

Abstract
p53 binding protein 1 (53BP1), a protein proposed to function as a transcriptional coactivator of the p53 tumor suppressor, has BRCT domains with high homology to the Saccharomyces cerevisiae Rad9p DNA damage checkpoint protein. To examine whether 53BP1 has a role in the cellular response to DNA damage, we probed its intracellular localization by immunofluorescence. In untreated primary cells and U2OS osteosarcoma cells, 53BP1 exhibited diffuse nuclear staining; whereas, within 5–15 min after exposure to ionizing radiation (IR), 53BP1 localized at discreet nuclear foci. We propose that these foci represent sites of processing of DNA double-strand breaks (DSBs), because they were induced by IR and chemicals that cause DSBs, but not by ultraviolet light; their peak number approximated the number of DSBs induced by IR and decreased over time with kinetics that parallel the rate of DNA repair; and they colocalized with IR-induced Mre11/NBS and γ-H2AX foci, which have been previously shown to localize at sites of DSBs. Formation of 53BP1 foci after irradiation was not dependent on ataxia-telangiectasia mutated (ATM), Nijmegen breakage syndrome (NBS1), or wild-type p53. Thus, the fast kinetics of 53BP1 focus formation after irradiation and the lack of dependency on ATM and NBS1 suggest that 53BP1 functions early in the cellular response to DNA DSBs.
Keywords: 53BP1, p53, DNA damage, ionizing radiation, cancer
Introduction
One of the most serious threats to the integrity of eukaryotic genomes is DNA double-strand breaks (DSBs). Such breaks can be produced from exogenous agents, such as ionizing radiation (IR), or from errors occurring during normal replication or recombination and elicit an evolutionarily conserved checkpoint response that induces cell cycle arrest and activation of DNA repair (Elledge 1996; Longhese et al. 1998; Weinert 1998; Boddy and Russell 1999; Caspari and Carr 1999). The first eukaryotic DNA damage checkpoint gene to be identified was RAD9 in Saccharomyces cerevisiae (Weinert and Hartwell 1988). The protein encoded by RAD9, along with proteins encoded by genes in the RAD24 epistasis group, including RAD17, RAD24, MEC3, and DDC1, is proposed to regulate, in response to DNA damage, activation and phosphorylation of Mec1p, a protein kinase required for subsequent phosphorylation and activation of the Rad53p/Spk1p and Chk1p kinases (Sanchez et al. 1996; Sun et al. 1996; de la Torre-Ruiz et al. 1998; Kondo et al. 1999). In turn, Rad53p/Spk1p and Chk1p regulate through phosphorylation proteins involved in cell cycle arrest and DNA repair (Cohen-Fix and Koshland 1997; Sidorova and Breeden 1997; Huang et al. 1998; Sanchez et al. 1999; Weinreich and Stillman 1999). The Mec1p and Rad53p/Spk1p components of the DNA damage checkpoint signaling pathway are also involved in the cellular response to DNA replication blocks. Activation of Mec1p by replication blocks does not require the RAD9 or RAD24 epistasis group genes, but is instead dependent on genes that have a role in DNA replication, such as DPB11, DNA–polymerase ε, DRC1, and others (Araki et al. 1995; Navas et al. 1996; Wang and Elledge 1999).
DNA damage checkpoint genes are highly conserved throughout evolution. Human homologues of S. cerevisiae RAD24, RAD17, MEC3, and DDC1 have been cloned and partially characterized (Lieberman et al. 1996; Bao et al. 1998; Freire et al. 1998; Volkmer and Karnitz 1999). There are two human homologues of MEC1, ataxia-telangiectasia mutated (ATM) (Savitsky et al. 1995) and ATR (ATM and Rad3-related) (Bentley et al. 1996; Cimprich et al. 1996). ATR is essential for development, but its precise role in the DNA damage response remains to be determined (Brown and Baltimore 2000). ATM, which is not essential for development, mediates the early response to DNA DSBs and its inactivation in patients with ataxia-telangiectasia (A-T) leads to checkpoint defects and chromosomal instability (Halazonetis and Shiloh 1999). The ATM protein product phosphorylates and activates Chk2/hCds1, the human homologue of the S. cerevisiae Rad53p/Spk1p kinase (Matsuoka et al. 1998; Blasina et al. 1999a; Brown et al. 1999; Chaturvedi et al. 1999), which in turn targets the p53 tumor suppressor protein and other proteins regulating cell cycle progression, such as Cdc25c (Matsuoka et al. 1998; Chehab et al. 2000; Hirao et al. 2000). Additionally, ATM phosphorylates NBS1 (Gatei et al. 2000; Lim et al. 2000; Wu et al. 2000; Zhao et al. 2000), a protein mutated in Nijmegen breakage syndrome (NBS) (Carney et al. 1998; Matsuura et al. 1998; Varon et al. 1998). NBS1 and the Mre11 and Rad50 proteins form a protein complex, which participates in DNA repair and in the DNA damage checkpoint response during S phase (Haber 1998; Petrini 1999; Paull and Gellert 1999; Lim et al. 2000) and which localizes to sites of DNA DSBs (Maser et al. 1997; Nelms et al. 1998).
One of the yeast DNA damage checkpoint genes, whose human equivalent is not known, is S. cerevisiae RAD9. RAD9 is an orthologue of Schizosaccharomyces pombe Crb2/Rhp9 and the protein products of these two genes share evolutionarily conserved BRCT domains at their COOH termini (Saka et al. 1997; Willson et al. 1997). BRCT domains may mediate protein–protein interactions and are found in many proteins involved in the cellular response to DNA damage, including BRCA1, NBS1, XRCC4, DNA ligase 4, and PARP (Bork et al. 1997; Callebaut and Mornon 1997; Zhang et al. 1998). Interestingly, p53 binding protein 1 (53BP1), a protein identified through its ability to bind p53 in a yeast two-hybrid screen (Iwabuchi et al. 1994), also has COOH-terminal BRCT domains. 53BP1 has been proposed to function as a transcriptional coactivator of p53 (Iwabuchi et al. 1998), but the presence of BRCT domains suggests that 53BP1 may also have a more direct role in the cellular response to DNA damage. In this study, we show that 53BP1 localizes rapidly to discreet foci within the nucleus of cells exposed to DNA DSB-inducing agents and propose that these foci represent sites of DSBs.
Materials and Methods
Antibodies
The 53BP1-reactive monoclonal antibodies were prepared using as antigen a recombinant protein consisting of the COOH-terminal 312 residues of human 53BP1 purified from Escherichia coli. Polyclonal antibodies recognizing Mre11 and NBS1 were obtained from Calbiochem; polyclonal (clone Y11) and monoclonal (clone B11) antibodies recognizing the hemagglutinin (HA) tag were obtained from Santa Cruz Biotechnology, Inc. and BabCo, respectively. A polyclonal antibody recognizing propromyelocytic leukemia (PML) (used at 1:400 dilution) was a gift from G. Maul (The Wistar Institute). The polyclonal antibody recognizing γ-H2AX (used at 1:800 dilution) was a gift from W. Bonner (National Institutes of Health, Bethesda, MD).
Cell Lines and Immunoblot Analysis
Fibroblasts from NBS patients were a gift from P. Concannon (Virginia Mason Research Center, Seattle, WA). All other cell lines were obtained from the American Type Culture Collection or have been described previously (Chehab et al. 2000). All cell lines were grown in DMEM medium, 10% FCS, penicillin (100 mg/ml)/streptomycin (100 mg/ml), and 2 mM glutamine (all from Life Technologies) at 37°C in a humidified 5% CO2 atmosphere. U2OS cells stably expressing various HA-tagged 53BP1 proteins were prepared by calcium phosphate transfection of cells with pSNV2 vectors (Chehab et al. 2000) containing the appropriate 53BP1 insert together with a plasmid conferring neomycin resistance. Colonies were selected with neomycin, pooled, and studied within one or two passages. Whole cell and nuclear extracts were prepared and examined by immunoblotting as described previously (Chehab et al. 2000).
