

Abstract
In nerve and muscle cells, the voltage-gated opening and closing of cation-selective ion channels is accompanied by the translocation of 12–14 elementary charges across the membrane's electric field. Although most of these charges are carried by residues in the S4 helix of the gating module of these channels, the precise nature of their physical movement is currently the topic of spirited debate. Broadly speaking, two classes of models have emerged: those that suggest that small-scale motions can account for the extensive charge displacement, and those that invoke a much larger physical movement. In the most recent incarnation of the latter type of model, which is based on structural and functional data from the archaebacterial K+ channel KvAP, a “voltage-sensor paddle” comprising a helix-turn-helix of S3–S4 translocates ∼20 Å through the bilayer during the gating cycle (Jiang, Y., A. Lee, J. Chen, V. Ruta, M. Cadene, B.T. Chait, and R. MacKinnon. 2003. Nature. 423:33–41; Jiang, Y., V. Ruta, J. Chen, A. Lee, and R. MacKinnon. 2003. Nature. 423:42–48.; Ruta, V., J. Chen, and R. MacKinnon. 2005. Cell. 123:463–475). We used two methods to test for analogous motions in the Shaker K+ channel, each examining the aqueous exposure of residues near S3. In the first, we employed a pore-blocking maleimide reagent (Blaustein, R.O., P.A. Cole, C. Williams, and C. Miller. 2000. Nat. Struct. Biol. 7:309–311) to probe for state-dependent changes in the chemical reactivity of substituted cysteines; in the second, we tested the state-dependent accessibility of a tethered biotin to external streptavidin (Qiu, X.Q., K.S. Jakes, A. Finkelstein, and S.L. Slatin. 1994. J. Biol. Chem. 269:7483–7488; Slatin, S.L., X.Q. Qiu, K.S. Jakes, and A. Finkelstein. 1994. Nature. 371:158–161). In both types of experiments, residues predicted to lie near the top of S3 did not exhibit any change in aqueous exposure during the gating cycle. This lack of state dependence argues against large-scale movements, either axially or radially, of Shaker's S3–S4 voltage-sensor paddle.
INTRODUCTION
The key process underlying the electrical activity of excitable tissue is the voltage-dependent opening and closing of tetrameric Na+, Ca2+, and K+ channels. This gating action is mediated by a voltage sensor whose movement is somehow coupled to an intracellular activation gate contained within the channel's S5–S6 pore domain (Yellen, 1998). Sequence analysis and early mutagenesis studies implicated the positively charged S4 segment as the likely voltage sensor, and more detailed gating measurements suggested that the first four charged residues in S4 account for most of the 12–14 charges per channel that are translocated across the membrane's electric field (Aggarwal and MacKinnon, 1996; Seoh et al., 1996). Despite significant advances in molecular and structural biology, however, the actual physical movements that give rise to this extensive charge displacement remain the topic of intense debate.
Further insight into the motions of the voltage sensor came from an analysis of the exposure of S4 residues to the extracellular or intracellular milieus. Several of the charged residues in S4, when substituted to cysteine, reacted with sulfhydryl-specific probes in a state-dependent fashion (Yang and Horn, 1995; Larsson et al., 1996; Yang et al., 1996; Yusaf et al., 1996). Assuming that S4 remains helical throughout the gating cycle, the pattern of reactivity suggested one of two possible physical models: (1) S4 moves only a small distance, but through a concentrated and mobile electric field within an aqueous crevice whose shape and accessibility change with the gating state; or (2) each S4 segment undergoes a large transverse movement during gating and is able to translocate approximately three charged residues through an electric field that need not be concentrated over a small area. Significant state-dependent changes in the environment of S4 residues were confirmed by additional thiol-specific probing (Baker et al., 1998; Wang et al., 1999) and fluorescence measurements (Mannuzzu et al., 1996; Gandhi et al., 2000), however, the possibility that S4 moves only small distances garnered further support over the next several years from histidine scanning studies (Starace et al., 1997; Starace and Bezanilla, 2001), electrostatic calculations (Islas and Sigworth, 2001), and resonance energy transfer measurements (Cha et al., 1999; Glauner et al., 1999).
Structural and functional data from KvAP, a prokaryotic voltage-gated K+ channel, prompted MacKinnon and colleagues to champion the notion of large-scale gating motions. X-ray crystal structures of the whole channel, as well as its isolated gating module, reveal S4 to be closely associated with a portion of S3 (S3b) in a helix-turn-helix motif, termed a “voltage-sensor paddle,” that is held together by hydrophobic interactions (Jiang et al., 2003a; Lee et al., 2005). To gain further insight into the possible motions that the voltage sensor might undergo during gating, Jiang et al. (2003b) complemented the structural data with a biotin-avidin functional assay (Qiu et al., 1994; Slatin et al., 1994). They found that at several positions in S3–S4, biotinylated cysteines bind to external avidin much more readily at depolarized voltages than at hyperpolarized voltages, and a biotin tethered to either of two residues in S4 (L121C and L122C) binds to avidin from either side of the bilayer. These measurements, as well as those from a more extensive mapping of KvAP residues using this approach (Ruta et al., 2005), led to the conclusion that the sizeable gating charge measured in voltage-gated channels arises from large (∼20 Å) transverse displacements of the paddle through the lipid bilayer as it shuttles back and forth during the gating cycle. A further assertion was that this greasy cation moves at the protein–lipid interface, largely surrounded by lipid, and that it forms salt bridges with charges in S2 or S3, as originally suggested by mutagenesis studies (Tiwari-Woodruff et al., 1997, 2000).
In a more recent effort, MacKinnon and colleagues determined the structure of the mammalian Shaker homologue Kv 1.2 in complex with a β subunit docked to its T1 domain (Long et al., 2005a,b). By using both lipids and detergents in the purification and crystallization process, they overcame some of the technical problems that limited interpretation of the KvAP structure; in the Kv 1.2 structure, the gating module was able to adopt a more native conformation and orientation. Here the voltage sensor paddle is tilted somewhat from the central axis, but it is more vertically oriented than the paddles in the KvAP structures, and, in agreement with a large body of accessibility data, the tops of S3 and S4 are positioned at or near the extracellular solution. Less clear is the precise composition of S3 and the S3–S4 loop since the electron density in these regions was not strong enough to permit a clear assignment of residues. Furthermore, since the structure of Kv 1.2 appears to be of an open (and possibly inactivated) channel, the paddle's location in the closed state remains an unanswered question.
Although a model in which charged residues move through lipid may not seem energetically favorable from an electrostatic perspective, this potentially could be offset by the gain in free energy imparted by the hydrophobic effect that would favor greasy residues of S3 and S4 maintaining contact with lipid (Freites et al., 2005; Hessa et al., 2005a,b). Such an effect may also explain how gating in diphtheria toxin channels is associated with the translocation of massive stretches of the protein that includes charged residues, possibly through a lipid environment (Oh et al., 1999; Finkelstein et al., 2000; Senzel et al., 2000; Gordon and Finkelstein, 2001). Nevertheless, several recent observations in mammalian voltage-gated K+ channels appear to be inconsistent with a large transmembrane motion of S3–S4: the gating modifier Hanatoxin remains bound to Kv 2.1's voltage sensor in both activated and resting states and remains within the bilayer's outer leaflet (Lee et al., 2003; Phillips et al., 2005), thiol-reactive compounds react with some S3 residues in Shaker equally well in both states (Gandhi et al., 2003; Gonzalez et al., 2005), and resonance energy transfer experiments detect only small motions (Chanda et al., 2005; Posson et al., 2005).
