

I. Chapter Overview
The first documented cases of acquired immunodeficiency syndrome (AIDS) were characterized by the presence of rare Kaposi’s sarcoma (KS) skin lesions. More than 10 years later, it was discovered that the causative agent of KS was a γ-herpesvirus, human herpesvirus-8 (HHV-8) (KS-associated herpesvirus, KSHV). It is now abundantly clear that cancers induced by viral agents [such as, Epstein-Barr virus (EBV) and human papillomavirus (HPV)] are exacerbated by human immunodeficiency virus (HIV) infection and subsequent immune suppression. For example, the incidence of and primary central nervous system (CNS) lymphoma (PCNSL) and Hodgkin’s and high grade B-cell non-Hodgkin’s lymphomas (NHL), nasopharyngeal carcinoma (NPC), anal, penile, oral, and invasive cervical carcinomas are much higher in AIDS patients. Also common in the AIDS-afflicted, are hematopoietic cancers, B- and T-cell lymphomas, myelosarcomas, lung cancers, and gastrointestinal tract cancers. The development of highly active antiretroviral therapy (HAART) has proved effective in inducing regression of PCNSL, NHL, KS, and other cancers caused by viruses, extending the life span and quality of life of AIDS patients. However, the general availability of HAART and other antiretrovirals in developing countries, where most HIV infections are reported, is still poor. Furthermore, several reports indicate that HAART is not effective in reversing HPV-induced cervical cancers, for unknown reasons. The development of the prophylactic HPV vaccine offers some hope that future generations can be protected against cervical and penile cancers. However, in countries with high rates of cervical cancers, such as in sub-Saharan Africa, the rate of HIV-positivity approaches 30%, antiretrovirals are scarce, and the HPV vaccine is not available, nor would it be effective for those already infected with HPVs. Thus, better methods of surveillance and management of these malignancies in HIV-positive individuals continues to be a need.
II. Introduction
Since the first report of AIDS over 25 years ago, it was noted that there is a close association between HIV infection and the development of a number of cancers (Levine, 2006; Noy, 2006; Pantanowitz et al., 2006; Wood and Harrington, 2005). They include KS, Hodgkin’s and high-grade B-cell NHL, anal, and invasive cervical carcinomas. In fact, AIDS was recognized in 1981 through an unusual increase in the number of cases of KS found among the young adult male having sex with male (MSM) population (Durack, 1981). Since then, increasing numbers of KS as well as other cancers were found in this population. Subsequently in 1982, the US Center for Disease Control and Prevention (CDC) included two malignancies, KS and PCNSL, as definitions for AIDS (1982). A third AIDS-associated malignancy, NHL, was also included as one of the AIDS-defining illnesses in 1987, and a fourth, the invasive cervical carcinoma was added in 1992. In addition to these four malignancies commonly found in AIDS patients, a number of other cancers have also increased substantially in the HIV-1-infected immunosuppressed population. These cancers include multiple myelomas, leukemia, and leiomyosarcomas in children, oral cavity cancers, lung cancers, and Hodgkin’s disease (Goedert et al., 1998; Grulich et al., 2002). Prior to the era of HAART, it has been estimated that up to 25% of all cancers in males under 45 years of age in the United States were associated with HIV and up to 30% of all HIV-infected individuals will develop cancer. Especially with NHL, it has been estimated that individuals with immune deficiency have between 10- and 150-fold higher risk in developing NHL (Biggar and Rabkin, 1996; Grulich and Vajdic, 2005). Moreover, the management and prognosis of these patients were poor, mainly due to the aggressiveness of the tumors on immunosuppression, an increase in hematological toxicity due to treatment, and complications due to opportunistic infections (Kaplan et al., 1997). Treatment outcomes were shown to be poor, regardless of the types of treatment, with the response rate for AIDS-related lymphomas (ARL) of about 50% and the medium survival rate time of between 5 and 8 months (Kaplan et al., 1991; Navarro and Kaplan, 2006).
With the introduction of antiretroviral treatment in the mid-1990s, the spectrum of AIDS-associated malignancies and the epidemiology of the disease has been completely changed. There was a substantial decline in incidence of KS. It has been shown in a Swiss cohort that the standardized incidence rates for KS was 25 for those on HAART versus 239 for those that were not on treatment (Clifford et al., 2005). Similarly, changes in the incidences of ARL were observed for individuals that were on HAART. A number of studies have shown a decrease in incidence and mortality in patients with ARL (Besson et al., 2001; Kirk et al., 2001; Lee and Hurwitz, 2000; Navarro and Kaplan, 2006). The same Swiss cohort study has shown an overall decrease of HNL incidence of about 76% on HAART treatment. Similarly, for PCNSL, the impact of HAART is even more dramatic than other systemic ARL; a combination of radiotherapy with HAART had led to improvement of survival rate (Hoffmann et al., 2001; Newell et al., 2004). HAART alone has also been shown to lead to a regression of PCNSL (McGowan and Shah, 1998). In spite of the effects of HAART on ARL, similar dramatic effects have not been observed with cervical and anal cancers, and the results have been controversial. A study by the Women’s Interagency HIV study group has shown an association between HAART and regression of cervical lesions (Minkoff et al., 2001), while other studies did not show such a correlation (Lillo et al., 2001; Schuman et al., 2003). Even with the successes of HAART on a number of AIDS-associated cancers, with the increase of the longevity of treated individuals and the prolonged effects of immunosuppression, it is likely that AIDS-associated malignancies will continue to be a major clinical manifestation of HIV-infected individuals. There is still a lack of widespread antiretroviral therapy in many developing countries where AIDS is epidemic and HIV continues to spread. Better tools for detection and better regimens for management of these malignancies are necessary. Moreover, a better understanding of the biology and the pathogenesis of these cancers is needed.
The mechanisms by which malignancies are induced in HIV-1 infected individuals are not exactly known. It is likely to vary with different types, but a common underlying course is a lack of immunological controls due to immunodeficiencies. In addition, several types of cancers have been linked to viral etiological agents. In fact, the three major types of cancers included as part of the AIDS-defining illnesses, such as KS, NHL, and cervical cancers, have been linked to infectious viral agents. KS primary effusion lymphomas (PEL) and Castleman’s disease have been linked to HHV-8 or KSHV. NHL, PCNSL, Hodgkin’s disease, and leiomyosarcoma were found to be associated with EBV. Cervical carcinoma and squamous cell neoplasm were linked to HPV. Substantial information is known about these tumors and their potential etiological agents. A number of mechanisms and viral genes were found to have transforming activities, and they may play a direct and indirect role in tumorigenesis. A better understanding on how infections by these viral agents can lead to tumors will lead to the development of methods to enhance the immune response to control these agents as well as development of therapeutic agents to treat these malignancies. The major focus of this chapter is to examine KSHV-, EBV-, and HPV-associated tumors and what is known about their potential mechanisms in cellular transformation and tumorigenesis.
III. Kaposi’s Sarcoma
KS was originally described by Moritz Kaposi in 1872 as an idiopathic, multiply pigmented sarcoma of the skin (Kaposi, 1872). The initiation of a KS lesion is called the patch stage of KS, and this stage is characterized by bluish red, well-demarcated, painless maculae, most often unilateral on the lower extremities. The lesion is composed of irregularly shaped vascular spaces present around preexisting vasculature. The lesion progresses into the plaque stage as these irregular spaces become lined with endothelial cells and proliferating spindle-shaped cells, presumed to be of endothelial origin. At this stage the lesion appears to be slightly elevated on the skin. The nodular stage can then develop and is characterized by a hard and solid appearance, brownish in color, and can be hyperkeratotic and ulcerative in nature. In advanced stages of KS, lesions are often bilateral and may involve the entire extremity as well as mucosal tissues and present with edema in the surrounding tissue. The nodular lesion is composed of bundles of proliferating spindle-shaped cells, extravasated lymphocytes and erythrocytes in an abundance of slit-like spaces. These lesions become raised on the skin and sometimes coalesce to form large nodular masses (Friedman-Kien and Saltzman, 1990; Iscovich et al., 2000).
There are four epidemiological types of KS recognized: classic, endemic, iatrogenic, and HIV/AIDS-associated KS. Classic KS is the disease originally described by Kaposi (Kaposi, 1872). It predominantly occurs in elderly persons, aged 50-80 years, in the Mediterranean and Eastern European regions or in persons of Jewish ancestry. It is seen primarily in men, with a male:female ratio of 10-15:1. It is infrequently detected in children and young adults (Friedman-Kien and Saltzman, 1990). Endemic KS was first characterized in African populations from Southern and Central Africa in the early 1960s (Cook, 1962; Lothe and Murray, 1962; Oettle, 1962). Endemic KS was predominantly seen in men, with male:female ratio of 10-17:1, a mean age of 40 years, and presented with multiple, nodular, and sometimes ulcerative lesions in a centrifugal distribution. Health was well maintained while the disease was indolent for several years and rapidly deteriorated as more aggressive lesion growth and dissemination occurred. Iatrogenic- or transplant-associated KS was first recognized in the 1960s and 1970s (Gange and Jones, 1978; Harwood et al., 1979; Kapadia and Krause, 1977; Klepp et al., 1978; Penn, 1979). It was associated with patients receiving immunosuppressive therapy, most often from organ transplant. KS lesions appeared from 2 months to 8 years after therapy. Iatrogenic KS had a male:female ratio of 2-3:1. The lesions were mostly localized to the skin and infrequently involved the visceral organs and often regressed when therapy was discontinued.
The fourth type of KS is the AIDS-KS. In contrast to the slow development of classic KS, lesions developed rapidly and often disseminated to several locations in the body. AIDS-KS not only involves the lower extremities and skin, but also the upper body, the head regions, and the lymph nodes. It can also disseminate to other organs, such as the spleen, the lungs, the liver, and gastrointestinal track (Hengge et al., 2002). AIDS-KS, due to its rapid dissemination, multiple organ involvement, and difficulty with treatment, can be a painful and debilitating disease (Friedman-Kien and Saltzman, 1990). Since the HIV-1/AIDS epidemic, the patterns and clinical manifestations of KS have dramatically changed, especially in Africa. Prior to the early 1980s, Kaposi sarcoma was a rare and indolent tumor of elderly adults, primarily men (Beral, 1991; Friedman-Kien and Saltzman, 1990; Kaposi, 1872). It was extremely rare in children; but with the AIDS epidemic, KS are often seen in young children, and when present, caused a rapidly disseminated disease that failed to respond to chemotherapy and led to death within 1-3 years (Bayley, 1991; Friedman-Kien and Saltzman, 1990).
A. Viral Etiology in KS
With the association of KS to AIDS, an infectious agent has been suspected in the development of KS, including HIV and cytomegalovirus (CMV). A report has shown that herpesvirus-like particles were found in short-term KS tissue culture, and were subsequently identified as CMV (Giraldo et al., 1980). However, the involvement of CMV in KS has never been confirmed. In 1994, herpesvirus-like sequences were isolated from biopsy material from an AIDS-KS patient using a subtractive PCR technique called representational difference analysis (Chang et al., 1994). This technique allows the preferential amplification of DNA sequences representative of affected tissue, which is absent in normal tissue from the same individual. The sequence was found to be homologous but not identical to any known herpesviruses. Thus, the virus was named KSHV or HHV-8 since it is the eighth known HHV. KSHV DNA sequences were found only in the KS tissue but not in normal skin tissues. Soon after KSHV DNA sequences were identified, the viral DNA was detected in biopsies from all clinical forms of KS, but was absent in normal tissue (Chang and Moore, 1996; Memar et al., 1995). KSHV is found in all KS lesions, and is mainly located in the vascular endothelial cells and perivascular spindle-shaped cells (Li et al., 1996).
To confirm the etiological role of KSHV in KS, the presence of the virus must be detected in patients prior to the appearance of the disease. Several studies have examined this by detecting viral DNA and seroconversion prior to KS development. In HIV-1-positive patients followed before and after onset of KS, it was observed that patients with KSHV viral DNA at study entry or any time prior to KS were significantly more likely to develop KS than those who were negative for viral DNA prior to KS (Moore et al., 1996c; Whitby et al., 1995). Similarly, in HIV-1-infected patients, those that developed KS were significantly more likely to be KSHV seropositive prior to the onset of KS than those that never developed KS (Gao et al., 1996a; Melbye et al., 1998). However, it is clear that not all KSHV-infected individuals will develop KS, thus KSHV infection plays a major role, but not sufficient, for the development of KS. It is likely that other cofactors, such as immunosuppression, are required for KS development.
In addition to KS, KSHV was found to be associated with two other lymphoproliferative disorders, PEL and multicentric Castleman’s disease (MCD) (Cesarman et al., 1995; Soulier et al., 1995). PEL is a very rare subtype of NHL, predominantly associated with HIV-infected individuals. PEL was first identified as a subset of body cavity-based lymphomas (BCBL), which were subsequently called PELs (Cesarman et al., 1995). This type of lymphoma is distinguished from others as having a distinctive morphology, bridging large cell immunoblastic lymphoma and anaplastic large cell lymphoma. PELs are extremely rare tumors, and estimated to be about 0.13% of all AIDS-related malignancies in AIDS patients in the United States (Mbulaiteye et al., 2002). PELs are unique as they were found to contain KSHV DNA and are most frequently found in male AIDS patients. Most PELs are coinfected with EBV and KSHV and lack c-myc gene rearrangements, and the role of EBV in this type of tumors will be described in a section. MCD, also known as multicentric angiofollicular lymphoid hyperplasia, is a very rare polyclonal B-cell lymphoproliferative disorder with vascular hyperplasia involving multiple lymphoid organs. This disease is both clinically and morphologically heterogeneous, and is defined using clinical and pathological characteristics (Soulier et al., 1995). Unlike KS, where KSHV sequences can be detected in almost all KS samples, the B cells in MCD are usually not infected. In situ hybridization studies showed that the KSHV-infected cells are mainly located in the mantel zone of the follicle and high levels of the viral homologue of the cellular cytokine vIL-6 can be detected, suggesting that uninfected cells are recruited and stimulated to grow in the affected areas (Katano et al., 2000). It is likely that KSHV may be playing an indirect role in the disease.
B. KSHV Epidemiology
KSHV belongs to the γ-herpesvirus subfamily, which can be further subdivided into two subgroups, gamma-1 or lymphocrytovirus, and gamma-2 or rhadinovirus. EBV is the prototype gamma-1 virus and the simian herpesvirus saimiri is the prototype gamma-2 herpesvirus (Roizmann et al., 1992). KSHV is classified as a gamma-2 rhadinovirus and is the first human virus of this subfamily identified (Moore et al., 1996b). The genome of KSHV is about 165 kb in length, with ∼140 kb of long unique DNA surrounded by two terminal repeat regions, 25-35 kb each (Russo et al., 1996). It encodes over 80-open reading frames and has significant homology to the Rhadinovirus genus of the γ-herpesvirus subfamily, all of which are known to infect lymphocytes (Moore et al., 1996c). KSHV is the only member of the rhadinovirus genus known to infect humans, but is closely related to another human γ-herpesvirus, EBV, which belongs to the lymphocryptovirus genus. A feature of γ-herpesviruses like KSHV is its ability to incorporate or pirate host genes such as cyclin D and growth factor IL-6 into their genome (Moore et al., 1996a), and these genes can then play a role in the replication, survival, and transformation function of the virus. Deciphering the functions of these viral genes will lead to a better understanding of viral pathogenesis and oncogenesis.
Unlike most other herpesviruses, KSHV infection does not seem to be widely distributed in most populations. The detection of KSHV infection relies on the presence of antibodies against either lytic and/or latent antigens and varies among the different tests that were used in different seroprevalence studies. There are several different methodologies for the detection of antibodies to KSHV. Most of these assays are based on immunofluorescence antigen (IFA), utilizing B-cell lymphoma cell lines known to be infected with KSHV as the antigen source or based on ELISA with recombinant immunogenic proteins or peptides of KSHV. The performance of these assays can be quite variable, and could account for the differences in seroprevalence reported in different studies (Pellett et al., 2003; Rabkin et al., 1998; Tedeschi et al., 2002). In North America and Northern and Western Europe, KSHV seroprevalence in adult general population or blood donors ranges from 0% to 8% (Challine et al., 2001; de Sanjose et al., 2002; Gambus et al., 2001; Gao et al., 1996b; Goedert et al., 1998; Lennette et al., 1996; Simpson et al., 1996). In these countries, the seroprevalence of KSHV in different risk groups mirrors the incidence of AIDS-KS, with a seroprevalence rate of between 25% and 50% among homosexual men. On the contrary, the reported seroprevalence of KSHV is high in the adult general population in regions of Brazil, French Guiana, the Mediterranean basin, and Central and Southern Africa, where it ranges from 10% to over 80%; these regions are considered endemic for KSHV (Cunha et al., 2005; Freitas et al., 2002; Kazanji et al., 2005; Klaskala et al., 2005; Mayama et al., 1998; Mbulaiteye et al., 2003; Olsen et al., 1998; Plancoulaine et al., 2000, 2004; Wilkinson et al., 1999). Central African countries, like the Republic of Congo, Uganda, and Zambia, also have the highest KSHV infection rates in the world (Gao et al., 1996b). Therefore, KSHV seroprevalence tracks very closely with KS, with the highest infection rate in geographic areas where classic or endemic forms of KS are more common.
C. Viral Oncogenesis
A common property shared between KSHV and several other members of the γ-herpesviruses, such as EBV, is their ability to cause proliferation of infected host cells and lead to neoplasm in the infected host. Infection of primary endothelial cells by KSHV has been shown to lead to morphologic and phenotypic changes of these cells that resemble the characteristics of KS spindle cells, suggesting that KSHV can lead to malignant transformation and plays a role in the pathogenesis of KS (Flore et al., 1998; Foglieni et al., 2005; Moses et al., 1999). KSHV has been shown to encode for a number of viral genes that may contribute to tumorigenesis. These genes include both unique viral genes and gene homologues of cellular genes. Some of these viral genes have transformation potential, such as the viral K1 gene kaposin and viral G protein-coupled receptor (vGPCR). Others are viral proteins that have homology to their cellular counterparts. These proteins may deregulate cell growth and lead to transformation. These genes include the viral IL-6, viral IL-10, viral cc-class chemokines, and viral FLICE-inhibitory protein (vFLIP). Yet there are other viral genes that are involved in maintaining viral latency such as the latency-associated nuclear antigen (LANA) and K15. These genes are involved in a number of strategies that the virus uses in sustaining viral infection in pathogenesis and in the development of malignancies. These mechanisms involved the stimulation of cell proliferation, activation of cellular gene expression, immune suppression, and modulation of immune surveillance. These viral genes may also participate indirectly via upregulation of other viral genes. A summary of the viral genes involved is shown in Table I.
TABLE I.
Potential KSHV Genes Involved in Tumorigenesis
| Viral gene | Function |
|---|---|
| K1 (KSHV open reading frame 1) | Signal transduction on receptor binding. Homologue of the herpesvirus saimiri STP transforming gene and is involved in the deregulation of NF-κB |
| K12 (Kaposin) | Two forms, Kaposin A and B. Kaposin A is a type II membrane protein; Kaposin B is involved in the MAPK signaling pathway |
| vGPCR (Viral G protein-coupled receptor) | Homologue of cellular IL-8 receptor and binds to CXC and CC chemokines. It stimulates MAPK pathway and leads to secretion of VEGF |
| vIL-6 (viral IL-6) | Homologue of cellular IL-6, it supports cell growth and protects the cells from undergoing apoptosis |
| K4, K4.1, and K6 (KSHV open reading frame 4, 4.1, and 6) | Viral homologues of cellular CC chemokines such as RANTES and MIP-1α. They induce signal transduction and enhance angiogenesis |
| vFLIP (viral FLICE-inhibitory protein or ORF71) | Homologue of cellular FLIP, it activates NF-κB pathway and protects cells from apoptosis |
| LANA (viral latency associated nuclear antigen) | Important in the maintenance of viral latency and binds to viral genome and a number of cellular factors, such as p53 |
The KSHV K1 gene is the first open reading frame of the viral genome. It encodes a transmembrane protein with a cytoplasmic domain containing a functional immunoreceptor tyrosine-based activation motif (ITAM) (Lagunoff and Ganem, 1997; Lee et al., 2003). ITAM motifs are known to be involved in signal transduction on ligand-receptor interaction. However, K1 signaling appears to be constitutive and may be responsible for the activation and proliferation of infected B lymphocytes by inducing phosphorylation of several cellular signal transduction proteins (Lagunoff et al., 1999; Lee et al., 1998a,b). The K1 gene was shown to have transforming activities; it transformed mouse cells in vitro and caused tumors in nude mice (Lee et al., 1998b). K1 was also found to be able to replace the transforming gene (STP) of the herpesvirus saimiri and caused immortalization of marmoset T lymphocytes (Lee et al., 1998b). Transgenic mice expressing K1 gene developed malignant plasmacytomas and these cells have elevated levels of NF-κB and other cellular transcription factors, further suggesting that deregulation of normal cellular functions by K1 may lead to the development of B-cell lymphomas (Prakash et al., 2002).
The viral kaposin or open reading frame K-12 has also been found to play a role in transformation. The kaposin gene is expressed during latency but can also be induced on lytic replication (Muralidhar et al., 1998, 2000; Sadler et al., 1999; Wang and Boshoff, 2005). This gene is most abundantly expressed during latency and has a complex translational pattern resulting in three different forms of the kaposin proteins, known as A, B, and C (Sadler et al., 1999). Kaposin A is a type II membrane protein, and it was shown to have transforming activities and can transform cells in vitro; the transformed cells caused tumors in nude mice (Kliche et al., 2001; Muralidhar et al., 1998, 2000). Its transforming activities were linked to its interaction with a guanine nucleotide exchange factor for ARF GTP ase known as cytokesin-1 and a domain known as the LXXLL motif on the protein seems to be important for transformation (Tomkowicz et al., 2005). Kaposin B appears to play a role in cytokine release; it binds to host cell protein kinase, such as mitogen-activated protein kinase (MAPK)-associated protein kinase 2, which plays an important role in the proinflammatory p28 MAPK signaling pathway, resulting in an enhancement of inflammatory cytokine secretions to enhance the development of KS (McCormick and Ganem, 2006). Currently, nothing is known about the function of Kaposin C.
