

Abstract
Administration of peroxisome proliferators to rodents causes proliferation of peroxisomes, induction of β-oxidation enzymes, hepatocelluar hypertrophy and hyperplasia, with chronic exposure ultimately leading to hepatocellular carcinomas. Many responses associated with peroxisome proliferators are nuclear receptor-mediated events involving peroxisome proliferators-activated receptor alpha (PPARα). A role for nuclear receptor-independent events has also been shown, with evidence of Kupffer cell-mediated free radical production, presumably through NAPDH oxidase, induction of redox-sensitive transcription factors involved in cytokine production and cytokine-mediated cell replication following acute treatment with peroxisome proliferators in rodents. Recent studies have demonstrated, by using p47phox-null mice which are deficient in NADPH oxidase, that this enzyme is not related to the phenotypic events caused by prolonged administration of peroxisome proliferators. In an effort to determine the timing of the transition from Kupffer cell- to PPARα-dependent modulation of peroxisome proliferator effects, gene expression was assessed in liver from Pparα-null, p47phox-null and corresponding wild-type mice following treatment with 4-chloro-6-(2,3-xylidino)-pyrimidynylthioacetic acid (WY-14,643) for 8 h, 24 h, 72 h, 1 wk, or 4 wks. WY-14,643-induced gene expression in p47phox-null mouse liver differed substantially from wild-type mice at acute doses and striking differences in baseline expression of immune related genes were evident. Pathway mapping of genes that respond to WY-14,643 in a time- and dose-dependent manner demonstrates suppression of immune response, cell death and signal transduction and promotion of lipid metabolism, cell cycle and DNA repair. Furthermore, these pathways were largely dependent on PPARα, not NADPH oxidase demonstrating a temporal shift in response to peroxisome proliferators. Overall, this study shows that NADPH oxidase-dependent events, while detectable following acute treatment, are transient and short-lived. To the contrary, a strong PPARα-specific gene signature was evident in mice that were continually exposed to WY-14,643.
Keywords: peroxisome proliferators, PPARα, Kupffer cells, toxicogenomics, microarrays
INTRODUCTION
Peroxisome proliferators (PPs) are a chemical class comprised of a wide range of industrial, pharmaceutical and endogenous compounds. These compounds have been the subject of debate for several decades because of their carcinogenicity in rodents (Lalwani et al., 1981) and uncertain risk to humans (Klaunig et al., 2003; Rusyn et al., 2006). A number of peroxisome proliferator-induced events leading up to carcinogenesis, including increased cell replication, oxidative damage and tumorigenesis itself require activation of nuclear receptor peroxisome proliferators-activated receptor alpha (PPARα) (Peters et al., 1997; Rusyn et al., 2004). This receptor is largely responsible for lipid metabolism through transcriptional regulation of fatty acid oxidation enzymes, apolipoproteins and transporters (Peters et al., 2005). Among species there are substantial structural differences in the DNA binding element, peroxisome proliferator response element (PPRE) along with differences in basal PPARα expression, with humans exhibiting a tenth of the levels observed in rodents (Palmer et al., 1998).
A wide range of nuclear receptor-independent effects of PPARa-agonists in rodent liver have been reported and some of these were attributed to activation of the Kupffer cells (Rusyn et al., 2006). Therefore, delineating the relative contribution of receptor-independent and -dependent molecular events is important for human health risk assessment of these agents, which is currently based largely on the inter-species differences in PPARα activation and signaling (Peters et al., 2005). Studies performed in vivo and in vitro demonstrated that peroxisome proliferators stimulate superoxide and cytokine production by these liver macrophages (Hasmall et al., 2000b). Studies using NADPH oxidase-deficient (p47phox-null) and Pparα–null mice demonstrated that acute effects of peroxisome proliferators seem to be largely mediated by oxidant production in Kupffer cells (Peters et al., 2000; Rusyn et al., 2001). WY-14,643-induced cell proliferation is abrogated in p47phox-null mice or when mice are pre-treated with NAPDH oxidase inhibitor, diphenyliodonium. Despite evidence demonstrating a role for Kupffer cells and mitogens in the early events in rodent liver associated with peroxisome proliferators, other studies conducted in TNFα-(Lawrence et al., 2001) and TNF receptor-(TNFR) null mice (Anderson et al., 2001) suggest that WY-14,643-induced cell proliferation may not be mediated by TNFα. It is clear, however, that PPARα mediates key events in rodent liver following sub-chronic and chronic treatment of peroxisome proliferators (Peters et al., 1997; Woods et al., 2007b).
