

Abstract
Fusion proteins containing the Src homology (SH)3 domains of Dictyostelium myosin IB (myoB) and IC (myoC) bind a 116-kD protein (p116), plus nine other proteins identified as the seven member Arp2/3 complex, and the α and β subunits of capping protein. Immunoprecipitation reactions indicate that myoB and myoC form a complex with p116, Arp2/3, and capping protein in vivo, that the myosins bind to p116 through their SH3 domains, and that capping protein and the Arp2/3 complex in turn bind to p116. Cloning of p116 reveals a protein dominated by leucine-rich repeats and proline-rich sequences, and indicates that it is a homologue of Acan 125. Studies using p116 fusion proteins confirm the location of the myosin I SH3 domain binding site, implicate NH2-terminal sequences in binding capping protein, and show that a region containing a short sequence found in several G-actin binding proteins, as well as an acidic stretch, can activate Arp2/3-dependent actin nucleation. p116 localizes along with the Arp2/3 complex, myoB, and myoC in dynamic actin-rich cellular extensions, including the leading edge of cells undergoing chemotactic migration, and dorsal, cup-like, macropinocytic extensions. Cells lacking p116 exhibit a striking defect in the formation of these macropinocytic structures, a concomitant reduction in the rate of fluid phase pinocytosis, a significant decrease in the efficiency of chemotactic aggregation, and a decrease in cellular F-actin content. These results identify a complex that links key players in the nucleation and termination of actin filament assembly with a ubiquitous barbed end–directed motor, indicate that the protein responsible for the formation of this complex is physiologically important, and suggest that previously reported myosin I mutant phenotypes in Dictyostelium may be due, at least in part, to defects in the assembly state of actin. We propose that p116 and Acan 125, along with homologues identified in Caenorhabditis elegans, Drosophila, mouse, and man, be named CARMIL proteins, for capping protein, Arp2/3, and myosin I linker.
Keywords: myosin I, Arp2/3 complex, capping protein, leucine-rich repeats, Dictyostelium
Introduction
Type I myosins are ubiquitous, nonfilamentous, actin-based mechanoenzymes that play important roles in several actin-dependent processes, including endocytosis and cell locomotion (Coluccio 1997; Mermall et al. 1998; Wu et al. 2000). The members of one myosin I subfamily, which include examples from Acanthamoeba, Dictyostelium, yeast, Aspergillus, rat, and human (Sellers 1999), contain an Src homology (SH)3 domain within their tail domain. These ∼55-residue protein modules are present in a diverse array of cytoskeletal and signaling proteins and mediate protein–protein interactions of moderate affinity and high specificity with proline-rich target sequences containing the core element PXXP (Kuriyan and Cowburn 1997). In the case of Dictyostelium myosin IB (myoB), yeast Myo3p, and yeast Myo5p, the presence of the SH3 domain has been shown to be critical for the function of the myosin in vivo (Anderson et al. 1998; Novak and Titus 1998; Evangelista et al. 2000; Lechler et al. 2000). Therefore, for these myosins, full expression of function appears to depend on the SH3 domain–dependent interaction with a target protein.
To date, three myosin I SH3 domain target proteins have been identified: Acan 125, verprolin (Vrp1p), and Las17p/Bee1p. The Acanthamoeba protein Acan 125, the first to be identified (Xu et al. 1995), binds to the SH3 domains of Acanthamoeba myosins IA and IC through PXXP motifs located near its COOH terminus, and coimmunoprecipitates with myosin IC (Xu et al. 1997; Lee et al. 1999; Zot et al. 2000). The cellular function of Acan 125, a member of the leucine-rich repeat family of proteins (Kobe and Deisenhofer 1995), is unknown. The yeast proteins verprolin and Las17/Bee1p bind to the SH3 domains of both Myo3p and Myo5p (Anderson et al. 1998; Evangelista et al. 2000; Lechler et al. 2000). Verprolin is proline-rich, binds actin monomers, and is required for endocytosis and the establishment of a polarized cortical actin cytoskeleton (Vaduva et al. 1997; Naqvi et al. 1998). Las17p/Bee1p (Li 1997) is the yeast homologue of WASp, the product of the Wiskott-Aldrich syndrome gene in humans (Derry et al. 1994), and is essential for assembly of the cortical actin cytoskeleton and for endocytosis (Li 1997; Naqvi et al. 1998). Both Las17p/Bee1p and WASp bind to the Arp2/3 complex, a highly conserved seven member complex containing the actin-related proteins Arp2 and Arp3 (Machesky et al. 1994), and accelerate Arp2/3-dependent nucleation of actin filament assembly (Machesky and Insall 1998; Machesky et al. 1999; Rohatgi et al. 1999; Winter et al. 1999; Yarar et al. 1999). Furthermore, verprolin interacts physically and genetically with Las17p/Bee1p (Naqvi et al. 1998). Similarly, the vertebrate homologue of verprolin, WIP, interacts physically and functionally with WASp (Ramesh et al. 1997; Vaduva et al. 1999; Moreau et al. 2000).
The purpose of the current study was to purify and characterize proteins that bind to the SH3 domains of Dictyostelium myoB (Jung et al. 1989) and myoC (Peterson et al. 1995). Our biochemical efforts resulted in the identification of p116, the Dictyostelium homologue of Acan 125. Like Acan 125, p116 binds to myoB and myoC through their SH3 domains. Surprisingly, p116 was also found to bind the seven member Arp2/3 complex, the central player in the de novo nucleation of actin filament assembly and in the formation of branched filament networks, and capping protein, the central player in the termination of actin filament assembly (Eddy et al. 1997; Carlier et al. 1999; Higgs and Pollard 1999; Machesky and Insall 1999; Welch 1999; Blanchoin et al. 2000; Cooper and Schafer 2000; Pantaloni et al. 2000). These interactions drive the formation of a cellular complex in which myoB and myoC are linked via their association with p116 to two key regulators of actin polymerization and three-dimensional organization. We show that p116, Arp2/3, myoB, and myoC are all concentrated in regions of active actin assembly, and that the normal functioning of these actin-rich domains depends on the presence of p116. Together, these results suggest that myoB and myoC support actin-dependent cellular processes not only through their roles as actin cross linkers and actin-based mechanoenzymes, but also through effects they may have on the assembly state and organization of actin. Such effects could arise from myosin I–dependent changes in the location, movement, and/or activity of the complex formed by the interaction of Arp2/3, capping protein, and myosin I with p116, which we have named the CARMIL protein, for capping protein, Arp2/3, and myosin I linker.
Materials and Methods
Identification of SH3 Domain Binding Proteins
The SH3 domains of Dictyostelium myoB (residues 1,057–1,111) and myoC (residues 1,126–1,181) were amplified by PCR using Pfu polymerase (600135; Stratagene) and the following oligonucleotides: 5′-CTAGGATCCACTGCAAAAGCACTCTACGATTATGAT-3′ and 5′-TGCGAATTCTTAATTATATTGTAAATAATTTGTTGG-3′ for myoB; 5′-CTAGGATCCCAATATATCGCTCTTTACGAGTACGAC-3′ and 5′-TGCGAATTCTTAAATTTGTTGAACATAATTTGAAGG-3′ for myoC. For the proline to leucine point mutation, the 3′ oligonucleotides for myoB and myoC were 5′-TGCGAATTCTTAATTATATTGTAAATAATTTGTTAGAGCCCAACCTTTTTG-3′ and 5′-TGCGAA-TTCTTAAATTTGTTGAACATAATTTGAAAGTAACATACCAA- TTTG-3′, respectively. The SH3 domain of Acanthamoeba myoC (residues 980–1,033) was amplified using the following oligonucleotides: 5′-CTAGGATCCCAGGCGCGTGCGCTGTATGACTTTGCG-3′ and 5′-TGCGAATTCTTAGATGAGTTCGACGTAGGACGCGGG-3′. All three products were digested with BamH1 and EcoRI, ligated into pGEX-KT, and transformed into Escherichia coli strain HB101. Overnight cultures were diluted 1:10 into 450 ml of fresh LB media containing 100 μg/ml ampicillin, grown at 37°C to OD 1.0 at 600 nm, and induced with IPTG (0.5 mM final). After a 3 h incubation, the cells were harvested, resuspended in 20 ml of PBS containing protease inhibitors (0.1 mM PMSF, 5 μg/ml AEBSF, 50 μg/ml leupeptin, 10 μg/ml aprotinin, and 10 μg/ml pepstatin A) and 0.1 mM DTT, and broken in a French press. Triton X-100 was added to a final concentration of 1%, at which point the lysate was mixed for 20 min at 4°C and centrifuged at 15,000 g for 15 min. The supernatant was then mixed with 1 ml of glutathione-Sepharose 4B (17-0756-01; Amersham Pharmacia Biotech) for 3 h at 4°C, at which point the resin was washed five times with PBS (15 ml and 5 min per wash, with rotation at 4°C). To generate the high-speed supernatant of lysed Dictyostelium, 10 g of wet cell pellet was broken by sonication in 50 ml of Dictyostelium lysis buffer (0.5× TBS containing 5 mM sodium pyrophosphate, 1 mM DTT, and protease inhibitors) and centrifuged at 90,000 g for 1 h at 4°C. Approximately 40 ml of supernatant was incubated on a rotating mixer with ∼1 ml of washed GST-SH3 domain resin for 3 h at 4°C. The resin was then washed five times in 1× TBS (10 ml and 5 min per wash with rotation 4°C). Bound proteins were eluted by incubation with 2 ml of 5× TBS for 30 min at 20°C. After centrifugation for 2 min at 15,000 g, the supernatant was mixed with 0.5 vol of SDS-PAGE sample buffer, resolved on 4–20% gradient gels, visualized by silver staining or Coomassie blue staining, and subjected to Western blot and microsequence analyses. For the latter, Coomassie blue–stained bands were excised from the gel, digested in situ with trypsin, and HPLC-purified tryptic peptides subjected to Edman degradation (Harvard Microsequencing Facility). For the far Western blot shown in Fig. 3 A, ∼1 mg of myoB or myoC SH3 domain fusion protein was incubated with 0.1 mg of Sulfo-NHS-LC biotin in 0.5 ml of PBS for 2 h at 4°C (21420; Pierce Chemical Co.). Unincorporated biotin was removed by chromatography on Sephadex G-25. The stoichiometry of biotin labeling, as determined using HABA reagent (28010; Pierce Chemical Co.), was ∼1.5 moles biotin per mole of fusion protein. Blots to be probed were blocked in TBST containing 0.2% gelatin, and incubated for 3 h at 4°C in TBST containing 0.2% gelatin and 10 μg/ml of biotinylated SH3 domain fusion protein. After four 5 min washes in TBST at 4°C, blots were incubated with streptavidin-conjugated alkaline phosphatase, washed, and developed using ECL reagents (RPN 2108; Amersham Pharmacia Biotech).
Figure 3.
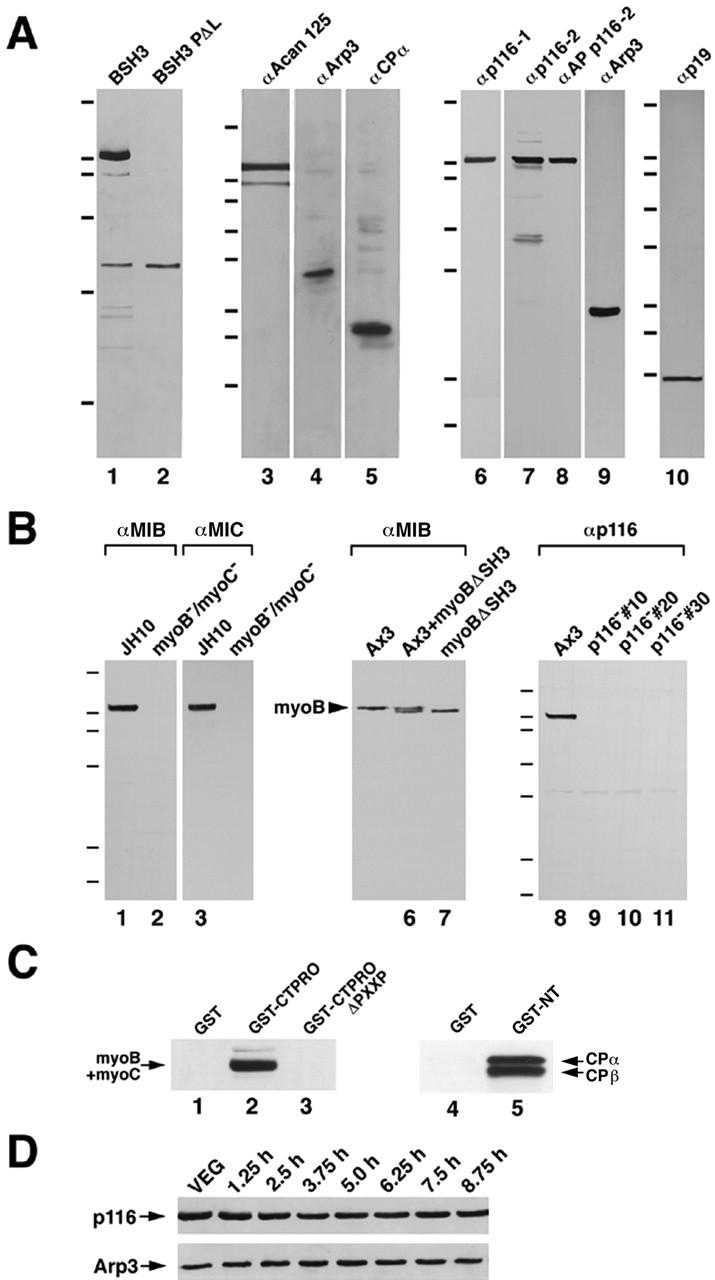
Far Western analysis of SH3 domain: p116 interaction, evidence for an Acanthamoeba complex, Western blot analyses of antibodies, mutant cell lines, identification of critical PXXP motifs, and developmental Westerns. (A) Lanes 1 and 2, blots of the material eluted from the myoB SH3 domain (lane 1) and the myoB PΔL SH3 domain (lane 2), and probed with biotinylated myoB SH3 domain. The hash marks here and elsewhere refer to the following molecular weight markers: 200, 116, 97, 66, 55, 37, 32, and 22 kD. Lanes 3–5, Western blots of the material eluted from the Acanthamoeba MIC SH3 domain and probed with antibodies to Acanthamoeba Acan 125 (lane 3), yeast Arp3 (lane 4), and Dictyostelium capping protein α (lane 5), using dilutions of 1:5,000, 1:3,000, and 1:3,000, respectively. Lanes 6–10, Western blots of Dictyostelium whole cell extracts probed with antibodies against Dictyostelium p116 (α-p116-1, lane 6; α-p116-2, lane 7; α-AP p116-2, lane 8), Dictyostelium Arp3 (lane 9), and Dictyostelium p19 (lane 10), using dilutions of 1:2,000, 1:20,000, 1:1,000, 1:20,000, and 1:10,000, respectively. (B) Lanes 1–4, Western blots of whole cell extracts from a myoB−/myoC− double mutant (lanes 2 and 4) and its parental strain JH10 (lanes 1 and 3), probed with affinity-purified antibodies to myoB (lanes 1 and 2) or myoC (lanes 3 and 4). Lanes 5–7, Western blot performed on whole cell extracts from strain Ax3 (lane 5), a myoB null cell line expressing myoB without its SH3 domain (myoBΔSH3) (lane 7), and an equal mixture of these two extracts (lane 6), and probed for myoB. Lanes 8–11, Western blot performed on whole cell extracts from three independent p116− cell lines (10, 20, and 30; lanes 9–11) and their parental strain Ax3 (lane 8), and probed with the anti-p116 antibody α-AP p116-2. (C) Lanes 1–3: Western blot of the eluates from glutathione beads loaded with unfused GST (lane 1), a GST fusion protein containing the COOH-terminal, proline-rich 203 residues of p116 (GST-CTPRO; lane 2), and GST-CTPRO in which the two PXXP motifs between residues 956–973 were deleted (GST-CTPROΔPXXP; lane 3) and incubated with a high-speed supernatant of lysed Dictyostelium. The eluates were probed with a mixture of antibodies to myoB and myoC (the arrow marks the position of their comigrating heavy chains). Lanes 4 and 5, Western blots of the eluates from glutathione beads loaded with unfused GST (lane 4) and a GST fusion protein containing the NH2-terminal 179 residues of p116 (GST-NT; lane 5), and incubated with a high speed supernatant of lysed Dictyostelium. The elutes were probed with a mixture of antibodies to the α and β subunits of capping protein. (D) Western blots of whole cell extracts from vegetative Ax3 cells (VEG), and cells starved on black filter supports for the indicated lengths of time (1.25–8.75 h) and probed with α-AP p116-2 (top) and α-Arp3 (bottom). The cells had reached ripple stage, the time of peak chemotactic aggregation, at ∼7 h.