The viral GPCR is a lytic viral gene and is a homologue of the cellular IL-8 receptor except that it is constitutively expressed. It binds to the CXC and CC families of chemokines (Arvanitakis et al., 1997; Cesarman et al., 1996; Gershengorn et al., 1998). KSHV GPCR has been shown to transform cell in vitro and promote immortalization of endothelial cells and tumor formation in the presence of KSHV latent genes, suggesting that both lytic and latent genes are important during the development of KS (Arvanitakis et al., 1997; Bais et al., 2003). The signal transduction property of GPCR is important for its transforming activities. It is known to stimulate the MAPK and PI3K pathways, leading to the stimulation of a large number of cellular genes that could enhance the proliferation of KSHV-infected cells. Activation of vGPCR has been associated with an increase in the secretion of vascular endothelial cell growth factors (VEGF) and VEGF receptors, which leads to an induction of the angiogenic response, to enhance the growth of immortalized KSHV-infected cells both in vitro and in vivo through a paracrine pathway mediated by vGPCR (Montaner et al., 2003; Sodhi et al., 2000; Yang et al., 2000).
In addition of vGPCR, there are other viral genes that have homology to cellular homologue genes, such as the viral IL-6, viral cc-chemokines (vCCLs), and vFLIP. KSHV vIL-6 has both sequence and functional homology to the cellular IL-6, but they differ in their ability to bind to cellular receptors. Cellular IL-6 requires both the α and the pg130 subunits for binding and signaling, whereas vIL-6 requires only the pg130 subunit (Molden et al., 1997). KSHV-infected PEL MCDD cells secrete vIL-6 to support the growth of the infected cells and also protect the cells from the antiviral effects mediated by the interferon pathway (Moore et al., 1996a; Nicholas et al., 1997). Thus, vIL-6 appears not only to have the ability to support the growth of infected cells, but also can protect infected cells from the effects of interferon. In addition to vIL-6, several viral genes, K4, K4.1, and K6, encode viral homologues of the cellular CC chemokines, such as RANTES and MIP-1α (Boshoff et al., 1997). These chemokines can induce signaling transduction, enhance angiogenesis, and contribute to the tumorigenesis process (Nakano et al., 2003). The KSHV FLIP gene ORF71 is a latent viral gene which encodes the FLIP protein and is structurally most homologous to the cellular FLIP. It facilitates lymphoma cell growth by activating NF-κB expression and its signaling pathway, and by conferring the infected cells resistance to apoptosis (Djerbi et al., 1999; Guasparri et al., 2004; Thome et al., 1997). In addition, vFLIP was shown to induce morphological changes in primary endothelial cells to become spindle-like in shape, similar to KS tumor cells (Grossmann et al., 2006). These together with the antiapoptotic functions of vFLIP reflect two features that are known to be the hallmark of KS.
Another KSHV latently expressed protein that may contribute to neoplasm is LANA. The KSHV LANA is a nuclear phosphoprotein that is important for the maintenance of viral latency (Dittmer et al., 1998). It has been shown to bind to the terminal repeat region of the viral genome and tethered the viral episome to the host chromosome so that it can be maintained during cellular mitotic replication and segregation (Ballestas et al., 1999; Cotter et al., 2001). LANA is a multifunctional viral protein and can bind to a number of cellular proteins. It can bind to tumor suppressors p53 and Rb and protect infected cells from apoptosis (Friborg et al., 1999; Radkov et al., 2000). This together with its ability to upregulate β-catenin expression promotes S-phase entry, modulates cell cycle pathways, and contributes to the development of neoplasm (Fujimuro et al., 2003).
IV. AIDS-Associated Lymphomas
As persons with HIV infection survive longer despite significant immunosuppression, more cases of malignancy are likely to appear. Although HIV infects T lymphocytes, AIDS-associated lymphomas are of B lymphoid origin in at least 95% of all cases described. As with other lymphomas, AIDS-associated lymphomas also fall into two broad categories: AIDS-associated Hodgkin’s disease and NHL. AIDS-associated NHLs are primarily encountered in patients with more advanced HIV infection, with a low CD4 count. Although Hodgkin’s disease is included in the HIV-associated lymphomas in the WHO classification, it will not be discussed in this chapter because the relation between HIV infection, AIDS, and Hodgkin’s disease is unclear (Carbone and Gloghini, 2005). Whether HIV infection promotes the development of Hodgkin’s disease or merely modifies its clinical progression is not yet known.
Mechanistic studies have revealed that potential factors contributing to lymphoma development. Three major factors promoting the development of lymphoma are HIV-induced immunosuppression, chronic antigenic stimulation, and cytokine overproduction. These alterations are associated with the development of oligoclonal B-cell expansions. The appearance of lymphomas is characterized by the presence of a monoclonal B-cell population displaying a variety of genetic lesions, including EBV infection, c-myc gene rearrangement, bcl-6 gene rearrangement, ras gene mutations, and p53 mutations/deletions. The number and type of genetic lesions varies according to the anatomic site and histopathology. Thus, it is apparent that more than one pathogenic mechanism is operational in the development and progression of AIDS-associated lymphomas (Carbone and Gloghini, 2005; Epeldegui et al., 2006). This chapter attempts to summarize the potential role of viral etiological factors, especially EBV, on the development of the malignancies.
A. EBV, Its Latency, and Its Role in AIDS-Associated Lymphomas
EBV is a ubiquitous human-herpesvirus that infects about 95% of the adult population worldwide. The majority of primary infections occur in early childhood and are generally asymptomatic. However, when primary infection is delayed until adolescence or adulthood, as often occurs in developed countries, it may cause infectious mononucleosis (IM), which is a self-limiting lymphoproliferative disorder characterized by increased numbers of EBV-infected B cells in peripheral blood and massive oligoclonal expansion of EBV-specific CD8+ T cells.
The biologic hallmark of the EBV-cell interaction is latency. EBV establishes several latencies on infection of target cells. Three types of latency have been described, each having its own distinct pattern of EBV gene expression. Type I latency is exemplified by Burkitt’s lymphoma (BL) tumors in vivo and earlier passages of cultured cell lines derived from BL biopsies. Epstein-Barr Nuclear Antigen 1 (EBNA-1) and small EBV-encoded, nonpolyadenylated nuclear RNAs (EBER-1 and -2) are expressed in this form of latency. Type II latency is exemplified by NPC and Hodgkin’s disease. EBNA-1, latent membrane protein 1 (LMP-1), LMP2A, and LMP2B proteins, as well as EBERs, are expressed in type II latency. EBV transforms adult primary B cells into continually growing lymphoblastoid cell lines (LCLs) and concomitantly establishes type III latency in vitro. Nine viral proteins are expressed, including six nuclear proteins (EBNA-1, EBNA-2, EBNA-3A, EBNA-3B, EBNA-3C, and EBNA-LP) and three integral membrane proteins (LMP-1, LMP-2A, and LMP-2B) plus EBERs (Kieff, 1996; Rickinson and Kieff, 1996). Extensive mechanistic studies on EBV transformation have identified several key viral genes that contribute to the viral transformation processes. They are LMP-1, LMP-2, EBNA-1, and EBNA-2.
B. Latent Membrane Protein 1 (LMP-1)
The role of LMP-1 in EBV transformation of primary B lymphocytes is well established. LMP-1 was initially identified as a viral oncoprotein on the basis of its ability to transform rodent cells. Fibroblasts constitutively expressing LMP-1 demonstrate reduced serum requirements, increased growth in soft agar, loss of contact inhibition, and tumorigenic potential in nude mice (Dawson et al., 1990; Fahraeus et al., 1990; Wang et al., 1985). Moreover, expression of LMP-1 as a transgene in mice under the control of the immunoglobulin promoter/enhancer results in increased frequency of B-cell lymphomas, indicating that this viral protein has oncogenic properties in vivo (Kulwichit Raab-Traub). In viral transformation assays with primary B cells, deletion of LMP-1 prevents the transformation of primary B cells (Izumi et al., 1997; Kaye et al., 1993), and inhibition of LMP-1 expression in EBV-transformed cells reverts the transformed phenotypes (Kilger et al., 1998). Thus, LMP-1 is required for EBV transformation of primary B cells in vitro in tissue culture system.
LMP-1 expression alone modulates cellular gene expression that is responsible for phenotypic and functional changes associated with EBV latency. These changes include the upregulation of adhesion molecules (LFA-1, ICAM-1, LFA-3), B-cell activation markers (CD23, CD30, CD40, CD71), transcriptional factors [signal transducer and activator of transcription 1 (STAT-1), -2, IRF-7], and antiapoptotic genes (Bcl-2, BclxL, Mcl1, A20). Thus, LMP-1 appears to be a central effector of altered cell growth, survival, adhesive, invasive, and even antiviral potential in EBV-infected cells (Fries et al., 1996; Miller et al., 1995; Wang et al., 1985, 1990; Yoshizaki et al., 1998).
Extensive studies have led to some insight into the molecular mechanisms underlying the function of LMP-1. LMP-1 is an integral membrane protein with six transmembrane-spanning domains and a long C-terminal domain located in the cytoplasm (Kieff, 1996; Liebowitz et al., 1986). LMP-1 acts as a constitutively active, receptor-like molecule that does not need the binding of a ligand (Gires et al., 1997). The six transmembrane domains mediate oligomerization of LMP-1 molecules in the plasma membrane, a prerequisite for LMP-1 function (Floettmann and Rowe, 1997; Gires et al., 1997). Two regions in the C-terminus of LMP-1 have been shown to initiate signaling processes, the C-terminal activator regions 1 (CTAR-1, amino acids 194-231) and 2 (CTAR-2, amino acids 332-386) (Huen et al., 1995; Mitchell and Sugden, 1995). In a more refined analysis, several kinds of functional domains have been identified. The PXQXT domain is located within the CTAR-1 and is involved in the interaction with tumor necrosis factor receptor (TNFR)-associated factors (TRAFs), and the binding of TRAFs to LMP-1 results in the induction of the NF-κB and AP-1 transcription factors. It is thus apparent that LMP-1 shares functional properties with members of the TNF-receptor superfamily, particularly CD40. Moreover, LMP-1 can partially restore the wildtype phenotype of mice deficient in CD40 (Devergne et al., 1996, 1998; Miller et al., 1997, 1998; Sandberg et al., 1997). LMP-1 also interacts with TNFR-associated death domain protein (TRADD) and receptor-interacting protein (RIP) at the C terminal (Devergne et al., 1998; Floettmann and Rowe, 1997; Izumi et al., 1997, 1999; Izumi and Kieff, 1997; Kaye et al., 1996). Interaction with these two molecules contributes the majority of the NF-κB activity induced by LMP-1. Also, c-Jun N-terminal kinase (JNK) is activated by CTAR-2. The domain for activation is mapped to most C-terminal amino acids and apparently overlaps the TRADD interaction domain. However, whether TRADD and TRAF2 are involved in the activation of JNK is disputed (Eliopoulos and Young, 1998; Eliopoulos et al., 1999; Kilger et al., 1998). In addition, two janus kinase 3 (JAK3) binding sites have been identified between CTAR-1 and CTAR-2. JAK3 binding to the sites is responsible for the activation of STAT-1(Gires et al., 1999). However, some other experimental evidence suggests an alternative mechanism (Brennan et al., 2001; Higuchi et al., 2002; Zhang et al., 2004b). In summary, the hijacking of these cellular signaling pathways by LMP-1 is likely to contribute to the pathogenesis of most EBV-associated disorders through the simultaneous or sequential activation of signals involved in the promotion of cell activation, growth, and survival.
C. Latent Membrane Protein 2
The LMP-2 protein contains multiple membrane spanning domains and cytoplasmic N- and C-terminal domains and forms aggregates in the membrane of EBV-infected B cells. The N-terminal domain can bind to the tyrosine kinases Lyn and Syk through their SH2 domains (Longnecker et al., 2000). These kinases are recruited to the BCR following antigen cross-linking, and their subsequent activation stimulates downstream events resulting in B-cell differentiation and proliferation. LMP-2A may work as a decoy protein sequestering Lyn and Syk to inhibit BCR signaling, which make LMP-2 an inhibitor of EBV lytic replication induced by BCR ligation (Longnecker, 2000; Longnecker et al., 2000). The property of LMP-2 may play a major role in mediating EBV persistence in vivo.
Unlike LMP-1 and EBNA-2, the LMP-2 protein is not essential for B-cell transformation in vitro. Nevertheless, the constant expression of LMP-2 in EBV-carrying memory B cells from healthy individuals suggests that LMP-2 probably plays an important role in viral persistence. LMP-2 in transgenic mice model has shown that LMP-2 provides survival signals that allow immature B cells to progress through developmental checkpoints and prevent cell death. This property may be related to the ability of LMP-2A to activate the serine-threonine kinase Akt. Akt is a multifunctional mediator of phosphatidylinositol 3-kinase (PI3-K) activity. Activation of the pathway results in the constitutive delivery of an antiapoptotic signal. Akt is also involved in the control of B-cell proliferation because chemical inhibition of PI3-K induced growth arrest of EBV-transformed B cells (Brennan et al., 2002).
D. Epstein-Barr Nuclear Antigen
The EBNA-1 protein is expressed in all EBV latency states and all EBV-associated tumors. The only exception is that EBNA-1 is hardly detectable in the circulating EBV-infected memory B cells. This alone suggests that the biologic properties of this protein are critical for EBV-mediated transformation (Kieff, 1996). EBV establishes itself efficiently in infected B lymphocytes, where it exists as 165 kb, circular episome which is duplicated once per cell cycle. Remarkably only EBNA-1 protein is required for the synthesis and partitioning of the viral episomes. EBNA-1 binds to two regions of the viral origin of replication (OriP), referred to as the family of repeats (FR) and the dyad symmetry (DS) element. FR is essential for episome maintenance, while DS is required for initiation of OriP-dependent DNA replication. EBNA-1 is also a transcriptional regulator that modulates the activity of the viral promoters: Wp and Cp and its own latent promoter Qp. Moreover, EBNA-1 is essential to drive transcription of EBV’s transforming genes after infection of primary B lymphocytes (Altmann et al., 2006). In addition, EBNA-1 can inhibit apoptosis in B cells that likely contributes to the persistence of EBV-infected cells and survival of EBV-transformed cells in vivo.
E. EBNA-2
The EBNA-2 protein is localized in the nucleus and is one of the first viral proteins expressed during EBV infection of primary B lymphocytes. In cooperation with EBNA-LP (also known as EBNA-5), EBNA-2 induces the transition of resting B cells from G0 to G1. EBNA-2 is a key regulator of viral gene expression, being able to stimulate transcription from the major latency BamHI-C promoter, which directs expression of all the EBNA genes, and the promoters of LMP-1 and LMP-2. In addition, EBNA-2 modulates the transcriptional activity of some cellular genes. Cellular C-fgr, c-myc, CD21, CD23, and EBI1/BLR2 are upregulated whereas the immunoglobulin heavy chain genes are repressed in lymphocytes. There is no evidence that EBNA-2 binds to DNA directly. Rather, its transcriptional activity is mainly mediated by its interaction with the DNA-binding cellular protein RBP-Jk (also called RBP-J, CBF1, KBF2, or CSL). EBNA-2 is essential for EBV-induced immortalization of B lymphocytes and complex formation with RBP-Jk is crucial for such activity. RBP-Jk is expressed ubiquitously and is an important component of the Notch signaling pathway that is involved in the regulation of lymphoid development. Notch proteins are a family of transmembrane receptors that on ligand binding undergo proteolytic cleavage of their intracellular domain (Notch1 IC). The cleaved and released Notch1 IC fragment is transported to the nucleus where it interacts with RBP-Jk and modulates the activity of target promoters. Although Notch1 IC and ENBA-2 share the ability to transactivate genes by interacting with RBP-Jk, the set of promoters regulated by Notch1 IC and EBNA-2 is overlapping but not identical.
Generally, transformation of a cell requires multiple molecular events (Cole and McMahon, 1999; Kelekar and Cole, 1986; Kohl and Ruley, 1987; Ralston, 1991; Shalloway et al., 1987; Weinberg, 1985, 1989). Several viral genes, such as LMP-1 and EBNA-2, are required for the transformation of primary B cells in vitro and are believed to drive EBV transformation process in vivo. EBV contributes to the cellular transformation processes through the activity of viral proteins that act cooperatively to modify cellular gene expression that involved in cell proliferation, apoptosis, angiogenesis, immune regulation, and signal transduction (Cahir-McFarland et al., 2000; Chen et al., 2003; Fries et al., 1996; Henderson et al., 1991; Miller et al., 1995; Wang et al., 1985, 1990; Yoshizaki et al., 1998; Zhang and Pagano, 1999; Zhang et al., 2004a,c).
V. AIDS-Associated NHL
AIDS-associated NHL is generally divided into three subtypes: PCNSL, PEL (“body cavity”), and systemic NHL (Knowles, 2003). The vast majority of AIDS-associated NHL is clinically aggressive B-cell-derived neoplasms. Approximately 80% arise systemically (nodal and/or extranodal), and the remaining 15-20% arise as PCNSL. A small proportion is BCBLs (Knowles, 2003). EBV apparently contributes to the development of these tumors in various fashions.
A. Primary Central Nervous System Lymphoma
PCNSL is a form of NHL arising within and confined to the CNS. It was first described by Bailey in 1929 as a perithelial sarcoma (Bailey, 1929). Subsequent classifications have included reticulum cell sarcoma and microglioma. Improvements in histopathology and immunohistochemical techniques definitively established the lymphoid nature of PCNSL. PCNSL accounts for up to 15% of NHLs in HIV-infected patients compared to only 1% of NHLs in the general population. The reported incidence of PCNSL in HIV-infected patients is 2-6% (at least 1000 times higher than in the general population) and has been as high as 10% in autopsy series. Although CNS involvement also occurs in AIDS-associated systemic lymphoma in the form of secondary spread of the tumor to the meninges, the disease is limited to the CNS in PCNSL (Cheung, 2004; Cingolani et al., 2005; Eichler and Batchelor, 2006; Gates and Kaplan, 2002; Sparano, 2003). Prior to the introduction of HAART, the incidence of PCNSL in the HIV-infected population was continuing to rise. However, the impact of these new drug regimens on the CD4 count may result in a decline in PCNSL, as the susceptibility to PCNSL is inversely proportional to the CD4 count (Sparano et al., 1999). In normal individuals, a small number of circulating B cells enter the CNS, and may do so in increased numbers as HIV infection advances (Cingolani et al., 2005; Ivers et al., 2004). EBV establishes latent, life-long infection in over 90% of adults. During the course of HIV infection, EBV-specific T cells progressively lose the capacity to produce interferon-gamma in response to EBV peptides. In addition, EBV-positive B lymphocytes occur more frequently in the CNS of HIV-infected individuals than in normal brains, which may set up a stage for EBV transformation of these infected cells.
EBV appears to play a major pathogenetic role in AIDS-associated PCNSL: (1) EBV genome within tumors is present in more than 95% of AIDS patients, but in only 0-20% (probably <5%) of immunocompetent patients. (2) More than half of AIDS PCNSL examined so far expressed at least EBNA-2, LMPs, and EBERs, a pattern referred to as type III latency and closely resembling that seen in transformation of primary B lymphocytes in vitro EBV (Cingolani et al., 2005; Ivers et al., 2004). Expression of type III latency genes leads to a variety of cellular effects, including upregulation of the genes that are involved in transformation, such Bcl-2 and IRF-7, and inactivation of the p53 and Rb tumor suppressor gene products. It is believed that the EBV triggers certain PCNSL in vivo in a process similar to transformation processes of primary B cells in vitro (Pagano, 1999).
B. Primary Effusion Lymphoma
PELs, also known as BCBL, are B-cell NHLs and most frequently occur in AIDS patients as lymphomatous effusions in the serous cavities without a detectable solid tumor mass. In the setting of AIDS, the clinical course for most of these lymphomas is extremely aggressive, with a mean survival from diagnosis of 5-7 months (Nador et al., 1996). While PELs are almost universally KSHV positive, the majority of PELs have concomitant EBV infection (reviewed in Dourmishev et al., 2003; Drexler et al., 1998; Moore and Chang, 2001). EBV apparently establishes levels type II latency in PELs with low of LMP-1 expression (Callahan et al., 1999; Fassone et al., 2000; Horenstein et al., 1997; Lacoste et al., 2000).
Both KSHV and EBV are oncogenic herpesviruses. It is thus interesting to examine if there are any interactions between the two viruses in PELs. Comparing to KSHV-only PELs, coinfection with EBV enhances the tumorigenecity of the dually infected PELs in severe combined immunodeficiency (SCID) mice model (Trivedi et al., 2004). The mechanism of the enhancement is currently unknown. However, LMP-1 might be involved in the enhancement because LMP-1 is expressed and its expression may be enhanced by both KSHV latent gene (LANA) and lytic gene (K-RTA). Although expression of LMP-1 at least in some of the PEL specimens strongly suggests the contribution of EBV to the development of the tumor, this enhancement was not apparent in clinical settings, possibly due to the fact that patients with PEL are usually at advanced HIV-infection stage.
At molecular levels, unique sets of cellular genes are expressed in dually infected, but not singly KSHV-infected PELs (Fan et al., 2005). KSHV reduces the expression of EBV EBNA-1 and represses EBV EBNA-2 activation (Krithivas et al., 2000). EBV inhibits KSHV lytic replication, in part, because of a regulatory loop in which KSHV lytic gene induces EBV LMP-1, and LMP-1, in turn, inhibits the lytic gene expression programs of KSHV (Xu et al., 2007). Like EBV EBNA-2, KSHV replication and transcriptional activator (K-RTA) bind to RBP-Jk (Liang et al., 2002), a key cellular target of the EBV latent transforming program (Zimber-Strobl and Strobl, 2001). Also, KSHV induces the expression of CD21, the cellular receptor for EBV, and thus facilitates EBV infection (Chang et al., 2005). All this data suggests that coordinated cellular transformation by the two viruses is a possibility. However, how these two viruses interact and affect each other and the pathobiology of PELs remains to be determined.
