

Abstract
Onconase (ONC) is a homolog of bovine pancreatic ribonuclease (RNase A) from the frog Rana pipiens. ONC displays antitumoral activity and is in advanced clinical trials for the treatment of cancer. Here, we report the first atomic structures of ONC·nucleic acid complexes: a T89N/E91A ONC·5′-AMP complex at 1.65 Å resolution and a wild-type ONC·d(AUGA) complex at 1.90 Å resolution. The latter structure and site-directed mutagenesis was used to reveal the atomic basis for substrate recognition and turnover by ONC. The residues in ONC that are proximal to the scissile phosphodiester bond (His10, Lys31, and His97) and uracil nucleobase (Thr35, Asp67, and Phe98) are conserved from RNase A and serve to generate a similar bell-shaped pH–kcat/KM profile for RNA cleavage. Glu91 of ONC forms two hydrogen bonds with the guanine nucleobase in d(AUGA), and Thr89 is in close proximity to that nucleobase. Installing a neutral or cationic residue at position 91 or an asparagine residue at position 89 virtually eliminated the 102-fold guanine:adenine preference of ONC. A variant that combined such substitutions, T89N/E91A ONC, actually preferred adenine over guanine. In contrast, installing an arginine residue at position 91 increased the guanine preference and afforded an ONC variant with the highest known kcat/KM value. These data indicate that ONC discriminates between guanine and adenine by using Coulombic interactions and a network of hydrogen bonds. The structure of the ONC·d(AUGA) complex was also used to probe other aspects of catalysis. For example, the T5R substitution, designed to create a favorable Coulombic interaction between ONC and a phosphoryl group in RNA, increased ribonucleolytic activity by twofold. No variant, however, was more toxic to human cancer cells than wild-type ONC. Together, these findings provide a cynosure for understanding catalysis of RNA cleavage in a system of high medicinal relevance.
Keywords: cytotoxin, ribonuclease, RNA, substrate specificity, X-ray crystallography
Introduction
The viability of organisms relies on the ability of proteins to recognize nucleic acids. In contrast, the ability of an enzyme to both recognize a nucleic acid and catalyze its cleavage can have deleterious consequences. For example, ribonucleases can be cytotoxic because cleaving RNA renders indecipherable its encoded information.1,2
Onconase (ONC; Figure 1(a)) is a ribonuclease found in the eggs and early embryos of the frog Rana pipiens. ONC is a homolog of bovine pancreatic ribonuclease (RNase A3), and the two proteins share 30% amino-acid sequence identity and a similar three-dimensional structure.4 ONC is in confirmatory Phase IIIb clinical trials for the treatment of malignant mesothelioma,5,6 and has been granted both orphan-drug and fast-track status by the US Food and Drug Administration. ONC also inhibits HIV-1 replication.7
Figure 1.
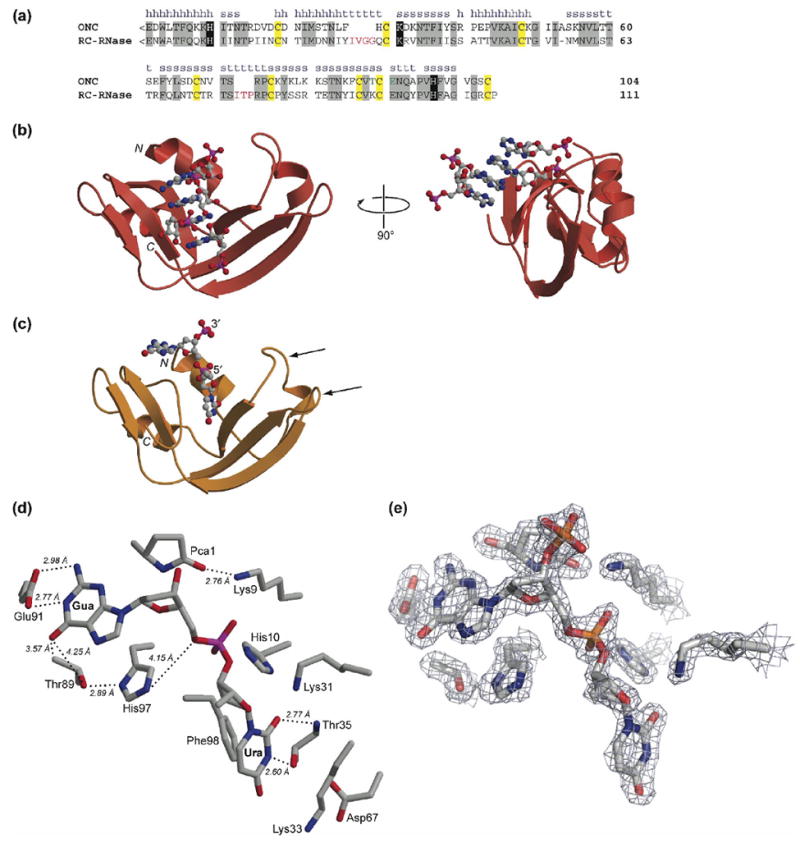
Primary and tertiary structure of ONC. (a) Amino-acid sequence alignment of ONC and RC-RNase. The secondary structure of ONC is labeled as h (α-helix), s (β-strand), or t (turn). Residues conserved between the two ribonucleases are in gray boxes. <E denotes a pyroglutamate residue. Key active-site residues are in black boxes. Cysteine residues that form intramolecular disulfide bonds are in yellow boxes. Residues that participate in the nucleobase recognition at the B2 subsite in ONC are in green. Extra amino acid residues in the loops of RC-RNase are in red. (b) Ribbon diagram of the three-dimensional structure of the T89N/E91A ONC·5′-AMP complex (PDB entry 2GMK). (c) Ribbon diagram of the three-dimensional structure of the ONC·d(AUGA) complex (PDB entry 2I5S). The two flanking adenosine nucleotides are not included in the diagram because of their low electron density. The two arrowheads indicate the two loops that were subjected to elongation herein. (d) Stick diagram of the active site and B1 and B2 subsites in the ONC·d(AUGA) complex. Thr35 forms two hydrogen bonds with the uracil nucleobase. Glu91 forms two hydrogen bonds with the guanine nucleobase. Thr89 is located close to the carbonyl oxygen of guanine. (e) Electron density map of the active site (σ-weighted 2Fo – Fc) contoured at 1.5σ over background. Images in panels (b)–(e) were created with the program MOLSCRIPT and rendered with the program RASTER3D.79
ONC is a unique ribonuclease. The protein exhibits remarkable conformational stability (Tm = 90 °C8). Four disulfide bonds and the absence of a cis prolyl peptide bond contribute to this attribute.8-11 ONC evades the cellular ribonuclease inhibitor protein (RI12), to which other ribonucleases bind with femtomolar affinity.13-19 The exceptional conformational stability and the RI-evading ability contribute to its cytotoxic activity.17,8,20
ONC is a poor catalyst. The ribonucleolytic activity of ONC is three to five orders of magnitude lower than that of RNase A, due in large part to low affinity for its substrate.21 Nonetheless, the catalytic activity of ONC is necessary for cytotoxicity.22
Homologs of RNase A bind a pyrimidine residue on the 5′ side of the scissile phosphodiester bond in a small, conserved nucleobase-binding site.23-26 ONC displays a distinct preference for a guanosine nucleoside on the 3′ side of the scissile phosphodiester bond.21 This guanine preference is also found in other frog ribonucleases,27 but not in mammalian homologs.28 The basis for this guanine preference in ONC is unknown. tRNA appears to be the major cellular substrate for ONC.29 A recent study revealed an unconventional cleavage sequence for ONC—the guanosine–guanosine phosphodiester bond in the variable loop or the D-arm in tRNA.30
Here, we report the first crystal structures of ONC·nucleic acid complexes. We use this structural information to address key issues in ONC catalysis. First, we determine the molecular basis for the nucleobase specificity of ONC through a systematic site-directed mutagenesis study. Next, we ask whether the low catalytic activity of ONC can be enhanced by a rational design approach. Finally, we seek to confirm the cellular target sequence of ONC in vitro using two novel fluorogenic substrates. We anticipate that the development of ONC as a cancer chemotherapeutic will benefit from the incipient understanding of its catalysis.
Results
Structural overview
The crystalline structures of the T89N/E91A ONC·5′-AMP and ONC·d(AUGA) complexes were solved to resolutions of 1.65 and 1.90 Å, respectively. Data collection, refinement, and model statistics are summarized in Table 1. The electron density was continuous for main-chain and side-chain atoms. Asymmetric units of the structures contain a single monomer with a chain-fold virtually identical to that of free ONC (PDB entry 1ONC4). Both structures exhibit the typical bilobal shape of the RNase A superfamily, with two anti-parallel β-sheets flanked by two α-helices. In addition, our structures contain the rare N-terminal pyroglutamate residue and C-terminal disulfide bond, which are conserved among the amphibian members of the superfamily.
Table 1.
Summary of data collection and refinement statisticsa
| T89N/E91A ONC·5′-AMP | Wild-type ONC·d(AUGA) | |
|---|---|---|
| Space group | P212121 | P21212 |
| Unit cell parameters (Å) | a=29.0, b=52.1, c=66.1 | a=129.2, b=26.1, c=32.5 |
| Data collection statistics | ||
| Resolution range (Å) | 66.14–1.65 (1.70–1.65) | 64.62–1.90 (1.95–1.90) |
| Number of reflections (measured/unique) | 336231/12645 | 91217/9270 |
| Completeness (%) | 99.9 (99.6) | 99.7 (95.9) |
| Rmergeb | 0.054 (0.663) | 0.125 (0.517) |
| Redundancy | 26.6 (5.1) | 9.8 (3.3) |
| Mean σ/I | 40.66 (3.32) | 12.30 (2.52) |
| Refinement statistics | ||
| Resolution range (Å) | 40.93–1.65 | 32.49–1.90 |
| Number of reflections (total/test) | 12597/612 | 9229/441 |
| Rcrystc/Rfreed | 0.165/0.217 | 0.178/0.240 |
| RMSD bonds (Å) | 0.010 | 0.013 |
| RMSD angles (deg) | 1.718 | 1.415 |
| Average B factor (Å2) | 19.35 | 16.60 |
| Number of water molecules | 196 | 155 |
| Ramachandran favored (%) | 98.1 | 100.0 |
| Ramachandran allowed (%) | 1.9 | 0.0 |
Values in parentheses are for the highest-resolution shell
Rmerge = ΣhΣi∣Ii–⟨I(h)⟩∣/ΣhΣiIi(h), where Ii(h) is the intensity of an individual measurement of the reflection and <I(h)> is the mean intensity of the reflection.
