

Abstract
Active neurons communicate to intracerebral arterioles in part through an elevation of cytosolic Ca2+ concentration ([Ca2+]i) in astrocytes, leading to the generation of vasoactive signals involved in neurovascular coupling. In particular, [Ca2+]i increases in astrocytic processes (“endfeet”), which encase cerebral arterioles, have been shown to result in vasodilation of arterioles in vivo. However, the spatial and temporal properties of endfoot [Ca2+]i signals have not been characterized, and information regarding the mechanism by which these signals arise is lacking. [Ca2+]i signaling in astrocytic endfeet was measured with high spatiotemporal resolution in cortical brain slices, using a fluorescent Ca2+ indicator and confocal microscopy. Increases in endfoot [Ca2+]i preceded vasodilation of arterioles within cortical slices, as detected by simultaneous measurement of endfoot [Ca2+]i and vascular diameter. Neuronal activity–evoked elevation of endfoot [Ca2+]i was reduced by inhibition of inositol 1,4,5-trisphosphate (InsP3) receptor Ca2+ release channels and almost completely abolished by inhibition of endoplasmic reticulum Ca2+ uptake. To probe the Ca2+ release mechanisms present within endfeet, spatially restricted flash photolysis of caged InsP3 was utilized to liberate InsP3 directly within endfeet. This maneuver generated large amplitude [Ca2+]i increases within endfeet that were spatially restricted to this region of the astrocyte. These InsP3-induced [Ca2+]i increases were sensitive to depletion of the intracellular Ca2+ store, but not to ryanodine, suggesting that Ca2+-induced Ca2+ release from ryanodine receptors does not contribute to the generation of endfoot [Ca2+]i signals. Neuronally evoked increases in astrocytic [Ca2+]i propagated through perivascular astrocytic processes and endfeet as multiple, distinct [Ca2+]i waves and exhibited a high degree of spatial heterogeneity. Regenerative Ca2+ release processes within the endfeet were evident, as were localized regions of Ca2+ release, and treatment of slices with the vasoactive neuropeptides somatostatin and vasoactive intestinal peptide was capable of inducing endfoot [Ca2+]i increases, suggesting the potential for signaling between local interneurons and astrocytic endfeet in the cortex. Furthermore, photorelease of InsP3 within individual endfeet resulted in a local vasodilation of adjacent arterioles, supporting the concept that astrocytic endfeet function as local “vasoregulatory units” by translating information from active neurons into complex InsP3-mediated Ca2+ release signals that modulate arteriolar diameter.
INTRODUCTION
Cytosolic Ca2+ concentration ([Ca2+]i) increases in perivascular astrocytic endfeet, specialized astrocytic processes that target the cerebral microcirculation and encase smooth muscle cells (SMCs) of intracerebral arterioles (Simard et al., 2003), have previously been identified as critical mediators of neurovascular coupling. These astrocytic processes have been recognized as a locus for the production of vasoactive factors, generated in response to neuronal activity, which are critical for coupling neuronal activity to dynamic, rapid, and spatially localized changes in cerebral blood flow (Zonta et al., 2003; Filosa et al., 2004; Mulligan and MacVicar, 2004; Filosa et al., 2006; Metea and Newman, 2006; Takano et al., 2006). Although production of the putative astrocyte-derived factors involved in this process of neurovascular coupling is dependent on increases in endfoot [Ca2+]i (Zonta et al., 2003; Takano et al., 2006), the mechanism by which endfoot [Ca2+]i increases are generated in response to neuronal activity, the spatiotemporal properties of these [Ca2+]i signals, and the effects on the cerebral vasculature of endfoot delimited [Ca2+]i signals generated through endogenous Ca2+ release pathways remain to be elucidated.
Activation of metabotropic glutamate receptors (mGluRs) located on astrocytic projections that surround synapses of glutamatergic neurons results in an increase in astrocytic [Ca2+]i that propagates through astrocytic processes, ultimately resulting in a [Ca2+]i increase in the endfoot (Cornell-Bell et al., 1990; Zonta et al., 2003). In addition to signaling from presynaptic neurons, local interneurons that innervate arterioles also make extensive contact with endfeet (Hamel, 2006); however, it is unclear whether cortical astrocytes can respond to stimuli derived from interneurons. Ca2+ release through both inositol 1,4,5-trisphosphate (InsP3) receptors (InsP3Rs) and ryanodine receptors (RyRs) has been implicated in the generation of astrocytic Ca2+ signals (Golovina and Blaustein, 1997, 2000; Verkhratsky et al., 1998; Beck et al., 2004; Perea and Araque, 2005). Golovina and Blaustein (1997, 2000) elegantly demonstrated the presence of spatially and functionally distinct InsP3 and ryanodine/caffeine-sensitive Ca2+ stores in primary cultured astrocytes, based on fluorescence measurements of endoplasmic reticulum Ca2+ load and changes in [Ca2+]i in response to agonist stimulation. Interestingly, these authors noted little overlap between regions of the endoplasmic reticulum that were sensitive to InsP3 and those sensitive to caffeine/ryanodine, indicating that regional specificity likely exists in the generation of astrocytic [Ca2+]i signals.
Several studies have shown that neuronal activity–induced increases in astrocytic [Ca2+]i can occur within spatially localized regions of astrocytic processes. Grosche et al. (1999) identified specific regions along processes of Bergmann glia, termed glial microdomains, in which spatially restricted [Ca2+]i increases were generated in response to stimulation of parallel fibers in the cerebellum. In addition, Araque et al. (2002) showed that hippocampal astrocytes in situ exhibit spatially defined regions of elevated [Ca2+]i in response to stimulation of cholinergic neurons. Although these studies investigated [Ca2+]i signals in nonperivascular astrocytic processes, the generation of local and spatially restricted [Ca2+]i increases is highly relevant in perivascular processes, from which Ca2+-dependent vasoactive factors are likely to be released into the endfoot-arteriolar space, rather than into the brain parenchyma.
One of the functional outcomes of astrocytic [Ca2+]i signaling is the regulation of cerebral arteriole diameter to modulate local cerebral blood flow. Astrocytes and cerebral arterioles form the functional gliovascular unit, with astrocytic endfeet and perivascular processes completely encasing the arterioles (Simard et al., 2003). The intimate association of endfeet and arteriolar SMCs (∼20 nm between endfoot and SMCs; Nagelhus et al., 2004) suggests that signaling mechanisms in endfeet are ideally positioned to exert profound changes on arteriolar function. Indeed, elevation of [Ca2+]i within individual endfeet in vivo, through photolysis of caged Ca2+, induces vasodilation of adjacent arterioles (Takano et al., 2006). However, similar studies in brain slices suggest that elevation of endfoot [Ca2+]i induces vasoconstriction of adjacent arterioles (Mulligan and MacVicar, 2004). Although these studies establish endfoot [Ca2+]i as an essential regulator of vascular function, the mechanisms by which endfoot [Ca2+]i signals arise and the spatiotemporal properties of these signals are not known. This knowledge is critical to a thorough understanding of neurovascular coupling.