C. Systemic AIDS-Associated NHL
Systemic AIDS-associated NHLs are aggressive B-cell lymphomas of high or intermediate grade and heterogeneous in nature. Approximately one-third can be classified as small noncleaved cell lymphomas, which are Burkitt or Burkitt-like lymphomas. The remaining two-thirds of the lymphomas are diffuse large cell lymphomas, which are immunoblastic lymphomas or large noncleaved cell lymphomas (Brockmeyer and Barthel, 1998). EBV infection and c-myc oncogene rearrangements are the two well-established factors in the pathogenesis of the systemic NHL. The diffuse large cell lymphomas frequently express EBV latency type III antigens including EBNA-2 and LMP-1 and -2, which have transforming activity in vitro are well established. EBV establishes type I latency expressing only EBNA-1 in BLs. However, the EBV genome can be detected in only 60% of the diffuse large cell lymphomas, and in around 30% of the AIDS-associated BLs. Because EBV is less frequently detected in systemic and lymphomas, and the increasing incidence type of this of cancer in HIV-infected patients, some additional common latent or chronic viral infections may be involved in the development of these tumors (Mueller, 1999; Shibata et al., 1993). In the setting of underlying HIV infection, systemic NHL truly behaves as an opportunistic neoplasm, overwhelming those immune mechanisms that may normally attempt to keep the cancer in check.
VI. HPV-Associated Cancers
A. Types of HPV-Induced Cancers
HPVs infect the stratified epithelia of skin or mucosa, where they cause benign warts. Of the 200 different types of HPVs (Cates and Dallabetta, 1999), the most common HPVs (types 2 and 4) are those that cause warts on the hands and feet of affected individuals (Howley, 1996). Anogenital tract HPVs, of which ∼40 have been identified, are divided into those which confer a “low risk” (types 6, 11, 42) or a “high risk” (types 16, 18, 31) for cervical cancer (Howley, 1996; Sakai et al., 1996; zur Hausen, 1999, 2000). Studies performed by Harold Zur Hausen’s laboratory provided the first definitive evidence that HPVs were present in genital cancers (Bosch et al., 1991; Durst et al., 1983; Gissmann et al., 1984; Schwarz et al., 1985; zur Hausen, 1999, 2000; zur Hausen et al., 1975, 1981). After more than 20 years of work, HPVs are now recognized as a necessary cause in 95% of invasive cervical cancers worldwide (Walboomers et al., 1999). Approximately 20 million US adults are infected with genital HPVs and there are 5.5 million new infections each year, representing a major public health concern (Cates and Dallabetta, 1999). In human cervical cancer cells, high-risk papillomavirus DNA is most often found integrated into the host chromosomes (Londesborough et al., 1996; Schwarz et al., 1985; Yee et al., 1985b).
HPVs are most commonly associated with cervical cancer, although, it is now known that many cancers are induced by HPV, including penile, anal, oral, and conjunctival cancers (Durst et al., 1983; Koutsky, 1997; Newton et al., 2002; Syrjanen, 2003; Waddell et al., 1996). High-risk HPVs have also been implicated recently in ∼30% of oral cancers (Gillison et al., 2000). In fact, HPVs are responsible for cancers in the tonsils, the palate, gums, tongue, and the larynx (Aaltonen et al., 2005; de Villiers et al., 1986; El-Mofty and Lu, 2003; Lopez Amado et al., 1996; Milde-Langosch et al., 1989; Mineta et al., 1998; Sinclair et al., 2005; Syrjanen, 2005; Yoshpe, 1995). High-risk HPVs have been further implicated in upper respiratory tract and lung cancers (Cheah and Looi, 1998; Clarke et al., 1991; de Villiers et al., 1986). Furthermore, evidence suggests that some digestive cancers are also HPV positive (Milde-Langosch et al., 1989; Nakano, 1994). HPVs are some of the most ubiquitous and stable viruses in nature, thus, it is not surprising that multiple tissues are targets of HPV-induced tumorigenesis.
Penile cancers are much less common than cancers of the cervix, for reasons that are not entirely clear (Gloeckler Ries et al., 2003). Despite the fact that men seldom show clinical signs, it is likely that many could be persistently infected and that the progression to penile cancer occurs under immunosuppressive conditions. Cancers of the vulva and vagina are also relatively rare compared to cervical cancers. The reason for these differences in incidence of cancers in different tissues is related to the cell-type infected. The cells of the cervical transformation zone at the squamous and columnar cell junction are the most susceptible to HPV-induced cell transformation (Jastreboff and Cymet, 2002; Jordan and Monaghan, 2004; Ponten and Guo, 1998). An analogous cell type apparently does not exist in men. However, despite lack of studies on the subject, it would be assumed that men are transmitters of HPVs (Baldwin et al., 2003; Dunne et al., 2006; Giuliano et al., 1999). Predictably, the incidence of penile cancers increases dramatically in individuals who are HIV positive (Aboulafia and Gibbons, 2001; Arany and Tyring, 1998; Laurence, 2003; Palefsky and Barrasso, 1996; Sirera et al., 2006; von Krogh et al., 1995), which likely reflects the overall increase incidence of HPV infections detected in women.
Anal cancer is a relatively rare disease, ∼80% of which are HPV positive (Gloeckler Ries et al., 2003; Hankey et al., 1999; Zippin and Lum, 1993). Anal cancer amounts to about 4% of all digestive tract cancers. The incidence of anal cancers in women is slightly higher than in men (Gloeckler Ries et al., 2003; Hankey et al., 1999; Zippin and Lum, 1993), which could be indicative of the overall higher rate of HPV infection among women. It appears that the incidence of anal cancers are rising in the past 10 years, reflecting changes in sexual behavior (http://seer.cancer.gov/) (Gloeckler Ries et al., 2003). The highest level of risk for anal cancer caused by HPV is associated with MSM (Piketty et al., 2004). The rate of anal cancer among HIV-negative heterosexual men is ∼1.3/100,000, while the rate of anal cancers in HIV-negative MSM is 35/100,000. Among MSM who are HIV positive, the rate of anal cancer is twofold higher than HIV-negative MSM (Fakhry and Gillison, 2006).
HPV infections of the conjunctiva of the eye are more common than previously appreciated, although resulting HPV-induced tumors of the conjunctiva are very rare (Mincione et al., 1992, 2006; Reszec and Sulkowski, 2005; Tabrizi et al., 1997; Waddell et al., 2003). Though the incidence of eye or eye-orbit tumors in the United States is extremely low (<1/100,000) (Gloeckler Ries et al., 2003), in African countries, conjunctival tumors are more common and are undoubtedly influenced by nutrition, additional disease burdens, but most obviously by the relatively high impact of HIV in Africa (Ateenyi-Agaba et al., 2006; Frisch et al., 2000; Goedert, 2000; Newton et al., 2002; Waddell et al., 1996, 2003).
Given the wide range of cancers caused by HPV, the recent development of an HPV vaccine provides some hope for providing protection against many of the cancers described earlier. However, vaccine efforts have concentrated on only two high-risk strains (HPV16 and HPV18) (Mao et al., 2006; Villa et al., 2005), while there are at least 15 known high-risk strains. We also have evidence that multiple regional HPV variants exist, particularly in Africa, for which, the extent of protection by the current vaccine is unknown (Calleja-Macias et al., 2004; Chan et al., 1992; Ong et al., 1993; Touze et al., 1998; Williamson et al., 1994; Xi et al., 1998). Furthermore, the distribution of high-risk HPVs varies from country to country (Calleja-Macias et al., 2004; Chan et al., 1992; De Vuyst et al., 2003; Munoz et al., 2004; Ong et al., 1993; Williamson et al., 1994; Xi et al., 1998). Therefore, it is important to take a long-range view of prevention of HPV-induced cancers by use of vaccination strategies that take into account variants.
VII. HPV—The Causative Agent
A. Papillomavirus Genome Structure
HPVs are a family of small, nonenveloped, double-strand DNA viruses that establish a persistent infection, which may remain subclinical in the skin or genital tract for up to 10-20 years, but can often cause acute warts. Papillomavirus genomes are small circular DNA of 8 kb, which encodes eight major proteins. As is typical for DNA viruses, the immediate early genes (E6 and E7) are involved with taking over the cell cycle (Fig. 1) (Howley, 1996; Howley et al., 1989; zur Hausen, 1999, 2000). Unlike more complex viruses like herpesviruses, HPVs use the strategy of replicating at low copy, and thus, do not carry their own polymerase gene. Instead, gene products encoded by E1 and E2 recruit cellular polymerase α to the viral origin (Frattini and Laimins, 1994; Howley, 1996; Howley et al., 1989; Sedman and Stenlund, 1995). The genome is simply organized into early and late genes, with only two capsid genes, L1 and L2.
FIGURE 1.
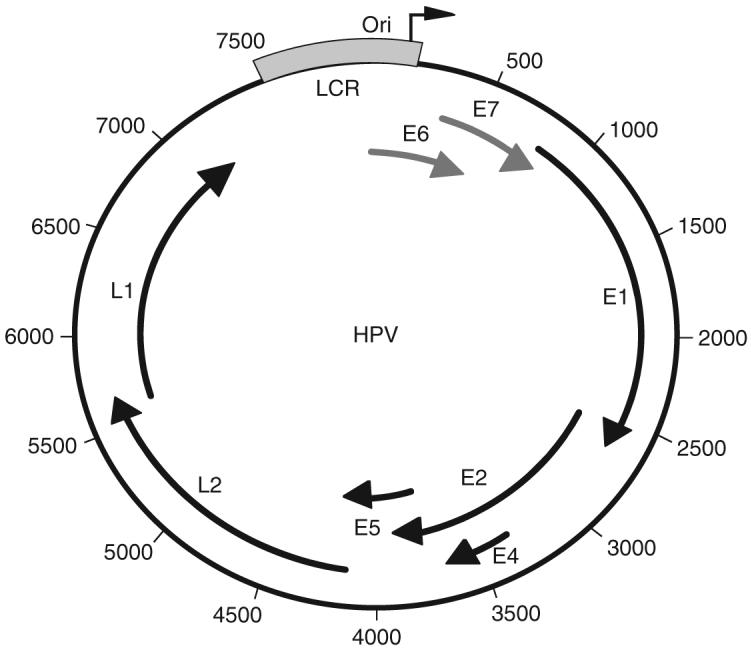
The HPV genome is a 7.9 kb double-stranded circular genome. The genome is controlled by a single keratin-dependent promoter element; the long control region (LCR; in blue). At the 3′ end of the LCR is the origin of replication (nucleotide position 1). E6, E7 (red), and E5 are viral oncogenes; E1 and E2 early genes encode replication proteins. The E4 ORF is actually expressed early and late in the viral life cycle. The late genes, L1 and L2, are the major and minor capsid genes, respectively. The viral protein functions are detailed in Table I.
The functions of the viral proteins are well established and are summarized below (Table II).
TABLE II.
| Designation | Function |
|---|---|
| E6 | Viral oncoprotein; functions by binding and degrading p53 tumor suppressor via activity of E6AP, ubiquitin ligase, resulting ubiquitin pathway-mediated degradation of p53, preventing apoptosis |
| E7 | Nuclear oncoprotein; functions by binding and degrading of pRb family tumor suppressor products. This causes release of E2F that induces expression of S-phase-related genes |
| E1 | An ATP-dependent DNA helicase; has double and single strand DNA-binding activities. Required for viral DNA replication; interacts with E2 protein and with DNA polymerase α. Significant structural homology to SV40 large Tantigen (Clertant and Seif, 1984; Mansky et al., 1997; Seif, 1984). Sequence-specific DNA binding by E1 is mediated by the papillomavirus E2 protein; association of E1 with E2 enhances the affinity of E1 for DNA (Frattini and Laimins, 1994; Sedman and Stenlund, 1995) |
| E2 | Binds a 12 base pair palindromic sequence: ACCG(A)4CGGT in the Ori and functions as a transcriptional activator; binds and recruits the E1 protein that stimulates initiation of DNA replication; E2 also functions in HPV genome maintenance by tethering newly synthesized DNAs to chromosomes during mitosis, which allows equal partitioning to daughter cells |
| E4 | Expressed as a late protein; blocks cell cycle at G2/M; induces collapse of cytokeratin network; possibly promoting virus release; poorly conserved across HPV types |
| E5 | A viral oncoprotein; binds EGF receptor; able to transform rodent cells in culture; membrane associated; functions include binding to β receptor for platelet-derived growth factor |
| L1 | Major capsid protein |
| L2 | Minor capsid protein |
B. The HPV Life Cycle
HPVs initiate their life cycle by gaining access to basal keratinocytes of the stratified epithelium; either skin or mucosa through a site of wounding (Fig. 2). Papillomavirus DNA replication is closely coupled to the process of keratinocyte differentiation in infected squamous epithelium (reviewed in Chow and Broker, 1994). In the basal and parabasal epithelial cells, HPV is maintained as a low-copy number episome (5-50 copies per cell) that under-goes regulated DNA replication under the control of viral and host proteins. As infected keratinocytes differentiate and enter the stratum spinosum layer of the epithelium, there is a coincident increase in concentration of E1 and E2 proteins (for review, see Shaw and Howley, 2001). Induction of vegetative replication is consistent with a mode switch from theta to rolling-circle replication mechanisms (Flores and Lambert, 1997). As a consequence of this rolling-circle DNA replication mechanism, multiple rounds of viral DNA synthesis occurs in a given S-phase of the host keratinocyte (Hoffmann et al., 2006), and an increase in copy number up to between 100 and 1000 copies per cell. Vegetative HPV DNA replication requires the virus-encoded E1 (a DNA helicase per ATPase) and E2 (a transcriptional trans-modulator) proteins, and initiates at the E1 binding site palindrome near the 5′ end of the viral long control region (Kuo et al., 1994).
FIGURE 2.

The HPV life cycle. Virions enter the stratified epithelium through a site of wounding, where they gain access to the mitotically active basal-layer keratinocytes. During the maintenance phase, expression of E6, E7, and E5 induces cell proliferation, and the viral genome is replicated extrachromosomally at low-copy number (5-50 copies per cell). As the cells differentiate, the expression level of E1, E2, and E4 increases in the spinous layer. A transition from theta to rolling-circle replication results in an increase in copy number up to 100-1000 copies per cell. Postamplification, high levels of L1 and L2 capsid genes are expressed and capsid assembly occurs in the granular and squamous layers of the stratified epithelium. Progeny virus is released by desquamation.
C. The HPV Capsid and the Vaccine
Capsid assembly of HPVs occurs in the more terminally differentiated layers of the stratified epithelium (Fig. 2). HPVs have icosahedral capsids arranged in a T = 7d lattice (Baker et al., 1991). The capsids are made up of 360 L1 molecules organized into 72 pentamers. Disulfide bond interactions between L1 molecules are important in particle assembly and disassembly (Li et al., 1998). There are 12 L2 minor capsid proteins that are associated with the inner surface of the L1 pentamers (Belnap et al., 1996). High resolution cryoelectron microscopic structures of bovine papillomavirus (BPV) have been achieved (Baker et al., 1991); less is known about HPV structure, assembly, and uncoating. L1 expression is sufficient to allow self-assembly of virus-like particles (VLPs) in the absence of other viral components (Casini et al., 2004). However, L2, when coexpressed with L1, intercalates into L1 VLPs and appears to be required for virion infectivity (Kawana et al., 2001; Stauffer et al., 1998). Though the precise role of L2 during infection is not clear, it may nucleate L1-pentamer formation. The simple, nonenveloped icosahedral HPV virions lend themselves to vaccine development. The recently developed quadrivalent vaccine targets two high-risk HPVs (16 and 18) and two low-risk HPVs (6 and 11). However, there is little evidence of significant cross-protection against the other 14 oncogenic HPVs. Furthermore, there are at least 40 genital HPVs. Thus, the development of vaccines that have a wider cross-protection or tailoring HPV vaccines for different regions of the world will become necessary.
D. Epidemiology of HPV and HIV/AIDS
Epidemiological evidence gathered over several years has determined that 15-20 of the 40 of the mucosal HPV types are associated with a higher risk of progression to cervical cancer (24, 29). The frequency of individual high-risk HPV types worldwide has been shown to vary in respect to major global regions such as Asia, Europe, North America, South America, and sub-Saharan Africa (5, 14, 23). The rate of genital HPVs in the United States, as detected by PCR of the L1 region, is ∼39.2% (Peyton et al., 2001). Estimates of the rates of HPV in Africa vary from 14% to 60% depending on the country, the coincident STDs, and the methods of detection (Czegledy et al., 1992; Gravitt et al., 2002; Hassen et al., 2003; Langley et al., 1996; Mayaud et al., 2001; Motti et al., 1996; Nzila et al., 1991; O’Farrell et al., 1989; Ong et al., 1993; Serwadda et al., 1999; St. Louis et al., 1993; Thomas et al., 2004; Waddell et al., 1996; Williamson et al., 2002). The rate of HPV infections in HIV-positive patients in Zambia are very high, though rather little data is available (Mosunjac et al., 2003; Patil et al., 1995). As a whole, sub-Saharan Africa has the among the highest rates of cervical cancer in the world (Bailie et al., 1996; Clarke and Chetty, 2002; Langley et al., 1996; ter Meulen et al., 1992; Williamson et al., 2002). The distribution of oncogenic HPVs in Africa differs from the United States and Europe. For example, studies done by Nubia Munoz have pointed out that in Nigeria, the most prevalent oncogenic HPV is HPV35, not HPV16, which is the most common in the United States and Europe (Thomas et al., 2004). Several studies have described HPV variants unique to Africa (Calleja-Macias et al., 2004; Chan et al., 1992; Ong et al., 1993; Williamson et al., 1994). This points to a need to further investigate HPV variants in terms of HPV pathogenesis and a potential need to expand the selection of vaccine targets.
The rates of HIV infection in urban area along a contiguous stretch from Uganda to Botswana and South Africa have continued to climb in recent years (Morison, 2001). Several studies have addressed HIV-related malignancies associated with HHV-8 (KSHV) and EBV, which are associated with increased morbidity (Ablashi et al., 1999; Contreras et al., 1997; Lazzi et al., 1998; Parkin et al., 2000; Sapp et al., 2001). Despite frequency variation, HPV16 infection has been shown to be more prevalent than any other high-risk HPV type in most regions of the world. However, HIV-positive populations have a much higher rate of HPV16 positive tumors than most HIV-negative populations (1, 2, 9, 18, 28). Thus, the incidence of high-risk HPV-malignancies is amplified by HIV immune suppression.
It is clear that impaired cell-mediated and humoral immunity influences the advancement of high-risk HPVs in HIV-positive individuals. Since the pool of memory and effector T cells can be dramatically shifted in HIV-positive individuals (even those who do not have AIDS), it would be expected that a certain degree of derepression of HPV replication would occur as well as a lack of adequate surveillance preneoplastic lesions. Several studies have shown a strong and consistent association between HIV and HPV coinfection and the development of cervical intraepithelial neoplasia (CIN) and genital cancer (7, 11, 13, 15). There is evidence to show that HIV-positive women have a significantly higher rate of CIN than their counterparts and are more likely to progress to invasive carcinoma than HIV-negative women (4, 12, 20). A recent study in Brazil has shown that a very high proportion of HIV-infected women is infected with HPV and often carries multiple HPV genotypes (15).
A relationship between the HIV and HPV pathogenesis has been investigated by several studies (Durante et al., 2003; Heard et al., 2004; Klencke and Palefsky, 2003; Massad et al., 2004; Palefsky, 1991, 2003; Piketty et al., 2003, 2004; Silverberg et al., 2002; Strickler et al., 2003; Williams et al., 1994). For example, a study by Silverberg et al. (2002) found that HIV-seropositive women were 3.2-fold more likely to present with genital warts than HIV-seronegative women. Malignancies as complications are an increasing cause of morbidity of HIV-infected individuals (Patil et al., 1995; Thomas, 2001). The EBV-induced malignancy, NHL, occurs at a rate of 2.9% in AIDS patients, ∼60 times the average in non-AIDS patients (Beral et al., 1991). Occurrences of AIDS-related KS still occur at elevated levels, but recent advances in detection and stage analysis has improved the prognosis for HIV-positive patients (Quinlivan et al., 2002). Likewise, an improved understanding of the HPV disease process, as it relates to HIV, is essential, since antiretroviral therapy in HIV-positive individuals has not been shown to effectively reverse HPV-related disease.
The association of malignancies, such as NHL and KS, has been recognized since the beginning of the HIV epidemic, and KS is the neoplasm most commonly found in people infected with HIV. These neoplasms are responsible for extensive morbidity and mortality. In Zambia and other sub-Saharan nations, cervical cancer is the most common cancer (Baay et al., 2004; Hawes et al., 2003; Xi et al., 1998, 2003). Although in Africa, public education campaigns about STDs and condoms have been instituted in urban areas, there has been little success in poorer rural areas (Agha and Kusanthan, 2003). Thus, combined with endemic HIV, a high prevalence of high-risk HPVs presents a great risk for progression of dysplasias to cancer.
Infection by high-risk HPVs, especially HPV16, can induce warts; low-grade dysplasias, CIN, CIN 1 and CIN 2 designations are reversible forms of precancerous lesions (Fig. 3). Integration of the HPV genome can lead to an increase of expression of E6 and E7 resulting in progression to CIN 3, which is irreversible. Accumulation of mutations in cellular genes results in permanent changes in cell character leading to invasive carcinoma in situ. Progression from a benign cervical lesion to invasive cervical cancer usually occurs years after infection.
FIGURE 3.

Progression from a benign cervical lesion to invasive cervical cancer. In the diagram, HPV-positive cells are depicted by yellow nuclei. Infection by oncogenic HPV types, especially HPV16, can cause formation of a benign wart, low or high-grade dysplasia- r. CIN 1 and CIN 2 designations are reversible forms of precancerous lesions and CIN 3 is irreversible. Carcinoma in situ occurs many years after an infection. This results from the effects of HPV genes, particularly those encoding E6 and E7, which are the two viral oncoproteins that are preferentially retained and expressed in cervical cancers by integration of the viral DNA into the host genome.