Immunofluorescence
Cells grown on coverslips were either mock-treated or were exposed to 50 J/m2 UV light or 0.2 mM etoposide for 24 h or 1 mM hydroxyurea (HU) for 24 h, or aphidicolin for 14 h, or 100 ng/ml neocarzinostatin for 4 h or between 0.5 and 12 Gy IR from a 137Cs source. In some experiments, 20 mM wortmannin or 2 mM caffeine was added to the cells 1 h before irradiation. At the indicated time points, the cells were fixed in 1% paraformaldehyde for 15 min followed by extraction on ice for 20 min in 0.2% Triton X-100/PBS. Coverslips were incubated with primary antibody for 1 h at room temperature, washed with PBS, and incubated with 1:200 dilution of secondary antibody (anti–mouse IgG conjugated to Texas red or anti–rabbit IgG conjugated to FITC; Vector Laboratories) for 30 min at room temperature. After washing, the cells were counterstained with DAPI for 2 min at room temperature, washed with PBS, mounted on glass slides, and inspected with a Leitz fluovert FU microscope. Images were acquired with an ORCA digital camera (Hamamatsu Photonics) and processed on an O2 computer using the image analysis tools that are part of the IRIX operating system (Silicon Graphics).
Results
53BP1 Has Similarity to Yeast DNA Damage Checkpoint Proteins
The S. cerevisiae Rad9p checkpoint protein and its functional orthologue Crb2p/Rhp9p in S. pombe show obvious amino acid sequence similarity only within their COOH-terminal BRCT domains. Within these domains, the amino acid identity is 25% and involves residues beyond those that are conserved in all BRCT domains (Fig. 1). Using the most current sequence database of the Caenorhabditis elegans genome (The C. elegans Sequencing Consortium 1998), we identified T05F1 as the gene whose open reading frame (ORF) has the highest amino acid sequence similarity to the BRCT domains of S. cerevisiae Rad9p and S. pombe Crb2p/Rhp9p. Within the BRCT domains, 26% of T05F1 ORF residues are identical to a Rad9p and/or Crb2p/Rhp9p residue at the corresponding position, suggesting that the T05F1 ORF may be their C. elegans orthologue. Analysis of the most current publicly available database of human sequences identified 53BP1 and the KIAA0170 ORF (Nagase et al. 1996) as the sequences with the highest and second highest similarity, respectively, to the T05F1 ORF sequence (Fig. 1). 37% of the 53BP1 residues are identical to a Rad9p and/or Crb2p/Rhp9p and/or T05F1 ORF residue at the corresponding position, versus 22% for KIAA0170. Thus, 53BP1 has significantly higher sequence similarity to the aligned sequences of S. cerevisiae Rad9p, S. pombe Crb2p/Rhp9p, and C. elegans T05F1 ORF than any other protein currently in the human sequence database, and may therefore be their human orthologue.
Figure 1.

Alignment of the amino acid sequences of the BRCT domains of S. cerevisiae Rad9p, S. pombe Crb2p/Rhp9p (Crb2p), C. elegans T05F1 ORF, human 53BP1, and human KIAA0170 ORF (KIAA). The boxes indicate residues of 53BP1 and KIAA0170 ORF that are identical with Rad9p, Crb2p/Rhp9p, or T05F1 ORF residues. Residue positions are indicated on either side of the amino acid sequences. The termination codons are indicated by asterisks. Shaded numbers within the sequences refer to sequences of the indicated length that are not shown, because they are present only within one member.
53BP1 Monoclonal Antibodies
To investigate whether 53BP1 participates in the cellular response to DNA damage, we obtained two independent high-affinity monoclonal antibodies, using as antigen the COOH-terminal 312 amino acids of human 53BP1 (residues 1661–1972), which encompass the predicted nuclear localization signal and the BRCT domains. Both antibody clones recognized with high specificity the COOH-terminal 312 amino acids of 53BP1, as determined by immunoblot analysis of U2OS osteosarcoma cell extracts containing ectopically expressed, HA-tagged, full-length or truncated 53BP1 proteins (Fig. 2 and data not shown). These antibodies also detected endogenous 53BP1 in nuclear extracts of U2OS cells either nonirradiated or exposed to 8 Gy IR with practically no crossreactivity to other nuclear proteins (Fig. 2). Endogenous 53BP1 migrated significantly slower than its predicted 217-kD molecular mass consistent with previous reports (Iwabuchi et al. 1998) and its levels remained unchanged after irradiation. There was only a hint of slower migration after irradiation, which does not allow us to comment as to whether 53BP1 is modified in response to DNA damage.
Figure 2.

Specificity of 53BP1-reactive monoclonal antibodies (MAb). Nuclear extracts of nontransfected cells and whole cell extracts from cells stably expressing HA-tagged full-length or truncated 53BP1 proteins were immunoblotted (IB) with a 53BP1-reactive monoclonal antibody (clone WI1) at a 1:20 dilution or an HA tag–reactive polyclonal antibody (PAb; clone Y11) used at a 1:1,000 dilution. Endog., endogenous.
53BP1 Forms Nuclear Foci in Response to Agents That Induce DNA DSBs
The availability of high-affinity antibodies allowed us to probe the intracellular localization of 53BP1 by immunofluorescence. In nonirradiated U2OS cells, 53BP1 showed diffuse staining of the nuclei with the exception of the nucleoli, which were not stained. After exposure to 8 Gy IR, 53BP1 relocalized to distinct nuclear foci. 53BP1 relocalization was evident with both antibody clones and no 53BP1 staining was observed in the absence of primary antibody (Fig. 3 A).
Figure 3.
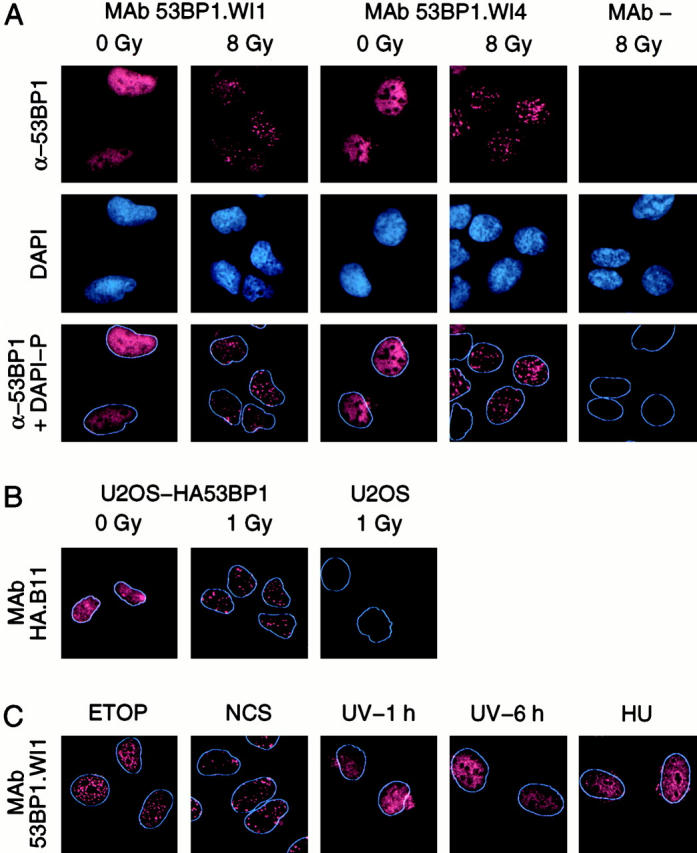
Relocalization of 53BP1 in response to IR. (A) Immunofluorescence of nonirradiated (0 Gy) and irradiated (8 Gy) U2OS cells with 53BP1-reactive monoclonal antibodies (clones WI1 and WI4) or with no primary antibody (MAb −). α-53BP1, 53BP1 immunofluorescence; DAPI, DAPI fluorescence marking the cell nuclei; α-53BP1 + DAPI-P, merging of the α-53BP1 immunofluorescence and processed DAPI image (DAPI-P) that shows only the periphery of the nucleus. (B) Relocalization of HA-tagged full-length 53BP1 in stably transfected U2OS cells using an HA tag–reactive monoclonal antibody (clone B11). (C) Relocalization of endogenous 53BP1 in nontransfected U2OS cells exposed to etoposide (ETOP), neocarzinostatin (NCS), UV light, or HU. The cells exposed to UV light were examined 1 or 6 h after irradiation.
