

Abstract
Recent studies have demonstrated that the MYB oncogene is frequently duplicated in human T cell acute lymphoblastic leukemia (T-ALL). We find that the human MYB locus is flanked by 257-bp Alu repeats and that the duplication is mediated somatically by homologous recombination between the flanking Alu elements on sister chromatids. Nested long-range PCR analysis indicated a low frequency of homologous recombination leading to MYB tandem duplication in the peripheral blood mononuclear cells of ∼50% of healthy individuals, none of whom had a MYB duplication in the germline. We conclude that Alu-mediated MYB tandem duplication occurs at low frequency during normal thymocyte development and is clonally selected during the molecular pathogenesis of human T-ALL.
T cell acute lymphoblastic leukemia (T-ALL) is a thymocyte malignancy that accounts for 10–15% of childhood and 25% of adult ALL cases. Molecular studies of recurrent chromosomal translocations in T-ALL have indicated a central role for the aberrant expression of transcription factors in the pathobiology of this disease. Dysregulated expression of TAL1, MYC, HOX11, and LMO2, for example, can lead to abnormal proliferation and differentiation arrest of T-lymphoid progenitors (1). More recently, activating mutations of the NOTCH1 gene were identified in >50% of human T-ALL cases (2). NOTCH1 encodes a transmembrane receptor with important functions in T cell development; activation of the receptor leads to proteolytic cleavage and release of the NOTCH intracellular domain, which transits to the nucleus and functions as a component of a transcriptional complex that regulates the expression of MYC and other key target genes (3–5).
The MYB transcription factor is the cellular counterpart of the v-Myb oncogene of the acutely transforming avian myeloblastosis virus, which causes a rapidly fatal monoblastic leukemia in chickens (6, 7). Myb is essential for hematopoietic, as well as T cell, development (8, 9), and when overexpressed in thymocytes, v-Myb causes T-ALL (10). Accordingly, retroviral insertion and transcriptional activation of the Myb locus represents one of the most frequent accelerating events in mouse models of T-ALL (11) (http://rtcgd.abcc.ncifcrf.gov/). In human cancers, MYB is known to be amplified in 2 colon cancer cell lines, 4 glioblastoma cell lines, and 29% of BRCA1-mutated primary breast cancer samples (12–14). MYB duplication and translocation was very recently demonstrated in human T-ALL (15, 16). We show that the most important mechanism underlying MYB copy number alteration in T-ALL occurs somatically by homologous recombination between Alu elements, providing a means by which an evolving T-ALL clone develops an increased dosage of MYB during leukemic transformation.
RESULTS AND DISCUSSION
The MYB gene is tandemly duplicated in human T-ALL
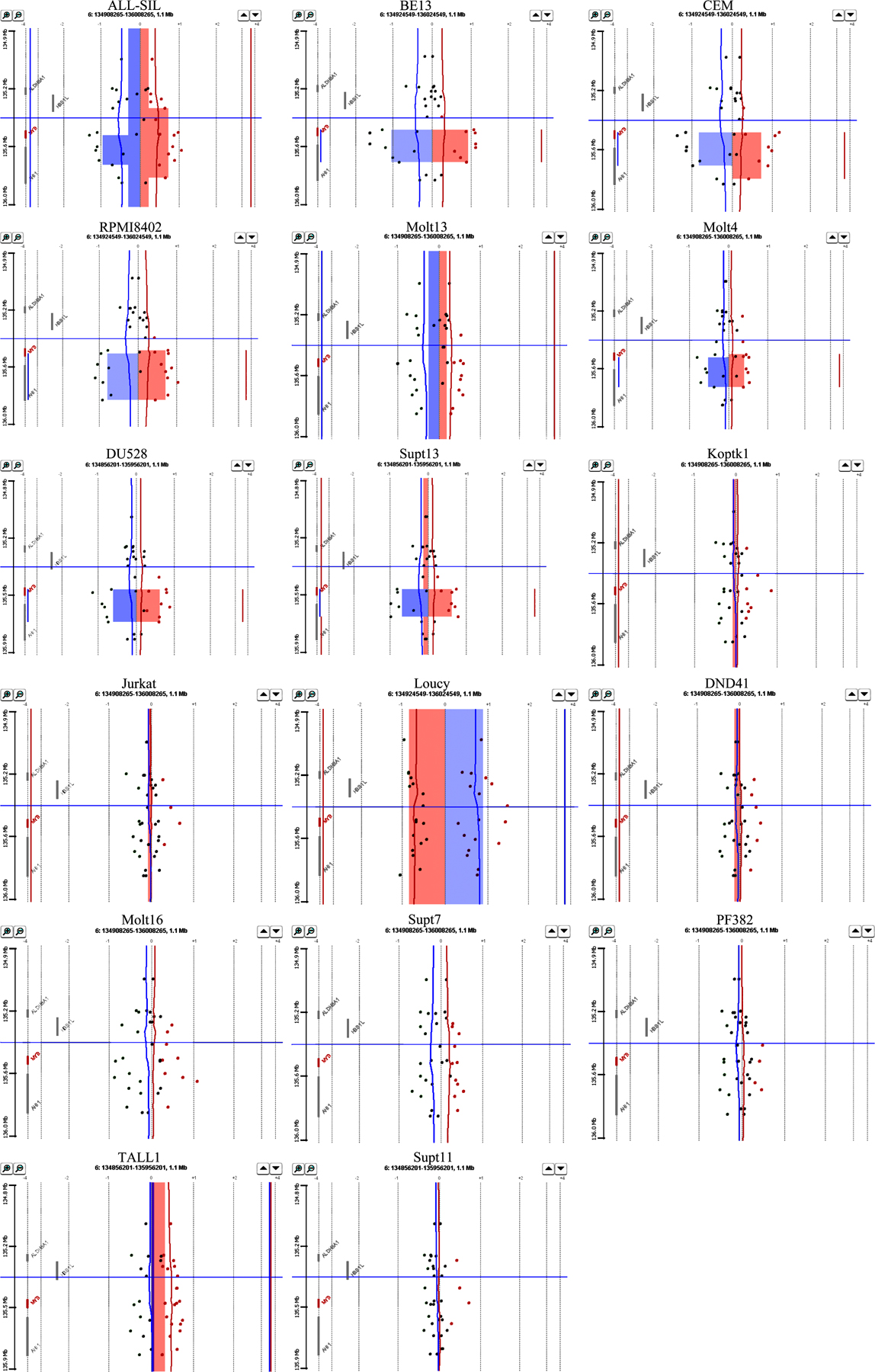
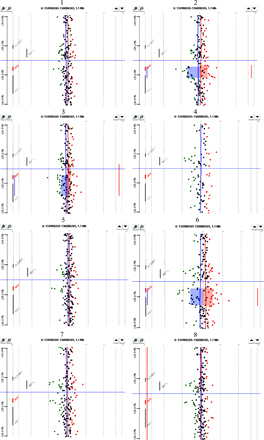
