

Abstract
The spatiotemporal characteristics of the Ca2+ release process in mouse skeletal muscle were investigated in enzymatically dissociated fibers from flexor digitorum brevis (FDB) muscles, using a custom-made two-photon microscope with laser scanning imaging (TPLSM) and spot detection capabilities. A two-microelectrode configuration was used to electrically stimulate the muscle fibers, to record action potentials (APs), and to control their myoplasmic composition. We used 125 μM of the low-affinity Ca2+ indicator Oregon green 488 BAPTA-5N (OGB-5N), and 5 or 10 mM of the Ca2+ chelator EGTA (pCa 7) in order to arrest fiber contraction and to constrain changes in the [Ca2+] close to the release sites. Image and spot data showed that the resting distribution of OGB-5N fluorescence was homogeneous along the fiber, except for narrow peaks (∼23% above the bulk fluorescence) centered at the Z-lines, as evidenced by their nonoverlapping localization with respect to di-8-ANEPPS staining of the transverse tubules (T-tubules). Using spot detection, localized Ca2+ transients evoked by AP stimulation were recorded from adjacent longitudinal positions 100 nm apart. The largest and fastest ΔF/F transients were detected at sites flanking the Z-lines and colocalized with T-tubules; the smallest and slowest were detected at the M-line, whereas transients at the Z-line showed intermediate features. Three-dimensional reconstructions demonstrate the creation of two AP-evoked Ca2+ release domains per sarcomere, which flank the Z-line and colocalize with T-tubules. In the presence of 10 mM intracellular EGTA, these domains are formed in ∼1.4 ms and dissipate within ∼4 ms, after the peak of the AP. Their full-width at half-maximum (FWHM), measured at the time that Ca2+ transients peaked at T-tubule locations, was 0.62 μm, similar to the 0.61 μm measured for di-8-ANEPPS profiles. Both these values exceed the limit of resolution of the optical system, but their similarity suggests that at high [EGTA] the Ca2+ domains in adult mammalian muscle fibers are confined to Ca2+ release sites located at the junctional sarcoplasmic reticulum (SR).
INTRODUCTION
It is generally accepted that skeletal muscle type excitation–contraction coupling involves complex mechanisms by which Ca2+ ions are released from the terminal cisternae of the SR in response to an action potential (AP) stimulation. It is currently believed that voltage transduction at the dihydropyridine receptors, located at the transverse tubule (T-tubule) membranes leads to the activation of the RyRs, located at junctional membranes of the SR, by means of a direct interaction between both channels (Rios and Pizarro, 1991). The resulting increase in free Ca2+ concentration ([Ca2+]) is in turn sensed by the regulatory protein troponin and this serves as a signal for activation of the contractile proteins.
In both frog and mammalian skeletal muscle, the amplitude and time course of AP-evoked average increases in [Ca2+] occurring throughout multisarcomeric regions of a muscle fiber (global Ca2+ transients) have been mostly measured using fluorescence microscopy and Ca2+ indicators (Vergara et al., 1991; Vergara and DiFranco, 1992; Delbono and Stefani, 1993; Kim and Vergara, 1998a; Baylor and Hollingworth, 2003; Woods et al., 2004; Woods et al., 2005). However, due to their inherent mix of fluorescence contributions from different regions of the sarcomere, the properties of global transients do not provide information about the existence of intrasarcomeric [Ca2+] gradients. Detailed studies about the spatiotemporal distribution of [Ca2+] changes within a sarcomere of frog skeletal muscle fibers was obtained for the first time with a confocal spot detection technique that demonstrated that Ca2+ sources were colocalized with the Z-lines (Escobar et al., 1994) where the triads, structural assemblies consisting of one T-tubule and two SR terminal cisternae, are located in this preparation (Peachey, 1965; Franzini-Armstrong, 1972). Subsequent refinements of the spot detection methodology allowing to record localized transients at consecutive positions 200 nm apart with spatial and temporal resolutions of 0.3 μm and 30 μs, respectively, confirmed the suggestion (Escobar et al., 1994) that in frog fibers the Ca2+ release sources, though centered at the Z-lines, were broadly distributed over a band that could comprise the entire length of the terminal cisternae of the SR (Vergara et al., 2001; DiFranco et al., 2002; Novo et al., 2003).
Comparable studies of the Ca2+ release domains in mammalian muscle, though only preliminarily reported (Vergara, J.L., M. DiFranco, and D. Novo. 2002. Biophys. J. 82:642a), are extremely important since there are structural and molecular differences between frog and mammalian fibers that might entail disparities in the Ca2+ release process. Electron microscopical evidence has shown that in unstretched mammalian fibers the triads are located at both sides of the Z-line at the A-I junction of the sarcomere (Revel, 1962; Franzini-Armstrong et al., 1988; Dulhunty, 1989). This evidence has been confirmed by in vivo fluorescence imaging of mammalian skeletal muscle fibers stained with styryl dyes (e.g., di-8-ANEPPS or RH-414), which show double rows of T-tubules per sarcomere as a distinctive feature (Lannergren et al., 1999; Woods et al., 2005; DiFranco et al., 2006), in contrast with the single row distribution in frog fibers (Escobar et al., 1994; Krolenko et al., 1995; Kim and Vergara, 1998a,b; Lannergren et al., 1999). In addition, mammalian muscle expresses two isoforms of the ryanodine receptor (RyR1 and RyR3). It has been reported that adult skeletal muscle displays a low RyR3/RyR1 ratio (Giannini et al., 1995; Flucher and Franzini-Armstrong, 1996); so it is believed that the Ca2+ release process depends mainly on the activity of RyR1, which is localized at junctional positions of the triads (Block et al., 1988; Flucher and Franzini-Armstrong, 1996; Franzini-Armstrong and Protasi, 1997). This is contrary to the case of frog skeletal muscle, where similar amounts of the RyR isoforms α and β (equivalent to RyR1 and RyR3, respectively) are found, and the β isoform is distributed in extrajunctional regions of the triads (Felder and Franzini-Armstrong, 2002).
The primary goal of the current experiments is to investigate the spatiotemporal properties of the AP-evoked Ca2+ release domains in mouse fast twitch skeletal fibers. To this end, we built a custom-made two-photon laser system with laser scanning imaging (Denk et al., 1990; Centonze and White, 1998; Wier et al., 2000; Nguyen et al., 2001) and stage scanning (spot detection) capabilities (Vergara et al., 2001; DiFranco et al., 2002).
Two-photon excitation of fluorescent molecules, achieved by using femtosecond pulsed lasers, provides an inherent optical sectioning that minimizes out-of-focus fluorescence contributions (Denk et al., 1990). We used the low-affinity calcium indicator Oregon green 488 BAPTA-5N (OGB-5N), which affords minimal distortion of the kinetic features of the localized Ca2+ transients (DiGregorio and Vergara, 1997; DiGregorio et al., 1999; DiFranco et al., 2002). In addition, by labeling the T-tubules with the fluorescent indicator di-8-ANEPPS (Kim and Vergara, 1998a,b; Woods et al., 2005), we mapped the sarcomeric distribution of Ca2+ release domains to that of the T-tubules. One of the main findings of this work is that, in AP stimulated fibers, two Ca2+ release domains per sarcomere can be observed, which flank the Z-lines, and are confined to the immediate surrounding of the junctional SR.
MATERIALS AND METHODS
Isolation of Muscle Fibers
Animals were handled according to the guidelines laid out by the local UCLA Animal Care Committee. Muscle fibers from the fast twitch muscle flexor digitorum brevis (FDB) of C57BL mice were enzymatically dissociated as described previously (Woods et al., 2004, 2005), but using 1,500 U/ml of collagenase IV (Worthington), instead of 262 U/ml of collagenase IV (Sigma-Aldrich), and transferred to custom-made, disposable, optical chambers (DiFranco et al., 2005). The average sarcomere length of the fibers was 2.36 ± 0.1 μm.
Solutions
The K-internal solution contained (in mM) 140 K-aspartate, 20 K-MOPS, 5 MgCl2, 5 Na2 creatine phosphate, 5 ATP-K2, 5 glucose, 5 glutathione, 5 or 10 K-EGTA, and 0.1 mg/ml creatine phosphokinase. Ca(OH)2 was added at a 2:1 ratio of total EGTA:total Ca2+ to yield an approximate [Ca2+] of 70 nM (Nagerl et al., 2000). The free [Mg2+] was calculated, using the Maxchelator program (Bers et al., 1994), to be 0.64 mM.
The external Tyrode solution contained (in mM) 150 NaCl, 2.5 KCl, 2 CaCl2, 1 MgCl2, 10 Na-MOPS, and 10 dextrose. N-benzyl-p-toluene sulphonamide (BTS), an inhibitor of the myosin II ATPase (Cheung et al., 2002), was added to the external solution at a concentration of 100 μM to prevent movement (Woods et al., 2004) when the [EGTA] in the internal solution was 5 mM.
All solutions had an osmolarity of ∼300 mOsmols, and the pH was adjusted to 7.2. Experiments were performed at 22°C. Chemicals were purchased from Sigma-Aldrich; calcium and potentiometric dyes were from Molecular Probes.
Two-photon Microscopy Setup
A schematic diagram of the two-photon laser system, based on an inverted epifluorescence microscope (model IX 71; Olympus America Inc.) is illustrated in Fig. 1. For two-photon measurements, the microscope was equipped with a Ti:Sapphire tunable laser (TiS; Chameleon; Coherent Inc.), an electro-optical modulator (EOM) with a liquid crystal controller (model 932; Newport Corporation) that permitted the beam intensity to vary with voltage commands, a scan head (SCH) with a set of xy orthogonal galvanometer-drivers with 3-mm dielectric coating mirrors (model 6210HB/67321H-1; Cambridge Technology Inc.), a scan lens (ScL) of 30-mm focal distance, and a tube lens (TL) of 75-mm focal distance (AC254-030B and AC254-075B, near IR achromat doublets; Thorlabs Inc.). The laser beam went through a lateral aperture in the fluorescence cube turret, was reflected by a 675DCSPXR dichroic mirror (DC; Chroma Technology Corp.) mounted in a custom-made cube (Olympus IX2-MFB-SP-R), and centered on the pupil of a 100× high-NA oil-immersion objective (OB; UPlanSApo 100×, 1.4 NA; Olympus), which focused it to a ∼0.5-μm spot on the preparation.