We hypothesize that astrocytic endfeet possess the necessary Ca2+ signaling machinery to generate [Ca2+]i increases independent of the remainder of the astrocyte and that these [Ca2+]i signals are generated primarily by Ca2+ release through endfoot resident InsP3R. In addition, we suggest that endfoot [Ca2+]i signals should be highly dynamic and thus capable of rapidly exerting local control over Ca2+-dependent signaling processes within endfeet. Indeed, our findings indicate that complex and dynamic [Ca2+]i signals are generated within astrocytic endfeet and result from InsP3-mediated Ca2+ release events within endfeet. Furthermore, release of InsP3 within a single endfoot is capable of inducing local vasodilation of an adjacent arteriole, supporting the concept that endfeet function as individual “vasoregulatory units” in the brain.
MATERIALS AND METHODS
Brain Slice Preparation
Cortical brain slices were prepared from 2–6-mo-old female FVB mice or 21–35-d-old Sprague Dawley rats (experiments in Fig. 5). Euthanasia was performed with an overdose of pentobarbital and rapid decapitation according to methods approved by the University of Vermont Office of Animal Care Management. The cortex was removed into artificial cerebrospinal fluid (aCSF) at 4°C and placed onto a vibratome (VT1000S; Leica) used to cut ∼200-μm-thick coronal slices. Slices were placed into 22°C aCSF equilibrated with 95% O2/5% CO2, pH ∼7.45. aCSF contained 125 mM NaCl, 3 mM KCl, 26 mM NaHCO3, 1.25 mM NaH2PO4, 2 mM CaCl2, 1 mM MgCl2, 10 mM glucose, and 400 μM l-ascorbic acid, added to reduce cell swelling associated with oxidative stress. All experiments were conducted at 35°C except those in Fig. 3, which were performed at 22°C to minimize the potential for extrusion of caged InsP3 from astrocytes.
Figure 5.

Astrocytic endfeet act as individual “vasoregulatory units” and modulate arteriolar diameter through activation of Ca2+-sensitive signaling mechanisms driven by Ca2+ release through InsP3R. (A) Photorelease of InsP3 in a single endfoot (arrow) abutting an arteriole in a rat cortical brain slice generated a rapid vasodilation in the region of the arteriole directly apposed to the endfoot. This dilation was restricted to an ∼30-μm length of the vessel. (B) Trace shows the diameter of the arteriole examined in A as a function of time. Note the rapid dilation of the arteriole in response to liberation of InsP3 in the endfoot. (C) Averaged data showing the diameter of arterioles adjacent to endfeet before and after photorelease of InsP3 in the endfeet. Error bars indicate mean ± SEM. *, P < 0.05.
Figure 3.

Spatially restricted photolysis of InsP3 within endfeet supports a role for InsP3R, but not RYR, in generating endfoot [Ca2+]i increases. (A) Exposure of endfeet to brief (∼1 ms) pulses of UV light liberated InsP3 and generated endfoot delimited [Ca2+]i increases, the properties of which were consistent over several release events. (B) Treatment of slices with 50 μM ryanodine (20-min incubation), to block RYR, did not alter endfoot [Ca2+]i increases induced by release of InsP3. (C) Treatment of slices with CPA resulted in a nearly complete block of Ca2+ release induced by photolysis of InsP3. Notice flash artifacts in both traces, confirming InsP3 was indeed released under both conditions. (D) Schematic of flash photolysis of caged InsP3 within individual astrocytic endfeet. In the image, the boundaries of a cerebral arteriole are shown with dotted lines. The local uncaging spot within the endfoot is shown by the black circle. (E) Mean data showing the effect of ryanodine, caffeine plus ryanodine, and CPA treatment on endfoot [Ca2+]i increases induced by local release of InsP3. These responses are compared with the mean time-matched control response. All experiments in this figure were performed at 22°C to minimize the potential for extrusion of caged InsP3 during the course of drug treatment. Error bars indicate mean ± SEM. **, P < 0.0001.
[Ca21]i Imaging and Arteriolar Diameter Measurements within Cortical Slices
[Ca2+]i imaging was performed using either a Solamere scanning confocal unit (QLC 100) and high-sensitivity, high-resolution intensified charge-coupled device camera (Stanford Photonics) attached to an upright microscope (E600FN; Nikon) with a 60× water-immersion objective (NA 1.0) or a Noran Oz laser scanning confocal attached to an inverted microscope (TE2000; Nikon) with a 60× oil-immersion objective. Slices were loaded with 10 μM fluo-4 AM (or fluo-5F in Fig. 4; Invitrogen) and 2.5 μg/ml pluronic acid in aCSF for 90 min at 22°C. Under these loading conditions, AM dyes load primarily into astrocytes, with little loading into SMCs and no loading into arteriolar endothelial cells (Zonta et al., 2003; Takano et al., 2006; Wang et al., 2006). Dye was excited at 488 nm with a krypton/argon laser, and fluorescence emission was collected above 495 nm. Images were acquired at 15–60 frames/s. Fractional fluorescence (F/Fo) was determined by dividing the fluorescence intensity (F) within a region of interest by a mean fluorescence value (Fo) determined from 50 images before stimulation. For arteriole diameter measurements within cortical slices, infrared differential interference contrast images were acquired at ∼5 frames/s with a charge-coupled device camera (Hamamatsu). Arteriolar internal (luminal) diameter was determined from the distance between two fixed points across the arteriole and directly adjacent to the identified endfoot. In experiments where arteriolar diameter was measured (presented in Figs. 1 and 5) the thromboxane A2 receptor agonist 9, 11-Dideoxy-11α, 9α-epoxymethanoprostaglandin F2α (U46619; 100 nM) was included to maintain vascular tone throughout the course of the experiment, such that vasodilation and vasoconstriction could be measured (Brown et al., 2002). The diameter recordings in Fig. 5 were performed in cortical brain slices from 21–35-d-old Sprague Dawley rats, as measurements of diameter changes in rats are more accurate and reproducible because of the larger diameter and, thus, larger absolute magnitude of the diameter changes of arterioles in the rat versus mouse.
Figure 4.

Perivascular astrocytic endfeet and processes generate spatially heterogeneous [Ca2+]i signals. (A) Laser scanning confocal microscopy image series of a fluo-5F AM loaded endfoot and perivascular processes within a cortical brain slice. In the image taken at the beginning of EFS (0 s), the location of the endfoot (dotted circle) and perivascular processes are outlined. The location of the arteriole is also shown, part of which was underneath the endfoot. In this example, EFS induced a [Ca2+]i wave in both perivascular processes, as well as a wave in the endfoot that traveled perpendicular to the waves in the processes. The direction of the [Ca2+]i waves is denoted by the arrows. Many areas of spatially localized [Ca2+]i increase were found, as well as a subregion of elevated [Ca2+]i within the endfoot that was maintained for an extended period of time after EFS. See Video 2 (available at http://www.jgp.org/cgi/content/full/jgp.200609650/DC1). These experiments were performed using the Ca2+-sensitive dye fluo-5F, which has a slightly lower affinity for Ca2+ than fluo-4 (K d = 388 nM vs. 215 nM for fluo-4 at 35°C), to aid in the detection of spatially localized regions of high [Ca2+]i. (B and C) Regions of spatially localized [Ca2+]i increase within a single endfoot in response to EFS. In this example (representative of at least six experiments), in which the images were cropped to include only the endfoot and immediately adjacent regions of the brain slice, three regions of Ca2+ release were found within the endfoot, as denoted by the arrows. These regions acted as initiation sites for the global endfoot [Ca2+]i increase. As seen in the traces in C, each region of interest, identified by the boxes in the 0 sec image, responded with distinct release kinetics.