While conducting high-resolution microarray-based comparative genomic hybridization (array CGH) analysis of full-complexity DNA derived from 17 established T-ALL cell lines, as well as bone marrow samples from 8 cases of newly diagnosed T-ALL, we observed a highly localized increase in copy number within the 6q23 region, which encompasses the MYB gene and part of the nearby downstream AHI1 gene in 6 cell lines and 2 clinical samples (Fig. 1, A–C; and Figs. S1 and S2, available at http://www.jem.org/cgi/content/full/jem.20071637/DC1).
Figure 1.

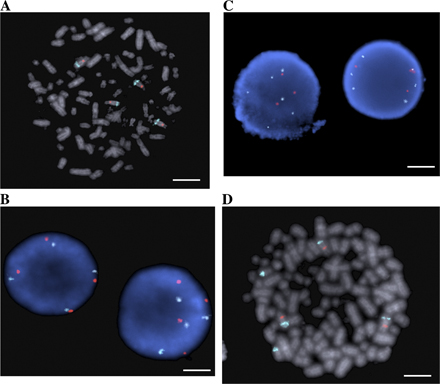
The MYB gene is tandemly duplicated in human T-ALL cell lines and patient samples. (A) Ideogram of chromosome 6 showing location of the MYB gene on q23. (B) Array CGH performed on DNAs from 17 T-ALL cell lines with Human Genome CGH 44K Microarrays (Agilent). A localized region on 6q23 surrounding the MYB locus is shown to have increased copy number in six of the cell lines. A 2.3-Mb region of chromosome 6 is shown. The top is centromeric, and the bottom is telomeric. Part of the AHI1 gene directly downstream of MYB is also amplified. NA indicates a probe in an intergenic region. Red indicates increased copy, blue indicates decreased copy, and the intensity of the color reflects the level of increase or decrease. (C) DNAs from leukemic cells in the diagnostic bone marrow of eight T-ALL patients were similarly analyzed by array CGH using Human Genome CGH 244K Microarrays, with an increased MYB copy number identified in two cases. A 500-kb region of chromosome 6 is shown. (D) Fiber-FISH on a T-ALL (TALL1) cell line with a diploid MYB copy number. The fosmid encompassing most of the MYB gene is labeled in green; a fosmid immediately 3′ of the MYB coding sequence is labeled in red. (E) Fiber-FISH on the Supt13 cell line showing a duplication of both fosmids spanning the entire MYB locus oriented in tandem on the same DNA fiber.
To determine the mechanisms of increased MYB copy number in T-ALL, we used fluorescent in situ hybridization (FISH) with a commercially available probe for MYB. 2 of the 17 T-ALL cell lines examined showed increased copies of MYB relative to the copy number of the chromosome 6 centromere (Molt4 and Molt13; Table I and Fig. S3, available at http://www.jem.org/cgi/content/full/jem.20071637/DC1), representing translocations of chromosome 6q regions to other chromosomes. These cell lines did not show the localized increases in MYB copy number found with array CGH (Fig. 1 B, samples 7 and 8). In fact, none of the cell lines with a discrete increase in MYB copy number had abnormalities affecting this locus by interphase FISH (Fig. S3). Thus, we postulated that the localized increase of MYB copy number observed by array CGH must affect a region adjacent to one of the normal MYB alleles, resulting in a merged signal by FISH on interphase nuclei and on metaphase chromosome spreads. We therefore used fiber-FISH to hybridize 5′ and 3′ MYB fosmid probes onto stretched DNA fibers from interphase cells of seven different T-ALL cell lines. This strategy demonstrated that MYB is tandemly duplicated on one allele in all five human T-ALL cell lines analyzed with array CGH findings of localized increases in MYB copy number (Fig. 1, D and E; and Table I). Thus, the increased MYB copy number in T-ALL that we and others (15, 16) have observed using array CGH is caused by tandem duplication.