Figure 1.

Schematic diagram of the optical system for two-photon laser scanning imaging and two-photon spot detection. The blue line represents the white light path for bright-field illumination. The red line shows the path of the Ti:Sapphire tunable laser beam (TiS); its intensity is controlled with an electro optical modulator (EOM). In addition, the scan head (SCH), consisting of two orthogonal (xy) galvanometer-based scanners with infrared mirrors, projects the beam through a scan lens (ScL) and a tube lens (TL) to a lateral aperture in the cube turret of the microscope under shutter (SH) control. The green line depicts the emitted fluorescence path. There are two detectors that could be selected with a mirror (M) attached to a slider (S). Emission filters (EF) are in front of each detector. A CCD camera was used for acquisition of bright-field and TPLSM images. In two-photon spot detection mode, fluorescence was detected by an avalanche photodiode (APD) while the laser beam was maintained at a stationary position by the SCH.
The experimental chamber and the microelectrode head stages were mounted on a custom-made microscope stage as described previously (Vergara et al., 2001; DiFranco et al., 2002). For high-resolution displacements in two-photon spot detection (TPSpD) experiments, two nanotranslators (model TSE-820; EXFO Burleigh Instruments Inc.), driven by inchworm motors with a closed loop integral linear encoder, permitted fine positioning of the muscle fiber in the X and Y directions with 20-nm accuracy with respect to the stationary laser beam. A stepper motor (Z-axis; Prairie Technologies) was used for driving the focusing mechanism of the microscope (Z-axis) with 200 nm resolution. The exposure time of the Ti:Sapphire excitation was controlled with a shutter (SH) in the laser beam path. The long axis of the muscle fiber was aligned parallel to the X-axis of the stage. The fluorescence emission was detected with a cooled avalanche photodiode (APD, active area = 1 mm). The photo-current was amplified by a PhotoMax 200 (Dagan Corporation), low-pass filtered at 1 kHz with a 4-pole Bessel filter, and digitized at 33.33 kHz using a 16-bit acquisition board (AT-MIO-16XE-10; National Instruments).
For laser scanning imaging (TPLSM), the specimen remained stationary and the Ti:Sapphire beam was steered in the XY plane of the preparation by the scan head. Both, two-photon and bright-field images were acquired with a cooled CCD camera (C4742- 95-12NRB; Hamamatsu Corporation) and processed using the public domain software Image J (Image J 1.34s; http://rsb.info.nih.gov/ij/docs/) and the commercial program Paint Shop Pro (Corel Corp).
The command signals driving the galvanometers of the SCH, and those used to control the shutter opening were generated by a PCI-6711 (1 MHz) and an AT-MIO-16XE-10 boards (National Instruments). The Z-axis stepper motor was driven by PC-STEP-4CX board (National Instruments). The CCD camera and the image acquisition were controlled by an IMAQ PCI-1424 board (National Instruments). All the drivers were controlled by custom-written software in G-Language (LabVIEW, National Instruments).
The lateral (XY) and axial (Z) resolutions of the system were measured as described previously (Vergara et al., 2001; DiFranco et al., 2002). The lateral resolution was 388 ± 20 nm and 294 ± 18 nm for the APD and the CCD camera, respectively (mean ± SEM), and the axial resolution was 841 ± 18 nm and 492 ± 5 nm for the APD and the camera, respectively.
The microscope was also equipped with a standard epifluorescence attachment and a Hg-arc lamp for global illumination of the preparation. Fluorescence from illumination areas of 20∼30 μm in diameter was detected with a PIN diode (HR-008, UDT) as described elsewhere (Woods et al., 2005; DiFranco et al., 2006).
Electrophysiological Methods
Enzymatically dissociated fibers were transferred to an optical chamber containing external solution and impaled with two microelectrodes placed ∼50 μm apart, as described previously (Woods et al., 2004). The voltage recording microelectrode had a resistance of ∼30 MΩ (filled with 3M KCl) and the current injection electrode had a resistance of ∼20 MΩ (filled with K-internal solution). Action potentials were low pass filtered at 10 kHz with a Bessel filter and sampled at 33.33 kHz using the multiplexed input of the AT-MIO-16XE-10 board.
Measurement and Analysis of Localized Ca2+ Transients
Myoplasmic [Ca2+] changes were measured using the cell impermeant form of the Ca2+ indicator OGB-5N at a concentration in the K-internal solution of 125 μM. Fluorescence records were obtained not earlier than 30 min after impalement to allow for dye equilibration in the muscle fiber (Woods et al., 2004). For the measurement of [Ca2+] changes using TPSpD, the fiber was electrically stimulated and illuminated with two-photon excitation by selecting the Ti:Sapphire laser wavelength to 820 nm. AP-evoked fluorescence transients, filtered with a 515–545 nm bandpass (Chroma Technology Corp.), were recorded simultaneously with the APs, from fiber positions separated by 100-nm steps (unless otherwise stated) along the X direction. This process was repeated in 7-s intervals until more than four sarcomeres had been scanned (∼10 μm). The laser spot was focused approximately at the center of the medial plane of the fiber; thus, Ca2+ and di-8-ANEPPS fluorescence data were collected close to the geometrical axis of the fiber and along its direction. Care was taken to verify, by observing a magnified image of the fiber on a monitor screen, that movement was completely arrested either by 10 mM [EGTA] or by BTS (in the case of 5 mM [EGTA]) (Woods et al., 2004).
The fluorescence transients were either raw traces or normalized as ΔF/F = (F − Frest)/Frest and characterized by their amplitude (ΔF/F)peak, full duration at half-maximum (FDHM), delay time (period between the peak of the AP and the initiation of the rising phase of the fluorescence transient), and maximum rate of rise ([d(ΔF/F)/dt]max) (DiFranco et al., 2002). These parameters were calculated with a custom-made computer program written in Delphi (Borland Software Corporation).
The equilibrium dissociation constant (Kd) and the Fmax/Fmin ratio (R) of OGB-5N was determined in vitro as described elsewhere (Escobar et al., 1997; Nagerl et al., 2000). For the particular batch of OGB-5N (lot 15C1-1) used in this report, Kd = 45 ± 3 μM and R = 38 ± 8 (n = 4). To estimate the AP-evoked changes in free [Ca2+] and SR Ca2+ release flux underlying the OGB-5N ΔF/F transients recorded in the presence of high internal [EGTA], we used a single compartment kinetic model that took into account the presence of endogenous and exogenous Ca2+ buffers using published values for rate constants as described elsewhere (Woods et al., 2004, 2005).
Measurements of di-8-ANEPPS T-tubule Fluorescence
The muscle fibers were stained with the nonpermeant fluorescent potentiometric dye di-8-ANEPPS as previously described (DiFranco et al., 2005; Woods et al., 2005), and the fluorescence was measured either with the TPSpD in 100-nm steps (unless otherwise stated) or with the scanning imaging protocol described above. Fluorescence profiles were obtained from these data by plotting fluorescence intensity vs. spot position (or pixel position) and their FWHMs were estimated from Gaussian fits. To compare the resting distribution of OGB-5N fluorescence (Frest) with respect to the T-tubules' position, a dual-staining protocol was used. Fibers were first stained with di-8-ANEPPS and subsequently loaded with OGB-5N. TPLSM and TPSpD measurements were made 30–40 min after microelectrode impalement of the fiber, at locations 200 nm apart. In both cases, the following Ti:Sapphire excitation//fluorescence filter combinations were used: 820 nm and 515–545 nm for OGB-5N, and 940 nm and 568–618 nm for di-8-ANEPPS.
Statistics
Origin (Microcal Corporation) and GraphPad Prism (GraphPad Software Inc.) were used for data analysis. Values are presented as a mean ± SEM. The statistical tests were t test or ANOVA.