Figure 1.

Elevation of astrocytic endfoot [Ca2+]i precedes vasodilation of cerebral arterioles. Image series depicting the rise in endfoot [Ca2+]i and dilation of an adjacent cerebral arteriole in a cortical brain slice loaded with the Ca2+-sensitive dye fluo-4 AM. The location of the vessel lumen is denoted by the dotted lines. The time of each image in seconds after onset of EFS is shown, as is the inner diameter of the vessel at each time point. The temporal relationship between the endfoot [Ca2+]i increase (in black) and change in vessel diameter (in gray) is shown in the traces below the image series. See Video 1 (available at http://www.jgp.org/cgi/content/full/jgp.200609650/DC1).
Flash Photolysis of Caged InsP3 and Electrical Stimulation of Neuronal Activity
For experiments using caged InsP3 to induce endfoot [Ca2+]i increases, slices were loaded with 2 μM iso-Ins(1,4,5)P3-PM (Axxora) along with 10 μM fluo-4 AM (Invitrogen) and 2.5 μg/ml pluronic acid in aCSF for 90 min at 22°C. Flash photolysis was achieved by means of a xenon arc lamp–based UV flash unit (Rapp OptoElectronics) and custom-designed condenser (TILL Photonics) with adjustable spot diameter restricted to an ∼2–3-μm region of the slice wholly contained within an endfoot and calibrated using fluorescent microspheres of varying diameter. The intensity of the UV flash was adjusted to induce endfoot delimited [Ca2+]i increases. The mean duration of an uncaging pulse was ∼1 ms. Electrical field stimulation (EFS) was used to stimulate neuronal activity by applying a 50-Hz alternating square pulse of 0.3-ms duration for 3–5 s (80–100 V) via a pair of platinum wires placed parallel to the brain slice. The elevation of astrocytic [Ca2+]i by EFS was inhibited by tetrodotoxin (Filosa et al., 2004), a blocker of voltage-dependent sodium channels, indicating neuronal involvement.
Data Analysis
Data were analyzed using the appropriate t test and were considered statistically significant when P < 0.05. All data are expressed as mean ± SEM. [Ca2+]i imaging and arteriole diameter data were analyzed using custom-designed software created by A. Bonev.
Reagents
Caged InsP3 (iso-InsP3-PM) was obtained from Axxora. The thromboxane A2 receptor agonist 9, 11-Dideoxy-11α, 9α-epoxymethanoprostaglandin F2α (U46619), xestospongin C, and U73122 were obtained from Calbiochem. Ryanodine was obtained from LC Labs and Calbiochem. Fluo-4 AM, Fluo-5F AM, and pluronic acid were obtained from Invitrogen. Cyclopiazonic acid (CPA), caffeine, and all other chemicals were obtained from Sigma-Aldrich.
Online Supplemental Material
The online supplemental material (Fig. S1 and Videos 1 and 2) contain additional examples of the lack of effect of caffeine on astrocytic [Ca2+]i signals and videos of the recordings presented in Fig. 1 and Fig. 4 A. Online supplemental material is available at http://www.jgp.org/cgi/content/full/jgp.200609650/DC1.
RESULTS
The spatiotemporal properties of astrocytic endfoot [Ca2+]i signals in situ were investigated using mouse cortical brain slices and high temporal and spatial resolution confocal [Ca2+]i imaging. We previously studied the interplay between astrocyte and SMC [Ca2+]i signals during neurovascular coupling (Filosa et al., 2004) and identified astrocytic endfeet encasing cerebral arterioles based on the following: (1) location and structure (endfeet and perivascular astrocytic processes abut and encase the SMCs of cerebral arterioles in a very characteristic manner); (2) preferential loading with AM dyes (astrocytes and their endfeet display a characteristic loading with the Ca2+-sensitive dye fluo-4 and exhibit the highest fluorescence intensity of all structures within a slice); (3) astrocytes exhibit a high potassium conductance (Kuffler et al., 1966; Filosa et al., 2006) and do not fire action potentials in response to depolarizing pulses; (4) endfeet are noncontractile in response to elevated [Ca2+]i, thus differentiating them from SMCs and pericytes; (5) the [Ca2+]i of endfeet and astrocytes increases in response to the mGluR agonist t-ACPD; and (6) astrocytes and their endfeet exhibit slow, spontaneous [Ca2+]i oscillations (Beck et al., 2004). Furthermore, the structure and localization of endfeet in this study is consistent with that found for individual astrocytes selectively loaded with the fluorescent dye Lucifer yellow (Zonta et al., 2003) and those expressing GFP under the control of an astrocyte-specific promoter (Mulligan and MacVicar, 2004).
Neuronally Evoked [Ca2+]i Increases in Astrocytic Endfeet In Situ Involve Ca2+ Release from InsP3R
[Ca2+]i increases in astrocytic endfeet have been identified as critical mediators of neurovascular coupling. To investigate this further, endfoot [Ca2+]i and arteriolar diameter changes were monitored simultaneously in mouse cortical brain slices loaded with the Ca2+-sensitive dye fluo-4 AM. Neuronal synaptic activity was induced through EFS, and the effect of this activity on endfoot [Ca2+]i and arteriolar diameter was measured. As illustrated in Fig. 1, EFS induced rapid increases in endfoot [Ca2+]i, which were followed by dilation of the adjacent arteriole under conditions in which arterioles were preconstricted with 100 nM of the thromboxane A2 receptor agonist U46619 to mimic vascular tone found in vivo (Brown et al., 2002). It was found that endfoot [Ca2+]i increased first, followed closely by dilation of the arteriole, which returned to prestimulus diameter as endfoot [Ca2+]i decreased to basal levels (Video 1, available at http://www.jgp.org/cgi/content/full/jgp.200609650/DC1). In ∼30% of experiments, we observed that the soma and endfoot of a particular astrocyte were aligned in the same focal plane; thus, the astrocytic [Ca2+]i increase induced by EFS could be visualized in both the soma and endfoot in these instances. These findings are in agreement with several published studies implicating astrocytes in the control of the cerebral vasculature (Zonta et al., 2003; Filosa et al., 2004, 2006; Metea and Newman, 2006).
Previous work conducted on cultured astrocytes has suggested that astrocytic [Ca2+]i increases are generated by activation of the InsP3–phospholipase C pathway (Golovina and Blaustein, 2000). To determine the molecular mechanisms by which endfoot [Ca2+]i increases are generated in response to neuronal stimulation, we used pharmacological inhibitors of InsP3-sensitive intracellular Ca2+ release channels and endoplasmic reticulum Ca2+ pumps (sarcoplasmic/endoplasmic reticulum Ca2+ ATPase [SERCA]). All subsequent experiments in which endfoot [Ca2+]i was measured were performed in the absence of U46619 to minimize movement of the endfeet because of vasodilation. EFS was capable of inducing multiple increases in endfoot [Ca2+]i that were consistent in terms of the peak [Ca2+]i change as well as the latency of the response (Fig. 2 A). The peak fractional fluorescence change (F/Fo) induced by the second EFS, which served as the time-matched control for experiments performed in the presence of pharmacological inhibitors, was 85.9 ± 0.7% of the first response (n = 6). The latency of the [Ca2+]i increase or time from neuronal activation (beginning of EFS) to 25% peak [Ca2+]i increase was 2.3 ± 0.2 s (n = 14) for the first response and was not significantly different between the first and second paired responses (P = 0.15; n = 6 pairs). To investigate the contribution of Ca2+ release from InsP3R to the generation of endfoot [Ca2+]i signals, cortical slices were incubated for 20 min with 10 μM of the InsP3R inhibitor xestospongin C. In the presence of xestospongin, the response induced by EFS was 63.4 ± 7.9% of the first response (Fig. 2, B and C; n = 6; P = 0.018, compared with time-matched control). In addition, treatment with the phospholipase C inhibitor U73122, to prevent InsP3 production, also resulted in a significant decrease in the EFS-induced [Ca2+]i response (63.9 ± 9.6% of first response; n = 6; P = 0.04, compared with time-matched control; Fig. 2 C). These results indicate a role for Ca2+ release from InsP3R in generating endfoot [Ca2+]i signals.
