early survival of the injected myoblasts. A great difference in the content of satellite cells has already been observed between slow and fast twitch muscles (Schmalbruch and Hellhammer, 1977; Kelly, 1978; Gibson and Schultz, 1982). The type of satellite cells isolated from these muscles may also possibly display a differential fate after transplantation. In fact, it has been reported that transplantation of L6 rat myoblasts expressing type II myosin heavy chain isoform (MyHCs) in vitro predominantly fuse together into myotubes or with host myofibers expressing the same myosin isoform (Pin et al., 1997). In contrast, a very low percentage of these L6 myoblasts have been found capable of fusing with myofibers expressing slow MyHC (type 1) when injected into soleus, plantaris, and medial gastrocnemius (Pin et al., 1997). Although C2C12 and cloned satellite cells have been found capable of fusing with all fiber types encountered (Hughes and Blau, 1992), both populations of cells have been capable of expressing both fast and slow MyHCs in vitro, and may represent a population of multipotential myoblast stem cells (Edom et al., 1994; MacIntyre and Merrifield, 1997).
We have observed that the PP6 muscle-derived cells have the ability to fuse with myofibers expressing both the slow and fast myosin isoforms in contrast to myoblasts obtained from isolated single myofibers, which either fused together or with host myofibers showing the absence of slow MyHC (see Fig. 4). The inability of myoblasts obtained from isolated single myofibers to fuse with myofibers expressing slow MyHC may be involved in the differential survival of the injected myoblasts, since the injected muscle (gastrocnemius and soleus) contains a mixture of myofibers expressing fast and slow myosin isoforms. The injected myoblasts that are less able to fuse with some specific types of host myofibers will likely display a poor survival at the injection site. These results suggest that the types of injected myoblasts and host muscle fibers (type 1 or 2) may represent a major determinant in the cell survival after transplantation.
Similarly, all the myotubes formed by the fusion of mdx cell line myoblasts in the injected muscle exhibited slow MyHC (not illustrated). However, the mdx cell line was found incapable of efficiently fusing with host myofibers expressing slow or fast MyCHs. The inability of some populations of myoblast to fuse efficiently with host myofibers may represent another factor involved in the poor survival of the injected cells.
Finally, it is unlikely that the poor survival of the injected cells (fiber myoblast, mdx myoblast cell line, and early preplate, PP1 and PP2), is due to inflammation and immune rejection related to the first generation adenovirus, since in some myoblast populations (PP6) the cells were infected with the same version of adenovirus and displayed a complete cell survival after injection. Furthermore, a similar poor survival of the injected myoblasts (48 h after injection) have been described by other research groups that were not using a first-generation adenovirus as a marker to follow the early fate of the injected cells (Beauchamps et al., 1994; Fan et al., 1996; Guerrette et al., 1997).
Even though different populations of muscle-derived cells and the IL-1Ra–expressing myoblasts display a better survival after implantation in skeletal muscle (48 h), long-term persistence of the injected cells appears to be hindered by immune responses. We have observed that most of the muscle cell populations display a reduction in the amount of β-galactosidase expression between days 2 and 5 after injection (see Fig. 2). Furthermore, we have observed that the number of LacZ-expressing myofibers decreases over time in the injected muscle, even when IL-1Ra–expressing myoblasts are used. We have further investigated this issue and observed an infiltration of CD4+- and CD8+- activated lymphocytes in the injected muscle that colocalize with the transduced myofibers, showing that engineered myoblasts still trigger an immune response (not illustrated). The presence of immune responses against adenovirally transduced allogenic myoblasts has already been described (Floyd et al., 1997; Floyd et al., 1998). We are, consequently, investigating whether engineering specific populations of muscle-derived cells with immunosuppressive substances may prevent both the poor survival and the immune response related to myoblast transfer.
In conclusion, the development of approaches to improve cell survival after myoblast transfer may improve both the success of myoblast transplantation and the ex vivo gene transfer mediated by myoblasts. Since the ex vivo gene transfer has been capable of improving the efficiency of viral gene transfer to mature skeletal muscle, the development of an approach to enhance the survival of the injected myoblast will further improve the delivery of viral vectors in mature myofibers. This study should help in the development of strategies based on cell and gene therapy to efficiently deliver genes for inherited diseases such as Duchenne Muscular Dystrophy, and for treating inadequate and deficient muscle healing after muscle injuries.