Techniques that rely on aqueous accessibility to small probes as a surrogate for movement have several limitations. Large motions may go undetected if they do not lead to changes in accessibility, small motions can yield large changes in reactivity, and the presence of water-filled crevices can further cloud interpretation. Although resonance energy transfer techniques that measure distances between donor and acceptor molecules can overcome some of these issues, this method also has technical limitations, including the size of the fluorophores, their attachment via flexible linkers, and preservation of orientation factors (Horn, 2000). In this paper we explore the range of voltage sensor motion in the Shaker potassium channel by examining the kinetics of two different chemical reactions: covalent tethering of maleimido-quaternary ammonium (QA) compounds to cysteines, and binding of streptavidin to biotinylated residues. Our approach offers two main advantages: (1) unlike methanethiosulfonate (MTS) reagents whose kinetics are determined only by cysteine reactivity, kinetics of tethered block are influenced both by reactivity and changes in distance to the pore that would occur during gating, and (2) the use of a larger probe like streptavidin can address concerns about the presence of aqueous crevices that allow access to smaller probes. Our results argue that residues near the top of Shaker's S3 segment do not move large distances either vertically, or toward or away from the pore during gating.
MATERIALS AND METHODS
Molecular Biology
Shaker B lacking the N-type inactivation sequence (Δ6–46) (Schwarz et al., 1988; Hoshi et al., 1990) was cloned into pBluescript KS (Stratagene); our “wild-type” construct also contained the point mutation F425G in the channel's external vestibule, as well as the T449F mutation to increase TEA blocking affinity (MacKinnon and Yellen, 1990; Heginbotham and MacKinnon, 1992; Kavanaugh et al., 1992). All mutations described were made using PCR-based methods and were confirmed by sequencing through the cloning cassette. For the biotin-streptavidin experiments described below, we used as our background a triple-cys(−) construct (C245V:C286V:C462A) in order to avoid potential interference from labeling at these positions. Mutant cRNA was transcribed in vitro from linearized plasmids using T7 RNA-polymerase (Promega), aliquoted, and stored at −80°C.
Electrophysiology and Data Analysis
Stage V–VI oocytes were harvested, defolliculated, injected with cRNA, and maintained at 17°C in an ND96-gentamicin solution containing DTT to prevent cysteine oxidation, as previously described (Blaustein, 2002). 1–5 d following injection, oocytes were transferred to a homemade low volume (75–100 μl) chamber containing a 0.3 mM CaCl2 ND96 solution without DTT or gentamicin, impaled with glass electrodes (Garner; KG-33) having resistances of 0.3–1.0 MΩ filled with 3 M KCl, 5 mM EGTA, and 10 mM HEPES, pH 7.6, and examined using a two-electrode voltage clamp amplifier (OC-725B; Warner Instruments). The voltage clamp amplifier was interfaced to a Digidata 1322A acquisition board (Axon Instruments) and a Windows-based computer running Axon's pClamp software (v 8.0). Electrical contact to the bath solution was made via 200 mM NaCl agar bridges. Maleimide-Gly7-TEA (abbreviated in future use as Gly7TEA; Fig. 1 C) was synthesized as previously described (Blaustein et al., 2000). The concentration of a stock solution of known volume was determined by quantitative amino acid analysis (Yale HHMI/Keck Biopolymer Facility). This solution was then aliquoted, lyophilized, stored dry at −20°C, and dissolved immediately before use. Gly7TEA dissolved in recording solution was added to the chamber via addition of 20 μl of a 5× stock solution to 80 μl of solution in the chamber, followed immediately by rapid withdrawal and readdition of 20 μl of the resultant solution to achieve 90% mixing within 5 s.
Figure 1.

(A) Transmembrane topology of a Shaker K+ channel subunit highlighting residues discussed in the text. (B) Alignment of Shaker and KvAP from Jiang et al. (2003a) showing the difference in length of the two S3–S4 linkers. Residues discussed in the text are in bold; arrows indicate portion of Shaker's S3–S4 loop removed via mutagenesis. (C) Chemical structure of maleimido-Gly7TEA (abbreviated as Gly7TEA).
Charge–voltage (Q-V) relationships in our WT Shaker channel and 334C mutant were determined by measuring gating currents in whole oocytes via two-electrode voltage clamp. Ionic current was eliminated by recording in the presence of 1 μM Agitoxin2 (provided by C. Miller, Brandeis University, Waltham, MA) and replacing the Na+ and K+ in the external solution with 95 mM NMDG. Oocytes were stepped from a potential of −120 to +40 mV in 10-mV increments for the “on” gating currents, and then back to −120 mV for the “off” currents. For each oocyte, the nonlinear components of the “on” and “off” capacitance currents were integrated (Aggarwal and MacKinnon, 1996), and then averaged and normalized to generate a Q-V for that oocyte (Lee et al., 2003; Kitaguchi et al., 2004). Normalized Q-V's for each oocyte were then averaged and fitted to the Boltzmann function Qnorm = (1 + exp(−zF(V − V1/2)/RT)−1.
Kinetics of Tethering Gly7TEA
Gly7TEA kinetic data were analyzed in the context of the model for maleimido-QA tethering previously developed (Blaustein, 2002). The four tethering reactions in a channel bearing four cysteine targets are represented by a kinetic scheme containing five states, depicting channels that have not reacted (C 0), channels that have undergone one tethering reaction (C 1), etc. (Scheme I, Blaustein, 2002):Scheme 1 is described by a set of linked first-order differential equations whose solutions C 0(t), C 1(t)…C 4(t) are sums of exponential functions with the pseudo first-order rate constants k 1…k 4 appearing in the exponents. Although the observed normalized current I(t) was expressed in Blaustein (2002) as the sum of those concentrations weighted by the fraction of unblocked channels in each state, it can also be written as
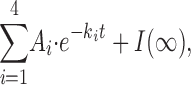 |
where I(∞) is the remaining fraction of current after all channels have reacted, k 1…k 4 are functions of the intrinsic second-order rate constant for the cysteine-maleimide reaction (k), the effective local concentration of Gly7TEA's maleimide end at its cysteine target when its QA end occupies the pore (Mt), the concentration of free Gly7TEA (Bf), the Kd for reversible block of the QA end of Gly7TEA at the pore, and each Ai is a function of k 1…k 4, Mt, Bf, and Kd (but independent of k). Covalent tethering of Gly7TEA is accelerated by an affinity label effect in which reversible pore block by its QA moiety serves to concentrate its maleimide end near its cysteine target. This effect is greatest for the first of the four tethering reactions in each channel (i.e., k 1 ≫ k 2 > k 3 > k 4, and A 1 ≫ A 2 > A 3 > A 4) since the initial tethering reaction creates a cloud of concentrated QA that inhibits the subsequent binding of the QA end of a Gly7TEA free in solution. The time course of the tethering reaction therefore appears biphasic and well fitted by a double-exponential function, with a fast time constant and weight dominated largely by 1/k 1 and A 1, respectively, and a slower time constant whose reciprocal lies somewhere between k 2 and k 4, and whose weight reflects the sum of A 2, A 3, and A 4.
(SCHEME 1).

Tethering rates of Gly7TEA to target cysteines in the open and closed state were compared by applying pulse protocols that varied the relative amounts of time that channels were held depolarized or hyperpolarized (Larsson et al., 1996). In our “short-pulse” protocols the voltage sensors were held in the activated position 1–2% of the time by applying a 50–100-ms depolarizing test pulse to +20 mV every 5 s from a holding potential of −110 mV. In the “long-pulse” protocols channels were open for 33% of the time by applying a 1-s pulse to +20 mV every 3 s. Currents were filtered at 1 kHz and sampled at 10 kHz. Time courses of inhibition were obtained by plotting isochronal currents (1 ms before the end of the depolarizing pulse) with time, and were fitted to double-exponential functions using Origin's (v 7, Microcal Software) implementation of the Levenberg-Marquhart algorithm.