Isolation of the p116 Gene
The gene for p116 was identified using four pooled degenerate oligonucleotides (5′-GARGARAGYCAAGCICAAAAYGARGC-3′; 5′-GARGARTCICAAGCICAAAAYGARGC-3′; 5′-CAAGCICAAAAYGARGCIAGYGGWGCIAC-3′; and 5′-CAAGCICAAAAYGARGCIT-CIGGWGCIAC-3′) corresponding to residues EESQAQNEA and QAQNEASGAT present in the longer peptide sequence obtained from p116 (Fig. 2). The PAGE-purified oligonucleotides were end labeled using T4 polynucleotide kinase (18004-010; GIBCO BRL) and γ[P32]ATP (35020; ICN Biomedicals), and purified by gel filtration on Sephadex G25 superfine. To create the genomic sub library enriched for EcoR1 fragments of ∼5 kb, EcoR1-digested Dictyostelium genomic DNA was resolved in a 0.9% agarose gel, and the DNA in the size range of 4.6–5.4 kb was collected by electroelution. These fragments were ligated into EcoR1-digested λ Zap II (236211; Stratagene), the ligation packaged using Gigapack III gold packaging extracts (200202; Stratagene), and the library plated using strain XL1-Blue MRF. Plaque lift-offs were prehybridized at 30°C in a solution containing 6× SSPE, 5× Denhardt's, 0.5% SDS, and 0.2 mg/ml salmon sperm DNA, and hybridized at 30°C in a solution containing 30% formamide, 5× SSPE, 2× Denhardt's, 0.1% SDS, 0.2 mg/ml salmon sperm DNA, and the pooled, end-labeled oligonucleotides. Filters were washed in 6× SSPE containing 0.1% SDS at 50°C. Bluescript plasmids were excised using helper phage RE704. The 4.8-kb EcoR1 insert was sequenced on both strands using a combination of restriction fragments and primer extension reactions. The precise boundaries of the four introns were obtained by sequencing reverse transcription (RT)-PCR products that spanned their splice donor and acceptor sites. Northern blots of total RNA from vegetative Dictyostelium and Southern blots of EcoRI-digested Dictyostelium genomic DNA were probed with the labeled oligonucleotides exactly as described above. General DNA sequence analyses were performed using the program GCG (University of Wisconsin Genetics Computer Group sequence analysis software). Data base searches were performed using the programs BLAST and FASTA. Dot matrix comparisons were performed using the DOT PLOT menu of GCG.
Figure 2.

Peptide microsequences. Shown are the sequences obtained from single peptides derived from p47, p41, p35, p21, p19, and p16, and two from p116. Each is aligned with the deduced protein sequence of a cDNA present in the Japanese Dictyostelium cDNA sequencing project database (the protein's identity and accession number are shown). Exceptions are the p35 peptide sequence, which is aligned with the sequence of capping protein α (Hug et al. 1995), and the two p116 peptide sequences, where no homologues were found.
Creation of MyoB−/MyoC− Double Mutants, P116− Mutants, and Cells Expressing MyoB without an SH3 Domain
To create myoB−/myoC− double mutants, plasmid pBsr2 (gift of Dr. Kazuo Sutoh, University of Tokyo, Tokyo, Japan), which confers resistance to blasticidin S, was linearized with HindIII, converted to blunt ends by T4 polymerase fill-in, and treated with phosphatase, creating plasmid A. A 3′ portion of the genomic sequence of myoB (nucleotides 2,330–3,086) was amplified by PCR using Pfu polymerase and the following oligonucleotides: 5′-CTAGCTGGTCAATGGCAATTGGG-3′ and 5′-CATAAGCTTGGTAAACCAGAAGCGATTG-3′. The purified product, which contains a nested HindIII site at the 3′ end, was cloned in the 5′ to 3′ orientation into plasmid A, creating plasmid B. Plasmid B was linearized with XbaI, converted to blunt ends by T4 polymerase fill-in, and treated with phosphatase, creating plasmid C. A 5′ portion of the genomic sequence of myoB (nucleotides 532–1,247) was amplified by PCR using the following oligonucleotides: 5′-GCATCTAGAGCATATAGAGGTAAACATGC-3′ and 5′-GTAGCATACTTGCAGCCAATTC-3′. The purified product, which contains a nested XbaI site at the 5′ end, was cloned into plasmid C in the 5′ to 3′ orientation, creating the final disruption vector (plasmid D). Plasmid D was digested with XbaI and HindIII, releasing a linear disruption fragment in which the 5′ and 3′ myoB coding fragments bracket the Bsr resistance cassette. After purification by proteinase K treatment and phenol/chloroform extraction, the mixture of vector backbone and disruption fragment was introduced by electroporation into a representative myoC− line created previously in axenic strain JH10 (Jung et al. 1996). Transformed cells were selected in 10 μg/ml blasticidin (150477; ICN Biomedicals), and independent clones purified by serial dilution. MyoB− knockout lines were identified by Western blot analysis and confirmed by Southern blot analysis.
The exact same strategy was used to create p116− cell lines. The 5′ and 3′ genomic fragments with nested XbaI and HindIII sites, respectively, were generated by PCR using the following oligonucleotides: 5′-TGCTCTAGAGAAACCAAGTCAAAACAAAGTAAAG-3′ and 5′-AGCACCTAATAAACATCTAATAGTATC-3′ for the 5′ fragment (nucleotides 1,316–1,766); 5′-ACCCTCAAGAATATGCCAACACCT-3′ and 5′-GCATAAGCTTAGTTGATCTTGGAGCAACAACAGG-3′ for the 3′ fragment (nucleotides 3,321–4,427). The digested disruption vector was introduced by electroporation into axenic strain Ax3. Whole cell extracts from independent, purified, blasticidin-resistant transformants were screened by Western blot analysis using p116 antibody α-p116-1 to identify knockout lines. Homologous recombination was confirmed by Southern blot analyses using probes to the blasticidin cassette, the 5′ genomic fragment used in the disruption construct, and the portion of the p116 gene lost from the genome as a consequence of the double-crossover, gene replacement event.
The creation of cells lines expressing myoB without its SH3 domain will be presented in detail elsewhere. In brief, the myoB null cell line null 6 (Jung and Hammer 1990) was transformed with the integrating plasmid pDEX containing the coding sequence for all but the COOH-terminal 54 amino acids of the myoB heavy chain. Several stable transformants were selected that expressed the truncated heavy chain at ∼1.5 times normal vegetative cell levels, as determined by quantitative Western blotting, which included a correction for the loss of epitopes due to the absence of the SH3 domain.
Generation of Antibodies to Dictyostelium p116, Arp3, and p19 (p21-Arc), and to Acanthamoeba Acan 125
Antibody α-p116-1 was generated using the Dictyostelium 116-kD protein present in myoB SH3 domain fusion protein column eluates. In brief, SDS-PAGE slices containing ∼70 μg of p116 were frozen, crushed, mixed with Freunds complete adjuvant, and injected subcutaneously into 15 sites on one rabbit. This animal was boosted using incomplete adjuvant at 4 wk, and bled at 8 wk. Antibody α-p116-2 was raised against a GST fusion protein containing p116 residues 847 to 1,050, which was generated by PCR using Pfu polymerase and the following oligonucleotides: 5′-TCAGGATCCGAAGAATCTCAAGCTCAAAATGAAGC-3′ and 5′-CAGGAATTCTTAATTTTCGGTCGGTGGTCTTGGTC-3′. This fragment was digested with BamHI and EcoRI and cloned into pGEX 2TK, and the fusion protein, referred to below as “GST-CT PRO,” was purified by chromatography on glutathione-Sepharose 4B. Rabbits were immunized as described above, except that 400 μg of protein was used per injection. To generate antibody α-APp116-2, α-p116-2 serum was incubated with a p116 null cell extract bound to nitrocellulose exactly as described previously (Jung et al. 1993). Antibody to Dictyostelium Arp3 was raised against a GST fusion protein containing Arp3 residues 169–312 (Murgia et al. 1995) generated by RT-PCR using Pfu polymerase, total Dictyostelium RNA, a 3' RACE kit (18373-019; GIBCO BRL), and the following oligonucleotides: 5′-ATCGGATCCGATAGTGGTGATGGTGTAAC-TCAT-3′, and 5′-GATGAATTCTTAACGACAATCGATTGGACA-GGATTG-3′. The PCR product was cloned into pGEX 4T-1 and rabbits immunized with the purified fusion protein as described for α-p116-2. Antibody to Dictyostelium p19 (p21-Arc) was raised against a GST fusion protein containing the entire coding sequence of p19, obtained by RT-PCR using oligonucleotides 5′-TCAGGATCCATGGTTTATCACTCACAATTCAAC-3′ and 5′-CAGGAATTCTTATAAGGCTTTGTTTAAGAATTTTCTC-3′. Antibody to Acanthamoeba Acan 125 was raised against a GST fusion protein containing Acan 125 residues 221–452, obtained by RT-PCR as described above, except that total Acanthamoeba RNA was used, and the oligonucleotides were 5′-TCAGGATCCCTCCACTTCAACAACTACTTCATCGGC-3′ and 5′-CTCGAATTCTTA-GCCGATGATGGCCTTGATGACATC-3′. The insoluble fusion protein was purified from a detergent resistant pellet of lysed bacteria by preparative SDS PAGE and electro elution (model 422; Bio-Rad Laboratories).
Immunofluorescence Light Microscopy
Vegetative cells adhered to coverslips were fixed for 5 min in 99% methanol/1% formaldehyde at −15°C and stained exactly as described previously (Jung et al. 1993). Crude rabbit sera against Dictyostelium Arp3 and p19 were diluted 1:300, whereas rabbit sera against Dictyostelium myoC and p116, which were first purified by incubation with their corresponding null cell extract (see above and Jung et al. 1996), were diluted 1:30 (representing an ∼1:500 dilution of the crude sera). The FITC-labeled goat anti–rabbit and LRSC-labeled goat anti–mouse secondary antibodies were from Jackson ImmunoResearch Laboratories (111-095-144 and 115-085-146, respectively) and were used at a dilution of 1:100. Neither showed any reactivity in the absence of primary antibody, or any cross species reactivity. The antiactin monoclonal antibody C4 (1378996; Boehringer) was diluted 1:300. The anticoronin monoclonal antibody (gift of Eugenio De Hostos, University of California at San Francisco, San Francisco, CA) was diluted 1:50. For staining of starved cells undergoing chemotactic aggregation, suspension-grown cells at late log were suspended at a density of 3 × 10 cells/ml in starvation buffer (SB) (20 mM potassium phosphate, pH 6.7, 2 mM MgCl2, and 0.2 mM CaCl2), and 50 μl was spotted in the center of a coverslip. Coverslips were incubated in a humidified chamber at 20°C until cells were actively streaming, at which point the cells were briefly overlaid with an agar sheet, fixed, and stained as described previously (Fukui et al. 1989; Jung et al. 1993). Confocal images were obtained on a ZEISS 410 LSM using a 63× Plan-Apo objective and 0.2–0.7-μm steps. Projected images and sections rendered in three dimensions were created using ZEISS and Metamorph (Universal Imaging) software.
Immunoprecipitations
Vegetative Dictyostelium cells were incubated for 10 min at 4°C in lysis buffer (100 mM NaCl, 20 mM Tris, pH 7.5, 2 mM ATP, pH 7.0, 1% (vol/vol) NP-40, and protease inhibitors) and centrifuged at 100,000 g for 15 min at 4°C. Approximately 20 ml of lysis buffer was used per gram of cell pellet. Similar results were obtained when cells were lysed by sonication in buffer lacking NP-40 and containing isotonic sucrose (0.25 M). Approximately 1 ml of high speed supernatant was mixed for 3 h at 4°C with 40 μl of protein A agarose (15918-014; GIBCO BRL) that had previously been incubated with 40 μl of primary antibody against Dictyostelium myoB (Jung and Hammer 1990), myoC (Jung et al. 1996), or p116 (α-p116-1) in 0.5 ml of 1X TBS for 3 h at 4°C. Beads were washed five times with 1× TBS (2 ml and 5 min per wash, with rotation at 4°C), boiled for 5 min in SDS-PAGE sample buffer, and the supernatants used for Western blot analyses. Blots were probed with antibodies to Dictyostelium myoB, myoC, p116 (α-p116-1), and capping protein α (gift of Dr. John Cooper, Washington University, St. Louis, MO), and to yeast Arp3 (gift of Dr. Kathy Gould, Vanderbilt University, Nashville, TN), at dilutions of 1:20,000, 1:30,000, 1:2,000, 1:5,000, and 1:3,000, respectively.
Identification in p116 of the Binding Sites for Myosin I and Capping Protein
To identify the myosin I binding site, two adjacent PXXP motifs present between residues 956 and 973 in p116 were deleted from the GST fusion protein GST-CT PRO (see above) by overlap extension PCR using Pfu polymerase and the following oligonucleotides: 5′-TCAGGATCCGAAGAATCTCAAGCTCAAAATGAAGC-3′ and 5′-GGTTTTGATTTA-AGTGGTGTAACATCTGGTGGCGCACCTCCACCCATATTTC-3′ for the 5′ fragment, and 5′-GAAATATGGGTGGAGGTGCGCCACCAGATGTTACACCACTTAAATCAAAACC-3′ and 5′-CAGGAATTCTTAATTTTCGGTCGGTGGTCTTGGTC-3′ for the 3′ fragment. The complete insert obtained using the outside primers and a mixture of the 5′ and 3′ fragments was digested with BamH1 and EcoR1 and cloned into pGEX 2TK. To test for the ability to interact with myosin I in whole cell extracts, glutathione-Sepharose resins loaded with the deletion-containing fusion protein (referred to below as “GST-CT PRO PXXP−”), the undeleted version, or GST alone (0.8 ml resin) were incubated at 4°C for 4 h with 8 ml of a high speed supernatant of lysed Dictyostelium. The preparation of this supernatant, the washing of the resin, and the elution of bound proteins were performed as described above for the identification of SH3 domain binding proteins. Eluates were then probed for the presence of myoB and myoC by Western blotting. To search for the binding site for capping protein, a fusion protein spanning residues 1–179 of p116 (referred to below as “GST-NT”) was generated using pGEX 2TK, Pfu polymerase, and the following oligonucleotides: 5′-TCAGGATCCATGTCAGAAGAAATATCACCAAATG-3′ and 5′-GTCGAATTCTTA-AGAGGAGATAATATTTGTCATATCCC-3′. Glutathione-Sepharose resins loaded with GST-NT or GST alone were incubated with a high-speed supernatant of lysed Dictyostelium and processed as described above. Eluates were probed for the presence of the α and β subunits of capping protein.
