

Abstract
Viruses have evolved a myriad of evasion strategies focused on undermining chemokine-mediated immune surveillance, exemplified by the mouse γ-herpesvirus 68 M3 decoy receptor. Crystal structures of M3 in complex with C chemokine ligand 1/lymphotactin and CC chemokine ligand 2/monocyte chemoattractant protein 1 reveal that invariant chemokine features associated with G protein–coupled receptor binding are primarily recognized by the decoy C-terminal domain, whereas the N-terminal domain (NTD) reconfigures to engage divergent basic residue clusters on the surface of chemokines. Favorable electrostatic forces dramatically enhance the association kinetics of chemokine binding by M3, with a primary role ascribed to acidic NTD regions that effectively mimic glycosaminoglycan interactions. Thus, M3 employs two distinct mechanisms of chemical imitation to potently sequester chemokines, thereby inhibiting chemokine receptor binding events as well as the formation of chemotactic gradients necessary for directed leukocyte trafficking.
Chemokines orchestrate the migration of leukocytes under homeostatic and inflammatory conditions by signaling through G protein–coupled receptors (GPCRs) expressed on trafficking immune cells. The chemokine family includes ∼50 members with 19 known receptors and is divided into four subclasses based on the arrangement of N-terminal cysteines (1, 2). Circulating leukocytes infiltrate tissues in response to cell-surface chemokine gradients established through association with glycosaminoglycans (GAGs). These ubiquitous sulfated polysaccharides influence multiple cytokines (3) and specifically modulate chemokines by stabilizing gradients under shear flow, protecting them from proteolysis, and regulating their biological activity (4). GAG binding also induces oligomerization of chemokines, which may be necessary for leukocyte recruitment in vivo (5, 6).
Large DNA viruses have evolved strategies to avoid detection and clearance by the host (7), including evasion of the chemokine network, an important component of the immune response to viral infection (8). Chemokines coordinate innate and adaptive immunity by modulating cellular recruitment, leukocyte activation, and polarization of the immune response (9). Herpesviruses and poxviruses encode numerous proteins to subvert chemokine signaling, including altered chemokine and GPCR variants, as well as secreted decoy receptors that function as chemokine scavengers capable of inhibiting chemokine signaling and chemotaxis (10). Members of the Orthopoxvirus and Leporipoxvirus genera encode a conserved 35-kD decoy receptor, viral chemokine-binding protein, that selectively binds CC chemokines and has been extensively investigated biophysically (11–15). Recent work has shown that some Orthopoxviruses encode additional chemokine inhibitors, termed smallpox virus–encoded chemokine receptor domains (16), which are encoded either alone or C-terminally fused to secreted TNF decoy receptors and are capable of sequestering select members of the CC and CXC chemokine families. In contrast, the expression of chemokine decoy receptors by herpesviruses appears to be less generalized. Examples include the glycoprotein G variants encoded by ruminant α-herpesviruses that bind CC and CXC chemokines (17), and UL21.5 from human cytomegalovirus that specifically binds CC chemokine ligand (CCL) 5 (18). All of the virally encoded chemokine decoy receptors identified to date are encoded by novel sequences unrelated to any host proteins and exhibit a variety of chemokine-binding profiles that may reflect differences in viral tropism or pathogenesis.
Mouse γ-herpesvirus 68 (MHV68) is a natural pathogen of rodents, closely related to Kaposi's sarcoma-associated herpesvirus and EBV, and serves as a model system to study herpesvirus pathogenesis and latency (19). The M3 protein encoded by MHV68 is the product of an immediate-early transcript and is abundantly secreted during acute infection (20). M3 disrupts chemokine signaling and chemotaxis in vitro (21), and the phenotype of an M3-knockout virus is consistent with an immune evasion function during central nervous system infection (22). M3 is the only decoy receptor discovered thus far that binds members of all four chemokine classes (21, 23); however, M3 displays selectivity within the CXC chemokine class, which may be functionally relevant for MHV68 pathogenesis. Mouse chemokines shown to be up-regulated during MHV68 infection include lymphotactin, monocyte chemoattractant protein 1, RANTES, macrophage inflammatory protein (MIP) 1α, MIP-1β, eotaxin, IFN-inducible protein (IP) 10, MIP-2, and KC (24, 25). Of these chemokines, only MIP-2 and KC are not bound by M3 with high affinity. Strikingly, MHV68 also encodes a chemokine GPCR, MHV68-encoded open reading frame 74, which signals in response to MIP-2 and KC but is antagonized by IP-10 (26). Thus, the chemokine-binding properties of M3 may be tailored to work cooperatively with other viral proteins involved in pathogenesis.
We previously determined the crystal structure of M3 alone and in complex with the P8A variant of CCL2 (27), which revealed significant details about how CC chemokine sequestration is enabled by the decoy receptor. In this paper, we describe the crystal structures of M3 in complex with C chemokine ligand (XCL) 1, as well as wild-type CCL2. These structures show that M3 engages two out of four chemokine classes with the same overall binding geometry and stoichiometry. Comparative analysis reveals that the M3 C-terminal domain (CTD) engages conserved chemokine structural elements associated with GPCR binding, whereas the acidic N-terminal domain (NTD) exhibits dramatic electrostatic complementarity with chemokines, contacting divergent basic clusters involved in GAG association. Based on these observations, we have undertaken kinetic and mutational analyses to assess the role of electrostatics in M3 chemokine binding. We have also developed competition assays to address whether M3 is capable of disrupting chemokine interactions with GAGs. Collectively, our data reveal that the M3 decoy receptor enables immune evasion through dual GAG and GPCR mimicry, effectively preventing the establishment of chemokine gradients and the activation of endogenous receptor signaling.
RESULTS
Structures of M3 in complex with XCL1 and CCL2
To ascertain the structural features of M3 that facilitate promiscuous recognition of distinct chemokine classes, the structures of M3 in complex with XCL1 and CCL2 were determined and compared. XCL1 uniquely contains only a single disulfide, herein referred to as the invariant disulfide, which is present in all four chemokine classes. Otherwise, both chemokines exhibit the characteristic chemokine-fold topology consisting of an extended N-loop ending in a short 310-helix, followed by three β strands connected by 30s and 40s loops and a C-terminal α-helix (28, 29). Interestingly, the sequence of XCL1 is substantially longer than CCL2 (92 vs. 75 residues, respectively), although the last 20 residues beyond the C-terminal helix have not been structurally defined.
The crystal structure of M3 in complex with XCL1 was determined to a resolution of 2.6 Å (Fig. 1 A and Table I). M3 is a two-domain protein that exists as a constitutive antiparallel homodimer, with two chemokine-binding clefts formed between adjacent NTDs and CTDs at opposite ends of the dimer. The M3–XCL1 model comprises a 2:2 complex, with an asymmetrical M3 dimer caused by crystal contacts involving the A'-A–loop (residues 251–253) and the C”, D, and E strands (residues 326–353) of the M3 A chain. XCL1 is deeply buried within the binding cleft, which sequesters ∼1,080 Å2 of chemokine surface area (19% total). XCL1 contributes 15 residues and M3 17 residues to >130 contacts at the interface (Table II), including 10 H-bonds, 3 of which form an antiparallel β strand between K8 and T10 of XCL1, and L273 and A275 of the M3 AB-loop. The two binding sites are strikingly similar, as reflected by the near equivalence of total buried surface area (BSA) equaling 2,156 Å2 at the XCL1 (D) interface, and 2,181 Å2 for the XCL1 (E) interface, with no major differences in the number of contacts at each site (Table II).
Figure 1.

Crystal structures of M3–XCL1 and M3–CCL2 complexes. Structures of (A) M3–XCL1 and (B) M3–CCL2. (left) Each complex displays a 2:2 stoichiometry, with M3 chains labeled A and B and chemokines labeled D and E. (middle) Space-fill models of XCL1 (E) and CCL2 (E). Sidechains of contact residues are highlighted in magenta. Shared chemokine features are circled, with residues in the basic cluster (dashed line) labeled in blue; also shown is the N-terminal segment (dotted line), with residues forming the antiparallel β strand labeled in black, as well as the hydrophobic cluster (bold dotted line). Cysteines of the CC and C motifs are labeled on CCL2 (C11, C12, and C52; note that C36 is not visible) and XCL1 (C11 and C48), respectively. The single disulfide in XCL1 is structurally equivalent to the second disulfide (C12–C52) in CCL2 and is referred to as the invariant disulfide. Conserved sidechain contacts are italicized, and GAG-binding residues are indicated by asterisks (references 35, 38). (right) The chemokine contact surface is highlighted in magenta, with 2.5–3.5-Å (short-range) contacts in a darker shade and 3.5–5-Å contacts in a gradient from magenta to white. M3 sidechain contacts are shown in stick form and labeled, with differential contacts highlighted with yellow labels and noncontacting residues shown in yellow for each structure. (C) Structure-based sequence alignment of XCL1 (1–65) and CCL2 (1–70). All residues that contact M3 NTD or CTD are highlighted in cyan and blue, respectively, with disulfide-forming cysteines in black. Conserved sidechain contacts are indicated by gray triangles and GAG-binding residues with asterisks. (D) Conformational rearrangement of M3 NTD with the Cα trace of M3–CCL2 in gray and M3–XCL1 superimposed in blue, cyan, and magenta. (E) RMSD (all atoms) between M3 in complex with XCL1 and CCL2 is highlighted on the trace of M3–XCL1 as a gradient from white to red (from 0.5 to ≥3 Å). Figures were prepared using Ribbons (reference 67) and GRASP (reference 68) software, as previously described, and the chemokine E chain interface is shown as the reference in all figures.