Rcryst = Σh∥Fobs∣–∣Fcalc∥/Σh∣Fcalc∣, where Fobs and Fcalc are the observed and calculated structure factor amplitudes, respectively.
Rfree was calculated as Rcryst using 5.0% of the randomly selected unique reflections that were omitted from structure refinement.
Structure of the T89N/E91A ONC·5′-AMP complex
In the crystalline T89N/E91A ONC·5′-AMP complex, four 5′-AMP molecules are bound to each enzyme molecule in a nonproductive mode (Figure 1(b)). The nucleobases of the 5′-AMP molecules form a stack that makes contact with multiple enzyme molecules in the crystal lattice. One of the four 5′-AMP molecules has its 5′-phosphate group bound in the active site of the enzyme. Another 5′-AMP molecule is located at the interface between two enzyme molecules. The remaining two 5′-AMP molecules show no significant interaction with the enzyme and have relatively high B-factors.
Structure of the wild-type ONC·d(AUGA) complex
In the crystalline ONC·d(AUGA) complex, one nucleic acid molecule is bound to one enzyme molecule (Figure 1(c)). There was no electron density above the noise level for the 3′ adenosine. The electron density for the 5′ adenosine was less than that of a typical water molecule, was therefore not included in the model. The disorder in these two flanking nucleosides suggests that they do not interact significantly with the enzyme.
The conformation of the Glu91 and His97 side chains in the ONC·d(AUGA) complex differed dramatically from those in free ONC (PDB entry 1ONC4). The χ2 dihedral angle of His97 was investigated in detail and the residue was assigned to a conformation that had the most uniform B-values around the imidazole ring. This conformation, in which Nδ1 forms a 2.89-Å hydrogen bond with Oγ1 of Thr89 (Figure 1(d,e)), is the one supported best by the diffraction data. The alternative conformation, which puts Nδ1 close to O5′ of guanosine, had refined B-factors around the imidazole ring that varied by nearly twofold, with the B-factors for the nitrogen atoms being too large and those of the carbon atoms being too small relative to surrounding residues.
Effect of pH on catalysis
RNase A cleaves RNA molecules by concerted general acid–base catalysis using the active-site residues His12 and His119.31-34 In this mechanism, both a protonated and an unprotonated histidine residue are required for efficient catalysis. As a result, the pH–kcat/KM profile of RNase A is a classic bell-shaped curve with a maximal catalytic activity near pH 6.35,36 These histidine residues are conserved in ONC and other amphibian homologs, suggesting that the catalytic mechanism of RNase A could be operative within the amphibian subfamily (Figure 2). A recent study by Liao and coworkers showed, however, that the pKa values of the catalytic histidine residues in RC-RNase 3, an amphibian homolog from the frog Rana catesbeiana, were significantly lower (4.26 and 5.96) than those manifested in RNase A.37 Because of these low pKa values, the catalytic activity of RC-RNase 3 is maximal near pH 5.38
Figure 2.
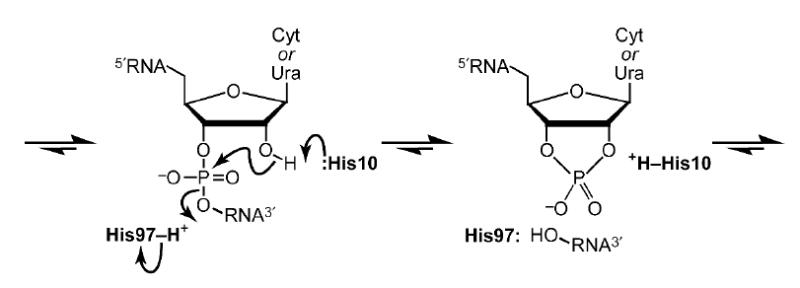
Putative mechanism of catalysis of RNA cleavage by ONC. In this transphosphorylation reaction, His10 acts as a base, and His97 acts as an acid.
To ensure that assays of the enzymatic activity of ONC were performed herein at an optimal pH, values of kcat/KM for the cleavage of 6-FAM–dArUdGdA–6-TAMRA by ONC were determined in buffers of various pH. The pH–kcat/KM profile for ONC has a symmetric bell shape with pKa values of 5.84 ± 0.05 and 6.77 ± 0.04 (Figure 3), which are similar to those of RNase A.39-41 Accordingly, assays were performed in a buffer of pH 6.0, which enables nearly maximal activity and allows for direct comparison with data on RNase A.
Figure 3.
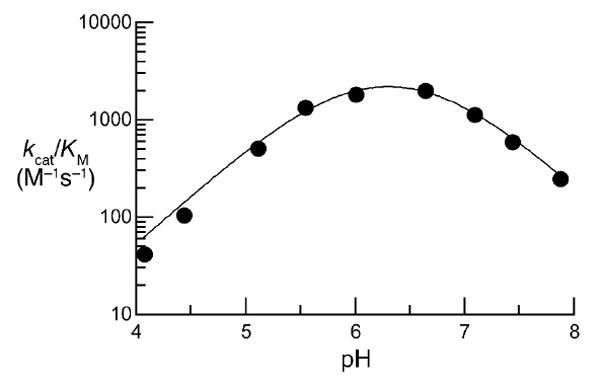
pH–kcat/KM profile for the cleavage of 6-FAM–dArUdGdA–6-TAMRA by ONC. Assays were performed at 23 °C in 1.0 mM buffer containing NaCl (1.0 M). Determination of kcat/KM values was performed in triplicate. Data were fitted to equation (1) by non-linear least squares regression analysis to give pKa values of 5.84 ± 0.05 and 6.77 ± 0.04.
Residues in the B1 subsite
RNase A and its homologs bind to RNA substrates in a cleft, leading to interactions that extend beyond the scissile bond. In RNase A, two subsites for a nucleobase (B1 and B2)42,25,43,26 and four subsites for a phosphoryl group (P−1, P0, P1, and P2)44-46 have been characterized in detail.3 The enzyme catalyzes the cleavage of the P–O5′ bond of a substrate bound in the B1–P1–B2 subsites.
In the structure of the ONC·d(AUGA) complex, several residues of ONC are in close contact with the nucleobase moieties of d(AUGA). Lys33, Thr35, Asp67, and Phe98 are proximal to the uracil nucleobase (Figure 1(d)). Oγ1 and backbone amide nitrogen atom of Thr35 form two hydrogen bonds with N4 and O2, respectively, of that nucleobase. The distance between Nζ of Lys 33 and O4 of the uracil base is 3.73 Å. Oδ1 of Asp67 forms a hydrogen bond with Oγ1 of Thr35. Each of these residues except Lys33 is conserved in the RNase A superfamily, and hence constitute the B1 subsite in ONC.
Residues in the B2 subsite
In the structure of crystalline ONC·d(AUGA) complex, Glu91 forms two hydrogen bonds with the guanine nucleobase of d(AUGA) (Figure 1(d)). In addition, Thr89 is located in close proximity to this nucleobase. We hypothesized that these two residues constitute the B2 subsite of ONC, where B2 refers to the nucleobase on the 3′ side of the scissile bond. To reveal the molecular basis for the guanine preference, we used site-directed mutagenesis to replace Thr89 and Glu91 with residues that differ in their net charge and ability to form hydrogen bonds (Table 2), and determined whether these variants showed an altered nucleobase preference.
Table 2.