Figure 2.

Neuronally evoked increases in endfoot [Ca2+]i involve Ca2+ release from an intracellular Ca2+ store, likely through activation of InsP3R. (A) EFS was used to induce neuronal activity, allowing for the generation of multiple consistent increases in endfoot [Ca2+]i. (B) Representative traces illustrating that EFS-induced increases in endfoot [Ca2+]i were significantly decreased after a 20-min incubation with 10 μM of the InsP3R antagonist xestospongin C. (C) Mean data showing the effect of RYR, InsP3R, and phospholipase C inhibition on the generation of EFS-induced [Ca2+]i increases. Drug-treated responses are compared with the mean time-matched control response. (D) Representative traces illustrating that EFS-induced increases in endfoot [Ca2+]i were significantly decreased after a 20-min incubation with 30 μM of the SERCA pump inhibitor CPA to deplete the internal Ca2+ store. (E) Mean data showing the peak [Ca2+]i increase elicited under control conditions and after incubation with CPA. Responses generated in the presence of CPA are compared with the mean time-matched control response. Error bars indicate mean ± SEM. *, P < 0.05; **, P < 0.0001.
The lack of complete inhibition of the [Ca2+]i signal by xestospongin and U73122 could be due to limited penetration into the brain slice or to participation of additional Ca2+ release channels, such as RYR, in the response. To investigate whether RYRs are involved in generating EFS-induced endfoot [Ca2+]i signals, we attempted to inhibit RYRs using ryanodine. In the presence of 50 μM ryanodine (20-min incubation), EFS-induced endfoot [Ca2+]i increases were 84.3 ± 6.9% of the first response (Fig. 2 C; n = 9; P = 0.9, compared with time-matched control), suggesting that RYRs are not involved in generating endfoot [Ca2+]i increases in response to EFS. To further probe the requirement for intracellular Ca2+ release, as opposed to Ca2+ influx, in generating endfoot [Ca2+]i responses, the integrity of the intracellular Ca2+ store was compromised with 30 μM of the selective SERCA pump inhibitor CPA (20-min incubation). Treatment with CPA virtually eliminated the EFS-induced [Ca2+]i increase (Fig. 2, D and E), reducing the peak [Ca2+]i increase to 10.3 ± 3.0% of control (n = 6; P < 0.0001, compared with time-matched control) and suggesting that intracellular Ca2+ stores constitute the major source of Ca2+ responsible for astrocytic [Ca2+]i increases.
Spatially Restricted Photolysis of InsP3 within Endfeet Supports a Role for Endfoot Localized InsP3R, but Not RYR, in Generating Endfoot [Ca2+]i Increases
The experiments presented in Fig. 2 rely on neuronal activity to induce increases in astrocytic endfoot [Ca2+]i. This process thus requires the generation of a [Ca2+]i increase in the astrocytic cell body followed by an increase in the endfoot, and as such, it is not possible to differentiate whether the [Ca2+]i increase occurs as a result of diffusion of Ca2+ from the astrocytic cell body or as a result of Ca2+ released from InsP3R within the endfoot itself. In addition, xestospongin C, U73122, and CPA could potentially alter neuronal [Ca2+]i signaling, such that the efficacy of EFS is compromised. To overcome these potential complications, as well as to directly probe the [Ca2+]i signaling components resident within endfeet, we used spatially restricted photolysis of caged InsP3 to release InsP3 directly within endfeet. This technique allows us to elicit Ca2+ release from InsP3R located within endfeet (assuming InsP3Rs are expressed in endfeet) without involving the remainder of the astrocyte or neurons.
As illustrated in Fig. 3 D, endfeet abutting arterioles were identified and aligned to a previously calibrated spot within the imaging window, into which UV light was focused (see Materials and methods). The diameter of the UV spot was adjusted between 2 and 3 μm (Fig. 3 D), a diameter sufficiently small to be wholly encompassed by an endfoot (mean visible dimensions of endfeet in uncaging experiments = 9 × 3 μm; n = 21). In addition, the intensity of UV light was adjusted such that endfoot delimited [Ca2+]i increases were elicited. Similar to astrocytic [Ca2+]i increases induced by EFS, endfoot [Ca2+]i increases induced by flash photolysis of caged InsP3 were consistent in terms of the peak [Ca2+]i increase elicited and the latency of the response over multiple uncaging events (Fig. 3 A). The peak [Ca2+]i increase elicited by the second photorelease of InsP3, which also served as the time-matched control for experiments conducted in the presence of pharmacological inhibitors, was 69.5 ± 7.5% of the first response (Fig. 3 A; n = 10). In addition, the peak [Ca2+]i increases elicited by EFS and uncaging did not differ significantly (first response peak F/Fo for EFS = 1.99 ± 0.12 vs. 2.16 ± 0.15 from uncaging; 15 ≤ n ≤ 20; P = 0.4). Importantly, we saw no evidence of the InsP3-induced [Ca2+]i increase spreading out of the endfoot, such as a secondary [Ca2+]i increase in the cell body (23 experiments), and no [Ca2+]i increase was generated by the UV flash in the absence of caged InsP3 (n = 11). Endfoot [Ca2+]i increases induced by uncaging occurred more rapidly than EFS-induced responses (1.00 ± 0.17 s from the time of uncaging to 25% peak [Ca2+]i increase vs. 2.3 ± 0.2 s for EFS; n = 10; P < 0.0001) and were not significantly different between the first and second uncaging events (P = 0.2; n = 8 pairs). These findings suggest that [Ca2+]i increases in astrocytic endfeet are generated as a result of regenerative Ca2+ release from InsP3Rs located within the endfoot.