Cervical cancer and precancerous lesions (CIN) are now the most common cancer-related affliction affecting women in sub-Saharan Africa and other developing countries in the world. The rates of cervical cancer in Africa are fourfold higher in than in North America and Europe.
E. The Mechanism of HPV-Induced Transformation and Cancer Progression
High-risk HPVs (types 16, 18, and 31 for example) are often found integrated into the host genome (Yee et al., 1985a). The integrated state of the viral genome is not supportive of the viral life cycle, but can confer a growth advantage to cells due to increased expression of E6 and E7 (Jeon et al., 1995). The common feature in cancers is the expression of E6 and E7 genes which functionally inactivate p53 and Rb, respectively (Durst et al., 1987; Howley et al., 1989; Munger et al., 1989) (Fig. 4). In oncogenic HPV strains, E6 and E7 oncoproteins can block the negative growth signaling pathways of the cell via interactions with p53 and pRB tumor suppressor proteins. As a result, high-risk HPV-infected cells proliferation become disregulated and then, transformation develops. The full-length HPV E6 genes encode a 160-amino acids protein, which contains two domains including zinc binding Cys-X-X-Cys motifs. High-risk HPV E6 proteins both have antiapoptotic activities and can interfere with the antiproliferative functions of p53, the cellular tumor suppressor. For this to occur, E6 first forms a complex with a cellular ubiquitin-protein ligase E6AP, the E6/E6AP complex then acts as a p53-specific ubiquitin-protein ligase to accelerate degradation of p53. E6 is also known to induce expression of human telomerase (htert), leading to the functional outcome of increased life span of infected keratinocytes. Upregulation of htert is also hallmark of a number of human cancers. Although the mechanism by which E6 suppresses cell death is established, relatively little is known about the localization of E6 proteins and its related splice products and how this relates to these functional interactions. Furthermore, the localization of E6AP in normal and E6-expressing cells is essentially unknown. Also, unknown is whether interactions and localization changes between E6 and htert alter E6 function.
FIGURE 4.
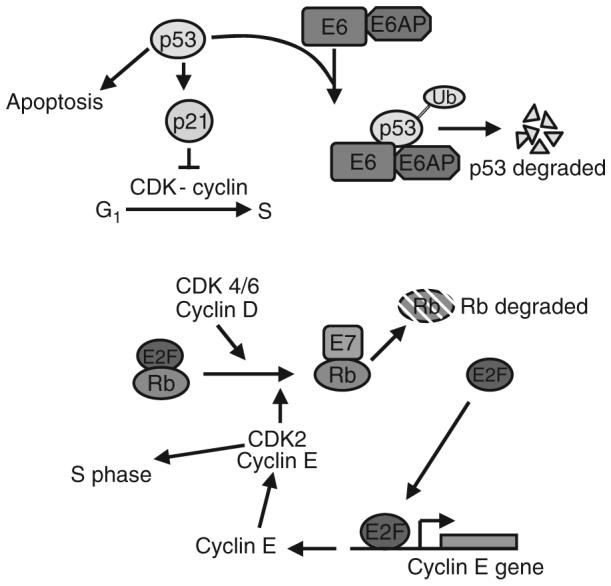
Diagram of the role of E6 and E7 in disregulation of the cell cycle. Expression of E6 leads to recruitment of E6AP (a ubiquitin ligase). This complex causes degradation of p53, which then inhibits the p21-dependent block of the G1 to S transition. Similarly, E7 binding to Rb displaces E2F, resulting in Rb’s degradation. E2F can then activate expression of cyclin E and other S-phase related gene products.
E7 binds Rb family member proteins resulting in their displacement from E2F and eventual degradation. Release of E2F allows it to freely activate S-phase related genes responsible for the G1 to S transition. Just as E6 causes inactivation of p53, allowing unchecked DNA synthesis, E7, by releasing E2F, activates the expression of genes required for cellular DNA synthesis. Even if the cellular DNA is damaged, the lack of p53 allows the cell to survive through an E7-induced S-phase and replicate the viral genome.
The rate of advancement of HPV lesions, from benign hyperplasia to carcinoma in situ, is affected by additional factors, which includes immunocompetence. HIV status, directly affects immune status which determines susceptibility to secondary infections, including HPV. In addition, progression of HPV tumors are affected by HIV status since surveillance of cancer cells is impaired. It is well established that cofactors in addition to immune status, such as alcohol, drugs, smoking, oral contraceptives, and hormone levels influence HPV infection and progression of HPV-induced cancers (Fig. 5). The ability of E6 and E7 of high-risk HPVs to inactivate p53 and pRb directly correlates with the probability to develop tumors. HPV coinfection, variants, genome integration, as well as other STDs affect the propensity for HPV-induced cancer to occur and progress.
FIGURE 5.

The rate of advancement of HPV lesions, from benign hyperplasia to carcinoma in situ, is affected by additional factors, which includes immunocompetence. HIV status, alcohol, drugs, smoking, oral contraceptives, and hormone levels influence HPV infection and progression of HPV-induced cancers. High-risk HPVs, HPV coinfection, variants, genome integration, and infection of other STDs affect the propensity for HPV-induced cancer to occur and progress.
HPV-related diseases are common causes of morbidity and mortality, both in the United States and worldwide. During the year of 2006, the American Cancer Society (ACS) estimated that there were 9710 new cases of cervical carcinoma and 3700 cervical cancer deaths in the United States. The ACS estimates that there were 4660 new anal cancers with 660 deaths and 1470 new penile cancers with 280 deaths (ACS, 2006). There were ∼6000 new cases of oral and pharyngeal cancers with 1400 deaths are attributable to HPV infection. In developing countries, the second greatest cancer cause of death among women is cervical cancer. Still, there are few effective antiviral therapies for prevention or treatment of HPV-related diseases. Furthermore, while the quadrivalent VLP HPV vaccine has the longterm potential to reduce HPV-induced cancers by 70%, population-based studies indicate that, until all girls are immunized prior to the onset of sexual activity, the vaccine will prevent only 30-50% of cervical malignancies. More of a concern is the lack of availability of the prophylactic vaccine in countries which are afflicted with high rates of cervical cancer. Thus, we will continue to face a great deal of cervical cancer morbidity and mortality in the years to come. We are far from eliminating the need for treatments for HPV infection and HPV-induced anogenital dysplasias and cancers. Hence, the continuing need for research into papillomavirus pathogenesis especially in the context of the ongoing HIV crisis.
VIII. Conclusions
AIDS malignancies have been a major complication of the HIV disease course, and this is likely to continue in HIV-infected individuals. In the era of HAART therapy, the survival rate of the HIV-infected individuals has increased dramatically mainly because of the suppression of HIV viral load and the restoration of the immune response. However, even though HAART appears to be effective, still only leads to partial immune reconstitution. Prolonged immunosuppression will likely lead to a resurgence of AIDS-associated cancers. This coupled with the fact that there are still over 40 million individuals living with HIV today, many of whom are located in regions of the world where HAART is still not widely available, such as the African continent. It is expected that AIDS-associated cancers will continue to pose a major challenge globally for many years to come. As described earlier in this chapter, many of the cancers associated with immunosuppressed individuals are those that were found to have viral etiology. Other than the development and refinement of effective vaccines against these viruses, as in the case of HPV, there is a need for a better understanding on the role of oncogenic viral cofactor in the disease, the potential mechanisms, the viral genes and the host immune response that are involved. This knowledge will lead to the development of better strategies that could prevent infection and the malignant transformation by these potentially oncogenic viruses.
Acknowledgments
This publication was made possible, in part, by support from the following NIH grants: PHS award CA76958, NCRR COBRE grant RR15635, and INBRE grant P20 RR016469. The authors also wish to acknowledge Ms. Dianna Wright for help with the preparation of this chapter.
References
- Aaltonen LM, Cajanus S, Back L, Nieminen P, Paavonen J, Ranki A. Extralaryngeal HPV infections in male patients with adult-onset laryngeal papillomatosis. Eur. Arch. Otorhinolaryngol. 2005;262(9):708–712. doi: 10.1007/s00405-004-0811-3. [DOI] [PubMed] [Google Scholar]
- Ablashi D, Chatlynne L, Cooper H, Thomas D, Yadav M, Norhanom AW, Chandana AK, Churdboonchart V, Kulpradist SA, Patnaik M, Liegmann K, Masood R, et al. Seroprevalence of human herpesvirus-8 (HHV-8) in countries of Southeast Asia compared to the USA, the Caribbean and Africa. Br. J. Cancer. 1999;81(5):893–897. doi: 10.1038/sj.bjc.6690782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aboulafia DM, Gibbons R. Penile cancer and human papilloma virus (HPV) in a human immunodeficiency virus (HIV)-infected patient. Cancer Invest. 2001;19(3):266–272. doi: 10.1081/cnv-100102554. [DOI] [PubMed] [Google Scholar]
- ACS . Cancer facts and figures. American Cancer Society; 2006. www.cancer.org. [Google Scholar]
- Agha S, Kusanthan T. Equity in access to condoms in urban Zambia. Health Policy Plan. 2003;18(3):299–305. doi: 10.1093/heapol/czg036. [DOI] [PubMed] [Google Scholar]
- Altmann M, Pich D, Ruiss R, Wang J, Sugden B, Hammerschmidt W. Transcriptional activation by EBV nuclear antigen 1 is essential for the expression of EBV’s transforming genes. Proc. Natl. Acad. Sci. USA. 2006;103(38):14188–14193. doi: 10.1073/pnas.0605985103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arany I, Tyring SK. Systemic immunosuppression by HIV infection influences HPV transcription and thus local immune responses in condyloma acuminatum. Int. J. STD AIDS. 1998;9(5):268–271. doi: 10.1258/0956462981922197. [DOI] [PubMed] [Google Scholar]
- Arvanitakis L, Geras-Raaka E, Varma A, Gershengorn MC, Cesarman E. Human herpesvirus KSHV encodes a constitutively active G-protein-coupled receptor linked to cell proliferation. Nature. 1997;385(6614):347–350. doi: 10.1038/385347a0. [DOI] [PubMed] [Google Scholar]
- Ateenyi-Agaba C, Weiderpass E, Tommasino M, Smet A, Arslan A, Dai M, Katongole-Mbidde E, Hainaut P, Snijders PJ, Franceschi S. Papillomavirus infection in the conjunctiva of individuals with and without AIDS: An autopsy series from Uganda. Cancer Lett. 2006;239(1):98–102. doi: 10.1016/j.canlet.2005.07.024. [DOI] [PubMed] [Google Scholar]
- Baay MF, Kjetland EF, Ndhlovu PD, Deschoolmeester V, Mduluza T, Gomo E, Friis H, Midzi N, Gwanzura L, Mason PR, Vermorken JB, Gundersen SG. Human papillomavirus in a rural community in Zimbabwe: The impact of HIV co-infection on HPV genotype distribution. J. Med. Virol. 2004;73(3):481–485. doi: 10.1002/jmv.20115. [DOI] [PubMed] [Google Scholar]
- Bailey P. Intracranial sarcomatous tumors of leptomeningeal origin. Arch. Surg. 1929;18:1359–1402. [Google Scholar]
- Bailie RS, Selvey CE, Bourne D, Bradshaw D. Trends in cervical cancer mortality in South Africa. Int. J. Epidemiol. 1996;25(3):488–493. doi: 10.1093/ije/25.3.488. [DOI] [PubMed] [Google Scholar]
- Bais C, Van Geelen A, Eroles P, Mutlu A, Chiozzini C, Dias S, Silverstein RL, Rafii S, Mesri EA. Kaposi’s sarcoma associated herpesvirus G protein-coupled receptor immortalizes human endothelial cells by activation of the VEGF receptor-2/KDR. Cancer Cell. 2003;3(2):131–143. doi: 10.1016/s1535-6108(03)00024-2. [DOI] [PubMed] [Google Scholar]
- Baker TS, Newcomb WW, Olson NH, Cowsert LM, Olson C, Brown JC. Structures of bovine and human papillomaviruses. Analysis by cryoelectron microscopy and three-dimensional image reconstruction. Biophys. J. 1991;60(6):1445–1456. doi: 10.1016/S0006-3495(91)82181-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baldwin SB, Wallace DR, Papenfuss MR, Abrahamsen M, Vaught LC, Kornegay JR, Hallum JA, Redmond SA, Giuliano AR. Human papillomavirus infection in men attending a sexually transmitted disease clinic. J. Infect. Dis. 2003;187(7):1064–1070. doi: 10.1086/368220. [DOI] [PubMed] [Google Scholar]
- Ballestas ME, Chatis PA, Kaye KM. Efficient persistence of extrachromosomal KSHV DNA mediated by latency-associated nuclear antigen. Science. 1999;284(5414):641–644. doi: 10.1126/science.284.5414.641. [DOI] [PubMed] [Google Scholar]
- Bayley AC. Occurrence, clinical behaviour and management of Kaposi’s sarcoma in Zambia. Cancer Surv. 1991;10:53–71. [PubMed] [Google Scholar]
- Belnap DM, Olson NH, Cladel NM, Newcomb WW, Brown JC, Kreider JW, Christensen ND, Baker TS. Conserved features in papillomavirus and polyomavirus capsids. J. Mol. Biol. 1996;259(2):249–263. doi: 10.1006/jmbi.1996.0317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beral V. Epidemiology of Kaposi’s sarcoma. Cancer Surv. 1991;10:5–22. [PubMed] [Google Scholar]
- Beral V, Peterman T, Berkelman R, Jaffe H. AIDS-associated non-Hodgkin lymphoma. Lancet. 1991;337(8745):805–809. doi: 10.1016/0140-6736(91)92513-2. [DOI] [PubMed] [Google Scholar]
- Besson C, Goubar A, Gabarre J, Rozenbaum W, Pialoux G, Chatelet FP, Katlama C, Charlotte F, Dupont B, Brousse N, Huerre M, Mikol J, et al. Changes in AIDS-related lymphoma since the era of highly active antiretroviral therapy. Blood. 2001;98(8):2339–2344. doi: 10.1182/blood.v98.8.2339. [DOI] [PubMed] [Google Scholar]
- Biggar RJ, Rabkin CS. The epidemiology of AIDS—related neoplasms. Hematol. Oncol. Clin. North Am. 1996;10(5):997–1010. doi: 10.1016/s0889-8588(05)70380-4. [DOI] [PubMed] [Google Scholar]
- Bosch FX, Durst M, Schwarz E, Boukamp P, Fusenig NE, zur Hausen H. The early genes E6 and E7 of cancer associated human papilloma viruses as targets of tumor suppression? Behring Inst. Mitt. 1991;89:108–121. [PubMed] [Google Scholar]
- Boshoff C, Endo Y, Collins PD, Takeuchi Y, Reeves JD, Schweickart VL, Siani MA, Sasaki T, Williams TJ, Gray PW, Moore PS, Chang Y, Weiss RA. Angiogenic and HIV-inhibitory functions of KSHV-encoded chemokines. Science. 1997;278(5336):290–294. doi: 10.1126/science.278.5336.290. [DOI] [PubMed] [Google Scholar]
- Brennan P, Floettmann JE, Mehl A, Jones M, Rowe M. Mechanism of action of a novel latent membrane protein-1 dominant negative. J. Biol. Chem. 2001;2001:1195–1203. doi: 10.1074/jbc.M005461200. [DOI] [PubMed] [Google Scholar]
- Brennan P, Mehl AM, Jones M, Rowe M. Phosphatidylinositol 3-kinase is essential for the proliferation of lymphoblastoid cells. Oncogene. 2002;21(8):1263–1271. doi: 10.1038/sj.onc.1205182. [DOI] [PubMed] [Google Scholar]
- Brockmeyer N, Barthel B. Clinical manifestations and therapies of AIDS associated tumors. Eur. J. Med. Res. 1998;3(3):127–147. [PubMed] [Google Scholar]
- Cahir-McFarland ED, Davidson DM, Schauer SL, Duong J, Kieff E. NF-kappaB inhibition causes spontaneous apoptosis in Epstein-Barr virus-transformed lymphoblastoid cells. Proc. Natl. Acad. Sci. USA. 2000;97:6055–6060. doi: 10.1073/pnas.100119497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Callahan J, Pai S, Cotter M, Robertson ES. Distinct patterns of viral antigen expression in Epstein-Barr virus and Kaposi’s sarcoma-associated herpesvirus coinfected body-cavity-based lymphoma cell lines: Potential switches in latent gene expression due to coinfection. Virology. 1999;262(1):18–30. doi: 10.1006/viro.1999.9876. [DOI] [PubMed] [Google Scholar]
- Calleja-Macias IE, Kalantari M, Huh J, Ortiz-Lopez R, Rojas-Martinez A, Gonzalez-Guerrero JF, Williamson AL, Hagmar B, Wiley DJ, Villarreal L, Bernard HU, et al. Genomic diversity of human papillomavirus-16, 18, 31, and 35 isolates in a Mexican population and relationship to European, African, and Native American variants. Virology. 2004;319(2):315–323. doi: 10.1016/j.virol.2003.11.009. [DOI] [PubMed] [Google Scholar]
- Carbone A, Gloghini A. AIDS-related lymphomas: From pathogenesis to pathology. Br. J. Haematol. 2005;130(5):662–670. doi: 10.1111/j.1365-2141.2005.05613.x. [DOI] [PubMed] [Google Scholar]
- Casini GL, Graham D, Heine D, Garcea RL, Wu DT. in vitro papillomavirus capsid assembly analyzed by light scattering. Virology. 2004;325(2):320–327. doi: 10.1016/j.virol.2004.04.034. [DOI] [PubMed] [Google Scholar]
- Cates W, Jr., Dallabetta G. The staying power of sexually transmitted diseases. Lancet. 1999;354(Suppl SIV62) doi: 10.1016/s0140-6736(99)90405-1. [DOI] [PubMed] [Google Scholar]
- Cesarman E, Chang Y, Moore PS, Said JW, Knowles DM. Kaposi’s sarcoma-associated herpesvirus-like DNA sequences in AIDS-related body-cavity-based lymphomas. N. Engl. J. Med. 1995;332(18):1186–1191. doi: 10.1056/NEJM199505043321802. [DOI] [PubMed] [Google Scholar]
- Cesarman E, Nador RG, Bai F, Bohenzky RA, Russo JJ, Moore PS, Chang Y, Knowles DM. Kaposi’s sarcoma-associated herpesvirus contains G protein-coupled receptor and cyclin D homologs which are expressed in Kaposi’s sarcoma and malignant lymphoma. J. Virol. 1996;70(11):8218–8223. doi: 10.1128/jvi.70.11.8218-8223.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Challine D, Roudot-Thoraval F, Sarah T, Laperche L, Boisson B, Mauberquez S, Dubernet F, Rigot P, Lefrere F, Mercier B, Brossard Y, Rouet F, et al. Seroprevalence of human herpes virus 8 antibody in populations at high or low risk of transfusion, graft, or sexual transmission of viruses. Transfusion. 2001;41(9):1120–1125. doi: 10.1046/j.1537-2995.2001.41091120.x. [DOI] [PubMed] [Google Scholar]
- Chan SY, Ho L, Ong CK, Chow V, Drescher B, Durst M, ter Meulen J, Villa L, Luande J, Mgaya HN. Molecular variants of human papillomavirus type 16 from four continents suggest ancient pandemic spread of the virus and its coevolution with humankind. J. Virol. 1992;66(4):2057–2066. doi: 10.1128/jvi.66.4.2057-2066.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang Y, Moore PS. Kaposi’s sarcoma (KS)-associated herpesvirus and its role in KS. Infect. Agents Dis. 1996;5(4):215–222. [PubMed] [Google Scholar]
- Chang Y, Cesarman E, Pessin MS, Lee F, Culpepper J, Knowles DM, Moore PS. Identification of herpesvirus-like DNA sequences in AIDS-associated Kaposi’s sarcoma. Science. 1994;266(5192):1865–1869. doi: 10.1126/science.7997879. [DOI] [PubMed] [Google Scholar]
- Chang H, Gwack Y, Kingston D, Souvlis J, Liang X, Means RE, Cesarman E, Hutt-Fletcher L, Jung JU. Activation of CD21 and CD23 gene expression by Kaposi’s sarcoma-associated herpesvirus RTA. J. Virol. 2005;79(8):4651–4663. doi: 10.1128/JVI.79.8.4651-4663.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheah PL, Looi LM. Biology and pathological associations of the human papillomaviruses: A review. Malays. J. Pathol. 1998;20(1):1–10. [PubMed] [Google Scholar]
- Chen H, Hutt-Fletcher L, Cao L, Hayward SD. A positive autoregulatory loop of LMP1 expression and STAT activation in epithelial cells latently infected with Epstein-Barr virus. J. Virol. 2003;77(7):4139–4148. doi: 10.1128/JVI.77.7.4139-4148.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheung TW. AIDS-related cancer in the era of highly active antiretroviral therapy (HAART), a model of the interplay of the immune system, virus, and cancer. “On the offensive—the Trojan Horse is being destroyed”—Part B: Malignant lymphoma. Cancer Invest. 2004;22(5):787–798. doi: 10.1081/cnv-200032792. [DOI] [PubMed] [Google Scholar]