Microarray technology has served as a valuable tool for gathering mechanistic information regarding toxicants molecular targets and temporal progression of toxicity leading to specific disease states. Genomic studies investigating the effects of peroxisome proliferators in rodent liver have provided critical insight into the molecular mechanisms responsible for liver-specific effects of peroxisome proliferators in rodents and supported a non-genotoxic mechanism of their action (Cherkaoui-Malki et al., 2001; Hamadeh et al., 2002; Currie et al., 2005). However, the gap in our knowledge remains on the temporal relationship between peroxisome proliferator-modulated effects and molecular mediators of these effects, especially at the early time points where PPARα-independent events are also known to occur. To address this, gene expression analysis was conducted in livers from p47phox-null, Pparα-null and wild type mice treated from 8 h to 4 wks with a potent peroxisome proliferator, 4-chloro-6-(2,3-xylidino)-pyrimidynylthioacetic acid (WY-14,643). We show that NADPH oxidase-dependent events, while detectable following acute treatment, are transient and short-lived. To the contrary, a strong PPARα-specific gene signature was evident in mice that were continually exposed to WY-14,643.
METHODS
Animals, Diets and Tissue Collection
p47phox-null male mice [C57BL/6J background; (Jackson et al., 1995)], Pparα-null male mice [SV129 background; (Lee et al., 1995)], and corresponding wild type counterparts (6–8 weeks of age at the beginning of treatment) were used in these experiments. All animals used for this study were housed in sterilized cages in a facility with a 12-hr night/day cycle. Temperature and relative humidity were held at 22 ± 2°C and 50 ± 5%, respectively. The UNC Division of Laboratory Animal Medicine maintains these animal facilities, and veterinarians were always available to ensure animal health. All animals were given humane care in compliance with NIH and institutional guidelines and studies were performed according to protocols approved by the appropriate institutional review board. Prior to experiments, animals were maintained on standard lab chow diet and purified water ad libitum. WY-14,643 was obtained from Aldrich (Milwaukee, WI). Acute doses were administered by a single oral gavage of 0 (control), 5 or 50 mg/kg of WY-14,643 in olive oil. Mice (n=3) were sacrificed 8 h, 24 h, or 72 h post dosing. Sub-chronic doses of WY-14,643 were administered in the diet ad libitum. NIH-07 was used as the base for the powdered diet containing either 0 (control), 50, or 500 ppm of WY-14,643. Mice (n=3) were sacrificed after either 1 week or 4 weeks of dietary treatment. Animals had free access to water throughout the study and the health status of the animals was monitored every other day. At sacrifice, mice were anesthetized with pentobarbital (100 mg/kg) and following exsanguination livers were removed and weighed. A section from the left lateral lobe was fixed in 10% formalin. The remaining tissue was snap frozen in liquid nitrogen. The samples were stored at −80°C until assayed.
RNA Isolation
While frozen a small fragment (approximately 30 mg) was removed for each liver sample and homogenized for 30 s in 600 μl RLT buffer (Qiagen, Valencia, CA) containing 1 % β-mercaptoethanol. The lysate was centrifuged for 3 min at 13000 rpm. From the resulting supernatant, total RNA was isolated using an RNeasy kit (Qiagen) as per the manufacturer’s protocol. Total RNA integrity and quantification were assessed using RNA 6000 nano assay LabChips® (Agilent Technologies, Santa Clara, CA) and analyzed on a 2100 Bioanalyzer (Agilent Technologies) as per the manufacturer’s protocol.
cDNA Preparation and Microarray Hybridization
Preparation of cDNA, labeling and hybridizations were performed using reagents from the low RNA input fluorescent linear amplification kit (Agilent Technologies) based on the manufacturer’s protocol. A pooled mouse RNA sample (Cogenics, RTP, NC) derived from equal amounts of RNA from kidney, spleen, lung, brain, and liver was used as a reference and prepared in parallel to the samples of interest. Samples were analyzed using an Agilent Mouse Oligo Microarray (~21,000 features, catalogue# G4121). The hybridized microarrays were washed and scanned using an Agilent G2565BA scanner. Data were extracted from the scanned image using Agilent Feature Extraction software version 6.1. Raw data is available from the UNC Microarray database (genome.unc.edu).