Table I.
Data collection and refinement summary for M3–chemokine complex structuresa
| M3–CCL2 | M3–XCL1 | |
|---|---|---|
| Data Set | ||
| Space group | P3121 | I212121 |
| Unit cell (Å) | a = b = 99.23, c = 243.5 | a = 85.44, b = 104.18, c = 290.85 |
| Wavelength (Å) | 1 | 1 |
| x-ray sourceb | APS 19-ID | APS 19-ID |
| Resolution (Å) overall (outer shell) | 2.5 (2.61–2.5) | 2.6 (2.76–2.6) |
| Observations/unique | 791,582/48,731 | 674,773/40,275 |
| Completeness (%) | 99.7 (100) | 99.3 (99.9) |
| Rsym (%) (I >0) | 13.9 (41.2) | 10 (49.2) |
| I/σ | 11.7 (4.1) | 16.1 (2.7) |
| Refinement summary c | ||
| Resolution (Å) | 20-2.5 | 20-2.6 |
| Reflections Rwork/Rfree (Fobs >0) | 45,669/2,258 | 33,788/1,771 |
| Molecules/ASU | 6 | 4 |
| No. protein residues/atoms/solvent | 1,299/10,113/546 | 865/6,720/192 |
| Rwork overall (outer shell) (%) | 23.2 (27.4) | 22 (37) |
| Rfree overall (outer shell) (%) | 29.9 (35.1) | 27.3 (42.2) |
| RMSD bond lengths (Å)/angles (°) | 0.006/1.4 | 0.007/1.4 |
| RMSD dihedral/improper (°) | 25.3/0.87 | 25.2/0.98 |
| Cross-validated Luzzati error (Å) | 0.47 | 0.46 |
| Ramachandran plot | ||
| Most favored/additional (%) | 86.8/12.9 | 83.2/16.4 |
| Generous/disallowed (%) | 0.3/0 | 0.4/0 |
| M3 domain B-values (Å2) | A-NTD = 40.4/A-CTD = 31.7 | A-NTD = 53.9/A-CTD = 55.6 |
| B-NTD = 40.8/B-CTD = 31 | B-NTD = 58.7/B-CTD = 44.5 | |
| X-NTD = 42/X-CTD = 31.2 | ||
| Chemokine B-values (Å2) | D = 40.4, E = 39.9, Y = 40.2 | D = 105.4, E = 78 |
Table II.
Structural analysis of M3–chemokine complexesa
| M3–CCL2 | M3–XCL1 | |
|---|---|---|
| Shape complementarityb | 0.65, 0.66, 0.69 | 0.63, 0.62 |
| BSAc (Å2) | ||
| Total | 2,084, 2,170, 2,134 | 2,156, 2,181 |
| M3 | 1,071, 1,112, 1,087 | 1,075, 1,090 |
| Chemokine | 1,013, 1,058, 1,049 | 1,081, 1,091 |
| M3 BSA (nonpolar/polar) (%) | 66/34, 64/36, 66/34 | 64/36, 68/32 |
| Chemokine BSA (nonpolar/polar) (%) | 58/42, 54/46, 59/41 | 57/43, 59/41 |
| Total contactsd (NTD/CTD) | 132 (56/76),133 (51/82),126 (50/76) | 136 (67/69), 135 (65/70) |
| H-bonds (NTD/CTD) (all same) | 12 (4/8) | 10 (4/6) |
We previously reported the structure of M3 in complex with the P8A mutant of CCL2 (27). Substitution of Pro8 with Ala has been clearly shown to disrupt CCL2 self-association (30), and we were concerned that this mutant chemokine might associate with M3 differently than wild-type CCL2. To address this issue, we now describe the 2.5-Å resolution structure of M3 in complex with wild-type CCL2. The model consists of a 2:2 complex (Fig. 1 A), plus another half complex completed by crystallographic twofold symmetry, thus providing two examples of the M3–CCL2 complex within the same crystal. Overall, the M3 structures with wild-type CCL2 and the P8A variant are almost identical, with a root mean square deviation (RMSD) of 0.50 Å (all atoms) between structures. Likewise, the two copies of the M3–CCL2 complex are quite similar, differing by an RMSD of 0.48 Å (all atoms), with comparable binding niches reflected by the total BSA in each complex (4,254 vs. 4,268 Å2; Table II). We find that M3 contacts precisely the same surface of CCL2 as the P8A variant and utilizes equivalent residues to make the same number of contacts (Table II). Neither the Pro8 nor Ala8 side chains make any significant contact with M3, and indeed, the chemokine N-terminal strands adopt the same conformation as they engage the M3 CTD. Importantly, all elements of GCPR mimicry that we described previously are conserved in the wild-type structure (27).
Similar M3 recognition features of XCL1 and CCL2
To determine the basis of promiscuous M3 engagement of diverse chemokines, the M3–XCL1 and M3–CCL2 interfaces were analyzed to identify similar features of chemokine recognition. Although XCL1 and CCL2 share only 24% sequence identity (70 aligned core residues), M3 binds to the same general surface of each chemokine with a similar number of contacts. The shape complementarity of each interface is also comparable, averaging 0.66 for CCL2 and 0.63 for XCL1 (Table II). Comparison of our structures reveals that M3 targets shared chemokine features that we have parsed into three distinct regions, the chemokine N-terminal segment, the hydrophobic seam, and the divergent basic cluster (Fig. 1, A and B, middle). M3 engages both chemokines by contacting their common N-terminal segment, which includes the invariant disulfide bond and adjacent residues. The M3 CTD makes several conserved contacts within this segment, including the packing of M3 P272 against the invariant disulfide, burial of hydrophobic chemokine residues (CCL2 Y13 and XCL1 V12) within a pocket formed by the M3 AB- and EF-loops, and formation of an antiparallel β strand between the M3 AB-loop and the N-terminal chemokine backbone. Central to each interface is a stretch of hydrophobic chemokine residues recognized by both M3 domains, termed the hydrophobic seam, situated in the N-loop region underneath the 40s loop. The two chemokines share major hydrophobic contacts in this region, specifically CCL2 residues I20 and I42, which are structurally analogous to XCL1 residues L19 and I38. Adjacent to the chemokine hydrophobic seam is a cluster of divergent basic residues in the N-loop, 310-helix, 40s loop, and C-terminal helix, which are engaged by the acidic M3 NTD. Electrostatic complementarity between acidic loops of the NTD and the basic cluster of each chemokine is a prominent feature of the shared interface. Specifically, 15 Glu and Asp residues of the s2b-s3, s4-s5, and s7-s8 loops of the M3 NTD are located opposite 9 (XCL1) and 6 (CCL2) Arg and Lys chemokine residues. Interestingly, although the basic cluster is a common feature of both chemokines, residues composing the cluster are variably positioned, and consequently, the only conserved interaction within this region is between XCL1 R23 and CCL2 R24 and the backbone carbonyl of M3 E81. Thus, M3 recognizes several conserved elements of chemokine structure that are shared between these CC and C family chemokines.
Unique M3 recognition features of XCL1 and CCL2
Given the low degree of sequence identity between XCL1 and CCL2, the structures were also analyzed for unique features of chemokine engagement. An analogous total of 20 CCL2 and XCL1 residues are contacted by M3, with only 1 unique residue position contacted in each (K38 in CCL2 and R43 in XCL1; Fig. 1 C). Remarkably, only five of these residues are conserved between the two chemokines, indicating that the M3-engaged interfaces are no more sequence conserved than the overall proteins. To bind these extraordinarily diverse interfaces, M3 uses 20 different residue sidechains, of which 7 are differentially used (Fig. 1, A and B, right). Many of the unique chemokine contacts are localized to the N-loop, a region of high sequence variation among chemokines because of its involvement in specific recognition by endogenous receptors (31). Although the extended N-loop adopts similar conformations in CCL2 and XCL1, distinct binding chemistries are used by M3 because of differences in primary sequences. For example, although CCL2 R18 reaches out and makes contact with the distally located M3 E129 and R170 sidechains, the structurally equivalent XCL1 residue Q17 instead hydrogen bonds with the more proximal M3 Q84. Near the chemokine N-terminal segment, CCL2 Y13 and XCL1 V12 are both sequestered in a CTD hydrophobic pocket, but the smaller XCL1 sidechain does not fill the pocket to the same degree as Y13 and, consequently, does not make contact with three M3 residues used by CCL2 (L273, P356, and T357; Fig. 1, A and B, right). This difference in hydrophobic contact correlates with an overall decrease in the M3 CTD shape complementarity value, which drops from 0.71 for CCL2 to 0.57 for XCL1. XCL1 also makes unique hydrophobic contacts with M3 T226 via L45 that replaces CCL2 K49. Thus, M3 is able to accommodate variable chemokine sequences through uniquely arrayed contact chemistries.