RESULTS
Fluorescence Imaging of Dual Stained FDB Fibers Using TPLSM and TPSpD
It has been previously reported that Ca2+ indicators are not distributed homogeneously in the myoplasm in both frog and mammalian muscle fibers (Klein et al., 1996; Shirokova et al., 1998; Hollingworth et al., 2000; DiFranco et al., 2002). We investigated the sarcomeric distribution of OGB-5N and compared it with the T-tubule localization in quiescent fibers, using both TPLSM and TPSpD. Fig. 2 A shows a bright-field image of a segment of a fiber that was loaded with OGB-5N and stained externally with di-8-ANEPPS. The image depicts the typical striated pattern that characterizes skeletal muscle fibers. Fig. 2 (B and C) shows di-8-ANEPPS and OGB-5N TPLSM fluorescence images, respectively obtained from the same focal plane as Fig. 2 A. As expected, in Fig. 2 B only the sarcolemma and the T-tubule membranes were stained by di-8-ANEPPS. Furthermore, the bands of T-tubule fluorescence run perpendicular to the sarcolemma and are organized in the typical double row-per-sarcomere pattern of mammalian skeletal muscle fibers (Lannergren et al., 1999; DiFranco et al., 2005; Woods et al., 2005) that results from the fact that triads (T-tubule-SR junctions) located at both sides of Z-lines are closer to each other than those flanking the M-line (Revel, 1962; Franzini-Armstrong et al., 1988). In contrast, the OGB-5N image in Fig. 2 C displays a single-banded pattern resulting from the presence of a line of increased resting fluorescence, which is localized between the pairs of T-tubules flanking the Z-lines, as illustrated in Fig. 2 D. To further illustrate the spatial distribution of di-8-ANEPPS and OGB-5N fluorescence along the fiber axis, as observed in TPLSM images, we calculated the average fluorescence intensities in identical areas longitudinally spanning approximately three sarcomeres (white rectangles in Fig. 2, B and C, respectively). The results for di-8-ANEPPS (red trace) and OGB-5N (green trace) are plotted as a function of the pixel position in Fig. 2 E. It can be observed that the di-8-ANEPPS fluorescence profile reflects the double row-per-sarcomere pattern from which sarcomere length (SL) can be estimated as the distance between every other fluorescence peak (one T-tubule row per peak). In this fiber, SL was 2.26 ± 0.03 μm. In addition, Fig. 2 E illustrates that there are two distinct distances between consecutive T-tubules: the separation between T-tubules flanking the Z-lines, hereafter called TSZ, and that between T-tubules at both side of M-lines, hereafter called TSM. For this fiber, TSZ and TSM were 0.79 ± 0.03 μm and 1.47 ± 0.03 μm, respectively. From pooled data from 19 sarcomeres (three fibers), we obtained values of 0.81 ± 0.1 μm and 1.47 ± 0.07 μm for TSZ and TSM, respectively. The ratio between fluorescence peaks (maxima) and valleys (minima) separated by TSM is 35.9 ± 2.5. In contrast with the double peak appearance of di-8-ANEPPS profiles, the OGB-5N fluorescence profile in Fig. 2 E shows single maxima, which are located precisely between the T-tubule double peaks separated by TSZ. This pattern was consistently observed in 38 sarcomeres from four double-stained muscle fibers. From pooled data, resting OGB-5N fluorescence peaks centered at the Z-lines have 23 ± 1% higher fluorescence than at the rest of the sarcomere, which permits their use as reliable Z-line markers in the absence of di-8-ANEPPS.
Figure 2.

TPLSM imaging and TPSpD of fluorescence from double stained mouse FDB skeletal muscle fiber. (A) Bright field image of a segment of an FDB skeletal muscle fiber. (B) TPLSM pseudocolor image of di-8-ANEPPS fluorescence in the same fiber as in A. The image was acquired 40 min after the fiber was impaled with the microelectrodes. Two-photon excitation wavelength was 940 nm and the fluorescence selected with a 568–618 nm bandpass filter. (C) TPLSM pseudocolor image of the same fiber as in A showing the distribution of the Ca2+ indicator OGB-5N. Two-photon excitation wavelength was 820 nm and the fluorescence selected with a 515–545-nm bandpass filter. The plane of focus was the same for images A–C. Images were 487 × 388 pixels. Acquisition time for fluorescence images was 3.6 s. The white calibration bar represents 10 μm and applies for images A–D. The images in B and C were processed to yield optimal contrast by adjusting the background intensity level (outside the muscle fiber) to zero and the maximal intensity (saturation) to 255. The pseudocolor rendition assigned 256 intensity levels of red for image (B) and of green for image C. (D) Superposition of images in B and C. The letter n in A–D denotes a nucleus of the muscle fiber. (E) Normalized fluorescence intensity profiles averaged in a region of interest and plotted as a function of the longitudinal pixel position. The red trace was obtained from the white box in B (di-8-ANEPPS distribution) and the green trace was obtained from the white box in C (OGB-5N distribution). The intensities were normalized to the maximum value within each box. (F) Normalized fluorescence intensity profiles obtained in the same fiber as in A–D, but using TPSpD in 200-nm steps. The red symbols (di-8-ANEPPS) correspond to position-dependent fluorescence intensities (integrated during 100-μs illumination periods) when the fiber was illuminated with a two-photon excitation wavelength of 940 nm and the fluorescence filtered through a 568–618-nm bandpass. The green symbols (OGB-5N) correspond to position-dependent fluorescence intensities (integrated during 35-ms illumination periods) excited at 820 nm and the fluorescence selected with a 515–545 bandpass filter. The interval between consecutive step advances was 5 s. The data for OGB-5N was corrected for 3% crosstalk from di-8-ANEPPS fluorescence observed at the lower excitation wavelength. The data for di-8-ANEPPS did not require correction. Both the OGB-5N and di-8-ANEPPS traces represent Gaussian fits to the data, and are shown normalized with respect to the maximum value obtained for each indicator. The [EGTA] was 10 mM.
Equivalent results were obtained using TPSpD as depicted in Fig. 2 F. Profiles of di-8-ANEPPS and OGB-5N fluorescence (red and green symbols, respectively), obtained in the same fiber, display a similar peak and valley distribution as those generated from TPLSM images (Fig. 2 E); double-peak profiles for di-8-ANEPPS and single peaks of OGB-5N fluorescence centered between consecutive di-8-ANEPPS peaks separated by TSZ. The values of TSZ and TSM obtained with the TPSpD method for this fiber were 0.85 ± 0.04 μm and 1.55 ± 0.1 μm, respectively, which are in the range of those calculated from pooled data (n = 16 sarcomeres from three fibers), 0.95 ± 0.05 μm and 1.43 ± 0.08 μm, respectively. These values are comparable to those obtained from CCD images (e.g., Fig. 2 B).
Positional Dependence of OGB-5N Fluorescence Transients Elicited by AP Stimulation
We used the TPSpD method to investigate the position dependence of localized fluorescence transients elicited by AP stimulation in FDB skeletal muscle fibers equilibrated with 125 μM OGB-5N and 10 mM [EGTA]. The TPSpD was initiated from a random position in the fiber. From this arbitrary location, we recorded a family of APs and evoked fluorescence transients at adjacent positions along the fiber axis. Fig. 3 A illustrates the results for a family of 31 transients (a span of 3 μm) plotted as a contour map in which the horizontal axis is time, the vertical axis is the spot position with respect to the starting point (position = 0), and the fluorescence is coded in pseudocolor, as shown in the palette inset. From this contour map it is possible to identify one position in which the resting OGB-5N fluorescence (Frest) is maximal. As shown previously (see Fig. 2, D–F), this position corresponds to the Z-line; the corresponding fluorescence record (Z-transient) is the red trace in Fig. 3 B. It can also be observed in Fig. 3 A that there are two positions flanking the Z-line where the amplitude of the evoked transients were maximal; we assumed those positions to be where the T-tubules are located (dotted lines in Fig. 3 A), and plotted the corresponding transients (T-transients) as black traces in Fig. 3 B. Finally, there are two positions, which we assumed to correspond to the M-line, where the amplitude of AP-evoked fluorescence transients (M-transients) was the smallest (dashed lines in Fig. 3 A, and blue traces in Fig. 3 B). The selected traces were obtained at the following positions: M-traces, 0.4 and 2.7 μm; Z-trace, 1.6 μm, T-traces, 1.1 and 2.0 μm.
Figure 3.

Spatial dependence of OGB-5N fluorescence transients obtained with TPSpD. (A) Contour map made by juxtaposing fluorescence transients acquired from 31 consecutive spot positions (100 nm apart) along the fiber axis (ordinate) and spanning approximately one and a half sarcomere. Z, T (dotted lines), and M (dashed lines) indicate the positions of the sarcomeric structures Z-line, T-tubule, and M-line, respectively. The fluorescence intensity is reported in arbitrary units (a.u.) displayed in pseudocolor as illustrated in the palette inset. (B) Fluorescence transients, corresponding to the Z (red), T (black), and M (blue) positions in the contour map. (C) ΔF/F fluorescence transients homologous to those shown in B. (D) Superimposed traces of the five APs recorded simultaneously with the evoked transients shown in B and C. Arrows indicate the time of stimulation. Black dashed lines indicate zero fluorescence in B, zero ΔF/F in C, and the resting Vm (−100 mV) in D.
As explained above, Fig. 3 B presents a set of single AP-evoked OGB-5N fluorescence transients obtained in different trials when the spot was located at selected positions of the sarcomere shown in Fig. 3 A (Z, in red; M, in blue; T, in black). Fig. 3 D shows the superposition of the APs recorded in association with the fluorescence transients at each position. In agreement with the results shown in Fig. 2 F, the data in Fig. 3 B show that the fluorescence level before stimulation (Frest) is larger (∼24%) for the Z-transient (red trace) than that for records obtained at other positions in the sarcomere (black and blue traces). It should be noted that the overall features of fluorescence records at intermediate locations spanning the length of the sarcomere vary monotonically from position to position (see below), attaining the particular depictions at each of the selected positions in Fig. 3 B. Furthermore, since Frest was not homogenous throughout the sarcomere but showed a local maximum at the Z-line, revealing an increased concentration of the indicator at this location and/or interaction with mitochondria (Lannergren et al., 1999), it was important to normalize the records and express them in terms of the conventional ΔF/F values. Fig. 3 C depicts the same family of transients of Fig. 3 B, but normalized in terms of ΔF/F. As expected, due to this normalization, the ΔF/F transients have a baseline value of zero. In addition, ΔF/F conversion unveils differences on the position dependence of the transients that could not be observed in raw transients because they were masked by the heterogeneous Frest. For example, while raw records show that the peak fluorescence reached at T and Z positions are almost equal (black and red traces, Fig. 3 B), due to the normalization, the amplitude of the ΔF/F transients observed at T positions (black trace, Fig. 3 C) are larger than that at the Z-line (red trace, Fig. 3 C). Interestingly, while ΔF/F normalization does not alter the kinetic properties of AP evoked transients, it allows for an equalized comparison of their position dependence. As shown in Fig. 3 C (see also Table I), Z and M transients have slower rising and decaying phases than the T transient.
TABLE I.