Acknowledgments
The authors thank Marcelle Pellerin for her technical assistance, and Dana Och and Megan Mowry for their secretarial assistance. The authors also wish to thank Dr. Jennifer Morgan and Dr. Terence Partridge for the immortalized myoblast cell line.
This work was supported by grants to Dr. Huard from The Parent Project (USA, Netherlands), Muscular Dystrophy Association (USA), Children's Hospital of Pittsburgh, National Institutes of Health (1 P60 AR44811-01), and a grant to Dr. P.D. Robbins from Public Health Service Grant (DK44935).
Abbreviations used in this paper
- AV
adenovirus
- DAP
dystrophin-associated protein
- DMD
Duchenne Muscular Dystrophy
- HSV-1
Herpes Simplex virus type 1
- IL-1Ra
interleukin-1 receptor antagonist protein
- MyHC
myosin heavy chain
- ONPG
o-nitrophenyl-β-d-galactopyranoside
- PP
preplate
Footnotes
Address all correspondence to Johnny Huard, Ph.D., Assistant Professor, Department of Orthopedic Surgery and Molecular Genetics & Biochemistry, Children's Hospital of Pittsburgh and University of Pittsburgh, Pittsburgh, Pennsylvania, 15261. Tel.: 412-692-7807. Fax: 412-692-7095. E-mail: jhuard+@pitt.edu
References
- Acsadi G, Dickson G, Love DR, Jani A, Walsh FS, Gurusinghe A, Wolff JA, Davies KE. Human dystrophin expression in mdx mice after intramuscular injection of DNA constructs. Nature. 1991;352:815–818. doi: 10.1038/352815a0. [DOI] [PubMed] [Google Scholar]
- Acsadi G, Jani A, Massie B, Simoneau M, Holland P, Blaschuk K, Karpati G. A differential efficiency of adenovirus-mediated in vivo gene transfer into skeletal muscle cells at different maturity. Hum Mol Genet. 1994a;3:579–584. doi: 10.1093/hmg/3.4.579. [DOI] [PubMed] [Google Scholar]
- Acsadi G, Jani A, Huard J, Blaschuk K, Massie B, Holland P, Lochmuller H, Karpati G. Cultured human myoblasts and myotubes show markedly different transducibility by replication-defective adenovirus recombinant. Gene Ther. 1994b;1:338–340. [PubMed] [Google Scholar]
- Acsadi G, Lochmuller H, Jani A, Huard J, Massie B, Prescott S, Simoneau M, Petrof BJ, Karpati G. Dystrophin expression in muscles of mdx mice after adenovirus-mediated in vivo gene transfer. Hum Gene Ther. 1996;7:129–140. doi: 10.1089/hum.1996.7.2-129. [DOI] [PubMed] [Google Scholar]
- Alamedine HS, Dehaupas M, Fardeau M. Regeneration of skeletal muscle fibers from autologous satellite cells multiplied in-vitro. Muscle Nerve. 1989;12:544–555. doi: 10.1002/mus.880120705. [DOI] [PubMed] [Google Scholar]
- Arahata K, Ishiura S, Ishiguro T, Tsukahara T, Suhara Y, Eguchi C, Ishihara T, Nonak I, Ozawa E, Sugita H. Immunostaining of skeletal and cardiac muscle surface membrane with antibody against Duchenne Muscular Dystrophy peptide. Nature. 1989;333:861–863. doi: 10.1038/333861a0. [DOI] [PubMed] [Google Scholar]
- Bandara G, Mueller GM, Galea-Lauri J, Tindal MH, Georgescu HI, Suchanek MK, Hung GL, Glorioso JC, Robbin PD, Evans CH. Intraarticular expression of biologically active interleukin-1 receptor antagonist protein by ex vivo gene transfer. Proc Natl Acad Sci USA. 1993;90:10764–10768. doi: 10.1073/pnas.90.22.10764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beauchamps JR, Morgan JE, Pagel CN, Partridge TA. Quantitative studies of efficacy of myoblast transplantation. Muscle Nerve. 1994;18(Suppl.):261. [Google Scholar]