M3 structural plasticity
To further ascertain how M3 is able to similarly engage two diverse chemokines, the conformational variation exhibited by the decoy receptor was assessed. We previously hypothesized that promiscuous ligand binding might be facilitated by the structural plasticity of the M3 chemokine-binding niches (27). Indeed, structural comparison of unliganded M3 with the M3–CCL2 (P8A) complex revealed significant conformational rearrangements associated with ligand binding, including loop remodeling and domain movements of up to 8°. Comparison between the M3–CCL2 and M3–XCL1 complexes indicates a more modest degree of plasticity, with an overall RMSD of 0.7 Å for all M3 atoms with only minor domain movements. The conformational variation that is exhibited can best be understood in the context of how the M3 NTD and CTD differentially interface each chemokine. Although the chemistry and geometry of the chemokine N-terminal segments engaged by the M3 CTD are highly similar, significant sequence and conformational variation in the basic N-loop, 310-helix, and 40s loop regions flanked by the acidic M3 NTD is readily apparent (Fig. 1 D). The M3 NTD adapts to these localized chemokine differences through sidechain and loop repositioning, along with an ∼2° rigid body rotation, which collectively result in a maximal Cα displacement of ∼2.5 Å at the top of the s2b-s3 loop. Thus, with XCL1 bound, the rearranged NTD is optimally positioned for M3 E80 and E81 to engage R23 and R43 of the XCL1 basic cluster. Additionally, the repositioned NTD effectively narrows the binding niche, thereby maintaining close contact with parts of the hydrophobic seam, which is organized in XCL1 to include L19, P20, and L45. Hence, although the M3 CTD maintains a rigid interface to contact chemokine N-terminal structural elements conserved between XCL1 and CCL2, the NTD capably tracks the unique display of chemokine basic and hydrophobic residues through concerted reconfiguration.
Electrostatic complementarity between M3 and chemokines
These structures reveal that for both CCL2 and XCL1, the flexible M3 structure adapts to optimize contacts with the basic cluster on an individual basis. This suggests that electrostatics may be an important element of promiscuous chemokine sequestration by the M3 NTD. To further assess the complementarity between M3 and chemokines, electrostatic potential maps were calculated for M3, XCL1, and the M3–XCL1 complex revealing that the positive potential of the XCL1 basic cluster is located directly opposite the negative potential of the M3 NTD acidic loops, and that both are significantly neutralized in the complex (Fig. 2 A). To demonstrate this charge neutralization biochemically, the M3 isoelectric point (pI) was experimentally determined (∼4.5) and was found to increase by >1 pH unit in complex with either chemokine (Fig. S1, available at http://www.jem.org/cgi/content/full/jem.20071677/DC1). Collectively, these data lend support for the hypothesis that electrostatics are important components of M3–chemokine interactions.
Figure 2.

Kinetic analysis of M3–XCL1 interactions. (A) Corey-Pauling-Koltun model of M3, XCL1, and the M3–XCL1 complex with an overlay of electrostatic potential maps from APBS (150 mM NaCl) in mesh, contoured at 0.7 kT/e and displayed using Chimera, as previously described (reference 69). Surface area buried in the complex is highlighted in green (XCL1) and yellow (M3), and both are labeled with experimental pI's. (B) Representative SPR sensorgrams (gray) and fits (red) for XCL1 binding to M3 as a function of NaCl. (C) Binding constants for the NaCl range investigated (from 200 mM to 1.5 M; Table III). (top) The on rate (kaapp) and off rate (kdapp) as a function of NaCl (mean ± SEM). (bottom) KD (from the ratio of on and off rates) and KD(eq) (from nonlinear fit to Req values; mean ± SEM).
M3–chemokine association is enhanced by electrostatics
To address the contribution of electrostatics to the interaction of M3 with chemokines, binding kinetics were examined by surface plasmon resonance (SPR) to determine apparent on and off rates. SPR analysis of chemokines binding to immobilized M3 revealed a common feature of relatively fast apparent on rates (≥107 M−1s−1) at 150 mM NaCl. To further investigate this phenomenon, XCL1 binding was assessed as a function of increasing ionic strength, which effectively screens electrostatic interactions that can enhance protein association rates (Fig. 2 B and Table III) (32). This series of measurements revealed that the M3–XCL1 apparent on rate (kaapp) is greatly reduced at high NaCl concentrations, decreasing by almost two orders of magnitude (70-fold) from 7 × 107 to 8 × 105 M−1s−1 (Fig. 2 C). This trend is also reflected in the dissociation constant (KD), which increases from 0.9 to 22 nM between 200 mM and 1.5 M NaCl. In contrast, the M3–XCL1 apparent off rate (kdapp) was relatively unperturbed over the range tested, varying only fivefold. A computational analysis of the M3–XCL1 structural interaction predicts a congruent trend in the calculated electrostatic interaction energy as a function of ionic strength, indicating that electrostatics contribute favorably to the free energy of interaction and, thus, enhance the binding affinity (Fig. S1 and Table I). It was also of interest to determine the on rate at lower salt concentrations; however, severe mass transport effects precluded analysis below 200 mM NaCl. Therefore, the experimental trend was extrapolated to obtain rough estimates for kaapp at low salt (Fig. S1 and Supplemental materials and methods, available at http://www.jem.org/cgi/content/full/jem.20071677/DC1). For example, extrapolation to zero salt leads to an estimated on rate of ∼4 × 1011 M−1s−1, suggesting that electrostatic forces would draw M3 and chemokines together at a rate much faster than diffusion alone (∼109 M−1s−1) (33). Importantly, the extrapolated on rate at physiological NaCl (150 mM) is ∼108 M−1s−1, which is two to three orders of magnitude faster than typical protein–protein interactions. Collectively, these kinetic and computational analyses clearly indicate that electrostatics facilitate fast M3–chemokine complex formation, and further, suggest that M3–XCL1 association may be among the most rapid protein–protein associations yet described (32, 34).
Table III.
NaCl dependence of M3–XCL1 binding kinetics
| NaCl (M) | KD(eq) (Req) (nM) | KD(kd/ka) (nM) | kaapp (M−1s−1) ×107 | kdapp (s−1) ×10−2 | χ2 avga |
|---|---|---|---|---|---|
| 0.2 | − | 0.9 (±0.2) | 6 (±1) | 5.6 (±0.2) | 21.61 |
| 0.25 | 2.6 (±0.4) | 1.8 (±0.4) | 5 (±2) | 9 (±3) | 2.15 |
| 0.3 | 4.6 (±0.5) | 3 (±0.5) | 3 (±1) | 9 (±2) | 0.49 |
| 0.35 | 6.9 (±0.8) | 4.6 (±0.6) | 2.2 (±0.7) | 10 (±2) | 0.35 |
| 0.4 | 9 (±0.9) | 7 (±1) | 1.6 (±0.4) | 10 (±2) | 0.4 |
| 0.5 | 11 (±1) | 9 (±1) | 0.9 (±0.2) | 9 (±1) | 0.3 |
| 0.6 | 16.2 (±0.8) | 12 (±1) | 0.33 (±0.04) | 3.9 (±0.6) | 0.49 |
| 0.7 | 18.8 (±0.9) | 17 (±2) | 0.24 (±0.02) | 4.2 (±0.5) | 0.69 |
| 1 | 21 (±2) | 17 (±2) | 0.19 (±0.03) | 3.1 (±0.3) | 0.56 |
| 1.25 | 28 (±2) | 19 (±0.8) | 0.13 (±0.01) | 2.5 (±2) | 0.37 |
| 1.5 | 26 (±6) | 22 (±2) | 0.083 (±0.003) | 1.9 (±0.1) | 0.52 |
Average χ2 for global kinetic fits at three ligand densities measured in triplicate.
M3 NTD acidic loops mediate electrostatic on-rate enhancement
Given that electrostatic complementarity is concentrated at the M3 NTD interface, the role of acidic NTD loops in enhancement of M3–chemokine apparent on rates was examined. A structure-based M3 variant, termed M3BBXB, was created by mutating the sequence 80EELGQ84 to 80SRRGR84 (Fig. 3 A). The mutant was designed with the goal of reducing electrostatic complementarity at the NTD interface by introducing Arg residues in the M3 s2b-s3 loop opposite basic chemokine sidechains in the N-loop and 40s loop. As anticipated, the M3BBXB mutant is less acidic than wild-type M3, with a pI shifted to ∼5, almost to the same degree as seen for wild-type M3 in complex with chemokine (Fig. S1). To assess whether this reduction of M3 surface charge results in a slower on rate for chemokine binding, kinetic analysis was performed for XCL1 binding to M3BBXB at 150 mM NaCl (Fig. 3 B). Indeed, the mutant displays a significantly reduced on rate, which is 1.8 × 106 M−1s−1, a decrease of ∼100-fold compared with wild-type estimates (Table IV). In contrast, the off rate for M3BBXB is relatively unchanged at 0.043 s−1 compared with ∼0.06 s−1 for wild-type (200 mM NaCl), corresponding to a half-life of ∼15 s for both. Together with the salt-dependent kinetic analysis, these results suggest that electrostatics facilitate on-rate enhancement but do not contribute as much to complex half-life for this chemokine. Furthermore, analysis of the M3BBXB mutant firmly establishes an important role for the acidic NTD loops in the electrostatic enhancement of M3–chemokine on rates.
Figure 3.

M3BBXB kinetics and M3 competition assay. (A) Positions mutated in M3BBXB and adjacent basic residues on XCL1 are shown in ball and stick form (oriented as in Fig. 1 A), and mutations are listed. Adjacent sections of the chemokine backbone are not shown, for clarity. (B) XCL1 binding to M3BBXB. Representative sensorgrams (gray) and Req values (plot, inset) are shown with corresponding fits (red and black, respectively; Table IV). (C) Competition assay to determine the solution affinity for M3 binding to XCL1 at 150 mM NaCl. Competition titration curve for coinjected M3 binding to XCL1 in competition with immobilized M3BBXB is shown with a corresponding fit to the data (Table IV).
Table IV.