Characteristic Parameters of AP-evoked Ca2+ Transients Recorded at T, Z, and M Spot Positions in Mouse Skeletal Fibers Perfused Internally with 10 mM EGTA
| Location | (ΔF/F)peak |
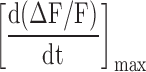
|
FDHM | Delay time | n |
|---|---|---|---|---|---|
| ms −1 | ms | ms | |||
| T | 1.35 ± 0.12 | 2.36 ± 0.24 | 1.45 ± 0.08 | 0.46 ± 0.05 | 63 |
| Z | 0.83 ± 0.09a | 1.44 ± 0.17a | 1.77 ± 0.06a | 0.54 ± 0.05 | 25 |
| M | 0.56 ± 0.05b | 1.11 ± 0.13b | 1.84 ± 0.13a | 0.62 ± 0.05a | 37 |
Data were obtained from seven fibers using TPSpD. n is the number of records at each location (rows) used to calculate the average parameter values (columns; see Materials and Methods).
P < 0.05 vs. values at T-position (Student's t test).
P < 0.001 vs. values at T-position (Student's t test).
Table I gives average values for kinetic parameters of ΔF/F transients recorded at T, Z, and M from several fibers with 10 mM EGTA. Salient features of the data in Table I are as follows. (a) The amplitude of transients (ΔF/F) in T (1.35 ± 0.12) is significantly larger (P < 0.05) than those in Z (0.83 ± 0.09) and more than double that of M (0.56 ± 0.05). (b) The kinetic properties of the rising phase were also strongly linked to the position of the spot. Values for [d(ΔF/F)/dt]max varied from T to M, from 2.36 ± 0.24 to 1.11 ± 0.13 ms−1. Interestingly, the value of this parameter at Z was 1.44 ± 0.17, which is closer to that obtained at the M position rather than at the T position. This is notable, considering that the distance between T and Z transients is relatively short (∼0.5 μm). (c) The mean FDHM calculated from transients recorded at T, Z, and M positions are 1.45 ± 0.08, 1.77 ± 0.06, and 1.84 ± 0.13 ms, respectively. It should be noted that while position-dependent differences among these values are statistically significant (P < 0.05), their dispersion is not as large as that observed for the changes in amplitude. For example, the FDHM at Z and M are only 22 and 27% longer, respectively, than at the T position. (d) There is a finite delay of ∼0.5 ms preceding the rising phase of the Ca2+ transient at the T-positions. Transients recorded at Z and M locations, have an extra delay of 0.08 ± 0.02 and 0.16 ± 0.03 ms, respectively. The extra delay to the M location is important considering that the distance between the M-line and the T-tubules is relatively short (∼0.7 μm).
Since global ΔF/F transients should approximately represent a weighted average of localized signals occurring along the sarcomeres, it was expected then to have smaller amplitude than those recorded at T locations with the TPSpD method. However, pooled data from global transients recorded in the same population of fibers in Table I yielded ΔF/F values of 1.45 ± 0.12, not statistically different from those of T-transients. A similar situation was found in 11 fibers equilibrated with 5 mM EGTA. In this case, ΔF/F for the T- and global transients were 1.92 ± 0.13 and 2.07 ± 0.12, respectively. While we do not have a definite explanation for this issue, it is interesting to note that the current ΔF/F values for global transients are approximately threefold larger than those of global OGB-5N transients reported previously under similar conditions, but using a proportionally less efficient batch of the dye (Woods et al., 2004).
AP-evoked [Ca2+] Changes at T-tubules and Z- and M-lines
The magnitude of the free [Ca2+] changes associated with localized fluorescence transients were estimated with a single compartment model as described previously for global Ca2+ transients (Woods et al., 2004, 2005). In seven fibers with 10 mM internal EGTA, the peak [Ca2+] calculated for ΔF/F transients at the T positions was 2.02 ± 0.18 μM (63 records). The respective values for the Z and M positions were 1.33 ± 0.12 μM (25 records) and 0.9 ± 0.07 μM (38 records), respectively. In 14 fibers with 5 mM EGTA, the peak [Ca2+] values were significantly (P < 0.05) higher: 2.84 ± 0.19 μM (126 records), 1.66 ± 0.13 μM (64 records), and 1.53 ± 0.12 μM (56 records) for the T, Z, and M positions, respectively. Interestingly, the Ca2+ release fluxes calculated from the same model for T-transients with 5 and 10 mM EGTA were 158 ± 11 and 164 ± 14 μM/ms, respectively, not significantly different from each other (P > 0.5). Finally, the peak free [Ca2+] calculated from global ΔF/F transients in the presence of 5 mM EGTA was 3.1 ± 0.2 μM, comparable to the 3.7 ± 0.3 μM reported previously for FDB fibers under similar conditions (Woods et al., 2004).
AP-evoked Ca2+ Release Domains
The spatiotemporal features of the changes in OGB-5N fluorescence that occur along the muscle fiber in response to AP stimulation can be visualized by plotting families of AP-evoked fluorescence transients as a function of the spot position (Fig. 4, A and B). Fig. 4 A shows a three-dimensional plot obtained by juxtaposing fluorescence records obtained, from a scan of approximately three sarcomeres along the fiber longitudinal axis (total length = 7.8 μm), using the TPSpD method. The plot allows us to visualize the topology of the dye distribution in the resting fiber (before stimulation, time <0) and the subsequent transient increase in fluorescence due to AP-evoked SR Ca2+ release. Fig. 4 B shows a contour map of the data, using the same pseudo-color rendition as Fig. 4 A. From both plots it is possible to identify, in each sarcomere, three spot positions where the resting OGB-5N fluorescence was maximal, which, according to their relative position with respect to the T-tubules, were likely to be at the Z-lines (see above). Such Z-line positions were at 1.5, 3.8, and 6.2 μm. In contrast, M-line positions were identified as middle points between consecutive Z-lines where the amplitudes of AP-evoked OGB-5N fluorescence transients are minimal (M-line positions are 2.6 and 5.1 μm). As noted before (Fig. 3, A and B), the increased OGB-5N Frest at the Z-lines creates a distortion in the visualization of the actual spatiotemporal distribution of the Ca2+-dependent signals. This is noticeable in that the OGB-5N fluorescence at the Z-lines is higher throughout the records, as evidenced by the lingering increased fluorescence after the termination of the release process (note the green band of fluorescence at the end of the Z-line records in Fig. 4 B). To verify that this distortion is linked to the heterogeneous dye distribution, we plotted the data using the ΔF/F normalization as shown in Fig. 4 (C and D) where the baseline is obviously zero. These plots demonstrate that ΔF/F normalization, besides homogenizing the fluorescence distribution throughout the three sarcomeres before AP stimulation (t = 0), also eliminated the residual fluorescence maxima at the Z-lines at the end of the records (compare the color rendition of Fig. 4, B and D, at 10 ms).
Figure 4.

Ca2+ release domains in mouse skeletal muscle fibers. (A and B) Three-dimensional plot and contour map, respectively, created by juxtaposing raw fluorescence transients acquired from 78 consecutive spot positions along the fiber axis and spanning approximately three sarcomeres. The fluorescence intensity is reported in arbitrary units (a.u.) displayed in pseudocolor as illustrated in the palette insert. (C and D) Three-dimensional plot and contour map of ΔF/F fluorescence transients calculated from the data in A and B, respectively. The fluorescence intensity is reported in ΔF/F units displayed in pseudocolor as illustrated in the palette insert. For A–D, the dashed lines indicate spot positions at the M-lines of the muscle fiber. (E) A rear perspective of three-dimensional plot in C. Same pseudocolor palette as in C. The data in A–E are displayed starting ∼2 ms before stimulation.
Most importantly, ΔF/F normalization illustrates that the TPSpD scanned segment of the muscle fiber undergoes the formation of six narrow domains of elevated ΔF/F (arranged in three pairs of peaks), which reached their maxima (on average) 2.7 ± 0.1 ms after stimulation (or 1.35 ms after the peak of the AP), and rapidly dissipated thereafter. Since these domains were obtained using a low-affinity Ca2+ indicator in the presence of high [EGTA], we operationally define them as Ca2+release domains because they depict the spatiotemporal dependence of [Ca2+] changes (evoked by an AP) in the vicinity of SR Ca2+ release sites (Pape et al., 1995; Novo et al., 2003). The dissipation of the Ca2+ release domains, calculated as the average time at which the spatial-dependent variance among ΔF/F transients vanishes (DiGregorio et al., 1999), is ∼5.3 ms after AP stimulation (or 3.95 ms after the peak of the AP). It should be noted that at this point, the [Ca2+] is still above its resting level (unpublished data).
The spatial distribution and time dependence of Ca2+ release domains can be readily appreciated in the contour map shown in Fig. 4 D. It can be seen that the domains flank the Z-line (indicated by Z) and have their maxima centered at the T-tubule locations; in contrast, at the M-lines, the [Ca2+] changes are minimal. It is also apparent in Fig. 4 (D and E) that the ΔF/F values at the Z-lines are lower than at the T-locations (peaks of Ca2+ release domains); this is in agreement with the implication that the Ca2+ release process takes place at triads symmetrically located at both sides of the Z-lines. As best illustrated in Fig. 4 E, the three pairs of Ca2+ release domains (one pair per sarcomere) are distributed at relatively constant intervals with their peaks in the following locations: pair 1, 0.9 and 2.0 μm; pair 2, 3.4 and 4.4 μm; and pair 3, 5.8 and 6.7 μm. Obviously, from these positions it was possible to estimate that (for this particular fiber) the center distance between dual peaks across the Z-line (equivalent to TSZ) was 1.0 ± 0.06 μm (n = 3) and the distance across the M-line (equivalent to TSM) was 1.4 μm (n = 2). Pooled spot detection data from 25 Ca2+ release domains recorded from five fibers under identical conditions, including the one in Fig. 4, showed that these values were 1.01 ± 0.02 and 1.35 ± 0.05 μm, for distances across the Z- and M-lines, respectively. Interestingly, these values are comparable to the TSZ and TSM values measured with di-8-ANNEPS (Fig. 2). Another interesting feature of the domains shown in Fig. 4 (C to E) is that the six observed peaks reached very similar amplitudes with respect to each other. Their average (ΔF/F)peak value was 1.70, and their standard deviation (σ) was 0.11 (in units of ΔF/F). In pooled data from 65 domains in seven fibers, σ was 0.13. It should be noted that this (interpeak) σ is comparable with that of the detection system's noise (∼0.11).