Values of kcat/KM, m/z, and IC50 for ONC and its variants
| kcat/KM | m/zc | |||||
|---|---|---|---|---|---|---|
| ONC | UpG (M−1s−1)a | UpA (M−1s−1)b | UpG/UpA | expected | observed | IC50 (μM)d |
| Wild-type | (4.2 ± 0.5)×104 | (3.8 ± 0.2)×102 | 1.1 × 102 | 11820 | 11820 | 0.2 |
| T89A | (1.5 ± 0.1)×104 | (4.7 ± 0.4)×102 | 32 | 11790 | 11793 | 0.4 |
| T89K | (4.1 ± 0.3)×104 | (7.0 ± 0.4)×101 | 5.9 × 102 | 11820 | 11824 | 1.0 |
| T89R | (1.4 ± 0.1)×105 | (1.1 ± 0.1)×102 | 1.3 × 103 | 11875 | 11868 | 0.8 |
| T89D | (1.2 ± 0.1)×103 | (1.1 ± 0.1)×102 | 11 | 11834 | 11840 | 2.0 |
| T89Q | (1.4 ± 0.1)×103 | (3.5 ± 0.2)×101 | 40 | 11847 | 11852 | ND |
| T89N | (2.6 ± 0.2)×103 | (1.2 ± 0.1)×103 | 2.2 | 11833 | 11826 | 0.7 |
| E91A | (3.9 ± 0.3)×103 | (1.6 ± 0.1)×103 | 2.4 | 11762 | 11750 | 1.5 |
| E91Q | (9.4 ± 0.3)×103 | (1.1 ± 0.1)×103 | 8.6 | 11819 | 11827 | 0.6 |
| E91N | (4.2 ± 0.3)×103 | (1.6 ± 0.3)×103 | 2.6 | 11811 | 11805 | ND |
| E91K | (2.4 ± 0.1)×103 | (3.8 ± 0.3)×103 | 0.63 | 11819 | 11828 | ND |
| T89N/E91A | (1.7 ± 0.3)×103 | (4.4 ± 0.2)×103 | 0.39 | 11775 | 11780 | 2.5 |
| T89K/E91A | (7.5 ± 0.3)×103 | (5.6 ± 0.2)×102 | 13 | 11788 | 11793 | 1.7 |
|
| ||||||
| T5K | (5.8 ± 0.1)×104 | NDe | ND | 11846 | 11852 | 0.4 |
| T5R | (8.3 ± 0.5)×104 | ND | ND | 11874 | 11881 | 1.2 |
| K33V | (4.4 ± 0.3)×104 | (3.7 ± 0.3)×102 | 1.2 × 102 | 11791 | 11795 | ND |
| K33V/T89K | (7.1 ± 0.7)×104 | (1.6 ± 0.2)×102 | 4.4 × 102 | 11818 | 11823 | ND |
| IVGGITP | (1.9 ± 0.2)×104 | (5.3 ± 0.4)×102 | 36 | 12447 | 12454 | 4.0 |
| L27I/F28Y IVGGITP | (3.4 ± 0.2)×104 | (6.0 ± 0.2)×102 | 57 | 12463 | 12470 | 4.0 |
Values of kcat/KM (± SE) are for cleavage of 6-FAM–dArUdGdA–6-TAMRA or 6-FAM–dArUdAdA–6-TAMRA in 0.10 M MES–NaOH buffer (pH 6.0) containing NaCl (0.10 M) at 23 ± 2 °C.
Values of m/z were determined by MALDI–TOF mass spectrometry.
Values of IC50 (± SE) are for incorporation of [methyl-3H]thymidine into the DNA of K-562 cells exposed to ONC or a variant, and were calculated with equation (3).
ND, not determined.
Variants with an increased preference for guanine
Insertion of a positive charge at position 89 increased the guanine preference of ONC for cleavage of UpG relative to UpA by 10-fold (Figure 4; Table 2). The T89R and T89K variants displayed a 103-fold preference for a guanine nucleobase over an adenine one. Furthermore, the value of kcat/KM for the cleavage of a guanine-containing substrate (UpG) by T89R ONC was increased by 3-fold from that of the wild-type enzyme.
Figure 4.
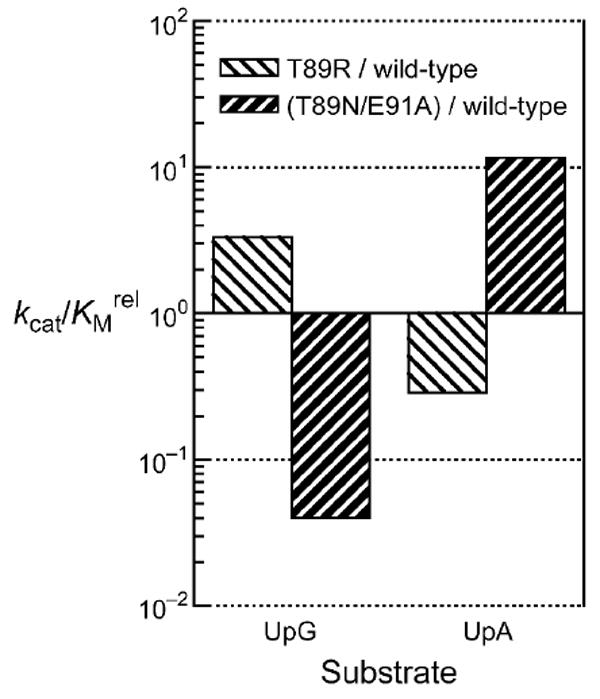
Effect of Thr89 and Glu91 on the substrate specificity of ONC. Bars indicate the effect of replacing Thr89 or Glu91 on the value of kcat/KM for the cleavage of 6-FAM–dArUdGdA–6-TAMRA (UpG) and 6-FAM–dArUdAdA–6-TAMRA (UpA). The guanine:adenine preference of T89R ONC is 3.3 × 103-fold greater than that of T89N/E91A ONC (Table 2).
Variants with a decreased preference for guanine
Elimination of the negative charge at position 91 not only diminished the preference of ONC for guanine versus adenenine, but also increased the value of kcat/KM for the cleavage of an adenine-containing substrate (UpA, Table 2). The E91A, E91Q, and E91N variants of ONC cleaved UpG 2- to 9-fold more quickly than UpA, indicative of substantially lower guanine preferences than that of the wild-type enzyme. The largest change in preference was observed with E91K ONC, which actually had a 1.6-fold preference for UpA over UpG. Each of these variants above had 3- to 10-fold greater UpA-cleaving activity than did the wild-type enzyme.
Replacing Thr89 with an asparagine residue increased the value of kcat/KM for UpA cleavage by 3-fold (Table 2). In addition, the T89N substitution decreased the guanine preference of ONC by 50-fold. An aspartate or glutamine residue at this position likewise decreased the guanine preference, though without increasing the kcat/KM value. Combining the T89N and E91A substitutions augmented their individual effect. T89N/E91A ONC had the highest catalytic activity toward UpA among all of the variants, with a greater than 12-fold increase from the wild-type enzyme (Figure 4; Table 2). Moreover, T89N/E91A ONC had a complete shift in nucleobase preference, favoring adenine over guanine by 2.6-fold.
Inhibition of catalysis
Catalysis of 6-FAM–dArUdGdA–6-TAMRA cleavage by wild-type ONC and its variants was inhibited by the nucleotides 5′-GMP and 5′-AMP (Table 3). Wild-type ONC was inhibited by 5′-GMP and 5′-AMP with Ki values of (6.7 ± 0.5) × 102 μM and (3.7 ± 0.3) × 103 μM, respectively. The Ki values of the T89K and T89R variants, which manifested an increased preference for the guanine nucleobase, did not differ markedly from that of wild-type ONC. On the contrary, variants with a decreased guanine preference showed relatively large changes in Ki values. Inhibition by 5′-AMP was 3-fold and 6-fold more pronounced for T89N ONC and T89N/E91A ONC, respectively. Inhibition by 5′-GMP was twofold less pronounced for E91A ONC.
Table 3.
Values of Ki for ONC and its variants
| ONC |
Ki (μM)a |
|
|---|---|---|
| 5′-GMP | 5′-AMP | |
| Wild-type | (6.7 ± 0.5) × 102 | (3.7 ± 0.3) × 103 |
| T89K | (5.3 ± 0.2) × 102 | (2.0 ± 0.2) × 103 |
| T89R | (4.3 ± 0.6) × 102 | (2.4 ± 0.4) × 103 |
| T89N | (1.0 ± 0.1) × 103 | (1.2 ± 0.1) × 103 |
| E91A | (1.3 ± 0.3) × 103 | (3.2 ± 0.4) × 103 |
| T89N/E91A | (9.9 ± 0.7) × 102 | (5.9 ± 0.2) × 102 |
Values of Ki (±SE) are for inhibition of catalysis of 6-FAM–dArUdGdA–6-TAMRA cleavage in 0.10 M MES–NaOH buffer (pH 6.0) containing NaCl (0.10 M) at 23 ± 2 °C.
Rational design of ONC variants with enhanced catalytic activity
Previous structural investigations have shown that the structure of ONC is less flexible than that of other ribonucleases.26,47 This rigidity could hinder facile interaction with the substrate. The structure of the ONC·d(AUGA) complex supports this hypothesis. The RMSD of the backbone Cα of ONC in the free and complexed form is just 0.4 Å,48 suggesting little conformational change upon substrate binding.
Global approach
We sought to increase the catalytic activity of ONC by enhancing its flexibility. Sica and coworkers have implicated the short loops of ONC as causing rigidity.49 Amphibian ribonucleases are grouped into two subfamilies.50 The members of the subfamily with two short loops connecting the α2-helix and β2-strand, and β4- and β5-strands (ONC, RC-RNase 2, RC-RNase 4, and RC-RNase 6) show low catalytic activity.38,51,52 The second subfamily, represented by RC-RNase and RC-RNase L1, contain loops with additional amino acid residues (IVGG and ITP, Figure 1(a)), and exhibit relatively high catalytic activity. The difference in catalytic activity between the two subfamilies reaches up to five orders of magnitude. We reasoned that installing the longer loops from RC-RNase onto ONC could enhance its catalytic activity. Accordingly, two ONC variants (IVGG/ITP and L27I/F28Y/IVGG/ITP ONC) were designed and tested for their ribonucleolytic activity. The L27I/F28Y substitutions were added to mimic the loop structure of RC-RNase more closely. Both variants did not, however, exhibit increased catalytic activity.
Local approach
A conspicuous feature of the ONC·d(AUGA) complex was the absence of significant electron density for the two flanking adenosine nucleosides in the nucleic acid (Figure 1(b)). This lack of electron density suggests a negligible interaction of these nucleosides with the enzyme. We asked whether the incorporation of residues that would promote “local” interaction with substrate could revive the catalytic activity of ONC. In RNase A, residues denoted as “P subsites” participate in substrate binding. These P-subsite residues are cationic lysine or arginine residues that can promote binding with the anionic phosphoryl groups in the nucleic acid backbone through favorable Coulombic interaction.44 Removal of any residues in the P subsites leads to a loss in catalytic activity and substrate binding.53,46 In the ONC·d(AUGA) complex structure, no residues other than the active-site lysine residues (Lys9 and Lys31; P1 subsite, Figure 1(d)) interact with phosphoryl groups in the nucleic acid. We hypothesized that installing a novel P subsite in ONC would increase its catalytic activity by enhancing substrate binding. A detailed structural comparison between the RNase A·d(ATAAG) complex (PDB entry 1RCN44) and the ONC complex pointed towards Thr5 in ONC as being a promising location for a novel P subsite. Consequently, we created the T5K and T5R variants and measured their catalytic activity. In this regard, the T5R substitution performed better than did the T5K substitution, conferring a twofold increase in catalytic activity.