- Chow LT, Broker TR. Papillomavirus DNA replication. Intervirology. 1994;37(34):150–158. doi: 10.1159/000150373. [DOI] [PubMed] [Google Scholar]
- Cingolani A, Fratino L, Scoppettuolo G, Antinori A. Changing pattern of primary cerebral lymphoma in the highly active antiretroviral therapy era. J. Neurovirol. 2005;11(Suppl 3):38–44. doi: 10.1080/13550280500511808. [DOI] [PubMed] [Google Scholar]
- Clarke B, Chetty R. Postmodern cancer: The role of human immunodeficiency virus in uterine cervical cancer. Mol. Pathol. 2002;55(1):19–24. doi: 10.1136/mp.55.1.19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clarke J, Terry RM, Lacey CJ. A study to estimate the prevalence of upper respiratory tract papillomatosis in patients with genital warts. Int. J. STD AIDS. 1991;2(2):114–115. doi: 10.1177/095646249100200207. [DOI] [PubMed] [Google Scholar]
- Clertant P, Seif I. A common function for polyoma virus large-T and papillomavirus E1 proteins? Nature. 1984;311(5983):276–279. doi: 10.1038/311276a0. [DOI] [PubMed] [Google Scholar]
- Clifford GM, Polesel J, Rickenbach M, Dal Maso L, Keiser O, Kofler A, Rapiti E, Levi F, Jundt G, Fisch T, Bordoni A, De Weck D, et al. Cancer risk in the Swiss HIV Cohort Study: Associations with immunodeficiency, smoking, and highly active antiretroviral therapy. J. Natl. Cancer Inst. 2005;97(6):425–432. doi: 10.1093/jnci/dji072. [DOI] [PubMed] [Google Scholar]
- Cole MD, McMahon SB. The Myc oncoprotein: A critical evaluation of transactivation and target gene regulation. Oncogene. 1999;18(19):2916–2924. doi: 10.1038/sj.onc.1202748. [DOI] [PubMed] [Google Scholar]
- Contreras A, Falkler WA, Jr., Enwonwu CO, Idigbe EO, Savage KO, Afolabi MB, Onwujekwe D, Rams TE, Slots J. Human herpesviridae in acute necrotizing ulcerative gingivitis in children in Nigeria. Oral Microbiol. Immunol. 1997;12(5):259–265. doi: 10.1111/j.1399-302x.1997.tb00389.x. [DOI] [PubMed] [Google Scholar]
- Cook J. The clinical features of Kaposi’s sarcoma in the East African Bantu. Acta Unio Int. Contra Cancrum. 1962;18:388–393. [PubMed] [Google Scholar]
- Cotter MA, 2nd, Subramanian C, Robertson ES. The Kaposi’s sarcoma-associated herpesvirus latency-associated nuclear antigen binds to specific sequences at the left end of the viral genome through its carboxy-terminus. Virology. 2001;291(2):241–259. doi: 10.1006/viro.2001.1202. [DOI] [PubMed] [Google Scholar]
- Cunha AM, Caterino-de-Araujo A, Costa SC, Santos-Fortuna E, Boa-Sorte NC, Goncalves MS, Costa FF, Galvao-Castro B. Increasing seroprevalence of human herpesvirus 8 (HHV-8) with age confirms HHV-8 endemicity in Amazon Amerindians from Brazil. J. Gen. Virol. 2005;86(Pt 9):2433–2437. doi: 10.1099/vir.0.81087-0. [DOI] [PubMed] [Google Scholar]
- Czegledy J, Rogo KO, Evander M, Wadell G. High-risk human papillomavirus types in cytologically normal cervical scrapes from Kenya. Med. Microbiol. Immunol. (Berl.) 1992;180(6):321–326. doi: 10.1007/BF00191552. [DOI] [PubMed] [Google Scholar]
- Dawson CW, Rickinson AB, Young LS. Epstein-Barr virus latent membrane protein inhibits human epithelial cell differentiation. Nature. 1990;344(6268):777–780. doi: 10.1038/344777a0. [DOI] [PubMed] [Google Scholar]
- de Sanjose S, Marshall V, Sola J, Palacio V, Almirall R, Goedert JJ, Bosch FX, Whitby D. Prevalence of Kaposi’s sarcoma-associated herpesvirus infection in sex workers and women from the general population in Spain. Int. J. Cancer. 2002;98(1):155–158. doi: 10.1002/ijc.10190. [DOI] [PubMed] [Google Scholar]
- Devergne O, Hatzivassiliou E, Izumi K, Kaye K, Kleijnen M, Kieff E, Mosialos G. Association of TRAF1, TRAF2, and TRAF3 with an Epstein-Barr virus LMP1 domain important for B lymphocyte transformation: Role in NF-κB activation. Mol. Cell Biol. 1996;16:7098–7108. doi: 10.1128/mcb.16.12.7098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Devergne O, McFarland EC, Mosialos G, Izumi KM, Ware CF, Kieff E. Role of the TRAF binding site and NF-kappaB activation in Epstein-Barr virus latent membrane protein 1-induced cell gene expression. J. Virol. 1998;72:7900–7908. doi: 10.1128/jvi.72.10.7900-7908.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Villiers EM, Weidauer H, Le JY, Neumann C, zur Hausen H. (Papilloma viruses in benign and malignant tumors of the mouth and upper respiratory tract) Laryngol. Rhinol. Otol. (Stuttg) 1986;65(4):177–179. [PubMed] [Google Scholar]
- De Vuyst H, Steyaert S, Van Renterghem L, Claeys P, Muchiri L, Sitati S, Vansteelandt S, Quint W, Kleter B, Van Marck E, Temmerman M. Distribution of human papillomavirus in a family planning population in Nairobi, Kenya. Sex Transm. Dis. 2003;30(2):137–142. doi: 10.1097/00007435-200302000-00009. [DOI] [PubMed] [Google Scholar]
- Dittmer D, Lagunoff M, Renne R, Staskus K, Haase A, Ganem D. A cluster of latently expressed genes in Kaposi’s sarcoma-associated herpesvirus. J. Virol. 1998;72(10):8309–8315. doi: 10.1128/jvi.72.10.8309-8315.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Djerbi M, Screpanti V, Catrina AI, Bogen B, Biberfeld P, Grandien A. The inhibitor of death receptor signaling, FLICE-inhibitory protein defines a new class of tumor progression factors. J. Exp. Med. 1999;190(7):1025–1032. doi: 10.1084/jem.190.7.1025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dourmishev LA, Dourmishev AL, Palmeri D, Schwartz RA, Lukac DM. Molecular genetics of Kaposi’s sarcoma-associated herpesvirus (human herpesvirus-8) epidemiology and pathogenesis. Microbiol. Mol. Biol. Rev. 2003;67(2):175–212. doi: 10.1128/MMBR.67.2.175-212.2003. table of contents. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drexler HG, Uphoff CC, Gaidano G, Carbone A. Lymphoma cell lines: in vitro models for the study of HHV-8+ primary effusion lymphomas (body cavity-based lymphomas) Leukemia. 1998;12(10):1507–1517. doi: 10.1038/sj.leu.2401160. [DOI] [PubMed] [Google Scholar]
- Dunne EF, Nielson CM, Stone KM, Markowitz LE, Giuliano AR. Prevalence of HPV infection among men: A systematic review of the literature. J. Infect. Dis. 2006;194(8):1044–1057. doi: 10.1086/507432. [DOI] [PubMed] [Google Scholar]
- Durack DT. Opportunistic infections and Kaposi’s sarcoma in homosexual men. N. Engl. J. Med. 1981;305(24):1465–1467. doi: 10.1056/NEJM198112103052408. [DOI] [PubMed] [Google Scholar]
- Durante AJ, Williams AB, Da Costa M, Darragh TM, Khoshnood K, Palefsky JM. Incidence of anal cytological abnormalities in a cohort of human immunodeficiency virus-infected women. Cancer Epidemiol. Biomark. Prev. 2003;12(7):638–642. [PubMed] [Google Scholar]
- Durst M, Gissmann L, Ikenberg H, zur Hausen H. A papillomavirus DNA from a cervical carcinoma and its prevalence in cancer biopsy samples from different geographic regions. Proc. Natl. Acad. Sci. USA. 1983;80(12):3812–3815. doi: 10.1073/pnas.80.12.3812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durst M, Croce CM, Gissmann L, Schwarz E, Huebner K. Papillomavirus sequences integrate near cellular oncogenes in some cervical carcinomas. Proc. Natl. Acad. Sci. USA. 1987;84(4):1070–1074. doi: 10.1073/pnas.84.4.1070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eichler AF, Batchelor TT. Primary central nervous system lymphoma: Presentation, diagnosis and staging. Neurosurg. Focus. 2006;21(5):E15. doi: 10.3171/foc.2006.21.5.16. [DOI] [PubMed] [Google Scholar]
- El-Mofty SK, Lu DW. Prevalence of human papillomavirus type 16 DNA in squamous cell carcinoma of the palatine tonsil, and not the oral cavity, in young patients: A distinct clinicopathologic and molecular disease entity. Am. J. Surg. Pathol. 2003;27(11):1463–1470. doi: 10.1097/00000478-200311000-00010. [DOI] [PubMed] [Google Scholar]
- Eliopoulos AG, Young LS. Activation of the cJun N-terminal kinase (JNK) pathway by the Epstein-Barr virus-encoded latent membrane protein 1 (LMP1) Oncogene. 1998;16:1731–1742. doi: 10.1038/sj.onc.1201694. [DOI] [PubMed] [Google Scholar]
- Eliopoulos AG, Blake SM, Floettmann JE, Rowe M, Young LS. Epstein-Barr virus-encoded latent membrane protein 1 activates the JNK pathway through its extreme C terminus via a mechanism involving TRADD and TRAF2. J. Virol. 1999;73:1023–1035. doi: 10.1128/jvi.73.2.1023-1035.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Epeldegui M, Widney DP, Martinez-Maza O. Pathogenesis of AIDS lymphoma: Role of oncogenic viruses and B cell activation-associated molecular lesions. Curr. Opin. Oncol. 2006;18(5):444–448. doi: 10.1097/01.cco.0000239882.23839.e5. [DOI] [PubMed] [Google Scholar]
- Fahraeus R, Rymo L, Rhim JS, Klein G. Morphological transformation of human keratinocytes expressing the LMP gene of Epstein-Barr virus. Nature. 1990;345:447–449. doi: 10.1038/345447a0. [DOI] [PubMed] [Google Scholar]
- Fakhry C, Gillison ML. Clinical implications of human papillomavirus in head and neck cancers. J. Clin. Oncol. 2006;24(17):2606–2611. doi: 10.1200/JCO.2006.06.1291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan W, Bubman D, Chadburn A, Harrington WJ, Jr., Cesarman E, Knowles DM. Distinct subsets of primary effusion lymphoma can be identified based on their cellular gene expression profile and viral association. J. Virol. 2005;79(2):1244–1251. doi: 10.1128/JVI.79.2.1244-1251.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fassone L, Bhatia K, Gutierrez M, Capello D, Gloghini A, Dolcetti R, Vivenza D, Ascoli V, Lo Coco F, Pagani L, Dotti G, Rambaldi A, et al. Molecular profile of Epstein-Barr virus infection in HHV-8-positive primary effusion lymphoma. Leukemia. 2000;14(2):271–277. doi: 10.1038/sj.leu.2401651. [DOI] [PubMed] [Google Scholar]
- Floettmann JE, Rowe M. Epstein-Barr virus latent membrane protein-1 (LMP1) C-terminus activation region 2 (CTAR2) maps to the far C-terminus and requires oligomerization for NF-κB activation. Oncogene. 1997;15:1851–1858. doi: 10.1038/sj.onc.1201359. [DOI] [PubMed] [Google Scholar]
- Flore O, Rafii S, Ely S, O’Leary JJ, Hyjek EM, Cesarman E. Transformation of primary human endothelial cells by Kaposi’s sarcoma-associated herpesvirus. Nature. 1998;394(6693):588–592. doi: 10.1038/29093. [DOI] [PubMed] [Google Scholar]
- Flores ER, Lambert PF. Evidence for a switch in the mode of human papillomavirus type 16 DNA replication during the viral life cycle. J. Virol. 1997;71(10):7167–7179. doi: 10.1128/jvi.71.10.7167-7179.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foglieni C, Scabini S, Belloni D, Broccolo F, Lusso P, Malnati MS, Ferrero E. Productive infection of HUVEC by HHV-8 is associated with changes compatible with angiogenic transformations. Eur. J. Histochem. 2005;49(3):273–284. doi: 10.4081/954. [DOI] [PubMed] [Google Scholar]
- Frattini MG, Laimins LA. Binding of the human papillomavirus E1 origin-recognition protein is regulated through complex formation with the E2 enhancer-binding protein. Proc. Natl. Acad. Sci. USA. 1994;91(26):12398–12402. doi: 10.1073/pnas.91.26.12398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freitas RB, Freitas MR, Linhares AC. Prevalence of human herpesvirus 8 antibodies in the population of Belem, Para, Brazil. Rev. Inst. Med. Trop. Sao Paulo. 2002;44(6):309–313. doi: 10.1590/s0036-46652002000600003. [DOI] [PubMed] [Google Scholar]
- Friborg J, Jr., Kong W, Hottiger MO, Nabel GJ. p53 inhibition by the LANA protein of KSHV protects against cell death. Nature. 1999;402(6764):889–894. doi: 10.1038/47266. [DOI] [PubMed] [Google Scholar]
- Friedman-Kien AE, Saltzman BR. Clinical manifestations of classical, endemic African, and epidemic AIDS-associated Kaposi’s sarcoma. J. Am. Acad. Dermatol. 1990;22(6 Pt 2):1237–1250. doi: 10.1016/0190-9622(90)70169-i. [DOI] [PubMed] [Google Scholar]
- Fries KL, Miller WE, Raab-Traub N. Epstein-Barr virus latent membrane protein 1 blocks p53-mediated apoptosis through the induction of the A20 gene. J. Virol. 1996;70:8653–8659. doi: 10.1128/jvi.70.12.8653-8659.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frisch M, Biggar RJ, Goedert JJ. Human papillomavirus-associated cancers in patients with human immunodeficiency virus infection and acquired immunodeficiency syndrome. J. Natl. Cancer Inst. 2000;92(18):1500–1510. doi: 10.1093/jnci/92.18.1500. [DOI] [PubMed] [Google Scholar]
- Fujimuro M, Wu FY, ApRhys C, Kajumbula H, Young DB, Hayward GS, Hayward SD. A novel viral mechanism for dysregulation of beta-catenin in Kaposi’s sarcoma-associated herpesvirus latency. Nat. Med. 2003;9(3):300–306. doi: 10.1038/nm829. [DOI] [PubMed] [Google Scholar]
- Gambus G, Bourboulia D, Esteve A, Lahoz R, Rodriguez C, Bolao F, Sirera G, Muga R, del Romero J, Boshoff C, Whitby D, Casabona J. Prevalence and distribution of HHV-8 in different subpopulations, with and without HIV infection, in Spain. AIDS. 2001;15(9):1167–1174. doi: 10.1097/00002030-200106150-00012. [DOI] [PubMed] [Google Scholar]
- Gange RW, Jones EW. Kaposi’s sarcoma and immunosuppressive therapy: An appraisal. Clin. Exp. Dermatol. 1978;3(2):135–146. doi: 10.1111/j.1365-2230.1978.tb01477.x. [DOI] [PubMed] [Google Scholar]
- Gao SJ, Kingsley L, Hoover DR, Spira TJ, Rinaldo CR, Saah A, Phair J, Detels R, Parry P, Chang Y, Moore PS. Seroconversion to antibodies against Kaposi’s sarcoma-associated herpesvirus-related latent nuclear antigens before the development of Kaposi’s sarcoma. N. Engl. J. Med. 1996a;335(4):233–241. doi: 10.1056/NEJM199607253350403. [DOI] [PubMed] [Google Scholar]
- Gao SJ, Kingsley L, Li M, Zheng W, Parravicini C, Ziegler J, Newton R, Rinaldo CR, Saah A, Phair J, Detels R, Chang Y, Moore PS. KSHV antibodies among Americans, Italians and Ugandans with and without Kaposi’s sarcoma. Nat. Med. 1996b;2(8):925–928. doi: 10.1038/nm0896-925. [DOI] [PubMed] [Google Scholar]
- Gates AE, Kaplan LD. AIDS malignancies in the era of highly active antiretroviral therapy. Oncology (Williston Park, N.Y.) 2002;16(5):657–665. discussion 665, 668-670. [PubMed] [Google Scholar]
- Gershengorn MC, Geras-Raaka E, Varma A, Clark-Lewis I. Chemokines activate Kaposi’s sarcoma-associated herpesvirus G protein-coupled receptor in mammalian cells in culture. J. Clin. Invest. 1998;102(8):1469–1472. doi: 10.1172/JCI4461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gillison ML, Koch WM, Capone RB, Spafford M, Westra WH, Wu L, Zahurak ML, Daniel RW, Viglione M, Symer DE, Shah KV, Sidransky D. Evidence for a causal association between human papillomavirus and a subset of head and neck cancers. J. Natl. Cancer Inst. 2000;92(9):709–720. doi: 10.1093/jnci/92.9.709. [DOI] [PubMed] [Google Scholar]
- Giraldo G, Beth E, Huang ES. Kaposi’s sarcoma and its relationship to cytomegalovirus (CMNV). III. CMV DNA and CMV early antigens in Kaposi’s sarcoma. Int. J. Cancer. 1980;26(1):23–29. doi: 10.1002/ijc.2910260105. [DOI] [PubMed] [Google Scholar]
- Gires O, Zimber-Strobl U, Gonnella R, Ueffing M, Marschall G, Zeidler R, Pich D, Hammerschmidt W. Latent membrane protein 1 of Epstein-Barr virus mimics a constitutively active receptor molecule. EMBO J. 1997;16:6131–6140. doi: 10.1093/emboj/16.20.6131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gires O, Kohlhuber F, Kilger E, Baumann M, Kieser A, Kaiser C, Zeidler R, Scheffer B, Ueffing M, Hammerschmidt W. Latent membrane protein 1 of Epstein-Barr virus interacts with JAK3 and activates STAT proteins. EMBO J. 1999;18:3064–3073. doi: 10.1093/emboj/18.11.3064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gissmann L, Boshart M, Durst M, Ikenberg H, Wagner D, zur Hausen H. Presence of human papillomavirus in genital tumors. J. Invest. Dermatol. 1984;83(Suppl 1):26S–28S. doi: 10.1111/1523-1747.ep12281143. [DOI] [PubMed] [Google Scholar]
- Giuliano AR, Papenfuss M, Schneider A, Nour M, Hatch K. Risk factors for high-risk type human papillomavirus infection among Mexican-American women. Cancer Epidemiol. Biomark. Prev. 1999;8(7):615–620. [PubMed] [Google Scholar]
- Gloeckler Ries LA, Reichman ME, Lewis DR, Hankey BF, Edwards BK. Cancer survival and incidence from the Surveillance, Epidemiology, and End Results (SEER) program. Oncologist. 2003;8(6):541–552. doi: 10.1634/theoncologist.8-6-541. [DOI] [PubMed] [Google Scholar]
- Goedert JJ. The epidemiology of acquired immunodeficiency syndrome malignancies. Semin. Oncol. 2000;27(4):390–401. [PubMed] [Google Scholar]
- Goedert JJ, Cote TR, Virgo P, Scoppa SM, Kingma DW, Gail MH, Jaffe ES, Biggar RJ. Spectrum of AIDS-associated malignant disorders. Lancet. 1998;351(9119):1833–1839. doi: 10.1016/s0140-6736(97)09028-4. [DOI] [PubMed] [Google Scholar]
- Gravitt PE, Kamath AM, Gaffikin L, Chirenje ZM, Womack S, Shah KV. Human papillomavirus genotype prevalence in high-grade squamous intraepithelial lesions and colposcopically normal women from Zimbabwe. Int. J. Cancer. 2002;100(6):729–732. doi: 10.1002/ijc.10538. [DOI] [PubMed] [Google Scholar]
- Grossmann C, Podgrabinska S, Skobe M, Ganem D. Activation of NF-kappaB by the latent vFLIP gene of Kaposi’s sarcoma-associated herpesvirus is required for the spindle shape of virus-infected endothelial cells and contributes to their proinflammatory phenotype. J. Virol. 2006;80(14):7179–7185. doi: 10.1128/JVI.01603-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grulich AE, Vajdic CM. The epidemiology of non-Hodgkin lymphoma. Pathology. 2005;37(6):409–419. doi: 10.1080/00313020500370192. [DOI] [PubMed] [Google Scholar]
- Grulich AE, Li Y, McDonald A, Correll PK, Law MG, Kaldor JM. Rates of non-AIDS-defining cancers in people with HIV infection before and after AIDS diagnosis. AIDS. 2002;16(8):1155–1161. doi: 10.1097/00002030-200205240-00009. [DOI] [PubMed] [Google Scholar]
- Guasparri I, Keller SA, Cesarman E. KSHV vFLIP is essential for the survival of infected lymphoma cells. J. Exp. Med. 2004;199(7):993–1003. doi: 10.1084/jem.20031467. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Hankey BF, Ries LA, Edwards BK. The surveillance, epidemiology, and end results program: A national resource. Cancer Epidemiol. Biomark. Prev. 1999;8(12):1117–1121. [PubMed] [Google Scholar]
- Harwood AR, Osoba D, Hofstader SL, Goldstein MB, Cardella CJ, Holecek MJ, Kunynetz R, Giammarco RA. Kaposi’s sarcoma in recipients of renal transplants. Am. J. Med. 1979;67(5):759–765. doi: 10.1016/0002-9343(79)90731-9. [DOI] [PubMed] [Google Scholar]
- Hassen E, Chaieb A, Letaief M, Khairi H, Zakhama A, Remadi S, Chouchane L. Cervical human papillomavirus infection in Tunisian women. Infection. 2003;31(3):143–148. doi: 10.1007/s15010-003-3112-7. [DOI] [PubMed] [Google Scholar]