The regular pattern of the three pairs of Ca2+ domains shown in Fig. 4 (D and E), illustrates an intrinsic symmetry of the Ca2+ release regions of the SR in each sarcomere and suggests that, as expected for two-photon excitation, during a longitudinal scan, the laser intensity required to obtain AP-evoked transients did not affect the ability of adjacent positions (separated by 100 nm) to release Ca2+. Furthermore, we have verified that at least 15 consecutive illumination-detection cycles, as described above, could be repeated, at 0.14 Hz, at the same fiber location without affecting the AP or the amplitude or kinetics of the Ca2+ transients (unpublished data). In other words, the intensity of the two-photon excitation required for the detection of Ca2+ transients with excellent signal-to-noise ratio was in turn low enough not to induce considerable phototoxicity in the fiber.
Estimation of Domain Size from Isochronal ΔF/F Profiles
To further investigate the spatiotemporal features of AP-evoked intrasarcomeric [Ca2+] changes, we estimated the FWHM of cross sections of Ca2+ release domains at various times after AP stimulation. To this end, isochronal profiles were fitted to Gaussian functions (DiGregorio et al., 1999; DiFranco et al., 2002). Fig. 5 A is a typical plot of profiles obtained at three different times (0.76, 0.91, and 1.36 ms) during the formation of the domains (rising phase of the ΔF/F transients) showed in the fiber of Fig. 4. The T-transients reached their peak values at 1.36 ms for the two adjacent sarcomeres sampled; at this point, the FWHM was 0.64 ± 0.03 μm (n = 4). For earlier profiles, at 0.76 and 0.91 ms, the FWHM values were 0.69 ± 0.06 and 0.65 ± 0.03 μm, respectively. Fig. 5 B shows three isochronal profiles (1.36, 1.96, and 2.26 ms) obtained from the same sarcomeres, but during the decaying phase of Ca2+ domains. In this case, the FWHM values were 0.72 ± 0.03 and 0.88 ± 0.12 (n = 4), respectively. It should be noted that during the rising phase of the transients (Fig. 5 A), the ΔF/F fluorescence at the M- and Z-valleys did not remain stationary at a very low level, but showed mild increases with time. Furthermore, during the falling phase of T-transients, the ΔF/F at the M- and Z-valleys continued increasing while the ΔF/F at the T-positions obviously decayed with time (Fig. 5 B). Interestingly, at 2.26 ms there are small differences between the observed ΔF/F at T-positions with respect to those at the M- and Z-valleys, even though the overall ΔF/F fluorescence is relatively high (>0.3). These observations are compatible with a simplified scheme whereby as time progresses the Ca2+ released at T-positions invades the M- and Z-regions of the sarcomere and, once the release stops, the gradients (generated during the release) progressively dissipate.
Figure 5.

Temporal evolution of the width of Ca2+ release domains. (A) Isochronal ΔF/F plots of four domains measured from the same fiber as in Fig. 4. ΔF/F values, averaged during 120 μs, are plotted as a function of the spot position at three different times after the peak of the AP: 0.76 ms (closed circles), 0.91 ms (open circles), and 1.36 ms (closed triangles). (B) Isochronal ΔF/F plots of the same domains as in A, but during the falling phase of the transients. The data was obtained at 1.36 ms (closed triangles), 1.96 ms (open circles), and 2.26 (closed circles) after the peak of the AP. Each continuous trace in A and B is the sum of four Gaussian curves fitted to the domains of every isochronal dataset. (C) Average FWHM (closed circles) and isochronal ΔF/F values from T-locations (open circles) plotted as a function of time for the four domains in A and B.
To compare and contrast the evolution in time of the width of the Ca2+ release domains with that of their amplitudes, we plotted (Fig. 5 C) the average FWHM and the average (ΔF/F)peak at the T-positions for the four domains in Fig. 5 (A and B) as a function of time. Interestingly, while the average ΔF/F values (open circles) changed significantly over time (P < 0.01), average FWHM measurements during the rising and falling phases of the domains (closed circles) did not vary significantly (P = 0.11). It should be noted that the continuous line fitted to the ΔF/F data is a segment of an average T-transient for the four domains shown. Thus, while the average transient grew and decayed in amplitude, the average width of the Ca2+ release domain did not change significantly. Pooled data from 16 domains recorded from four fibers under identical conditions (including the one above) confirm this statement (average P > 0.1).
Effect of the [EGTA] on the Properties of Localized Ca2+ Transients
The Ca2+ chelator EGTA has been used to investigate the spatiotemporal dependence of Ca2+-dependent phenomena (Pape et al., 1995; Neher, 1998; DiGregorio et al., 1999; Novo et al., 2003). Here, we study the effects of two concentrations of EGTA (5 and 10 mM) on the time course and spatial dependence of AP-induced Ca2+-dependent fluorescence transients. Fig. 6 presents plots of averaged values for kinetic parameters of AP-induced ΔF/F transients recorded at T, Z, and M positions from several fibers with 5 and 10 mM EGTA. These parameters were obtained from 126 (T), 64 (Z), and 56 (M) records in 14 fibers. Fig. 6 A shows that, as expected, the amplitude of ΔF/F transients in T, Z, and M is larger in fibers with 5 mM internal [EGTA] than with 10 mM [EGTA]. Average amplitude values for 5 mM [EGTA] were (from T to M) from 1.92 ± 0.13, 1.35 ± 0.09, and 1.11 ± 0.09, which are significantly larger (P < 0.05) than the respective values reported for 10 mM [EGTA] in Table I. Interestingly, the reduction in the (ΔF/F)peak with the higher [EGTA] is more prominent in the Z and M positions (39% and 50%, respectively) than at the T positions (30%). This is consistent with the predicted narrowing of the Ca2+ domains by EGTA considering that the process involves diffusion-reaction mechanisms within the sarcomere (Cannell and Allen, 1984; Novo et al., 2003). The influence of the [EGTA] on the rising kinetics of the transients were also strongly linked to the position of the spot. In 5 mM [EGTA], their maximal rates of rise were 3.30 ± 0.31, 2.42 ± 0.22, and 2.03 ± 0.21 ms−1 for the T, Z, and M spot positions, respectively (Fig. 6 B). As observed for the amplitude of the ΔF/F transients, values for [d(ΔF/F)/dt]max were reduced more in the higher [EGTA] at the Z and M positions (41% and 45%, respectively) than at the T position (29%). Fig. 6 C illustrates that also the FDHM of ΔF/F transients was critically affected by the [EGTA]. In 5 mM [EGTA], they were 1.77 ± 0.08, 2.29 ± 0.15, and 2.44 ± 0.18 ms for the T, Z, and M positions, respectively, which are significantly longer (P < 0.05) than those calculated in presence of 10 mM [EGTA] (see Table I). Finally, Fig. 6 D shows that the delay time of localized Ca2+ transients recorded in 5 mM [EGTA] were 0.46 ± 0.05 (T), 0.54 ± 0.05 (Z), and 0.62 ± 0.05 (M) ms. These values for Z and M are slightly longer than those observed in presence of 10 mM [EGTA] (see Table I), but neither of them differs significantly.
Figure 6.

Comparative properties of AP-evoked localized Ca2+ transients in 5 and 10 mM EGTA. A–D are bar plots of (ΔF/F)peak, [d(ΔF/F)/dt]max, FDHM, and delay time, respectively, for fluorescence transients detected at the T, Z, and M positions. Open and filled bars represent values obtained in the presence of 5 and 10 mM [EGTA], respectively. The * and ** indicate statistical significance with P < 0.05 and P < 0.001, respectively (Student's t test). The absence of symbols implies lack of statistical significance (P > 0.5).
Effects of the [EGTA] on the Properties of Ca2+ Release Domains
We studied whether the effects of varying the [EGTA] on the FWHM of Ca2+ domains was compatible with the notion that their dimensions are mostly determined by the size of the Ca2+ release site (DiGregorio et al., 1999; Novo et al., 2003). To this end, several fibers were stained with di-8-ANEPPS and scanned with the TPSpD in order to assess the spatial distribution of the T-tubules' staining, similarly as shown in Fig. 2 F (red trace). The FWHM of di-8-ANEPPS fluorescence profiles were calculated from Gaussian fits, yielding values of 0.61 ± 0.03 μm (49 domains in seven fibers). Comparatively, the FWHM of isochronal profiles of Ca2+ domains, measured at the time when T-transients peaked, were 0.69 ± 0.03 (32 domains in eight fibers) and 0.62 ± 0.02 (36 domains in nine fibers), for 5 and 10 mM internal [EGTA], respectively. These values are not significantly different from each other (P ∼ 0.1). Interestingly, the average FWHM of the Ca2+ domains in 10 mM [EGTA] approximates that measured for the T-tubules with di-8-ANEPPS (P ∼ 0.8). In addition, although the average FWHM of Ca2+ domains in 5 mM [EGTA] was apparently different from that of the T-tubule fluorescence, the values were still not statistically different (P ∼ 0.09).