- Bonilla EC, Samitt AF, Miranda AP, Hays G, Salviati S, Dimauro S, Kunkel LM, Hoffman EP, Rowland LP. Duchenne Muscular Dystrophy: deficiency of dystrophin at the muscle cell surface. Cell. 1988;54:447–452. doi: 10.1016/0092-8674(88)90065-7. [DOI] [PubMed] [Google Scholar]
- Booth DK, Floyd SS, Day CS, Glorioso JC, Kovesdi I, Huard J. Myoblast mediated ex vivo gene transfer to mature muscle. Tiss Eng. 1997;3:125–133. [Google Scholar]
- Bulfield G, Siller WG, Wright PAL, Moore KJ. X-chromosome-linked muscular dystrophy (mdx) in the mouse. Proc Natl Acad Sci USA. 1984;81:1189–1192. doi: 10.1073/pnas.81.4.1189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clemens PR, Kochanek S, Sunada Y, Chan S, Chen SS, Campbell KP, Caskey CT. In vivo muscle gene transfer of full-length dystrophin with an adenoviral vector that lacks all viral genes. Gene Ther. 1996;3:965–972. [PubMed] [Google Scholar]
- Dai Y, Schwarz EM, Gu D, Zhang WW, Sarvetnick N, Verma IM. Cellular and humoral immune responses to adenoviral vectors containing factor IX gene: tolerization of factor IX and vector antigens allow for long-term expression. Proc Natl Acad Sci USA. 1995;92:1401–1405. doi: 10.1073/pnas.92.5.1401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doherty PJ. Gene therapy and arthritis. J Rheumatol. 1995;22:1220–1223. [PubMed] [Google Scholar]
- Dunckley MG, Davies KE, Walsh FS, Morris GE, Dickson G. Retroviral-mediated transfer of a dystrophin minigene into mdx mouse myoblasts in vitro. FEBS Lett. 1992;296:128–134. doi: 10.1016/0014-5793(92)80363-l. [DOI] [PubMed] [Google Scholar]
- Dunckley MG, Wells DJ, Walsh FS, Dickson G. Direct retroviral-mediated transfer of a dystrophin minigene into mdx mouse muscle in vivo. Hum Mol Genet. 1993;2:717–723. doi: 10.1093/hmg/2.6.717. [DOI] [PubMed] [Google Scholar]
- Engelhardt JF, Ye X, Doranz B, Wilson JM. Ablation of E2A in recombinant adenoviruses improves transgene persistence and decreases inflammatory responses in mouse liver. Proc Natl Acad Sci USA. 1994;91:6196–6200. doi: 10.1073/pnas.91.13.6196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ervasti JM, Campbell KP. Membrane organization of the dystrophin-glycoprotein complex. Cell. 1991;66:1121–1131. doi: 10.1016/0092-8674(91)90035-w. [DOI] [PubMed] [Google Scholar]
- Edom F, Mouly V, Barbet JP, Fiszman MY, Butler-Browne GS. Clones of human satellite cells can express in vitro both fast and slow myosin heavy chains. Dev Biol. 1994;164:219–229. doi: 10.1006/dbio.1994.1193. [DOI] [PubMed] [Google Scholar]
- Fan Y, Maley M, Beilharz M, Grounds M. Rapid death of injected myoblasts in myoblast transfer therapy. Muscle Nerve. 1996;19:853–860. doi: 10.1002/(SICI)1097-4598(199607)19:7<853::AID-MUS7>3.0.CO;2-8. [DOI] [PubMed] [Google Scholar]
- Feero WG, Rosenblatt JD, Huard J, Watkins SC, Epperly M, Clemmens PC, Glorioso JC, Partridge TA, Hoffman EP. Single fibers as a model system for viral gene delivery to skeletal muscle: insights on maturation-dependant loss of fiber infectivity. Gene Ther. 1997;4:371–380. doi: 10.1089/hum.1997.8.4-371. [DOI] [PubMed] [Google Scholar]
- Floyd SS, Booth DK, van Deutekom JCT, Day CS, Huard J. Autologous myoblast transfer: a combination of myoblast transplantation and gene therapy. Basic Appl Myol. 1997;7:241–250. [Google Scholar]