Val37 in RC-RNase is involved in nucleobase recognition (B1 subsite).52 The corresponding residue in ONC is Lys33. K33V ONC was designed to test whether this substitution could increase nucleobase recognition in ONC. The catalytic activity of K33V ONC did not, however, differ from that of wild-type ONC.
Catalytic activity toward GpG
Excess wild-type ONC (10 μM) did not catalyze the cleavage of 6-FAM–dUrGdGdA–6-TAMRA or 6-FAM–dArGdGdA–6-TAMRA with a rate measurable by our assay. We estimate that the values of kcat/KM to be <1.0 M–1s–1. As a positive control we used RNase T1, which cleaves RNA at the 3′-side of guanosine residues.54 One unit of RNase T1 (which produces an increase of 0.0004 A260 units in 1 min in 1 mL at room temperature using GpA as substrate) catalyzed the cleavage of our two GpG substrates to completion within a minute (data not shown).
Cytotoxicity of ONC variants
All ONC variants with altered nucleobase preference were less toxic than the wild-type enzyme for K-562 cells (Table 2). Likewise, ONC variants designed to possess increased catalytic activity displayed less cytotoxic activity than did wild-type ONC.
Discussion
Molecular basis for the B2-subsite specificity of ONC
The contribution hydrogen bonds in an enzyme–substrate interface to catalysis has been established.55-59 In the structure of an ONC·nucleic acid complex, Oε1 and Oε2 of Glu91 form two hydrogen bonds with N1 and N2 of the guanine nucleobase (Figure 1(d)). In addition, Thr89 is located proximal to the guanine nucleobase. The distances between Oγ1 and Cγ2 of Thr89, and O6 of guanine are 3.57 and 4.25 Å, respectively. Similar interactions were observed in the RC-RNase·d(ACGA) complex.52 Specifically, Lys95 and Glu97 from RC-RNase establish an extensive network of hydrogen bonds with the nucleobase. Replacement of these residues with alanine resulted in a significant change in nucleobase preference.52 To understand the molecular basis underlying the nucleobase specificity of ONC, we took a more systematic approach—we replaced Thr89 and Glu91 with residues that vary in their ability to form hydrogen bonds or in their net charge (or both).
Catalysis of UpG cleavage by wild-type ONC is 110-fold more efficient than is the cleavage of UpA (Table 2). Each of the substitutions made at position 91 (alanine, glutamine, asparagine, and lysine) necessarily led to the loss of the two Glu91–guanine hydrogen bonds observed in the crystalline complex (Figure 1(d)). And, each of these variants exhibited a lower preference for guanine. The lysine substitution had the largest effect, actually producing a slight preference for adenine. It is worth noting that this specificity change originates from both a decrease in the catalytic activity toward the UpG substrate and an increase toward the UpA substrate.
To understand the substrate specificity on ONC in greater detail, we performed electrostatic potential calculations. The results are in agreement with previous calculations,60 showing that the electron density on N1 and N2 of guanine is low (Figure 5). Thus, the interaction of this region with the anionic side chain of Glu91 would be promoted by favorable Coulombic interaction. The electrostatic potential of adenine differs significantly from that of guanine. The electron density on the corresponding region of adenine is high, and the anionic side chain of Glu91 would produce unfavorable interactions. We conclude that the 110-fold preference of ONC for a guanine nucleobase can be explained, at least in part, by electrostatic interactions between Glu91 and a purine nucleobase.
Figure 5.
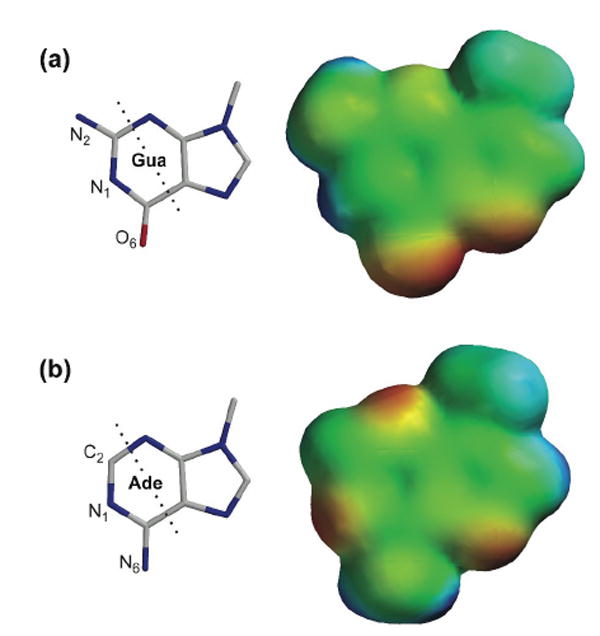
Structure and electron density diagram of (a) 9-methylguanine and (b) 9-methyladenine. Atoms to the left of the dashed lines interact with Thr89 and Glu91 of the B2 subsite of ONC (Figure 1(d)). Electron density of the nucleobases was calculated with the program SPARTAN (Wavefunction, Irvine, CA). Electron-rich regions are colored red; electron-poor regions are blue; neutral regions are green.
The structure of the ONC·d(AUGA) complex predicts that O6 of the guanine nucleobase would be located close to a cationic side chain at position 89. In the RC-RNase·d(ACGA) complex, a lysine residue at the corresponding position makes a hydrogen bond with O6 of guanine.52 Our data demonstrate that a residue bearing a positive charge at this position (lysine or arginine) strengthens the guanine preference of ONC, while a negative charge (aspartate) attenuates that preference. Because O6 on guanine is electron-rich, and N6 on adenine is electron-poor (Figure 2), this result suggests again that the substrate specificity of ONC originates from its electrostatic interaction with the nucleobases. It is not clear, however, why T89N ONC has increased catalytic activity for the UpA substrate, because this substitution does not involve a change in net charge. We have also shown that the increase in catalytic activity toward the UpA substrate by the single substitutions can be enhanced further by their combination. T89N/E91A ONC, a variant that would possess two compatible interfaces for adenine, displays an 11-fold enhanced catalytic activity toward the UpA substrate. Furthermore, this double variant now prefers adenine to guanine by 2.6-fold.
We used the inhibitory effect of 5′-GMP or 5′-AMP on the cleavage of 6-FAM–dArUdGdA–6-TAMRA to assess the effect of substitutions on ground-state binding (Table 2). In this analysis, we assume that the mononucleotides bind at the B2 and P1 subsites, and that this binding competitively inhibits the cleavage of the substrate. The inhibitory effect of 5′-AMP is somewhat enhanced in T89K ONC and T89R ONC, indicating that binding of adenine at the B2 subsite is slightly stronger in these variants, despite the adverse Coulombic effect of adding a positive charge. The binding of guanine became only slightly stronger after the substitutions. Thus, the major ramification of the extra positive charge at position 89 is the destabilization of transition-state binding of adenine as shown in the considerably decreased kcat/KM values (Table 2). The T89N substitution lowers the ground-state energy of the enzyme-adenine complex as indicated by the 3-fold lower value of Ki. The E91A substitution destabilizes the ground-state binding of guanine.
Guanine and adenine are part of nucleotides with a broad range of distinct biological functions. Discrimination between these two nucleobases, similar in shape and size, is critical for many enzymes (e.g., kinases, ATPases, and GTPases). These proteins usually exhibit a strong preference for their cognate nucleotide over the other. Statistical analyses of protein–nucleotide structures in the RCSB Protein Data Bank has suggested that Coulombic interactions and a network of hydrogen bonds are the two major determinants of selectivity.61,60 A hydrogen bond is primarily an electrostatic interaction,62 and a correlation between hydrogen bonds and the electrostatic potential distribution in the protein–ligand interface was indeed found in the statistical analyses. In accord with these findings, our study indicates that Glu91 of ONC uses both Coulombic interactions and hydrogen bonds to confer nucleobase specificity.
Rational design of ONC with enhanced catalytic activity
Implanting extra amino acid residues (IVGG and ITP) designed to affect the “global” breathing motion on ONC49 did not increase its catalytic activity (Table 2). This result implies that the factors causing the low catalytic activity of ONC reside outside of the loops. The greater catalytic activity and flexibility attained by the M23L substitution9,47,49 supports this notion. Met23 is located in a hydrophobic cavity created by the side chains of Ile22, Phe28, Lys31, Phe36, Cys68, and Tyr77. A bulky leucine residue at this position is thought to alter the position of residues that are critical for catalysis, especially that of Lys31. In this regard, it would be interesting to identify those amino acid residues responsible for rigidity in the β-sheet region by thorough sequence and structural comparison of amphibian ribonucleases. The “local” modifications (T5K, T5R, and K33V substitutions) did not result in a sizeable increase in catalytic activity. It is possible that the envisioned local interactions were not realized due to the low flexibility of ONC.
It is noteworthy that His97 in the ONC·d(AUGA) complex assumes a conformation distinct from that of corresponding histidine residues in other members of the RNase A superfamily. His97 is thought to function as an acid during catalysis by ONC (Figure 2), providing a proton to the displaced O5′. In the RNase A·d(ATAAG) and RC-RNase·d(ACGA) complexes, the corresponding histidine residue adopts a suitable conformation for the transfer of a proton from Nδ1 to O5′.44,52 The distance between Nδ1 and O5′ is 2.99 and 3.01 Å in the RNase A·d(ATAAG) and RC-RNase·d(ACGA) complex, respectively. In the ONC·d(AUGA) complex, however, the imidazole ring of His97 is rotated by nearly 180° about the χ2 dihedral angle, such that the Nδ1–O5′ distance is 5.35 Å (Figure 1(d)). In addition, Nδ1 forms a hydrogen bond with Oγ1 of Thr89, which is not conserved in either RNase A or RC-RNase. Thus in ONC, Nε2 of His97 rather than Nδ1 could be the proton source.