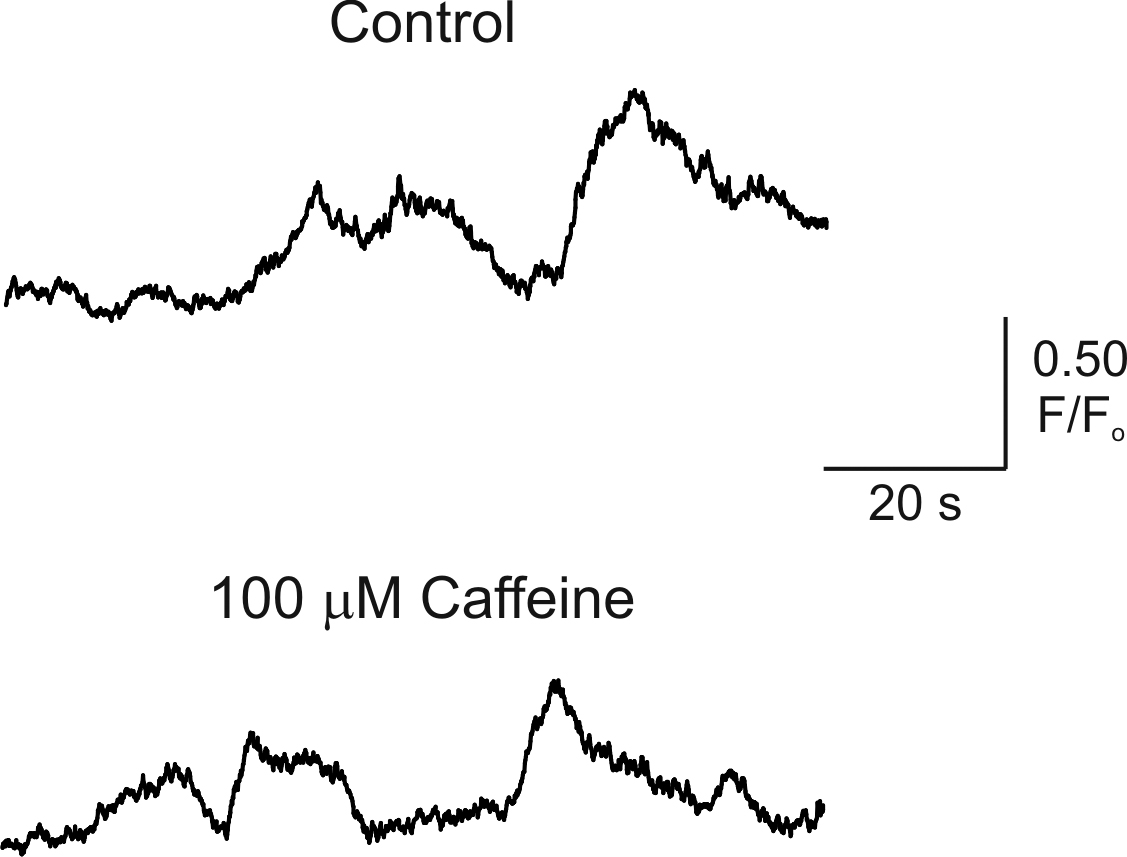
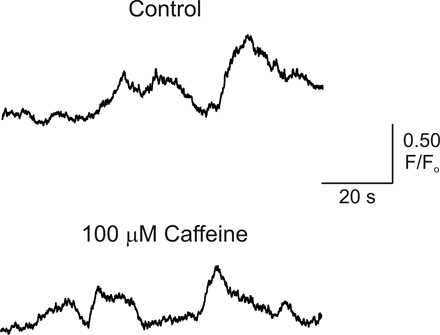
Studies of [Ca2+]i signaling in cultured astrocytes have suggested that Ca2+ release from InsP3R and RYR contribute to astrocytic [Ca2+]i signals (Golovina and Blaustein, 2000), although a contribution for Ca2+ release from RYR in the generation of astrocytic [Ca2+]i signals in situ has not been established (Beck et al., 2004). To determine whether photolysis of InsP3 results in Ca2+-induced Ca2+ release through RYR in endfeet, cortical slices were incubated with 50 μM ryanodine (20-min incubation) to block Ca2+ release from RYR. In the presence of ryanodine, peak [Ca2+]i increases induced by photorelease of InsP3 did not differ significantly from control responses (Fig. 3, B and E; 75.3 ± 9.5% of first response in ryanodine vs. 69.5 ± 7.5% of first response under control conditions; 7 ≤ n ≤ 10; P = 0.64). Because the effects of ryanodine on RYR are use dependent, such that ryanodine only binds to the open channel (Zucchi and Ronca-Testoni, 1997), we attempted to activate RYRs using caffeine, thereby increasing ryanodine binding to the channel. In the presence of 100 μM caffeine and 50 μM ryanodine (15–20-min incubation), InsP3-induced [Ca2+]i increases were 71.4 ± 9.8% of the first response (Fig. 3 E; n = 3; P = 0.9, compared with time-matched control), again suggesting a lack of involvement of RYR in generating endfoot [Ca2+]i increases. In addition, 100 μM caffeine alone failed to affect spontaneous astrocytic [Ca2+]i oscillations, which occur independently of neuronal signaling (Beck et al., 2004; Fig. S1, available at http://www.jgp.org/cgi/content/full/jgp.200609650/DC1). However, InsP3-induced endfoot [Ca2+]i increases were driven primarily by release of Ca2+ from intracellular stores, as treatment of slices with CPA to deplete the intracellular Ca2+ store significantly decreased the responsiveness of endfeet to InsP3 (Fig. 3, C and E; 13.7 ± 6.6% of first response for CPA treatment vs. 69.5 ± 7.5% under control conditions; 4 ≤ n ≤ 10; P < 0.0001). These findings suggest that endfeet possess the Ca2+ release machinery, in the form of InsP3R, to generate [Ca2+]i signals independently of the remainder of the astrocyte. These regenerative Ca2+ release signals likely underlie the generation of vasoactive substances involved in neurovascular coupling.
Perivascular Astrocytic Endfeet and Processes Generate Spatially Heterogeneous [Ca2+]i Signals
Traditionally, astrocytic [Ca2+]i increases involved in neuron–astrocyte–blood vessel signaling have been recognized as a wave that “travels” to the endfoot. Only a few studies have investigated the possibility that astrocytes contain spatially heterogeneous [Ca2+]i signaling domains, and no study to date has investigated the [Ca2+]i signaling dynamics of astrocytic endfeet. Thus, we investigated the spatial properties of neuronally mediated increases in [Ca2+]i in perivascular astrocytic endfeet and processes.
Perivascular astrocytic [Ca2+]i signals generated in response to neuronal activity exhibited nonhomogeneous complex behavior as observed by the following: (1) the presence of multiple distinct propagating [Ca2+]i waves within individual endfeet and perivascular processes; (2) heterogeneity in the [Ca2+]i increase in both endfeet and perivascular processes; (3) the presence of persistently elevated, spatially restricted regions of [Ca2+]i in endfoot regions directly apposed to arterioles; and (4) the presence of local Ca2+ release sites within endfeet. In the example shown in Fig. 4 A (also Video 2, available at http://www.jgp.org/cgi/content/full/jgp.200609650/DC1), in response to EFS, a propagating [Ca2+]i wave traveled down one perivascular process toward the endfoot (shown by arrow in 1-s snapshot). The mean rate of wave propagation in perivascular astrocytic processes studied in these experiments was 12.7 ± 0.8 μm/s (n = 3). This [Ca2+]i wave invaded the endfoot, which generated its own [Ca2+]i wave traveling perpendicular to the [Ca2+]i wave in the process (shown by arrow in 3-s snapshot). Regions of elevated [Ca2+]i within the endfoot were clearly visible before invasion of the endfoot by the [Ca2+]i wave (visible in 1-s snapshot), suggesting the presence of active Ca2+ release within the endfoot. In further support of active Ca2+ release within the endfoot, a spatially restricted region of elevated [Ca2+]i existed within the endfoot after decay of [Ca2+]i back to basal levels in the processes and most of the endfoot (15-s snapshot). On average, these regions of elevated [Ca2+]i occupied 37.7 ± 8.7% of the visible area of the endfoot 15 s after EFS (n = 3) and decayed back to basal levels within 45 s.