Microarray Data Analysis
Array quality was assessed using Agilent Feature Extraction software and genes with fewer than 70% present data across all arrays were excluded from further analysis. A total of 16,030 probes passed this data quality filter. Removal of control oligos, and RIKENs, for which little or no functional data was available reduced the list to 11,421 genes. These transcripts comprised our working data set. LOWESS normalization was performed to eliminate dye bias. A second normalization was performed to correct for basal differences in gene expression between SV129 and C57BL/6J mice or between wild-type and knockout mice on both backgrounds. This normalization involved dividing the Cy5/Cy3 ratio for a given gene by the mean Cy5/Cy3 ratio for the same gene from time and strain-matched controls. Missing data points were calculated using K-nearest neighbor imputation method (Troyanskaya et al., 2001). Average-linkage, hierarchical clustering was performed using Cluster software on median centered (by genes) data and visualization was facilitated by Treeview (Eisen et al., 1998).
Differentially expressed genes were identified using either Significance Analysis of Microarrays (SAM) or EDGE software (Tusher et al., 2001; Leek et al., 2006). SAM was performed in cases where statistical significance across only one variable (strain or dose) was being assessed. EDGE was used for identifying differentially expressed genes over time within a single condition or among several conditions. Q-values, which represent the false discover rate (FDR) of less than 0.05 for SAM and EDGE were selected as thresholds for differential expression. Once the list of significant genes was generated by EDGE, a t-statistic was calculated for each gene at each strain/time combination to determine statistical difference between high dose and control expression. For each strain/time combination, a list of differentially expressed genes (p<0.05) was used for functional analysis.
Functional Analysis of Significant Genes
EASE (Dennis, Jr. et al., 2003), GOMiner (Zeeberg et al., 2003), and High-Throughput GOMiner (Zeeberg et al., 2005) were used to determine biological function of differentially expressed genes, in the context of Gene Ontology (GO). EASE and GOMiner were utilized for pathway analysis of genes identified in two class SAM comparisons. A score (i.e., p value) of < 0.05 was selected as the cutoff for statistical significance. High-Throughput GOMiner was used for pathway analysis of significant genes lists generated from EDGE time-course analysis. A Q-value, representative of the FDR<0.05 and p<0.05 were the basis for statistical significance from this analysis. Finally, gene networks were prepared with Pathway Studio® 4.0 software (Ariadne Genomics, Rockville, MD) (Nikitin et al., 2003). The software uses Medscan natural language processing to gather information from all abstracts on PubMed and other public data sources, which is extracted to assemble molecular networks.
Real-Time PCR
Real-time PCR assays were performed using Taqman® (Applied Biosystems) microfluidics cards to probe over 300 genes of interest (Supplemental Table 1). RNA samples from animals treated with control or WY-14,643-containing diet for 4 weeks were used for this analysis. Preparation of cDNA from 25ng of RNA was performed using High capacity cDNA archive kit (Applied Biosystems), according to the manufacturer’s protocol. PCR mix was prepared used Taqman Universal PCR Master Mix was used to prepare sample-specific PCR mix and PCR was performed using the ABI Prism 7700 Sequence Detection system according to the manufacturer’s protocol (http://docs.appliedbiosystems.com/pebiodocs/04319399.pdf). Quantification of data involved calculating 2ΔΔCT. The ΔCt values for all genes were calculated relative to the average Ct value for four 18s probes. The ΔΔCt values were calculated using ΔCt values for WY-14,643-treated samples relative to mean ΔCt values for strain-matched controls.
RESULTS AND DISCUSSION
The aim of this study was to investigate WY-14,643-induced temporal changes in gene expression and to understand the inter-relationship between, and timing of PPAR□- and Kupffer cell-dependent molecular events in the mechanism of action of peroxisome proliferators in rodent liver. To address these questions, we evaluated the differences between modulation of gene expression by WY-14,643 in p47phox- and Ppar□-null mice, two mouse engineered models that were used previously to elucidate receptor-dependent and -independent pathways. This study provides an extensive time-course and dose-response of peroxisome proliferator-induced gene expression changes in mouse liver in the context of two important modes of action of these compounds: activation of PPAR□ and Kupffer cells. Gene expression data presented here clearly demonstrate that Kupffer cell–mediated molecular changes in mouse liver evoked by WY-14,643 are transient and short-lived, and are not observed with continuous treatment.
Basal differences in liver gene expression between wild type and p47phox-null mice
First, gene expression in control-fed wild-type and p47phox-null mice was compared. A two–class comparison using Significance Analysis of Microarrays (SAM) identified 2257 genes as differentially expressed between the strains across all time points (Figure 1). Immune response, fatty acid metabolism (specifically β-oxidation), glucose metabolism and amino acid metabolism were identified as the biological processes that were differentially expressed between wild type and p47phox-null mice.