M3 binding affinity and kinetics at 150 mM NaCl
| M3 variant–chemokine | KD(eq) (Req) (nM) | KD (kd/ka) (nM) | ka (M−1s−1) ×107 | kd (s−1) |
|---|---|---|---|---|
| M3–XCL1a | 0.5 (±0.07) | ∼0.6 | ∼10 | ∼0.06 |
| M3BBXB–XCL1 | 59 (±6) | 23 (±1) | 0.18 (±0.01) | 0.043 (±0.004) |
Estimates (∼) for on and off rate derived from extrapolation of salt-dependent kinetics, kinetic-based KD was determined from the ratio of these estimates, and equilibrium-based KD(eq) was determined by competition assay (referred to as KI in the text).
Assessment of M3 chemokine binding by SPR competition
We exploited the reduced on rate of the M3BBXB variant to develop an SPR assay that facilitates the measurement of wild-type M3 binding to chemokines in solution, thereby avoiding the mass transport surface effects that limited analysis at 150 mM NaCl. To this end, M3BBXB was immobilized on the sensor chip, and the solution affinity for XCL1 binding to wild-type M3 (inhibition dissociation constant [KI]) was obtained by titrating coinjected M3 that competes for XCL1 binding to immobilized M3BBXB (Fig. 3 C; a complete description of the assays is provided in Supplemental materials and methods). This competition assay yielded a solution affinity for XCL1 binding to M3 of KI = 500 (±70) pM (Table IV). By comparison, extrapolation of our salt-dependent data estimates an apparent on rate of ∼108 M−1s−1 and an off rate of ∼0.06 s−1, yielding a KD from the kinetic ratio of ∼600 pM. Thus, the M3–XCL1 affinity determined by competition at 150 mM NaCl is remarkably consistent with that extrapolated from our limited kinetic analysis.
M3 stoichiometrically inhibits chemokine–GAG interactions
The experiments described thus far have established that electrostatic interactions with the M3 NTD profoundly enhance chemokine on rates. Electrostatics are also an important component of chemokine–GAG interactions, exemplified by their salt dependence (35), the importance of N- and O-sulfation of GAGs (36), and the role of residues in the chemokine basic cluster (4). In support of this concept, previous studies have shown that the addition of M3 to heparin-binding assays reduces GAG binding for some chemokines (37). To address whether M3 could directly inhibit GAG interactions for diverse chemokines, an SPR competition assay was developed (Fig. 4). Initially, chemokine-binding affinity for immobilized heparin was measured, and chemokine was titrated with coinjected M3 to assay inhibition of heparin binding (Fig. 4). This analysis was ultimately performed for members of three out of the four classes of chemokines, as CX3CL1 exhibited very low affinity for heparin (unpublished data). Analysis of CCL2, XCL1, and CXC chemokine ligand (CXCL) 10 binding to heparin yielded KD estimates (Fig. 4 A and Table V) in agreement with previous papers (35, 38), as XCL1 and CXCL10 exhibited nanomolar affinity and CCL2 micromolar. Qualitative assessment of sensorgrams shows that XCL1 and CXCL10 display relatively slow multiphasic association kinetics and long-lived complexes in comparison with the rapid kinetics observed for CCL2, reflecting the diverse nature of these individual chemokine–GAG interactions. Results of the M3 competition assay show that M3 blocks CC, C, and CXC chemokine binding to GAGs in a stoichiometric manner, as M3 completely inhibits heparin binding for all chemokines at nearly a 1:1 molar ratio (Fig. 4 B). Furthermore, estimates for the M3 dissociation constant (KI) in competition with heparin are in the picomolar–nanomolar range (Table V), suggesting that M3 could effectively inhibit GAG-binding at physiologically relevant chemokine concentrations (39).
Figure 4.

Heparin-binding and M3 competition assays. (A, left) Representative SPR traces for chemokines binding to immobilized heparin. Note that chemokines display multiphasic-binding kinetics, which therefore precluded kinetic analysis using a simple bimolecular interaction model. (right) Req values for chemokine binding to heparin with corresponding nonlinear fits to obtain KD for each interaction (Table V). Equilibrium (Req) binding for CCL2 and CXCL10 was described well by a simple 1:1 interaction model; however, XCL1 displayed cooperative binding behavior and, thus, a more complex model was used (Supplemental materials and methods). (B) Competition titration curves with corresponding fits for coinjected M3 inhibition of XCL1, CCL2, and CXCL10 binding to immobilized heparin (Table V). (C) FACS analysis of chemokine binding to CHO-K1 (wild-type) and CHO-745 (GAG-deficient) cell lines. Staining of CHO-745 cells is shown in violet for XCL1, CCL2, CXCL10, and VEGF control, with staining in the absence (green) and presence (magenta) of M3 superimposed.
Table V.
Heparin-binding and M3 competition assay
| Heparin
|
M3 competition
|
|
|---|---|---|
| Chemokine | KD (Req) (nM) | KI (nM) |
| CCL2 | 1,200 (±200) | 18 (±7) |
| XCL1 | 90 (±10) | 2.3 (±0.7) |
| CXCL10 | 60 (±10) | 0.9 (±0.3) |
A cellular competition assay was also conducted to assess M3 inhibition under conditions that more closely reflect the GAG environment in vivo. Chinese hamster ovary (CHO)–K1 cells, rich in heparan sulfate and chondroitin sulfate, were stained with CCL2, XCL1, and CXCL10, as well as the control GAG-binding proteins vascular endothelial growth factor (VEGF) and soluble fibroblast growth factor receptor (FGFR; unpublished data), in the absence and presence of equal molar amounts of M3. The GAG-deficient CHO-745 line (40) was also stained as a control. The results clearly show that all chemokines and control proteins bind to GAGs on CHO-K1 cells. In the presence of M3, staining is specifically reduced for all three chemokines, with CCL2 and CXCL10 staining completely reduced to the level of the 745 cells. The incomplete inhibition observed for XCL1 appears to be an artifact of nonspecific biotinylation, as similar results were obtained for other chemokines labeled in the same manner (unpublished data). As expected, the other control GAG-binding proteins, VEGF (Fig. 4 C) and FGFR (not depicted), were completely unaffected by incubation with M3, confirming that GAG blockade by M3 is chemokine specific. Collectively, these complementary GAG competition assays demonstrate that M3 specifically blocks chemokine–GAG interactions in a stoichiometric manner, disrupting cell adhesion for chemokines that display widely different GAG affinities and binding modes.
DISCUSSION
GPCR mimicry by the M3 CTD
The M3–XCL1 and M3–CCL2 structures reveal that GPCR mimicry by the M3 CTD is conserved for at least two chemokine subclasses. The most evident element of structural mimicry is the hydrophobic packing of M3 P272 against the chemokine invariant disulfide (Fig. 1), which mimics the interaction observed for an N-terminal peptide of CXC chemokine receptor 1 in complex with CXCL8 (27, 41). M3 P272 packs against the invariant disulfide in precisely the same manner as P29 of CXC chemokine receptor 1, and given that proline is highly conserved at this position among chemokine receptors, this may represent a general mode in which the invariant disulfide is recognized by GPCRs. Interestingly, CXCL8 N-loop interactions with the CXCL1 peptide do not appear to preclude chemokine dimerization through the β1 strand. If the M3–chemokine binding mode is conserved for all classes, it is possible that CXCL8 could also bind to M3 as a dimer, and in fact, preliminary data for the M3–CXCL8 complex are consistent with this hypothesis. In addition, hydrophobic residues immediately after the invariant disulfide bond, CCL2 Y13 and XCL1 V12, are both sequestered within the CTD hydrophobic pocket. Virtually all high affinity M3 ligands harbor a Tyr, Phe, or aliphatic sidechain at this position, which also appears to be important for receptor recognition (42, 43). This suggests that the CTD sequesters another key position engaged by GPCRs in general. Likewise, the M3 AB-loop forms an antiparallel β strand with residues in the N-terminal segment that participate in receptor binding and signaling (39). Interestingly, the H-bonds within this strand are identically positioned for XCL1 and CCL2, unperturbed by differences in the disulfide bonding patterns of each chemokine. This suggests that the M3 AB-loop recognizes an N-terminal backbone conformation that is common to chemokines of different subclasses within a region important for GPCR recognition. In summary, the two complex structures presented in this study demonstrate that M3 engages several conserved chemokine structural features associated with GPCR binding, including the invariant disulfide, adjacent hydrophobic residues, and the common N-terminal segment.
Prominent role of electrostatics in M3–chemokine interactions
This study has also demonstrated that electrostatic interactions are a conserved element of chemokine recognition by the M3 NTD and play an important role in chemokine sequestration by this decoy receptor. The structures presented in this study reveal that electrostatic complementarity is a prominent feature of the M3 binding interface, with the acidic M3 NTD adapting to engage the divergent chemokine basic cluster in each case. The importance of electrostatics in M3–chemokine interactions is highlighted by our kinetic and mutational analyses showing that M3 rapidly associates with chemokines, with an on rate that is remarkably enhanced by electrostatic interactions with the acidic M3 NTD. Electrostatic complementarity may in fact be a common mechanistic strategy among pathogen-encoded chemokine decoy receptors. For example, the poxvirus viral chemokine-binding proteins have pI's nearly as acidic as M3, and chemokine basic cluster residues play an important role in binding (14, 15). Similarly, when M3 is compared with some of the fastest electrostatically enhanced interactions found in biology, it is evident that all of these systems share the common goal of rapid inhibition. Other proteins have evolved fast association kinetics to necessarily achieve instant blockade of harmful enzymatic processes or neutralization of toxic intermediates (32, 34). In the case of M3, rapid chemokine sequestration may be necessary to prevent the establishment and spread of gradients early during infection. M3 is abundantly secreted by virally infected cells but is still overshadowed by the high level of GAG expression in the extracellular matrix. The concentrated negative potential of the M3 NTD may therefore be required to serve as an electrostatic beacon, attracting chemokines over long range, increasing their rate of association, and allowing M3 to compete with GAGs that are in vast excess. Thus, by rapidly sequestering chemokines before gradients are established, M3 could mitigate the ensuing consequences of chemokine signaling that are essentially toxic to the virus.