- Hawes SE, Critchlow CW, Faye Niang MA, Diouf MB, Diop A, Toure P, Aziz Kasse A, Dembele B, Salif Sow P, Coll-Seck AM, Kuypers JM, Kiviat NB. Increased risk of high-grade cervical squamous intraepithelial lesions and invasive cervical cancer among African women with human immunodeficiency virus type 1 and 2 infections. J. Infect. Dis. 2003;188(4):555–563. doi: 10.1086/376996. [DOI] [PubMed] [Google Scholar]
- Heard I, Palefsky JM, Kazatchkine MD. The impact of HIV antiviral therapy on human papillomavirus (HPV) infections and HPV-related diseases. Antivir. Ther. 2004;9(1):13–22. [PubMed] [Google Scholar]
- Henderson S, Rowe M, Gregory C, Croom-Carter D, Wang F, Longnecker R, Kieff E, Rickinson A. Induction of bcl-2 expression by Epstein-Barr virus latent membrane protein 1 protects infected B cells from programmed cell death. Cell. 1991;65:1107–1115. doi: 10.1016/0092-8674(91)90007-l. [DOI] [PubMed] [Google Scholar]
- Hengge UR, Ruzicka T, Tyring SK, Stuschke M, Roggendorf M, Schwartz RA, Seeber S. Update on Kaposi’s sarcoma and other HHV8 associated diseases. Part 1: Epidemiology, environmental predispositions, clinical manifestations, and therapy. Lancet Infect. Dis. 2002;2(5):281–292. doi: 10.1016/s1473-3099(02)00263-3. [DOI] [PubMed] [Google Scholar]
- Higuchi M, Kieff E, Izumi KM. The Epstein-Barr virus latent membrane protein 1 putative Janus kinase 3 (JAK3) binding domain does not mediate JAK3 association or activation in B-lymphoma or lymphoblastoid cell lines. J. Virol. 2002;76(1):455–459. doi: 10.1128/JVI.76.1.455-459.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffmann C, Tabrizian S, Wolf E, Eggers C, Stoehr A, Plettenberg A, Buhk T, Stellbrink HJ, Horst HA, Jager H, Rosenkranz T. Survival of AIDS patients with primary central nervous system lymphoma is dramatically improved by HAART-induced immune recovery. AIDS. 2001;15(16):2119–2127. doi: 10.1097/00002030-200111090-00007. [DOI] [PubMed] [Google Scholar]
- Hoffmann R, Hirt B, Bechtold V, Beard P, Raj K. Different modes of human papillomavirus DNA replication during maintenance. J. Virol. 2006;80(9):4431–4439. doi: 10.1128/JVI.80.9.4431-4439.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horenstein MG, Nador RG, Chadburn A, Hyjek EM, Inghirami G, Knowles DM, Cesarman E. Epstein-Barr virus latent gene expression in primary effusion lymphomas containing Kaposi’s sarcoma-associated herpesvirus/human herpesvirus-8. Blood. 1997;90(3):1186–1191. [PubMed] [Google Scholar]
- Howley PM. Virology. 2nd edn. Lippincott-Raven Publishers; Philadelphia, PA: 1996. Papillomavirinae: The viruses and their replication; pp. 2045–2076. [Google Scholar]
- Howley PM, Munger K, Werness BA, Phelps WC, Schlegel R. Molecular mechanisms of transformation by the human papillomaviruses. Princess Takamatsu Symp. 1989;20:199–206. [PubMed] [Google Scholar]
- Huen DS, Henderson SA, Croom-Carter S, Rowe M. The Epstein-Barr virus latent membrane protein-1 (LMP1) mediates activation of NF-κB and cell surface phenotype via two effector regions in its carboxy-terminal cytoplasmic domain. Oncogene. 1995;10:549–560. [PubMed] [Google Scholar]
- Iscovich J, Boffetta P, Franceschi S, Azizi E, Sarid R. Classic kaposi sarcoma: Epidemiology and risk factors. Cancer. 2000;88(3):500–517. [PubMed] [Google Scholar]
- Ivers LC, Kim AY, Sax PE. Predictive value of polymerase chain reaction of cerebrospinal fluid for detection of Epstein-Barr virus to establish the diagnosis of HIV-related primary central nervous system lymphoma. Clin. Infect. Dis. 2004;38(11):1629–1632. doi: 10.1086/420934. [DOI] [PubMed] [Google Scholar]
- Izumi K, Kieff E. The Epstein-Barr virus oncogene product latent membrane protein 1 engages the tumor necrosis factor receptor-associated death domain protein to mediate B-lymphocyte growth transformation and activate NF-κB. Proc. Natl. Acad. Sci. 1997;94:12592–12597. doi: 10.1073/pnas.94.23.12592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Izumi KM, Kaye KM, Kieff ED. The Epstein-Barr virus LMP1 amino acid sequence that engages tumor necrosis factor receptor associated factors is critical for primary B lymphocyte growth transformation. Proc. Natl. Acad. Sci. USA. 1997;94:1447–1452. doi: 10.1073/pnas.94.4.1447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Izumi KM, McFarland EC, Ting AT, Riley EA, Seed B, Kieff ED. The Epstein-Barr virus oncoprotein latent membrane protein 1 engages the tumor necrosis factor receptor-associated proteins TRADD and receptor-interacting protein (RIP) but does not induce apoptosis or require RIP for NF-kappaB activation. Mol. Cell Biol. 1999;19:5759–5767. doi: 10.1128/mcb.19.8.5759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jastreboff AM, Cymet T. Role of the human papilloma virus in the development of cervical intraepithelial neoplasia and malignancy. Postgrad. Med. J. 2002;78(918):225–228. doi: 10.1136/pmj.78.918.225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeon S, Allen-Hoffmann BL, Lambert PF. Integration of human papilloma-virus type 16 into the human genome correlates with a selective growth advantage of cells. J. Virol. 1995;69(5):2989–2997. doi: 10.1128/jvi.69.5.2989-2997.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jordan LB, Monaghan H. Pathology of the cervix: Recent developments. Clin. Oncol. (R. Coll. Radiol.) 2004;16(4):248–254. doi: 10.1016/j.clon.2004.02.006. [DOI] [PubMed] [Google Scholar]
- Kapadia SB, Krause JR. Kaposi’s sarcoma after long-term alkylating agent therapy for multiple myeloma. South Med. J. 1977;70(8):1011–1013. doi: 10.1097/00007611-197708000-00036. [DOI] [PubMed] [Google Scholar]
- Kaplan LD, Kahn JO, Crowe S, Northfelt D, Neville P, Grossberg H, Abrams DI, Tracey J, Mills J, Volberding PA. Clinical and virologic effects of recombinant human granulocyte-macrophage colony-stimulating factor in patients receiving chemotherapy for human immunodeficiency virus-associated non-Hodgkin’s lymphoma: Results of a randomized trial. J. Clin. Oncol. 1991;9(6):929–940. doi: 10.1200/JCO.1991.9.6.929. [DOI] [PubMed] [Google Scholar]
- Kaplan LD, Straus DJ, Testa MA, Von Roenn J, Dezube BJ, Cooley TP, Herndier B, Northfelt DW, Huang J, Tulpule A, Levine AM, National Institute of Allergy and Infectious Diseases AIDS Clinical Trials Group Low-dose compared with standard-dose m-BACOD chemotherapy for non-Hodgkin’s lymphoma associated with human immunodeficiency virus infection. N. Engl. J. Med. 1997;336(23):1641–1648. doi: 10.1056/NEJM199706053362304. [DOI] [PubMed] [Google Scholar]
- Kaposi M. Idiopathic multiple pigmented sarcoma of the skin. Arch. Dermatol. Syphilis. 1872;4:265–273. [Google Scholar]
- Katano H, Sato Y, Kurata T, Mori S, Sata T. Expression and localization of human herpesvirus 8-encoded proteins in primary effusion lymphoma, Kaposi’s sarcoma, and multicentric Castleman’s disease. Virology. 2000;269(2):335–344. doi: 10.1006/viro.2000.0196. [DOI] [PubMed] [Google Scholar]
- Kawana Y, Kawana K, Yoshikawa H, Taketani Y, Yoshiike K, Kanda T. Human papillomavirus type 16 minor capsid protein l2 N-terminal region containing a common neutralization epitope binds to the cell surface and enters the cytoplasm. J. Virol. 2001;75(5):2331–2336. doi: 10.1128/JVI.75.5.2331-2336.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaye KM, Izumi KM, Kieff E. Epstein-Barr virus latent membrane protein 1 is essential for B-lymphocyte growth transformation. Proc. Natl. Acad. Sci. USA. 1993;90(19):9150–9154. doi: 10.1073/pnas.90.19.9150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaye K, Devergne O, Harada J, Izumi K, Yalamanchili R, Kieff E, Mosialos G. Tumor necrosis factor receptor associated factor 2 is a mediator of NF-κB activation by latent infection membrane protein 1, the Epstein-Barr virus transforming protein. Proc. Natl. Acad. Sci. USA. 1996;93:11085–11090. doi: 10.1073/pnas.93.20.11085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kazanji M, Dussart P, Duprez R, Tortevoye P, Pouliquen JF, Vandekerkhove J, Couppie P, Morvan J, Talarmin A, Gessain A. Serological and molecular evidence that human herpesvirus 8 is endemic among Amerindians in French Guiana. J. Infect. Dis. 2005;192(9):1525–1529. doi: 10.1086/491744. [DOI] [PubMed] [Google Scholar]
- Kelekar A, Cole MD. Tumorigenicity of fibroblast lines expressing the adenovirus E1a, cellular p53, or normal c-myc genes. Mol. Cell Biol. 1986;6(1):7–14. doi: 10.1128/mcb.6.1.7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kieff E. Epstein-Barr virus and its replication. In: Fields BN, Knipe DM, Howley PM, editors. Virology. 3rd edn. Lippinscott-Raven Publishers; Philadelphia, PA: 1996. pp. 2343–2396. [Google Scholar]
- Kilger E, Kieser A, Baumann M, Hammerschmidt W. Epstein-Barr virus-mediated B-cell proliferation is dependent upon latent membrane protein 1, which simulates an activated CD40 receptor. EMBO J. 1998;17:1700–1709. doi: 10.1093/emboj/17.6.1700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirk O, Pedersen C, Cozzi-Lepri A, Antunes F, Miller V, Gatell JM, Katlama C, Lazzarin A, Skinhoj P, Barton SE. Non-Hodgkin lymphoma in HIV-infected patients in the era of highly active antiretroviral therapy. Blood. 2001;98(12):3406–3412. doi: 10.1182/blood.v98.12.3406. [DOI] [PubMed] [Google Scholar]
- Klaskala W, Brayfield BP, Kankasa C, Bhat G, West JT, Mitchell CD, Wood C. Epidemiological characteristics of human herpesvirus-8 infection in a large population of antenatal women in Zambia. J. Med. Virol. 2005;75(1):93–100. doi: 10.1002/jmv.20242. [DOI] [PubMed] [Google Scholar]
- Klencke BJ, Palefsky JM. Anal cancer: An HIV-associated cancer. Hematol. Oncol. Clin. North Am. 2003;17(3):859–872. doi: 10.1016/s0889-8588(03)00039-x. [DOI] [PubMed] [Google Scholar]
- Klepp O, Dahl O, Stenwig JT. Association of Kaposi’s sarcoma and prior immunosuppressive therapy: A 5-year material of Kaposi’s sarcoma in Norway. Cancer. 1978;42(6):2626–2630. doi: 10.1002/1097-0142(197812)42:6<2626::aid-cncr2820420618>3.0.co;2-7. [DOI] [PubMed] [Google Scholar]
- Kliche S, Nagel W, Kremmer E, Atzler C, Ege A, Knorr T, Koszinowski U, Kolanus W, Haas J. Signaling by human herpesvirus 8 kaposin A through direct membrane recruitment of cytohesin-1. Mol. Cell. 2001;7(4):833–843. doi: 10.1016/s1097-2765(01)00227-1. [DOI] [PubMed] [Google Scholar]
- Knowles DM. Etiology and pathogenesis of AIDS-related non-Hodgkin’s lymphoma. Hematol. Oncol. Clin. North Am. 2003;17(3):785–820. doi: 10.1016/s0889-8588(03)00050-9. [DOI] [PubMed] [Google Scholar]
- Kohl NE, Ruley HE. Role of c-myc in the transformation of REF52 cells by viral and cellular oncogenes. Oncogene. 1987;2(1):41–48. [PubMed] [Google Scholar]
- Koutsky L. Epidemiology of genital human papillomavirus D. infection. Am. J. Med. 1997;102(5A):3–8. doi: 10.1016/s0002-9343(97)00177-0. [DOI] [PubMed] [Google Scholar]
- Krithivas A, Young B, Liao G, Greene D, Hayward SD. Human herpesvirus 8 LANA interacts with proteins of the mSin3 corepressor complex and negatively regulates Epstein-Barr virus gene expression in dually infected PEL cells. J. Virol. 2000;74(20):9637–9645. doi: 10.1128/jvi.74.20.9637-9645.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuo SR, Liu JS, Broker TR, Chow LT. Cell-free replication of the human papillomavirus DNA with homologous viral E1 and E2 proteins and human cell extracts. J. Biol. Chem. 1994;269(39):24058–24065. [PubMed] [Google Scholar]
- Lacoste V, Judde JG, Bestett G, Cadranel J, Antoine M, Valensi F, Delabesse E, Macintyre E, Gessain A. Virological and molecular characterisation of a new B lymphoid cell line, established from an AIDS patient with primary effusion lymphoma, harbouring both KSHV/HHV8 and EBV viruses. Leuk. Lymphoma. 2000;38(34):401–409. doi: 10.3109/10428190009087032. [DOI] [PubMed] [Google Scholar]
- Lagunoff M, Ganem D. The structure and coding organization of the genomic termini of Kaposi’s sarcoma-associated herpesvirus. Virology. 1997;236(1):147–154. doi: 10.1006/viro.1997.8713. [DOI] [PubMed] [Google Scholar]
- Lagunoff M, Majeti R, Weiss A, Ganem D. Deregulated signal transduction by the K1 gene product of Kaposi’s sarcoma-associated herpesvirus. Proc. Natl. Acad. Sci. USA. 1999;96(10):5704–5709. doi: 10.1073/pnas.96.10.5704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langley CL, Benga-De E, Critchlow CW, Ndoye I, Mbengue-Ly MD, Kuypers J, Woto-Gaye G, Mboup S, Bergeron C, Holmes KK, Kiviat NB. HIV-1, HIV-2, human papillomavirus infection and cervical neoplasia in high-risk African women. AIDS. 1996;10(4):413–417. doi: 10.1097/00002030-199604000-00010. [DOI] [PubMed] [Google Scholar]
- Laurence J. Repetitive and consistent cervicovaginal exposure to certain viral pathogens appears to protect against their sexual acquisition in some women: Potential mechanisms. J. Reprod. Immunol. 2003;58(1):79–91. doi: 10.1016/s0165-0378(02)00047-5. [DOI] [PubMed] [Google Scholar]
- Lazzi S, Ferrari F, Nyongo A, Palummo N, de Milito A, Zazzi M, Leoncini L, Luzi P, Tosi P. HIV-associated malignant lymphomas in Kenya (Equatorial Africa) Hum. Pathol. 1998;29(11):1285–1289. doi: 10.1016/s0046-8177(98)90258-1. [DOI] [PubMed] [Google Scholar]
- Lee JK, Hurwitz J. Isolation and characterization of various complexes of the minichromosome maintenance proteins of Schizosaccharomyces pombe. J. Biol. Chem. 2000;275(25):18871–18878. doi: 10.1074/jbc.M001118200. [DOI] [PubMed] [Google Scholar]
- Lee H, Guo J, Li M, Choi JK, DeMaria M, Rosenzweig M, Jung JU. Identification of an immunoreceptor tyrosine-based activation motif of K1 transforming protein of Kaposi’s sarcoma-associated herpesvirus. Mol. Cell Biol. 1998a;18(9):5219–5228. doi: 10.1128/mcb.18.9.5219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee H, Veazey R, Williams K, Li M, Guo J, Neipel F, Fleckenstein B, Lackner A, Desrosiers RC, Jung JU. Deregulation of cell growth by the K1 gene of Kaposi’s sarcoma-associated herpesvirus. Nat. Med. 1998b;4(4):435–440. doi: 10.1038/nm0498-435. [DOI] [PubMed] [Google Scholar]
- Lee BS, Connole M, Tang Z, Harris NL, Jung JU. Structural analysis of the Kaposi’s sarcoma-associated herpesvirus K1 protein. J. Virol. 2003;77(14):8072–8086. doi: 10.1128/JVI.77.14.8072-8086.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lennette ET, Blackbourn DJ, Levy JA. Antibodies to human herpesvirus type 8 in the general population and in Kaposi’s sarcoma patients. Lancet. 1996;348(9031):858–861. doi: 10.1016/S0140-6736(96)03240-0. [DOI] [PubMed] [Google Scholar]
- Levine AM. AIDS-related lymphoma. Semin. Oncol. Nurs. 2006;22(2):80–89. doi: 10.1016/j.soncn.2006.01.004. [DOI] [PubMed] [Google Scholar]
- Li JJ, Huang YQ, Cockerell CJ, Friedman-Kien AE. Localization of human herpes-like virus type 8 in vascular endothelial cells and perivascular spindle-shaped cells of Kaposi’s sarcoma lesions by in situ hybridization. Am. J. Pathol. 1996;148(6):1741–1748. [PMC free article] [PubMed] [Google Scholar]
- Li M, Beard P, Estes PA, Lyon MK, Garcea RL. Intercapsomeric disulfide bonds in papillomavirus assembly and disassembly. J. Virol. 1998;72(3):2160–2167. doi: 10.1128/jvi.72.3.2160-2167.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang Y, Chang J, Lynch SJ, Lukac DM, Ganem D. The lytic switch protein of KSHV activates gene expression via functional interaction with RBP-Jkappa (CSL), the target of the Notch signaling pathway. Genes Dev. 2002;16(15):1977–1989. doi: 10.1101/gad.996502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liebowitz D, Wang D, Kieff E. Orientation and patching of the latent infection membrane protein encoded by Epstein-Barr virus. J. Virol. 1986;58(1):233–237. doi: 10.1128/jvi.58.1.233-237.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lillo FB, Ferrari D, Veglia F, Origoni M, Grasso MA, Lodini S, Mastrorilli E, Taccagni G, Lazzarin A, Uberti-Foppa C. Human papillomavirus infection and associated cervical disease in human immunodeficiency virus-infected women: Effect of highly active antiretroviral therapy. J. Infect. Dis. 2001;184(5):547–551. doi: 10.1086/322856. [DOI] [PubMed] [Google Scholar]
- Londesborough P, Ho L, Terry G, Cuzick J, Wheeler C, Singer A. Human papillomavirus genotype as a predictor of persistence and development of high-grade lesions in women with minor cervical abnormalities. Int. J. Cancer. 1996;69(5):364–368. doi: 10.1002/(SICI)1097-0215(19961021)69:5<364::AID-IJC2>3.0.CO;2-3. [DOI] [PubMed] [Google Scholar]
- Longnecker R. Epstein-Barr virus latency: LMP2, a regulator or means for Epstein-Barr virus persistence? Adv. Cancer Res. 2000;79:175–200. doi: 10.1016/s0065-230x(00)79006-3. [DOI] [PubMed] [Google Scholar]
- Longnecker R, Merchant M, Brown ME, Fruehling S, Bickford JO, Ikeda M, Harty RN. WW- and SH3-domain interactions with Epstein-Barr virus LMP2A. Exp. Cell Res. 2000;257(2):332–340. doi: 10.1006/excr.2000.4900. [DOI] [PubMed] [Google Scholar]
- Lopez Amado M, Castro Lareo I, LozanoRamirez A, Caballero Torcuato L. Epidemiology of otorhinolaryngological squamous cell papillomas in a Spanish subpopulation during a 20 year period. Rev. Laryngol. Otol. Rhinol. (Bord) 1996;117(2):105–110. [PubMed] [Google Scholar]
- Lothe F, Murray JF. Kaposi’s sarcoma: Autopsy findings in the African. Acta Unio Int. Contra Cancrum. 1962;18:429–452. [PubMed] [Google Scholar]
- Mansky KC, Batiza A, Lambert PF. Bovine papillomavirus type 1 E1 and simian virus 40 large T antigen share regions of sequence similarity required for multiple functions. J. Virol. 1997;71(10):7600–7608. doi: 10.1128/jvi.71.10.7600-7608.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mao C, Koutsky LA, Ault KA, Wheeler CM, Brown DR, Wiley DJ, Alvarez FB, Bautista OM, Jansen KU, Barr E. Efficacy of human papillomavirus-16 vaccine to prevent cervical intraepithelial neoplasia: A randomized controlled trial. Obstet. Gynecol. 2006;107(1):18–27. doi: 10.1097/01.AOG.0000192397.41191.fb. [DOI] [PubMed] [Google Scholar]
- Massad LS, Schneider MF, Watts DH, Strickler HD, Melnick S, Palefsky J, Anastos K, Levine AM, Minkoff H. HPV testing for triage of HIV-infected women with papanicolaou smears read as atypical squamous cells of uncertain significance. J. Womens Health (Larchmt) 2004;13(2):147–153. doi: 10.1089/154099904322966128. [DOI] [PubMed] [Google Scholar]
- Mayama S, Cuevas LE, Sheldon J, Omar OH, Smith DH, Okong P, Silvel B, Hart CA, Schulz TF. Prevalence and transmission of Kaposi’s sarcoma-associated herpesvirus (human herpesvirus 8) in Ugandan children and adolescents. Int. J. Cancer. 1998;77(6):817–820. doi: 10.1002/(sici)1097-0215(19980911)77:6<817::aid-ijc2>3.0.co;2-x. [DOI] [PubMed] [Google Scholar]