To verify that the immunofluorescence signal could be attributed to 53BP1, we also examined the intracellular localization of HA-tagged full-length 53BP1, stably expressed in U2OS cells, using an HA tag–reactive monoclonal antibody. Like endogenous 53BP1, HA-tagged 53BP1 formed foci after irradiation. Again the immunofluorescence reactions were specific, since no HA-reactive signal was observed in nontransfected U2OS cells (Fig. 3 B).
In addition to IR, DNA DSBs can be induced by chemicals, such as etoposide and neocarzinostatin. These two agents induced relocalization of endogenous 53BP1 to discrete foci. However, UV light, which induces pyrimidine cross-linking, and HU, which inhibits DNA replication, did not lead to 53BP1 relocalization (Fig. 3 C). Thus, 53BP1 focus formation was specifically associated with agents that induce DSBs.
The absence of 53BP1 focus formation in cells treated with HU raises the possibility that 53BP1 does not form foci when cells are in S phase. To explore this possibility, the cells were treated with aphidicolin, a DNA polymerase inhibitor that arrests cells in S phase and leads to DNA DSBs. Under these conditions, 53BP1 foci formed (data not shown), suggesting that 53BP1 forms foci in response to DNA DSBs throughout the cell cycle.
Dose Dependence and Time Course of 53BP1 Focus Formation
The induction of 53BP1 foci by agents that induce DNA DSBs raises the possibility that these foci represent actual sites of DNA breaks. According to this hypothesis, the number of foci should match the number of DNA DSBs and the kinetics of focus dispersion over time should match the kinetics of DNA repair. We first studied the dose dependence of 53BP1 focus formation 15 min after exposure of U2OS cells to IR. 53BP1 foci were evident after exposure to 0.5 Gy IR and the number of foci increased linearly with the dose of IR, except at the higher doses, where the lack of linearity may reflect the difficulty in resolving foci that are very close to each other (Fig. 4A and Fig. B).
Figure 4.

Dose dependence and time course of 53BP1 relocalization. (A and B) Dose dependence: (A) immunofluorescence images of U2OS cells exposed to the indicated dose of IR and examined 15-min later for 53BP1 relocalization; (B) average number of 53BP1 foci per cell calculated by counting 50–100 cells per data point. (C–E) Time course: (C) immunofluorescence images of U2OS cells exposed to 1 Gy IR and examined at the indicated times after irradiation; (D) average number of 53BP1 foci per cell; (E) fraction of cells with foci. 50–100 cells were counted per data point. Bars indicate standard errors in three different experiments.
We next investigated the time course of 53BP1 focus formation and dispersion after exposure to 1 Gy-IR. 53BP1 foci were evident at the earliest time point examined, 5 min after irradiation. The average number of foci per cell peaked at the 30-min time point; by 2 h about half of the foci had dispersed; and thereafter the rate of dispersion slowed such that the number of foci per cell returned to baseline levels 12–24 h after irradiation (Fig. 4C and Fig. D). At their peak, there were, on average, ∼25 53BP1 foci per cell per Gy similar to the previously calculated number of ∼35 DNA DSBs per cell per Gy (Lobrich et al. 1993; Ruiz de Almodovar et al. 1994; Cedervall et al. 1995). Furthermore, the rate of 53BP1 focus dispersion over time paralleled the rate of DNA DSB repair, which shows biphasic kinetics with a fast component for repair of most breaks and a slow component for repair of the remaining breaks (Lobrich et al. 1995; Nunez et al. 1995; DiBiase et al. 2000).
The fraction of cells with 53BP1 foci after irradiation was also calculated. It peaked at ∼90–95% between 15 min and 2 h after irradiation and decreased to baseline at the 16-h time point (Fig. 4 E). These results provide a somewhat different perspective than average number of foci per cell and indicate that virtually the entire pool of irradiated cells responded by forming 53BP1 foci.
To further investigate whether 53BP1 foci represent sites of DNA DSBs, we examined whether wortmannin, a kinase inhibitor that slows repair of DNA DSBs (Boulton et al. 1996; Okayasa et al., 1998; DiBiase et al. 2000), affects 53BP1 focus formation and dispersion. Wortmannin was added 1 h before exposure of U2OS cells to 1 Gy IR and 53BP1 focus formation was examined over an 8-h period. Addition of wortmannin delayed the formation of 53BP1 foci, such that their number peaked 2 h, rather than 30 min, after exposure to IR. The peak average number of foci per cell also decreased to about half of the number seen in the absence of wortmannin and focus dispersion occurred with slower kinetics (Fig. 5 A). The effect of wortmannin on 53BP1 focus formation and dispersion was also evident when we plotted the percentage of control and wortmannin-treated cells with 53BP1 foci as a function of time after irradiation (Fig. 5 B). In contrast to wortmannin, caffeine, which abrogates the DNA damage cell cycle checkpoint (Lau and Pardee 1982; Schlegel and Pardee 1986), did not affect 53BP1 focus formation or dispersion (Fig. 5).
Figure 5.

Effect of wortmannin (Wort) and caffeine (Caff) on 53BP1 relocalization in response to irradiation. (A) Average number of 53BP1 foci per cell. (B) Fraction of cells with foci. Cells treated with 20 mM wortmannin, 2 mM caffeine, or untreated (Ctrl) cells were exposed to 1 Gy IR and 53BP1 relocalization was examined at the indicated times. 50–100 cells were counted per data point. Bars indicate standard errors in three different experiments.
Colocalization of 53BP1, γ-H2AX, and Mre11/NBS1 Foci
To further investigate the site of 53BP1 relocalization after DNA damage we examined whether 53BP1 colocalized with γ-H2AX and the Mre11/NBS1/Rad50 protein complex, which have been reported to localize to sites of DNA DSBs. H2AX is one of the three types of conserved H2A histones and contains within its COOH terminus a conserved serine residue (Ser139) that is phosphorylated rapidly after exposure of cells to IR (Rogakou et al. 1998, Rogakou et al. 1999). An antibody specific for γ-H2AX, the form of H2AX protein phosphorylated on Ser139, recognizes nuclear foci that form specifically at sites of DNA DSBs. We examined whether 53BP1 and γ-H2AX foci colocalize both at early (30-min) and late (8-h) time points after irradiation. At both time points there was remarkable colocalization (Fig. 6).
Figure 6.

Colocalization of 53BP1 with γ-H2AX and Mre11/NBS1 IR-induced foci. Nonirradiated (0 Gy) and irradiated (1 or 8 Gy) U2OS cells were stained by immunofluorescence with antibodies that recognize 53BP1 (clone WI1, 1:20 dilution), γ-H2AX (1:800 dilution of polyclonal antibody), Mre11 (1:1,000 dilution of polyclonal antibody), or NBS1 (1:1,000 dilution of polyclonal antibody) as indicated. As a control, cells were also stained with antibodies to PML (1:400 dilution of polyclonal antibody) to mark the nuclear domain 10 (ND10) nuclear structures.