Colocalization of Ca2+ Release Domains with the T-tubules
To further investigate the localization of the Ca2+ release domains in reference with the position of the T-tubules, we performed double staining experiments. At each position, di-8-ANEPPS fluorescence and OGB-5N transients were alternately recorded using the TPSpD method (see Materials and Methods). Results from an experiment using this protocol are shown in Fig. 7. The open circles represent a ΔF/F profile, including six adjacent Ca2+ release domains (from three sarcomeres), measured at the time when T-transients peaked. The filled circles represent the corresponding values of di-8-ANEPPS fluorescence recorded at identical positions. The lines represent the sum of six individual Gaussian fits to the OGB-5N (continuous line) and the di-8-ANEPPS data (dashed line). The center parameter (xc) of the Gaussian fits was used to evaluate the spatial correspondence between di-8-ANEPPS fluorescence peaks (T-tubules' location) and the centers of the Ca2+ domains. We found that the values of xc for di-8-ANEPPS profiles fall within 0.1 μm of the values for the isochronal profiles, thus providing an ultimate confirmation that the Ca2+ release domains are colocalized (within the resolution of the optical system) with the T-tubules.
Figure 7.

Colocalization of Ca2+ release domains with the T-tubules. Fluorescence from a dual stained fiber with di-8-ANEPPS and OGB-5N was measured with the TPSpD method using steps of 200 nm. The green trace is an isochronal profile (measured ∼1.4 ms after the peak of the AP) that spans six consecutive Ca2+ release domains. The data was corrected from cross-talk contributions of di-8-ANEPPS fluorescence when using the OGB-5N filter selection. Data points (open circles) for each Ca2+ domain were fitted to individual Gaussian curves; the green line represents the sum of the Gaussian curves thus generated. Di-8-ANEPPS fluorescence was measured at the same positions where Ca2+ transients were recorded. Individual Gaussian curves were fitted to data points (filled circles). Cross talk correction was not necessary. The red line represents the sum of the individual Gaussian curves. The value for the centers of the Gaussian fits (xc) for the calcium domains were 0.6, 1.4, 2.7, 3.6, 5.0, and 5.7 μm; for di-8-ANEPPS, the respective values were 0.6, 1.5, 2.8, 3.6, 5.1, and 5.8 μm.
DISCUSSION
Localized Fluorescence Signals in Mammalian Skeletal Muscle Fibers
In this article we describe the properties of AP-evoked Ca2+ release domains in mouse skeletal muscle fibers measured using a custom-built two-photon laser microscope system. This system affords important technical advances for the detection and identification of AP-evoked Ca2+ microdomains in this preparation since it was designed to combine the high spatiotemporal resolution of the spot detection method (Escobar et al., 1994; DiGregorio et al., 1999; DiFranco et al., 2002; Novo et al., 2003) with the obviously advantageous imaging capabilities of TPLSM (Denk et al., 1990; Centonze and White, 1998; Cahalan et al., 2002). In other words, by switching from laser scanning mode (e.g., using the SCH in Fig. 1) to stage scanning mode (with X and Y nanotranslator motors) it was possible to alternatively acquire high resolution images of the steady-state subsarcomeric distribution of fluorescent probes (e.g., Fig. 2) or position-dependent AP-evoked Ca2+ transients (e.g., Fig. 3) from the same live muscle fiber preparation. Furthermore, by using a near infrared pulsed laser (Ti:Sapphire), two-photon microscopy allows for localized fluorescence detection with minimal phototoxicity (Denk et al., 1990; Yuste and Denk, 1995; Cahalan et al., 2002). This latter feature is relevant for the current studies because we could demonstrate that 15 illuminating periods of ∼80 ms, required to record 15 individual AP-evoked Ca2+ transients, while maintaining the spot at the same location, did not induce detectable distortions in the signals (unpublished data). Consequently, Ca2+ transients recorded from neighboring positions (even as close as 20 nm apart) along a spot scan are expected to remain unaffected by potential photodynamic damage induced by laser illumination during the acquisition of data at previous locations. This problem is more likely to occur in single-photon systems (either laser scanning microscopes or confocal spot detection methods) because the regions of localized illumination (at effective excitation wavelengths) are not as narrowly constrained, particularly in the Z-axis of the microscope (depth of the preparation).
TPSpD of AP-evoked Ca2+ Transients in Intact Isolated Mammalian Muscle Fibers
Localized detection of rapidly evolving Ca2+ release domains was attained by stage displacement of muscle fibers with respect to a stationary two-photon illumination spot. This method relies on the fact that AP-evoked Ca2+ release is a robustly reproducible process (Escobar et al., 1994; Vergara et al., 2001; DiFranco et al., 2002). Furthermore, it demands a complete arrest of fiber shortening in order to avoid distortions in the position-dependent recording of localized Ca2+ transients. In the current study, we used millimolar [EGTA]s in the internal solution, which are known to constrain the free myoplasmic [Ca2+] changes to narrow regions circumscribing Ca2+ release (or entry) sites (Pape et al., 1995; DiGregorio et al., 1999; Novo et al., 2003). As expected, we found that 10 mM [EGTA] fully blocks AP-evoked movement in intact dissociated FDB fibers kept at slack length. When 5 mM [EGTA] was used, 100 μM BTS (Cheung et al., 2002) was added to the external Tyrode solution to completely arrest fiber movement, as previously demonstrated (Woods et al., 2004). Since OGB-5N is a fast tracking Ca2+ indicator which, when used in conjunction with 10 mM EGTA, reports accurately the underlying Ca2+ release flux (Song et al., 1998; Woods et al., 2004), it is possible to infer that transients recorded at the center of the Ca2+ release domains (T-transients) are representative waveforms for the AP-evoked physiological process of Ca2+ release in mammalian muscle fibers. Interestingly, the FDHM of T-transients at room temperature is 1.45 ± 0.08 ms (Table I), which is even shorter than that of the AP itself (∼2 ms, Fig. 3; also see Woods et al., 2005).
Our finding that under similar conditions the amplitude of global transients is comparable to that of T-transients, and not smaller, as expected for signals reporting average values of the Ca2+ distribution along the sarcomere is puzzling. A possible explanation is that global transients, recorded from an ample volume of the muscle fiber, might contain contributions from triads capable of larger Ca2+ release than those probed with the TPSpD system. This is compatible with the a priori choice, in these latter experiments, to scan the spot only within the medial axial plane of the fiber. The implications of these results can, and will be, investigated in the future.
The comparison of [Ca2+] changes at different locations is based on the accurate calculation of ΔF/F at each site (see Materials and Methods). This calculation is in turn based on the realistic assumption that at any location, the fluorescence before stimulation (at resting state) represents the concentration of OGB-5N molecules capable of readily binding Ca2+ions; as a consequence, smaller ΔF/Fs at one location represent reduced [Ca2+] changes at that site. Bearing this in mind, the presence of a band of higher resting fluorescence colocalized with the Z-line has been supposed to reflect an increased dye concentration at that location, probably resulting from dye binding to proteins specific to this region (e.g., α-actinin). Thus, we inferred that the reduced ΔF/F at the Z-line truly reflected smaller [Ca2+] changes there. A caveat to this result is that the association of selected Ca2+ indicators with representative myoplasmic proteins has been reported to decrease their Ca2+ binding affinity (Hove-Madsen and Bers, 1992; Harkins et al., 1993), a possibility that can result in reduced ΔF/F transients for similar [Ca2+] changes. Nevertheless, we have verified, using methods similar to those of the latter authors, that the Ca2+ binding affinity of OGB-5N is either not altered or increased in the presence of large concentrations (up to 80 mg/ml) of albumin and creatine phosphokinase, respectively (unpublished data). An alternative caution is the possibility that the Ca2+ dye might get trapped inside the mitochondria, which are known to be clustered around the Z-lines, and give an artificially high level of fluorescence. If this were the case, our ΔF/F calculations would be underestimated at these locations, leading to an exacerbation of the valleys between adjacent Ca2+ sources.
Two Localized Ca2+ Release Domains per Sarcomere
A critical question answered in these studies is the colocalization of the Ca2+ release sites with respect to anatomical structures in mammalian skeletal muscle fibers. We used di-8-ANEPPS, a nonpenetrating potentiometric dye, to specifically label the T-tubules, which are the central component of the triads. The di-8-ANEPPS fluorescence images (Fig. 2 B) showed double row of T-tubules per sarcomere, a distinctive feature of mammalian skeletal muscle fibers that has been documented previously (Revel, 1962; Franzini-Armstrong et al., 1988; Dulhunty, 1989; Lannergren et al., 1999; DiFranco et al., 2005; Woods et al., 2005). This is consistent with electron microscopy evidence showing that triads in unstretched mammalian fibers are located at both sides of the Z-line at the I-A junction region of the sarcomere (Revel, 1962; Franzini-Armstrong et al., 1988; Dulhunty, 1989). We investigated here the exact sarcomeric colocalization of Ca2+ domains with the T-tubules by dual staining fibers with di-8-ANEPPS and OGB-5N and verified that the central position of Ca2+ domains coincides with the peak of the di-8-ANEPPS bands (Fig. 7). In fact, we found that it was not necessary to perform every Ca2+ domain measurement in dual stained fibers because the resting distribution of OGB-5N fluorescence, by showing a clear peak at the Z-line (Fig. 2, C–F), provided an alternative anatomical reference. Consequently, although the majority of the experimental data were obtained from fibers stained with OGB-5N alone (Figs. 3–6 and Table I), the position dependence of Ca2+ transients always relied on the precise determination of the Z-line position (Figs. 4, 5, and 7).