Purification of Actin and the Arp2/3 Complex, and Actin Nucleation Assays
Actin was purified from Acanthamoeba castellanii according to Gordon et al. 1976, followed by gel filtration on HiPrep Sephacryl S200 (17-1195-01; Amersham Pharmacia Biotech). Monomeric actin was labeled with N-(1-pyrene)-iodoacetamide (P29; Molecular Probes) and stored lyophil-ized at −80°C. Before use it was resuspended and dialyzed in G buffer (0.1 mM CaCl2, 0.5 mM ATP, pH 7.0, 0.75 mM β-mercaptoethanol, and 3 mM imidazole, pH 7.5). The Arp2/3 complex was purified from Acanthamoeba as described by Kelleher et al. 1998 and stored at −70°C in complex storage buffer supplemented with 200 mM sucrose. The portion of p116 (residues 644–863) that contains a short sequence conserved among G-actin binding proteins (including verprolin), plus an acidic region was amplified by PCR using Pfu polymerase and the following oligonucleotides: 5′-TCAGGATCCATTTGGAAGGAAATCGATTCATGTATC-3′ and 5′-ACTGAATTCTTAATCTGGAATTGGGGTGGCACCACT-3′. The product was digested with BamH1 and EcoR1, cloned into pGEX 2TK (27-4587-01; Amersham Pharmacia Biotech), and the fusion protein, referred to below as “GST VA,” expressed and purified as described above. Actin nucleation assays were performed essentially as described by Higgs et al. 1999. In brief, G-actin was mixed with pyrene-labeled actin at a ratio of 20:1, converted to Mg++-actin, and polymerized at a final concentration of 4 μM by the addition of 10× polymerization buffer (500 mM KCL, 10 mM MgCl2, 10 mM EGTA, and 100 mM imidazole, pH 7.0). Additional proteins (e.g., Arp2/3, GST VA; see the legend to Fig. 5) were dialyzed into G buffer supplemented with 1 mM MgCl2 and added to the actin solution just before the initiation of polymerization. Fluorescence intensities were measured every second for 10 min using a PTI QuantaMaster fluorometer (Photon Technology International) and wavelengths of 365 and 407 nm for excitation and emission, respectively.
Figure 5.
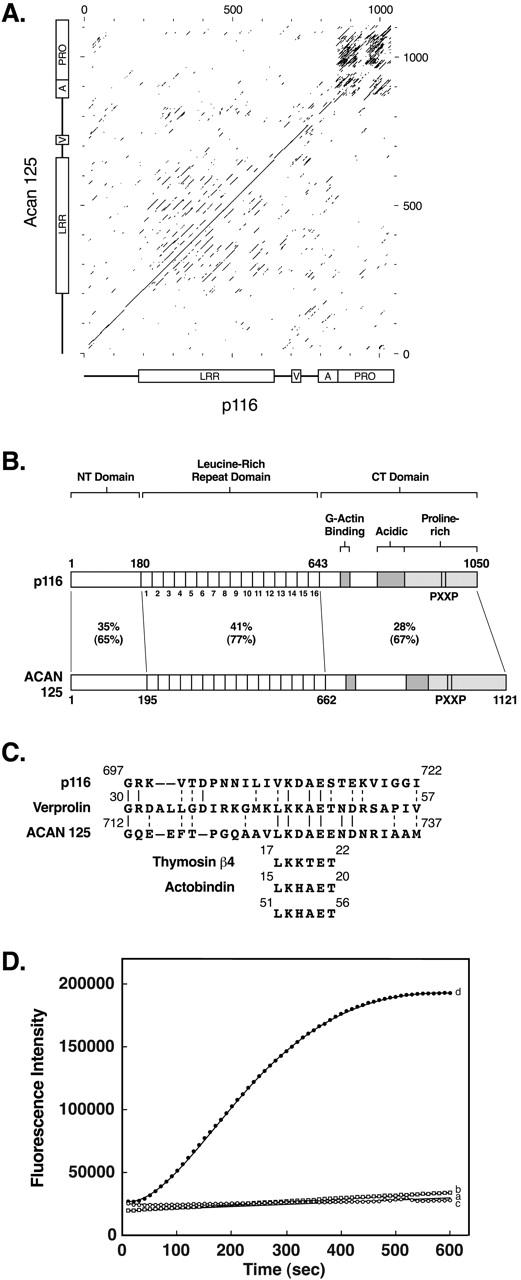
Dot matrix comparison, schematic of the domain organization of p116 and Acan 125, alignment of the verprolin-like sequences, and acceleration of Arp2/3-dependent actin nucleation by the VA domain of p116. (A) Dot matrix comparison between Dictyostelium p116 and Acanthamoeba Acan 125 (window size, 30; stringency, 11). The dark rectangular region in the upper right corner is due to the alignment of their repetitive proline-rich sequences. (B) Schematic of the tripartite domain organization of p116 and Acan 125, showing the percent identity and the percent similarity (identities plus conservative substitutions in parentheses) for each domain. The positions of the 16 LRRs, the verprolin-like sequence that in verprolin has been implicated in binding G-actin, the acidic region, the proline-rich domain, the two PXXP motifs known to be critical for the interaction between Acan 125 and the SH3 domain of Acanthamoeba myosin IC, and the two PXXP motifs deleted from p116 in this study, are indicated. (C) Alignment of a portion of p116 and the homologous sequence in Acan 125 with a region of yeast verprolin that contributes to the binding of G-actin, and with six-residue sequences present in thymosin β4 and actobindin that also contribute to binding monomeric actin. (D) Actin nucleation assays were performed using 4 μM G-actin (5% pyrene actin) with or without the Arp2/3 complex and GST VA (see Materials and Methods). Shown are the rates of polymerization for actin alone (trace a, no symbols), actin plus 50 nM Arp2/3 complex (trace b, squares), actin plus 3 μM GST VA (trace c, open circles), and actin plus 50 nM Arp2/3 complex and 3 μM GST VA (trace d, closed circles). Similar results were obtained using two different preparations of proteins. Addition of 3 μM unfused GST did not accelerate Arp2/3-dependent actin nucleation (data not shown).
Estimation of Cellular F-Actin Content
The amount of F-actin in cells was estimated by a modification of previous methods (Podolski and Steck 1990; Watts and Howard 1994). In brief, 2.5 × 107 log phase cells were seeded in a tissue culture dish (3003; Falcon) in SB and incubated until streams were evident (∼6 h for wild-type strain Ax3; ∼10 h for p116 null cells). Cells were lysed by the addition of 2 ml of ice cold buffer A (50 mM KCL, 10 mM imidazole. pH 7.5, 3.3 mM Tris-acetate, pH 7.5, 2.2 mM magnesium acetate, 1 mM EGTA, 0.5 mM ATP, pH 7.0, 1% Triton X-100, and protease inhibitors), followed by incubation on ice for 5 min. Triton-insoluble cytoskeletons were collected by centrifugation at 1,000 g for 2 min at 4°C. To estimate the amount of actin in this pellet, which should be predominantly in filamentous form, samples were resolved by SDS-PAGE and the amount of Coomassie blue–stained material present at the position of actin quantitated by densitometry (ChemImager 4400; Alpha Innotech). A second estimate of cellular F-actin content was obtained by lysing cells on ice in buffer A containing 20 μM FITC-labeled phalloidin (P5282; Sigma-Aldrich), washing the resulting Triton-insoluble cytoskeletons twice with ice-cold buffer A, dissolving the pellets in 50 mM sodium phosphate buffer, pH 9.2, and measuring the fluorescence intensities using wavelengths of 485 and 520 nm for excitation and emission, respectively. Essentially identical results were obtained using these two methods. The values presented in the text are those obtained using fluorescence measurements, and are given for p116 null cells as the percent of the value obtained for the parental strain Ax3.
Cell Biological Assays and General Methods
To perform streaming assays, midlog phase cells (∼4 × 106/ml) were pelleted at 500 g for 5 min, washed in SB, resuspended in SB at 1.2 × 106/ml, and seeded in 60 mm tissue culture dishes (3002; Falcon) (2.5 ml/dish). Essentially all of the cells (both control and p116−) adhered to the dish. Images were obtained using a Nikon ZMU stereo microscope coupled to a CCD camera (model C2400; Hamamatsu) and an optical disc recorder (TQ3031F; Panasonic). The rate of fluid phase pinocytosis was determined on cells in suspension using FITC-labeled dextran and centrifugation through PEG 8000 cushions exactly as described previously (Jung et al. 1996). Other general methods were performed as described previously (Jung et al. 1996).
Results
The SH3 Domains of MyoB and MyoC Interact In Vitro with a Complex Mixture of Polypeptides Comprised of the Seven-Member Arp2/3 Complex, Capping Protein, and a Protein of 116 kD
To look for soluble proteins that interact with the SH3 domains of myoB and myoC, these domains were expressed as GST fusion proteins, incubated with high speed supernatants of lysed Dictyostelium, washed extensively at physiologic ionic strength, and eluted with high salt. Eluates were then resolved by SDS-PAGE and visualized by silver staining (Fig. 1). Relative to controls (beads alone, lane 2; beads coupled to GST only, lane 3), the myoB SH3-domain fusion protein bound nine polypeptides with molecular masses of 116, 47, 44, 41, 35, 33, 21, 19, and 16 kD (lane 4, see arrowheads). Introduction into the myoB SH3 domain of a proline to leucine (PΔL) point mutation, which is function blocking when present in the SH3 domain of the Caenorhabditis elegans adaptor protein SEM 5 (Stern et al. 1993), completely abrogated the interaction with these nine polypeptides (lane 5), suggesting that the interactions are specific. Similar results were obtained with the myoC SH3-domain fusion protein (lane 6) and its corresponding point mutant (lane 7).
Figure 1.

Isolation of SH3 domain binding proteins. Shown are silver-stained SDS-PAGE gels (4–20%) of molecular weight standards (lane 1), proteins eluted from glutathione beads (lane 2) and beads loaded with unfused GST (lane 3), and proteins eluted from beads loaded with the following GST fusion proteins: GST-myoB SH3 domain (lane 4), GST-myoB SH3 domain containing the PΔL point mutation (lane 5), GST-myoC SH3 domain (lane 6), GST-myoC SH3 domain containing the PΔL mutation (lane 7), and GST-myoB SH3 domain (lane 8). The asterisk next to lane 2 marks the position of the silver-stained band that reacted with an antiactin monoclonal antibody. The arrowheads adjacent to lanes 4 and 6 point to the nine apparent polypeptides that bound to the myoB and myoC SH3 domain fusion proteins. The identities of the 10 proteins that correspond to these nine bands are shown to the right of lane 8. Using a 15% SDS-PAGE gel and authentic Dictyostelium profilin as a standard, we found that the isolated complex is devoid of profilin. Westerns performed using an antibody to Dictyostelium SCAR (Bear et al. 1998; gift of Dr. Karl Saxe, Emory University, Atlanta, GA) show that this Arp2/3 binding protein is also not present.
All nine polypeptides described above were subjected to protein microsequencing. Unambiguous sequences were obtained for p116, p47, p41, p35, p21, p19, and p16 (Fig. 2). Blast searches failed to detect any homologues to the sequences of two peptides obtained from p116, but identified the 35-kD band as the α subunit of capping protein, and revealed that the remaining five sequenced proteins correspond to the Dictyostelium homologues of the Arp2/3 complex components Arp3 (47-kD band), p41-Arc (41-kD band), p21-Arc (19-kD band), p20-Arc (21-kD band) and p16-Arc (16-kD band) (Fig. 2 and Fig. 1, lane 8). Given that a tight association exists between all seven members of the Arp2/3 complex (Higgs and Pollard 1999), and that the β subunit of capping protein should be present along with the α subunit, we conclude that the remaining two unaccounted for bands at 44 and 33 kD correspond to Arp2 and a mixture of the ∼34-kD Arp2/3 complex component p34-Arc and the ∼33-kD β subunit of capping protein (Fig. 1, lane 8). This latter conclusion is supported by the fact that an antibody specific for the β subunit of capping protein (Hug et al. 1995) recognized the ∼33-kD band (data not shown). Therefore these data indicate that the nine bands correspond to ten different polypeptides: the α and β subunits of capping protein, the seven members of the Arp2/3 complex, and an unknown protein of 116 kD (Fig. 1, lane 8).
Given that capping protein and all seven Arp2/3 complex components lack a consensus SH3 domain binding motif, we hypothesized that p116 alone interacts directly with the myosin I SH3 domains. To test this idea, the material in Fig. 1, lane 8 was transferred to nitrocellulose and probed with biotinylated myoB SH3-domain fusion protein. Development of the blot with strepavidin/alkaline phosphatase revealed that the SH3 domain did indeed interact to a significant extent with just p116 (Fig. 3 A, lane 1; lane 2, which contained the material bound to the myoB SH3 PΔL column, serves as a control). This result, and identical results obtained using the myoC SH3 domain as a probe (data not shown), indicate that the proline-rich binding site for the two myosin I SH3 domains resides within p116, and suggest that p116 in turn binds capping protein and the Arp2/3 complex. Consistent with this idea, the abundance of p116 in many individual preparations always dictated the abundance of capping protein and the Arp2/3 complex. When the yield of p116 was high, the yield of capping protein and Arp2/3 could be high, sometimes approaching apparent stoichiometry with p116, but when the yield of p116 was low, the yield of capping protein and Arp2/3 was always low.
Although not evident from the gel in Fig. 1, these complexes do not contain profilin (data not shown, but see legend to Fig. 1). Furthermore, we never observed bands other than the nine shown in Fig. 1 appearing consistently or in amounts remotely approaching stoichiometry with p116. Finally, Western blots indicated that some or all of the band at 45 kD in control and SH3-domain column eluates (see asterisk in Fig. 1, lane 2) is conventional actin (data not shown). This small amount of actin could not be responsible for the presence of capping protein and Arp2/3 in the SH3 domain column eluates because all four controls (Fig. 1, lanes 2, 3, 5, and 7) contain the same amount of immunoreactive and silver-stained material at 45 kD, and yet lack all of the complex components. To summarize, these biochemical studies indicate that Arp2/3, capping protein, an unknown protein of 116 kD, and, by inference, myosin I, can be isolated as a complex at physiologic ionic strength, and suggest that p116 serves as the scaffold for assembly of the complex.
Immunoprecipitation Reactions Support the Existence of the Complex In Vivo, Confirm That Myosin I Interacts with p116 Through Its SH3 Domain, and Indicate that p116 Serves as the Scaffold for Assembly of the Complex
To look for the existence of the complex between myosin I, Arp2/3, capping protein, and p116 in vivo, we performed immunoprecipitation experiments using wild-type and mutant cell extracts, antibodies to Dictyostelium myoB (Jung and Hammer 1990), myoC (Jung et al. 1996), and capping protein (Hug et al. 1995), an antibody to yeast Arp3 (gift of Dr. Kathy Gould, Vanderbilt University, Nashville, TN), and an antibody raised against p116 eluted from gels of the purified complex (α-p116-1; see Fig. 3 A, lane 6).