Table I.
Summary of results for 17 T-ALL cell lines
| Numbera | Cell line | MYB status by array CGH |
MYB status by MLPA |
Long-range PCR |
Breakpoint subtypeb c |
Interphase FISH | Fiber-FISHc |
|---|---|---|---|---|---|---|---|
| 1 | ALL-SIL | Increased copy | Increased copy | Negative-larger region amplified |
NA | No increase relative to centromere |
Tandem duplication |
| 2 | BE13 | Increased copy | Increased copy | Positive | 1,2,3 | No increase relative to centromere |
Tandem duplication |
| 3 | CEM | Increased copy | Increased copy | Positive | 6 | No increase relative to centromere |
Tandem duplication |
| 4 | RPMI8402 | Increased copy | Increased copy | Positive | 3,4,5 | No increase relative to centromere |
ND |
| 5 | Supt13 | Increased copy | Increased copy | Positive | 2,3 | No increase relative to centromere |
Tandem duplication |
| 6 | DU528 | Increased copy | Increased copy | Positive | 2,3,4,5 | No increase relative to centromere |
ND |
| 7 | Molt4 | Increased copy | Increased copy | Negative | NA | Increased MYB
relative to centromere |
ND |
| 8 | Molt13 | Increased copy | Increased copy | Negative | NA | Increased MYB
relative to centromere |
Tandem duplication |
| 9 | TALL1 | Inconclusive | Deletion | Positive | 4 | MYB Translocation | Normal |
| 10 | PF382 | No increase | No increase | Negative | NA | ND | ND |
| 11 | Supt7 | No increase | No increase | Positive | ND | ND | ND |
| 12 | Molt16 | No increase | Increased copy | Negative | NA | No increase relative to centromere |
ND |
| 13 | DND41 | No increase | Inconclusive | Positive | 1,2,3,4,5 | No increase relative to centromere |
ND |
| 14 | Loucy | 6q deletion | Deletion | Negative | NA | Deletion | ND |
| 15 | Jurkat | No increase | No increase | Negative | NA | No increase relative to centromere |
Normal |
| 16 | Koptk1 | No increase | No increase | Positive | 3,4,5 | No increase relative to centromere |
ND |
| 17 | Supt11 | No increase | No increase | Negative | NA | No increase relative to centromere |
ND |
Numbers correspond to labels in Fig. S1 and Fig. S4 B.
Based on subtypes shown in Fig. 1 D.
ND, not done; NA, not applicable.
Tandem duplication of MYB is mediated by Alu elements flanking the MYB gene
To clone the breakpoints between the duplicated copies of MYB in T-ALL and define the mechanism underlying MYB tandem duplication, we performed long-range PCR. From the duplicated genomic region on chromosome 6, as defined by others (15, 16), and from our own array CGH data on T-ALL cell lines and patient samples (Tables I and II and Fig. 1), we designed a forward primer 54 kb 3′ of the end of the MYB coding sequence and a reverse primer at the 5′ end of the MYB gene (primers 1 and 6; Fig. 2 A). This strategy generated an amplified band of ∼10 kb from T-ALL cell lines (Fig. 2 A). Two more rounds of nested PCR with forward primers progressively 3′ of the MYB gene and reverse primers further 5′ of the MYB gene yielded a 1-kb product that was sequenced (primers 3, 4, 5, 7, and 8; Fig. 2 A). Comparison of the sequence of this product with the normal human genome sequence identified a junction between the duplicated copies of MYB. Analysis of 15 T-ALL samples and cell lines showed that this junction always occurred within a 257-bp region of sequence homology that occurs both 3.5 kb 5′ and 73.5 kb 3′ of MYB. Analysis of this sequence using RepeatMasker (http://www.repeatmasker.org) revealed that it constitutes an Alu element. Alu repeats are short, repetitive DNA sequences of <500 bp that are estimated to account for 10% of the human genome (17). The Alu repeats on either side of the MYB gene are 76% identical, implicating homologous recombination as the underlying mechanism of the tandem duplication (Fig. 2, B and C) (18). Sequence differences between the upstream and downstream Alu elements enabled us to group the junctions of T-ALL cell lines into five classes (Fig. 2 D) (1–5). In each class, the downstream Alu element had merged directly with the upstream Alu region of the duplicated gene in a manner that is most consistent with a synthesis-dependent strand annealing (SDSA) event. In brief, the sequences from the downstream Alu element from one sister chromatid invade the upstream Alu element on the other sister, accompanied by DNA synthesis that proceeds past the downstream homologous sequence. The event is completed by strand annealing of the two ends that now contain identical sequence information (Fig. 3). We demonstrate that Alu elements on either side of the MYB gene mediate its somatic tandem duplication in a significant subset of T-ALL cell lines and patients. Previous reports have identified a larger region of duplication in some samples, which encompasses more than the MYB locus (15, 16). We have also seen that the duplicated region can be larger, and, in these cases, we cannot detect the duplication using our long-range PCR method (e.g., cell line 1 in Fig. 1 B). In these cases, the duplication either occurs by a different mechanism or is mediated by another region of homology.