GAG mimicry by the M3 NTD
The SPR competition and CHO cell-surface assays reported in this paper demonstrate that M3 blocks GAG interactions for the chemokines CCL2, XCL1, and CXCL10. We have shown that electrostatic interactions between the divergent basic cluster found on chemokines and the acidic M3 NTD facilitate this blockade. GAGs are flexible linear polysaccharides heterogeneously decorated with acidic sulfate and carboxylate groups that engage chemokine basic clusters in a highly diverse manner (4). A comparison of available mutational data for CCL2 (35, 44) and XCL1 (38) with our complex structures reveals that the majority of residues implicated in GAG binding are also engaged by M3 (Fig. 5, A and B). Specifically, the M3 NTD directly contacts four out of six residues identified by mutational analysis of CCL2, and two out of four residues of XCL1, with a prominent role played by E80 and E81 in the M3 s2b-s3 loop. The contacted chemokine residues are located in the N-loop, 310-helix, and 40s loop, whereas the noncontacted residues are located in the β1 strand and C-terminal helix. Importantly, we note that our SPR and cell-surface binding assays demonstrate the disruption of CCL2 and XCL1 interactions with GAGs despite the fact that M3 does not make contact with all of the surface epitopes identified for these chemokines.
Figure 5.

Dual GPCR and GAG inhibition by M3. (A) Surface representation of CCL2 is shown, with previously identified GAG-binding residues labeled (references 35, 44). Four out of the six residues defined by mutational analysis as creating the GAG-binding epitope of CCL2 are directly contacted by M3 and are colored blue (Arg18, Lys19, Arg24, and Lys49), whereas the noncontacted residues are colored gray (Lys58 and His66). The M3 NTD s2b-s3 loop is displayed in cyan, with acidic contact residues E80 and E81 shown in stick form. (B) M3 contacts two out of the four previously identified GAG-binding residues of XCL1 (R23 and R43, blue; K25 and R70, gray; reference 38). (C) The structure of human CXCL10 (reference 70) is shown, with the four conserved GAG-binding residues identified by mutational analysis in mouse IP-10 highlighted in blue (R22, K26, K46, and K47; reference 45). (D) The structure of CCL5 is shown, with GAG-binding residues established by structural and mutational analysis highlighted in blue (R44, K45, and R47; references 46, 47). (E) Proposed model for the disruption of chemokine gradients by M3 during MHV68 infection. (F) Schematic of M3 NTD-mediated disruption of chemokine interactions with cell-surface GAGs (green) and CTD-mediated disruption of chemokine interactions with GPCRs (violet). The electrostatic potential is indicated by red (+) and blue (−) on chemokines and GAGs, respectively.
Because most chemokines harbor a basic cluster located in roughly the same position as CCL2 and XCL1, the coincidence of GAG-binding residues with M3 NTD contacts may in fact be a more general phenomenon. In this study, we have shown that M3 capably disrupts the interaction of CXCL10 with GAGs, and that three out of four residues implicated in GAG binding (45) are located in the 310-helix and 40s loop, forming a basic cluster that maps to approximately the same location as where CCL2 and XCL1 basic residues are contacted by the M3 NTD (Fig. 5 C). Computational docking studies indicate that M3 could readily contact CXCL10 R22, K46, and K47 but would unlikely contact K26 located on the β1 strand. Nevertheless, as for CCL2 and XCL1, we suggest that partial engagement of the basic cluster is sufficient for complete blockade. Functional and structural mapping of GAG binding to another MHV68 up-regulated chemokine, CCL5, provides further support for the notion of M3 GAG-binding mimicry (Fig. 5 D). The M3-contacted residues K49 of CCL2 and R43 of XCL1 are located in the same 40s loop region as the BBXB motif residues of CCL5 (46). Furthermore, the crystal structure of a CCL5–heparin disaccharide complex reveals that the 40s loop BBXB residues directly contact sulfate and carboxylate moieties of the carbohydrate (47). Analogously, the 40s loop basic residues of XCL1 and CCL2 interact with carbonyl and carboxylate moieties from the M3 s2b-s3 loop. Thus, M3 acts as a functional GAG mimic for chemokines, using similar chemical means (absent sulfation) to competitively disrupt chemotactic gradient formation, thereby preventing inflammation at sites of MHV68 infection (Fig. 5 E).
Collaborative nature of the two-domain M3 binding interface
In this paper, we have demonstrated that M3 can stoichiometrically block GAG interactions for three chemokines with similar albeit not identically located basic clusters that apparently facilitate distinct modes of heparin binding, as revealed by our SPR analysis. The M3–XCL1 and M3–CCL2 structures reveal how M3 is able to block such diverse interactions by interfacial reconfiguration that optimally engages the unique GAG-binding residues of each chemokine. Notably, M3 is not effective against other GAG-binding proteins, including VEGF and soluble FGFR. This is most likely a result of dual chemokine recognition by the two-domain M3 interface (Fig. 5 F), which effectively ensures that GAG blockade is chemokine specific.
M3 is the only decoy receptor known to recognize chemokines with a two-domain interface and, accordingly, displays the broadest chemokine-binding profile, engaging members of all four classes with high affinity (10). Thus, an important advantage of two-domain recognition may be promiscuous binding. We argue in this paper that GPCR and GAG mimicry can be primarily attributed to functions of the M3 CTD and NTD, respectively (Fig. 5 F). This parsing of roles allows M3 to recognize conserved chemokine structural elements through the CTD and, at the same time, imparts structural plasticity to the interface for adaptation of the NTD to divergent chemokine features. With collaboration between the two domains of the interface, it is possible for each to make a unique contribution to the overall affinity of M3–chemokine interactions. Our mutational analysis supports the notion that the NTD contributes long-range electrostatic interactions to rapidly steer chemokines into the binding niche, where short-range hydrophobic and H-bond interactions subsequently form with the CTD, ultimately resulting in a long-lived complex. Indeed, the M3BBXB variant exhibits a dramatically attenuated ability to recruit XCL1 compared with wild-type M3 (100-fold on-rate reduction) but is capable of stable sequestration with equivalent complex half-lives. It will be of great interest in the future to assess whether short-range interactions contributed by the CTD play a role in determining the half-life for specific M3–chemokine complexes. In the context of the extracellular milieu, where these components are not likely to be in equilibrium, complex stability may in fact dictate whether a given chemokine will be effectively targeted by M3.
Chemokines play a critical role in leukocyte recruitment, and therefore, antagonism of the chemokine system represents a promising antiinflammatory strategy relevant not only to viruses but also to medical therapeutics. The high degree of redundancy among individual ligands and receptors has classically presented an inherent difficulty in targeting the chemokine network. The M3 decoy receptor is an ideal agent in this regard, because it exhibits the most broad-spectrum inhibition of any known chemokine decoy receptor and is further capable of blocking both GPCR and GAG interactions. Not surprisingly, M3 has shown promise as an antiinflammatory therapeutic in several models, including tumor rejection (48), vascular injury (49), aortic allograft rejection (50), and CCL21-induced lymphocytic infiltration of pancreatic islets (51). More recent experiments have shown that islet-specific M3 expression can also prevent inflammatory recruitment, islet destruction, and subsequent diabetes in mouse insulitis models (52). This study describes in precise detail the structural basis for chemokine inhibition by the M3 viral decoy receptor, revealing the importance of dual mimicry of both GPCRs and GAGs in the establishment of broad-spectrum antiinflammatory activity.
MATERIALS AND METHODS
DNA constructs, protein expression, and purification.
Full-length, untagged M3 protein was expressed in baculovirus-infected insect cells (SF9) and purified as previously described (27). The M3BBXB (80EELGQ/SRRGR84) variant was created by quick-change site-directed mutagenesis (courtesy of V. van Berkel, Washington University, St. Louis, MO), expressed using baculovirus, and elutes from size-exclusion chromatography with the same (dimeric) profile as wild-type M3. Full-length synthetic human XCL1 (residues 1–92) and Escherichia coli–produced human CCL2 (M64I) used in crystallization (courtesy of T. Handel, University of California, San Diego, La Jolla, CA) were made as previously described (30, 53). For cell staining, CCL2 (M64I) and CXCL10 were cloned into a customized pET28 vector with a thrombin-cleavable C-terminal tag designed for site-specific biotinylation (courtesy of M. Miley, Washington University, St. Louis, MO). Proteins were expressed in E. coli as inclusion bodies, were refolded under oxidizing conditions, and were purified by ion exchange. The C-terminal tag was removed from CCL2 and CXCL10 before SPR analysis.
x-ray crystallography.