To look for the association of p116 with myosin I in vivo, immunoprecipitations were made using wild-type cell extracts and antibodies to either myoB (Fig. 4 A, lanes 1 and 2) or myoC (lanes 3 and 4). These immunoprecipitates contained not only the corresponding myosin I heavy chain (lanes 1 and 3; the band at ∼116 kD in lane 1 is a breakdown product of myoB), but also p116 (lanes 2 and 4). Similarly, immunoprecipitations made using wild-type cell extracts and the antibody to p116 (lanes 5–7) contained not only p116 (lane 5), but also myoB (lane 6) and myoC (lane 7). To demonstrate that the interaction between myosin I and p116 is SH3 domain–dependent, extracts of cells that express myoB lacking its SH3 domain, which were made by complementing a myoB null cell line with a truncated version of myoB missing the COOH-terminal 54 amino acids (Fig. 3 B, lanes 5–7 and Materials and Methods regarding this mutant), were used. As predicted, myoB was present in immunoprecipitates made from these cells using an antibody to myoB (Fig. 4 A, lane 8), but p116 was not (lane 9). Together, these results indicate that p116 interacts with myoB and myoC in vivo, and that these interactions require the presence of the myosin I SH3 domain.
Figure 4.

Immunoprecipita-tions. Shown throughout are ECL-based Western blots performed on the final immunoprecipitate. (A) Wild-type (WT) cell extracts (lanes 1–7) were immunoprecipitated (IP) with antibodies to myoB (lanes 1 and 2), myoC (lanes 3 and 4), and p116 (lanes 5–7), and the immunoprecipitates probed (Western) with antibodies to myoB (lanes 1 and 6), myoC (lanes 3 and 7), and p116 (lanes 2, 4, and 5). Extracts from cells expressing myoB without its SH3 domain (BSH3−; lanes 8 and 9) were immunoprecipitated with an antibody to myoB (lanes 8 and 9), and the immunoprecipitate probed with antibodies to myoB (lane 8) and p116 (lane 9). (B) Wild-type cell extracts (lanes 1–3) were immunoprecipitated with antibodies to myoB (lane 1), myoC (lane 2), and p116 (lane 3), and the immunoprecipitates probed with an antibody to yeast Arp3 (lanes 1–3). Extracts from myoB−/myoC− double mutants (B−/C−) (lane 4) were immunoprecipitated with an antibody to p116 (lane 4), and the immunoprecipitate probed with an antibody to yeast Arp3 (lane 4). Extracts from cells expressing myoB without its SH3 domain (lane 5) were immunoprecipitated with an antibody to myoB (lane 5), and the immunoprecipitate probed with an antibody to yeast Arp3 (lane 5). (C) Wild-type extracts (lanes 1–4) were immunoprecipitated with preimmune serum (PI) (lane 1) and antibodies to myoB (lane 2), myoC (lane 3), and p116 (lane 4), and the immunoprecipitates probed with an antibody to the α subunit of capping protein (lanes 1–4). Extracts from myoB−/myoC− cells (lane 5) were immunoprecipitated with an antibody to p116 (lane 5), and the immunoprecipitate probed with an antibody to capping protein α (lane 5). Extracts from cells expressing myoB without its SH3 domain (lane 6) were immunoprecipitated with an antibody to myoB (lane 6), and the immunoprecipitate probed with the antibody to capping protein α (lane 6). We note that immunoprecipitates made from p116− cells using antibodies to myoB or myoC were devoid of Arp3 and capping protein, consistent with the idea that the association of Arp2/3 and capping protein with myosin I is via their interaction with p116 (data not shown).
To look for the association of the Arp2/3 complex with myosin I and p116 in vivo, immunoprecipitations were made using wild-type cell extracts and antibodies to myoB, myoC, or p116 (Fig. 4 B, lanes 1–3, respectively). Arp3 was present in all three immunoprecipitates. Furthermore, immunoprecipitates made using the antibody to p116 and extracts from cells lacking both the myoB and myoC heavy chains, which were created by targeted disruption of both genes (Fig. 3 B, lanes 1–4, and Materials and Methods regarding this mutant), contained Arp3 (Fig. 4 B, lane 4). Conversely, antibody to myoB did not immunoprecipitate Arp3 from extracts of cells expressing myoB without its SH3 domain (lane 5). Together, these results indicate that Arp2/3 is associated with myosin I and p116 in vivo, and that its presence in this complex is mediated by an interaction with p116, not myosin I.
To look for the association of capping protein with myosin I and p116 in vivo, immunoprecipitations were made using wild-type cell extracts and either preimmune serum or antibodies to myoB, myoC, or p116 (Fig. 4 C, lanes 1–4, respectively). The α subunit of capping protein was present in all but the immunoprecipitate made using preimmune serum (lanes 1–4). Furthermore, immunoprecipitates made using extracts of cells lacking myoB and myoC, and the antibody to p116, contained capping protein (lane 5). Conversely, immunoprecipitates made using cells expressing myoB without its SH3 domain and the antibody to myoB did not contain capping protein (lane 6). Together, these results indicate that capping protein is associated with myosin I and p116 in vivo, and that its presence in this complex is mediated by an interaction with p116, not myosin I.
Cloning of the Gene Encoding p116 Indicates that It Is the Dictyostelium Homologue of Acan 125
We used degenerate oligonucleotides to the longer of the two p116 peptide sequences to search for the gene encoding p116. These oligonucleotides recognized a single ∼5-kb EcoRI band in digests of Dictyostelium genomic DNA, as well as a single mRNA of a size (∼2,700 nucleotides) consistent with a 116-kD polypeptide (data not shown). Therefore, these oligonucleotide probes were used to screen a genomic DNA sub library created in λZAP using EcoRI-digested Dictyostelium genomic DNA in the size range of ∼4.6–5.4 kb. The 4.8-kb EcoRI insert present in several purified phage clones obtained was sequenced on both strands (EMBL/GenBank/DDBJ accession no. AF388524), revealing 3,150 nucleotides of protein coding sequence interrupted by four introns, whose precise boundaries were confirmed by the sequencing of RT-PCR products (data not shown). The complete ORF contains 1,050 amino acids with a calculated molecular mass of 115.8 kD, and possesses both peptides obtained from the microsequencing of p116 (data not shown), indicating that this genomic clone does encode the 116-kD polypeptide present in the SH3 domain column eluates. Despite the fact that the two peptides obtained from p116 were not obviously present in Acan 125, and that antibodies to both proteins do not cross react (data not shown), p116 is indeed the Dictyostelium homologue of Acan 125, as can be seen in a dot matrix comparison between the two (Fig. 5 A), where a line of identity spans virtually the entire matrix. Direct pair wise comparisons (data not shown) indicate that the two proteins are 32% identical and 65% similar.
P116 and Acan 125 Exhibit the Same Domain Structure and Bind Myosin I in a Similar Fashion
Fig. 5 B shows a schematic of the domain structure of p116 and Acan 125 (Xu et al. 1997). Like Acan 125, the central 456 amino acids of p116 are comprised of 16 copies of a ∼29-residue α-β structural unit known as a leucine-rich repeat (LRR). LRRs contain a loose consensus sequence dominated by leucines, form amphipathic β-α structural units, and mediate protein–protein interactions, either by serving as the ligand binding site itself, or by increasing the affinity and/or specificity of binding at a separate site (Kobe and Deisenhofer 1995). p116 and Acan 125 are 41% identical and 77% similar in this domain. NH2-terminal of the LRR domain in both proteins are ∼190 amino acids that, while conserved (35% identical, 65% similar), are largely unremarkable in composition. By contrast, the COOH-terminal 189 residues of p116 and 198 residues of Acan 125 are remarkable in being proline-rich (24% in p116, 26% in Acan 125; Fig. 5 B). Runs of five or six prolines in a row, which represent profilin binding sites in proteins like WASp and VASP (Zeile et al. 1998), are not present in either p116 or Acan 125, however, consistent with the fact that our complex lacks profilin.
As reported by Xu et al. 1997, the proline-rich domain of Acan 125 contains two closely spaced PXXP motifs (residues 980–983 and 989–992) that fit the consensus for SH3 domain target sequences. Consistent with this, a fusion protein containing the COOH-terminal 344 residues of Acan 125 was found to bind to the isolated SH3 domain of Acanthamoeba myosin IC, and this interaction was abrogated by an 18-residue deletion spanning the two PXXP motifs (Xu et al. 1997). We find that the COOH-terminal proline-rich domain of p116 also contains two closely spaced PXXP motifs (residues 959–962 and 968–971; Fig. 5 B). Moreover, a GST fusion protein containing the proline-rich COOH terminus of p116 (residues 847–1050; GST-CT PRO) pulls down myoB and myoC from high-speed supernatants of lysed Dictyostelium (Fig. 3 C, lane 2), but not when residues 956–973, which span the two PXXP motifs, have been deleted from the fusion protein (Fig. 3 C, lane 3). Furthermore, the wild-type fusion protein fails to pull down myoB from extracts of cells expressing the truncated version of the protein lacking the SH3 domain (data not shown).
Identification of an Arp2/3 Activation Domain and the Capping Protein Interaction Site in p116
Given that p116 and Acan 125 (see below) bind the Arp2/3 complex, we searched their sequences for evidence of the domains that in WASp/SCAR proteins are responsible for binding Arp2/3 (the acidic or A domain) and for accelerating Arp2/3-dependent actin nucleation (the WH2 domain, in combination with the A domain and, possibly, an intervening cofilin-like sequence) (Higgs and Pollard 1999; Welch 1999). Neither p116 nor Acan 125 possesses a highly concentrated patch of acidic residues containing a trytophan, which together comprise the WASp/SCAR A domain. Furthermore, neither protein contains a cofilin-like sequence. Finally, neither protein contains a stretch of sequence with strong similarity throughout to the ∼18-residue WH2 domain, which participates in the binding of G-actin by WASp/SCAR proteins (Machesky and Insall 1998; Miki and Takenawa 1998; Egile et al. 1999; Higgs et al. 1999). Nevertheless, analysis of the ∼200 residues of p116 and Acan 125 located between their LRR and COOH-terminal proline-rich domains revealed two sequence features that could be functionally related to the WH2 and A domains. The first is a 26-residue sequence in p116 (residues 697–722), and the corresponding sequence in Acan 125 (residues 712–737), both of which can be aligned with residues 30–57 in verprolin (Fig. 5B and Fig. C). This portion of verprolin has been implicated in the binding of G-actin (Vaduva et al. 1997), and overlaps the portion of the protein (residues 30–46) with significant homology to the WH2 domain of WASp/SCAR (Higgs and Pollard 1999). Furthermore, contained within the verprolin-like sequences of p116 and Acan 125 is a six-residue motif that verprolin shares with several other G-actin-binding proteins, including thymosin β4 and actobindin (Vancompernolle et al. 1991; Vaduva et al. 1997) (Fig. 5 C). Cross-linking studies and mutational analyses have shown that this six-residue sequence forms part of an actin monomer-binding site in these latter two proteins (Vancompernolle et al. 1991; Van Troys et al. 1996).
The second sequence feature, which lies COOH-terminal of the verprolin-like sequence, is a region of 72 residues in p116 (792–863) and 55 residues in Acan 125 (871–925), both of which exhibit a strong net negative charge (Fig. 5 B). Specifically, these regions possess net charges of −10 for p116 (14 D/E, 4 K/R) and −16 for Acan 125 (17 D/E, 1 K/R).
To determine whether these p116/Acan 125 sequences function like the WH2/A domains of WASp/SCAR to accelerate Arp2/3-dependent actin nucleation (Machesky and Insall 1998; Higgs et al. 1999; Machesky et al. 1999; Winter et al. 1999), actin assembly assays were performed in the presence of the Arp2/3 complex, with and without GST VA, a GST fusion protein containing the verprolin-like and acidic sequences of p116 (residues 644–863; Fig. 5 D). As seen by others, Arp2/3 alone had little effect on actin nucleation (trace b; trace a is actin alone). Similarly, addition of GST VA alone had no obvious effect on the rate of actin assembly (trace c). However, the addition of both together resulted in a significant acceleration of actin polymerization (trace d). Although these results indicate that the VA domain of p116 activates Arp2/3-dependent actin nucleation, the concentration of GST VA required to obtain this level of activation (low μM) is significantly higher than that needed for the isolated WH2/A domain of WASp/SCAR (Higgs and Pollard 1999). Therefore, either the ability of intact p116 to activate Arp2/3-dependent actin nucleation is relatively modest, or analysis of the isolated VA domain underestimates the activating potential of the intact protein (see Discussion).
In an effort to identify the binding site in p116 for capping protein, the NH2-terminal 179 residues of p116, which spans the region from the ATG to the start of the LRR domain, was expressed as a GST fusion protein (GST NT) and incubated with a high speed supernatant of lysed Dictyostelium. Western blots of the material eluted from this resin revealed the presence of the α and β subunits of capping protein (Fig. 3 C, lane 5). By contrast, the material eluted from GST alone (Fig. 3 C, lane 4), GST VA, or GST CT PRO (data not shown) did not contain capping protein. These results are consistent with the presence of a capping protein interaction site within the NH2-terminal 179 residues of p116.
Acan 125 Also Interacts with the Arp2/3 Complex and Capping Protein
The identification of p116 as a homologue of Acan 125 implies that Acan 125 should also interact with the Arp2/3 complex and capping protein, in addition to myosin I. To test for this, we performed biochemical experiments like those in Fig. 1 using the SH3 domain of Acanthamoeba myosin IC and Acanthamoeba cell lysates. Western blots (Fig. 3 A, lanes 3–5) indicated that the eluate contained not only Acan 125 (lane 3), as expected, but also Arp 3 (lane 4) and the α subunit of capping protein (lane 5). These results are consistent with the idea that a portion of cellular Acan 125 is present in a complex similar to the one identified here for p116.
Homologues of p116 and Acan 125 Are Present in Metazoans
A previous search for metazoan homologues to Acan 125 (Xu et al. 1997) revealed a Caenorhabditis elegans ORF of ∼1,000 residues (EMBL/GenBank/DDBJ accession no. Z71264) that showed significant similarity to Acan 125 primarily over the NH2-terminal ∼640 amino acids of both proteins. Our analysis of this C. elegans sequence using an updated sequence file (EMBL/GenBank/DDBJ accession no. CAA95828.1) reveals a 998-residue polypeptide that can be aligned with p116 and Acan 125 from NH2 to COOH terminus (data not shown). Our searches also identified a Drosophila melanogaster ORF of 1,369 amino acids (EMBL/GenBank/DDBJ accession no. AE003840), which exhibits significant sequence similarity to p116 and Acan 125 over much of the protein (data not shown). Importantly, searches performed using this D. melanogas-ter sequence identified putative homologues in humans (EMBL/GenBank/DDBJ accession no. CAA18156.1 and BAA90912.1) and mouse (EMBL/GenBank/DDBJ accession no. AA407423.1 and AA118567.1). The human ORF exhibits 24% identity and 40% similarity to residues 647–1196 of the D. melanogaster sequence.