Two types of gating motions influence the kinetics of Gly7TEA tethering. Vertical movement of a target cysteine into and out of the bilayer affects only its reactivity (k); time courses of inhibition obtained with long- and short-pulse protocols would differ only with respect to the taus of their fits, with fast and slow components changing by the same degree since each k 1…k 4 is proportional to k. The associated weights will not change since the Ai's are independent of k. The effect of a pure radial motion is more complex and depends on both the distance from the cysteine to the pore and the extent that this changes during gating. (In our discussion of movements towards or away from [or distances to or from] the pore, we are referring to the channel's external TEA binding site at the outer mouth of the pore rather than the vertical axis of symmetry that is coincident with the permeation pathway.) Polymer theory dictates that this parameter influences the affinity label effect by changing the effective maleimide concentration at the cysteine when Gly7TEA's QA group is bound to the pore (Flory, 1969). Therefore, if a cysteine moved closer to the pore during depolarization, tethering would proceed more rapidly when measured with our long-pulse protocol than with our short-pulse protocol. The functional relationship of the ki's and Ai's on Mt predicts that the taus for the long-pulse fits would be faster, and more heavily weighted. The extent of the shift in weights depends on the tethered concentration of Gly7TEA's QA group at the pore, with higher concentrations leading to more pronounced shifts.
To estimate the effect of a 5-Å radial movement of S3 on the kinetics of Gly7TEA tethering under our long-pulse protocol, we consider the case in which the top of S3 is 30 Å from the channel's external TEA binding site in the depolarized state (duration = 1 s), and 35 Å away in the hyperpolarized state (duration = 2 s; see Fig. 7, A and B) (Blaustein et al., 2000; Posson et al., 2005). Kinetics resulting from application of the short-pulse protocol and from 5 Å movements away from the pore during depolarization are determined analogously. k 1…k 4 are calculated according to Eqs. 1–4 in Blaustein (2002) using a value of 50 μM for Bf (as in our experiments), a value of 30 M−1s−1 for k (estimated by fits of our Gly7TEA data to the model), and a value of 1.5 mM for the Kd for reversible block of the QA end of Gly7TEA at the pore, determined experimentally. The other parameter used to calculate the rate constants is Mt, the effective local concentration of Gly7TEA's maleimide end at the top of S3 when its QA end occupies the pore, and it is this quantity that varies as S3 moves toward or away from the pore. To determine its value in each case, we approximate Gly7TEA as a polyglycine molecule and model its behavior using polymer statistical methods. P(r), the concentration of one end of the polymer at a distance r from its other end, is calculated from the exact solution of the random flight method applied to the freely jointed chain (see Ch. 8 of Flory, 1969):
 |
where P(r) is in M, r is in Å, l is the length of each repeating segment, n is the number of segments, and 1.66 × 103 is a factor that converts to molar units. This equation is more accurate than the Gaussian approximation at predicting the behavior of a polymer near its extended length, a regime of interest here. A value of 8.6 Å is used for l; this “statistical segment,” which comprises ∼2.3 glycines, provides a more realistic approximation for polyglycine in the freely jointed model (Flory, 1969). A value of 5 is used for n, which yields a polymer whose extended length of 43 Å is nearly that of Gly7TEA. For the 5-Å movement described above, P(r) is determined via numerical integration for r = 30 and 35 Å using Mathcad software (v. 11). P(30) and P(35) are then each used for the value of Mt in the kinetic model to yield a set of four rate constants, written in shorthand notation as k 30 for the constants during the 1-s depolarized portion of the pulse, and k 35 for those during the 2-s hyperpolarizing phase (see Fig. 7 B). Switching back and forth between these two different sets of rate constants with each pulse requires that the values of C 0(t)…C 4(t) at the end of the depolarizing portion of the pulse (determined using k 30) serve as the initial values for C 0(t)…C 4(t) for the hyperpolarizing portion of the pulse (determined using k 35), and vice versa at the start of the next pulse. This necessitated modification of C 0(t)…C 4(t) in Blaustein (2002) to allow for arbitrary initial conditions, as the differential equations describing Scheme 1 were originally solved using the initial condition that all channels are unreacted at t = 0 (C 0(t) = 1 and C 1(t)…C 4(t) = 0). Since the four rate constants for the inactivated state are not equal to the corresponding four rate constants in the activated state, simulating I(t) in this fashion does not yield a simple sum of four exponentials. The simulated data are, however, well fitted by double-exponential functions since they exhibit a fast phase dominated by contributions from the faster rate constants in each state, and a slower phase that reflects a mix of the slower rate constants in each state.
Figure 7.

Kinetic simulations of a 5-Å movement of the top of S3 toward (A–C) or away (D–F) from the pore during depolarization. Movement of S3 along the surface of a hemisphere 35 Å from the pore, depicted as a solid circular arc in A and D, would preserve radial distance. An inward movement during depolarization would involve a displacement of S3 to the surface of a hemisphere at 30 Å, depicted as a dotted circular arc in A, and an outward movement would involve a displacement to the dotted hemisphere at 40 Å in D. (B and E) Templates of pulse protocols used for the simulations. Upper protocols have 50-ms depolarizations followed by 4.95-s hyperpolarizations; bottom protocols are 1-s depolarizations followed by 2-s hyperpolarizations. k 30 represents a set of four rate constants determined at 30 Å as described in the text; k 35 and k 40 are similarly determined at 35 and 40 Å, respectively. (C and F) Simulated time courses. Black curves are generated by application of short-pulse protocols; gray curves from long-pulse protocols. Each time course is generated by switching back and forth between the two sets of rate constants depicted in each protocol template in B and E.
Biotinylation and Inhibition with Streptavidin
Oocytes expressing the D336C mutation in the triple-cys(−) construct described above were maintained in ND-96 containing 1% DTT at 17°C, rinsed in DTT-free ND-96, and biotinylated with 1 mM N-biotinylaminoethyl methanethiosulfonate (MTSEA-biotin; Toronto Research Chemicals) or 1 mM N-(β-d-glucopyranosyl)-N′-[(2-methanethiosulfonyl) ethyl] urea (MTS-glucose; Toronto Research Chemicals) under depolarizing conditions using a high K+ buffer (52 mM KCl, 48 mM NaCl, 1 mM MgCl2, 0.3 mM CaCl2, 10 mM HEPES) for 1 h at room temperature. Under these conditions, channels are fully biotinylated as judged from a lack of any subsequent inhibition upon exposure to Gly7TEA. They were then devitellinized in a hypertonic solution (296 mM NaCl, 1 mM MgCl2, 0.3 mM CaCl2, 10 mM HEPES), transferred into 2% agar-coated dishes filled with ND-96, and allowed to recover for 2–3 h at 17°C. We found that devitellinizing the oocyte led to a more robust and reproducible effect of streptavidin, and the recovery period eliminated leak currents that are present if we record immediately after devitellinization. Currents through biotinylated channels were measured by two-electrode voltage clamp as described above, with minor modification to the voltage protocol. In the short-pulse protocol, oocytes were held at −90 mV and subjected to a 100-ms depolarizing pulse to +20 mV every 10 s; in the long-pulse protocol, oocytes received 1-s pulses every 10 s (this construct exhibited more rapid C-inactivation, likely due to the C462A mutation, and necessitated the longer interpulse interval to allow adequate recovery). Tetrameric streptavidin (Calbiochem/EMD Biosciences) was added by pipetting a 1 mg/ml (in ND-96) solution into the chamber and rapidly mixing to a final concentration of 200 μg/ml (3.3 μM). Time courses obtained with the short-pulse protocol were generated by plotting the isochronal currents at 99 ms (1 ms prior at the end of each depolarized pulse) as a function of time. For comparison with long-pulse protocol experiments (Fig. 6 C), these latter currents were also obtained 99 ms into each pulse. To quench the streptavidin reaction at desired time points, d-biotin (Sigma-Aldrich) was rapidly added to a final concentration of 500 μM; these experiments were done using long pulses, and isochronal currents at 999 ms were used in generating the time courses of Fig. 6 D.
Figure 6.