Figure 1. Immune response and fatty acid metabolism are affected in liver of p47phox-null mice.

Differentially expressed genes were identified between untreated wild type and p47phox-null mice across all time points. Biological pathway analysis identified (A) immune response and (B) fatty acid metabolism as significantly enriched categories. Hierarchical clustering of the genes contributing to these biological processes is shown.
Divergent expression of genes involved in innate immunity (Figure 1A) was expected given the major role that Kupffer cell NADPH oxidase plays in the liver’s defense response (Li and Diehl, 2003). A number of lipid mediators of inflammation exhibited higher basal levels of expression in p47phox-null mice. These include phosopholipase A2 (Pla2g4a), which plays a role in arachidonic acid release and eiconsanoid biosynthesis, prostaglandin endoperoxide synthase 1 (Ptgs1), also known as cyclooxygenase 1 (Cox1), lipoprotein lipase (Lpl), and NADPH-dependent enzyme leukotriene B4 dehydrogenase (Ltb4dh). p47phox-null mice also exhibited elevated levels of Toll-like receptor 4 (Tlr4), which is a well-known mediator of lipopolysaccharide (LPS)-induced inflammation. Complement 3 (C3) and fibronectin 1 (Fn1), which both promote phagocytosis, were also suppressed in p47phox-null mice.
Interestingly, striking differences in basal gene expression for fatty acid metabolism genes, a large number of which are PPARα–regulated was also observed between wild-type and p47phox-null mice (Figure 1B). Several studies have drawn a link between NADPH oxidase and lipid metabolism through reactive oxygen species-related low density lipoprotein (LDL) oxidation (Bey and Cathcart, 2000; Teissier et al., 2004). The oxidized LDL metabolites are PPARα ligands and can promote transcription of PPARα target genes. Based on this proposed mechanism of cross-talk between PPARα and liver macrophages, basal levels of PPARα-active molecules in the liver may be lower in NADPH oxidase-deficient mice compared to wild types, in line with the observations from this study of the lower basal mRNA levels of PPARα-target genes in p47phox-null animals.
WY-14,643-induced differences in gene expression between p47phox-null and wild type mice
It has been shown previously that p47phox-null mice are protected from acute liver-specific effects of peroxisome proliferators (Rusyn et al., 2000). To elucidate the temporal role of Kupffer cell-mediated pathways in response to peroxisome proliferator treatment, acute (8 hrs) and sub-chronic (4 wks) changes in gene expression were assessed using p47phox-null mice (Figure 2). Specifically, to determine the Kupffer cell-specific molecular signature in response to WY-14,643, we identified genes that were differentially expressed between control and WY-14,643 samples in the wild type, but not in p47phox-null mice. From 444 genes selected in this analysis, four clusters are evident. There are genes that were up- (gene set A), or down- (gene set C) regulated in response to treatment with WY-14,643 for 8 hrs, but not for 4 wks. In addition, a large number of genes exhibited sustained (present in both 8 hr and 4 wk samples) up- (gene set B), or down- (gene set D) regulation in response to WY-14,643 treatment.
Figure 2. p47phox-specific effects of WY-14,643 in mouse liver.