P116 Localizes Along with MyoB, MyoC, and the Arp2/3 Complex in Dynamic Actin-rich Cellular Extensions, Including Macropinocytic Crowns and the Leading Edge of Locomoting Cells
We next sought to determine the intracellular localization of p116, and to see if it, myosin I, and the Arp2/3 complex localize to the same cellular structures. We used previously characterized antibodies to myoB and myoC (Jung and Hammer 1990; Jung et al. 1996; Fig. 3 B, lanes 1–4), newly generated antibodies to the Dictyostelium Arp2/3 complex components Arp3 and p19 (p21-Arc; Fig. 3 A, lanes 9 and 10, respectively), and a second new antibody to p116 (α-p116-2), which was raised against the proline-rich domain of p116 and purified by absorption against p116 null cell extracts (α-AP-p116-2; Fig. 3 A, lanes 7 and 8, respectively). Given that myoB and myoC in Dictyostelium, and the Arp2/3 complex in other cell types, have been shown to localize to regions of active actin assembly (Uyeda and Titus 1997; Higgs and Pollard 1999), cells were sometimes double-stained for actin or for the actin-binding protein coronin, which accumulates in actin-rich structures (De Hostos 1999). We also examined both vegetative and starved cells because p116 and Arp3 (Fig. 3 D), as well as myoB and myoC (Jung et al. 1996), are expressed at both stages, and because cells at these two stages differ significantly in terms of shape, speed, and the site where actin assembly is most pronounced (Varnum et al. 1985; Spudich 1987). Vegetative cells are relatively round, slow moving (∼1 μm/min), and exhibit dramatic actin assembly primarily on their dorsal surface, where cuplike membrane extensions rich in F-actin and coronin are continuously being extended upwards (De Hostos 1999). These structures, known variously as cups, crowns, or amoebastomes, are macropinocytic structures that rise up, close, and retreat into the cell, bringing with them large fluid-filled macropinosomes (Hacker et al. 1997). Starved cells undergoing chemotactic migration towards extracellular cAMP are highly elongated, fast moving (∼12 μm/min), and exhibit dramatic actin assembly primarily in the pseudopods and lamellopods at their leading edge.
With regard to vegetative cells, Fig. 6 A shows a Z series of a cell that had been double stained for Arp3 and actin. Striking colocalization between the two (yellow) is seen almost exclusively in dorsal confocal sections, where both are concentrated throughout a cup-shaped macropinocytic crown. Very similar images were obtained by double staining vegetative cells for Arp3 and coronin, a bona fide marker for these dorsal macropinocytic extensions (De Hostos 1999), and by using the antibody to p19 instead of Arp3 (data not shown). Examination of the ventral confocal sections in Fig. 6 A, and the single ventral confocal section shown for the two cells in Fig. 6 B (top), indicates that the Arp2/3 complex, while present within the ventral actin–rich lamellae of vegetative cells (marked by arrows in upper “Actin” panel), is not highly concentrated there. This may reflect the fact that the requirement for actin assembly in these lamellae, which are not driving rapid cell migration, is significantly less than in dorsal crowns, which are driving constitutive endocytosis. Interestingly, Arp3 is often concentrated in bright spots that accumulate just inside the ventral lamellae (see arrowheads in Fig. 6 B, top, “Arp3” panel). The nature and function of these spots is currently unknown.
Figure 6.
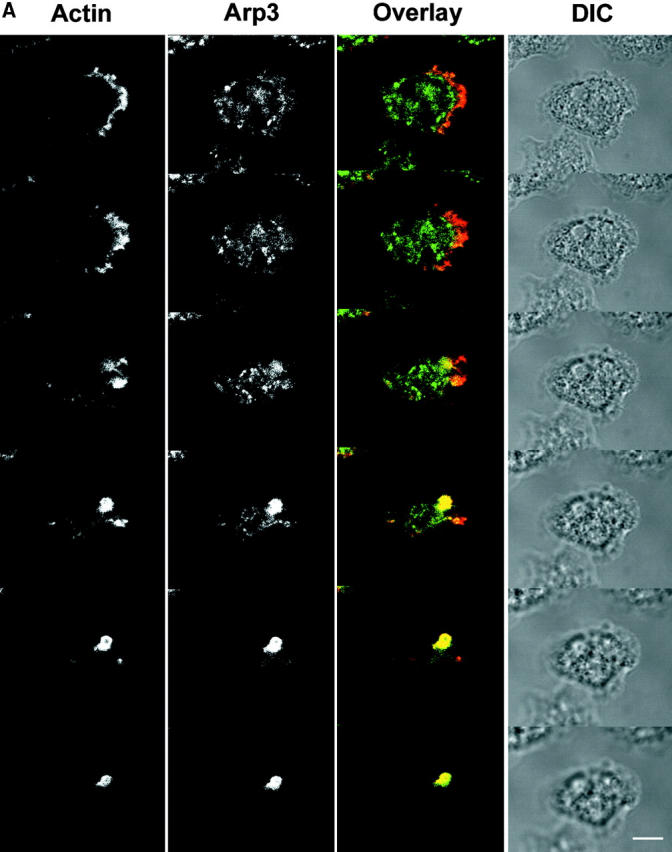

Localization of the Arp2/3 complex and p116 in vegetative cells. (A) Z series (0.7 μm sections) of a vegetative cell double stained for actin and Arp3 (top, ventral; bottom, dorsal). The corresponding overlaid (actin, red; Arp3, green; colocalization, yellow) and DIC images are also shown. (B) A single, ventral, 1 μm-confocal section through vegetative cells double stained for actin and Arp3 (top) or actin and p116 (bottom). The corresponding overlaid (red, actin; green, Arp3 or p116; colocalization, yellow) and DIC images are also shown. The arrows in the two Actin panels point to the actin-rich ventral lamella, whereas the arrowheads in the Arp3 and p116 panels point to the prominent spots of staining seen for both antibodies just inside the lamella. Bars: (A) 4.6; (B) 7.2 μm.
Previous work by Novak and Titus 1998 showed that myoB is concentrated in macropinocytic crowns. We now find the same result for myoC (Fig. 7 A; arrowheads point to a myoC-positive crown). Furthermore, p116 is also present in these macropinocytic structures, as can be seen for two representative cells in Fig. 7 B, where the prominent macropinocytic crowns evident in the DIC images (see arrowheads) are strongly stained for p116 using antibody α-APp116-2. Similar results were obtained using antibody α-p116-1, and p116 null cells showed negligible staining with both antibodies, as expected (data not shown). Interestingly, p116, like Arp3, is not particularly concentrated in the ventral actin–rich lamellae of vegetative cells (Fig. 6 B, bottom). Furthermore, p116, like Arp3, is often present in prominent spots that accumulate just inside these lamellae (see arrowheads in Fig. 6 B, bottom, “p116” panel). Whether these p116-rich spots are the same as the Arp3-rich spots described above remains to be determined.
Figure 7.
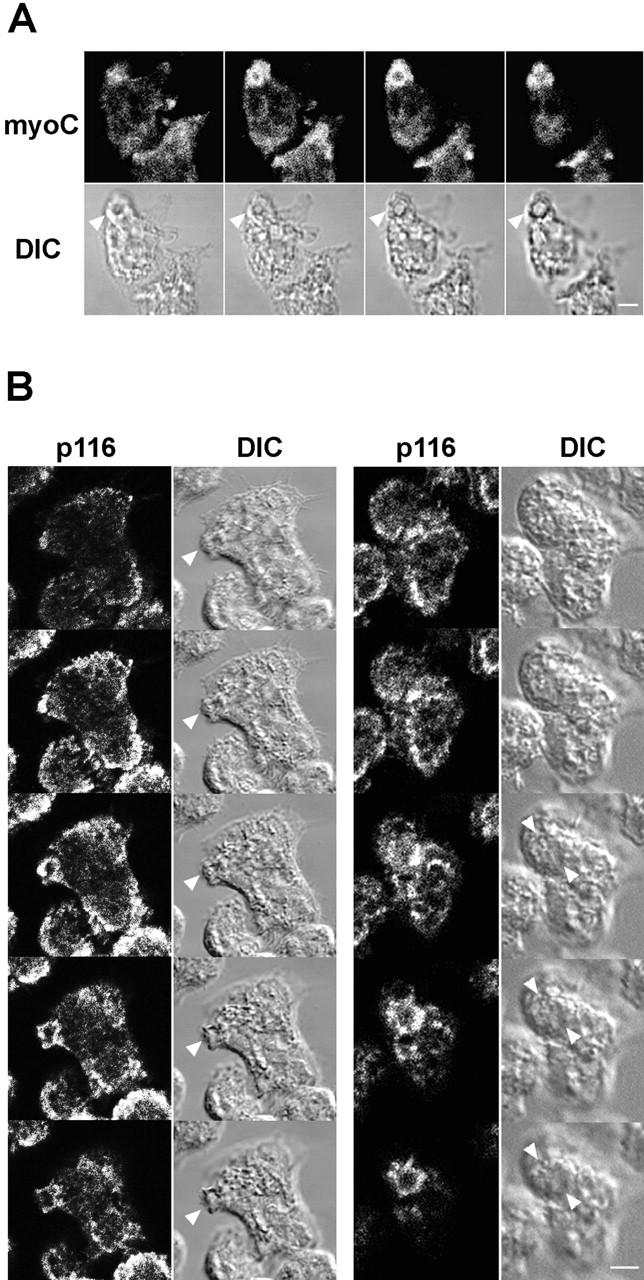
Localization of MyoC and p116 in vegetative cells. (A) Z series (1-μm sections) of a vegetative cell stained for myoC (left, ventral; right, dorsal). Arrowheads in the corresponding DIC images mark the prominent myoC-positive crown. (B) Two sets of Z series (1-μm sections) of vegetative cells stained for p116 using α-AP-p116-2 (top, ventral; bottom, dorsal). Arrowheads in the corresponding DIC images mark the dorsal p116-positive crowns. Bars: (A) 2.2; (B) 4.4 μm.
With regard to starved cells, Fig. 8 A shows a projected image of a field of such cells that were double stained for actin and Arp3. These cells, which were undergoing chemotactic migration towards the upper left upon fixation, exhibit a striking concentration of actin and Arp3 within the pseudopods and lamellopods at their leading edge. Similar results were obtained using the antibody to p19 (data not shown). Previous work by Fukui et al. 1989 and Jung et al. 1996 has shown that myoB and myoC are also concentrated in these leading edge pseudopods. Fig. 8 B shows that p116 is concentrated there as well. Examination of the individual confocal sections that make up this projected image indicated that actin and p116 colocalize throughout the leading edge.
Figure 8.
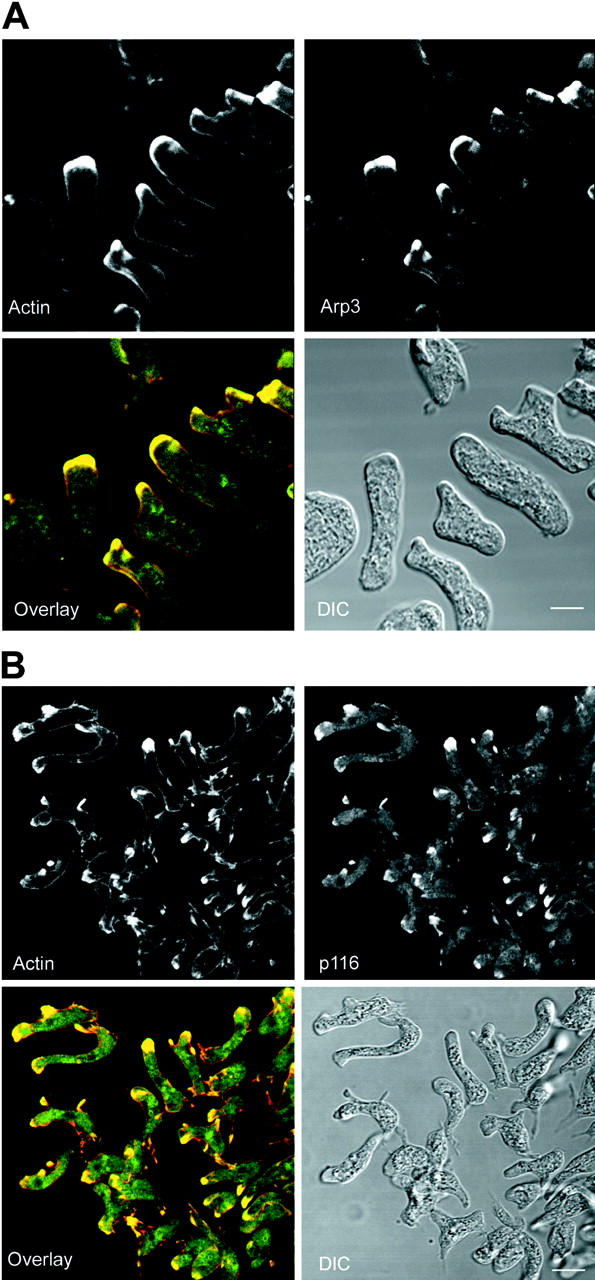
Localization of the Arp2/3 complex and p116 in starved, chemotaxing cells. (A) A projected image of a field of cells undergoing chemotactic aggregation that were double stained for actin and Arp3 (the cells are moving towards the upper left). The corresponding overlaid (actin, red; Arp3, green; colocalization, yellow) and DIC images are also shown. (B) A projected image of a field of cells undergoing chemotactic aggregation that were double stained for actin and p116 (the cells are moving towards the left). The corresponding overlaid (actin, red; p116, green; colocalization, yellow) and DIC images are also shown. Bars: (A) 6.5; (B) 12 μm.
Cells Lacking p116 Exhibit Defects in Fluid Phase Endocytosis, the Formation of Macropinocytic Crowns and the Efficiency of Chemotactic Aggregation, and a Reduction in Cellular F-Actin Content
We created p116− cell lines in an effort to gauge the physiologic importance of the protein. Ax3 cells were transformed with a linear gene disruption fragment comprised of the selectable marker blasticidin flanked by portions of the p116 gene (see Materials and Methods). Approximately 20% of ∼60 purified blasticidin-resistant transformants were found by Western blot analysis to lack p116. Three independent clones were chosen for further study (clones 10, 20, and 30). Fig. 3 B, lanes 8–11, show that these clones are completely devoid of p116 protein, and Southern blot analyses indicated that the linear disruption fragment had indeed disrupted the p116 locus, and only this locus (data not shown). All three clones gave essentially identical results in the behavioral assays described below.
Given that p116 localizes to macropincytic crowns, that macropinocytosis appears to account for most of fluid phase endocytosis in axenically grown Dictyostelium (Hacker et al. 1997), and that axenically grown p116 null cells exhibit a 35% reduction in growth rate relative to the parental cell line (doubling times: ∼8.5 h for Ax3, ∼12 h for p116−), we compared the rate of uptake of the fluid phase marker FITC-dextran in control and p116− cells. The rate of accumulation of this marker within cells during the first ∼60 min after its addition to the medium accurately reports the rate of fluid phase endocytosis because the time interval between endocytosis and exocytosis of nondigestable markers in Dictyostelium, which lack a rapid recycling component, is ∼60 min (Klein and Satre 1986; Jung et al. 1996). Fig. 9 A shows that all three p116− cell lines exhibit an ∼ 45% reduction in the rate of fluid phase endocytosis relative to the parental cell line. Mutants also appear to have a smaller intracellular endocytic compartment, as the amount of cell-associated dextran at steady state, which is attained in ∼75 to 90 min, and represents the point of equivalence between marker uptake and exocytosis, is about half the value of control cells.
Figure 9.