- Mayaud P, Gill DK, Weiss HA, Uledi E, Kopwe L, Todd J, ka-Gina G, Grosskurth H, Hayes RJ, Mabey DC, Lacey CJ. The interrelation of HIV, cervical human papillomavirus, and neoplasia among antenatal clinic attenders in Tanzania. Sex Transm. Infect. 2001;77(4):248–254. doi: 10.1136/sti.77.4.248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mbulaiteye SM, Biggar RJ, Goedert JJ, Engels EA. Pleural and peritoneal lymphoma among people with AIDS in the United States. J. Acquir. Immune Defic. Syndr. 2002;29(4):418–421. doi: 10.1097/00126334-200204010-00014. [DOI] [PubMed] [Google Scholar]
- Mbulaiteye SM, Pfeiffer RM, Whitby D, Brubaker GR, Shao J, Biggar RJ. Human herpesvirus 8 infection within families in rural Tanzania. J. Infect. Dis. 2003;187(11):1780–1785. doi: 10.1086/374973. [DOI] [PubMed] [Google Scholar]
- McCormick C, Ganem D. Phosphorylation and function of the kaposin B direct repeats of Kaposi’s sarcoma-associated herpesvirus. J. Virol. 2006;80(12):6165–6170. doi: 10.1128/JVI.02331-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGowan JP, Shah S. Long-term remission of AIDS-related primary central nervous system lymphoma associated with highly active antiretroviral therapy. AIDS. 1998;12(8):952–954. [PubMed] [Google Scholar]
- Melbye M, Cook PM, Hjalgrim H, Begtrup K, Simpson GR, Biggar RJ, Ebbesen P, Schulz TF. Risk factors for Kaposi’s-sarcoma-associated herpesvirus(KSHV/HHV-8) seropositivity in a cohort of homosexual men, 1981-1996. Int. J. Cancer. 1998;77(4):543–548. doi: 10.1002/(sici)1097-0215(19980812)77:4<543::aid-ijc12>3.0.co;2-7. [DOI] [PubMed] [Google Scholar]
- Memar OM, Rady PL, Tyring SK. Human herpesvirus-8: Detection of novel herpesvirus-like DNA sequences in Kaposi’s sarcoma and other lesions. J. Mol. Med. 1995;73(12):603–609. doi: 10.1007/BF00196354. [DOI] [PubMed] [Google Scholar]
- Milde-Langosch KM, Loning T, Meichsner M, Henke RP. HPV infections in tumors of the upper respiratory and digestive tract. In situ hybridization and dot-blot hybridization with biotinylated DNA probes) Acta Histochem. Suppl. 1989;37:103–108. [PubMed] [Google Scholar]
- Miller WE, Earp HS, Raab-Traub N. The Epstein-Barr virus latent membrane protein1 induces expression of the epidermal growth factor receptor. J. Virol. 1995;69:4390–4398. doi: 10.1128/jvi.69.7.4390-4398.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller WE, Mosialos G, Kieff E, Raab-Traub N. Epstein-Barr virus LMP1 induction of the EGFR is mediated through a TRAF signaling pathway distinct from NF-κB activation. J. Virol. 1997;71:586–594. doi: 10.1128/jvi.71.1.586-594.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller W, Cheshire J, Raab-Traub N. Interaction of tumor necrosis factor receptor-associated factor signaling proteins with the latent membrane protein 1 PXQXT motif is essential for induction of epidermal growth factor receptor expression. Mol. Cell Biol. 1998;18:2835–2844. doi: 10.1128/mcb.18.5.2835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minchiotti S, Masucci L, Serapiao Dos Santos M, Perrella E, Graffeo R, Lambiase A, Bonini S. Conjunctival papilloma and human papillomavirus: Identification of HPV types by PCR. Eur. J. Ophthalmol. 2006;16(3):473–477. doi: 10.1177/112067210601600320. [DOI] [PubMed] [Google Scholar]
- Mincione GP, Taddei GL, Wolovsky M, Calzolari A, Mincione F. Detection of human papillomavirus (HPV) DNA type 6/11 in a conjunctival papilloma by in situ hybridization with biotinylated probes. Pathologica. 1992;84(1092):483–488. [PubMed] [Google Scholar]
- Mineta H, Ogino T, Amano HM, Ohkawa Y, Araki K, Takebayashi S, Miura K. Human papilloma virus (HPV) type 16 and 18 detected in head and neck squamous cell carcinoma. Anticancer Res. 1998;18(6B):4765–4768. [PubMed] [Google Scholar]
- Minkoff H, Ahdieh L, Massad LS, Anastos K, Watts DH, Melnick S, Muderspach L, Burk R, Palefsky J. The effect of highly active antiretroviral therapy on cervical cytologic changes associated with oncogenic HPV among HIV-infected women. AIDS. 2001;15(16):2157–2164. doi: 10.1097/00002030-200111090-00011. [DOI] [PubMed] [Google Scholar]
- Mitchell T, Sugden B. Stimulation of NF-κB-mediated transcription by mutant derivatives of the latent membrane protein of Epstein-Barr virus. J. Virol. 1995;69(5):2968–2976. doi: 10.1128/jvi.69.5.2968-2976.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molden J, Chang Y, You Y, Moore PS, Goldsmith MA. A Kaposi’s sarcoma-associated herpesvirus-encoded cytokine homolog (vIL-6) activates signaling through the shared gp130 receptor subunit. J. Biol. Chem. 1997;272(31):19625–19631. doi: 10.1074/jbc.272.31.19625. [DOI] [PubMed] [Google Scholar]
- Montaner S, Sodhi A, Molinolo A, Bugge TH, Sawai ET, He Y, Li Y, Ray PE, Gutkind JS. Endothelial infection with KSHV genes in vivo reveals that vGPCR initiates Kaposi’s sarcomagenesis and can promote the tumorigenic potential of viral latent genes. Cancer Cell. 2003;3(1):23–36. doi: 10.1016/s1535-6108(02)00237-4. [DOI] [PubMed] [Google Scholar]
- Moore P, Chang Y. Kaposi’s sacrcoma-associated herpesvirus. In: Knipe DM, editor. Virology. Lippincott Williams & Wilkins; Philadelphia, PA: 2001. pp. 2803–2833. [Google Scholar]
- Moore PS, Boshoff C, Weiss RA, Chang Y. Molecular mimicry of human cytokine and cytokine response pathway genes by KSHV. Science. 1996a;274(5293):1739–1744. doi: 10.1126/science.274.5293.1739. [DOI] [PubMed] [Google Scholar]
- Moore PS, Gao SJ, Dominguez G, Cesarman E, Lungu O, Knowles DM, Garber R, Pellett PE, McGeoch DJ, Chang Y. Primary characterization of a herpesvirus agent associated with Kaposi’s sarcomae. J. Virol. 1996b;70(1):549–558. doi: 10.1128/jvi.70.1.549-558.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore PS, Kingsley LA, Holmberg SD, Spira T, Gupta P, Hoover DR, Parry JP, Conley LJ, Jaffe HW, Chang Y. Kaposi’s sarcoma-associated herpesvirus infection prior to onset of Kaposi’s sarcoma. AIDS. 1996c;10(2):175–180. doi: 10.1097/00002030-199602000-00007. [DOI] [PubMed] [Google Scholar]
- Morison L. The global epidemiology of HIV/AIDS. Br. Med. Bull. 2001;58:7–18. doi: 10.1093/bmb/58.1.7. [DOI] [PubMed] [Google Scholar]
- Moses AV, Fish KN, Ruhl R, Smith PP, Strussenberg JG, Zhu L, Chandran B, Nelson JA. Long-term infection and transformation of dermal microvascular endothelial cells by human herpesvirus 8. J. Virol. 1999;73(8):6892–6902. doi: 10.1128/jvi.73.8.6892-6902.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mosunjac MB, Tadros T, Beach R, Majmudar B. Cervical schistosomiasis, human papilloma virus (HPV), and human immunodeficiency virus (HIV): A dangerous coexistence or coincidence? Gynecol. Oncol. 2003;90(1):211–214. doi: 10.1016/s0090-8258(03)00191-4. [DOI] [PubMed] [Google Scholar]
- Motti PG, Dallabetta GA, Daniel RW, Canner JK, Chiphangwi JD, Liomba GN, Yang L, Shah KV. Cervical abnormalities, human papillomavirus, and human immunodeficiency virus infections in women in Malawi. J. Infect. Dis. 1996;173(3):714–717. doi: 10.1093/infdis/173.3.714. [DOI] [PubMed] [Google Scholar]
- Mueller N. Overview of the epidemiology of malignancy in immune deficiency. J. Acquir. Immune Defic. Syndr. 1999;21(Suppl 1):S5–S10. [PubMed] [Google Scholar]
- Munger K, Phelps WC, Bubb V, Howley PM, Schlegel R. The E6 and E7 genes of the human papillomavirus type 16 together are necessary and sufficient for transformation of primary human keratinocytes. J. Virol. 1989;63(10):4417–4421. doi: 10.1128/jvi.63.10.4417-4421.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munoz N, Bosch FX, Castellsague X, Diaz M, de Sanjose S, Hammouda D, Shah KV, Meijer CJ. Against which human papillomavirus types shall we vaccinate and screen? The international perspective. Int. J. Cancer. 2004;111(2):278–285. doi: 10.1002/ijc.20244. [DOI] [PubMed] [Google Scholar]
- Muralidhar S, Pumfery AM, Hassani M, Sadaie MR, Kishishita M, Brady JN, Doniger J, Medveczky P, Rosenthal LJ. Identification of kaposin (open reading frame K12) as a human herpesvirus 8 (Kaposi’s sarcoma-associated herpesvirus) transforming gene. J. Virol. 1998;72(6):4980–4988. doi: 10.1128/jvi.72.6.4980-4988.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muralidhar S, Veytsmann G, Chandran B, Ablashi D, Doniger J, Rosenthal LJ. Characterization of the human herpesvirus 8 (Kaposi’s sarcoma-associated herpesvirus) oncogene, kaposin (ORF K12) J. Clin. Virol. 2000;16(3):203–213. doi: 10.1016/s1386-6532(99)00081-5. [DOI] [PubMed] [Google Scholar]
- Nador RG, Cesarman E, Chadburn A, Dawson DB, Ansari MQ, Sald J, Knowles DM. Primary effusion lymphoma: A distinct clinicopathologic entity associated with the Kaposi’s sarcoma-associated herpes virus. Blood. 1996;88(2):645–656. [PubMed] [Google Scholar]
- Nakano K. Characteristics of human papillomavirus (HPV) infection in papilloma of the head and neck—detection of HPV according to clinical features and type specificity in the head and neck. Nippon Jibiinkoka Gakkai Kaiho. 1994;97(8):1381–1392. doi: 10.3950/jibiinkoka.97.1381. [DOI] [PubMed] [Google Scholar]
- Nakano K, Isegawa Y, Zou P, Tadagaki K, Inagi R, Yamanishi K. Kaposi’s sarcoma-associated herpesvirus (KSHV)-encoded vMIP-I and vMIP-II induce signal transduction and chemotaxis in monocytic cells. Arch. Virol. 2003;148(5):871–890. doi: 10.1007/s00705-002-0971-7. [DOI] [PubMed] [Google Scholar]
- Navarro WH, Kaplan LD. AIDS-related lymphoproliferative disease. Blood. 2006;107(1):13–20. doi: 10.1182/blood-2004-11-4278. [DOI] [PubMed] [Google Scholar]
- Newell ME, Hoy JF, Cooper SG, DeGraaff B, Grulich AE, Bryant M, Millar JL, Brew BJ, Quinn DI. Human immunodeficiency virus-related primary central nervous system lymphoma: Factors influencing survival in 111 patients. Cancer. 2004;100(12):2627–2636. doi: 10.1002/cncr.20300. [DOI] [PubMed] [Google Scholar]
- Newton R, Ziegler J, Ateenyi-Agaba C, Bousarghin L, Casabonne D, Beral V, Mbidde E, Carpenter L, Reeves G, Parkin DM, Wabinga H, Mbulaiteye S, et al. The epidemiology of conjunctival squamous cell carcinoma in Uganda. Br. J. Cancer. 2002;87(3):301–308. doi: 10.1038/sj.bjc.6600451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicholas J, Ruvolo VR, Burns WH, Sandford G, Wan X, Ciufo D, Hendrickson SB, Guo HG, Hayward GS, Reitz MS. Kaposi’s sarcoma-associated human herpesvirus-8 encodes homologues of macrophage inflammatory protein-1 and interleukin-6. Nat. Med. 1997;3(3):287–292. doi: 10.1038/nm0397-287. [DOI] [PubMed] [Google Scholar]
- Noy A. Update in HIV lymphoma. Curr. Opin. Oncol. 2006;18(5):449–455. doi: 10.1097/01.cco.0000239883.23839.ac. [DOI] [PubMed] [Google Scholar]
- Nzila N, Laga M, Thiam MA, Mayimona K, Edidi B, Van Dyck E, Behets F, Hassig S, Nelson A, Mokwa K, et al. HIV and other sexually transmitted diseases among female prostitutes in Kinshasa. AIDS. 1991;5(6):715–721. doi: 10.1097/00002030-199106000-00011. [DOI] [PubMed] [Google Scholar]
- O’Farrell N, Hoosen AA, Kharsany AB, van den Ende J. Sexually transmitted pathogens in pregnant women in a rural South African community. Genitourin Med. 1989;65(4):276–280. doi: 10.1136/sti.65.4.276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oettle AG. Geographical and racial differences in the frequency of Kaposi’s sarcoma as evidence of environmental or genetic causes. Acta Unio Int. Contra Cancrum. 1962;18:330–363. [PubMed] [Google Scholar]
- Olsen SJ, Chang Y, Moore PS, Biggar RJ, Melbye M. Increasing Kaposi’s sarcoma-associated herpesvirus seroprevalence with age in a highly Kaposi’s sarcoma endemic region, Zambia in 1985. AIDS. 1998;12(14):1921–1925. doi: 10.1097/00002030-199814000-00024. [DOI] [PubMed] [Google Scholar]
- Ong CK, Chan SY, Campo MS, Fujinaga K, Mavromara-Nazos P, Labropoulou V, Pfister H, Tay SK, Meulen J, Villa LL, et al. Evolution of human papillomavirus type 18: An ancient phylogenetic root in Africa and intratype diversity reflect coevolution with human ethnic groups. J. Virol. 1993;67(11):6424–6431. doi: 10.1128/jvi.67.11.6424-6431.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pagano JS. Epstein-Barr virus: The first human tumor virus and its role in cancer. Proc. Assoc. Am. Physicians. 1999;111:573–580. doi: 10.1046/j.1525-1381.1999.t01-1-99220.x. [DOI] [PubMed] [Google Scholar]
- Palefsky J. Human papillomavirus infection among HIV-infected individuals. Implications for development of malignant tumors. Hematol. Oncol. Clin. North Am. 1991;5(2):357–370. [PubMed] [Google Scholar]
- Palefsky JM. Cervical human papillomavirus infection and cervical intraepithelial neoplasia in women positive for human immunodeficiency virus in the era of highly active antiretroviral therapy. Curr. Opin. Oncol. 2003;15(5):382–388. doi: 10.1097/00001622-200309000-00007. [DOI] [PubMed] [Google Scholar]
- Palefsky JM, Barrasso R. HPV infection and disease in men. Obstet. Gynecol. Clin. North Am. 1996;23(4):895–916. doi: 10.1016/s0889-8545(05)70281-3. [DOI] [PubMed] [Google Scholar]
- Pantanowitz L, Schlecht HP, Dezube BJ. The growing problem of non-AIDS-defining malignancies in HIV. Curr. Opin. Oncol. 2006;18(5):469–478. doi: 10.1097/01.cco.0000239886.13537.ed. [DOI] [PubMed] [Google Scholar]
- Parkin DM, Garcia-Giannoli H, Raphael M, Martin A, Katangole-Mbidde E, Wabinga H, Ziegler J. Non-Hodgkin lymphoma in Uganda: A case-control study. AIDS. 2000;14(18):2929–2936. doi: 10.1097/00002030-200012220-00015. [DOI] [PubMed] [Google Scholar]
- Patil P, Elem B, Zumla A. Pattern of adult malignancies in Zambia (1980-1989) in light of the human immunodeficiency virus type 1 epidemic. J. Trop. Med. Hyg. 1995;98(4):281–284. [PubMed] [Google Scholar]
- Pellett PE, Wright DJ, Engels EA, Ablashi DV, Dollard SC, Forghani B, Glynn SA, Goedert JJ, Jenkins FJ, Lee TH, Neipel F, Todd DS, et al. Multicenter comparison of serologic assays and estimation of human herpesvirus 8 seroprevalence among US blood donors. Transfusion. 2003;43(9):1260–1268. doi: 10.1046/j.1537-2995.2003.00490.x. [DOI] [PubMed] [Google Scholar]
- Penn I. Kaposi’s sarcoma in organ transplant recipients: Report of 20 cases. Transplantation. 1979;27(1):8–11. doi: 10.1097/00007890-197901000-00003. [DOI] [PubMed] [Google Scholar]
- Peyton CL, Gravitt PE, Hunt WC, Hundley RS, Zhao M, Apple RJ, Wheeler CM. Determinants of genital human papillomavirus detection in a US population. J. Infect. Dis. 2001;183(11):1554–1564. doi: 10.1086/320696. [DOI] [PubMed] [Google Scholar]
- Piketty C, Darragh TM, Da Costa M, Bruneval P, Heard I, Kazatchkine MD, Palefsky JM. High prevalence of anal human papillomavirus infection and anal cancer precursors among HIV-infected persons in the absence of anal intercourse. Ann. Intern. Med. 2003;138(6):453–459. doi: 10.7326/0003-4819-138-6-200303180-00008. [DOI] [PubMed] [Google Scholar]
- Piketty C, Darragh TM, Heard I, Da Costa M, Bruneval P, Kazatchkine MD, Palefsky JM. High prevalence of anal squamous intraepithelial lesions in HIV-positive men despite the use of highly active antiretroviral therapy. Sex Transm. Dis. 2004;31(2):96–99. doi: 10.1097/01.OLQ.0000109515.75864.2B. [DOI] [PubMed] [Google Scholar]
- Plancoulaine S, Abel L, van Beveren M, Tregouet DA, Joubert M, Tortevoye P, de The G, Gessain A. Human herpesvirus 8 transmission from mother to child and between siblings in an endemic population. Lancet. 2000;356(9235):1062–1065. doi: 10.1016/S0140-6736(00)02729-X. [DOI] [PubMed] [Google Scholar]
- Plancoulaine S, Abel L, Tregouet D, Duprez R, van Beveren M, Tortevoye P, Froment A, Gessain A. Respective roles of serological status and blood specific antihuman herpesvirus 8 antibody levels in human herpesvirus 8 intrafamilial transmission in a highly endemic area. Cancer Res. 2004;64(23):8782–8787. doi: 10.1158/0008-5472.CAN-04-2000. [DOI] [PubMed] [Google Scholar]
- Ponten J, Guo Z. Precancer of the human cervix. Cancer Surv. 1998;32:201–229. [PubMed] [Google Scholar]
- Prakash O, Tang ZY, Peng X, Coleman R, Gill J, Farr G, Samaniego F. Tumorigenesis and aberrant signaling in transgenic mice expressing the human herpesvirus-8 K1 gene. J. Natl. Cancer Inst. 2002;94(12):926–935. doi: 10.1093/jnci/94.12.926. [DOI] [PubMed] [Google Scholar]
- Quinlivan EB, Zhang C, Stewart PW, Komoltri C, Davis MG, Wehbie RS. Elevated virus loads of Kaposi’s sarcoma-associated human herpesvirus 8 predict Kaposi’s sarcoma disease progression, but elevated levels of human immunodeficiency virus type 1 do not. J. Infect. Dis. 2002;185(12):1736–1744. doi: 10.1086/340652. [DOI] [PubMed] [Google Scholar]
- Rabkin CS, Schulz TF, Whitby D, Lennette ET, Magpantay LI, Chatlynne L, Biggar RJ, HHV-8 Interlaboratory Collaborative Groups Interassay correlation of human herpesvirus 8 serologic tests. J. Infect. Dis. 1998;178(2):304–309. doi: 10.1086/515649. [DOI] [PubMed] [Google Scholar]
- Radkov SA, Kellam P, Boshoff C. The latent nuclear antigen of Kaposi sarcoma-associated herpesvirus targets the retinoblastoma-E2F pathway and with the oncogene Hras transforms primary rat cells. Nat. Med. 2000;6(10):1121–1127. doi: 10.1038/80459. [DOI] [PubMed] [Google Scholar]
- Ralston R. Complementation of transforming domains in E1a/myc chimaeras. Nature. 1991;353(6347):866–868. doi: 10.1038/353866a0. [DOI] [PubMed] [Google Scholar]
- Reszec J, Sulkowski S. The expression of P53 protein and infection of human papilloma virus in conjunctival and eyelid neoplasms. Int. J. Mol. Med. 2005;16(4):559–564. [PubMed] [Google Scholar]
- Center for Infectious Diseases Council of State and Territorial Epidemiologists AIDS Program. MMWR Morb. Mortal Wkly. Rep. 1987;36(Suppl 1):1S–15S. Revision of the CDC surveillance case definition for acquired immunodeficiency syndrome. [PubMed] [Google Scholar]
- Rickinson AB, Kieff E. Epstein-Barr Virus. In: Fields BN, Knipe DM, Howley PM, editors. Virology. 3rd edn. Lippinscott-Raven Publishers; Philadelphia, PA: 1996. pp. 2397–2446. [Google Scholar]
- Roizmann B, Desrosiers RC, Fleckenstein B, Lopez C, Minson AC, Studdert MJ. The family herpesviridae: An update. The herpesvirus study group of the international committee on taxonomy of viruses. Arch. Virol. 1992;123(34):425–449. doi: 10.1007/BF01317276. [DOI] [PubMed] [Google Scholar]