Kinetics of inhibition of biotinylated D336C channels by streptavidin. (A) Pulse protocols are as described in Fig. 4, except that depolarizations are every 10 s to allow for recovery from inactivation, and the hyperpolarized voltage was −90 mV in each case. (B) Currents through channels that have been biotinylated with MTSEA-biotin. Top traces in each set are before addition of 3.3 μM streptavidin; bottom traces are after ∼25 min of exposure. (C) Kinetics of inhibition. Isochronal currents at 99 ms were normalized to the values before streptavidin exposure, averaged from several experiments, and plotted against time. Error bars are ± SEM. Short-pulse protocol (n = 6), circles; long-pulse protocol (n = 8), triangles. Control experiments (three upper time courses) generated using the long-pulse protocol: diamonds, D336C with 1 mM MTSEA-biotin added to chamber; inverted triangles, streptavidin applied to D336 channels; squares, streptavidin applied to D336C channels that were prereacted with 1 mM MTS-glucose. (D) Biotin quenching using long-pulse protocols. 500 μM d-biotin added 60 s before addition of streptavidin (diamonds), and at 30 (squares), 60 (pentagons), and 120 s (circles) after streptavidin exposure. These time courses are from isochronal currents at 999 ms and are superimposed on the long-pulse data (triangles) obtained from the same experiments as in (C) except isochronal currents at 999 ms are plotted.
RESULTS
We substituted cysteine for three residues (singly) near the extracellular end of S3 (Fig. 1 A), and examined the chemical reactivity of each to Gly7TEA, a flexible 45-Å-long thiol-specific reagent (Fig. 1 C) that acts as a tethered pore blocker (Blaustein et al., 2000). Since a permanent cloud of concentrated quaternary ammonium (QA) is created at the channel's pore within a millisecond of covalent tethering, the time course of current inhibition induced by Gly7TEA reflects the kinetics of the second-order maleimide–cysteine tethering reaction, which in turn reflects the accessibility of the cysteine's thiol (Blaustein, 2002). We assayed for state-dependent changes in reactivity by comparing rates of inhibition in the presence of Gly7TEA using pulse protocols that differ in their relative times in the depolarized and hyperpolarized states; 33% of the time depolarized with our long-pulse protocol compared with 1–2% of the time with our short-pulse protocol (see Materials and methods and Larsson et al., 1996). Gating current measurements of the extent of charge translocation demonstrate that at the hyperpolarizing (−90 or −110 mV) and depolarizing (+20 mV) voltages used in our protocols, Shaker's four voltage sensors would be predominantly in either the deactivated or activated state, respectively; introduction of a cysteine residue near S3 does not significantly alter the voltage dependence of voltage sensor movement (Fig. 2).
Figure 2.

Normalized Q-V relationships for WT Shaker (circles; n = 3 oocytes) and Shaker E334C (squares; n = 4 oocytes). Qnorm was determined as described in the text. Data points are mean ± SEM. Smooth curves are fits to single Boltzmann functions with V 1/2 and z values of −36.5 mV and 2.5 for WT, and −38.2 mV and 2.8 for E334C.
Exposure of oocytes expressing Shaker D336C to 50 μM Gly7TEA results in irreversible inhibition of ∼60% of the current (Fig. 3), in agreement with previous measurements at this residue using higher concentrations of Gly7TEA (Blaustein et al., 2000). An important feature of this reaction is that the observed Gly7TEA tethering rate does not depend on the relative amounts of time that the channel spends at −110 or +20 mV. This lack of state-dependent reactivity is not unique to D336C; the rates of tethering of Gly7TEA to two neighboring cysteine substitutions, E334C and E335C, are also independent of the gating state of the channel (Table I). Exposure of these residues to shorter maleimido-QA compounds, which irreversibly inhibit a smaller fraction of the current than Gly7TEA, yields similar results (unpublished data). We did not observe significant inhibition (>20%) by Gly7TEA at several cysteine substitutions predicted to lie deeper into (i.e., toward the N terminus of) S3 (326–333).
Figure 3.

Kinetics of Gly7TEA tethering to D336C. (A) Templates of the pulse protocols used to assay for state-dependent tethering. In each case, oocytes are depolarized to +20 mV and hyperpolarized to −110 mV. Top, “short-pulse” protocol in which 50-ms depolarizations are applied every 5 s. Bottom, “long-pulse” protocol in which 1-s depolarizations are applied every 3 s. (B) Top, currents in response to short-pulse protocol. Upper trace is before addition of 50 μM Gly7TEA; bottom trace is after ∼23 min of exposure. Bottom, currents in response to long-pulse protocol with top and bottom traces as described above. (C) Kinetics of tethered block. Isochronal currents taken 1 ms before end of each pulse were normalized to their values before Gly7TEA exposure and plotted against time (short-pulse, circles; long pulse, triangles). Solid gray curves through the data are fits to double-exponential functions with taus (and weights) of 25 s (70%) and 212 s (30%) for circles, and 26 s (66%) and 190 s (34%) for triangles.
TABLE I.
Summary of Tethering Kinetics Data
| Residue | [Gly7TEA] | Protocol | τf | Wf | τs | Ws | Block | n |
|---|---|---|---|---|---|---|---|---|
| μM | s | % | s | % | % | |||
| E334C | 100 | 100 ms/5 s | 36 ± 3 | 36 ± 2 | 627 ± 128 | 64 ± 2 | 43 ± 3 | 4 |
| 1 s/3 s | 33 ± 4 | 38 ± 7 | 657 ± 32 | 62 ± 7 | 48 ± 5 | 5 | ||
| E335Ca | 100 | 100 ms/5 s | 37 ± 5 | 37 ± 7 | 182 ± 27 | 63 ± 7 | 48 ± 5 | 5 |
| 1 s/3 s | 34 ± 6 | 27 ± 7 | 267 ± 36 | 73 ± 7 | 44 ± 4 | 5 | ||
| D336C | 50 | 50 ms/5 s | 24 ± 1 | 67 ± 6 | 191 ± 11 | 33 ± 6 | 57 ± 2 | 6 |
| 1 s/3 s | 27 ± 1 | 63 ± 2 | 193 ± 12 | 37 ± 2 | 59 ± 0 | 6 | ||
| E334C Δ336–357 |
100 | 100 ms/5 s | 30 ± 2 | 55 ± 3 | 318 ± 33 | 45 ± 3 | 65 ± 2 | 5 |
| 1 s/3 s | 36 ± 5 | 56 ± 4 | 304 ± 59 | 44 ± 4 | 61 ± 3 | 5 | ||
| A359C | 400 | 50 ms/5 s | 106 ± 15 | 27 ± 3 | 1067 ± 133 | 73 ± 3 | 86 ± 3 | 4 |
| 1 s/3 s | 46 ± 5 | 51 ± 5 | 230 ± 17 | 49 ± 5 | 91 ± 0 | 4 |
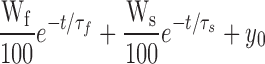 |
Differences in τs and the weights for E335C are not statistically significant when analyzed by t test.
The loop connecting S3 to S4 is considerably longer in Shaker than in most other mammalian Kv channels (Mathur et al., 1997). Indeed, sequence alignment of KvAP and Shaker suggests that Shaker's loop is longer by 22 amino acids, although the actual number depends on whether the four negatively charged residues 333–336 in Shaker are part of its S3 helix. To test whether linker length might account for the difference between our results and those of Jiang et al. (2003b), we examined the kinetics of Gly7TEA tethering in a Shaker construct lacking this corresponding 22–amino acid stretch (Shaker Δ336–357; see Fig. 1 B). Related deletions in the S3–S4 loop Shaker have previously been studied and, although they exhibit alterations in activation and deactivation kinetics, they behave fairly normally (Mathur et al., 1997; Gonzalez et al., 2000, 2001; Sørensen et al., 2000). E334C in this loop-deleted construct reacts well with Gly7TEA and, as is the case with the linker intact, its kinetics of tethering to Gly7TEA are not state dependent (Fig. 4 and Table I).
Figure 4.

Kinetics of Gly7TEA tethering to E334C Δ336–357. (A) Pulse protocols are as described in Fig. 3, except that the short-pulse protocol in these experiments employed a 100-ms depolarizing pulse to allow full activation since this construct exhibits slower activation kinetics. (B) Top, currents in response to short-pulse protocol. Top trace is before addition of 100 μM Gly7TEA; bottom trace is after ∼24 min of exposure. Bottom, currents in response to long pulse protocol with top and bottom traces as described above. (C) Kinetics of tethered block. Time courses were generated as described in Fig. 3 (short-pulse, circles; long pulse, triangles). Solid gray curves through the data are fits to double-exponential functions with taus (and weights) of 29 s (55%) and 303 s (45%) for circles, and 33 s (56%) and 318 s (44%) for triangles.