Like γ-H2AX foci, Mre11/NBS1 foci have also been mapped to sites of DNA DSBs. However, unlike γ-H2AX, the Mre11/NBS1 foci are evident only at late time points after exposure of cells to radiation generated by 137Cs (Maser et al. 1997; Nelms et al. 1998). We therefore examined for colocalization only at 8 h after irradiation. At this time point, U2OS cells exposed to 8 Gy IR showed colocalization of 53BP1 and Mre11/NBS1 foci (Fig. 6). This is consistent with the colocalization of 53BP1 and γ-H2AX foci at this time point, since γ-H2AX and Mre11/NBS1 foci have also been reported to colocalize (Paull et al. 2000). As a control, there was no colocalization of 53BP1 with the PML protein (Fig. 6). PML localizes at foci, referred to as nuclear domain 10 (ND10) structures (Maul 1998), whose number and distribution does not change in response to DNA damage.
53BP1 Focus Formation Does Not Require ATM or NBS1
Because ATM and NBS1 participate in the cellular responses to DNA damage, we investigated whether 53BP1 focus formation was ATM or NBS1 dependent. 53BP1 focus formation was examined in AT1BR and AT5BI cells, which are nontransformed fibroblasts derived from patients with A-T, and in NBS 780816 and 880823 cells, which are nontransformed fibroblasts from patients with NBS. There was no difference in 53BP1 focus formation after irradiation between the A-T or NBS fibroblasts and AG1522 fibroblasts, which are derived from a normal individual (Fig. 7). A time course analysis further indicated that in the cells with mutant ATM or NBS1 the number of 53BP1 foci per cell peaked 30 min after irradiation, similar to cells with wild-type ATM and NBS1 (data not shown).
Figure 7.

53BP1 focus formation in cells deficient in ATM, NBS1, or DNA-PK. 53BP1 relocalization was studied in nontransformed fibroblasts from a normal individual (AG1522), from patients with A-T (AT1BR and AT5BI), and from patients with NBS (NBS7, NBS780816; and NBS8, NBS880823) and in MO59K and MO59J glioblastoma cells.
To determine whether DNA-dependent protein kinase (DNA-PK), a protein kinase implicated in DNA repair, was required for 53BP1 focus formation, we examined the MO59K and MO59J glioblastoma cells. The MO59K cells have wild-type DNA-PK, whereas MO59J cells, which are derived from MO59K, are DNA-PK deficient. MO59K cells showed a normal response, namely 53BP1 focus formation after irradiation. However, the MO59J cells contained a high number of 53BP1 foci even in the absence of exposure to IR (Fig. 7). The presence of 53BP1 foci indicates that DNA-PK is not required for 53BP1 foci to form; however, the high variability in the basal number of 53BP1 foci from cell to cell made it impossible to determine whether MO59J cells exhibit a normal induction of 53BP1 focus formation after irradiation.
Discussion
In eukaryotes, DNA damage induces an evolutionarily conserved response that leads to activation of cell cycle checkpoints and stimulation of DNA repair (Elledge 1996; Longhese et al. 1998; Weinert 1998; Boddy and Russell 1999; Caspari and Carr 1999). Although several of the proteins that participate in the DNA damage response have been identified, the entire spectrum of proteins that function in the cellular response to DNA damage is most likely not known. Here we identify 53BP1 as a protein that participates in the DNA damage response. After exposure of cells to IR or other agents that induce DNA DSBs, 53BP1 localized to discrete foci within the nucleus. 53BP1 foci were evident 5 min after irradiation, by 15 min the entire cell population had developed foci, and by 30 min the average number of 53BP1 foci per cell had peaked. Thereafter, the number of 53BP1 foci decreased over time and returned to baseline levels about 16 h after irradiation.
Several observations suggest that the 53BP1 foci represent sites of DNA DSB processing. First, 53BP1 foci were induced by agents that cause DNA DSBs, but not by agents that cause DNA damage other than DSBs or by agents that block DNA replication. Second, the number of 53BP1 foci was proportional to the dose of IR and at its peak, 30 min after irradiation, matched the number of DNA DSBs induced by IR (Lobrich et al. 1993; Ruiz de Almodovar, 1994; Cedervall et al. 1995). Third, the time course of 53BP1 focus dispersion paralleled the kinetics of DNA DSB repair (Lobrich et al. 1995; Nunez et al. 1995; DiBiase et al. 2000). Additionally, as discussed below, wortmannin, which slows the rate of DNA DSB repair (Boulton et al. 1996; Okayasu et al. 1998; DiBiase et al. 2000), slowed 53BP1 focus formation and dispersion; and 53BP1 foci colocalized with the IR-induced γ-H2AX and Mre11/NBS1/Rad50 foci, which have been shown to represent sites of DNA DSB processing (Maser et al. 1997; Nelms et al. 1998; Rogakou et al. 1998, Rogakou et al. 1999; Paull et al. 2000).
Wortmannin, an inhibitor of kinases in the phosphatidylinositol 3-kinase family, delayed the formation and dispersion of 53BP1 foci after irradiation. In the presence of wortmannin, the number of 53BP1 foci per cell peaked at 2 h, rather than at 30 min, after irradiation and did not reach baseline levels by the 24-h time point. Because wortmannin slows DNA DSB repair, our results establish a correlation between the kinetics of DNA DSB repair and 53BP1 relocalization. Wortmannin inhibits at least three kinases involved in the cellular response to DNA damage: DNA-PK, ATM, and ATR (Rosenzweig et al. 1997; Banin et al. 1998; Sarkaria et al. 1998). One or more of these kinases may facilitate recruitment of 53BP1 to sites of DNA DSBs. In cells from patients with A-T, 53BP1 foci formed rapidly (Fig. 7 and data not shown), suggesting that ATM is not involved in 53BP1 relocalization or that there is redundancy with other kinases, such as ATR and/or DNA-PK. The roles of ATR and DNA-PK are more difficult to resolve. Human cells lacking ATR are not available. MO59J cells lack detectable levels of DNA-PK (Galloway et al. 1999), but have many 53BP1 foci even in the absence of exposure to IR, which has precluded us from obtaining reliable data regarding the kinetics of 53BP1 focus formation after irradiation (Fig. 7). Nevertheless, we speculate that DNA-PK is the responsible kinase, because caffeine, which inhibits ATM and ATR, but not DNA-PK (Blasina et al. 1999b; Hall-Jackson et al. 1999; Sarkaria et al. 1999; Zhou et al. 2000), did not affect 53BP1 focus formation or dispersion in response to IR.
Interestingly, the 53BP1 foci are very reminiscent of the recently described IR-induced nuclear foci detected using an antibody specific for phosphorylated histone H2AX (Rogakou et al. 1999; Paull et al. 2000). The kinetics of formation and dispersion and the average number per cell of γ-H2AX and 53BP1 foci are virtually identical and γ-H2AX foci form in cells from A-T and NBS patients, as observed here for 53BP1. Furthermore, the γ-H2AX and 53BP1 foci colocalized in irradiated cells. The physiological significance of IR-induced H2AX phosphorylation remains to be determined. An attractive model is that H2AX phosphorylation serves to recruit proteins, such as 53BP1, involved in the cellular response to DNA damage (Paull et al. 2000). The sequence surrounding Ser139 of H2AX resembles the consensus substrate site for DNA-PK, ATR, and ATM, further raising the possibility that wortmannin delays 53BP1 focus formation by inhibiting H2AX phosphorylation.