Table II.
Summary of results for 25 T-ALL patient samples
| Numbera | MYB status by array CGHb | MYB status by MLPA | Long-Range PCR for tandem duplication |
Breakpoint subtypec |
|---|---|---|---|---|
| 1 | No increase | Deletion | Positive | 1, 2, 3, 4 |
| 2 | Increased copy | Increased copy | Negative | NA |
| 3 | Inconclusive | No increase | Positive | ND |
| 4 | No increase | No increase | Negative | NA |
| 5 | No increase | No increase | Positive | ND |
| 6 | Increased copy | Increased copy (not in remission) |
Negative | NA |
| 7 | No increase | No increase | Negative | NA |
| 8 | No increase | No increase | Positive | ND |
| 9 | ND | No increase | Negative | NA |
| 10 | ND | No increase | Positive | 1, 2, 3, 4 |
| 11 | ND | No increase | Negative | NA |
| 12 | ND | No increase | Positive | 5 |
| 13 | ND | No increase | Positive | 1, 2, 3, 4, 5 |
| 14 | ND | Increased copy (not in remission) |
Positive | 1, 2, 3, 4 |
| 15 | ND | No increase | Negative | NA |
| 16 | ND | No increase | Negative | NA |
| 17 | ND | No increase | Positive | 1,2,3 |
| 18 | ND | ND | Negative | NA |
| 19 | ND | ND | Positive | ND |
| 20 | ND | ND | Negative | NA |
| 21 | ND | No increase | Negative | NA |
| 22 | ND | ND | Positive | 1, 2, 3, 4, 5 |
| 23 | ND | No increase | Negative | NA |
| 24 | ND | Increased copy | Negative | NA |
| 25 | ND | Increased copy | Negative | NA |
Samples 1–8 correspond to samples 1–8 in Fig. S4 C.
ND, not done; NA, not applicable.
Based on subtypes shown in Fig. 1 D.
Figure 2.

MYB duplication is mediated by Alu elements on chromosome 6. (A) Detection of the MYB tandem duplication by long-range PCR. Primers were designed based on the fosmids used in the fiber-FISH experiments (Fig. S3). Primers 1, 6, and 7 were used in long-range PCR to detect the MYB duplication. Primer 2 is a control primer for long-range PCR. Primers 3, 4, 5, and 8 are nested primers used to generate a smaller product that was sequenced. Arrows inside boxes indicate Alu elements and their orientation. Insets show PCR results on representative cell lines. (left) Primers 1 and 6 were used in reaction 1; primers 1 and 7 were used in reaction 2; and primers 1 and 2 were used in reaction 3. (right) After three rounds of nested PCR, a band of 1 kb (reaction 3) was obtained in the CEM cell line, but not in normal thymocyte DNA. Primers 3 and 8 were used in reaction 1; primers 4 and 8 in reaction 2; and primers 5 and 8 in reaction 3. (B) Sequence in upper case is 116 kb downstream of MYB in the AHI1 locus. (C) Sequence in lower case is 3.5 kb upstream of MYB. (D) Sequences 1–5 represent the 5 different junctions between the 2 copies of MYB detected in T-ALL cell lines and primary samples. One additional breakpoint (sequence 6) was identified in normal lymphoblastoid cell line DNA. Underlined sequences represent the proposed crossover region. Fig. S3 is available at http://www.jem.org/cgi/content/full/jem.20071637/DC1.
Figure 3.

Proposed mechanism of MYB tandem duplication. (A) A DNA double-strand break occurs in (or near) the Alu element downstream of MYB. (B) The broken DNA strand invades the sister chromatid and pairs with the Alu element upstream of MYB. (C) DNA repair synthesis continues through the MYB gene, and the newly synthesized strand anneals to the broken chromatid by strand annealing. (D) The result is duplication of the sequence between the Alu elements. The sequence of the Alu element between the two copies of MYB (gray box) is a hybrid of the two repeats on either side of MYB, depending on where the strand anneals.
As shown in Table I, we detected MYB tandem duplication by long-range PCR, but not by multiplex ligation-dependent probe amplification (MLPA) or array CGH in 4 T-ALL cell lines: TALL1, Supt7, DND41, and Koptk1. We also detected MYB tandem duplication by long-range PCR in 11 of 25 (44%) primary T-ALL samples (Table II), which is a higher frequency than that found by MLPA in these samples. Thus, nested long-range PCR detects the tandem duplication in a minority of cells, whereas MLPA and array CGH are quantitative assays that detect MYB tandem duplication only when it occurs in a majority of the cells. Collectively, our results indicate that MYB tandem duplication occurs relatively often in T-ALL patients, but is clonally selected in only approximately one fifth of the cases during evolution of the disease, presumably because some malignant clones have increased MYB copies that have translocated to other chromosomes (as in Molt4 and Molt13; Table I and Fig. S3, available at http://www.jem.org/cgi/content/full/jem.20071637/DC1) or already express sufficient MYB protein levels by virtue of their differentiation state to optimally synergize with other oncogenic events, such as mutation of the NOTCH1 gene (Fig. S4). This conclusion is supported by MLPA analysis showing the absence of MYB tandem duplication in remission samples from two patients whose malignant clones were positive for MYB duplication at diagnosis (Table II).