To verify that Gly7TEA can serve as a reliable probe for assaying state-dependent changes in solvent accessibility, we tested it on A359C, a residue near S4 known to change its exposure during gating. A359C reacts with methanethiosulfonate ethyltriethylammonium (MTSET) much more rapidly at depolarized potentials than at hyperpolarized potentials (Larsson et al., 1996; Baker et al., 1998). Reaction of A359C with Gly7TEA is also state dependent, proceeding more rapidly at depolarized potentials (Fig. 5), however, in contrast to the single-exponential kinetics displayed by MTSET, Gly7TEA reacts with biphasic kinetics, exhibiting taus (and weights) of 122 s (23%) and 999 s (77%) using a short-pulse protocol, and 50 s (58%) and 254 s (42%) using a long-pulse protocol. A359C reacts with Gly7TEA more slowly than 334C-336C, even at depolarized potentials, since ∼10-fold higher concentrations are needed to obtain comparable rates of inhibition. (A consequence of using a higher concentration of Gly7TEA [400 μM] in these experiments is that we observe an initial rapid inhibition of ∼20% due to reversible block by the QA end of untethered Gly7TEA. At the end of the time course, however, that reversible component contributes negligibly since tethering has created an effective local concentration of QA at the pore of ∼20 mM.)
Figure 5.

Kinetics of Gly7TEA tethering to A359C. (A) Pulse protocols are as described in Fig. 3. (B) Top, currents in response to short-pulse protocol. Top trace is before addition of 400 μM Gly7TEA; bottom trace is after ∼78 min of exposure. Bottom, currents in response to long-pulse protocol. Top trace is before addition of 400 μM Gly7TEA; bottom trace is after ∼35 min of exposure. (C) Kinetics of tethered block. Time courses were generated as described in Fig. 3 (short-pulse, circles; long pulse, triangles). Solid gray curves through the data are fits to double-exponential functions with taus (and weights) of 50 s (58%) and 254 s (42%) for triangles, and 122 s (23%) and 999 s (77%) for circles.
We explored another possible reason for the difference between our results with Shaker and those obtained with KvAP: the nature of the probes used to assay accessibility. Jiang et al. (2003b) used avidin, which is a very large probe (∼40 Å diameter), whereas our data above were obtained using a maleimide reagent whose smaller size might allow it to reach regions inaccessible to avidin. We therefore employed an approach similar to that of Jiang et al.; we biotinylated Shaker at position D336C using an ∼16-Å-long MTSEA-biotin, exposed it to streptavidin, and looked for state-dependent changes in accessibility (biotinylation alone does not alter the level of steady-state current through these channels ; Fig. 6 C). As shown in Fig. 6 (B and C), streptavidin added to the extracellular solution results in a progressive inhibition of current with each pulse, with no change in the “leak” current at −90 mV. We performed initial experiments with avidin, which also inhibits; however, streptavidin yielded more consistent results. This may be due to its more physiologic isoelectric point (pI = 5–6) compared with avidin (pI = 10), and the fact that it is not glycosylated (Green, 1990). Exposure to streptavidin for 25–30 min leads to an ∼50% inhibition of current that does not recover upon washout, with a t 1/2 of inhibition of ∼370 s. Importantly, the rate of inhibition is independent of the relative amounts of time spent by the channel at −90 or +20 mV. There is also no recovery of current if the oocyte is maintained for 5 min at −90 mV, either before or after washout of streptavidin, suggesting that channels have not reversibly entered a C-inactivated state. We observed similar inhibition (unpublished data) using an ∼29-Å-long PEO-maleimido-biotin; this compound leaves a much longer linker after exposure to streptavidin (∼13 vs. ∼0 Å). We also performed several types of control experiments to ensure that the observed inhibition is due to a specific interaction between streptavidin in solution and biotin tethered to residue 336C. Streptavidin has no effect on “wild-type” channels that have been exposed to MTSEA-biotin, or on D336C channels that have been prereacted with MTS-glucose instead of MTSEA-biotin (Fig. 6 C). We do occasionally observe a small amount of “nonspecific” inhibition (10–15% inhibition after 45 min) when we apply streptavidin to D336C channels that have not been exposed to any thiol-reactive reagents. Although we do not fully understand its mechanism, we suspect that cysteine oxidation is involved since it is not seen in the presence of a reducing agent, or if D336C channels have been prereacted with MTS-glucose, as described above.
In contrast to maleimido-QA compounds whose mechanisms of inhibition have been well studied (Blaustein et al., 2000; Blaustein, 2002), little is known about the mechanism by which avidin or streptavidin inhibits current when tethered to a channel's voltage sensor. Jiang et al. (2003b) postulated that avidin tethered to KvAP's voltage sensor promotes inactivation by locking the voltage sensor paddles in the activated position, but this has not been demonstrated experimentally, nor is it known which process is rate limiting: avidin binding or inactivation. If, for example, avidin rapidly bound to a biotinylated channel after the first depolarizing pulse, the time course of inhibition could reflect the kinetics of cumulative inactivation rather than those of voltage-sensor solvent exposure. To test this possibility, we added a large excess of biotin to our streptavidin reactions at different time points. If inhibition requires continual exposure to streptavidin, then addition of excess biotin should quench the reaction, whereas if all of the streptavidin bound immediately, then subsequent addition of biotin should have little effect. Our finding that biotin does quench the reaction (Fig. 6 D) argues that whatever its mechanism, the rate of inhibition reflects the exposure of residue 336 to extracellular streptavidin.
DISCUSSION
We measured the kinetics of Gly7TEA tethering to three consecutive cysteines substituted near the C-terminal end of Shaker's S3 segment (334–336) and found that each cysteine reacts equally well with Gly7TEA at depolarized and hyperpolarized voltages. The most straightforward interpretation of these data is that residues 334–336 do not experience a significant change in chemical environment during the gating cycle and therefore do not move a large distance axially into or within the ∼15 Å water-containing region occupied by the phospholipid headgroups. Although this region is polar and could potentially support thiol-maleimide chemistry, the steep gradient in polarity that a sulfhydryl would experience as it moved into or within this interfacial region makes it unlikely that its reactivity would not change (White and Wimley, 1998). Steric constraints and local electrostatic potentials can also influence thiol reactivity, but an extensive movement in the absence of any change in reactivity would require that these forces change during gating, and in a way that would exactly offset the change in ionization exerted by the gradient in polarity.
Crystallographic data from KvAP and its isolated gating module formed the structural basis for a model in which S4 remains tightly coupled to S3b throughout the gating cycle. Indeed, the minimal length of KvAP's S3–S4 linker makes it hard to envisage how S4 might move into and out of the bilayer without dragging S3 with it. But Shaker has a much longer S3–S4 linker; could it provide enough slack to allow Shaker's S4 to uncouple from S3 and move independently? The combination of a lack of state-dependent exposure of S3 with the observed change in S4's reactivity is consistent with such a mechanism; however, our failure to unearth any state dependence when we removed most of that linker argues against it, as does the preservation of gating charge in other loop-deletion constructs (Gonzalez et al., 2000).
Although our view of the structure of the lipid bilayer has been brought into sharper focus over the past two decades (Wiener et al., 1991; Wiener and White, 1992a,b), atomic level details of the interaction between lipids and membrane proteins are only just beginning to emerge. Recent molecular dynamics calculations may shed light on this issue, particularly as it relates to voltage sensor structure. Simulations of an isolated KvAP-like S4 peptide embedded in a lipid bilayer reveal significant distortions of the bilayer in the vicinity of the S4 arginines (Freites et al., 2005). If these results can be generalized to the S3b–S4 paddle, then this structure might actually drag lipid and water with it if it moved down toward the core of the bilayer. A small probe like Gly7TEA might not be ideally suited for measuring physical displacements in this case, since it might not be capable of reporting a more global change in environment during gating. This potential shortcoming, as well as the results of Jiang et al. (2003b), motivated us to probe with a much larger molecule like streptavidin, whose ∼40 Å diameter makes it less sensitive to small-scale bilayer perturbations.