Regions of spatially localized [Ca2+]i increases within an individual endfoot are further illustrated in the representative example in Fig. 4 (B and C). In this endfoot, three spatially distinct regions of elevated [Ca2+]i were generated after EFS. The response was initiated at two sites within the endfoot, with the appearance of a third site at the opposite end of the endfoot. After the generation of a global [Ca2+]i increase within the endfoot, [Ca2+]i decreased last in the original sites of initiation. We saw no evidence of Ca2+ sparks or similar transient, highly localized Ca2+ release events that would suggest the presence of RYR within endfeet, even in the presence of the RYR activator caffeine (Fig. S1). Thus, the temporal properties of these [Ca2+]i signals (global [Ca2+]i increase was generated in ∼2 s after appearance of the initial [Ca2+]i increase), the polarity of the response ([Ca2+]i increase initiates and ends at the same location), and the lack of a ryanodine-sensitive release component are consistent with the assertion that endfoot [Ca2+]i increases are driven by Ca2+ release through InsP3Rs that are likely organized into heterogeneous Ca2+ release sites within endfeet.
Ca2+ Release from Endfoot InsP3R Is Sufficient to Activate Signaling Mechanisms that Dilate Adjacent Arterioles
The organization of the [Ca2+]i signaling machinery within astrocytic endfeet likely underlies the ability of astrocytes to rapidly modulate local cerebral blood flow in response to neuronal activity. To determine whether [Ca2+]i signals that are spatially restricted to a single endfoot and occur as a result of Ca2+ release through InsP3R are coupled to a vascular response, changes in the diameter of adjacent arterioles in response to the photolysis of caged InsP3 in endfeet were measured in brain slices. As shown in Fig. 5, photolysis of caged InsP3 in a single endfoot abutting an arteriole generated vasodilation of the arteriole, which remained localized to the region of the vessel in the immediate vicinity of the endfoot. As noted previously, we saw no evidence of a retrograde increase in [Ca2+]i in the astrocytic cell body. In response to these endfoot delimited [Ca2+]i signals, arterioles dilated to an average of 118 ± 6% of prestimulus diameter (n = 6; P = 0.03), and this dilation was restricted to an ∼30-μm length of the vessel. This InsP3-induced vasodilation occurred rapidly, with the dilation reaching 50% of the maximum within 1.6 ± 0.3 s (n = 4) of the uncaging event. The rapidity with which the vasodilation occurred after uncaging correlates well with the latency in the endfoot [Ca2+]i increase noted previously (1.00 ± 0.17 s from time of uncaging to 25% peak [Ca2+]i increase) and suggests that the vasodilation is indeed induced by the rise in endfoot [Ca2+]i that occurs as a result of activation of endfoot InsP3R. Overall, these findings support a model of neurovascular coupling in which highly dynamic, InsP3-mediated [Ca2+]i signaling events in astrocytic endfeet exert local control over vascular function.
Astrocytic Endfeet Generate [Ca2+]i Signals in Response to Putative Neurotransmitters Released by Local Interneurons in Contact with Endfeet
In addition to being covered with astrocytic endfoot processes, cerebral arterioles receive innervation from intrinsic neurons. Cauli et al. (2004) elegantly showed that depolarization of an individual γ-aminobutyric acid (GABA) interneuron can dilate or constrict an adjacent arteriole, depending on whether vasoactive intestinal peptide (VIP) or somatostatin (SOM) are released from the interneuron. Interestingly, these neurons also make extensive contact with endfoot processes (Hamel, 2006), which express receptors for several of the neurotransmitters released by these neurons (Porter and McCarthy, 1997). This raises the intriguing possibility that, in addition to glutamatergic input, astrocytes may receive an additional layer of information from local interneurons that might serve to fine-tune the endfoot [Ca2+]i signal and further modulate the vascular response, allowing for a more exquisite matching of vascular diameter changes to the metabolic needs of the neuronal system.
To investigate the effect of putative interneuron neurotransmitters on endfoot [Ca2+]i in cortical astrocytes, fluo-4 AM loaded cortical brain slices were stimulated with either SOM or VIP in the presence of 1 μM tetrodotoxin to limit neuronal activity, and the resultant effect of agonist treatment on endfoot [Ca2+]i monitored with confocal microscopy (Fig. 6). Rapid local application (pipette placed within ∼30 μm of endfoot) of SOM and VIP (1 μM each) in the vicinity of an endfoot induced large amplitude increases in astrocytic endfoot [Ca2+]i (mean peak [Ca2+]i increase, 2.27 ± 0.21 F/Fo units for SOM and 2.45 ± 0.46 units for VIP; 5 ≤ n ≤ 7). 1 μM neuropeptide Y was less effective in eliciting endfoot [Ca2+]i increases, with small (<1.4 F/Fo units) increases detected in only two out of five endfeet studied. These findings support the concept that endfoot [Ca2+]i changes can be elicited by neurotransmitters released from interneurons in contact with endfeet and illuminate a possible additional level of control over the generation of endfoot [Ca2+]i signals.
Figure 6.

Generation of astrocytic endfoot [Ca2+]i increases in response to treatment with vasoactive neuropeptides. (A and B) Fluo-4 AM loaded cortical slices were superfused with either SOM or VIP by means of a Picospritzer and locally (within 30 μm of endfoot) placed micropipette, and the resultant changes in astrocytic [Ca2+]i were measured with confocal microscopy. All agonist treatments were performed in the presence of 1 μM tetrodotoxin, which was included in the aCSF solution to limit neuronal activity.
DISCUSSION
Increases in [Ca2+]i in astrocytic endfeet, which encase cerebral arterioles, have been shown to underlie the generation of vasoactive factors involved in coupling neuronal activity to rapid, spatially localized changes in cerebral blood flow (Zonta et al., 2003; Filosa et al., 2004, 2006; Mulligan and MacVicar, 2004; Metea and Newman, 2006; Takano et al., 2006). Zonta et al. (2003) described this process in rat cortical brain slices and found that induction of neuronal activity led to an astrocytic [Ca2+]i signal that was dependent on activation of astrocytic mGluRs. This [Ca2+]i signal led to vasodilation of cerebral arterioles adjacent to astrocytic endfeet, likely through astrocytic production and release of prostaglandin E2, a response that could be mimicked by mechanical activation of an individual astrocyte. Extending these observations, Takano et al. (2006) found that photolysis of caged Ca2+ in individual astrocytic endfeet in vivo led to vasodilation of adjacent cerebral arterioles. This vasodilatory response was attributed to the Ca2+ and phospholipase A2–dependent generation of a cyclooxygenase product. Thus, elevation of endfoot [Ca2+]i is a critical link coupling neuronal activity to local vasodilation.