In addition to 53BP1 and γ-H2AX, the protein complex containing Mre11, NBS1, and Rad50 localizes at discrete foci after exposure of cells to IR. These foci represent sites of processing of DNA DSBs, as suggested by the direct role of Mre11/NBS1/Rad50 in DNA DSB processing and by the intracellular distribution of these proteins to IR-exposed regions in cells irradiated through an IR-impermeable grid (Maser et al. 1997; Haber 1998; Nelms et al. 1998; Paull and Gellert 1999; Petrini 1999; Lim et al. 2000). Similar to 53BP1, the Mre11/NBS1/Rad50 foci form after exposure to IR, but not in response to UV light or HU. Furthermore, the Mre11/NBS1/Rad50 foci colocalized with the 53BP1 foci 8 h after irradiation, suggesting that at this time point, Mre11/NBS1/Rad50 and 53BP1 coexist in the same nuclear structure. Since Mre11/NBS1/Rad50 foci develop 8 h after irradiation, after most 53BP1 foci have already dispersed, it appears that the few 53BP1 foci that persist for several hours after exposure to IR are the ones that accumulate Mre11/NBS1/Rad50. However, we cannot exclude the possibility that lower amounts of Mre11/NBS1/Rad50, undetectable by immunofluorescence, are present within all 53BP1 foci even at early time points, since in cells exposed to high dose radiation from a synchrotron source, Mre11/NBS1/Rad50 relocalization was evident as early as 30 min after irradiation (Nelms et al. 1998). Irrespective of when Mre11/NBS1/Rad50 accumulate at the sites of DNA DSBs, 53BP1 recruitment to these sites can proceed in the absence of Mre11/NBS1/Rad50 recruitment, as evidenced by the presence of 53BP1 foci in irradiated cells from NBS patients, in which Mre11/NBS1/Rad50 foci do not form (Carney et al. 1998).
The relocalization of 53BP1 in response to agents that induce DNA DSBs has functional implications. 53BP1 was originally identified because it interacted with the p53 tumor suppressor protein in a yeast two-hybrid assay (Iwabuchi et al. 1994). More recently 53BP1 was proposed to function as a transcriptional coactivator for p53 (Iwabuchi et al. 1998), even though it differs from classical coactivators in that in vitro 53BP1 binds to the p53 sequence-specific DNA-binding domain, rather than the transactivation domain (Thukral et al. 1994). None of our experiments address directly the function of 53BP1 as a transcriptional coactivator. However, we note that 53BP1 focus formation after irradiation did not require wild-type p53, since it was observed in cells that lack p53 or that express mutant p53 proteins. In preliminary experiments, we have also been unable to detect an interaction between p53 and 53BP1 in vivo either by coimmunoprecipitation or by colocalization using immunofluorescence.
While further studies are needed to address the significance of the interaction between p53 and 53BP1 in vitro, our results suggest a new function for 53BP1 in the cellular response to DNA DSBs. The relocalization of 53BP1 after irradiation is much more dramatic than that previously observed for any other protein, including Mre11, NBS1, Rad50, Rad51, Rad54, BLM, and BRCA1, in terms of the early response after irradiation, the specificity for agents that induce DNA DSBs, and the involvement of the entire population of irradiated cells (Haaf et al. 1995; Maser et al. 1997; Scully et al. 1997; Gharibyan and Youssoufian 1999; Tan et al. 1999). The only other protein that shows an equally strong response is γ-H2AX; however, in this case there is probably no net H2AX relocalization, but rather H2AX phosphorylation (Rogakou et al. 1998, Rogakou et al. 1999). The relocalization of 53BP1 to putative sites of DNA DSBs suggests a role in DNA DSB processing and/or DNA damage signaling. In support of a role in DNA damage signaling, 53BP1 has the highest similarity of all human sequences in the public databases to the C. elegans T05F1 ORF, which in turn is the C. elegans sequence with the highest similarity to the S. cerevisiae Rad9p and S. pombe Crb2p/Rhp9p sequences. RAD9 was the first checkpoint gene to be identified; it is required for modification and activation of the checkpoint kinases Mec1p and Rad53p/Spk1p in response to irradiation, and also has a role in DNA repair (Weinert and Hartwell 1988; Sanchez et al. 1996; Sun et al. 1996; de la Torre-Ruiz and Lowndes 2000). 53BP1 displays some functional similarities to Rad9p. 53BP1 foci formed in response to IR, but not HU, and similarly Rad9p and its S. pombe orthologue Crb2p/Rhp9p are required for checkpoint activation in response to IR, but not HU (Navas et al. 1996; Willson et al. 1997). Also, 53BP1 relocalizes as early as 5 min after irradiation, which is fast enough for 53BP1 to be involved in modification and activation of the ATM and Chk2/hCds1 kinases, which are activated a few minutes after exposure of cells to IR and are the human homologues of Mec1p and Rad53p/Spk1p, respectively (Banin et al. 1998; Canman et al. 1998; Matsuoka et al. 1998). It will be important to determine whether 53BP1 is the functional orthologue of Rad9p or has some other role in the cellular response to DNA DSBs.
Acknowledgments
We thank K. Iwabuchi (Kanazawa Medical University, Uchinada, Japan) for the full-length 53BP1 cDNA, P. Concannon for the NBS fibroblasts, G. Maul for the PML antibody, C. Harris for the NCS, and W. Bonner for the γ-H2AX antibody. We also thank G. Rovera, F. Rauscher III, D. Scolnick, E. Stavridi, and R. DiTullio (The Wistar Institute) for support and helpful discussions.
Financial support was provided by the National Cancer Institute, National Institutes of Health (grant CA76367).
Footnotes
Abbreviations used in this paper: A-T, ataxia-telangiectasia; ATM, A-T mutated protein; ATR, ATM- and Rad3-related protein; 53BP1, p53 binding protein 1; DNA-PK, DNA-dependent protein kinase; DSB, double-strand break; HA, hemagglutinin; HU, hydroxyurea; IR, ionizing radiation; NBS, Nijmegen breakage syndrome; ORF, open reading frame; PML, promyelocytic leukemia.
References
- Araki H., Leem S.H., Phongdara A., Sugino A. Dpb11, which interacts with DNA polymerase II(epsilon) in Saccharomyces cerevisiae, has a dual role in S-phase progression and at a cell cycle checkpoint. Proc. Natl. Acad. Sci. USA. 1995;92:11791–11795. doi: 10.1073/pnas.92.25.11791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banin S., Moyal L., Shieh S., Taya Y., Anderson C.W., Chessa L., Smorodinsky N.I., Prives C., Reiss Y., Shiloh Y., Ziv Y. Enhanced phosphorylation of p53 by ATM in response to DNA damage. Science. 1998;281:1674–1677. doi: 10.1126/science.281.5383.1674. [DOI] [PubMed] [Google Scholar]
- Bao S., Shen X., Shen K., Liu Y., Wang X.F. The mammalian Rad24 homologous to yeast Saccharomyces cerevisiae Rad24 and Schizosaccharomyces pombe Rad17 is involved in DNA damage checkpoint. Cell Growth Differ. 1998;9:961–967. [PubMed] [Google Scholar]