- Floyd SS, Clemens PR, Ontell MR, Kochanek S, Day CS, Yang J, Hauschka S, Balkir L, Morgan JE, Moreland MS, et al. Ex vivo gene transfer using adenovirus mediated full-length dystrophin delivery to dystrophic muscles. Gene Ther. 1998;5:19–30. doi: 10.1038/sj.gt.3300549. [DOI] [PubMed] [Google Scholar]
- Gibson MC, Schultz E. The distribution of satellite cells and their relationship to specific fiber types in soleus and extensor digitorum longus muscles. . Anat Rec. 1982;202:329. doi: 10.1002/ar.1092020305. [DOI] [PubMed] [Google Scholar]
- Guerette B, Asselin I, Skuk D, Entman M, Tremblay JP. Control of inflammatory damage by anti-LFA-1: Increase success of myoblast transplantation. Cell Transplant. 1997;6:101–107. doi: 10.1177/096368979700600203. [DOI] [PubMed] [Google Scholar]
- Gussoni E, Pavlath PK, Lancot AM, Sharma K, Miller RG, Steinman L, Blau RM. Normal dystrophin transcripts detected in DMD patients after myoblast transplantation. Nature. 1992;356:435–438. doi: 10.1038/356435a0. [DOI] [PubMed] [Google Scholar]
- Gussoni E, Blau HM, Kunkel LM. The fate of individual myoblasts after transplantation into muscles of DMD patients. Nat Med. 1997;3:970–977. doi: 10.1038/nm0997-970. [DOI] [PubMed] [Google Scholar]
- Haecker SE, Stedman HH, Balice-Gordon RJ, Smith DB, Greelish JP, Mitchell MA, Wells A, Sweeney HL, Wilson JM. In vivo expression of full-length human dystrophin from adenoviral vectors deleted of all viral genes. Hum Gene Ther. 1996;7:1907–1914. doi: 10.1089/hum.1996.7.15-1907. [DOI] [PubMed] [Google Scholar]
- Hoffman EP, Brown J, Kunkel LM. Dystrophin: the protein product of the Duchenne Muscular Dystrophy locus. Cell. 1987;51:919–928. doi: 10.1016/0092-8674(87)90579-4. [DOI] [PubMed] [Google Scholar]
- Huard J, Labrecque C, Dansereau G, Robitaille L, Tremblay JP. Dystrophin expression in myotubes formed by the fusion of normal and dystrophic myoblasts. Muscle Nerve. 1991;14:178–182. doi: 10.1002/mus.880140213. [DOI] [PubMed] [Google Scholar]
- Huard J, Bouchard JP, Roy R, Malouin F, Dansereau G, Labrecque C, Albert N, Richards CL, Lemieux B, Tremblay JP. Human myoblast transplantation: preliminary results of four cases. Muscle Nerve. 1992a;15:550–560. doi: 10.1002/mus.880150504. [DOI] [PubMed] [Google Scholar]
- Huard J, Roy R, Bouchard JP, Malouin F, Richards CL, Tremblay JP. Human myoblast transplantation between immunohistocompatible donors and recipients produces immune reactions. Transpl Proc. 1992b;24:3049–3051. [PubMed] [Google Scholar]
- Huard J, Guerette B, Verreault S, Tremblay G, Roy R, Lille S, Tremblay JP. Human myoblast transplantation in immunodeficient and immunosuppressed mice: Evidence of rejection. Muscle Nerve. 1994a;17:224–234. doi: 10.1002/mus.880170214. [DOI] [PubMed] [Google Scholar]
- Huard J, Verreault S, Roy R, Tremblay M, Tremblay JP. High efficiency of muscle regeneration following human myoblast clone transplantation in SCID mice. J Clin Invest. 1994b;93:586–599. doi: 10.1172/JCI117011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huard J, Acsadi G, Jani A, Massie B, Karpati G. Gene transfer into skeletal muscles by isogenic myoblasts. Hum Gene Ther. 1994c;5:949–958. doi: 10.1089/hum.1994.5.8-949. [DOI] [PubMed] [Google Scholar]
- Huard J, Goins B, Glorioso JC. Herpes simplex virus type 1 vector mediated gene transfer to muscle. Gene Ther. 1995;2:1–9. [PubMed] [Google Scholar]