The single-peaked pattern reported here for the resting OGB-5N fluorescence in mammalian fibers (Fig. 2) is different than the double banded pattern observed in frog fibers, where a wide band was centered at the M-line and a much thinner peak at the Z-line (Vergara et al., 2001; DiFranco et al., 2002). Interestingly, the OGB-5N distribution in frog fibers was similar to that found for Fluo-3 (Tsugorka et al., 1995; Klein et al., 1996; Hollingworth et al., 2000), a correspondence between dyes that is seemingly maintained for mammalian fibers (Shirokova et al., 1998). However, it has been suggested that the single Fluo-4 fluorescence bands observed in skinned mammalian fibers were centered at the M-lines (Kirsch et al., 2001). This is surprising given the similarity between Fluo-3 and Fluo-4, which is greater than that between OGB-5N and Fluo-3. What is important is that these authors (Kirsch et al., 2001) used the M-line as a reference point to infer that the location of dual sites of elementary release were off-centered with respect to Z-lines; notably, their reference point could have been reversed. Our data clearly demonstrate that AP-evoked Ca2+ release domains are symmetrically located flanking the resting OGB-5N peaks (Z-lines) and that the shortest center-to-center distance between domains (∼1 μm) is comparable to the TSZ value measured with di-8-ANEPPS (∼0.8 μm) but significantly smaller than the TSM distance across the M-line (∼1.5 μm). This is appealing because, even in unstretched mammalian muscle fibers, it is possible to observe (Fig. 4) an asymmetrical pattern in which pairs of domains flanking the Z-lines are closer to each other than pairs across the M-lines. This pattern is markedly different than that observed in amphibian muscle fibers where the domains are evenly spaced along the fiber axis with a spacing identical to the SL of the fiber (Escobar et al., 1994; Hollingworth et al., 2000; Vergara et al., 2001; DiFranco et al., 2002). Most notably, by putting together the results reported here with previous amphibian data we may conclude that functional Ca2+ release domains in both taxa (mammals and amphibians) are uniquely centered where the triads are observed with electron microscopy, namely at the Z-lines in frog fibers (Peachey, 1965; Zampighi et al., 1975), and in off-centered pairs flanking the Z-lines in mammalian fibers (Revel, 1962; Franzini-Armstrong et al., 1988; Dulhunty, 1989).
We have recently demonstrated in mammalian fibers stained with di-8-ANEPPS that lengthening the sarcomeres by stretch results in increases of TSM without changing TSZ significantly (DiFranco et al., 2005, 2006). Thus, it could be predicted that stretching would segregate the pairs of Ca2+ release domains away from regions of actomyosin overlapping, an effect possibly contributing to mechanical uncoupling. Preliminarily data obtained with single-photon confocal microscopy (Vergara et al., 2002) support this contention. We are currently extending these studies by using TPSpD and mathematical modeling to investigate whether spatial segregation of Ca2+ release sites, afforded by different degrees of stretching, significantly alters the sarcomeric Ca2+ distribution. Furthermore, to date we have found no evidence that the Ca2+ release domains are systematically skewed in a way compatible with the contention that there are intrinsic asymmetries in SR release sites. Nevertheless, this is an open question, which would require a higher optical resolution to be answered.
The Dimensions of Ca2+ Release Domains
Although in vitro bead calibrations showed that the lateral resolution of the two-photon system is ∼0.4 μm, we observed the FWHM of stained T-tubules to be ∼0.6 μm. This discrepancy is important; given their elliptical cross section with a long diameter of <50 nm (Revel, 1962), they should have been reported as blurred objects with an FWHM of ∼0.4 μm. A plausible reason for the divergence arises from the tortuous geometry of the T-system as illustrated by electron micrographs of mammalian fibers in which the T-tubules wander at regions of the sarcomere flanking the Z-line (AI bands) in an apparently random fashion (e.g., see Fig. 4 in Revel, 1962). Thus, in our case the T-tubules could fold back and forth hundreds of nanometers within the spot. It is also possible that the fiber represents an anisotropic and complex optical media, which could alter the point-spread function of the microscope. Nevertheless, in vitro bead calibrations in sucrose-agar solutions were not different from those in water (unpublished data).
The T-tubule measurements have important meaning for the estimation of the actual width of the Ca2+ release domains observed with 10 mM internal [EGTA]. Namely, the ∼0.6 μm FWHM sets a practical lower limit for how small functional Ca2+ release sources would be observed with the two-photon spot system. It is interesting to note that the FWHM of Ca2+ release domains measured early during the rising phase of the individual Ca2+ transients was also ∼0.6 μm (Fig. 5, A and C). Taken together, these results show that in adult mammalian fibers the Ca2+ release sources are probably confined to the T-tubules' location. This is reinforced by the observation that the Ca2+ release domains did not expand significantly in time (Fig. 5 C), as it would be expected in the presence of high [EGTA], due to the constraining effect of this chelator on the size of Ca2+ release domains around the Ca2+ release source (Pape et al., 1995; Neher, 1998; DiGregorio et al., 1999; Novo et al., 2003). These results are compatible with the strict localization of RyR1 at the junctional SR and the absence of Ca2+ release channels at extrajunctional regions of the SR in mammalian fibers (Flucher and Franzini-Armstrong, 1996; Felder and Franzini-Armstrong, 2002). The situation is different than what was found in amphibian muscle fibers where Ca2+ domains are centered at the T-tubules, but displays broader distributions than what is expected for junctional SR (Vergara et al., 2001; DiFranco et al., 2002; Novo et al., 2003). These latter results were explained on the basis of a broad release region encompassing the entire terminal cisternae of the SR, as purportedly supported by the presence of extrajunctional Ca2+ release channels (RyRβ) in this preparation (Dulhunty et al., 1992; Felder and Franzini-Armstrong, 2002).
In contrast to the case of amphibian muscle fibers, calcium sparks are not physiological events in mammalian skeletal muscle fibers (Shirokova et al., 1998). They are detectable only under nonphysiological conditions, e.g., when the sarcolemma is removed and the fibers are exposed to “intracellular” SO4 2−-based solutions (Kirsch et al., 2001; Zhou et al., 2003). Under these circumstances, mammalian sparks have been reported to be long lasting events lasting up to 60 ms, with a spatially broad distribution (2–5 μm width) spanning at least a full sarcomere (Zhou et al., 2003). Furthermore, evidence demonstrating clear colocalization of the center of the sparks with the triad(s) involved in their generation is lacking. Consequently, it is difficult to directly compare the properties of spark events recorded in such disparate conditions with AP-evoked Ca2+ release domains that are formed and dissipate within ∼10 ms, as reported here under quasi-physiological conditions. Nevertheless, the results reported in this paper allow us to realistically state that mammalian Ca2+ sparks, with properties like those reported in the past, are not good candidates to be considered as unitary events underlying the Ca2+ release process at the triads in response to AP stimulation.
Acknowledgments
The authors thank Dr. Tom Otis for sharing the TPLSM facilities with us, and Dr. Christopher Woods and Ms. Joana Capote for reading the manuscript.
This work was supported by National Institutes of Health grants AR25201 and AR47664, a Grant in Aid from the Muscular Dystrophy Association, and internal UCLA funds from the Stein Oppenheimer Endowment Award to J.L. Vergara. Additional support was obtained from a Fulbright International Postdoctoral Fellowship (P. Ñeco), Project PI042004-072 from Gobierno de Canarias and Fundación Doctor Morales, Spain (J. Gómez).
Olaf S. Andersen served as editor.
J. Gómez and P. Ñeco contributed equally to this work.
Abbreviations used in this paper: AP, action potential; BTS, N-benzyl-p-toluene sulphonamide; FDB, flexor digitorum brevis; FWHM, full-width at half-maximum; SCH, scan head; SL, sarcomere length; TPLSM, two-photon laser scanning microscope; TPSpD, two-photon spot detection.