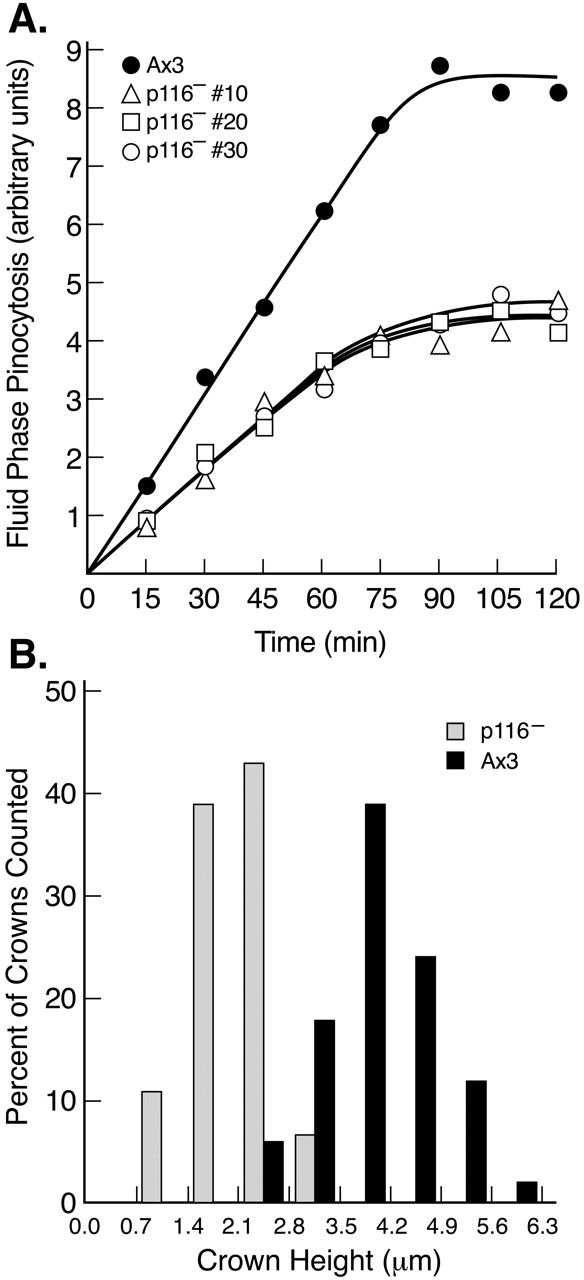
Rate of fluid phase endocytosis, and crown size, in wild-type and p116− cells. (A) Kinetics of accumulation of the fluid phase pinocytic marker FITC-dextran (arbitrary units) within three independent p116− cell lines and the parental line Ax3 (see key). Each value is the mean of duplicate samples. (B) Histogram of the height of crowns in Ax3 and p116− cells (see key). The values, which were taken from confocal Z series of cells stained for actin, were binned in 0.7-μm intervals and are presented as a percent of total crowns counted (64 in Ax3, 58 in p116−). Crowns were counted only if they rose almost vertically from the dorsal surface of cells. The bottom of such crowns typically exhibited a spherical, solid disc of actin staining. Above this the crown had the appearance of a ring of fluorescence in each section. Similar results were obtained using cells stained for coronin.
To identify a structural basis for the reduced rate of macropinocytosis, control and p116− cells in growth media were fixed, stained for actin, optically sectioned at 0.2-μm intervals, and the sections rendered in three dimensions to visualize the shape and size of macropinocytic crowns. Fig. 10, A and B, show control cells at 30 and 90 degrees of tilt. These cells typically have one or two large and well-formed macropinocytic cups on their dorsal surfaces (see arrowheads). By contrast, p116− cells possess much less robust crowns on their dorsal surface (Fig. 10C and Fig. D, see arrowheads; to view an image series that pro-gresses gradually from 30 to 90 degrees of tilt, go to www.nhlbi.nih.gov/labs/cellbiology/index.htm). To quantify this apparent difference, we measured the height of dorsal crowns in confocal images obtained using 0.2–0.7 μm steps, binned the heights in 0.7-μm intervals, and graphed the values as a percentage of crowns counted (Fig. 9 B). As one would predict from the fact that this approach captures crowns at all stages of formation, the values obtained exhibit a gaussian distribution. Importantly, this distribution for p116− cells is shifted relative to control cells towards an obviously shorter height. Indeed, p116− cells exhibit a mean crown height of 2.4 μm, versus 4.3 μm for Ax3 cells. Furthermore, no crowns of ≥3.5 μm were found in p116− cells, whereas 78% of crowns in Ax3 cells were ≥3.5 μm in height. Therefore, although p116− cells can still form crowns, they do so less effectively than wild-type cells, and there is a striking defect in the formation of large well-formed macropinocytic cups when p116 is absent. We suggest that this structural defect underlies the decreased rate of endocytosis exhibited by p116− cells.
Figure 10.
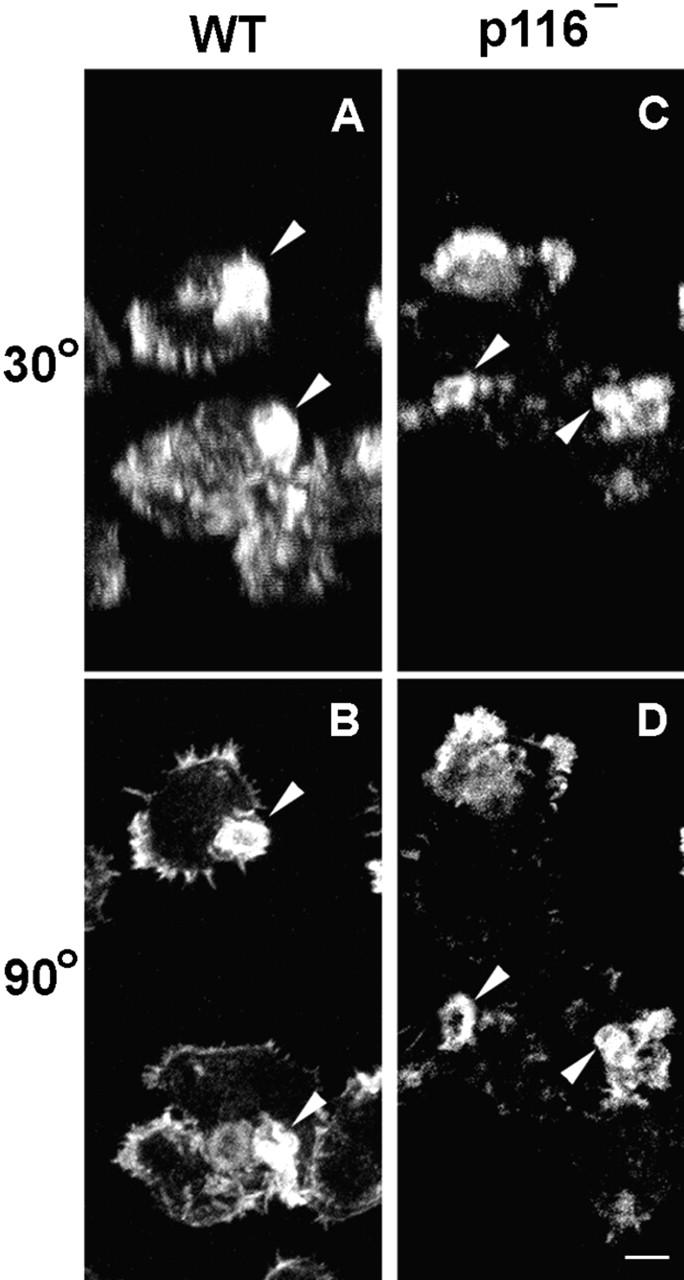
The appearance of crowns in wild-type and p116− cells. Vegetative Ax3 and p116− cells were stained for actin, sectioned in 0.2-μm intervals, and the sections rendered in three dimensions. Shown are images at 30 and 90 degrees of tilt for Ax3 (A and B) and p116− (C and D) cells. Arrowheads point to the dorsal crowns. The hollow center of these crowns is especially obvious when viewed from above (i.e., 90 degrees; see, for example, the crown marked by the lower arrowhead in B), whereas their sides are most evident when viewed from an angle that is somewhat above 0 degrees (i.e., 30 degrees; see, for example, the same crown mentioned above, but now in A). To view an image series that progresses gradually from 30 to 90 degrees of tilt, go to www.nhlbi.nih.gov/labs/cellbiology/index.htm. Bar, 3.4 μm.
Given the striking concentration of p116 within leading edge pseudopods, we also compared the ability of control and p116− cells to undergo starvation-induced chemotactic aggregation in streaming assays. Fig. 11 shows the typical result obtained when an initial cell density of 105 cells/cm2 was used. p116− cells began to aggregate ∼3 h after control cells (compare Fig. 11H and A), and by 13 h, when control cells had nearly completed streaming and the formation of large, robust aggregates (Panel D), p116− cells had formed only small, loosely organized clumps (Fig. 11I). Although this difference was accentuated at lower starting cell densities, p116− cells lagged behind control cells and formed much smaller streams and aggregates even at higher initial cell densities (e.g., ∼3 × 105 cells/cm2; data not shown). Therefore, although p116− cells can still undergo chemotactic aggregation, they do so much less efficiently than wild-type cells, and form only small aggregates (Fig. 11J). At least part of this defect may be due to a reduction in cellular F-actin content, as aggregating (∼10 h starved) p116− cells contain 77 ± 2.3% as much F-actin as aggregating (∼6 h starved) control cells (n = 8). This reduction could be due to the loss of that portion of Arp2/3-dependent actin nucleation that is stimulated by p116, to an increase in the level of active capping protein, or to a combination of both (see Discussion).
Figure 11.
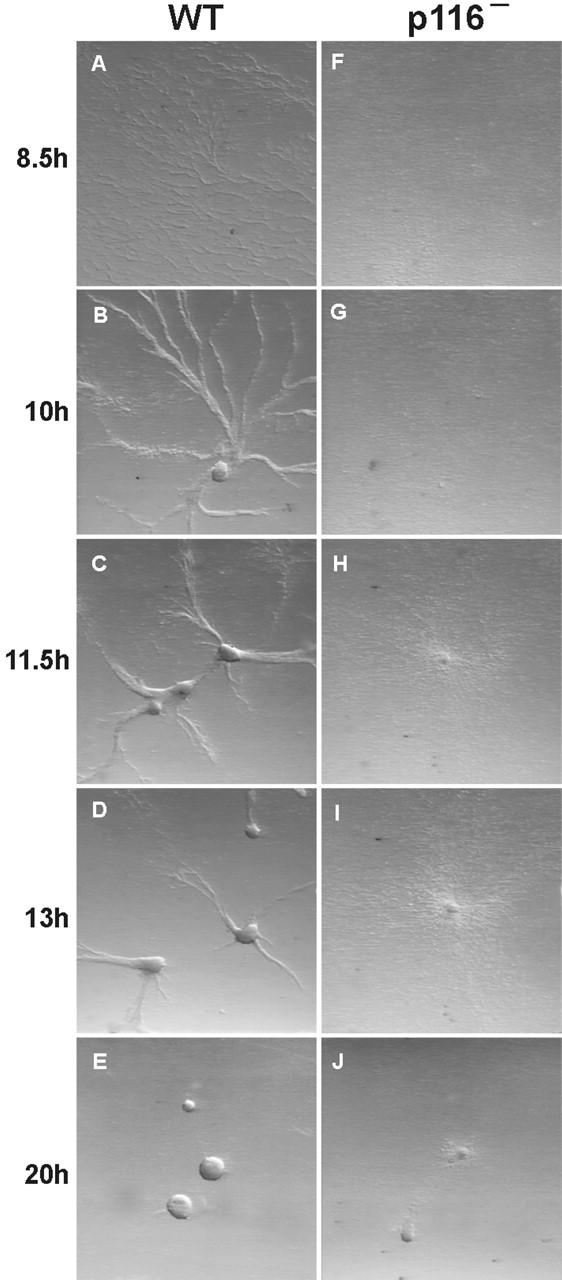
Streaming assays. In these assays, vegetative cells are plated at moderate densities in the presence of a buffered salt solution. After ∼8 h of starvation, normal cells begin to undergo chemotactic migration towards certain cells in the population that have begun to secrete cAMP. Cells initially migrate individually, but soon express surface adhesion proteins, causing them to migrate towards the source of extracellular cAMP as elongated aggregates (“streams”) (Spudich 1987). Shown are Ax3 cells (A–E) and p116− cells (F–J) undergoing this starvation-induced chemotactic streaming and aggregation. The numbers to the left indicate the time in hours between when the cells were plated in SB (0 h) and when each pair of images was taken.
Discussion
Identification and Organization of the Complex Containing CARMIL, Myosin I, Arp2/3, and Capping Protein
The main goal of this study was to identify through biochemical means proteins that bind to the SH3 domains of Dictyostelium myoB and myoC. Using these domains as affinity matrices, we isolated p116, the Dictyostelium homologue of Acan 125 (Xu et al. 1995). Our SH3 domain column eluates also contained on a consistent basis nine other polypeptides, which we identified as the seven members of the Arp2/3 complex and the α and β subunits of capping protein. Immunoprecipitation reactions and other experiments provided evidence that at least some fraction of cellular myoB and myoC is present in a complex with p116, Arp2/3, and capping protein in vivo, and that p116 serves as a scaffold for assembly of the complex, binding myosin I, capping protein, and Arp2/3 at independent sites. Given its central role in complex formation, we propose the name CARMIL for p116, which stands for capping protein, Arp2/3, myosin I linker.
In terms of how the complex is organized, Fig. 12 presents a working model based on our efforts to identify the sites in CARMIL where myosin I, capping protein, and the Arp2/3 complex bind. First, deletion analyses indicated that the myosin I SH3 domain binds to CARMIL through one or two closely spaced PXXP motifs present within CARMIL's COOH-terminal, proline-rich domain. These latter results agree with those of Xu et al. 1997 and Zot et al. 2000, who have shown that one or both of two PXXP motifs present in the analogous region of Acan 125 are necessary and sufficient to drive its interaction with myosin I. Second, the ability of a fusion protein containing the N-terminal 179 residues of CARMIL to bind capping protein present in cell lysates implicates this portion of CARMIL in its interaction with capping protein. Finally, the ability of a fusion protein containing the verprolin-like and acidic sequences of CARMIL to accelerate Arp2/3-dependent actin nucleation implicates the region between the LRR domain and COOH-terminal proline-rich domain in binding the Arp2/3 complex.
Figure 12.

Working model for the complex between myosin I, CARMIL, Arp2/3, and capping protein. In this working model, CARMIL serves as the scaffold for assembly of the complex. CARMIL, and the most conspicuous domains within it (NT, NH2-terminal domain; LRR, leucine-rich repeat domain; V, verprolin-like sequence; A, acidic domain; Pro-Rich, proline-rich COOH-terminal domain), are shown approximately to scale, whereas the remaining proteins are not. The binding of G-actin is inferred from the sequence of CARMIL and the activation data. The large black arrow signifies that myosin I might move the complex towards the barbed end of actin filaments, making this complex the “cargo”. The model also suggests that the LRR domain may be the site where upstream signaling molecules bind (e.g., Ras-related GTPases), based on the fact that the LRR domains in yeast adenylate cyclase (Suzuki et al. 1990) and the mouse protein rsp-1 (Masuelli and Cutler 1996) bind ras-related GTPases. If this were also the case for the LRR domains of p116 and Acan 125, it would provide a mechanism through which the activities of the complex could be regulated by signal transduction pathways known to regulate actin assembly in other contexts. The fact that we obtained complexes from the SH3 column in which both capping protein and the Arp2/3 complex appear to be approximately stoichiometric with CARMIL makes it very unlikely that their interaction with CARMIL is mutually exclusive. With regard to dependent binding, the fact that we sometimes obtained complexes with much more capping protein than Arp2/3 argues that capping protein does not bind to CARMIL through Arp2/3. Having said this, we never obtained complexes in which the amount of Arp2/3 complex exceeded that of capping protein. Based on this observation alone, we cannot exclude the possibility that Arp2/3 binds to CARMIL through capping protein. However, this seems unlikely based on our demonstration that different parts of CARMIL are responsible for binding capping protein and for activating Arp2/3, and based on the fact that no one has reported that capping protein binds the pseudobarbed end of the Arp2/3 complex.