M3–CCL2 crystallized in the space group P3121 (a = b = 99.23, c = 243.5), and M3–XCL1 crystallized in the space group I212121 (a = 85.44, b = 104.18, c = 290.85). Crystallographic data were collected in the beamline 19-ID at the Advanced Photon Source. Diffraction intensities were integrated and scaled using the HKL2000 program suite, as previously described (54). The phase problem was solved by molecular replacement in AMoRe, as previously described (55), using M3–CCL2 (P8A) (available from the Protein Data Bank under accession no. 1ML0) as a search model. For M3–XCL1, prime-and-switch phasing was used to reduce model-phase bias in the initial maps with RESOLVE, as previously described (56). Model building was undertaken with O, as previously described (57), and refinement against 2.6-Å resolution data was performed using the Crystallography and Nuclear Magnetic Resonance System (CNS), as previously described (58), with a final R-value of 22% and a free R-value of 27.3% (5% test set). The atomic model consists of a 2:2 complex related by noncrystallographic symmetry (NCS), including residues 12–382 of M3 (chains A and B) and residues 7–67 and 7–72 of XCL1 (chains D and E, respectively). The first 11 residues of M3 were not located, nor were the first 6 and last 26 for XCL1 D, and the first 6 and last 21 for XCL1 E. For the M3–CCL2 complex, refinement was performed to a resolution of 2.3 Å, with a final R-value of 23.2% and a free R-value of 29.9% (5% test set). The atomic model consists of three M3 molecules (residues 12–382) and three CCL2 molecules (residues 8–71), representing one 2:2 complex (NCS) and a half complex completed by the crystallographic twofold symmetry axis (CS).
Structure analysis.
Structure analysis was performed for the 2:2 M3–XCL1 complex and for the NCS and CS complexes of M3–CCL2. LSQKAB (59) was used to calculate RMSD between M3 Cα atoms of AB (NCS) and X (CS) of M3–CCL2 and M3–CCL2 (P8A). RMSD between M3 (all atoms) in complex with XCL1 and CCL2 was calculated using CNS, as previously described (58). For RMSD on a per-residue basis, values were calculated for the two (XCL1) and three (CCL2) interfaces and subsequently averaged. SC, as previously described (60), was used to calculate the shape complementarity for each binding interface. NACCESS, as previously described (61), was used to analyze the BSA. HBPLUS, as previously described (62), was used to enumerate atomic contacts (≤4 Å) and H-bonds.
Electrostatic analysis.
The ln(ka) was plotted versus 1/1 + κa over the range of salt concentrations studied to yield an estimate for the M3–XCL1 kaapp at 150 mM NaCl of ∼108 M−1s−1 (Supplemental materials and methods). In addition, to test the role of protein–ion binding in M3–XCL1 interactions, measurements were conducted under the same ionic strength using either NaCl or MgCl2 (Fig. S1 and Table S1, available at http://www.jem.org/cgi/content/full/jem.20071677/DC1). The program APBS was used to calculate potential maps and electrostatic interaction energies (ΔGelec) for the M3–XCL1 interaction (Supplemental materials and methods).
SPR experiments.
SPR experiments were conducted using a biosensor (Biacore 2000; GE Healthcare). All experiments were conducted at 25°C under conditions of 20 mM Hepes (pH 7.4), 150 mM NaCl, and 0.005% Triton X-100, unless otherwise indicated. A detailed description of all SPR experiments is provided in Supplemental materials and methods.
For M3–chemokine interaction analysis, M3 variants were immobilized on a Biacore CM5 sensor chip in 10 mM sodium acetate, pH 4.1, using standard amine coupling. To measure M3–XCL1 binding kinetics as a function of NaCl, M3 was immobilized on the sensor chip and 80 μl/min XCL1 was injected. Data were collected over the range from 200 mM to 1.5 M NaCl. To measure M3BBXB binding to XCL1, the mutant was immobilized and 40 μl/min XCL1 was injected, and for wild-type M3 binding by competition, XCL1 was preequilibrated with M3 and injected over the M3BBXB chip in the same manner.
To measure chemokine binding to heparin and M3 competition, heparin sensor chips were prepared by neutravidin capture of biotinylated heparin. Neutravidin (Thermo Fisher Scientific) was coupled to a CM5 chip in 10 mM sodium citrate, pH 4.5, using standard amine chemistry. 15-kD heparin-biotin was injected at 5 mg/ml in Hepes running buffer with 300 mM NaCl and bound to a level of ∼150–300 RU. For heparin binding, chemokines were injected at 20 μl/min CCL2, and 80 μl/min XCL1 and CXCL10, and for M3 competition assays, chemokines were preequilibrated with M3 and injected over the heparin chip in the same manner. The resulting estimates for XCL1 and CXCL10 affinity by this assay are greater than CCL2, which is unexpected, because CCL2 is predicted to be much higher affinity. A likely explanation is that the KI for CCL2 is beyond the limits of detection of the competition assay when conducted in the micromolar range (CCL2), and therefore, the KI represents a lower limit for the true dissociation constant in this case.
SPR competition assay.
It was of interest to develop an SPR competition assay based on an analysis of Req values, which are not subject to the same caveats as SPR-derived kinetic constants (63), which have been used in previously described assays (64). The competition assay was derived in a similar manner as reported for a fluorescence-based assay (Supplemental materials and methods) (65). Competition titration curves were analyzed using the following equations, assuming a 1:1 interaction for both the receptor on the chip and the solution competitor, with A = total analyte, M = receptor on chip, M*=competitor in solution, and affinities for the chip-bound receptor and the solution-phase competitor denoted as KM and KM*, respectively:
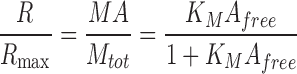 |
(1) |
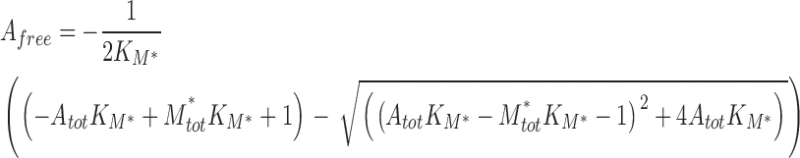 |
(2) |
Competition titration curves were fit to eqs. 1 and 2 (referred to as eqs. 1 and 6 in Supplemental materials and methods) using the program Scientist (Micromath) to yield KI (1/KM*) for M3 binding to chemokines in competition with the chip-bound receptor. The assay was conducted in the same manner for the M3BBXB or heparin competition assays, where M denotes M3BBXB or heparin on the chip and M* is M3 (competitor) in solution.
Flow cytometry.
XCL1 and VEGF (PeproTech) were nonspecifically biotinylated with Sulfo-NHS-LC-Biotin (Thermo Fisher Scientific), and CCL2 and CXCL10 were site-specifically biotinylated using BirA ligase (courtesy of M. Fremont, Washington University, St. Louis, MO) according to standard protocol (Avidity). Streptavidin-PE was purchased from BD Biosciences. CHO-K1 and -745 cell lines were a gift from L. Zhang (Washington University, St. Louis, MO) and cultured as previously described (66). To analyze chemokine binding to GAGs on CHO cells, 5 × 105 cells were stained with 1–2 μg of each chemokine in 200 μl PBS with 10% FBS. After washing, the cells were stained with 1 μg streptavidin-PE. Flow cytometry was conducted on a flow cytometer (FACScan; BD Biosciences). For competition experiments, the cells were incubated with chemokines in the presence of equal molar amounts of M3.
Coordinates.
The coordinates of M3–CCL2 and M3–XCL1 are available from the Protein Data Bank under accession nos. 2NZ1 and 2NYZ, respectively.
Online supplemental material.
Supplemental materials and methods includes a detailed description of SPR experiments, with derivation of the equations used in binding analysis, and a detailed analysis and discussion of M3–XCL1 electrostatic interactions. Fig. S1 depicts isoelectric focusing experiments for M3 variants and M3–chemokine complexes, results of a computational analysis of M3–XCL1 interactions, and a comparison of salt-dependent binding kinetics for M3–XCL1 conducted in the presence of NaCl versus MgCl2. Table S1 provides data from the analysis of ionic strength effects on M3–XCL1 binding. Online supplemental material is available at http://www.jem.org/cgi/content/full/jem.20071677/DC1.
Supplemental Material
Acknowledgments
We thank Timothy Lohman for guidance in the development of the SPR competition assay, and Todd Dolinsky, Nathan Baker, and David Sept for electrostatic discussions. We thank Tracy Handel, Victor van Berkel, Lijuan Zhang, Michael Miley, and Marcel Fremont for sharing reagents. We also thank Chris Nelson, Tom Brett, Skip Virgin, and Timothy Lohman for discussion and critical reading of the manuscript.
This work was supported by National Institutes of Health grants AI05142604, GM62414–04 (D.H. Fremont), and U54-AI05716003.
The authors declare that they have no competing financial interests.
Abbreviations used: BSA, buried surface area; CCL, CC chemokine ligand; CHO, Chinese hamster ovary; CTD, C-terminal domain; CXCL, CXC chemokine ligand; FGFR, fibroblast growth factor receptor; GAG, glycosaminoglycan; GPCR, G protein–coupled receptor; IP, IFN-inducible protein; KD, dissociation constant; KI, inhibition dissociation constant; MHV68, mouse γ-herpesvirus 68; MIP, macrophage inflammatory protein; NTD, N-terminal domain; pI, isoelectric point; RMSD, root mean square deviation; SPR, surface plasmon resonance; VEGF, vascular endothelial growth factor; XCL, C chemokine ligand.