References
- Baylor, S.M., and S. Hollingworth. 2003. Sarcoplasmic reticulum calcium release compared in slow-twitch and fast-twitch fibres of mouse muscle. J. Physiol. 551:125–138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bers, D.M., C.W. Patton, and R. Nuccitelli. 1994. A practical guide to the preparation of Ca2+ buffers. Methods Cell Biol. 40:3–29. [DOI] [PubMed] [Google Scholar]
- Block, B.A., T. Imagawa, K.P. Campbell, and C. Franzini-Armstrong. 1988. Structural evidence for direct interaction between the molecular components of the transverse tubule/sarcoplasmic reticulum junction in skeletal muscle. J. Cell Biol. 107:2587–2600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cahalan, M.D., I. Parker, S.H. Wei, and M.J. Miller. 2002. Two-photon tissue imaging: seeing the immune system in a fresh light. Nat. Rev. Immunol. 2:872–880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cannell, M.B., and D.G. Allen. 1984. Model of calcium movements during activation in the sarcomere of frog skeletal muscle. Biophys. J. 45:913–925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Centonze, V.E., and J.G. White. 1998. Multiphoton excitation provides optical sections from deeper within scattering specimens than confocal imaging. Biophys. J. 75:2015–2024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheung, A., J.A. Dantzig, S. Hollingworth, S.M. Baylor, Y.E. Goldman, T.J. Mitchison, and A.F. Straight. 2002. A small-molecule inhibitor of skeletal muscle myosin II. Nat. Cell Biol. 4:83–88. [DOI] [PubMed] [Google Scholar]
- Delbono, O., and E. Stefani. 1993. Calcium transients in single mammalian skeletal muscle fibres. J. Physiol. 463:689–707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denk, W., J.H. Strickler, and W.W. Webb. 1990. Two-photon laser scanning fluorescence microscopy. Science. 248:73–76. [DOI] [PubMed] [Google Scholar]
- DiFranco, M., J. Capote, and J.L. Vergara. 2005. Optical imaging and functional characterization of the transverse tubular system of mammalian muscle fibers using the potentiometric indicator di-8-ANEPPS. J. Membr. Biol. 208:141–153. [DOI] [PubMed] [Google Scholar]
- DiFranco, M., J. Capote, and J.L. Vergara. 2006. Optical imaging and functional characterization of the transverse tubular system of mammalian muscle fibers using the potentiometric indicator di-8-ANEPPS. J. Membr. Biol. In press. [DOI] [PubMed]
- DiFranco, M., D. Novo, and J.L. Vergara. 2002. Characterization of the calcium release domains during excitation-contraction coupling in skeletal muscle fibres. Pflugers Arch. 443:508–519. [DOI] [PubMed] [Google Scholar]
- DiGregorio, D.A., A. Peskoff, and J.L. Vergara. 1999. Measurement of action potential-induced presynaptic calcium domains at a cultured neuromuscular junction. J. Neurosci. 19:7846–7859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DiGregorio, D.A., and J.L. Vergara. 1997. Localized detection of action potential-induced presynaptic calcium transients at a Xenopus neuromuscular junction. J. Physiol. 505:585–592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dulhunty, A.F. 1989. Feet, bridges, and pillars in triad junctions of mammalian skeletal muscle: their possible relationship to calcium buffers in terminal cisternae and T-tubules and to excitation-contraction coupling. J. Membr. Biol. 109:73–83. [DOI] [PubMed] [Google Scholar]
- Dulhunty, A.F., P.R. Junankar, and C. Stanhope. 1992. Extra-junctional ryanodine receptors in the terminal cisternae of mammalian skeletal muscle fibres. Proceedings of the Royal Society of London. Series B: Biological Sciences. 247:69–75. [DOI] [PubMed] [Google Scholar]
- Escobar, A.L., J.R. Monck, J.M. Fernandez, and J.L. Vergara. 1994. Localization of the site of Ca2+ release at the level of a single sarcomere in skeletal muscle fibres. Nature. 367:739–741. [DOI] [PubMed] [Google Scholar]
- Escobar, A.L., P. Velez, A.M. Kim, F. Cifuentes, M. Fill, and J.L. Vergara. 1997. Kinetic properties of DM-nitrophen and calcium indicators: rapid transient response to flash photolysis. Pflugers Arch. 434:615–631. [DOI] [PubMed] [Google Scholar]
- Felder, E., and C. Franzini-Armstrong. 2002. Type 3 ryanodine receptors of skeletal muscle are segregated in a parajunctional position. Proc. Natl. Acad. Sci. USA. 99:1695–1700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flucher, B.E., and C. Franzini-Armstrong. 1996. Formation of junctions involved in excitation-contraction coupling in skeletal and cardiac muscle. Proc. Natl. Acad. Sci. USA. 93:8101–8106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franzini-Armstrong, C. 1972. Studies of the triad. 3. Structure of the junction in fast twitch fibers. Tissue Cell. 4:469–478. [DOI] [PubMed] [Google Scholar]
- Franzini-Armstrong, C., D.G. Ferguson, and C. Champ. 1988. Discrimination between fast- and slow-twitch fibres of guinea pig skeletal muscle using the relative surface density of junctional transverse tubule membrane. J. Muscle Res. Cell Motil. 9:403–414. [DOI] [PubMed] [Google Scholar]
- Franzini-Armstrong, C., and F. Protasi. 1997. Ryanodine receptors of striated muscles: a complex channel capable of multiple interactions. Physiol. Rev. 77:699–729. [DOI] [PubMed] [Google Scholar]
- Giannini, G., A. Conti, S. Mammarella, M. Scrobogna, and V. Sorrentino. 1995. The ryanodine receptor/calcium channel genes are widely and differentially expressed in murine brain and peripheral tissues. J. Cell Biol. 128:893–904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harkins, A.B., N. Kurebayashi, and S.M. Baylor. 1993. Resting myoplasmic free calcium in frog skeletal muscle fibers estimated with fluo-3. Biophys. J. 65:865–881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hollingworth, S., C. Soeller, S.M. Baylor, and M.B. Cannell. 2000. Sarcomeric Ca2+ gradients during activation of frog skeletal muscle fibres imaged with confocal and two-photon microscopy. J. Physiol. 526:551–560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hove-Madsen, L., and D.M. Bers. 1992. Indo-1 binding to protein in permeabilized ventricular myocytes alters its spectral and Ca binding properties. Biophys. J. 63:89–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim, A.M., and J.L. Vergara. 1998. a. Fast voltage gating of Ca2+ release in frog skeletal muscle revealed by supercharging pulses. J. Physiol. 511:509–518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim, A.M., and J.L. Vergara. 1998. b. Supercharging accelerates T-tubule membrane potential changes in voltage clamped frog skeletal muscle fibers. Biophys. J. 75:2098–2116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirsch, W.G., D. Uttenweiler, and R.H. Fink. 2001. Spark- and ember-like elementary Ca2+ release events in skinned fibres of adult mammalian skeletal muscle. J. Physiol. 537:379–389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein, M.G., H. Cheng, L.F. Santana, Y.H. Jiang, W.J. Lederer, and M.F. Schneider. 1996. Two mechanisms of quantized calcium release in skeletal muscle. Nature. 379:455–458. [DOI] [PubMed] [Google Scholar]
- Krolenko, S.A., W.B. Amos, and J.A. Lucy. 1995. Reversible vacuolation of the transverse tubules of frog skeletal muscle: a confocal fluorescence microscopy study. J. Muscle Res. Cell Motil. 16:401–411. [DOI] [PubMed] [Google Scholar]
- Lannergren, J., J.D. Bruton, and H. Westerblad. 1999. Vacuole formation in fatigued single muscle fibres from frog and mouse. J. Muscle Res. Cell Motil. 20:19–32. [DOI] [PubMed] [Google Scholar]
- Nagerl, U.V., D. Novo, I. Mody, and J.L. Vergara. 2000. Binding kinetics of calbindin-D(28k) determined by flash photolysis of caged Ca(2+). Biophys. J. 79:3009–3018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neher, E. 1998. Vesicle pools and Ca2+ microdomains: new tools for understanding their roles in neurotransmitter release. Neuron. 20:389–399. [DOI] [PubMed] [Google Scholar]
- Nguyen, Q.T., N. Callamaras, C. Hsieh, and I. Parker. 2001. Construction of a two-photon microscope for video-rate Ca(2+) imaging. Cell Calcium. 30:383–393. [DOI] [PubMed] [Google Scholar]
- Novo, D., M. DiFranco, and J.L. Vergara. 2003. Comparison between the predictions of diffusion-reaction models and localized Ca2+ transients in amphibian skeletal muscle fibers. Biophys. J. 85:1080–1097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pape, P.C., D.S. Jong, and W.K. Chandler. 1995. Calcium release and its voltage dependence in frog cut muscle fibers equilibrated with 20 mM EGTA. J. Gen. Physiol. 106:259–336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peachey, L.D. 1965. The sarcoplasmic reticulum and transverse tubules of the frog's sartorius. J. Cell Biol. 25:209–231. [DOI] [PubMed] [Google Scholar]
- Revel, J.P. 1962. The sarcoplasmic reticulum of the bat cricothyroid muscle. J. Cell Biol. 12:571–588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rios, E., and G. Pizarro. 1991. Voltage sensor of excitation-contraction coupling in skeletal muscle. Physiol. Rev. 71:849–908. [DOI] [PubMed] [Google Scholar]
- Shirokova, N., J. Garcia, and E. Rios. 1998. Local calcium release in mammalian skeletal muscle. J. Physiol. 512:377–384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song, L.S., J.S. Sham, M.D. Stern, E.G. Lakatta, and H. Cheng. 1998. Direct measurement of SR release flux by tracking ‘Ca2+ spikes’ in rat cardiac myocytes. J. Physiol. 512:677–691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsugorka, A., E. Rios, and L.A. Blatter. 1995. Imaging elementary events of calcium release in skeletal muscle cells. Science. 269:1723–1726. [DOI] [PubMed] [Google Scholar]
- Vergara, J., and M. DiFranco. 1992. Imaging of calcium transients during excitation-contraction coupling in skeletal muscle fibers. Adv. Exp. Med. Biol. 311:227–236. [DOI] [PubMed] [Google Scholar]
- Vergara, J., M. DiFranco, D. Compagnon, and B.A. Suarez-Isla. 1991. Imaging of calcium transients in skeletal muscle fibers. Biophys. J. 59:12–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vergara, J.L., M. DiFranco, and D. Novo. 2001. Dimensions of calcium release domains in frog skeletal muscle fibers. Proceedings of SPIE. 4259:133–143. [Google Scholar]
- Wier, W.G., C.W. Balke, J.A. Michael, and J.R. Mauban. 2000. A custom confocal and two-photon digital laser scanning microscope. Am. J. Physiol. Heart Circ. Physiol. 278:H2150–H2156. [DOI] [PubMed] [Google Scholar]
- Woods, C.E., D. Novo, M. Difranco, J. Capote, and J.L. Vergara. 2005. Propagation in the transverse tubular system and voltage dependence of calcium release in normal and mdx muscle fibres. J. Physiol. 568:867–880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woods, C.E., D. Novo, M. DiFranco, and J.L. Vergara. 2004. The action potential-evoked sarcoplasmic reticulum calcium release is impaired in mdx mouse muscle fibres. J. Physiol. 557:59–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuste, R., and W. Denk. 1995. Dendritic spines as basic functional units of neuronal integration. Nature. 375:682–684. [DOI] [PubMed] [Google Scholar]
- Zampighi, G., J. Vergara, and F. Ramon. 1975. On the connection between the transverse tubules and the plasma membrane in frog semitendinosus skeletal muscle. Are caveolae the mouths of the transverse tubule system? J. Cell Biol. 64:734–740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou, J., G. Brum, A. Gonzalez, B.S. Launikonis, M.D. Stern, and E. Rios. 2003. Ca2+ sparks and embers of mammalian muscle. Properties of the sources. J. Gen. Physiol. 122:95–114. [DOI] [PMC free article] [PubMed] [Google Scholar]