- Bentley N.J., Holtzman D.A., Flaggs G., Keegan K.S., DeMaggio A., Ford J.C., Hoekstra M., Carr A.M. The Schizosaccharomyces pombe rad3 checkpoint gene. EMBO (Eur. Mol. Biol. Organ.) J. 1996;15:6641–6651. [PMC free article] [PubMed] [Google Scholar]
- Blasina A., de Weyer I.V., Laus M.C., Luyten W.H., Parker A.E., McGowan C.H. A human homologue of the checkpoint kinase Cds1 directly inhibits Cdc25 phosphatase Curr. Biol. 9 1999. 1 10a [DOI] [PubMed] [Google Scholar]
- Blasina A., Price B.D., Turenne G.A., McGowan C.H. Caffeine inhibits the checkpoint kinase ATM Curr. Biol. 9 1999. 1135 1138b [DOI] [PubMed] [Google Scholar]
- Boddy M.N., Russell P. DNA replication checkpoint control. Front. Biosci. 1999;4:D841–D848. doi: 10.2741/boddy. [DOI] [PubMed] [Google Scholar]
- Bork P., Hofmann K., Bucher P., Neuwald A.F., Altschul S.F., Koonin E.V. A superfamily of conserved domains in DNA damage-responsive cell cycle checkpoint proteins. FASEB J. 1997;11:68–76. [PubMed] [Google Scholar]
- Boulton S., Kyle S., Yalcintepe L., Durkacz B.W. Wortmannin is a potent inhibitor of DNA double strand break but not single strand break repair in Chinese hamster ovary cells. Carcinogenesis. 1996;17:2285–2290. doi: 10.1093/carcin/17.11.2285. [DOI] [PubMed] [Google Scholar]
- Brown A.L., Lee C.H., Schwarz J.K., Mitiku N., Piwnica-Worms H., Chung J.H. A human Cds1-related kinase that functions downstream of ATM protein in the cellular response to DNA damage. Proc. Natl. Acad. Sci. USA. 1999;96:3745–3750. doi: 10.1073/pnas.96.7.3745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown E.J., Baltimore D. ATR disruption leads to chromosomal fragmentation and early embryonic lethality. Genes Dev. 2000;14:397–402. [PMC free article] [PubMed] [Google Scholar]
- Callebaut I., Mornon J.P. From BRCA1 to RAP1a widespread BRCT module closely associated with DNA repair. FEBS Lett. 1997;400:25–30. doi: 10.1016/s0014-5793(96)01312-9. [DOI] [PubMed] [Google Scholar]
- Canman C.E., Lim D.S., Cimprich K.A., Taya Y., Tamai K., Sakaguchi K., Appella E., Kastan M.B., Siliciano J.D. Activation of the ATM kinase by ionizing radiation and phosphorylation of p53. Science. 1998;281:1677–1679. doi: 10.1126/science.281.5383.1677. [DOI] [PubMed] [Google Scholar]
- Carney J.P., Maser R.S., Olivares H., Davis E.M., Le M., Beau J.R., Yates L., III, Hays W.F., Morgan, Petrini J.H. The hMre11/hRad50 protein complex and Nijmegen breakage syndromelinkage of double-strand break repair to the cellular DNA damage response. Cell. 1998;93:477–486. doi: 10.1016/s0092-8674(00)81175-7. [DOI] [PubMed] [Google Scholar]
- Caspari T., Carr A.M. DNA structure checkpoint pathways in Schizosaccharomyces pombe . Biochimie. 1999;81:173–181. doi: 10.1016/s0300-9084(99)80050-9. [DOI] [PubMed] [Google Scholar]
- Cedervall B., Wong R., Albright N., Dynlacht J., Lambin P., Dewey W.C. Methods for the quantification of DNA double-strand breaks determined from the distribution of DNA fragment sizes measured by pulsed-field gel electrophoresis. Radiat. Res. 1995;143:8–16. [PubMed] [Google Scholar]
- Chaturvedi P., Eng W.K., Zhu Y., Mattern M.R., Mishra R., Hurle M.R., Zhang X., Annan R.S., Lu Q., Faucette L.F. Mammalian Chk2 is a downstream effector of the ATM-dependent DNA damage checkpoint pathway. Oncogene. 1999;18:4047–4054. doi: 10.1038/sj.onc.1202925. [DOI] [PubMed] [Google Scholar]
- Chehab N.H., Malikzay A., Appel M., Halazonetis T.D. Chk2/hCds1 functions as a DNA damage checkpoint in G1 by stabilizing p53. Genes Dev. 2000;14:278–288. [PMC free article] [PubMed] [Google Scholar]
- Cimprich K.A., Shin T.B., Keith C.T., Schreiber S.L. cDNA cloning and gene mapping of a candidate human cell cycle checkpoint protein. Proc. Natl. Acad. Sci. USA. 1996;93:2850–2855. doi: 10.1073/pnas.93.7.2850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen-Fix O., Koshland D. The anaphase inhibitor of Saccharomyces cerevisiae Pds1p is a target of the DNA damage checkpoint pathway. Proc. Natl. Acad. Sci. USA. 1997;94:14361–14366. doi: 10.1073/pnas.94.26.14361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de la Torre-Ruiz M., Lowndes N.F. The Saccharomyces cerevisiae DNA damage checkpoint is required for efficient repair of double strand breaks by non-homologous end joining. FEBS Lett. 2000;467:311–315. doi: 10.1016/s0014-5793(00)01180-7. [DOI] [PubMed] [Google Scholar]
- de la Torre-Ruiz M.A., Green C.M., Lowndes N.F. RAD9 and RAD24 define two additive, interacting branches of the DNA damage checkpoint pathway in budding yeast normally required for Rad53 modification and activation. EMBO (Eur. Mol. Biol. Organ.) J. 1998;17:2687–2698. doi: 10.1093/emboj/17.9.2687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DiBiase S.J., Zeng Z.C., Chen R., Hyslop T., Curran W.J., Jr., Iliakis G. DNA-dependent protein kinase stimulates an independently active, nonhomologous, end-joining apparatus. Cancer Res. 2000;60:1245–1253. [PubMed] [Google Scholar]
- Elledge S.J. Cell cycle checkpointspreventing an identity crisis. Science. 1996;274:1664–1672. doi: 10.1126/science.274.5293.1664. [DOI] [PubMed] [Google Scholar]
- Freire R., Murguia J.R., Tarsounas M., Lowndes N.F., Moens P.B., Jackson S.P. Human and mouse homologs of Schizosaccharomyces pombe rad1(+) and Saccharomyces cerevisiae RAD17linkage to checkpoint control and mammalian meiosis. Genes Dev. 1998;12:2560–2573. doi: 10.1101/gad.12.16.2560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galloway A.M., Spencer C.A., Anderson C.W., Allalunis-Turner M.J. Differential stability of the DNA-activated protein kinase catalytic subunit mRNA in human glioma cells. Oncogene. 1999;18:1361–1368. doi: 10.1038/sj.onc.1202433. [DOI] [PubMed] [Google Scholar]
- Gatei M., Young D., Cerosaletti K.M., Desai-Mehta A., Spring K., Kozlov S., Lavin M.F., Gatti R.A., Concannon P., Khanna K. ATM-dependent phosphorylation of nibrin in response to radiation exposure. Nat. Genet. 2000;25:115–119. doi: 10.1038/75508. [DOI] [PubMed] [Google Scholar]
- Gharibyan V., Youssoufian H. Localization of the Bloom syndrome helicase to punctate nuclear structures and the nuclear matrix and regulation during the cell cyclecomparison with the Werner's syndrome helicase. Mol. Carcinog. 1999;26:261–273. doi: 10.1002/(sici)1098-2744(199912)26:4<261::aid-mc5>3.0.co;2-a. [DOI] [PubMed] [Google Scholar]
- Haaf T., Golub E.I., Reddy G., Radding C.M., Ward D.C. Nuclear foci of mammalian Rad51 recombination protein in somatic cells after DNA damage and its localization in synaptonemal complexes. Proc. Natl. Acad. Sci. USA. 1995;92:2298–2302. doi: 10.1073/pnas.92.6.2298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haber J.E. The many interfaces of Mre11. Cell. 1998;95:583–586. doi: 10.1016/s0092-8674(00)81626-8. [DOI] [PubMed] [Google Scholar]