The following observations suggest that endfoot [Ca2+]i increases are highly dynamic and heterogeneous events driven primarily by Ca2+ release from InsP3R expressed in endfeet: (1) the peak amplitude of EFS-induced [Ca2+]i increases was decreased by InsP3R and phospholipase C inhibition and responses were virtually eliminated by depletion of the intracellular Ca2+ store; (2) photolysis of caged InsP3 within endfeet generated an endfoot delimited [Ca2+]i increase, with no evidence of [Ca2+]i elevation in regions of the astrocyte outside the endfoot; (3) InsP3 and EFS-induced endfoot [Ca2+]i increases were not altered by block of RYR; and (4) endfeet and perivascular processes generated nonuniform increases in [Ca2+]i and multiple distinct [Ca2+]i waves in response to neuronal activity. Furthermore, the current findings are the first to show that [Ca2+]i increases that are spatially restricted to endfeet and generated by Ca2+ release through InsP3R are sufficient to induce vasodilation of adjacent arterioles. In addition, we find no evidence for a contribution of Ca2+-induced Ca2+ release through RYR in the generation of endfoot [Ca2+]i increases in response to neuronal activity or photolysis of caged InsP3. Our findings do not rule out the possibility that RYRs contribute to the astrocytic [Ca2+]i signal in the soma. In addition, it is possible that RYRs may generate Ca2+ release events that affect local physiological responses without contributing significantly to global endfoot [Ca2+]i changes. Several studies have shown that [Ca2+]i increases in the astrocytic soma or processes can be blocked by inhibition of the InsP3–phospholipase C pathway and induced by photolysis of caged InsP3 (Golovina and Blaustein, 1997, 2000; Beck et al., 2004; Fiacco and McCarthy, 2004); the current results extend these findings to create an understanding of the processes underlying the generation of [Ca2+]i signals in endfeet.
Spatial heterogeneity in astrocytic [Ca2+]i signaling has been observed in primary cultured rat cortical astrocytes (Golovina and Blaustein, 1997) and Bergmann glial cells (Grosche et al., 1999) and rat hippocampal astrocytes (Araque et al., 2002) in situ. However, no study has yet investigated the spatial characteristics of endfoot [Ca2+]i signals. This information is vital because the spatial organization of endfoot [Ca2+]i signals likely underlies the specific activation of Ca2+-sensitive targets, such as large conductance Ca2+-sensitive potassium (BK) channels (Price et al., 2002; Filosa et al., 2006), which require micromolar [Ca2+]i for activation (Perez et al., 2001). Based on observations of regions of local Ca2+ release within individual endfeet (Fig. 4), as well as regions of persistently elevated [Ca2+]i within a fraction of the endfoot volume (Fig. 4 A), our findings support the assertion that local [Ca2+]i signaling domains are present in endfeet and are likely due to clustering of release channels within endfeet. Although the exact localization of Ca2+-sensitive proteins, such as BK channels or phospholipase A2, in the endfeet is not known, it is likely that these proteins are organized along endfoot membranes in contact with arterioles, an arrangement that would allow for privileged communication between endfeet and the vasculature, as has been argued for the expression of aquaporin 4 (Simard and Nedergaard, 2004). Thus, the spatial organization of local [Ca2+]i signals in endfeet, coupled with the polarized expression of effector proteins (Price et al., 2002), likely underlies the rapid and specific activation of Ca2+-sensitive signaling processes involved in modulating arteriolar diameter.
In the cortex, astrocytic [Ca2+]i increases generated in response to neuronal activity typically arise from the release of glutamate from presynaptic neurons and activation of mGluR on astrocytes (Kirischuk et al., 1999; Zonta et al., 2003). In addition, ionotropic NMDA, AMPA (α-amino-3-hydroxy-5-methylisoxazole-4-propionic acid), and kainate receptors expressed by astrocytes may allow for the influx of Ca2+ in response to glutamate binding (Clark and Barbour, 1997; Lalo et al., 2006). Our results illustrate an additional mechanism by which [Ca2+]i increases can arise in cortical astrocytes, namely, through release of neurotransmitters from γ-aminobutyric acid (GABA) interneurons in contact with endfeet. Specifically, we tested the neurotransmitters SOM and VIP, as these have been shown to induce vascular responses when perfused onto cerebral arterioles in brain slices and are present in interneurons that induce changes in arteriolar diameter when depolarized (Cauli et al., 2004). These authors showed opposing effects of these neurotransmitters on cerebral arterioles, with VIP inducing dilation and SOM inducing constriction of vessels. Interestingly, elevation of endfoot [Ca2+]i has also been shown to be capable of inducing both vasodilation (Zonta et al., 2003; Filosa et al., 2006; Metea and Newman, 2006; Takano et al., 2006) and vasoconstriction (Mulligan and MacVicar, 2004) of cerebral arterioles. Although the work of Cauli et al. (2004) clearly illustrates a direct effect of interneurons on arterioles, the current findings suggest that the effects of direct vascular innervation from interneurons might be fine-tuned by additional signals from endfeet, with the interaction of both signaling mechanisms eliciting specific degrees and/or periods of vascular diameter change.
Neurovascular coupling must be rapid and spatially localized, such that an increase in blood flow is delivered to the site of increased metabolic demand (Iadecola, 2004). Thus, mechanisms must exist to spatially restrict increases in blood flow in response to neuronal activity. Based on the local vasodilating influence of InsP3-induced [Ca2+]i increases in individual astrocytic endfeet (Fig. 5), our findings suggest that individual endfeet exert control over the regulation of arteriolar diameter within a spatially restricted region of the arteriole that extends only a short distance from the main endfoot process. This finding implies that the extent of spread of the increase in blood flow will be determined by the number of endfeet activated, which in turn is a function of the extent of neuronal activity. Thus, endfeet serve as individual “vasoregulatory units,” the sum total of which defines the region in which blood flow changes are generated.
Our findings suggest that astrocytic endfeet serve as active participants in the process of neurovascular coupling and are capable of generating dynamic and spatially specific [Ca2+]i signals and exerting local control over arteriolar diameter. These findings further illuminate the critical role that astrocytes serve in coupling neuronal activity to changes in local cerebral blood flow and highlight the ability of endfoot [Ca2+]i signals to specifically activate Ca2+-sensitive processes that ultimately lead to spatially restricted alterations in cerebral arteriolar diameter.
Supplemental Material
Acknowledgments
We thank Drs. D. Hill-Eubanks and S. Earley for critical evaluation of the manuscript and K. Brunelle and S. Cawley for technical assistance.
This work was supported by the National Heart, Lung, and Blood Institute of the National Institutes of Health through a postdoctoral fellowship to S.V. Straub (HL83768), a postdoctoral training grant to M.K. Wilkerson (HL07944), and a grant to M.T. Nelson (HL44455), and by the Totman Trust for Medical Research.
Olaf S. Andersen served as editor.
Abbreviations used in this paper: aCSF, artificial cerebrospinal fluid; [Ca2+]i, cytosolic Ca2+ concentration; CPA, cyclopiazonic acid; EFS, electrical field stimulation; InsP3, inositol 1,4,5-trisphosphate; InsP3R, InsP3 receptor; mGluR, metabotropic glutamate receptor; RyR, ryanodine receptor; SERCA, sarcoplasmic/endoplasmic reticulum Ca2+ ATPase; SMC, smooth muscle cell; SOM, somatostatin; VIP, vasoactive intestinal peptide.