MYB tandem duplication in normal lymphocytes and peripheral blood cells
To determine whether MYB duplication occurs in normal lymphocytes, we performed long-range PCR on 96 DNA samples extracted from lymphoblastoid cell lines from the HapMap panel of individuals. We detected a band representing MYB tandem duplication in 45 of 96 samples (46.9%), and sequencing of all the products revealed that the breakpoints in normal samples were of the 5 types previously identified in T-ALL cell lines (Fig. 2 D), with the identification of one additional breakpoint (no. 6). Further analysis with MLPA did not detect an increase in MYB copy number in any of the 36 HapMap lymphoblastoid cell lines that were analyzed, implying that the duplication occurs infrequently, if at all, as a germline copy number variation, but rather is a somatic event affecting a small fraction of normal lymphocytes. To determine whether RAG expression is necessary for Alu-mediated MYB duplication, we have analyzed the AML cell lines HL60 and Meg01. We have detected the MYB gene duplication in these two lines, and sequencing of the PCR products revealed that the duplication was Alu-mediated, as in the T-ALL cell lines and samples. Therefore, RAG expression is not required for Alu-mediated MYB gene duplication.
We also analyzed 10 peripheral blood and 37 buccal swab samples from healthy individuals for the MYB tandem duplication by long-range PCR and MLPA. We detected the duplication in 2 of 10 blood samples and none of 37 buccal samples by long-range PCR. However, none of the blood or buccal samples was positive for MYB tandem duplication by MLPA. These results demonstrate that MYB tandem duplication occurs spontaneously in normal blood cells and is not an artifact of generating lymphoblastoid cell lines from normal cells. They also indicate that circulating blood mononuclear cells are more prone than epithelial cells to undergo MYB tandem duplication through Alu-mediated homologous recombination (Fisher's exact test, P = 0.042). Blood cells may be more likely to undergo this recombination event because they are more prone to double-strand breaks that initiate the Alu-mediated recombination.
Recent studies in mice have demonstrated that distinct levels of MYB are required at different stages of hematopoietic development for differentiation and proliferation (19, 20). Therefore, it is not surprising that a relatively modest increase in MYB expression, produced as a result of tandem duplication or translocation of a single copy, would be clonally selected in some T-ALLs during their molecular pathogenesis. Intriguingly, in several of our primary T-ALL samples with evidence by nested long-range PCR of rare cells with MYB tandem duplication, this subset of cells did not have a growth advantage, and the MYB copy number of the majority of cells was normal by CGH and MLPA. Thus, if a particular T-ALL clone already expresses sufficiently high levels of MYB for transformation because of its differentiation state, then the increased levels provided by MYB duplication may not provide an advantage for clonal selection.
We and others (15, 16) have observed that MYB duplication occurs often in T-ALL cases with activating mutations in NOTCH1. Knockout studies in the mouse, as well as MYB small interfering RNA experiments in T-ALL cell lines, demonstrated that MYB plays an important role in T cell differentiation (15, 21–23). Furthermore, γ-secretase inhibitors were shown to synergize with MYB small interfering RNA to inhibit T-ALL cell growth (15). These results are consistent with studies in the zebrafish showing that Notch and Myb act in parallel pathways to specify hematopoietic stem cell fate in the AGM (24) and indicate that NOTCH activation and MYB duplication synergize not only in normal blood cell development, but also in malignant T cell transformation.
Our study introduces a new mechanism by which an evolving T-ALL clone can increase the dosage of a critical oncogene during leukemic transformation. Homologous recombination between Alu elements in meiotic germ cells leading to deletions and duplications are responsible for several genetic diseases, including glycogen storage disease type II, Lesch-Nyhan syndrome, and Ehler-Danlos syndrome (25). Alu elements have also been implicated in mediating somatic genetic alterations in cancer, such as MLL translocations and partial tandem duplications in acute myelogenous leukemia (26, 27), BRCA1 gene deletions in breast cancer (28), and TRE rearrangement in Ewing's sarcoma (29). Alu elements have also been found near the LMO2 and LCK chromosomal breakpoints in T-ALL (30, 31), suggesting that they may also play role in these chromosomal translocations. Because Alu elements make up a large percentage of the human genome, it is likely that the copy number of many other oncogenes might be increased, or tumor suppressors deleted, through the recombination mechanism that we describe. The emergence of technologies for global comparative genomic hybridization and single-nucleotide polymorphism analysis in human cancers should enable efficient detection of these pathogenic copy number alterations, especially when the analyses are paired with analysis of normal germline DNA from the same individual.