References
- 1.Murphy, P.M. 2002. International Union of Pharmacology. XXX. Update on chemokine receptor nomenclature. Pharmacol. Rev. 54:227–229. [DOI] [PubMed] [Google Scholar]
- 2.Rollins, B.J. 1997. Chemokines. Blood. 90:909–928. [PubMed] [Google Scholar]
- 3.Linhardt, R.J., and T. Toida. 2004. Role of glycosaminoglycans in cellular communication. Acc. Chem. Res. 37:431–438. [DOI] [PubMed] [Google Scholar]
- 4.Lortat-Jacob, H., A. Grosdidier, and A. Imberty. 2002. Structural diversity of heparan sulfate binding domains in chemokines. Proc. Natl. Acad. Sci. USA. 99:1229–1234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Proudfoot, A.E., T.M. Handel, Z. Johnson, E.K. Lau, P. LiWang, I. Clark-Lewis, F. Borlat, T.N. Wells, and M.H. Kosco-Vilbois. 2003. Glycosaminoglycan binding and oligomerization are essential for the in vivo activity of certain chemokines. Proc. Natl. Acad. Sci. USA. 100:1885–1890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Johnson, Z., M.H. Kosco-Vilbois, S. Herren, R. Cirillo, V. Muzio, P. Zaratin, M. Carbonatto, M. Mack, A. Smailbegovic, M. Rose, et al. 2004. Interference with heparin binding and oligomerization creates a novel anti-inflammatory strategy targeting the chemokine system. J. Immunol. 173:5776–5785. [DOI] [PubMed] [Google Scholar]
- 7.Alcami, A., and U.H. Koszinowski. 2000. Viral mechanisms of immune evasion. Immunol. Today. 21:447–455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lusso, P. 2000. Chemokines and viruses: the dearest enemies. Virology. 273:228–240. [DOI] [PubMed] [Google Scholar]
- 9.Luster, A.D. 1998. Chemokines–chemotactic cytokines that mediate inflammation. N. Engl. J. Med. 338:436–445. [DOI] [PubMed] [Google Scholar]
- 10.Alcami, A. 2003. Viral mimicry of cytokines, chemokines and their receptors. Nat. Rev. Immunol. 3:36–50. [DOI] [PubMed] [Google Scholar]
- 11.Graham, K.A., A.S. Lalani, J.L. Macen, T.L. Ness, M. Barry, L.Y. Liu, A. Lucas, I. Clark-Lewis, R.W. Moyer, and G. McFadden. 1997. The T1/35kDa family of poxvirus-secreted proteins bind chemokines and modulate leukocyte influx into virus-infected tissues. Virology. 229:12–24. [DOI] [PubMed] [Google Scholar]
- 12.Carfi, A., C.A. Smith, P.J. Smolak, J. McGrew, and D.C. Wiley. 1999. Structure of a soluble secreted chemokine inhibitor vCCI (p35) from cowpox virus. Proc. Natl. Acad. Sci. USA. 96:12379–12383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Seet, B.T., R. Singh, C. Paavola, E.K. Lau, T.M. Handel, and G. McFadden. 2001. Molecular determinants for CC-chemokine recognition by a poxvirus CC-chemokine inhibitor. Proc. Natl. Acad. Sci. USA. 98:9008–9013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Arnold, P.L., and D.H. Fremont. 2006. Structural determinants of chemokine binding by an Ectromelia virus-encoded decoy receptor. J. Virol. 80:7439–7449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhang, L., M. Derider, M.A. McCornack, S.C. Jao, N. Isern, T. Ness, R. Moyer, and P.J. LiWang. 2006. Solution structure of the complex between poxvirus-encoded CC chemokine inhibitor vCCI and human MIP-1beta. Proc. Natl. Acad. Sci. USA. 103:13985–13990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Alejo, A., M.B. Ruiz-Arguello, Y. Ho, V.P. Smith, M. Saraiva, and A. Alcami. 2006. A chemokine-binding domain in the tumor necrosis factor receptor from variola (smallpox) virus. Proc. Natl. Acad. Sci. USA. 103:5995–6000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bryant, N.A., N. Davis-Poynter, A. Vanderplasschen, and A. Alcami. 2003. Glycoprotein G isoforms from some alphaherpesviruses function as broad-spectrum chemokine binding proteins. EMBO J. 22:833–846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wang, D., W. Bresnahan, and T. Shenk. 2004. Human cytomegalovirus encodes a highly specific RANTES decoy receptor. Proc. Natl. Acad. Sci. USA. 101:16642–16647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Virgin, H.W., IV, P. Latreille, P. Wamsley, K. Hallsworth, K.E. Weck, A.J. Dal Canto, and S.H. Speck. 1997. Complete sequence and genomic analysis of murine gammaherpesvirus 68. J. Virol. 71:5894–5904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.van Berkel, V., K. Preiter, H.W. Virgin IV, and S.H. Speck. 1999. Identification and initial characterization of the murine gammaherpesvirus 68 gene M3, encoding an abundantly secreted protein. J. Virol. 73:4524–4529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.van Berkel, V., J. Barrett, H.L. Tiffany, D.H. Fremont, P.M. Murphy, G. McFadden, S.H. Speck, and H.W. Virgin IV. 2000. Identification of a gammaherpesvirus selective chemokine binding protein that inhibits chemokine action. J. Virol. 74:6741–6747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.van Berkel, V., B. Levine, S.B. Kapadia, J.E. Goldman, S.H. Speck, and H.W. Virgin IV. 2002. Critical role for a high-affinity chemokine-binding protein in gamma-herpesvirus-induced lethal meningitis. J. Clin. Invest. 109:905–914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Parry, C.M., J.P. Simas, V.P. Smith, C.A. Stewart, A.C. Minson, S. Efstathiou, and A. Alcami. 2000. A broad spectrum secreted chemokine binding protein encoded by a herpesvirus. J. Exp. Med. 191:573–578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Weinberg, J.B., M.L. Lutzke, S. Efstathiou, S.L. Kunkel, and R. Rochford. 2002. Elevated chemokine responses are maintained in lungs after clearance of viral infection. J. Virol. 76:10518–10523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sarawar, S.R., R.D. Cardin, J.W. Brooks, M. Mehrpooya, R.A. Tripp, and P.C. Doherty. 1996. Cytokine production in the immune response to murine gammaherpesvirus 68. J. Virol. 70:3264–3268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Verzijl, D., C.P. Fitzsimons, M. Van Dijk, J.P. Stewart, H. Timmerman, M.J. Smit, and R. Leurs. 2004. Differential activation of murine herpesvirus 68- and Kaposi's sarcoma-associated herpesvirus-encoded ORF74 G protein-coupled receptors by human and murine chemokines. J. Virol. 78:3343–3351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Alexander, J.M., C.A. Nelson, V. van Berkel, E.K. Lau, J.M. Studts, T.J. Brett, S.H. Speck, T.M. Handel, H.W. Virgin IV, and D.H. Fremont. 2002. Structural basis of chemokine sequestration by a herpesvirus decoy receptor. Cell. 111:343–356. [DOI] [PubMed] [Google Scholar]
- 28.Lubkowski, J., G. Bujacz, L. Boque, P.J. Domaille, T.M. Handel, and A. Wlodawer. 1997. The structure of MCP-1 in two crystal forms provides a rare example of variable quaternary interactions. Nat. Struct. Biol. 4:64–69. [DOI] [PubMed] [Google Scholar]
- 29.Kuloglu, E.S., D.R. McCaslin, J.L. Markley, and B.F. Volkman. 2002. Structural rearrangement of human lymphotactin, a C chemokine, under physiological solution conditions. J. Biol. Chem. 277:17863–17870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Paavola, C.D., S. Hemmerich, D. Grunberger, I. Polsky, A. Bloom, R. Freedman, M. Mulkins, S. Bhakta, D. McCarley, L. Wiesent, et al. 1998. Monomeric monocyte chemoattractant protein-1 (MCP-1) binds and activates the MCP-1 receptor CCR2B. J. Biol. Chem. 273:33157–33165. [DOI] [PubMed] [Google Scholar]
- 31.Baggiolini, M., B. Dewald, and B. Moser. 1997. Human chemokines: an update. Annu. Rev. Immunol. 15:675–705. [DOI] [PubMed] [Google Scholar]
- 32.Schreiber, G., and A.R. Fersht. 1996. Rapid, electrostatically assisted association of proteins. Nat. Struct. Biol. 3:427–431. [DOI] [PubMed] [Google Scholar]
- 33.Selzer, T., and G. Schreiber. 1999. Predicting the rate enhancement of protein complex formation from the electrostatic energy of interaction. J. Mol. Biol. 287:409–419. [DOI] [PubMed] [Google Scholar]
- 34.Radic, Z., P.D. Kirchhoff, D.M. Quinn, J.A. McCammon, and P. Taylor. 1997. Electrostatic influence on the kinetics of ligand binding to acetylcholinesterase. Distinctions between active center ligands and fasciculin. J. Biol. Chem. 272:23265–23277. [DOI] [PubMed] [Google Scholar]
- 35.Lau, E.K., C.D. Paavola, Z. Johnson, J.P. Gaudry, E. Geretti, F. Borlat, A.J. Kungl, A.E. Proudfoot, and T.M. Handel. 2004. Identification of the glycosaminoglycan binding site of the CC chemokine, MCP-1: implications for structure and function in vivo. J. Biol. Chem. 279:22294–22305. [DOI] [PubMed] [Google Scholar]