In terms of the affinities of these interactions, Lee et al. 1999 and Zot et al. 2000 have measured binding constants in the range of 20–150 nM for the interaction between Acan 125 and the SH3 domain of myosin I. These values, together with the cellular concentrations of myosin I and Acan 125 (∼1 and ∼2 μM, respectively), suggest that type I myosins and the CARMIL protein may be largely associated in vivo, barring some type of regulation (Lee et al. 1999). Although we have not measured affinities for the interaction of CARMIL with capping protein and Arp2/3, the fact that the amount of Arp2/3 in SH3 domain column eluates sometimes approached apparent stoichiometry with CARMIL, and that the amount of capping protein almost always appeared to be approximately stoichiometric with CARMIL, suggests that the affinities of Arp2/3 and capping protein for CARMIL are fairly high. Having said this, we note that the concentration of GST VA required to activate Arp2/3-dependent actin nucleation is much higher than that required for GST fusion proteins containing the VCA domain of WASp (Higgs and Pollard 1999). Although this may mean that CARMIL's ability to activate Arp2/3 is relatively modest, it is also possible that the activity of GST VA significantly underestimates the activating potential of the intact protein. For example, the optimal conformation of the VA domain for activation of Arp2/3 may be dependent on its presence in the intact protein. Relevant to this, LRR domains have been shown to increase the affinity of adjacent sites for their ligands (Kobe and Deisenhofer 1995). Resolution of this question must await characterization of purified CARMIL.
Localization of CARMIL, Phenotype of CARMIL Null Cells, and Implications for Myosin I Mutant Phenotypes
Previous studies have shown that myoB and myoC in Dictyostelium (Uyeda and Titus 1997), the Arp2/3 complex in other cell types (Higgs and Pollard 1999; Machesky and Insall 1999), and capping protein in Acanthamoeba (Cooper et al. 1984) are all concentrated in dynamic, actin-rich cellular extensions. We find that CARMIL is concentrated along with myoB, myoC, and Arp2/3 within two such regions in Dictyostelium: crown-shaped macropinocytic extensions on the dorsal surface of vegetative cells, and psuedopods at the leading edge of starved cells undergoing chemotactic migration. Although this apparent colocalization does not prove the existence of the complex, it is consistent with the possibility that these proteins interact physically at least part of the time. The localization data also suggest that CARMIL plays a role in the structure and function of dynamic, actin-rich extensions associated with endocytosis and chemotaxis. Consistent with this, cells that lack CARMIL exhibit reduced rates of fluid phase endocytosis, a defect in the formation of the dorsal crowns responsible for this fluid uptake, and a reduction in the efficiency of chemotactic aggregation. Therefore, we conclude that CARMIL is physiologically important, that the behavioral defects exhibited by CARMIL null cells correlate with the localization of the protein to crowns and the leading edge, where myoB, myoC, and the Arp2/3 complex also localize, and that CARMIL plays a significant role in the structure and function of these actin-rich cellular extensions.
Novak and Titus 1997, Novak and Titus 1998 have shown that the ability of Dictyostelium myoB to rescue the defect in endocytosis exhibited by myoB/myoC double mutants, and the deleterious effects of myoB overexpression on pinocytosis and cell migration, are all eliminated by deletion of the myosin's SH3 domain. Similarly, the SH3 domains of the two yeast type I myosins are required for these myosins to exert most of their positive effects on actin cytoskeletal organization (Anderson et al. 1998; see below). Therefore, the in vivo functions of these type I myosins appear to be critically dependent on their ability to interact with the targets of their SH3 domains. We now suggest, based on our demonstration here, that the target for the SH3 domains of Dictyostelium myoB and myoC also interacts with Arp2/3 and capping protein, that the phenotypes of myoB−/myoC− mutants, which include reduced rates of pinocytosis and cell migration (Ostap and Pollard 1996a), might be due at least in part to defects in the assembly state and organization of actin. These defects could arise from alterations in the localization, activity, and/or movement of the complex caused by the loss of myosin I. This role for myosin I, together with its role in generating tension within the actin cortex (Dai et al. 1999) as a result of its ability to cross link actin filaments and collapse isotropic actin mesh works (Fujisaki et al. 1985), and its role in membrane/organelle movements (Neuhaus and Soldati 2000), may be largely responsible for the defects exhibited by myosin I mutants in Dictyostelium.
Biochemical Properties of the CARMIL Protein
CARMIL possesses at least three biochemical functions. First, it binds the Arp2/3 complex and, based on analysis of an internal fragment, has at least a modest ability to activate Arp2/3-dependent actin nucleation. Second, it binds capping protein. The critical question regarding this interaction is whether capping protein retains its ability to cap the barbed ends of actin filaments when bound to CARMIL. If this does not occur, then CARMIL's main function as regards capping protein may be to regulate the functional levels of the protein. Given the cellular concentration of CARMIL (∼2 μM, versus ∼1 μM for capping protein; Lee et al. 1999), and the likelihood that CARMIL has a relatively high affinity for capping protein (see above), CARMIL could be a very significant competitor with actin filament barbed ends for free capping protein. Depending on the local concentrations of reactants, and on possible regulation of CARMIL–capping protein interaction, CARMIL could drive both the uncapping of filaments and the accelerated capping of filaments by the binding and release of capping protein, respectively. However, the situation will be quite different if the complex of CARMIL and capping protein can cap, and, by extension, if CARMIL can bind to capping protein present at the ends of capped filaments. If these interactions occur, then the complex could, by virtue of its ability to also recruit and activate the Arp2/3 complex, drive the formation of a new filament off of a capped barbed end. This could represent a novel mechanism for nucleating actin filaments (see also Pantaloni et al. 2000). Resolution of these and other important questions, such as how the stimulation of Arp2/3-dependent actin nucleation by CARMIL is regulated, should go a long way towards answering the more general question of why this complex binds both the principal nucleator and terminator of actin filament assembly.
Finally, CARMIL binds myosin I. Given that type I myosins are bona fide, barbed end–directed motors, the myosin I bound to CARMIL may function to translocate the complex towards the barbed end of existing actin filaments (Fig. 12), thereby serving to concentrate CARMIL and the Arp2/3 complex at the interface between the actin-rich cortex and the plasma membrane. Actin polymerization in this zone would most effectively drive protrusion. The added ability of type I myosins to move membranes relative to actin (Zot et al. 1992) might allow them, in conjunction with their ability to target machinery regulating actin assembly, to facilitate the growth of filaments directly abutting the plasma membrane. However, for all of these mechanisms of action, the fact that myosin I cannot function at low density as a processive motor (Ostap and Pollard 1996b) must be taken into consideration (see Machesky 2000).
Generality of Our Results
Several recent papers have revealed important physical and functional connections between the two type I myosins in yeast and the Arp2/3 complex. First, Anderson et al. 1998 reported that the SH3 domains of Myo3p and Myo5p bind to verprolin. The subsequent demonstration that verprolin interacts physically and genetically with Las17p/Bee1p (Naqvi et al. 1998), together with the fact that Las17p/Bee1p, like its mammalian homologue WASp, binds to and activates the Arp2/3 complex (Machesky and Insall 1998; Winter et al. 1999), provided an indirect connection between the yeast type I myosins and the Arp2/3 complex. This connection has been strengthened by the recent work of Evangelista et al. 2000 and Lechler et al. 2000, both of whom showed that the SH3 domains of Myo3p and Myo5p interact with Las17p/Bee1p as well with verprolin, and that an acidic sequence at the COOH terminus of Myo3p and Myo5p, which is homologous to the acidic, Arp2/3-binding A domain of Las17p/Bee1p and other WASp/SCAR proteins, binds the Arp2/3 complex. Loss of the acidic motifs of either Las17p/Bee1p or the type I myosins did not cause a dramatic cellular phenotype, whereas loss of both led to striking defects in growth, actin organization, and F-actin content (Evangelista et al. 2000). Similarly, the ability of actin to polymerize in the cortex of permeabilized yeast cells was normal when supplemental extracts contain either acidic motif, but dramatically impaired when extracts lacked both motifs (Lechler et al. 2000). These results suggest that the A domains of myosin I and Las17p/Bee1p function redundantly in the activation of the Arp2/3 complex (see also the recent results for the fission yeast myosin I, Myo1p; Lee et al. 2000). Given that the mechanism of this activation appears to require a WH2/V domain–dependent G-actin binding activity as well as an acidic domain (Machesky and Insall 1998; Egile et al. 1999; Higgs et al. 1999), and that the yeast type I myosins possess only the latter, it seems likely that the activation pathway used by these myosins requires their SH3 domain–dependent interaction with verprolin. In this way the WH2/V domain of verprolin could act in trans with the acidic domain of the type I myosins to activate the Arp2/3 complex.
To date, the only type I myosins that have been found to possess a COOH-terminal acidic domain are those from fungi (Evangelista et al. 2000), suggesting that the direct interaction between type I myosins and the Arp2/3 complex is fungal-specific. Furthermore, connections between type I myosins and proteins that interact with the Arp2/3 complex either directly or indirectly have been made only in yeast, raising the question of how general these interactions are. Here we show that type I myosins in Dictyostelium are also connected to the Arp2/3 complex, albeit by a different mechanism that involves the CARMIL protein. Moreover, the Dictyostelium complex contains capping protein as well as Arp2/3. Interestingly, these two components, together with cofilin, have recently been shown to be sufficient to drive actin-based motility (Loisel et al. 1999). We note that CARMIL represents the first cytoskeletal protein other than actin that has been shown to bind capping protein. We also note that CARMIL represents a novel Arp2/3 activator since it exhibits no sequence similarity with ActA (Welch et al. 1998), cortactin (Uruno et al. 2001; Weaver et al. 2001), or WASp/SCAR family proteins (Welch 1999) beyond a short sequence shared by many proteins that bind G-actin. Finally, our efforts to identify metazoan homologues of CARMIL confirmed and extended those of Xu et al. 1997 regarding a C. elegans homologue, identified a D. melanogaster homologue, and, using this latter sequence for searches, identified CARMIL homologues in mouse and human. These results, and the fact that metazoans also contain type I myosins with SH3 domains (Sellers 1999), suggest that the complex we have identified may be present in many eukaryotic organisms.
Acknowledgments
We thank John Cooper, Kathy Gould, and Eugenio De Hostos for antibodies, Blair Bowers for help with microscopy, and Edward D. Korn for comments on the manuscript.
Footnotes
Abbreviations used in this paper: LRR, leucine-rich repeat; myo, myosin I; RT, reverse transcription; SB, starvation buffer; SH, Src homology.
References
- Anderson B.L., Boldogh I., Evangelista M., Boone C., Greene L.A., Pon L.A. The Src homology domain 3 (SH3) of a yeast type I myosin, myo5p, binds to verprolin and is required for targeting to sites of actin polarization. J. Cell Biol. 1998;141:1357–1370. doi: 10.1083/jcb.141.6.1357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bear J.E., Rawls J.F., Saxe C.L., III. SCAR, a WASP-related protein, isolated as a suppressor of receptor defects in late Dictyostelium development. J. Cell Biol. 1998;142:1325–1335. doi: 10.1083/jcb.142.5.1325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blanchoin L., Amann K.J., Higgs H.N., Marchand J.B., Kaiser D.A., Pollard T.D. Direct observation of dendritic actin filament networks nucleated by Arp2/3 complex and WASp/Scar proteins. Nature. 2000;404:1007–1011. doi: 10.1038/35010008. [DOI] [PubMed] [Google Scholar]
- Carlier M.-F., Ducruix A., Pantaloni D. Signaling to actinthe Cdc42-N-WASP-Arp2/3 connection. Chem. Biol. 1999;6:R235–R240. doi: 10.1016/s1074-5521(99)80107-0. [DOI] [PubMed] [Google Scholar]
- Coluccio L.M. Myosin I. Amer. J. Physiol. 1997;273:C347–C359. doi: 10.1152/ajpcell.1997.273.2.C347. [DOI] [PubMed] [Google Scholar]
- Cooper J.A., Schafer D.A. Control of actin assembly and disassembly at filament ends. Curr. Opin. Cell Biol. 2000;12:97–103. doi: 10.1016/s0955-0674(99)00062-9. [DOI] [PubMed] [Google Scholar]
- Cooper J.A., Blum J.D., Pollard T.D. Acanthamoeba castellanii capping proteinproperties, mechanism of action, immunological crossreactivity, and localization. J. Cell Biol. 1984;99:217–225. doi: 10.1083/jcb.99.1.217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Hostos E.L. The coronin family of actin-associated proteins. Trends Cell Biol. 1999;9:345–350. doi: 10.1016/s0962-8924(99)01620-7. [DOI] [PubMed] [Google Scholar]
- Derry J.M., Ochs H.D., Francke U. Isolation of a novel gene mutated in Wiskott-Aldrich Syndrome. Cell. 1994;78:635–644. doi: 10.1016/0092-8674(94)90528-2. [DOI] [PubMed] [Google Scholar]
- Dai J., Ting-Beal H.P., Hochmuth R.M., Sheetz M.P., Titus M.A. Myosin I contributes to the generation of resting cortical tension. Biophys. J. 1999;77:1168–1176. doi: 10.1016/s0006-3495(99)76968-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eddy R.J., Han J., Condeelis J.S. Capping protein terminates but does not initiate chemoattractant-induced actin assembly in Dictyostelium . J. Cell Biol. 1997;139:1243–1253. doi: 10.1083/jcb.139.5.1243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Egile C., Loisel T.P., Laurent V., Li R., Pantaloni D., Sansonetti P.J., Carlier M.-F. Activation of the Cdc42 effector N-WASp by the Shigella flexneri IcsA protein promotes actin nucleation by Arp2/3 complex and bacterial actin-based motility. J. Cell Biol. 1999;146:1319–1332. doi: 10.1083/jcb.146.6.1319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evangelista M., Klebl B.M., Tony A.H.Y., Webb B.A., Leeuw T., Leberer E., Whiteway M., Thomas D.Y., Boone C. A role for myosin-I actin assembly through interactions with Vrp1p, Bee1p, and the Arp2/3 complex. J. Cell Biol. 2000;148:353–362. doi: 10.1083/jcb.148.2.353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujisaki H., Albanesi J.P., Korn E.D. Experimental evidence for the contractile activities of Acanthamoeba myosin I-A and myosin I-B. J. Biol. Chem. 1985;260:1183–1189. [PubMed] [Google Scholar]