References
- Araque, A., E.D. Martin, G. Perea, J.I. Arellano, and W. Buno. 2002. Synaptically released acetylcholine evokes Ca2+ elevations in astrocytes in hippocampal slices. J. Neurosci. 22:2443–2450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beck, A., R.Z. Nieden, H.P. Schneider, and J.W. Deitmer. 2004. Calcium release from intracellular stores in rodent astrocytes and neurons in situ. Cell Calcium. 35:47–58. [DOI] [PubMed] [Google Scholar]
- Brown, L.A., B.J. Key, and T.A. Lovick. 2002. Inhibition of vasomotion in hippocampal cerebral arterioles during increases in neuronal activity. Auton. Neurosci. 95:137–140. [DOI] [PubMed] [Google Scholar]
- Cauli, B., X.K. Tong, A. Rancillac, N. Serluca, B. Lambolez, J. Rossier, and E. Hamel. 2004. Cortical GABA interneurons in neurovascular coupling: relays for subcortical vasoactive pathways. J. Neurosci. 24:8940–8949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark, B.A., and B. Barbour. 1997. Currents evoked in Bergmann glial cells by parallel fibre stimulation in rat cerebellar slices. J. Physiol. 502:335–350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cornell-Bell, A.H., S.M. Finkbeiner, M.S. Cooper, and S.J. Smith. 1990. Glutamate induces calcium waves in cultured astrocytes: long-range glial signaling. Science. 247:470–473. [DOI] [PubMed] [Google Scholar]
- Fiacco, T.A., and K.D. McCarthy. 2004. Intracellular astrocyte calcium waves in situ increase the frequency of spontaneous AMPA receptor currents in CA1 pyramidal neurons. J. Neurosci. 24:722–732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Filosa, J.A., A.D. Bonev, and M.T. Nelson. 2004. Calcium dynamics in cortical astrocytes and arterioles during neurovascular coupling. Circ. Res. 95:e73–e81. [DOI] [PubMed] [Google Scholar]
- Filosa, J.A., A.D. Bonev, S.V. Straub, A.L. Meredith, M.K. Wilkerson, R.W. Aldrich, and M.T. Nelson. 2006. Local potassium signaling couples neuronal activity to vasodilation in the brain. Nat. Neurosci. 9:1397–1403. [DOI] [PubMed] [Google Scholar]
- Golovina, V.A., and M.P. Blaustein. 1997. Spatially and functionally distinct Ca2+ stores in sarcoplasmic and endoplasmic reticulum. Science. 275:1643–1648. [DOI] [PubMed] [Google Scholar]
- Golovina, V.A., and M.P. Blaustein. 2000. Unloading and refilling of two classes of spatially resolved endoplasmic reticulum Ca2+ stores in astrocytes. Glia. 31:15–28. [DOI] [PubMed] [Google Scholar]
- Grosche, J., V. Matyash, T. Moller, A. Verkhratsky, A. Reichenbach, and H. Kettenmann. 1999. Microdomains for neuron-glia interaction: parallel fiber signaling to Bergmann glial cells. Nat. Neurosci. 2:139–143. [DOI] [PubMed] [Google Scholar]
- Hamel, E. 2006. Perivascular nerves and the regulation of cerebrovascular tone. J. Appl. Physiol. 100:1059–1064. [DOI] [PubMed] [Google Scholar]
- Iadecola, C. 2004. Neurovascular regulation in the normal brain and in Alzheimer's disease. Nat. Rev. Neurosci. 5:347–360. [DOI] [PubMed] [Google Scholar]
- Kirischuk, S., F. Kirchhoff, V. Matyash, H. Kettenmann, and A. Verkhratsky. 1999. Glutamate-triggered calcium signalling in mouse bergmann glial cells in situ: role of inositol-1,4,5-trisphosphate- mediated intracellular calcium release. Neuroscience. 92:1051–1059. [DOI] [PubMed] [Google Scholar]
- Kuffler, S.W., J.G. Nicholls, and R.K. Orkand. 1966. Physiological properties of glial cells in the central nervous system of amphibia. J. Neurophysiol. 29:768–787. [DOI] [PubMed] [Google Scholar]
- Lalo, U., Y. Pankratov, F. Kirchhoff, R.A. North, and A. Verkhratsky. 2006. NMDA receptors mediate neuron-to-glia signaling in mouse cortical astrocytes. J. Neurosci. 26:2673–2683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Metea, M.R., and E.A. Newman. 2006. Glial cells dilate and constrict blood vessels: a mechanism of neurovascular coupling. J. Neurosci. 26:2862–2870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mulligan, S.J., and B.A. MacVicar. 2004. Calcium transients in astrocyte endfeet cause cerebrovascular constrictions. Nature. 431:195–199. [DOI] [PubMed] [Google Scholar]
- Nagelhus, E.A., T.M. Mathiisen, and O.P. Ottersen. 2004. Aquaporin-4 in the central nervous system: cellular and subcellular distribution and coexpression with KIR4.1. Neuroscience. 129:905–913. [DOI] [PubMed] [Google Scholar]
- Perea, G., and A. Araque. 2005. Glial calcium signaling and neuron-glia communication. Cell Calcium. 38:375–382. [DOI] [PubMed] [Google Scholar]
- Perez, G.J., A.D. Bonev, and M.T. Nelson. 2001. Micromolar Ca2+ from sparks activates Ca2+-sensitive K+ channels in rat cerebral artery smooth muscle. Am. J. Physiol. Cell Physiol. 281:C1769–C1775. [DOI] [PubMed] [Google Scholar]
- Porter, J.T., and K.D. McCarthy. 1997. Astrocytic neurotransmitter receptors in situ and in vivo. Prog. Neurobiol. 51:439–455. [DOI] [PubMed] [Google Scholar]
- Price, D.L., J.W. Ludwig, H. Mi, T.L. Schwarz, and M.H. Ellisman. 2002. Distribution of rSlo Ca2+-activated K+ channels in rat astrocyte perivascular endfeet. Brain Res. 956:183–193. [DOI] [PubMed] [Google Scholar]
- Simard, M., and M. Nedergaard. 2004. The neurobiology of glia in the context of water and ion homeostasis. Neuroscience. 129:877–896. [DOI] [PubMed] [Google Scholar]
- Simard, M., G. Arcuino, T. Takano, Q.S. Liu, and M. Nedergaard. 2003. Signaling at the gliovascular interface. J. Neurosci. 23:9254–9262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takano, T., G.F. Tian, W. Peng, N. Lou, W. Libionka, X. Han, and M. Nedergaard. 2006. Astrocyte-mediated control of cerebral blood flow. Nat. Neurosci. 9:260–267. [DOI] [PubMed] [Google Scholar]
- Verkhratsky, A., R.K. Orkand, and H. Kettenmann. 1998. Glial calcium: homeostasis and signaling function. Physiol. Rev. 78:99–141. [DOI] [PubMed] [Google Scholar]
- Wang, X., N. Lou, Q. Xu, G.F. Tian, W.G. Peng, X. Han, J. Kang, T. Takano, and M. Nedergaard. 2006. Astrocytic Ca2+ signaling evoked by sensory stimulation in vivo. Nat. Neurosci. 9:816–823. [DOI] [PubMed] [Google Scholar]
- Zonta, M., M.C. Angulo, S. Gobbo, B. Rosengarten, K.A. Hossmann, T. Pozzan, and G. Carmignoto. 2003. Neuron-to-astrocyte signaling is central to the dynamic control of brain microcirculation. Nat. Neurosci. 6:43–50. [DOI] [PubMed] [Google Scholar]
- Zucchi, R., and S. Ronca-Testoni. 1997. The sarcoplasmic reticulum Ca2+ channel/ryanodine receptor: modulation by endogenous effectors, drugs and disease states. Pharmacol. Rev. 49:1–51. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}