We found that externally applied streptavidin inhibits Shaker channels that have been biotinylated near S3, and the rate of this inhibition is independent of the gating state of the channel. Since the MTSEA-biotin reagent that we used leaves essentially no distance between its cysteine target and the biotin–streptavidin complex, the lack of state dependence argues that the biotinylated cysteine remains exposed to the extracellular solution throughout the gating cycle. This is in contrast to results obtained with avidin in KvAP where a biotinylated cysteine at the top of S3 is not accessible to extracellular avidin in the closed state (Jiang et al., 2003b). As Jiang et al. argue, given the length of the biotin reagent used, this implies that the top of S3 in KvAP is buried in the membrane in the closed state, and remains at least ∼10 Å from the extracellular solution during this portion of the gating cycle.
The extent to which the biotin-streptavidin and Gly7TEA experiments constrain the possible axial motions of the voltage sensor hinges on the exact make-up of Shaker's S3 transmembrane segment. If Shaker residues 334 and 335 form part of its S3 helix, as suggested by the alignment of KvAP and Shaker (Fig. 1 B), then any such motion would have to be very small. However, the C-terminal portions of S3 in Shaker and KvAP are not well conserved, and this may limit the degree to which we can extrapolate from the KvAP structure to that of Shaker. For example, if residues 334–336 are disordered and extend far enough into the S3–S4 loop, larger movements might still be compatible with our data. Unfortunately, the structure of Kv 1.2 does not shed light on this question since the residues comprising its S3 helix were not able to be resolved. Although further indirect probing will provide additional insight into gating motions, resolution of the discrepancy between our results and those in KvAP will also likely require high resolution crystal structures of mammalian and bacterial channels in both open and closed configurations.
The overall rates of inhibition that we observe with streptavidin are one to three orders of magnitude slower (when compared at comparable concentrations) than those seen with purified biotinylated KvAP and diphtheria toxin channels reconstituted into lipid bilayers (Senzel et al., 2000; Ruta et al., 2005). Since KvAP and diphtheria toxin were studied in an idealized system lacking other molecules, it is not surprising that capture rates are faster than in oocytes whose surfaces contain glycosylated proteins that could potentially interfere with streptavidin binding. Capture rates of streptavidin to biotin-conjugated BSA immobilized on a BIAcor chip, a lipid-free system, are even faster, displaying association rate constants two orders of magnitude greater than in the bilayer system (Qureshi et al., 2001), suggesting that partitioning of biotin into the lipid may affect its aqueous accessibility in the more biological systems. An additional factor that makes it difficult to compare our rates of capture to those obtained in other systems is that our time courses of inhibition are not single-exponential decays as expected for a second-order reaction of biotin with streptavidin under pseudo-first order conditions. Knowledge of the molecular mechanism of inhibition by tethered QA ligands allowed us to conclude that the multiexponential time courses exhibited by those compounds arise from a combination of an affinity label effect coupled with multiple reactions in a given channel. Although multiple reactions of streptavidin in a tetrameric channel bearing four biotins may also be responsible for the biphasic nature of its inhibition, we do not know how many streptavidin molecules must bind to inhibit, or whether the first streptavidin reaction proceeds more rapidly than subsequent reactions. Accurate interpretation of fits of these time courses and rates will therefore only be possible when streptavidin's mechanism of inhibition has been elucidated.
Although our discussion of Gly7TEA tethering kinetics has focused on the lack of axial motion exhibited by S3, as pointed out in Materials and methods, the lack of any state dependence of any of the parameters (taus and associated weights) of the tethering kinetics for Gly7TEA near S3 also imposes a radial constraint on S3′s motions: if the top of S3 does move, it must do so in a way that does not significantly change its distance to the channel's external TEA binding site. To estimate the range of motions that we could detect, we modeled Gly7TEA using a polyglycine polymer of similar length and examined the difference in tethering kinetics resulting from our short- and long-pulse protocols. Fig. 7 (A–C) shows the simulated effects of S3 moving from a distance of 35 Å from the pore in the hyperpolarized state to 30 Å away in the depolarized state, a range of distances that correspond to those measured in this region previously (Blaustein et al., 2000; Posson et al., 2005). Our model generates a set of four rate constants at each distance (see Materials and methods): k 35 is our shorthand for the constants at 35 Å, and k 30 denotes those at 30 Å. Since the effective maleimide concentration is greater when S3 is closer to the pore, the k 30 rate constants and the resultant tethering rate at 30 Å are larger than the corresponding constants in the k 35 set and the tethering rate at 35 Å. Simulation of the short-pulse protocol yields a time course (Fig. 7 C, black curve) that is well described by a double-exponential function with taus (and weights) of 27 s (57%) and 120 s (43%). As expected, simulated kinetics resulting from the long-pulse protocol are faster since S3 is closer to the pore for a greater fraction of the time (Fig. 7 C, gray curve). The approximately twofold increase in rate predicted by the model, with taus of 12 s (57%) and 60 s (43%), should be readily detectible experimentally since it falls well within the 10–15% standard error of our measurements. A 5-Å movement away from the pore during depolarization produces the opposite effect: the long-pulse protocol generates slower kinetics than the short-pulse protocol, since S3 spends a greater fraction of its time farther from the pore (Fig. 7, D–F). Fits of the simulations yield taus (and weights) of 28 s (57%) and 125 s (43%) for short pulses (Fig. 7 F, black curve) and 40 s (57%) and 169 s (43%) for long pulses (Fig. 7 F, gray curve). This ∼40% slowing is less dramatic than the twofold acceleration effect seen above, and is probably at the edge of detectability. Such a limitation would not allow us, for example, to detect the 0.7–1.1-Å outward movement of E335C demonstrated by Posson et al. (2005) using luminescence energy transfer. Could a radial motion that accelerates (or slows) tethering be coincident with an axial motion that decreases (or increases) exposure in such a way that these two effects exactly cancel each other to produce state-independent tethering kinetics? This is unlikely since the large change in reactivity that would follow a change in exposure would need to be offset by a large radial motion to preserve the taus of the fits. In such a case, we would also see significant changes in the weights of the fits.
We emphasize that our analysis does not argue that the top of S3 must remain motionless during gating but rather that any motion must preserve its distance to the pore, i.e., the motion must be along the surface of a hemisphere whose origin is at the outer mouth of the channel. Such a constraint would likely rule out a pure rotation of S3, but it would allow a more complex movement of S3 involving a vertical translation combined with a movement toward the central axis of the channel. However, neither we nor others have detected vertical motions that lead to large changes in accessibility (Gandhi et al., 2003; Gonzalez et al., 2005).
To bolster our argument that Gly7TEA is a suitable probe for measuring state-dependent changes in solvent exposure of S3 residues, we performed a “positive control” by examining the rate of Gly7TEA tethering to A359C, a residue near the top of S4 that was previously shown to react with MTSET more rapidly in the open state than in the closed state (Larsson et al., 1996; Baker et al., 1998). Since the short distance between the QA moiety of MTSET and its reactive disulfide does not enable this compound to behave as an affinity label, its reaction kinetics can only reveal changes in cysteine exposure during gating. In contrast, Gly7TEA kinetics are determined both by exposure and distance to the pore of its target cysteine, and analysis of the state dependence of Gly7TEA tethering near at 359C can therefore provide further insight into the motion of S4. For example, to account for our observation that the shift in taus is accompanied by a large shift in weights to the faster component, A359C must do more than simply increase its exposure during depolarization. Our model dictates that we must also invoke a state-dependent change in the effective local concentration near A359C of Gly7TEA's maleimide end when its free QA end is blocking the pore (Mt) (see Materials and methods and Blaustein, 2002). Since the only way for this to occur is if the distance from the tethering point to the pore changed during gating, our data are best explained if the increase in reactivity of A359C in the open state is also accompanied by a radial movement toward the pore. How large a movement is necessary to account for our findings? If in modeling the state-dependent kinetics we use a range of 5–20-fold for the increase in intrinsic reactivity of Gly7TEA, based on our own data and those of others using MTSET, then Mt would have to increase by 5–10-fold at depolarized voltages to yield a shift in the taus and weights comparable to what we observe. If we further assume that A359C is 30–35 Å from the pore, and use the polymer statistical approach detailed in Materials and methods, this would correspond to an inward movement of 2–5 Å. Although the large number of assumptions required to generate this estimate limits its accuracy, it is worth noting that Posson et al. (2005) found that the neighboring residue L361C also moves inward upon depolarization, although to a lesser extent. Our data still leave open the question of whether the proposed inward movement of A359C results from a rotation of S4, a change in its tilt, or some combination of these or other motions. A possible trajectory consistent with our S3 data would be one in which S4 pivots around S3; such a motion could still be compatible with an S3–S4 paddle, but as discussed above, not with a model that involves large vertical translocations.