Under physiological conditions, His97 could adopt a more conventional conformation. Twenty-five years ago, the corresponding histidine residue in RNase A was refined in two alternative conformations, indicative of its intrinsic flexibility.63,64 This histidine residue in RC-RNase exhibits conformational flexibility upon ligand binding.52 Likewise, the χ1 dihedral angle of His97 differs by nearly 180° in unliganded ONC (PDB entry 1ONC) and the ONC·d(AUGA) complex. Regardless of this apparent flexibility, both His10 and His97 appear to participate in the catalysis of RNA cleavage (Figure 2), as the pH–kcat/KM profile of ONC retains the bell shape that is renown in the RNase A superfamily (Figure 3)35-37 and is known to arise from the titration of the two active-site histidine residues.65
In vivo cleavage sites for ONC
tRNA has shown to be a predominant cellular target for ONC.29 Still, it remains a matter of debate whether cellular tRNA is the sole substrate for ONC in the cell.66 Members of the RNase A superfamily usually cleave single-stranded region of RNA that is not base-paired. How tRNA, which is extensively base-paired, becomes a target for ONC is unknown. Moreover, calculations predict that most of the tRNA in mammalian mitochondria and Escherichia coli exists as a ternary complex with elongation factor Tu (EF-Tu) and GTP,67 which would limit accessibility to ONC even further. A recent report shed some light on this issue. Through an extensive sequence analysis on the cleavage site in tRNA, Suhasini & Sirdeshimukh demonstrated that ONC preferentially targets the variable loop or the D-arm in tRNA.30 These regions of tRNA lack base pairing and are thus candidates for cleavage by ONC. In our ONC·d(AUGA) structure, ONC makes minimal contacts with substrate (only one P and two B subsites), implying that a compact single-stranded region in RNA is sufficient for an ONC substrate. In addition, the structure of the EF-Tu·GMP·tRNAPhe complex (PDB entry 1TTT68) confirms that the variable loop and the D-arm regions are not blocked by EF-Tu. Remarkably, the authors found that the predominant cleavage site is a guanosine–guanosine phosphodiester bond. In the structure of the ONC·d(AUGA) complex, the narrow B1 subsite composed of Lys33, Thr35, and Phe98 makes intimate contact with the small uracil nucleobase (Figure 1(d)), and would likely occlude the large guanine nucleobase. To determine whether ONC catalyzes the cleavage of a guanosine–guanosine phosphodiester bond in vitro, we developed two novel substrates (6-FAM–dUrGdGdA–6-TAMRA and 6-FAM–dArGdGdA–6-TAMRA). The inability of ONC to cleave either substrate leads us to conclude that the B1 subsite of ONC is too constricted to accommodate a large guanine nucleobase in an unstructured RNA molecule, at least in its anti orientation. We conclude that as yet unidentified elements of tRNA structure are involved in the cleavage of the guanosine–guanosine phosphodiester bond in tRNA.
Conclusions
We have determined the crystalline structure of two ONC·nucleic acid complexes at resolutions of 1.90 and 1.65 Å. Guided by these structures, we have revealed the atomic basis for substrate recognition and turnover by ONC. We have discovered that ONC utilizes Coulombic interactions (especially from Glu91) and a hydrogen bonding network to mediate substrate specificity, and have demonstrated that rational amino-acid substitutions can alter this specificity. Finally, we have probed structural attributes responsible for the low catalytic activity of ONC and the unusual cleavage of a guanosine–guanosine phosphodiester bond in tRNA.
Materials and Methods
Materials
Human RI (as RNasin®) was from Promega (Madison, WI). RNase T1 was from Amibon (Austin, TX). 6-Carboxyfluorescein–dArUdAdA–6-carboxytetramethylrhodamine (6-FAM–dArUdAdA–6-TAMRA), 6-FAM–dArUdGdA–6-TAMRA, 6-FAM–dUrGdGdA–6-TAMRA, and 6-FAM–dArGdGdA–6-TAMRA were from Integrated DNA Technologies (Coralville, IA). 2-(N-Morpholino)ethanesulfonic acid (MES), adenosine 5′-monophosphate (5′-AMP), and guanosine 5′-monophosphate (5′-GMP) were from Sigma Chemical (St. Louis, MO). MES was purified further by anion-exchange chromatography prior to its use so as to eliminate contaminating oligo(vinylsulfonic acid), which is a potent inhibitor of ribonucleases.69 [methyl-3H]Thymidine was from PerkinElmer Life Sciences (Boston, MA). Phosphate-buffered saline (PBS) contained (in 1 liter) 0.20 g of KCl, 0.20 g of KH2PO4, 8.0 g of NaCl, and 2.16 g of Na2HPO4·7H2O. All other chemicals and reagents were of commercial grade or better, and were used without further purification.
K-562 cells, which derive from a continuous human chronic myelogenous leukemia line, were from the American Type Culture Collection (Manassas, VA). Cell culture medium and supplements were from Invitrogen (Carlsbad, CA).
Biophysical measurements
Mass was measured by matrix-assisted laser desorption ionization–time-of-flight (MALDI–TOF) mass spectrometry using a Voyager-DE-PRO Biospectrometry Workstation Applied Biosystems, Foster City, CA) and a 3,5-dimethoxy-4-hydroxycinnamic acid (sinapinic acid) matrix (Sigma Chemical). Fluorescence measurements were performed with a QuantaMaster 1 Photon Counting Fluorometer equipped with sample stirring (Photon Technology International, South Brunswick, NJ). Radioactivity was quantitated with a Microbeta TriLux liquid scintillation and luminescence counter (PerkinElmer, Wellesley, MA).
Production of ONC and its variants
Wild-type ONC and its variants were produced in E. coli strain BL21(DE3) as described previously.21 The observed molecular mass of each purified protein was within 0.1% of its expected molecular mass (Table 2). The yield of each purified protein was 20 mg per liter of culture.
Crystal preparation
Crystals of T89N/E91A ONC were grown at 20 °C by the hanging-drop method from a solution containing protein (21.4 mg/ml) in water mixed with an equivalent volume of reservoir solution, which was 90 mM bis(2-hydroxyethyl)amino-tris(hydroxymethyl)methane (BIS-TRIS) buffer, pH 6.5, containing polyethylene glycol monomethyl ether (MEPEG) 2K (30.6% w/v) and 5′-AMP (50 mM). Crystals were soaked in a reservoir solution supplemented with ethylene glycol (5% v/v), and then flash-frozen in a stream of cryogenic N2(g).
Crystals of the wild-type ONC·d(AUGA) complex were grown at 20 °C by the hanging-drop method from a solution containing protein (10 mg/ml) and nucleic acid (3.0 mM) in water mixed with an equivalent volume of reservoir solution, which was 0.10 M 4-(2-hydroxyethyl)piperazine-1-ethanesulfonic acid (HEPES) buffer, pH 7.5, containing polyethylene glycol (PEG) 3350 (25% v/v). Crystals were soaked in reservoir solutions supplemented with increasing amounts of ethylene glycol (up to 20% v/v), and then flash-frozen in a stream of cryogenic N2(g).
X-ray data collection
X-ray diffraction data for the T89N/E91A ONC·5′-AMP and the wild-type ONC·d(AUGA) complex were collected with a Bruker AXS Proteum R CCD detector and Microstar rotating-anode generator using copper Kα radiation. All data were processed and scaled with the programs SAINT and SADABS from the Proteum software suite (Bruker, Madison, WI).
Structure refinement
Structures of the T89N/E91A ONC·5′-AMP and the wild-type ONC·d(AUGA) complex were solved by molecular replacement with apo-ONC (PDB entry 1ONC4) as a model using the program MOLREP.70 The structures were completed using alternate cycles of manual building in COOT71 and refinement in REFMAC5.72 The stereochemical quality of the final models was assessed with MolProbity.73 The final coordinates were deposited in the RCSB Protein Data Bank74 with accession numbers 2GMK and 2I5S for the T89N/E91A ONC·5′-AMP and wild-type ONC·d(AUGA) complexes, respectively.
Assays of catalytic activity
Ribonucleolytic activity of wild-type ONC and its variants was measured with a hypersensitive assay based on the cessation of fluorescence quenching as described in previous studies.21 To assess the B2-subsite specificity of ONC, two fluorogenic substrates that contain distinct nucleobase sequences were used. Preference for the interaction with the guanine nucleobase was measured by using 6-FAM–dArUdGdA–6-TAMRA, and the interaction with the adenine nucleobase was measured by using 6-FAM–dArUdAdA–6-TAMRA. We also assessed the catalytic activity of wild-type ONC toward two novel ONC substrates, each containing a single cleavable guanosine–guanosine phosphodiester bond: 6-FAM–dUrGdGdA–6-TAMRA and 6-FAM–dArGdGdA–6-TAMRA.
pH–kcat/KM profile
The effect of pH on the value of kcat/KM for the cleavage of 6-FAM–dArUdGdA–6-TAMRA by wild-type ONC was determined in 2.00 mL of 1.0 mM buffer containing NaCl (1.0 M), 6-FAM–dArUdGdA–6-TAMRA (50–500 nM), and wild-type ONC (50 nM–1.0 μM). Buffers were sodium formate–HCl (pH 4.07), sodium acetate–HCl (pH 4.43–5.54), BIS-TRIS–NaOH (pH 6.0–6.64), MOPS–NaOH (pH 7.09–7.43), and TRIS–HCl (pH 7.87). The pH values of buffers were determined with a Φ40 pH meter from Beckman instruments (Fullerton, CA). Buffer concentration was kept low to ensure an identical salt environment, which is known to affect catalysis by ribonucleases.53,75 No change in the pH values of reaction mixtures was detectable during the course of the reaction. Higher concentration of the substrate was used at acidic pH (350–500 nM) than at basic pH (50 nM) because the protonation of the fluorescein moiety of the substrate decreases its fluorescence intensity. To obtain values of K1 and K2, which are the apparent macroscopic acid dissociation constants of two functional groups important in catalysis, data were fitted by non-linear least squares regression analysis with the program PRISM 4 (GraphPad Software, San Diego, CA) to equation (1):
| (1) |
where (kcat/KM)max is the pH-independent specificity constant.