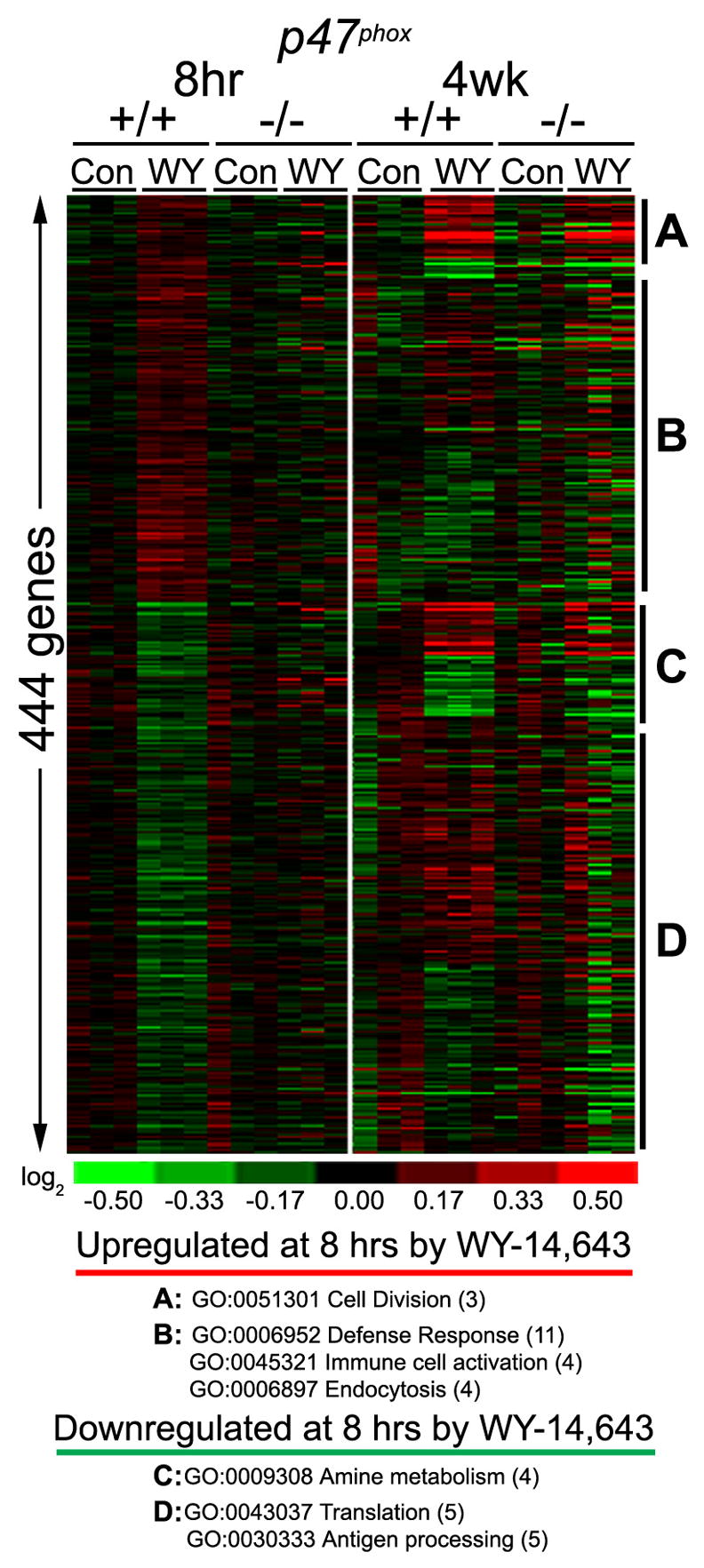
Gene expression data from p47phox-null and wild type mice treated with WY-14,643 for 8 hrs (50 mg/kg, i.g.) or 4 weeks (500 ppm, diet) was used to identify differentially expressed transcripts. The analysis produced a list of 444 genes which are shown as the heat map. Clusters of genes with concordant changes in expression were used to identify enriched (p<0.05) biological processes. (A and C) The genes that exhibit an early response to WY-14,643 and remain induced with continued treatment, but only the initial induction appears to be mediated by NADPH-oxidase. (B and D) The genes that exhibit WY-14,643-induced response which appears to be mediated by NAPDH oxidase- mediated at acute and sub-chronic time points.
Pathway mapping of the genes from each classification was performed using GOMiner. Gene set A was enriched with transcripts involved in cell division, which supports previous studies in which DNA synthesis or cell proliferation in liver is abrogated in response to acute peroxisome proliferator treatment as a result of inactivation of NADPH oxidase or Kupffer cells (Rose et al., 1999; Rusyn et al., 2000). Within gene set B, increases in defense response, immune cell activation and endocytosis were most pronounced acute responses to WY-14,643 that diminish with long-term treatment. This gene expression signature is supported by Kupffer cell activation by peroxisome proliferators and increased phagocytosis by these cells (Bojes and Thurman, 1996). The link between Kupffer cells and pathways identified in gene sets C and D corroborates a previous report of suppression of amino acid metabolism by peroxisome proliferators (Kersten et al., 2001).
Interestingly, induction of cell division genes at later time points was independent of NADPH oxidase (Figure 2, gene set A). In chronic dietary feeding studies in which mice were fed WY-14,643 (0.1% w/w) for up to 5 months, p47phox-null mice exhibited peroxisome proliferator-induced liver effects similar to those in wild type mice (Woods et al., 2007a). Hepatomegaly, increased cell proliferation and oxidative DNA damage, important modes of action in nongenotoxic carcinogenesis, were observed in wild type and p47phox-null mice fed WY-14,643. This shows that involvement of Kupffer cells in WY-14,643-induced parenchymal cell proliferation and oxidative stress in rodent liver is an acute phenomenon that may not be relevant to long-term effects of peroxisome proliferators. While these results suggest that induction of cell proliferation in rodent liver appears to be tightly linked to PPARα-mediated signaling in the hepatocyte, the exact mechanism of such interaction is not presently known. Several hypothesis suggesting a link via mitogen activated kinases, such as p38 (Roberts et al., 2001), and/or Ras-mediated signaling (Wheeler et al., 2003) have been recently brought forward. Furthermore, it is plausible that other, non-PPARα-mediated events in hepatocytes and/or other cell types in liver may be important for induction for the proliferative response in rodent liver.