MATERIALS AND METHODS
Patient materials.
Cryopreserved lymphoblasts or lymphoblast lysates ALL were collected with informed consent and institutional review board approval from patients with newly diagnosed T-ALL who were treated with Dana-Farber Cancer Institute Acute Lymphoblastic Leukemia Consortium protocols 95-001 or 00-001. Genomic DNA was extracted with the PUREGENE kit according to the manufacturer's protocol (Gentra Systems).
Microarray-based CGH.
Genomic DNA was fragmented and random-prime labeled, as previously described (32). Labeled DNAs were hybridized to microarrays containing 44K (cell lines) or 244K (primary samples) 60-mer oligonucleotides (Agilent Technologies), as previously described (33). Microarray data have been deposited in the GEO database under accession no. GSE7615.
FISH.
FISH was performed with a commercially available probe mix containing a MYB (6q23) probe labeled with spectrum aqua and a centromeric probe for chromosome 6 labeled with spectrum orange (Vysis, Inc.). Slides and probe were co-denatured at 75°C, hybridized overnight at 37°C, washed, counterstained with 4′,6-diamidino-2-phenylindole, and analyzed under a fluorescence microscope (Carl Zeiss, Inc.). For each cell line, a minimum of 100 nuclei and any identifiable metaphases were scored. The orange chromosome 6 signals and the aqua LSI MYB signals were counted simultaneously in each nucleus/metaphase. MYB was considered to be amplified if the cells showed more aqua signals than orange signals.
Fiber-FISH.
Cells were lysed overnight using PUREGENE Cell Lysis Solution (Gentra Systems). The DNA was stretched on poly-l-lysine–coated slides, and the DNA fibers were fixed in 100% ethanol. To generate FISH probes, fosmids G248P89100B2 and G248P89828A1 were labeled with digoxigenin and biotin, respectively, using BioPrime Array CGH Labeling Module (Invitrogen). DNA fibers and labeled FISH probes were co-denatured for 3 min at 95°C and hybridized overnight in a humidified chamber at 37°C. The haptens were detected using anti–digoxigenin-fluorescein/Fab fragments (Roche) and Strepavidin/Alexa Fluor 594 conjugate (Invitrogen). Slides were analyzed under a fluorescence microscope (model BX51AI; Olympus). A normal allele showed green dots followed by red dots. A tandem duplication was seen as a direct repeat of the normal signal pattern.
MLPA.
MLPA probe mix containing all probes was added to 150 ng denatured genomic DNA. A universal primer pair was used to amplify all ligated probes in a multiplex PCR assay. The PCR product was analyzed by sequence-type electrophoresis and samples were compared with two control samples. A difference in relative peak area indicated a copy number change of the DNA sequence targeted by the probe. A probe was considered amplified if the ratio sample/control was >1.3.
Long-range PCR.
Reactions were performed with 500 ng of genomic DNA and the LA PCR kit version 2.1 (Takara Mirus Bio) according to the manufacturer's specifications. Primer sequences are available upon request.
Online supplemental material.
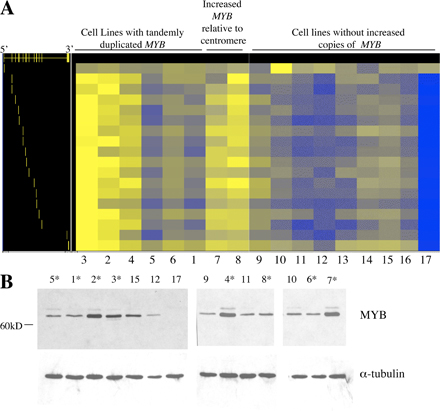
Figs. S1 and S2 show array CGH data on T-ALL cell lines and patient samples analyzed by CGH Analytics (Agilent Technologies). Fig. S3 shows MYB FISH on T-ALL cell lines. Lines with tandemly duplicated MYB do not show increased copies of MYB by this method; however, two T-ALL cell lines with extra copies of MYB dispersed to other chromosomes were identified. Fig. S4 shows that MYB duplication results in increased levels of MYB RNA and protein levels in T-ALL cell lines. The online version of this article is available at http://www.jem.org/cgi/content/full/jem.20071637/DC1.
Supplemental Material
Acknowledgments
We would like to thank James Haber for helpful discussions and John Gilbert for editing and general comments.
This work was performed and supported in part by the Center for Applied Cancer Science of the Robert A. and Renee E. Belfer Foundation Institute for Innovative Cancer Science, a National Cancer Institute grant CA11560, and a Leukemia and Lymphoma Society Translational grant 6161-05 (C. Lee), National Institutes of Health (NIH) grants CA109901 and CA68484-11 (A.T. Look), and a German Research Foundation Fellowship Award Tc-57/1-1 (J. Tchinda). J. O'Neil is supported by a Harvard Medical School Hematology Training Grant. R.A. DePinho is an American Cancer Society Research Professor and an Ellison Medical Foundation Senior Scholar. R.S. Maser is a Damon-Runyon Postdoctoral Fellow. R. Rothstein is supported by NIH grants GM050237, GM067055, and CA115853.