Inhibition of catalytic activity
Inhibition of ribonucleolytic activity was measured using 6-FAM–dArUdGdA–TAMRA as a substrate. Inhibition by 5′-AMP or 5′-GMP was assessed at 25 °C in 2.0 mL of 50 mM imidazole–HCl buffer, pH 6.0, containing NaCl (0–0.25 M), 6-FAM–dArUdAdA–6-TAMRA (60 nM), and ONC (1–5 nM), as described previously.76,77 We assumed that the nucleobases of 5′-AMP and 5′-GMP bind exclusively at the B2 subsite, because the narrow B1 subsite cannot accommodate a purine. This assumption is supported by our finding that ONC does not cleave our novel substrates containing a guanosine–guanosine phosphodiester bond (vide supra).
In the crystalline T89N/E91A ONC·5′-AMP complex, four 5′-AMP molecules are bound in a mode that is not productive in that the active site is occupied by phosphoryl groups from two 5′-AMP molecules and no adenine nucleobase is proximal to the B2 subsite (i.e., Asn89 and Ala91). The formation of such a nonproductive complex is unlikely during our inhibition assay. 5′-AMP molecules in the crystalline T89N/E91A ONC·5′-AMP complex are shared by multiple enzyme molecules in the crystal lattice, which is comprised of an extensive array of enzymes and nucleotides. Such protein–nucleotide interactions are unlikely to form in dilute aqueous solution.
Fluorescence (F) was measured with 493 and 515 nm as the excitation and emission wavelengths, respectively. The value of ΔF/Δt was measured for 3 min after the addition of ONC. Next, an aliquot of inhibitor (I) dissolved in the assay buffer was added, and ΔF/Δt was measured in the presence of the inhibitor for 3 min. The concentration of inhibitor in the assay was doubled repeatedly at 3-min intervals. Excess wild-type RNase A was then added to the mixture to ensure that <10% of the substrate had been cleaved prior to completion of the inhibition assay. Apparent changes in ribonucleolytic activity due to dilution were corrected by comparing values to those from an assay in which aliquots of buffer were added. To obtain values of Ki, data were fitted by non-linear least squares regression analysis with the program DELTAGRAPH 5.5 (Red Rock Software, Salt Lake City, UT) to equation (2)76,77:
| (2) |
where (ΔF/Δt)0 is the ribonucleolytic activity prior to the addition of the inhibitor.
Assays of cytotoxic activity
The effect of wild-type ONC, its variants, and RNase A on cell proliferation was determined as described in previous studies.21 Cytotoxicity data were analyzed with the programs SIGMAPLOT (SPSS Science, Chicago, IL) and DELTAGRAPH 5.5. Each data point represents the mean (±SE) of at least three experiments performed in triplicate. To obtain values of IC50, data were fitted by non-linear least squares regression analysis with the program DELTAGRAPH 5.5 to equation (3)78:
| (3) |
where S is the percent of total DNA synthesis during the 4-h pulse as compared to that of a PBS control.
Acknowledgments
We are grateful to J. B. Binder for help with calculations of the electron density of nucleobases, E. L. Myers and T. J. Rutkoski for contributive discussions, and members of the Center for Eukaryotic Structural Genomics, including L. Meske and A. Hibbard for help with crystallization and E. Bitto and J. G. McCoy for help with data collection and processing. J.E.L. was supported by a Steenbock Predoctoral Fellowship from the Department of Biochemistry. This work was supported by grant CA73808 (NIH) and Protein Structure Initiative P50 GM064598 and U54 GM074901 (NIH).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Abbreviations used: 5′-AMP, adenosine 5′-monophosphate; 5′-GMP, guanosine 5′-monophosphate; 6-FAM, 6-carboxyfluorescein; 6-TAMRA, 6-tetraethylaminerhodamine; BIS-TRIS, bis(2-hydroxyethyl)amino-tris(hydroxymethyl)methane; CCD, charge-coupled device; DTT, dithiothreitol; <E, pyroglutamic acid; EDTA, ethylenediaminetetraacetic acid; EF-Tu, elongation factor Tu; FBS, fetal bovine serum; HEPES, 4-(2-hydroxyethyl)piperazine-1-ethanesulfonic acid; HIV, human immunodeficiency virus; I, inhibitor; IPTG, isopropyl-1-thio-β-d-galactopyranoside; MALDI–TOF, matrix-assisted laser desorption ionization–time-of-flight; MEPEG, polyethylene glycol monomethyl ether; MES, 2-(N-morpholino)-ethanesulfonic acid; ONC, Onconase® (a registered trademark of Alfacell, Inc.), also known as “ranpirnase”; PBS, phosphate-buffered saline; PCA, pyroglutamic acid; PDB, protein data bank; PEG, polyethylene glycol; RC-RNase, ribonuclease from Rana catesbeiana; RI, ribonuclease inhibitor protein; RMSD, root-mean-square deviation; RNase A, unglycosylated bovine pancreatic ribonuclease; RNase T1, microbial ribonuclease from Aspergillus oryzae; Tris, 2-amino-2-(hydroxymethyl)-1,3-propanediol; UpA, 6-carboxyfluorescein–dArUdAdA–6-carboxytetramethylrhodamine; UpG, 6-carboxyfluorescein–dArUdGdA–6-carboxytetramethylrhodamine.
References
- 1.Leland PA, Raines RT. Cancer chemotherapy—ribonucleases to the rescue. Chem Biol. 2001;8:405–413. doi: 10.1016/s1074-5521(01)00030-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Tafech A, Bassett T, Sparanese D, Lee CH. Destroying RNA as a therapeutic approach. Curr Med Chem. 2006;13:863–881. doi: 10.2174/092986706776361021. [DOI] [PubMed] [Google Scholar]
- 3.Raines RT. Ribonuclease A. Chem Rev. 1998;98:1045–1065. doi: 10.1021/cr960427h. [DOI] [PubMed] [Google Scholar]
- 4.Mosimann SC, Ardelt W, James MNG. Refined 1.7 Å X-ray crystallographic structure of P-30 protein, an amphibian ribonuclease with anti-tumor activity. J Mol Biol. 1994;236:1141–1153. doi: 10.1016/0022-2836(94)90017-5. [DOI] [PubMed] [Google Scholar]
- 5.Mikulski SM, Costanzi JJ, Vogelzang NJ, McCachren S, Taub RN, Chun H, Mittelman A, Panella T, Puccio C, Fine R, Shogen K. Phase II trial of a single weekly intravenous dose of ranpirnase in patients with unresectable malignant mesothelioma. J Clin Oncol. 2002;20:274–281. doi: 10.1200/JCO.2002.20.1.274. [DOI] [PubMed] [Google Scholar]
- 6.Pavalkis N, Vogelzang NJ. Ranpirnase—an antitumour ribonuclease: Its potential role in malignant mesothelioma. Expert Opin Biol Ther. 2006;6:391–399. doi: 10.1517/14712598.6.4.391. [DOI] [PubMed] [Google Scholar]
- 7.Youle RJ, Wu YN, Mikulski SM, Shogen K, Hamilton RS, Newton D, D’Alessio G, Gravell M. RNase inhibition of human immunodeficiency virus infection of H9 cells. Proc Natl Acad Sci USA. 1994;91:6012–6016. doi: 10.1073/pnas.91.13.6012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Leland PA, Staniszewski KE, Kim B, Raines RT. A synapomorphic disulfide bond is critical for the conformational stability and cytotoxicity of an amphibian ribonuclease. FEBS Lett. 2000;477:203–207. doi: 10.1016/s0014-5793(00)01804-4. [DOI] [PubMed] [Google Scholar]
- 9.Notomista E, Catanzano F, Graziano G, Dal Piaz F, Barone G, D’Alessio G, Di Donato A. Onconase: An unusually stable protein. Biochemistry. 2000;39:8711–8718. doi: 10.1021/bi000415x. [DOI] [PubMed] [Google Scholar]
- 10.Notomista E, Catanzano F, Graziano G, Di Gaetano S, Barone G, Di Donato A. Contribution of chain termini to the conformational stability and biological activity of onconase. Biochemistry. 2001;40:9097–9103. doi: 10.1021/bi010741s. [DOI] [PubMed] [Google Scholar]
- 11.Arnold U, Schulenburg C, Schmidt D, Ulbrich-Hofmann R. Contribution of structural peculiarities of onconase to its high stability and folding kinetics. Biochemistry. 2006;45:3580–3587. doi: 10.1021/bi0525223. [DOI] [PubMed] [Google Scholar]
- 12.Dickson KA, Haigis MC, Raines RT. Ribonuclease inhibitor: Structure and function. Prog Nucleic Acid Res Mol Biol. 2005;80:349–374. doi: 10.1016/S0079-6603(05)80009-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lee FS, Shapiro R, Vallee BL. Tight-binding inhibition of angiogenin and ribonuclease A by placental ribonuclease inhibitor. Biochemistry. 1989;28:225–230. doi: 10.1021/bi00427a031. [DOI] [PubMed] [Google Scholar]