- Huard J, Feero WG, Watkins SC, Hoffman EP, Rosenblatt DJ, Glorioso JC. The basal lamina is a physical barrier to herpes simplex virus-mediated gene delivery to mature muscle fibers. J Virol. 1996;70:8117–8123. doi: 10.1128/jvi.70.11.8117-8123.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huard J, Akkaraju G, Watkins SC, Pike-Cavalcoli M, Glorioso JC. LacZ gene transfer to skeletal muscle using a replication defective herpes simplex virus type 1 mutant vector. Hum Gene Ther. 1997;4:439–452. doi: 10.1089/hum.1997.8.4-439. [DOI] [PubMed] [Google Scholar]
- Hughes SM, Blau HM. Muscle fiber pattern is independent of cell lineage in postnatal rodent development. Cell. 1992;68:659–671. doi: 10.1016/0092-8674(92)90142-y. [DOI] [PubMed] [Google Scholar]
- Ibraghinov-Beskrovnaya O, Ervasti JM, Leville CJ, Slaughter CA, Senett SW, Campbell KP. Primary structure of dystrophin-associated linking dystrophin to the extracellular matrix. Nature. 1992;355:696–702. doi: 10.1038/355696a0. [DOI] [PubMed] [Google Scholar]
- Karpati G, Ajdukovic D, Arnold D, Gledhill RB, Guttmann R, Holland P, Koch PA, Shoubridge E, Spence D, Vanasse M. Myoblast transfer in Duchenne Muscular Dystrophy. Ann Neurol. 1993;34:8–17. doi: 10.1002/ana.410340105. [DOI] [PubMed] [Google Scholar]
- Katsumi A, Emi N, Abe A, Hasegawa Y, Ito M, Saito H. Humoral and cellular immunity to an encoded protein induced by direct DNA injection. Hum Gene Ther. 1994;5:1335–1339. doi: 10.1089/hum.1994.5.11-1335. [DOI] [PubMed] [Google Scholar]
- Kelly AM. Satellite cells and myofiber growth in the rat soleus and extensor digitorum longus muscles. Dev Biol. 1978;65:1–10. doi: 10.1016/0012-1606(78)90174-4. [DOI] [PubMed] [Google Scholar]
- Kessler PD, Podsakoff GM, Chen X, McQuiston SA, Colosi PC, Matelis LA, Kurtzman GJ, Byrne BJ. Gene delivery to skeletal muscle results in sustained expression and systemic delivery of a therapeutic protein. Proc Natl Acad Sci USA. 1996;93:14082–14087. doi: 10.1073/pnas.93.24.14082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kinoshita I, Vilquin JT, Guerette B, Asselin I, Roy R, Tremblay JP. Very efficient myoblast allotransplantation in mice under FK506 immunosuppression. Muscle Nerve. 1994;17:1407–1415. doi: 10.1002/mus.880171210. [DOI] [PubMed] [Google Scholar]
- Kochanek S, Clemens PR, Mitani K, Chen HH, Chan S, Caskey CT. A new adenoviral vector: Replacement of all viral coding sequences with 28 Kb of DNA independently expressing both full-length dystrophin and β-galactosidase. Proc Natl Acad Sci USA. 1996;93:5731–5736. doi: 10.1073/pnas.93.12.5731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar-Singh R, Chamberlain JS. Encapsidated adenovirus minichromosomes allow delivery and expression of a 14 Kb dystrophin cDNA to muscle cells. Hum Mol Genet. 1996;5:913–921. doi: 10.1093/hmg/5.7.913. [DOI] [PubMed] [Google Scholar]
- Law, P.K., T.G. Goodwin, Q. Fang, M. Chen, H.J. Li, A. Florendo, D. Kirby, T. Bertorini, H. Herred, and G. Golden. 1991. Pioneering development of myoblast transfer therapy. In Muscular Dystrophy Research. C. Angelini, G.A. Darrieli, and D. Fontanan, editors. Elsevier Science Inc., New York. 109–116.
- MacIntyre, N., and P. Merrifield. 1997. Fibre-type potential of mouse myoblast cell lines cultured in vitro. Differentiation. In press.