The authors have no conflicting financial interests.
J. O'Neil and J. Tchinda and C. Lee and A.T. Look contributed equally to this paper.
References
- 1.Look, A.T. 1997. Oncogenic transcription factors in the human acute leukemias. Science. 278:1059–1064. [DOI] [PubMed] [Google Scholar]
- 2.Weng, A.P., A.A. Ferrando, W. Lee, J.P. Morris, L.B. Silverman, C. Sanchez-Irizarry, S.C. Blacklow, A.T. Look, and J.C. Aster. 2004. Activating mutations of NOTCH1 in human T cell acute lymphoblastic leukemia. Science. 306:269–271. [DOI] [PubMed] [Google Scholar]
- 3.Sharma, V.M., J.A. Calvo, K.M. Draheim, L.A. Cunningham, N. Hermance, L. Beverly, V. Krishnamoorthy, M. Bhasin, A.J. Capobianco, and M.A. Kelliher. 2006. Notch1 contributes to mouse T-cell leukemia by directly inducing the expression of c-myc. Mol. Cell. Biol. 26:8022–8031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Weng, A.P., J.M. Millholland, Y. Yashiro-Ohtani, M.L. Arcangeli, A. Lau, C. Wai, C. Del Bianco, C.G. Rodriguez, H. Sai, J. Tobias, et al. 2006. c-Myc is an important direct target of Notch1 in T-cell acute lymphoblastic leukemia/lymphoma. Genes Dev. 20:2096–2109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Palomero, T., W.K. Lim, D.T. Odom, M.L. Sulis, P.J. Real, A. Margolin, K.C. Barnes, J. O'Neil, D. Neuberg, A.P. Weng, et al. 2006. NOTCH1 directly regulates c-MYC and activates a feed-forward-loop transcriptional network promoting leukemic cell growth. Proc. Natl. Acad. Sci. USA. 103:18261–18266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Dalla-Favera, R., G. Franchini, S. Martinotti, F. Wong-Staal, R.C. Gallo, and C.M. Croce. 1982. Chromosomal assignment of the human homologues of feline sarcoma virus and avian myeloblastosis virus onc genes. Proc. Natl. Acad. Sci. USA. 79:4714–4717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Klempnauer, K.H., T.J. Gonda, and J.M. Bishop. 1982. Nucleotide sequence of the retroviral leukemia gene v-myb and its cellular progenitor c-myb: the architecture of a transduced oncogene. Cell. 31:453–463. [DOI] [PubMed] [Google Scholar]
- 8.Clarke, M.F., J.F. Kukowska-Latallo, E. Westin, M. Smith, and E.V. Prochownik. 1988. Constitutive expression of a c-myb cDNA blocks Friend murine erythroleukemia cell differentiation. Mol. Cell. Biol. 8:884–892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Pearson, R., and K. Weston. 2000. c-Myb regulates the proliferation of immature thymocytes following beta-selection. EMBO J. 19:6112–6120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Badiani, P.A., D. Kioussis, D.M. Swirsky, I.A. Lampert, and K. Weston. 1996. T-cell lymphomas in v-Myb transgenic mice. Oncogene. 13:2205–2212. [PubMed] [Google Scholar]
- 11.Lund, A.H., G. Turner, A. Trubetskoy, E. Verhoeven, E. Wientjens, D. Hulsman, R. Russell, R.A. DePinho, J. Lenz, and M. van Lohuizen. 2002. Genome-wide retroviral insertional tagging of genes involved in cancer in Cdkn2a-deficient mice. Nat. Genet. 32:160–165. [DOI] [PubMed] [Google Scholar]
- 12.Alitalo, K., R. Winqvist, C.C. Lin, A. de la Chapelle, M. Schwab, and J.M. Bishop. 1984. Aberrant expression of an amplified c-myb oncogene in two cell lines from a colon carcinoma. Proc. Natl. Acad. Sci. USA. 81:4534–4538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Welter, C., W. Henn, B. Theisinger, H. Fischer, K.D. Zang, and N. Blin. 1990. The cellular myb oncogene is amplified, rearranged and activated in human glioblastoma cell lines. Cancer Lett. 52:57–62. [DOI] [PubMed] [Google Scholar]
- 14.Kauraniemi, P., I. Hedenfalk, K. Persson, D.J. Duggan, M. Tanner, O. Johannsson, H. Olsson, J.M. Trent, J. Isola, and A. Borg. 2000. MYB oncogene amplification in hereditary BRCA1 breast cancer. Cancer Res. 60:5323–5328. [PubMed] [Google Scholar]
- 15.Lahortiga, I., K. De Keersmaecker, P. Van Vlierberghe, C. Graux, B. Cauwelier, F. Lambert, N. Mentens, H.B. Beverloo, R. Pieters, F. Speleman, et al. 2007. Duplication of the MYB oncogene in T cell acute lymphoblastic leukemia. Nat. Genet. 39:593–595. [DOI] [PubMed] [Google Scholar]
- 16.Clappier, E., W. Cuccuini, A. Kalota, A. Crinquette, J.M. Cayuela, W.A. Dik, A.W. Langerak, B. Montpellier, B. Nadel, P. Walrafen, et al. 2007. The C-MYB locus is involved in chromosomal translocation and genomic duplications in human T-cell acute leukemia (T-ALL) - the translocation defining a new T-ALL subtype in very young children. Blood. 110:1251–1261. [DOI] [PubMed] [Google Scholar]