- Halazonetis T.D., Shiloh Y. Many faces of ATMeighth international workshop on ataxia-telangiectasia. Biochim. Biophys. Acta. 1999;1424:R45–R55. doi: 10.1016/s0304-419x(99)00023-2. [DOI] [PubMed] [Google Scholar]
- Hall-Jackson C.A., Cross D.A., Morrice N., Smythe C. ATR is a caffeine-sensitive, DNA-activated protein kinase with a substrate specificity distinct from DNA-PK. Oncogene. 1999;18:6707–6713. doi: 10.1038/sj.onc.1203077. [DOI] [PubMed] [Google Scholar]
- Hirao A., Kong Y.Y., Matsuoka S., Wakeham A., Ruland J., Yoshida H., Liu D., Elledge S.J., Mak T.W. DNA damage-induced activation of p53 by the checkpoint kinase Chk2. Science. 2000;287:1824–1827. doi: 10.1126/science.287.5459.1824. [DOI] [PubMed] [Google Scholar]
- Huang M., Zhou Z., Elledge S.J. The DNA replication and damage checkpoint pathways induce transcription by inhibition of the Crt1 repressor. Cell. 1998;94:595–605. doi: 10.1016/s0092-8674(00)81601-3. [DOI] [PubMed] [Google Scholar]
- Iwabuchi K., Bartel P.L., Li B., Marraccino R., Fields S. Two cellular proteins that bind to wild-type but not mutant p53. Proc. Natl. Acad. Sci. USA. 1994;91:6098–6102. doi: 10.1073/pnas.91.13.6098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwabuchi K., Li B., Massa H.F., Trask B.J., Date T., Fields S. Stimulation of p53-mediated transcriptional activation by the p53-binding proteins, 53BP1 and 53BP2. J. Biol. Chem. 1998;273:26061–26068. doi: 10.1074/jbc.273.40.26061. [DOI] [PubMed] [Google Scholar]
- Kondo T., Matsumoto K., Sugimoto K. Role of a complex containing Rad17, Mec3, and Ddc1 in the yeast DNA damage checkpoint pathway. Mol. Cell. Biol. 1999;19:1136–1143. doi: 10.1128/mcb.19.2.1136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lau C.C., Pardee A.B. Mechanism by which caffeine potentiates lethality of nitrogen mustard. Proc. Natl. Acad. Sci. USA. 1982;79:2942–2946. doi: 10.1073/pnas.79.9.2942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lieberman H.B., Hopkins K.M., Nass M., Demetrick D., Davey S. A human homolog of the Schizosaccharomyces pombe rad9+ checkpoint control gene. Proc. Natl. Acad. Sci. USA. 1996;93:13890–13895. doi: 10.1073/pnas.93.24.13890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim D.S., Kim S.T., Xu B., Maser R.S., Lin J., Petrini J.H., Kastan M.B. ATM phosphorylates p95/nbs1 in an S-phase checkpoint pathway. Nature. 2000;404:613–617. doi: 10.1038/35007091. [DOI] [PubMed] [Google Scholar]
- Lobrich M., Ikpeme S., Haub P., Weber K.J., Kiefer J. DNA double-strand break induction in yeast by X-rays and alpha-particles measured by pulsed-field gel electrophoresis. Int. J. Radiat. Biol. 1993;64:539–546. doi: 10.1080/09553009314551751. [DOI] [PubMed] [Google Scholar]
- Lobrich M., Rydberg B., Cooper P.K. Repair of x-ray-induced DNA double-strand breaks in specific Not I restriction fragments in human fibroblastsjoining of correct and incorrect ends. Proc. Natl. Acad. Sci. USA. 1995;92:12050–12054. doi: 10.1073/pnas.92.26.12050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Longhese M.P., Foiani M., Muzi-Falconi M., Lucchini G., Plevani P. DNA damage checkpoint in budding yeast. EMBO (Eur. Mol. Biol. Organ.) J. 1998;17:5525–5528. doi: 10.1093/emboj/17.19.5525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maser R.S., Monsen K.J., Nelms B.E., Petrini J.H. hMre11 and hRad50 nuclear foci are induced during the normal cellular response to DNA double-strand breaks. Mol. Cell. Biol. 1997;17:6087–6096. doi: 10.1128/mcb.17.10.6087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuoka S., Huang M., Elledge S.J. Linkage of ATM to cell cycle regulation by the Chk2 protein kinase. Science. 1998;282:1893–1897. doi: 10.1126/science.282.5395.1893. [DOI] [PubMed] [Google Scholar]
- Matsuura S., Tauchi H., Nakamura A., Kondo N., Sakamoto S., Endo S., Smeets D., Solder B., Belohradsky B.H., Der Kaloustian V.M. Positional cloning of the gene for Nijmegen breakage syndrome. Nat. Genet. 1998;19:179–181. doi: 10.1038/549. [DOI] [PubMed] [Google Scholar]
- Maul G.G. Nuclear domain 10, the site of DNA virus transcription and replication. Bioessays. 1998;20:660–667. doi: 10.1002/(SICI)1521-1878(199808)20:8<660::AID-BIES9>3.0.CO;2-M. [DOI] [PubMed] [Google Scholar]
- Nagase T., Seki N., Ishikawa K., Tanaka A., Nomura N. Prediction of the coding sequences of unidentified human genes. V. The coding sequences of 40 new genes (KIAA0161-KIAA0200) deduced by analysis of cDNA clones from human cell line KG-1. DNA Res. 1996;3:43–53. doi: 10.1093/dnares/3.1.43. [DOI] [PubMed] [Google Scholar]
- Nelms B.E., Maser R.S., MacKay J.F., Lagally M.G., Petrini J.H. In situ visualization of DNA double-strand break repair in human fibroblasts. Science. 1998;280:590–592. doi: 10.1126/science.280.5363.590. [DOI] [PubMed] [Google Scholar]
- Nunez M.I., Villalobos M., Olea N., Valenzuela M.T., Pedraza V., McMillan T.J., Ruiz de Almodovar J.M. Radiation-induced DNA double-strand break rejoining in human tumour cells. Br. J. Cancer. 1995;71:311–316. doi: 10.1038/bjc.1995.62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okayasu R., Suetomi K., Ullrich R.L. Wortmannin inhibits repair of DNA double-strand breaks in irradiated normal human cells. Radiat. Res. 1998;149:440–445. [PubMed] [Google Scholar]
- Paull T.T., Gellert M. Nbs1 potentiates ATP-driven DNA unwinding and endonuclease cleavage by the Mre11/Rad50 complex. Genes Dev. 1999;13:1276–1288. doi: 10.1101/gad.13.10.1276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paull T.T., Rogakou E.P., Yamazaki V., Kirchgessner C.U., Gellert M., Bonner W.M. A critical role for histone H2AX in recruitment of repair factors to nuclear foci after DNA damage. Curr. Biol. 2000;10:886–895. doi: 10.1016/s0960-9822(00)00610-2. [DOI] [PubMed] [Google Scholar]
- Petrini J.H. The mammalian Mre11-Rad50-nbs1 protein complexintegration of functions in the cellular DNA-damage response. Am. J. Hum. Genet. 1999;64:1264–1269. doi: 10.1086/302391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogakou E.P., Pilch D.R., Orr A.H., Ivanova V.S., Bonner W.M. DNA double-stranded breaks induce histone H2AX phosphorylation on serine 139. J. Biol. Chem. 1998;273:5858–5868. doi: 10.1074/jbc.273.10.5858. [DOI] [PubMed] [Google Scholar]
- Rogakou E.P., Boon C., Redon C., Bonner W.M. Megabase chromatin domains involved in DNA double-strand breaks in vivo. J. Cell Biol. 1999;146:905–916. doi: 10.1083/jcb.146.5.905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenzweig K.E., Youmell M.B., Palayoor S.T., Price B.D. Radiosensitization of human tumor cells by the phosphatidylinositol3-kinase inhibitors wortmannin and LY294002 correlates with inhibition of DNA-dependent protein kinase and prolonged G2-M delay. Clin. Cancer Res. 1997;3:1149–1156. [PubMed] [Google Scholar]