- 14.Lee FS, Vallee BL. Binding of placental ribonuclease inhibitor to the active site of angiogenin. Biochemistry. 1989;28:3556–3561. doi: 10.1021/bi00434a061. [DOI] [PubMed] [Google Scholar]
- 15.Vicentini AM, Kieffer B, Matthies R, Meyhack B, Hemmings BA, Stone SR, Hofsteenge J. Protein chemical and kinetic characterization of recombinant porcine ribonuclease inhibitor expressed in Saccharomyces cerevisiae. Biochemistry. 1990;29:8827–8834. doi: 10.1021/bi00489a046. [DOI] [PubMed] [Google Scholar]
- 16.Shapiro R, Vallee BL. Interaction of human placental ribonuclease with placental ribonuclease inhibitor. Biochemistry. 1991;30:2246–2255. doi: 10.1021/bi00222a030. [DOI] [PubMed] [Google Scholar]
- 17.Boix E, Wu Y, Vasandani VM, Saxena SK, Ardelt W, Ladner J, Youle RJ. Role of the N terminus in RNase A homologues: Differences in catalytic activity, ribonuclease inhibitor interaction and cytotoxicity. J Mol Biol. 1996;257:992–1007. doi: 10.1006/jmbi.1996.0218. [DOI] [PubMed] [Google Scholar]
- 18.Haigis MC, Kurten EL, Raines RT. Ribonuclease inhibitor as an intracellular sentry. Nucleic Acids Res. 2003;31:1024–1032. doi: 10.1093/nar/gkg163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Johnson RJ, McCoy JG, Bingman CA, Phillips GN, Jr, Raines RT. Inhibition of human pancreatic ribonuclease by the human ribonuclease inhibitor protein. J Mol Biol. 2007;368:434–449. doi: 10.1016/j.jmb.2007.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dickson KA, Dahlberg CL, Raines RT. Compensating effects on the cytotoxicity of ribonuclease A variants. Arch Biochem Biophys. 2003;415:172–177. doi: 10.1016/s0003-9861(03)00214-5. [DOI] [PubMed] [Google Scholar]
- 21.Lee JE, Raines RT. Contribution of active-site residues to the function of onconase, a ribonuclease with antitumoral activity. Biochemistry. 2003;42:11443–11450. doi: 10.1021/bi035147s. [DOI] [PubMed] [Google Scholar]
- 22.Wu Y, Mikulski SM, Ardelt W, Rybak SM, Youle RJ. A cytotoxic ribonuclease. Study of the mechanism of onconase cytotoxicity. J Biol Chem. 1993;268:10686–10693. [PubMed] [Google Scholar]
- 23.Rushizky GW, Knight CA, Sober HA. Studies on the preferential specificity of pancreatic ribonuclease as deduced from partial digests. J Biol Chem. 1961;236:2732–2737. [PubMed] [Google Scholar]
- 24.Wlodawer A, Miller M, Sjolin L. Active site of RNase: Neutron diffraction study of a complex with uridine vanadate, a transition-state analog. Proc Natl Acad Sci USA. 1983;80:3628–3631. doi: 10.1073/pnas.80.12.3628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.delCardayré SB, Raines RT. Structural determinants of enzymatic processivity. Biochemistry. 1994;33:6031–6037. doi: 10.1021/bi00186a001. [DOI] [PubMed] [Google Scholar]
- 26.Kelemen BR, Schultz LW, Sweeney RY, Raines RT. Excavating an active site: The nucleobase specificity of ribonuclease A. Biochemistry. 2000;39:14487–14494. doi: 10.1021/bi001862f. [DOI] [PubMed] [Google Scholar]
- 27.Okabe Y, Katayama N, Iwama M, Watanabe H, Ohgi K, Irie M, Nitta K, Kawauchi H, Takayanagi Y, Oyama F, Titani K, Abe Y, Oakzaki T, Inokuchi N, Koyama T. Comparative base specificity, stability, and lectin activity of two lectins from eggs of Rana catesbeiana and R. japonica and liver ribonuclease from R. catesbeiana. J Biochem (Tokyo) 1991;109:786–790. doi: 10.1093/oxfordjournals.jbchem.a123457. [DOI] [PubMed] [Google Scholar]
- 28.Witzel H, Barnard EA. Mechanism and binding sites in the ribonuclease reaction. II. Kinetic studies on the first step of the reaction. Biochem Biophys Res Commun. 1962;7:295–299. doi: 10.1016/0006-291x(62)90194-8. [DOI] [PubMed] [Google Scholar]
- 29.Saxena SK, Sirdeshmukh R, Ardelt W, Mikulski SM, Shogen K, Youle RJ. Entry into cells and selective degradation of tRNAs by a cytotoxic member of the RNase A family. J Biol Chem. 2002;277:15142–15146. doi: 10.1074/jbc.M108115200. [DOI] [PubMed] [Google Scholar]
- 30.Suhasini AN, Sirdeshmukh R. Transfer RNA cleavages by onconase reveal unusual cleavage sites. J Biol Chem. 2006;281:12201–12209. doi: 10.1074/jbc.M504488200. [DOI] [PubMed] [Google Scholar]
- 31.Findlay D, Herries DG, Mathias AP, Rabin BR, Ross CA. The active site and mechanism of action of bovine pancreatic ribonuclease. Nature. 1961;190:781–784. doi: 10.1038/190781a0. [DOI] [PubMed] [Google Scholar]
- 32.Findlay D, Herries DG, Mathias AP, Rabin BR, Ross CA. The active site and mechanism of action of bovine pancreatic ribonuclease. 7. The catalytic mechanism. Biochem J. 1962;85:152–153. doi: 10.1042/bj0850152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Roberts GC, Dennis EA, Meadows DH, Cohen JS, Jardetzky O. The mechanism of action of ribonuclease. Proc Natl Acad Sci USA. 1969;62:1151–1158. doi: 10.1073/pnas.62.4.1151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Thompson JE, Venegas FD, Raines RT. Energetics of catalysis by ribonucleases: Fate of the 2′,3′-cyclic intermediate. Biochemistry. 1994;33:7408–7414. doi: 10.1021/bi00189a047. [DOI] [PubMed] [Google Scholar]
- 35.Herries DG, Mathias AP, Rabin BR. The active site and mechanism of action of bovine pancreatic ribonuclease. 3. The pH-dependence of the kinetic parameters for the hydrolysis of cytidine 2¢,3¢-phosphate. Biochem J. 1962;85:127–134. doi: 10.1042/bj0850127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.del Rosario EJ, Hammes GG. Kinetic and equilibrium studies of the ribonuclease-catalyzed hydrolysis of uridine 2′,3′-cyclic phosphate. Biochemistry. 1969;8:1884–1889. doi: 10.1021/bi00833a017. [DOI] [PubMed] [Google Scholar]
- 37.Lou YC, Huang YC, Pan YR, Chen C, Liao YD. Roles of N-terminal pyroglutamate in maintaining structural integrity and pKa values of catalytic histidine residues in bullfrog ribonuclease 3. J Mol Biol. 2006;355:409–421. doi: 10.1016/j.jmb.2005.10.069. [DOI] [PubMed] [Google Scholar]
- 38.Liao YD, Huang HC, Leu YJ, Wei CW, Tang PC, Wang SC. Purification and cloning of cytotoxic ribonucleases from Rana catesbeiana (bullfrog) Nucleic Acids Res. 2000;28:4097–4104. doi: 10.1093/nar/28.21.4097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Markley JL. Correlation proton magnetic resonance studies at 250 MHz of bovine pancreatic ribonuclease. I. Reinvestigation of the histidine peak assignment. Biochemistry. 1975;14:3546–3553. doi: 10.1021/bi00687a006. [DOI] [PubMed] [Google Scholar]
- 40.Quirk DJ, Raines RT. His⋯Asp catalytic dyad of ribonuclease A: Histidine pKa values in the wild-type, D121N, and D121A enzymes. Biophys J. 1999;76:1571–1579. doi: 10.1016/S0006-3495(99)77316-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Park C, Schultz LW, Raines RT. Contribution of the active site histidine residues of ribonuclease A to nucleic acid binding. Biochemistry. 2001;40:4949–4956. doi: 10.1021/bi0100182. [DOI] [PubMed] [Google Scholar]
- 42.Tarragona-Fiol A, Eggelte HJ, Harbron S, Sanchez E, Taylorson CJ, Ward JM, Rabin BR. Identification by site-directed mutagenesis of amino acids in the B2 subsite of bovine pancreatic ribonuclease A. Protein Eng. 1993;6:901–906. doi: 10.1093/protein/6.8.901. [DOI] [PubMed] [Google Scholar]
- 43.delCardayré SB, Raines RT. A residue to residue hydrogen bond mediates the nucleotide specificity of ribonuclease A. J Mol Biol. 1995;252:328–336. doi: 10.1006/jmbi.1995.0500. [DOI] [PubMed] [Google Scholar]
- 44.Fontecilla-Camps JC, de Llorens R, le Du MH, Cuchillo CM. Crystal structure of ribonuclease A·d(ApTpApApG) complex. J Biol Chem. 1994;269:21526–21531. doi: 10.2210/pdb1rcn/pdb. [DOI] [PubMed] [Google Scholar]
- 45.Nogués MV, Moussaoui M, Boix E, Vilanova M, Ribó M, Cuchillo CM. The contribution of noncatalytic phosphate-binding subsites to the mechanism of bovine pancreatic ribonuclease A. Cell Mol Life Sci. 1998;54:766–774. doi: 10.1007/s000180050205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Fisher BM, Grilley JE, Raines RT. A new remote subsite in ribonuclease A. J Biol Chem. 1998;273:34134–34138. doi: 10.1074/jbc.273.51.34134. [DOI] [PubMed] [Google Scholar]