- Matsumura K, Ervasti JM, Ohlendieck K, Kahl SD, Campbell KP. Association of dystrophin-related protein with dystrophin-associated proteins in mdx mouse muscle. Nature. 1992;360:588–591. doi: 10.1038/360588a0. [DOI] [PubMed] [Google Scholar]
- Matsumura K, Campbell KP. Dystrophin-glycoprotein complex: Its role in the molecular pathogenesis of muscular dystrophies. Muscle Nerve. 1994;17:2–15. doi: 10.1002/mus.880170103. [DOI] [PubMed] [Google Scholar]
- Mendell JR, Kissel JT, Amato AA, King W, Signore L, Prior TW, Sahenk Z, Benson S, Mcandrew PE, Rice R. Myoblast transfer in the treatment of Duchenne Muscular Dystrophy. N Eng J Med. 1995;333:832–838. doi: 10.1056/NEJM199509283331303. [DOI] [PubMed] [Google Scholar]
- Menke A, Jokush H. Decreased osmotic stability of dystrophin less muscle cells from the mdx mice. Nature. 1991;349:69–71. doi: 10.1038/349069a0. [DOI] [PubMed] [Google Scholar]
- Merly F, Huard C, Asselin I, Robbins PD, Tremblay J. Anti-inflammatory effect of transforming growth factor-β1 in myoblast transplantation. Transplantation. 1998;65:793–799. doi: 10.1097/00007890-199803270-00005. [DOI] [PubMed] [Google Scholar]
- Morgan JE, Hoffman EP, Partridge TA. Normal myogenic cells from newborn mice restore normal histology to degenerating muscle of the mdx mouse. J Cell Biol. 1990;111:2437–2449. doi: 10.1083/jcb.111.6.2437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morgan JE, Beauchamps JR, Pagel CN, Peckham M, Ataliotis P, Jat PS, Noble NM, Farmer K, Partridge TA. Myogenic cell lines derived from transgenic mice carrying a thermolabile T antigen: A model system for the derivation of tissues-specific and mutation-specific cell lines. Dev Biol. 1994;162:486–498. doi: 10.1006/dbio.1994.1103. [DOI] [PubMed] [Google Scholar]
- Ozawa E, Yoshida M, Suzuki A, Mizuna Y, Hagiwara Y, Noguchi S. Dystrophin-associated proteins in muscular dystrophy. Hum Mol Genet. 1995;4:1711–1716. doi: 10.1093/hmg/4.suppl_1.1711. [DOI] [PubMed] [Google Scholar]
- Partridge TA, Morgan JE, Coulton GR, Hoffman EP, Kunkel LM. Conversion of mdx myofibers from dystrophin negative to positive by injection of normal myoblasts. Nature. 1989;337:176–179. doi: 10.1038/337176a0. [DOI] [PubMed] [Google Scholar]
- Partridge TA. Myoblast transfer: A possible therapy for inherited myopathies. Muscle Nerve. 1991;14:197–212. doi: 10.1002/mus.880140302. [DOI] [PubMed] [Google Scholar]
- Pin C, Merrifield PA. Developmental potential of rat L6 myoblasts in vivo following injection into regenerating muscles. Dev Biol. 1997;188:147–166. doi: 10.1006/dbio.1997.8624. [DOI] [PubMed] [Google Scholar]
- Quantin B, Perricaudet LD, Tajbakhsh S, Mandell JL. Adenovirus as an expression vector in muscle cells in vivo. Proc Natl Acad Sci USA. 1992;89:2581–2584. doi: 10.1073/pnas.89.7.2581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ragot T, Vincent M, Chafey P, Vigne E, Gilgenkrantz H, Couton B, Cartaud J, Briand P, Kaplan JC, Perricaudet M, Kahn A. Efficient adenovirus mediated gene transfer of a human minidystrophin gene to skeletal muscle of mdx mice. Nature. 1993;361:647–650. doi: 10.1038/361647a0. [DOI] [PubMed] [Google Scholar]
- Rando TA, Blau HM. Primary mouse myoblast purification, characterization, and transplantation for cell-mediated gene therapy. J Cell Biol. 1994;125:1275–1287. doi: 10.1083/jcb.125.6.1275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reed Clark, K., T.J. Sferra, and P.R. Johnson. Recombinant adeno-associated viral vectors mediated long-term transgene expression in muscle. Hum Gene Ther. 1997;8:659–669. doi: 10.1089/hum.1997.8.6-659. [DOI] [PubMed] [Google Scholar]
- Rosenblatt DJ, Lunt A, Parry DJ, Partridge TA. Culturing of satellite cells from living single muscle fibers explants. In vitro. Dev Biol. 1995;31:773–779. doi: 10.1007/BF02634119. [DOI] [PubMed] [Google Scholar]
- Salvatori G, Ferrari G, Messogiorno A, Servidel S, Colette M, Tonalli P, Giarassi R, Cosso G, Mavillo F. Retroviral vector-mediated gene transfer into human primary myogenic cells lead to expression in muscle fibers in vivo. Hum Gene Ther. 1993;4:713–723. doi: 10.1089/hum.1993.4.6-713. [DOI] [PubMed] [Google Scholar]
- Sambrook, J., E.F. Fritsch, and T. Maniatis. 1989. Molecular Cloning. A Laboratory Manual. Cold Spring Harbor Laboratory Press, Plainview, NY. 16.66– 16.67.