- Ruiz de Almodovar J.M., Steel G.G., Whitaker S.J., McMillan T.J. A comparison of methods for calculating DNA double-strand break induction frequency in mammalian cells by pulsed-field gel electrophoresis. Int. J. Radiat. Biol. 1994;65:641–649. doi: 10.1080/09553009414550751. [DOI] [PubMed] [Google Scholar]
- Saka Y., Esashi F., Matsusaka T., Mochida S., Yanagida M. Damage and replication checkpoint control in fission yeast is ensured by interactions of Crb2, a protein with BRCT motif, with Cut5 and Chk1. Genes Dev. 1997;11:3387–3400. doi: 10.1101/gad.11.24.3387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanchez Y., Desany B.A., Jones W.J., Liu Q., Wang B., Elledge S.J. Regulation of RAD53 by the ATM-like kinases MEC1 and TEL1 in yeast cell cycle checkpoint pathways. Science. 1996;271:357–360. doi: 10.1126/science.271.5247.357. [DOI] [PubMed] [Google Scholar]
- Sanchez Y., Bachant J., Wang H., Hu F., Liu D., Tetzlaff M., Elledge S.J. Control of the DNA damage checkpoint by Chk1 and Rad53 protein kinases through distinct mechanisms. Science. 1999;286:1166–1171. doi: 10.1126/science.286.5442.1166. [DOI] [PubMed] [Google Scholar]
- Sarkaria J.N., Tibbetts R.S., Busby E.C., Kennedy A.P., Hill D.E., Abraham R.T. Inhibition of phosphoinositide 3-kinase related kinases by the radiosensitizing agent wortmannin. Cancer Res. 1998;58:4375–4382. [PubMed] [Google Scholar]
- Sarkaria J.N., Busby E.C., Tibbetts R.S., Roos P., Taya Y., Karnitz L.M., Abraham R.T. Inhibition of ATM and ATR kinase activities by the radiosensitizing agent, caffeine. Cancer Res. 1999;59:4375–4382. [PubMed] [Google Scholar]
- Savitsky K., Bar-Shira A., Gilad S., Rotman G., Ziv Y., Vanagaite L., Tagle D.A., Smith S., Uziel T., Sfez S. A single ataxia telangiectasia gene with a product similar to PI-3 kinase. Science. 1995;268:1749–1753. doi: 10.1126/science.7792600. [DOI] [PubMed] [Google Scholar]
- Schlegel R., Pardee A.B. Caffeine-induced uncoupling of mitosis from the completion of DNA replication in mammalian cells. Science. 1986;232:1264–1266. doi: 10.1126/science.2422760. [DOI] [PubMed] [Google Scholar]
- Scully R., Chen J., Ochs R.L., Keegan K., Hoekstra M., Feunteun J., Livingston D.M. Dynamic changes of BRCA1 subnuclear location and phosphorylation state are initiated by DNA damage. Cell. 1997;90:425–435. doi: 10.1016/s0092-8674(00)80503-6. [DOI] [PubMed] [Google Scholar]
- Sidorova J.M., Breeden L.L. Rad53-dependent phosphorylation of Swi6 and down-regulation of CLN1 and CLN2 transcription occur in response to DNA damage in Saccharomyces cerevisiae . Genes Dev. 1997;11:3032–3045. doi: 10.1101/gad.11.22.3032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun Z., Fay D.S., Marini F., Foiani M., Stern D.F. Spk1/Rad53 is regulated by Mec1-dependent protein phosphorylation in DNA replication and damage checkpoint pathways. Genes Dev. 1996;10:395–406. doi: 10.1101/gad.10.4.395. [DOI] [PubMed] [Google Scholar]
- Tan T.L., Essers J., Citterio E., Swagemakers S.M., de Wit J., Benson F.E., Hoeijmakers J.H., Kanaar R. Mouse Rad54 affects DNA conformation and DNA-damage-induced Rad51 foci formation. Curr. Biol. 1999;9:325–328. doi: 10.1016/s0960-9822(99)80142-0. [DOI] [PubMed] [Google Scholar]
- The C. elegans Sequencing Consortium Genome sequence of the nematode C. elegansa platform for investigating biology. Science. 1998;282:2012–2018. doi: 10.1126/science.282.5396.2012. [DOI] [PubMed] [Google Scholar]
- Thukral S.K., Blain G.C., Chang K.K., Fields S. Distinct residues of human p53 implicated in binding to DNA, simian virus 40 large T antigen, 53BP1, and 53BP2. Mol. Cell. Biol. 1994;14:8315–8321. doi: 10.1128/mcb.14.12.8315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varon R., Vissinga C., Platzer M., Cerosaletti K.M., Chrzanowska K.H., Saar K., Beckmann G., Seemanova E., Cooper P.R., Nowak N.J. Nibrin, a novel DNA double-strand break repair protein, is mutated in Nijmegen breakage syndrome. Cell. 1998;93:467–476. doi: 10.1016/s0092-8674(00)81174-5. [DOI] [PubMed] [Google Scholar]
- Volkmer E., Karnitz L.M. Human homologs of Schizosaccharomyces pombe rad1, hus1, and rad9 form a DNA damage-responsive protein complex. J. Biol. Chem. 1999;274:567–570. doi: 10.1074/jbc.274.2.567. [DOI] [PubMed] [Google Scholar]
- Wang H., Elledge S.J. DRC1, DNA replication and checkpoint protein 1, functions with DPB11 to control DNA replication and the S-phase checkpoint in Saccharomyces cerevisiae . Proc. Natl. Acad. Sci. USA. 1999;96:3824–3829. doi: 10.1073/pnas.96.7.3824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinert T. DNA damage and checkpoint pathwaysmolecular anatomy and interactions with repair. Cell. 1998;94:555–558. doi: 10.1016/s0092-8674(00)81597-4. [DOI] [PubMed] [Google Scholar]
- Weinert T.A., Hartwell L.H. The RAD9 gene controls the cell cycle response to DNA damage in Saccharomyces cerevisiae . Science. 1988;241:317–322. doi: 10.1126/science.3291120. [DOI] [PubMed] [Google Scholar]
- Weinreich M., Stillman B. Cdc7p-Dbf4p kinase binds to chromatin during S phase and is regulated by both the APC and the RAD53 checkpoint pathway. EMBO (Eur. Mol. Biol. Organ.) J. 1999;18:5334–5346. doi: 10.1093/emboj/18.19.5334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willson J., Wilson S., Warr N., Watts F.Z. Isolation and characterization of the Schizosaccharomyces pombe rhp9 genea gene required for the DNA damage checkpoint but not the replication checkpoint. Nucleic Acids Res. 1997;25:2138–2146. doi: 10.1093/nar/25.11.2138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu X., Ranganathan V., Weisman D.S., Heine W.F., Ciccone D.N., O'Neill T.B., Crick K.E., Pierce K.A., Lane W.S., Rathbun G., Livingston D.M., Weaver D.T. ATM phosphorylation of Nijmegen breakage syndrome protein is required in a DNA damage response. Nature. 2000;405:477–482. doi: 10.1038/35013089. [DOI] [PubMed] [Google Scholar]
- Zhang X., Morera S., Bates P.A., Whitehead P.C., Coffer A.I., Hainbucher K., Nash R.A., Sternberg M.J., Lindahl T., Freemont P.S. Structure of an XRCC1 BRCT domaina new protein-protein interaction module. EMBO (Eur. Mol. Biol. Organ.) J. 1998;17:6404–6411. doi: 10.1093/emboj/17.21.6404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao S., Weng Y.C., Yuan S.S., Lin Y.T., Hsu H.C., Lin S.C., Gerbino E., Song M.H., Zdzienicka M.Z., Gatti R.A. Functional link between ataxia-telangiectasia and Nijmegen breakage syndrome gene products. Nature. 2000;405:473–477. doi: 10.1038/35013083. [DOI] [PubMed] [Google Scholar]
- Zhou B.B., Chaturvedi P., Spring K., Scott S.P., Johanson R.A., Mishra R., Mattern M.R., Winkler J.D., Khanna K.K. Caffeine abolishes the mammalian G(2)/M DNA damage checkpoint by inhibiting ataxia-telangiectasia-mutated kinase activity. J. Biol. Chem. 2000;275:10342–10348. doi: 10.1074/jbc.275.14.10342. [DOI] [PubMed] [Google Scholar]