- 47.Gorbatyuk VY, Tsai CK, Chang CF, Huang TH. Effect of N-terminal and Met23 mutations on the structure and dynamics of onconase. J Biol Chem. 2004;279:5772–5780. doi: 10.1074/jbc.M311233200. [DOI] [PubMed] [Google Scholar]
- 48.Shindyalov IN, Bourne PE. Protein structure alignment by incremental combinatorial extension (CE) of the optimal path. Protein Eng. 1998;11:739–747. doi: 10.1093/protein/11.9.739. [DOI] [PubMed] [Google Scholar]
- 49.Merlino A, Mazzarella L, Carannante A, Di Fiore A, Di Donato A, Notomista E, Sica F. The importance of dynamic effects on the enzyme activity: X-ray structure and molecular dynamics of onconase mutants. J Biol Chem. 2005;280:17953–17960. doi: 10.1074/jbc.M501339200. [DOI] [PubMed] [Google Scholar]
- 50.Rosenberg HF, Zhang J, Liao YD, Dyer KD. Rapid diversification of RNase A superfamily ribonucleases from the bullfrog. Rana catesbeiana J Mol Evol. 2001;53:31–38. doi: 10.1007/s002390010188. [DOI] [PubMed] [Google Scholar]
- 51.Hsu CH, Liao YD, Pan YR, Chen LW, Wu SH, Leu YJ, Chen C. Solution structure of the cytotoxic RNase 4 from oocytes of bullfrog Rana catesbeiana. J Mol Biol. 2003;326:1189–1201. doi: 10.1016/s0022-2836(02)01472-9. [DOI] [PubMed] [Google Scholar]
- 52.Leu YJ, Chern SS, Wang SC, Hsiao YY, Amiraslanov I, Liaw YC, Liao YD. Residues involved in the catalysis, base specificity, and cytotoxicity of ribonuclease from Rana catesbeiana based upon mutagenesis and x-ray crystallography. J Biol Chem. 2003;278:7300–7309. doi: 10.1074/jbc.M206701200. [DOI] [PubMed] [Google Scholar]
- 53.Fisher BM, Ha J-H, Raines RT. Coulombic forces in protein–RNA interactions: Binding and cleavage by ribonuclease A and variants at Lys7, Arg10, and Lys66. Biochemistry. 1998;37:12121–12132. doi: 10.1021/bi980743l. [DOI] [PubMed] [Google Scholar]
- 54.Steyaert J. A decade of protein engineering on ribonuclease T1—atomic dissection of the enzyme-substrate interactions. Eur J Biochem. 1997;247:1–11. doi: 10.1111/j.1432-1033.1997.t01-1-00001.x. [DOI] [PubMed] [Google Scholar]
- 55.Carlow DC, Short SA, Wolfenden R. Complementary truncations of a hydrogen bond to ribose involved in transition-state stabilization by cytidine deaminase. Biochemistry. 1998;37:1199–1203. doi: 10.1021/bi971731n. [DOI] [PubMed] [Google Scholar]
- 56.Loverix S, Winqvist A, Stromberg R, Steyaert J. Mechanism of RNase T1: Concerted triester-like phosphoryl transfer via a catalytic three-centered hydrogen bond. Chem Biol. 2000;7:651–658. doi: 10.1016/s1074-5521(00)00005-3. [DOI] [PubMed] [Google Scholar]
- 57.Yin Y, Sampson NS, Vrielink A, Lario PI. The presence of a hydrogen bond between asparagine 485 and the π system of FAD modulates the redox potential in the reaction catalyzed by cholesterol oxidase. Biochemistry. 2001;40:13779–13787. doi: 10.1021/bi010843i. [DOI] [PubMed] [Google Scholar]
- 58.Kicska GA, Tyler PC, Evans GB, Furneaux RH, Shi W, Fedorov A, Lewandowicz A, Cahill SM, Almo SC, Schramm VL. Atomic dissection of the hydrogen bond network for transition-state analogue binding to purine nucleoside phosphorylase. Biochemistry. 2002;41:14489–14498. doi: 10.1021/bi026636f. [DOI] [PubMed] [Google Scholar]
- 59.Kim KW, Wang Z, Busby J, Tsuruda T, Chen M, Hale C, Castro VM, Svensson S, Nybo R, Xiong F, Wang M. The role of tyrosine 177 in human 11β-hydroxysteroid dehydrogenase type 1 in substrate and inhibitor binding: An unlikely hydrogen bond donor for the substrate. Biochim Biophys Acta. 2006;1764:824–830. doi: 10.1016/j.bbapap.2006.02.008. [DOI] [PubMed] [Google Scholar]
- 60.Basu G, Sivanesan D, Kawabata T, Go N. Electrostatic potential of nucleotide-free protein is sufficient for discrimination between adenine and guanine-specific binding sites. J Mol Biol. 2004;342:1053–1066. doi: 10.1016/j.jmb.2004.07.047. [DOI] [PubMed] [Google Scholar]
- 61.Nobeli I, Laskowski RA, Valdar WS, Thornton JM. On the molecular discrimination between adenine and guanine by proteins. Nucleic Acids Res. 2001;29:4294–4309. doi: 10.1093/nar/29.21.4294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Jeffrey GA. An Introduction to Hydrogen Bonding. Oxford University Press; New York: 1997. [Google Scholar]
- 63.Borkakoti N, Moss DS, Palmer RA. Ribonuclease A—least-squares refinement of the structure at 1.45 Å resolution. Acta Crystallogr B Struct Sci. 1982;38:2210–2217. [Google Scholar]
- 64.Howlin B, Moss DS, Harris GW. Segmented anisotropic refinement of bovine ribonuclease-A by the application of the rigid-body TLS model. Acta Cryst A. 1989;45:851–861. doi: 10.1107/s0108767389009177. [DOI] [PubMed] [Google Scholar]
- 65.Jackson DY, Burnier J, Quan C, Stanley M, Tom J, Wells JA. A designed peptide ligase for total synthesis of ribonuclease A with unnatural catalytic residues. Science. 1994;266:243–247. doi: 10.1126/science.7939659. [DOI] [PubMed] [Google Scholar]
- 66.Ardelt B, Ardelt W, Darzynkiewicz Z. Cytotoxic ribonucleases and RNA interference (RNAi) Cell Cycle. 2003;2:22–24. doi: 10.4161/cc.2.1.232. [DOI] [PubMed] [Google Scholar]
- 67.Cai YC, Bullard JM, Thompson NL, Spremulli LL. Interaction of mitochondrial elongation factor Tu with aminoacyl-tRNA and elongation factor Ts. J Biol Chem. 2000;275:20308–20314. doi: 10.1074/jbc.M001899200. [DOI] [PubMed] [Google Scholar]
- 68.Nissen P, Kjeldgaard M, Thirup S, Polekhina G, Reshetnikova L, Clark BFC, Nyborg J. Crystal structure of the ternary complex of Phe-tRNAPhe, EF-Tu, and a GTP analog. Science. 1995;270:1464–1472. doi: 10.1126/science.270.5241.1464. [DOI] [PubMed] [Google Scholar]
- 69.Smith BD, Soellner MB, Raines RT. Potent inhibition of ribonuclease A by oligo(vinylsulfonic acid) J Biol Chem. 2003;278:20934–20938. doi: 10.1074/jbc.M301852200. [DOI] [PubMed] [Google Scholar]
- 70.Vagin A, Teplyakov A. MOLREP: An automated program for molecular replacemet. J Appl Crystallogr. 1997;30:1022–1025. [Google Scholar]
- 71.Emsley P, Cowtan K. Coot: Model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 72.Murshudov GN, Vagin AA, Dodson EJ. Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr D Biol Crystallogr. 1997;53:240–255. doi: 10.1107/S0907444996012255. [DOI] [PubMed] [Google Scholar]
- 73.Lovell SC, Davis IW, Arendall WB, III, de Bakker PIW, Word JM, Prisant MG, Richardson JS, Richardson DC. Structure validation by Cα geometry: φ, ψ and Cβ deviation. Proteins. 2003;50:437–450. doi: 10.1002/prot.10286. [DOI] [PubMed] [Google Scholar]
- 74.Berman HM, Westbrook J, Feng Z, Gilliland G, Bhat TN, Weissig H, Shindyalov IN, Bourne PE. The Protein Data Bank. Nucleic Acids Res. 2000;28:235–242. doi: 10.1093/nar/28.1.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Park C, Raines RT. Quantitative analysis of the effect of salt concentration on enzymatic catalysis. J Am Chem Soc. 2001;123:11472–11479. doi: 10.1021/ja0164834. [DOI] [PubMed] [Google Scholar]
- 76.Kelemen BR, Klink TA, Behlke MA, Eubanks SR, Leland PA, Raines RT. Hypersensitive substrate for ribonucleases. Nucleic Acids Res. 1999;27:3696–3701. doi: 10.1093/nar/27.18.3696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Park C, Kelemen BR, Klink TA, Sweeney RY, Behlke MA, Eubanks SR, Raines RT. Fast, facile, hypersensitive assays for ribonucleolytic activity. Methods Enzymol. 2001;341:81–94. doi: 10.1016/s0076-6879(01)41146-3. [DOI] [PubMed] [Google Scholar]
- 78.Haigis MC, Kurten EL, Abel RL, Raines RT. KFERQ sequence in ribonuclease A-mediated cytotoxicity. J Biol Chem. 2002;277:11576–11581. doi: 10.1074/jbc.M112227200. [DOI] [PubMed] [Google Scholar]
- 79.Merritt EA, Murphy MEP. Raster3D Version 2.0. A program for photorealistic molecular graphics. Acta Crystallogr D Biol Crystallogr. 1994;50:869–873. doi: 10.1107/S0907444994006396. [DOI] [PubMed] [Google Scholar]