Acknowledgments
We thank Merritt Maduke, Dan Cox, and Spike Horn for helpful discussions and for critically reviewing the manuscript, Stephen White for his expertise on lipid bilayers and for sharing results prior to publication, and Gary Yellen for his biophysical acumen.
This work was supported by National Institutes of Health grants R01HL68985 (R. Blaustein and V. Ketty), T32HL069770 (A. Ivy and R. Darman), and American Heart Association Postdoctoral Fellowship 0225647T (R. Darman).
Olaf S. Andersen served as editor.
R.B. Darman and A.A. Ivy contributed equally to this work.
A.A. Ivy's present address is Abbott Laboratories, Abbott Park, IL 60064.
V. Ketty's present address is BD Biosciences, Bedford, MA 01730.
Abbreviations used in this paper: MTS, methanethiosulfonate; MTSET, methanethiosulfonate ethyltriethylammonium; QA, quaternary ammonium.
References
- Aggarwal, S.K., and R. MacKinnon. 1996. Contribution of the S4 segment to gating charge in the Shaker K+ channel. Neuron. 16:1169–1177. [DOI] [PubMed] [Google Scholar]
- Baker, O.S., H.P. Larsson, L.M. Mannuzzu, and E.Y. Isacoff. 1998. Three transmembrane conformations and sequence-dependent displacement of the S4 domain in Shaker K+ channel gating. Neuron. 20:1283–1294. [DOI] [PubMed] [Google Scholar]
- Blaustein, R.O. 2002. Kinetics of tethering quaternary ammonium compounds to K+ channels. J. Gen. Physiol. 120:203–216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blaustein, R.O., P.A. Cole, C. Williams, and C. Miller. 2000. Tethered blockers as molecular ‘tape measures’ for a voltage-gated K+ channel. Nat. Struct. Biol. 7:309–311. [DOI] [PubMed] [Google Scholar]
- Cha, A., G.E. Snyder, P.R. Selvin, and F. Bezanilla. 1999. Atomic scale movement of the voltage-sensing region in a potassium channel measured via spectroscopy. Nature. 402:809–813. [DOI] [PubMed] [Google Scholar]
- Chanda, B., O.K. Asamoah, R. Blunck, B. Roux, and F. Bezanilla. 2005. Gating charge displacement in voltage-gated ion channels involves limited transmembrane movement. Nature. 436:852–856. [DOI] [PubMed] [Google Scholar]
- Finkelstein, A., K.J. Oh, L. Senzel, M. Gordon, R.O. Blaustein, and R.J. Collier. 2000. The diphtheria toxin channel-forming T-domain translocates its own NH2-terminal region and the catalytic domain across planar phospholipid bilayers. Int. J. Med. Microbiol. 290:435–440. [DOI] [PubMed] [Google Scholar]
- Flory, P.J. 1969. Statistical Mechanics of Chain Molecules. Hanser Publishers, New York. 432 pp.
- Freites, J.A., D.J. Tobias, G. von Heijne, and S.H. White. 2005. Interface connections of a transmembrane voltage sensor. Proc. Natl. Acad. Sci. USA. 102:15059–15064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gandhi, C.S., E. Clark, E. Loots, A. Pralle, and E.Y. Isacoff. 2003. The orientation and molecular movement of a K+ channel voltage-sensing domain. Neuron. 40:515–525. [DOI] [PubMed] [Google Scholar]
- Gandhi, C.S., E. Loots, and E.Y. Isacoff. 2000. Reconstructing voltage sensor-pore interaction from a fluorescence scan of a voltage-gated K+ channel. Neuron. 27:585–595. [DOI] [PubMed] [Google Scholar]
- Glauner, K.S., L.M. Mannuzzu, C.S. Gandhi, and E.Y. Isacoff. 1999. Spectroscopic mapping of voltage sensor movement in the Shaker potassium channel. Nature. 402:813–817. [DOI] [PubMed] [Google Scholar]
- Gonzalez, C., F.J. Morera, E. Rosenmann, O. Alvarez, and R. Latorre. 2005. S3b amino acid residues do not shuttle across the bilayer in voltage-dependent Shaker K+ channels. Proc. Natl. Acad. Sci. USA. 102:5020–5025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez, C., E. Rosenman, F. Bezanilla, O. Alvarez, and R. Latorre. 2000. Modulation of the Shaker K+ channel gating kinetics by the S3–S4 linker. J. Gen. Physiol. 115:193–208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez, C., E. Rosenman, F. Bezanilla, O. Alvarez, and R. Latorre. 2001. Periodic perturbations in Shaker K+ channel gating kinetics by deletions in the S3–S4 linker. Proc. Natl. Acad. Sci. USA. 98:9617–9623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gordon, M., and A. Finkelstein. 2001. The number of subunits comprising the channel formed by the T domain of diphtheria toxin. J. Gen. Physiol. 118:471–480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Green, N.M. 1990. Avidin and streptavidin. Methods Enzymol. 184:51–67. [DOI] [PubMed] [Google Scholar]
- Heginbotham, L., and R. MacKinnon. 1992. The aromatic binding site for tetraethylammonium ion on potassium channels. Neuron. 8:483–491. [DOI] [PubMed] [Google Scholar]
- Hessa, T., H. Kim, K. Bihlmaier, C. Lundin, J. Boekel, H. Andersson, I. Nilsson, S.H. White, and G. von Heijne. 2005. a. Recognition of transmembrane helices by the endoplasmic reticulum translocon. Nature. 433:377–381. [DOI] [PubMed] [Google Scholar]
- Hessa, T., S.H. White, and G. von Heijne. 2005. b. Membrane insertion of a potassium-channel voltage sensor. Science. 307:1427. [DOI] [PubMed] [Google Scholar]
- Horn, R. 2000. A new twist in the saga of charge movement in voltage-dependent ion channels. Neuron. 25:511–514. [DOI] [PubMed] [Google Scholar]
- Hoshi, T., W.N. Zagotta, and R.W. Aldrich. 1990. Biophysical and molecular mechanisms of Shaker potassium channel inactivation. Science. 250:533–538. [DOI] [PubMed] [Google Scholar]
- Islas, L.D., and F.J. Sigworth. 2001. Electrostatics and the gating pore of Shaker potassium channels. J. Gen. Physiol. 117:69–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang, Y., A. Lee, J. Chen, V. Ruta, M. Cadene, B.T. Chait, and R. MacKinnon. 2003. a. X-ray structure of a voltage-dependent K+ channel. Nature. 423:33–41. [DOI] [PubMed] [Google Scholar]
- Jiang, Y., V. Ruta, J. Chen, A. Lee, and R. MacKinnon. 2003. b. The principle of gating charge movement in a voltage-dependent K+ channel. Nature. 423:42–48. [DOI] [PubMed] [Google Scholar]
- Kavanaugh, M.P., R.S. Hurst, J. Yakel, M.D. Varnum, J.P. Adelman, and R.A. North. 1992. Multiple subunits of a voltage-dependent potassium channel contribute to the binding site for tetraethylammonium. Neuron. 8:493–497. [DOI] [PubMed] [Google Scholar]
- Kitaguchi, T., M. Sukhareva, and K.J. Swartz. 2004. Stabilizing the closed S6 gate in the Shaker Kv channel through modification of a hydrophobic seal. J. Gen. Physiol. 124:319–332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larsson, H.P., O.S. Baker, D.S. Dhillon, and E.Y. Isacoff. 1996. Transmembrane movement of the Shaker K+ channel S4. Neuron. 16:387–397. [DOI] [PubMed] [Google Scholar]
- Lee, H.C., J.M. Wang, and K.J. Swartz. 2003. Interaction between extracellular Hanatoxin and the resting conformation of the voltage-sensor paddle in Kv channels. Neuron. 40:527–536. [DOI] [PubMed] [Google Scholar]