- 17.Batzer, M.A., and P.L. Deininger. 2002. Alu repeats and human genomic diversity. Nat. Rev. Genet. 3:370–379. [DOI] [PubMed] [Google Scholar]
- 18.Weinstock, D.M., C.A. Richardson, B. Elliott, and M. Jasin. 2006. Modeling oncogenic translocations: distinct roles for double-strand break repair pathways in translocation formation in mammalian cells. DNA Repair (Amst.). 5:1065–1074. [DOI] [PubMed] [Google Scholar]
- 19.Sakamoto, H., G. Dai, K. Tsujino, K. Hashimoto, X. Huang, T. Fujimoto, M. Mucenski, J. Frampton, and M. Ogawa. 2006. Proper levels of c-Myb are discretely defined at distinct steps of hematopoietic cell development. Blood. 108:896–903. [DOI] [PubMed] [Google Scholar]
- 20.Emambokus, N., A. Vegiopoulos, B. Harman, E. Jenkinson, G. Anderson, and J. Frampton. 2003. Progression through key stages of haemopoiesis is dependent on distinct threshold levels of c-Myb. EMBO J. 22:4478–4488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lieu, Y.K., A. Kumar, A.G. Pajerowski, T.J. Rogers, and E.P. Reddy. 2004. Requirement of c-myb in T cell development and in mature T cell function. Proc. Natl. Acad. Sci. USA. 101:14853–14858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Allen, R.D. III, T.P. Bender, and G. Siu. 1999. c-Myb is essential for early T cell development. Genes Dev. 13:1073–1078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bender, T.P., C.S. Kremer, M. Kraus, T. Buch, and K. Rajewsky. 2004. Critical functions for c-Myb at three checkpoints during thymocyte development. Nat. Immunol. 5:721–729. [DOI] [PubMed] [Google Scholar]
- 24.Burns, C.E., D. Traver, E. Mayhall, J.L. Shepard, and L.I. Zon. 2005. Hematopoietic stem cell fate is established by the Notch-Runx pathway. Genes Dev. 19:2331–2342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Deininger, P.L., and M.A. Batzer. 1999. Alu repeats and human disease. Mol. Genet. Metab. 67:183–193. [DOI] [PubMed] [Google Scholar]
- 26.Zhang, Y., N. Zeleznik-Le, N. Emmanuel, N. Jayathilaka, J. Chen, P. Strissel, R. Strick, L. Li, M.B. Neilly, T. Taki, et al. 2004. Characterization of genomic breakpoints in MLL and CBP in leukemia patients with t(11;16). Genes Chromosomes Cancer. 41:257–265. [DOI] [PubMed] [Google Scholar]
- 27.Strout, M.P., G. Marcucci, C.D. Bloomfield, and M.A. Caligiuri. 1998. The partial tandem duplication of ALL1 (MLL) is consistently generated by Alu-mediated homologous recombination in acute myeloid leukemia. Proc. Natl. Acad. Sci. USA. 95:2390–2395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Puget, N., O.M. Sinilnikova, D. Stoppa-Lyonnet, C. Audoynaud, S. Pages, H.T. Lynch, D. Goldgar, G.M. Lenoir, and S. Mazoyer. 1999. An Alu-mediated 6-kb duplication in the BRCA1 gene: a new founder mutation? Am. J. Hum. Genet. 64:300–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Onno, M., T. Nakamura, J. Hillova, and M. Hill. 1992. Rearrangement of the human tre oncogene by homologous recombination between Alu repeats of nucleotide sequences from two different chromosomes. Oncogene. 7:2519–2523. [PubMed] [Google Scholar]
- 30.Dong, W.F., Y. Xu, Q.L. Hu, D. Munroe, J. Minowada, D.E. Housman, and M.D. Minden. 1995. Molecular characterization of a chromosome translocation breakpoint t(11;14)(p13;q11) from the cell line KOPT-K1. Leukemia. 9:1812–1817. [PubMed] [Google Scholar]
- 31.Tycko, B., S.D. Smith, and J. Sklar. 1991. Chromosomal translocations joining LCK and TCRB loci in human T cell leukemia. J. Exp. Med. 174:867–873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Brennan, C., Y. Zhang, C. Leo, B. Feng, C. Cauwels, A.J. Aguirre, M. Kim, A. Protopopov, and L. Chin. 2004. High-resolution global profiling of genomic alterations with long oligonucleotide microarray. Cancer Res. 64:4744–4748. [DOI] [PubMed] [Google Scholar]
- 33.Maser, R.S., B. Choudhury, P.J. Campbell, B. Feng, K.K. Wong, A. Protopopov, J. O'Neil, A. Gutierrez, E. Ivanova, I. Perna, et al. 2007. Chromosomally unstable mouse tumours have genomic alterations similar to diverse human cancers. Nature. 447:966–971. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}