- Russo JJ, Bohenzky RA, Chien MC, Chen J, Yan M, Maddalena D, Parry JP, Peruzzi D, Edelman IS, Chang Y, Moore PS. Nucleotide sequence of the Kaposi sarcoma-associated herpesvirus (HHV8) Proc. Natl. Acad. Sci. USA. 1996;93(25):14862–14867. doi: 10.1073/pnas.93.25.14862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sadler R, Wu L, Forghani B, Renne R, Zhong W, Herndier B, Ganem D. A complex translational program generates multiple novel proteins from the latently expressed kaposin (K12) locus of Kaposi’s sarcoma-associated herpesvirus. J. Virol. 1999;73(7):5722–5730. doi: 10.1128/jvi.73.7.5722-5730.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakai H, Yasugi T, Benson JD, Dowhanick JJ, Howley PM. Targeted mutagenesis of the human papillomavirus type 16 E2 transactivation domain reveals separable transcriptional activation and DNA replication functions. J. Virol. 1996;70(3):1602–1611. doi: 10.1128/jvi.70.3.1602-1611.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sandberg M, Hammerschmidt W, Sugden B. Characterizarion of LMP1’s association with TRAF1, TRAF2, and TRAF3. J. Virol. 1997;71:4649–4656. doi: 10.1128/jvi.71.6.4649-4656.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sapp M, Perez-Ordonez B, Brenneman F, Imrie K, Morava-Protzner I, Lim MS. EBV-associated perianal Hodgkin’s disease in an HIV-positive individual. Am. J. Hematol. 2001;66(1):42–45. doi: 10.1002/1096-8652(200101)66:1<42::AID-AJH1006>3.0.CO;2-V. [DOI] [PubMed] [Google Scholar]
- Schuman P, Ohmit SE, Klein RS, Duerr A, Cu-Uvin S, Jamieson DJ, Anderson J, Shah KV. Longitudinal study of cervical squamous intraepithelial lesions in human immunodeficiency virus (HIV)-seropositive and at-risk HIV-seronegative women. J. Infect. Dis. 2003;188(1):128–136. doi: 10.1086/375783. [DOI] [PubMed] [Google Scholar]
- Schwarz E, Freese UK, Gissmann L, Mayer W, Roggenbuck B, Stremlau A, zur Hausen H. Structure and transcription of human papillomavirus sequences in cervical carcinoma cells. Nature. 1985;314(6006):111–114. doi: 10.1038/314111a0. [DOI] [PubMed] [Google Scholar]
- Sedman J, Stenlund A. Co-operative interaction between the initiator E1 and the transcriptional activator E2 is required for replicator specific DNA replication of bovine papillomavirus in vivo and in vitro. EMBO J. 1995;14(24):6218–6228. doi: 10.1002/j.1460-2075.1995.tb00312.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seif I. Sequence homology between the large tumor antigen of polyoma viruses and the putative E1 protein of papilloma viruses. Virology. 1984;138(2):347–352. doi: 10.1016/0042-6822(84)90359-3. [DOI] [PubMed] [Google Scholar]
- Serwadda D, Wawer MJ, Shah KV, Sewankambo NK, Daniel R, Li C, Lorincz A, Meehan MP, Wabwire-Mangen F, Gray RH. Use of a hybrid capture assay of self-collected vaginal swabs in rural Uganda for detection of human papillomavirus. J. Infect. Dis. 1999;180(4):1316–1319. doi: 10.1086/315026. [DOI] [PubMed] [Google Scholar]
- Shalloway D, Johnson PJ, Freed EO, Coulter D, Flood WA., Jr. Transformation of NIH 3T3 cells by cotransfection with c-src and nuclear oncogenes. Mol. Cell Biol. 1987;7(10):3582–3590. doi: 10.1128/mcb.7.10.3582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaw KV, Howley PM. Papillomavirinae: The viruses and their replication. In: Knipe DM, Howley PM, Griffin DE, Lamb RA, Martin MA, editors. Fields Virology. 4th edn. Lippincott Williams & Wilkins; Philadelphia, PA: 2001. [Google Scholar]
- Shibata D, Weiss LM, Hernandez AM, Nathwani BN, Bernstein L, Levine AM. Epstein-Barr virus-associated non-Hodgkin’s lymphoma in patients infected with the human immunodeficiency virus. Blood. 1993;81(8):2102–2109. [PubMed] [Google Scholar]
- Silverberg MJ, Ahdieh L, Munoz A, Anastos K, Burk RD, Cu-Uvin S, Duerr A, Greenblatt RM, Klein RS, Massad S, Minkoff H, Muderspach L, et al. The impact of HIV infection and immunodeficiency on human papillomavirus type 6 or 11 infection and on genital warts. Sex Transm. Dis. 2002;29(8):427–435. doi: 10.1097/00007435-200208000-00001. [DOI] [PubMed] [Google Scholar]
- Simpson GR, Schulz TF, Whitby D, Cook PM, Boshoff C, Rainbow L, Howard MR, Gao SJ, Bohenzky RA, Simmonds P, Lee C, de Ruiter A, et al. Prevalence of Kaposi’s sarcoma associated herpesvirus infection measured by antibodies to recombinant capsid protein and latent immunofluorescence antigen. Lancet. 1996;348(9035):1133–1138. doi: 10.1016/S0140-6736(96)07560-5. [DOI] [PubMed] [Google Scholar]
- Sinclair KA, Woods CR, Kirse DJ, Sinal SH. Anogenital and respiratory tract human papillomavirus infections among children: Age, gender, and potential transmission through sexual abuse. Pediatrics. 2005;116(4):815–825. doi: 10.1542/peds.2005-0652. [DOI] [PubMed] [Google Scholar]
- Sirera G, Videla S, Pinol M, Canadas MP, Llatjos M, Ballesteros AL, Garcia-Cuyas F, Castella E, Guerola R, Tural C, Rey-Joly C, Clotet B. High prevalence of human papillomavirus infection in the anus, penis and mouth in HIV-positive men. AIDS. 2006;20(8):1201–1204. doi: 10.1097/01.aids.0000226963.10342.f4. [DOI] [PubMed] [Google Scholar]
- Sodhi A, Montaner S, Patel V, Zohar M, Bais C, Mesri EA, Gutkind JS. The Kaposi’s sarcoma-associated herpes virus G protein-coupled receptor up-regulates vascular endothelial growth factor expression and secretion through mitogen-activated protein kinase and p38 pathways acting on hypoxia-inducible factor 1alpha. Cancer Res. 2000;60(17):4873–4880. [PubMed] [Google Scholar]
- Soulier J, Grollet L, Oksenhendler E, Cacoub P, Cazals-Hatem D, Babinet P, d’Agay MF, Clauvel JP, Raphael M, Degos L. Kaposi’s sarcoma-associated herpesvirus-like DNA sequences in multicentric Castleman’s disease. Blood. 1995;86(4):1276–1280. [PubMed] [Google Scholar]
- Sparano JA. Human immunodeficiency virus associated lymphoma. Curr. Opin. Oncol. 2003;15(5):372–378. doi: 10.1097/00001622-200309000-00005. [DOI] [PubMed] [Google Scholar]
- Sparano JA, Anand K, Desai J, Mitnick RJ, Kalkut GE, Hanau LH. Effect of highly active antiretroviral therapy on the incidence of HIV-associated malignancies at an urban medical center. J. Acquir. Immune Defic. Syndr. 1999;21(Suppl 1):S18–S22. [PubMed] [Google Scholar]
- St. Louis ME, Icenogle JP, Manzila T, Kamenga M, Ryder RW, Heyward WL, Reeves WC. Genital types of papillomavirus in children of women with HIV-1 infection in Kinshasa, Zaire. Int. J. Cancer. 1993;54(2):181–184. doi: 10.1002/ijc.2910540203. Louis. [DOI] [PubMed] [Google Scholar]
- Stauffer Y, Raj K, Masternak K, Beard P. Infectious human papillomavirus type 18 pseudovirions. J. Mol. Biol. 1998;283(3):529–536. doi: 10.1006/jmbi.1998.2113. [DOI] [PubMed] [Google Scholar]
- Strickler HD, Palefsky JM, Shah KV, Anastos K, Klein RS, Minkoff H, Duerr A, Massad LS, Celentano DD, Hall C, Fazzari M, Cu-Uvin S, et al. Human papillomavirus type 16 and immune status in human immunodeficiency virus-seropositive women. J. Natl. Cancer Inst. 2003;95(14):1062–1071. doi: 10.1093/jnci/95.14.1062. [DOI] [PubMed] [Google Scholar]
- Syrjanen S. Human papillomavirus infections and oral tumors. Med. Microbiol. Immunol. (Berl.) 2003;192(3):123–128. doi: 10.1007/s00430-002-0173-7. [DOI] [PubMed] [Google Scholar]
- Syrjanen S. Human papillomavirus (HPV) in head and neck cancer. J. Clin. Virol. 2005;32(Suppl 1):S59–S66. doi: 10.1016/j.jcv.2004.11.017. [DOI] [PubMed] [Google Scholar]
- Tabrizi SN, McCurrach FE, Drewe RH, Borg AJ, Garland SM, Taylor HR. Human papillomavirus in corneal and conjunctival carcinoma. Aust. N. Z. J. Ophthalmol. 1997;25(3):211–215. doi: 10.1111/j.1442-9071.1997.tb01394.x. [DOI] [PubMed] [Google Scholar]
- Tedeschi R, Dillner J, De Paoli P. Laboratory diagnosis of human herpesvirus 8 infection in humans. Eur. J. Clin. Microbiol. Infect. Dis. 2002;21(12):831–844. doi: 10.1007/s10096-002-0836-8. [DOI] [PubMed] [Google Scholar]
- ter Meulen J, Eberhardt HC, Luande J, Mgaya HN, Chang-Claude J, Mtiro H, Mhina M, Kashaija P, Ockert S, Yu X, Meinhardt G, Gissmann L, et al. Human papillomavirus (HPV) infection, HIV infection and cervical cancer in Tanzania, east Africa. Int. J. Cancer. 1992;51(4):515–521. doi: 10.1002/ijc.2910510403. [DOI] [PubMed] [Google Scholar]
- Thomas JO. Acquired immunodeficiency syndrome-associated cancers in sub-Saharan Africa. Semin. Oncol. 2001;28(2):198–206. doi: 10.1016/s0093-7754(01)90092-2. [DOI] [PubMed] [Google Scholar]
- Thomas JO, Herrero R, Omigbodun AA, Ojemakinde K, Ajayi IO, Fawole A, Oladepo O, Smith JS, Arslan A, Munoz N, Snijders PJ, Meijer CJ, et al. Prevalence of papillomavirus infection in women in Ibadan, Nigeria: A population-based study. Br. J. Cancer. 2004;90(3):638–645. doi: 10.1038/sj.bjc.6601515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thome M, Schneider P, Hofmann K, Fickenscher H, Meinl E, Neipel F, Mattmann C, Burns K, Bodmer JL, Schroter M, Scaffidi C, Krammer PH, et al. Viral FLICE-inhibitory proteins (FLIPs) prevent apoptosis induced by death receptors. Nature. 1997;386(6624):517–521. doi: 10.1038/386517a0. [DOI] [PubMed] [Google Scholar]
- Tomkowicz B, Singh SP, Lai D, Singh A, Mahalingham S, Joseph J, Srivastava S, Srinivasan A. Mutational analysis reveals an essential role for the LXXLL motif in the transformation function of the human herpesvirus-8 oncoprotein, kaposin. DNA Cell Biol. 2005;24(1):10–20. doi: 10.1089/dna.2005.24.10. [DOI] [PubMed] [Google Scholar]
- Touze A, El Mehdaoui S, Sizaret PY, Mougin C, Munoz N, Coursaget P. The L1 major capsid protein of human papillomavirus type 16 variants affects yield of virus-like particles produced in an insect cell expression system. J. Clin. Microbiol. 1998;36(7):2046–2051. doi: 10.1128/jcm.36.7.2046-2051.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trivedi P, Takazawa K, Zompetta C, Cuomo L, Anastasiadou E, Carbone A, Uccini S, Belardelli F, Takada K, Frati L, Faggioni A. Infection of HHV-8+ primary effusion lymphoma cells with a recombinant Epstein-Barr virus leads to restricted EBV latency, altered phenotype, and increased tumorigenicity without affecting TCL1 expression. Blood. 2004;103(1):313–316. doi: 10.1182/blood-2003-05-1710. [DOI] [PubMed] [Google Scholar]
- Update on acquired immune deficiency syndrome (AIDS)— United States. MMWR Morb. Mortal Wkly. Rep. 1982;31(37):507–508. 513–504. [PubMed] [Google Scholar]
- Update: National Breast and Cervical Cancer Early Detection Program July 1991-July 1992. MMWR Morb. Mortal Wkly. Rep. 1992;41(40):739–743. [PubMed] [Google Scholar]
- Villa LL, Costa RL, Petta CA, Andrade RP, Ault KA, Giuliano AR, Wheeler CM, Koutsky LA, Malm C, Lehtinen M, Skjeldestad FE, Olsson SE, et al. Prophylactic quadrivalent human papillomavirus (types 6, 11, 16, and 18) L1 virus-like particle vaccine in young women: A randomised double-blind placebo-controlled multi-centre phase II efficacy trial. Lancet Oncol. 2005;6(5):271–278. doi: 10.1016/S1470-2045(05)70101-7. [DOI] [PubMed] [Google Scholar]
- von Krogh G, Wikstrom A, Syrjanen K, Syrjanen S. Anal and penile condylomas in HIV-negative and HIV-positive men: Clinical, histological and virological characteristics correlated to therapeutic outcome. Acta Derm. Venereol. 1995;75(6):470–474. doi: 10.2340/0001555575470474. [DOI] [PubMed] [Google Scholar]
- Waddell KM, Lewallen S, Lucas SB, Atenyi-Agaba C, Herrington CS, Liomba G. Carcinoma of the conjunctiva and HIV infection in Uganda and Malawi. Br. J. Ophthalmol. 1996;80(6):503–508. doi: 10.1136/bjo.80.6.503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waddell K, Magyezi J, Bousarghin L, Coursaget P, Lucas S, Downing R, Casabonne D, Newton R. Antibodies against human papillomavirus type 16 (HPV-16) and conjunctival squamous cell neoplasia in Uganda. Br. J. Cancer. 2003;88(12):2002–2003. doi: 10.1038/sj.bjc.6600950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walboomers JM, Jacobs MV, Manos MM, Bosch FX, Kummer JA, Shah KV, Snijders PJ, Peto J, Meijer CJ, Munoz N. Human papillomavirus is a necessary cause of invasive cervical cancer worldwide. J. Pathol. 1999;189(1):12–19. doi: 10.1002/(SICI)1096-9896(199909)189:1<12::AID-PATH431>3.0.CO;2-F. [DOI] [PubMed] [Google Scholar]
- Wang HW, Boshoff C. Linking Kaposi virus to cancer-associated cytokines. Trends Mol. Med. 2005;11(7):309–312. doi: 10.1016/j.molmed.2005.06.002. [DOI] [PubMed] [Google Scholar]
- Wang D, Leibowitz D, Kieff E. An EBV membrane protein expressed in immortalized lymphocytes transforms established rodent cells. Cell. 1985;43:831–840. doi: 10.1016/0092-8674(85)90256-9. [DOI] [PubMed] [Google Scholar]
- Wang F, Gregory C, Sample C, Rowe M, Liebowitz D, Murray R, Rickinson A, Kieff E. Epstein-Barr virus latent membrane protein (LMP1) and nuclear proteins 2 and 3C are effectors of phenotypic changes in B lymphocytes: EBNA-2 and LMP1 cooperatively induce CD23. J. Virol. 1990;64(5):2309–2318. doi: 10.1128/jvi.64.5.2309-2318.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinberg RA. The action of oncogenes in the cytoplasm and nucleus. Science. 1985;230(4727):770–776. doi: 10.1126/science.2997917. [DOI] [PubMed] [Google Scholar]
- Weinberg RA. Oncogenes, antioncogenes, and the molecular bases of multistep carcinogenesis. Cancer Res. 1989;49(14):3713–3721. [PubMed] [Google Scholar]
- Whitby D, Howard MR, Tenant-Flowers M, Brink NS, Copas A, Boshoff C, Hatzioannou T, Suggett FE, Aldam DM, Denton AS, et al. Detection of Kaposi sarcoma associated herpesvirus in peripheral blood of HIV-infected individuals and progression to Kaposi’s sarcoma. Lancet. 1995;346(8978):799–802. doi: 10.1016/s0140-6736(95)91619-9. [DOI] [PubMed] [Google Scholar]
- Wilkinson D, Sheldon J, Gilks CF, Schulz TF. Prevalence of infection with human herpesvirus 8/Kaposi’s sarcoma herpesvirus in rural South Africa. S. Afr. Med. J. 1999;89(5):554–557. [PubMed] [Google Scholar]
- Williams AB, Darragh TM, Vranizan K, Ochia C, Moss AR, Palefsky JM. Anal and cervical human papillomavirus infection and risk of anal and cervical epithelial abnormalities in human immunodeficiency virus-infected women. Obstet. Gynecol. 1994;83(2):205–211. [PubMed] [Google Scholar]
- Williamson AL, Brink NS, Dehaeck CM, Ovens S, Soeters R, Rybicki EP. Typing of human papillomaviruses in cervical carcinoma biopsies from Cape Town. J. Med. Virol. 1994;43(3):231–237. doi: 10.1002/jmv.1890430307. [DOI] [PubMed] [Google Scholar]
- Williamson AL, Marais D, Passmore JA, Rybicki E. Human papillomavirus (HPV) infection in Southern Africa: Prevalence, immunity, and vaccine prospects. IUBMB Life. 2002;53(45):253–258. doi: 10.1080/15216540212654. [DOI] [PubMed] [Google Scholar]
- Wood C, Harrington W., Jr. AIDS and associated malignancies. Cell Res. 2005;15(1112):947–952. doi: 10.1038/sj.cr.7290372. [DOI] [PubMed] [Google Scholar]
- Xi LF, Critchlow CW, Wheeler CM, Koutsky LA, Galloway DA, Kuypers J, Hughes JP, Hawes SE, Surawicz C, Goldbaum G, Holmes KK, Kiviat NB. Risk of anal carcinoma in situ in relation to human papillomavirus type 16 variants. Cancer Res. 1998;58(17):3839–3844. [PubMed] [Google Scholar]
- Xi LF, Toure P, Critchlow CW, Hawes SE, Dembele B, Sow PS, Kiviat NB. Prevalence of specific types of human papillomavirus and cervical squamous intraepithelial lesions in consecutive, previously unscreened, West-African women over 35 years of age. Int. J. Cancer. 2003;103(6):803–809. doi: 10.1002/ijc.10876. [DOI] [PubMed] [Google Scholar]
- Xu D, Coleman T, Zhang J, Fagot A, Kotalik C, Zhao L, Trevidi P, Jones C, Zhang L. Epstein-Barr virus inhibits Kaposis sarcoma-associated herpesvirus lytic replication in primary effusion lymphomas. 2007 doi: 10.1128/JVI.02743-06. (submitted for publication) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang TY, Chen SC, Leach MW, Manfra D, Homey B, Wiekowski M, Sullivan L, Jenh CH, Narula SK, Chensue SW, Lira SA. Transgenic expression of the chemokine receptor encoded by human herpesvirus 8 induces an angioproliferative disease resembling Kaposi’s sarcoma. J. Exp. Med. 2000;191(3):445–454. doi: 10.1084/jem.191.3.445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yee C, Krishnan-Hewlett I, Baker CC, Schlegel R, Howley PM. Presence and expression of human papillomavirus sequences in human cervical carcinoma cell lines. Am. J. Pathol. 1985a;119(3):361–366. [PMC free article] [PubMed] [Google Scholar]
- Yee C, Krishnan-Hewlett I, Baker CC, Schlegel R, Howley PM. Presence and expression of human papillomavirus sequences in human cervical carcinoma cell lines. Am. J. Pathol. 1985b;119(3):361–366. [PMC free article] [PubMed] [Google Scholar]
- Yoshizaki T, Sato H, Furukawa M, Pagano JS. The expression of matrix metalloproteinase 9 is enhanced by Epstein-Barr virus latent membrane protein 1. Proc. Natl. Acad. Sci. USA. 1998;95:3621–3626. doi: 10.1073/pnas.95.7.3621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshpe NS. Oral and laryngeal papilloma: A pediatric manifestation of sexually transmitted disease? Int. J. Pediatr. Otorhinolaryngol. 1995;31(1):77–83. doi: 10.1016/0165-5876(94)01104-6. [DOI] [PubMed] [Google Scholar]
- Zhang L, Pagano JS. Interferon regulatory factor 2 represses the Epstein-Barr virus BamH I Q latency promoter in type III latency. Mol. Cell Biol. 1999;19:3216–3223. doi: 10.1128/mcb.19.4.3216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Das SC, Kotalik C, Pattnaik AK, Zhang L. The latent membrane protein 1 of Epstein-Barr virus establishes an antiviral state via induction of interferon-stimulated genes. J. Biol. Chem. 2004a;279(44):46335–46342. doi: 10.1074/jbc.M403966200. [DOI] [PubMed] [Google Scholar]
- Zhang L, Hong K, Zhang J, Pagano J. Multiple signal transducers and activators of transcription (STATs) are induced by EBV LMP-1. Virology. 2004b;323:141–152. doi: 10.1016/j.virol.2004.03.007. [DOI] [PubMed] [Google Scholar]
- Zhang L, Zhang J, Lambert Q, Der CJ, Del Valle L, Miklossy J, Khalili K, Zhou Y, Pagano JS. Interferon regulatory factor 7 is associated with Epstein-Barr virus-transformed central nervous system lymphoma and has oncogenic properties. J. Virol. 2004c;78(23):12987–12995. doi: 10.1128/JVI.78.23.12987-12995.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zimber-Strobl U, Strobl LJ. EBNA2 and Notch signalling in Epstein-Barr virus mediated immortalization of B lymphocytes. Semin. Cancer Biol. 2001;11(6):423–434. doi: 10.1006/scbi.2001.0409. [DOI] [PubMed] [Google Scholar]
- Zippin C, Lum D. Study of completeness of the surveillance, epidemiology and end results (SEER) program case ascertainment by hospital size and casefinding source. Health Rep. 1993;5(1):87–90. [PubMed] [Google Scholar]
- zur Hausen H. Papillomaviruses in human cancers. Proc. Assoc. Am. Physic. 1999;111(6):581–587. doi: 10.1046/j.1525-1381.1999.99723.x. [DOI] [PubMed] [Google Scholar]
- zur Hausen H. Papillomaviruses causing cancer: Evasion from host-cell control in early events in carcinogenesis. J. Natl. Cancer Inst. 2000;92(9):690–698. doi: 10.1093/jnci/92.9.690. [DOI] [PubMed] [Google Scholar]
- zur Hausen H, Gissmann L, Steiner W, Dippold W, Dreger I. Human papilloma viruses and cancer. Bibl. Haematol. 1975;(43):569–571. doi: 10.1159/000399220. [DOI] [PubMed] [Google Scholar]
- zur Hausen H, de Villiers EM, Gissmann L. Papillomavirus infections and human genital cancer. Gynecol. Oncol. 1981;12(2 Pt 2):S124–128. doi: 10.1016/0090-8258(81)90067-6. [DOI] [PubMed] [Google Scholar]