- Lee, S.Y., A. Lee, J. Chen, and R. MacKinnon. 2005. Structure of the KvAP voltage-dependent K+ channel and its dependence on the lipid membrane. Proc. Natl. Acad. Sci. USA. 102:15441–15446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Long, S.B., E.B. Campbell, and R. Mackinnon. 2005. a. Crystal structure of a mammalian voltage-dependent Shaker family K+ channel. Science. 309:897–903. [DOI] [PubMed] [Google Scholar]
- Long, S.B., E.B. Campbell, and R. Mackinnon. 2005. b. Voltage sensor of Kv1.2: structural basis of electromechanical coupling. Science. 309:903–908. [DOI] [PubMed] [Google Scholar]
- MacKinnon, R., and G. Yellen. 1990. Mutations affecting TEA blockade and ion permeation in voltage-activated K+ channels. Science. 250:276–279. [DOI] [PubMed] [Google Scholar]
- Mannuzzu, L.M., M.M. Moronne, and E.Y. Isacoff. 1996. Direct physical measure of conformational rearrangement underlying potassium channel gating. Science. 271:213–216. [DOI] [PubMed] [Google Scholar]
- Mathur, R., J. Zheng, Y. Yan, and F.J. Sigworth. 1997. Role of the S3–S4 linker in Shaker potassium channel activation. J. Gen. Physiol. 109:191–199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oh, K.J., L. Senzel, R.J. Collier, and A. Finkelstein. 1999. Translocation of the catalytic domain of diphtheria toxin across planar phospholipid bilayers by its own T domain. Proc. Natl. Acad. Sci. USA. 96:8467–8470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phillips, L.R., M. Milescu, Y. Li-Smerin, J.A. Mindell, J.I. Kim, and K.J. Swartz. 2005. Voltage-sensor activation with a tarantula toxin as cargo. Nature. 436:857–860. [DOI] [PubMed] [Google Scholar]
- Posson, D.J., P. Ge, C. Miller, F. Bezanilla, and P.R. Selvin. 2005. Small vertical movement of a K+ channel voltage sensor measured with luminescence energy transfer. Nature. 436:848–851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiu, X.Q., K.S. Jakes, A. Finkelstein, and S.L. Slatin. 1994. Site-specific biotinylation of colicin Ia. A probe for protein conformation in the membrane. J. Biol. Chem. 269:7483–7488. [PubMed] [Google Scholar]
- Qureshi, M.H., J.C. Yeung, S.C. Wu, and S.L. Wong. 2001. Development and characterization of a series of soluble tetrameric and monomeric streptavidin muteins with differential biotin binding affinities. J. Biol. Chem. 276:46422–46428. [DOI] [PubMed] [Google Scholar]
- Ruta, V., J. Chen, and R. MacKinnon. 2005. Calibrated measurement of gating-charge arginine displacement in the KvAP voltage-dependent K+ channel. Cell. 123:463–475. [DOI] [PubMed] [Google Scholar]
- Schwarz, T.L., B.L. Tempel, D.M. Papazian, Y.N. Jan, and L.Y. Jan. 1988. Multiple potassium-channel components are produced by alternative splicing at the Shaker locus in Drosophila. Nature. 331:137–142. [DOI] [PubMed] [Google Scholar]
- Senzel, L., M. Gordon, R.O. Blaustein, K.J. Oh, R.J. Collier, and A. Finkelstein. 2000. Topography of diphtheria Toxin's T domain in the open channel state. J. Gen. Physiol. 115:421–434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seoh, S.A., D. Sigg, D.M. Papazian, and F. Bezanilla. 1996. Voltage-sensing residues in the S2 and S4 segments of the Shaker K+ channel. Neuron. 16:1159–1167. [DOI] [PubMed] [Google Scholar]
- Slatin, S.L., X.Q. Qiu, K.S. Jakes, and A. Finkelstein. 1994. Identification of a translocated protein segment in a voltage-dependent channel. Nature. 371:158–161. [DOI] [PubMed] [Google Scholar]
- Sørensen, J.B., A. Cha, R. Latorre, E. Rosenman, and F. Bezanilla. 2000. Deletion of the S3–S4 linker in the Shaker potassium channel reveals two quenching groups near the outside of S4. J. Gen. Physiol. 115:209–222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Starace, D.M., and F. Bezanilla. 2001. Histidine scanning mutagenesis of basic residues of the S4 segment of the Shaker K+ channel. J. Gen. Physiol. 117:469–490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Starace, D.M., E. Stefani, and F. Bezanilla. 1997. Voltage-dependent proton transport by the voltage sensor of the Shaker K+ channel. Neuron. 19:1319–1327. [DOI] [PubMed] [Google Scholar]
- Tiwari-Woodruff, S.K., M.A. Lin, C.T. Schulteis, and D.M. Papazian. 2000. Voltage-dependent structural interactions in the Shaker K+ channel. J. Gen. Physiol. 115:123–138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tiwari-Woodruff, S.K., C.T. Schulteis, A.F. Mock, and D.M. Papazian. 1997. Electrostatic interactions between transmembrane segments mediate folding of Shaker K+ channel subunits. Biophys. J. 72:1489–1500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, M.H., S.P. Yusaf, D.J. Elliott, D. Wray, and A. Sivaprasadarao. 1999. Effect of cysteine substitutions on the topology of the S4 segment of the Shaker potassium channel: implications for molecular models of gating. J. Physiol. 521(Pt 2):315–326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White, S.H., and W.C. Wimley. 1998. Hydrophobic interactions of peptides with membrane interfaces. Biochim. Biophys. Acta. 1376:339–352. [DOI] [PubMed] [Google Scholar]
- Wiener, M.C., G.I. King, and S.H. White. 1991. Structure of a fluid dioleoylphosphatidylcholine bilayer determined by joint refinement of x-ray and neutron diffraction data. I. Scaling of neutron data and the distributions of double bonds and water. Biophys. J. 60:568–576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiener, M.C., and S.H. White. 1992. a. Structure of a fluid dioleoylphosphatidylcholine bilayer determined by joint refinement of x-ray and neutron diffraction data. II. Distribution and packing of terminal methyl groups. Biophys. J. 61:428–433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiener, M.C., and S.H. White. 1992. b. Structure of a fluid dioleoylphosphatidylcholine bilayer determined by joint refinement of x-ray and neutron diffraction data. III. Complete structure. Biophys. J. 61:434–447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang, N., A.L. George, and R. Horn. 1996. Molecular basis of charge movement in voltage-gated sodium channels. Neuron. 16:113–122. [DOI] [PubMed] [Google Scholar]
- Yang, N., and R. Horn. 1995. Evidence for voltage-dependent S4 movement in sodium channels. Neuron. 15:213–218. [DOI] [PubMed] [Google Scholar]
- Yellen, G. 1998. The moving parts of voltage-gated ion channels. Q. Rev. Biophys. 31:239–295. [DOI] [PubMed] [Google Scholar]
- Yusaf, S.P., D. Wray, and A. Sivaprasadarao. 1996. Measurement of the movement of the S4 segment during the activation of a voltage-gated potassium channel. Pflugers Arch. 433:91–97. [DOI] [PubMed] [Google Scholar]