- Fukui Y., Lynch T.J., Brzeska H., Korn E.D. Myosin I is located at the leading edge of locomoting Dictyostelium amoebae. Nature. 1989;341:328–331. doi: 10.1038/341328a0. [DOI] [PubMed] [Google Scholar]
- Gordon D.J., Eisenberg E., Korn E.D. Characterization of cytoplasmic actin isolated from Acanthamoeba castellanii by a new method. J. Biol. Chem. 1976;251:4778–4780. [PubMed] [Google Scholar]
- Hacker U., Albrecht R., Maniak M. Fluid-phase uptake by macropinocytosis in Dictyostelium . J. Cell Sci. 1997;110:105–112. doi: 10.1242/jcs.110.2.105. [DOI] [PubMed] [Google Scholar]
- Higgs H.N., Pollard T.D. Regulation of actin polymerization by Arp2/3 complex and WASp/Scar proteins. J. Biol. Chem. 1999;274:32531–32534. doi: 10.1074/jbc.274.46.32531. [DOI] [PubMed] [Google Scholar]
- Higgs H.N., Blanchoin L., Pollard T.D. Influence of the C terminus of Wiskott-Aldrich syndrome protein (WASp) and the Arp2/3 complex on actin polymerization. Biochemistry. 1999;38:15212–15222. doi: 10.1021/bi991843+. [DOI] [PubMed] [Google Scholar]
- Hug C., Jay P.Y., Reddy I., McNally J.G., Bridgman P.C., Elson E.L., Cooper J.A. Capping protein levels influence actin assembly and cell motility in Dictyostelium . Cell. 1995;81:591–600. doi: 10.1016/0092-8674(95)90080-2. [DOI] [PubMed] [Google Scholar]
- Jung G., Hammer J.A., III. Generation and characterization of Dictyostelium cells deficient in a myosin I heavy chain isoform. J. Cell Biol. 1990;110:1955–1964. doi: 10.1083/jcb.110.6.1955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung G., Saxe C.L., III, Kimmel A.R., Hammer J.A., III. Dictyostelium discoideum contains a gene encoding a myosin I heavy chain. Proc. Natl. Acad. Sci. USA. 1989;86:6186–6190. doi: 10.1073/pnas.86.16.6186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung G., Fukui Y., Martin B., Hammer J.A., III. Sequence, expression pattern, intracellular localization, and targeted disruption of the Dictyostelium myosin-ID heavy chain isoform. J. Biol. Chem. 1993;268:14981–14990. [PubMed] [Google Scholar]
- Jung G., Wu X., Hammer J.A., III. Dictyostelium mutants lacking multiple classic myosin I isoforms reveal combinations of shared and distinct functions. J. Cell Biol. 1996;133:305–323. doi: 10.1083/jcb.133.2.305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelleher J.F., Mullins R.D., Pollard T.D. Purification and assay of the Arp2/3 complex from Acanthamoeba castellanii . Methods Enzymol. 1998;298:42–51. doi: 10.1016/s0076-6879(98)98007-7. [DOI] [PubMed] [Google Scholar]
- Klein G., Satre M. Kinetics of fluid phase pinocytosis in Dictyostelium discoideum amoebae. Biochem. Biophys. Res. Comm. 1986;138:1140–1152. doi: 10.1016/s0006-291x(86)80402-8. [DOI] [PubMed] [Google Scholar]
- Kobe B., Deisenhofer J. Proteins with leucine-rich repeats Curr. Opin Struct.Biol. 51995. 409 416 [DOI] [PubMed] [Google Scholar]
- Kuriyan J., Cowburn D. Modular peptide recognition domains in eukaryotic signaling. Annu. Rev. Biophys. Biomol. Struct. 1997;26:259–288. doi: 10.1146/annurev.biophys.26.1.259. [DOI] [PubMed] [Google Scholar]
- Lechler T., Shevchenko A., Shevchenko A., Li R. Direct involvement of yeast type I myosins in Cdc42-dependent actin polymerization. J. Cell Biol. 2000;148:363–373. doi: 10.1083/jcb.148.2.363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee W.-L., Ostap E.M., Zot H.G., Pollard T.D. Organization and ligand binding properties of the tail of Acanthamoeba myosin-IA. J. Biol. Chem. 1999;49:35159–35171. doi: 10.1074/jbc.274.49.35159. [DOI] [PubMed] [Google Scholar]
- Lee W.-L., Bezanilla M., Pollard T.D. Fission yeast myosin-I, Myo1p, stimulates actin assembly by Arp2/3 complex and shares functions with WASp. J. Cell Biol. 2000;151:789–799. doi: 10.1083/jcb.151.4.789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li R. Bee1, a yeast protein with homology to Wiskott-Aldrich syndrome protein, is critical for the assembly of cortical actin cytoskeleton. J. Cell Biol. 1997;136:649–658. doi: 10.1083/jcb.136.3.649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loisel T.P., Boujemaa R., Pantaloni D., Carlier M.-F. Reconstitution of actin-based motility of Listeria and Shigella using pure proteins. Nature. 1999;401:613–616. doi: 10.1038/44183. [DOI] [PubMed] [Google Scholar]
- Machesky L.M. The tail of two myosins. J. Cell Biol. 2000;148:219–221. doi: 10.1083/jcb.148.2.219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Machesky L.M., Insall R.H. Scar1 and the related Wiskott-Aldrich syndrome protein, WASP, regulate the actin cytoskeleton through the Arp2/3 complex. Curr. Biol. 1998;8:1347–1356. doi: 10.1016/s0960-9822(98)00015-3. [DOI] [PubMed] [Google Scholar]
- Machesky L.M., Insall R.H. Signaling to actin dynamics. J. Cell Biol. 1999;146:267–272. doi: 10.1083/jcb.146.2.267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Machesky L.M., Atkinson S.J., Ampe C., Vandekerckhove J., Pollard T.D. Purification of a cortical complex containing 2 unconventional actins from Acanthamoeba by affinity-chromatography on profilin-agarose. J. Cell Biol. 1994;127:107–115. doi: 10.1083/jcb.127.1.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Machesky L.M., Mullins R.D., Higgs H.N., Kaiser D.A., Blanchoin L., May R.C., Hall M.E., Pollard T.D. Scar, a WASp-related protein, activates nucleation of actin filaments by the Arp2/3 complex. Proc. Natl. Acad. Sci. USA. 1999;96:3739–3744. doi: 10.1073/pnas.96.7.3739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masuelli L., Cutler M.L. Increased expression of the Ras suppressor Rsu-1 enhances Erk-2 activation and inhibits Jun kinase activation. Mol. Cell. Biol. 1996;16:5466–5476. doi: 10.1128/mcb.16.10.5466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mermall V., Post P.L., Mooseker M.S. Unconventional myosins in cell movement, membrane traffic, and signal transduction. Science. 1998;279:527–533. doi: 10.1126/science.279.5350.527. [DOI] [PubMed] [Google Scholar]
- Miki H., Takenawa T. Direct binding of the verprolin-homology domain in N-WASP to actin is essential for cytoskeletal organization. Biochem. Biophys. Res. Commun. 1998;243:73–78. doi: 10.1006/bbrc.1997.8064. [DOI] [PubMed] [Google Scholar]
- Moreau V., Frischknecht F., Reckman I., Vincentelli R., Rabut G., Stewart D., Way M. A complex of N-Wasp and WIP integrates signaling cascades that lead to actin polymerization. Nat. Cell Biol. 2000;2:441–448. doi: 10.1038/35017080. [DOI] [PubMed] [Google Scholar]
- Murgia I., Maciver S.K., Morandini P. An actin-related protein from Dictyostelium discoideum is developmentally regulated and associated with mitochondria. FEBS Letts. 1995;360:235–241. doi: 10.1016/0014-5793(95)00111-l. [DOI] [PubMed] [Google Scholar]
- Naqvi S.N., Zahn R., Mitchell D.A., Stevenson B.J., Munn A.L. The WASp homologue Las17p functions with the WIP homologue End5p/verprolin and is essential for endocytosis in yeast. Curr. Biol. 1998;8:959–962. doi: 10.1016/s0960-9822(98)70396-3. [DOI] [PubMed] [Google Scholar]
- Neuhaus E.M., Soldati T. A myosin I is involved in membrane recycling from early endosomes. J. Cell Biol. 2000;150:1013–1026. doi: 10.1083/jcb.150.5.1013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Novak K.D., Titus M.A. Myosin I overexpression impairs cell migration. J. Cell Biol. 1997;136:633–647. doi: 10.1083/jcb.136.3.633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Novak K.D., Titus M.A. The myosin I SH3 domain and TEDS rule phosphorylation site are required for in vivo function. Mol. Biol. Cell. 1998;9:75–88. doi: 10.1091/mbc.9.1.75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ostap E.M., Pollard T.D. Overlapping functions of myosin-I isoforms? J. Cell Biol. 133 1996. 221 224a [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ostap E.M., Pollard T.D. Biochemical kinetic characterization of the Acanthamoeba myosin I ATPase J. Cell Biol 132 1996. 1053 1060b [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pantaloni D., Boujemaa R., Didry D., Gounon P., Carlier M.-F. The Arp2/3 complex branches filament barbed endsfunctional antagonism with capping proteins. Nat. Cell Biol. 2000;2:385–390. doi: 10.1038/35017011. [DOI] [PubMed] [Google Scholar]
- Peterson M.D., Novak K.D., Reedy M.C., Ruman J.I., Titus M.A. Molecular genetic analysis of myoC, a Dictyostelium myosin I. J. Cell Sci. 1995;108:1093–1103. doi: 10.1242/jcs.108.3.1093. [DOI] [PubMed] [Google Scholar]
- Podolski J.L., Steck T.L. Length distribution of F-actin in Dictyostelium discoideum . J. Biol. Chem. 1990;265:1312–1318. [PubMed] [Google Scholar]
- Ramesh N., Anton I.M., Hartwig J.H., Geha R.S. WIP, a protein associated with Wiskott-Aldrich syndrome protein, induces actin polymerization and redistribution in lymphoid cells. Proc. Natl. Acad. Sci. USA. 1997;94:14671–14676. doi: 10.1073/pnas.94.26.14671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rohatgi R., Ma L., Miki H., Lopez M., Kirchhausen T., Takenawa T., Kirschner M.W. The interaction between N-WASP and the Arp2/3 complex links Cdc42-dependent signals to actin assembly. Cell. 1999;97:221–231. doi: 10.1016/s0092-8674(00)80732-1. [DOI] [PubMed] [Google Scholar]
- Sellers, J.R. 1999. Myosins. P. Sheterline, editor. Oxford University Press, Oxford, UK. 237 pp.
- Spudich, J.A. 1987. Dictyostelium discoideum: molecular approaches to cell biology. Methods in Cell Biology. Vol. 28. Academic Press, New York. 516 pp. [PubMed]
- Stern M.J., Marengere L.E., Daly R.J., Lowenstein E.J., Kokel M., Batzer A., Olivier P., Pawson T., Schlessinger J. The human GRB2 and Drosophila Drk genes can functionally replace the Caenorhabditis elegans signaling gene sem-5. Mol. Biol. Cell. 1993;4:1175–1188. doi: 10.1091/mbc.4.11.1175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki N., Choe H.-R., Nishida Y., Yamawaki-Kataoka Y., Ohnishi S., Takmoaki T., Kataoka T. Leucine-rich repeats and carboxyl terminus are required for interaction of yeast adenylate cyclase with ras proteins. Proc. Natl. Acad. Sci. USA. 1990;87:8711–8715. doi: 10.1073/pnas.87.22.8711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uruno T., Liu J., Zhang P., Fan Y., Egile C., Li R., Mueller S. C., Zhan X. Activation of the Arp2/3 complex-mediated actin polymerization by cortactin. Nat. Cell Biol. 2001;3:259–266. doi: 10.1038/35060051. [DOI] [PubMed] [Google Scholar]
- Uyeda T.Q.P., Titus M.A. The myosins in Dictyostelium . In: Maeda Y., Inouye K., Takeuchi I., editors. Dictyostelium: A Model System for Cell and Developmental Biology. University Academy Press; Tokyo, Japan: 1997. pp. 43–64. [Google Scholar]
- Vaduva G., Martin N.C., Hopper A.K. Actin-binding verprolin is a developmental protein required for the morphogenesis and function of the yeast actin cytoskeleton. J. Cell Biol. 1997;139:1821–1833. doi: 10.1083/jcb.139.7.1821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaduva G., Matinez-Quiles N., Anton I.M., Martin N.C., Geha R.S., Hopper A.K., Ramesh N. The human WASP-interacting protein, WIP, activates the cell polarity pathway in yeast. J. Biol. Chem. 1999;274:17103–17108. doi: 10.1074/jbc.274.24.17103. [DOI] [PubMed] [Google Scholar]
- Vancompernolle K., Vandekerckhove J., Bubb M.R., Korn E.D. The interfaces of actin and Acanthamoeba actobindin - Identification of a new actin-binding motif. J. Biol. Chem. 1991;266:15427–15431. [PubMed] [Google Scholar]
- Van Troys M., Dewitte D., Goethals M., Carlier M.-F., Vandekerckhove J., Ampe C. The actin binding site of thymosin B4 mapped by mutational analysis. EMBO J. 1996;15:201–210. [PMC free article] [PubMed] [Google Scholar]
- Varnum B., Edwards K., Soll D.R. The developmental regulation of single cell motility in Dictyostelium discoideum . Dev. Biol. 1985;113:218–227. doi: 10.1016/0012-1606(86)90124-7. [DOI] [PubMed] [Google Scholar]
- Watts R.G., Howard T.H. Role of tropomyosin, α-actinin, and actin binding protein 280 in stabilizing F-actin in basal and chemotactic factor activated neutrophils. Cell Motil. Cytoskeleton. 1994;28:155–164. doi: 10.1002/cm.970280207. [DOI] [PubMed] [Google Scholar]
- Weaver A.M., Karginov A.V., Kinley A.W., Weed S.A., Li Y., Parsons J.T., Cooper J.A. Cortactin promotes and stabilizes Arp2/3-induced actin filament network formation. Curr. Biol. 2001;11:370–374. doi: 10.1016/s0960-9822(01)00098-7. [DOI] [PubMed] [Google Scholar]
- Welch M.A. The world according to Arpregulation of actin nucleation by the Arp2/3 complex. Trends Cell Biol. 1999;9:423–427. doi: 10.1016/s0962-8924(99)01651-7. [DOI] [PubMed] [Google Scholar]
- Welch M.D., Rosenblatt J., Skoble J., Portnoy D.A., Mitchison T.J. Interaction of human Arp2/3 complex and the Listeria monocytogenes ActA protein in actin filament nucleation. Science. 1998;281:105–108. doi: 10.1126/science.281.5373.105. [DOI] [PubMed] [Google Scholar]
- Winter D., Lechler T., Li R. Activation of the yeast Arp2/3 complex by Bee1p, a WASP-family protein. Curr. Biol. 1999;9:501–504. doi: 10.1016/s0960-9822(99)80218-8. [DOI] [PubMed] [Google Scholar]
- Wu X., Jung G., Hammer J.A., III. Functions of unconventional myosins. Curr. Opin. Cell Biol. 2000;12:42–51. doi: 10.1016/s0955-0674(99)00055-1. [DOI] [PubMed] [Google Scholar]
- Xu P., Zot A.S., Zot H.G. Identification of Acan 125 as a myosin-I-binding protein present with myosin-I on cellular organelles of Acanthamoeba . J. Biol. Chem. 1995;270:25316–25319. doi: 10.1074/jbc.270.43.25316. [DOI] [PubMed] [Google Scholar]
- Xu P., Mitchelhill K.I., Kobe B., Kemp B.E., Zot H.G. The myosin-I binding protein Acan125 binds the SH3 domain and belongs to the superfamily of leucine-rich repeat proteins. Proc. Natl. Acad. Sci. USA. 1997;94:3685–3690. doi: 10.1073/pnas.94.8.3685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yarar D., To W., Abo A., Welch M.D. The Wiskott-Aldrich syndrome protein directs actin-based motility by stimulating actin nucleation with the Arp2/3 complex. Curr. Biol. 1999;9:555–558. doi: 10.1016/s0960-9822(99)80243-7. [DOI] [PubMed] [Google Scholar]
- Zeile W.L., Condit R.C., Lewis J.I., Purich D.L., Southwick F.S. Vaccinia locomotion in host cellsevidence for the universal involvement of actin-based motility sequences ABM-1 and ABM-2. Proc. Natl. Acad. Sci. USA. 1998;95:13917–13922. doi: 10.1073/pnas.95.23.13917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zot H.G., Doberstein S.K., Pollard T.D. Myosin-I moves actin filaments on a phospholipid substrateimplications for membrane targeting. J. Cell Biol. 1992;116:367–376. doi: 10.1083/jcb.116.2.367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zot H.G., Bhaskara V., Liu L. Acan 125 binding to the SH3 domain of Acanthamoeba myosin-IC. Arch. Biochem. Biophys. 2000;375:161–164. doi: 10.1006/abbi.1999.1648. [DOI] [PubMed] [Google Scholar]