- Schmalbruch H, Hellhammer U. The number of nuclei in adult rat muscles with special reference to satellite cells. Anat Rec. 1977;189:169–175. doi: 10.1002/ar.1091890204. [DOI] [PubMed] [Google Scholar]
- Sincinsky P, Geng Y, Ryderr-Cook A, Barnard E, Darlinson M, Barnard P. The molecular basis of muscular dystrophy in the mdx mouse: A point mutation. Science. 1989;244:1578–1580. doi: 10.1126/science.2662404. [DOI] [PubMed] [Google Scholar]
- Smith TAG, Mehaffey MG, Kayda DB, Saunders JM, Yei S, Trapnell BC, McClell A, Kaleko M. Adenovirus mediated expression of therapeutic plasma levels of human factor 1X in mice. Nat Genet. 1993;5:397–402. doi: 10.1038/ng1293-397. [DOI] [PubMed] [Google Scholar]
- Sugita H, Arahata K, Ishiguro T, Tsukahara T, Suhara Y, Eguchi C, Ishihara T, Nonaka I, Ozawa E. Negative immunostaining of Muscular Dystrophy (DMD) and mdx muscle surface membrane with antibody against synthetic peptide fragment predicted from DMD cDNA. Proc Jpn Acad. 1988;64:37–39. [Google Scholar]
- Tremblay JP, Malouin F, Roy R, Huard J, Bouchard JP, Satoh A, Richards CL. Results of a blind clinical study of myoblast transplantations without immunosuppressive treatment in young boys with Duchenne Muscular Dystrophy. Cell Transpl. 1993;2:99–112. doi: 10.1177/096368979300200203. [DOI] [PubMed] [Google Scholar]
- Tremblay JP, Guerette B. Myoblast Transplantation: a brief review of the problems and of some solutions. Basic Appl Myol. 1997;7:221–230. [Google Scholar]
- van Deutekom JCT, Floyd SS, Booth DK, Oligino T, Krisky D, Marconi P, Glorioso JC, Huard J. Implications of maturation for viral gene delivery to skeletal muscle. Neuromuscular Disorders. 1998a;8:135–148. doi: 10.1016/s0960-8966(98)00019-4. [DOI] [PubMed] [Google Scholar]
- van Deutekom JCT, Hoffman EP, Huard J. Muscle maturation: Implications for gene therapy. Mol Med Today. 1998b;4:214–220. doi: 10.1016/s1357-4310(98)01231-3. [DOI] [PubMed] [Google Scholar]
- Vincent M, Ragot T, Gilgenkrantz H, Couton D, Chafey P, Gregoire A, Briand P, Kaplan JC, Kahn A, Perricaudet M. Long-term correction of mouse dystrophic degeneration by adenovirus-mediated transfer of a mini dystrophin gene. Nat Genet. 1993;5:130–134. doi: 10.1038/ng1093-130. [DOI] [PubMed] [Google Scholar]
- Watkins SC, Hoffman EP, Slayter HS, Kunkel LM. Immunoelectron microscopic localization of dystrophin in myofibers. Nature. 1988;333:863–866. doi: 10.1038/333863a0. [DOI] [PubMed] [Google Scholar]
- Wolff JA, Ludtke JJ, Ascadi G, Williams P, Jani A. Long-term persistence of plasmid DNA and foreign gene expression in mouse muscle. Hum Mol Genet. 1992;1:363–369. doi: 10.1093/hmg/1.6.363. [DOI] [PubMed] [Google Scholar]
- Xiao X, Li J, Samulski RJ. Efficient long-term gene transfer into muscle tissue of immunocompetent mice by adeno-associated virus vector. J Virol. 1996;70:8098–8108. doi: 10.1128/jvi.70.11.8098-8108.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y, Nunes FA, Berencsi K, Furth EE, Gonczol E, Wilson JM. Cellular immunity to viral antigens limits E1-deleted adenoviruses for gene therapy. Proc Natl Acad Sci USA. 1994;91:4407–4411. doi: 10.1073/pnas.91.10.4407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zubryzcka-Gaarn EE, Bulman DE, Karpati G, Burghes B, Belfall, Klamut HJ, Talbot J, Hodges RS, Ray PN, Worton RG. The Duchenne Muscular Dystrophy gene is localized in the sarcolemma of human skeletal muscle. Nature. 1988;333:466–469. doi: 10.1038/333466a0. [DOI] [PubMed] [Google Scholar]