- 36.Spillmann, D., D. Witt, and U. Lindahl. 1998. Defining the interleukin-8-binding domain of heparan sulfate. J. Biol. Chem. 273:15487–15493. [DOI] [PubMed] [Google Scholar]
- 37.Webb, L.M., V.P. Smith, and A. Alcami. 2004. The gammaherpesvirus chemokine binding protein can inhibit the interaction of chemokines with glycosaminoglycans. FASEB J. 18:571–573. [DOI] [PubMed] [Google Scholar]
- 38.Peterson, F.C., E.S. Elgin, T.J. Nelson, F. Zhang, T.J. Hoeger, R.J. Linhardt, and B.F. Volkman. 2004. Identification and characterization of a glycosaminoglycan recognition element of the C chemokine lymphotactin. J. Biol. Chem. 279:12598–12604. [DOI] [PubMed] [Google Scholar]
- 39.Fernandez, E.J., and E. Lolis. 2002. Structure, function, and inhibition of chemokines. Annu. Rev. Pharmacol. Toxicol. 42:469–499. [DOI] [PubMed] [Google Scholar]
- 40.Esko, J.D., J.L. Weinke, W.H. Taylor, G. Ekborg, L. Roden, G. Anantharamaiah, and A. Gawish. 1987. Inhibition of chondroitin and heparan sulfate biosynthesis in Chinese hamster ovary cell mutants defective in galactosyltransferase I. J. Biol. Chem. 262:12189–12195. [PubMed] [Google Scholar]
- 41.Skelton, N.J., C. Quan, D. Reilly, and H. Lowman. 1999. Structure of a CXC chemokine-receptor fragment in complex with interleukin-8. Structure. 7:157–168. [DOI] [PubMed] [Google Scholar]
- 42.Hemmerich, S., C. Paavola, A. Bloom, S. Bhakta, R. Freedman, D. Grunberger, J. Krstenansky, S. Lee, D. McCarley, M. Mulkins, et al. 1999. Identification of residues in the monocyte chemotactic protein-1 that contact the MCP-1 receptor, CCR2. Biochemistry. 38:13013–13025. [DOI] [PubMed] [Google Scholar]
- 43.Nardese, V., R. Longhi, S. Polo, F. Sironi, C. Arcelloni, R. Paroni, C. DeSantis, P. Sarmientos, M. Rizzi, M. Bolognesi, et al. 2001. Structural determinants of CCR5 recognition and HIV-1 blockade in RANTES. Nat. Struct. Biol. 8:611–615. [DOI] [PubMed] [Google Scholar]
- 44.Chakravarty, L., L. Rogers, T. Quach, S. Breckenridge, and P.E. Kolattukudy. 1998. Lysine 58 and histidine 66 at the C-terminal alpha-helix of monocyte chemoattractant protein-1 are essential for glycosaminoglycan binding. J. Biol. Chem. 273:29641–29647. [DOI] [PubMed] [Google Scholar]
- 45.Campanella, G.S., E.M. Lee, J. Sun, and A.D. Luster. 2003. CXCR3 and heparin binding sites of the chemokine IP-10 (CXCL10). J. Biol. Chem. 278:17066–17074. [DOI] [PubMed] [Google Scholar]
- 46.Proudfoot, A.E., S. Fritchley, F. Borlat, J.P. Shaw, F. Vilbois, C. Zwahlen, A. Trkola, D. Marchant, P.R. Clapham, and T.N. Wells. 2001. The BBXB motif of RANTES is the principal site for heparin binding and controls receptor selectivity. J. Biol. Chem. 276:10620–10626. [DOI] [PubMed] [Google Scholar]
- 47.Shaw, J.P., Z. Johnson, F. Borlat, C. Zwahlen, A. Kungl, K. Roulin, A. Harrenga, T.N. Wells, and A.E. Proudfoot. 2004. The X-ray structure of RANTES: heparin-derived disaccharides allows the rational design of chemokine inhibitors. Structure. 12:2081–2093. [DOI] [PubMed] [Google Scholar]
- 48.Rice, J., B. de Lima, F.K. Stevenson, and P.G. Stevenson. 2002. A gamma-herpesvirus immune evasion gene allows tumor cells in vivo to escape attack by cytotoxic T cells specific for a tumor epitope. Eur. J. Immunol. 32:3481–3487. [DOI] [PubMed] [Google Scholar]
- 49.Pyo, R., K.K. Jensen, M.T. Wiekowski, D. Manfra, A. Alcami, M.B. Taubman, and S.A. Lira. 2004. Inhibition of intimal hyperplasia in transgenic mice conditionally expressing the chemokine-binding protein M3. Am. J. Pathol. 164:2289–2297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Liu, L., E. Dai, L. Miller, B. Seet, A. Lalani, C. Macauley, X. Li, H.W. Virgin IV, C. Bunce, P. Turner, et al. 2004. Viral chemokine-binding proteins inhibit inflammatory responses and aortic allograft transplant vasculopathy in rat models. Transplantation. 77:1652–1660. [DOI] [PubMed] [Google Scholar]
- 51.Jensen, K.K., S.C. Chen, R.W. Hipkin, M.T. Wiekowski, M.A. Schwarz, C.C. Chou, J.P. Simas, A. Alcami, and S.A. Lira. 2003. Disruption of CCL21-induced chemotaxis in vitro and in vivo by M3, a chemokine-binding protein encoded by murine gammaherpesvirus 68. J. Virol. 77:624–630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Martin, A.P., J.M. Alexander-Brett, C. Canasto-Chibuque, A. Garin, J.S. Bromberg, D.H. Fremont, and S.A. Lira. 2007. The chemokine binding protein M3 prevents diabetes induced by multiple low doses of streptozotocin. J. Immunol. 178:4623–4631. [DOI] [PubMed] [Google Scholar]
- 53.Marcaurelle, L.A., L.S. Mizoue, J. Wilken, L. Oldham, S.B. Kent, T.M. Handel, and C.R. Bertozzi. 2001. Chemical synthesis of lymphotactin: a glycosylated chemokine with a C-terminal mucin-like domain. Chemistry. 7:1129–1132. [DOI] [PubMed] [Google Scholar]
- 54.Otwinowski, Z., and W. Minor. 1997. Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 276:307–326. [DOI] [PubMed] [Google Scholar]
- 55.Navaza, J., and P. Saludjian. 1997. AMoRe: An automated molecular replacement program package. Methods Enzymol. 276:581–594. [DOI] [PubMed] [Google Scholar]
- 56.Terwilliger, T.C. 2004. Using prime-and-switch phasing to reduce model bias in molecular replacement. Acta Crystallogr. D Biol. Crystallogr. 60:2144–2149. [DOI] [PubMed] [Google Scholar]
- 57.Jones, T.A., J.Y. Zou, S.W. Cowan, and M. Kjeldgaard. 1991. Improved methods for binding protein models in electron density maps and the location of errors in these models. Acta Crystallogr. A. 47:110–119. [DOI] [PubMed] [Google Scholar]
- 58.Brunger, A.T., P.D. Adams, G.M. Clore, W.L. DeLano, P. Gros, R.W. Grosse-Kunstleve, J.S. Jiang, J. Kuszewski, M. Nilges, N.S. Pannu, et al. 1998. Crystallography & NMR system: A new software suite for macromolecular structure determination. Acta Crystallogr. D Biol. Crystallogr. 54:905–921. [DOI] [PubMed] [Google Scholar]
- 59.Kabsch, W. 1976. A solution for the best rotation to relate two sets of vectors. Acta Crystallogr. A. 32:922–923. [Google Scholar]
- 60.Lawrence, M.C., and P.M. Colman. 1993. Shape complementarity at protein/protein interfaces. J. Mol. Biol. 234:946–950. [DOI] [PubMed] [Google Scholar]
- 61.Hubbard, S.J., S.F. Campbell, and J.M. Thornton. 1991. Molecular recognition. Conformational analysis of limited proteolytic sites and serine proteinase protein inhibitors. J. Mol. Biol. 220:507–530. [DOI] [PubMed] [Google Scholar]
- 62.McDonald, I.K., and J.M. Thornton. 1994. Satisfying hydrogen bonding potential in proteins. J. Mol. Biol. 238:777–793. [DOI] [PubMed] [Google Scholar]
- 63.Schuck, P. 1997. Reliable determination of binding affinity and kinetics using surface plasmon resonance biosensors. Curr. Opin. Biotechnol. 8:498–502. [DOI] [PubMed] [Google Scholar]
- 64.Nieba, L., A. Krebber, and A. Pluckthun. 1996. Competition BIAcore for measuring true affinities: large differences from values determined from binding kinetics. Anal. Biochem. 234:155–165. [DOI] [PubMed] [Google Scholar]
- 65.Jason Wong, C., A.L. Lucius, and T.M. Lohman. 2005. Energetics of DNA end binding by E. coli RecBC and RecBCD helicases indicate loop formation in the 3′-single-stranded DNA tail. J. Mol. Biol. 352:765–782. [DOI] [PubMed] [Google Scholar]
- 66.Zhang, L., D.L. Beeler, R. Lawrence, M. Lech, J. Liu, J.C. Davis, Z. Shriver, R. Sasisekharan, and R.D. Rosenberg. 2001. 6-O-sulfotransferase-1 represents a critical enzyme in the anticoagulant heparan sulfate biosynthetic pathway. J. Biol. Chem. 276:42311–42321. [DOI] [PubMed] [Google Scholar]
- 67.Carson, M. 1997. Ribbons. Methods Enzymol. 277:493–502. [PubMed] [Google Scholar]
- 68.Nicholls, A., K.A. Sharp, and B. Honig. 1991. Protein folding and association: insights from the interfacial and thermodynamic properties of hydrocarbons. Proteins. 11:281–296. [DOI] [PubMed] [Google Scholar]
- 69.Pettersen, E.F., T.D. Goddard, C.C. Huang, G.S. Couch, D.M. Greenblatt, E.C. Meng, and T.E. Ferrin. 2004. UCSF Chimera–a visualization system for exploratory research and analysis. J. Comput. Chem. 25:1605–1612. [DOI] [PubMed] [Google Scholar]
- 70.Swaminathan, G.J., D.E. Holloway, R.A. Colvin, G.K. Campanella, A.C. Papageorgiou, A.D. Luster, and K.R. Acharya. 2003. Crystal structures of oligomeric forms of the IP-10/CXCL10 chemokine. Structure. 11:521–532. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.