Temporal changes in liver gene expression demonstrate a robust PPARα-dependent signature following WY-14,643 treatment
To evaluate the timing of PPARα- and Kupffer cell-mediated events, time-course gene expression data from WY-14,643-fed mouse liver was analyzed using EDGE. Temporal and dose-dependent effects of WY-14,643 are evident following hierarchical clustering based on the genes significantly changing in p47phox wild type (C57BL/6J) mice (Supplementary Figure 1). While the initial timing of most profound effects on gene expression varied among groups, a uniformly robust response was observed in both wild type strains and p47phox-null mice with 1 or 4 wk of continued WY-14,643 treatment. The large majority of the changes were PPARα-dependent with few genes being affected by WY-14,643 in Pparα-null mice (see below), a response that has been demonstrated in other gene array studies (Anderson et al., 2004a; Anderson et al., 2004b). Pathway analysis of the significantly different genes identified a number of biological processes that were perturbed by peroxisome proliferators in a dose- and time-dependent manner (Figure 3). Individual significant genes within each process are listed in Supplemental Table 2–5.
Figure 3. Time- and dose-dependent changes in biological pathways affected by WY-14,643 in mouse liver.

Significant genes from EDGE analysis (Figure 3) were used to identify enriched biological processes affected by WY-14,643 treatment. Supervised hierarchical clustering was used to organize treatments and strains (columns) and biological processes (rows). Processes that were down- (top half) and up- (bottom half) regulated were analyzed separately. Heat map shading reflects the significance (based on false discovery rate statistics, FDR) for a given pathway. The pathways are ordered within each category according to Gene Ontology hierarchy.
Among the processes that were down-regulated by WY-14,643 treatment in liver are immune response, cytolysis, electron transport and signal transduction. Up-regulation of pathways traditionally associated with peroxisome proliferators, including lipid metabolism, cell division, and response to endogenous stimulus (DNA repair) was also observed. Only acyl-coA metabolism (and its associated GO Term categories) exhibits a strong acute response that is sustained with continued feeding. As shown in Figure 3, the most robust signature for the majority of affected biological pathways is present at 1 wk and/or 4 wk. This may suggest that some of the early effects of peroxisome proliferators (i.e., induced cell proliferation), which historically have been considered to be transient acute/sub-acute phenotypes, may be sustained. Indeed, when cell proliferation was measured in the liver of wild type or p47phox-null mice, a robust elevation in BrdU labeling index was found to persist for up to 5 months of treatment (Woods et al., 2007a). Interestingly, temporal pathway analysis also revealed an early PPARα-independent cell replication signature (see below). The gene expression changes observed at 4 wks were confirmed using RT-PCR (Supplemental Figure 2) in which increased expression of genes related to fatty acid metabolism, DNA repair, and protein catabolism was observed and transcript levels for genes involved in signal transduction were down regulated in all strains except for Pparα-null mice.
Gene expression profiling reveals PPARα-mediated immunosuppression by WY-14,643
Liver-specific immunosuppression by peroxisome proliferators has been previously observed (Devchand et al., 1996). Our study found that treatment with WY-14,643 led to down-regulation of immune response and response to stimulus in liver in a manner that is dependent on PPARα, but not NADPH oxidase (Figure 4, top). This effect was most pronounced at 1 and 4 weeks after treatment, but the earliest onset may occur as early as 24 hrs post-dosing (Figure 3). Decrease in complement activation system was associated with reduced transcript levels of complements 1 (C1), C2, C5, C8, C9 and serine protease mannan-binding lectin serine peptidase (Masp1), effects similar to those reported by others (Wong and Gill, 2002; Anderson et al., 2004a; Currie et al., 2005). In addition to serving as important mediators of innate and adaptive immune response, complements play a major role in cell death byopsonizing apoptotic cells which are later cleared by phagocytes (Fishelson et al., 2001; Flierman and Daha, 2007). The membrane attack complex of complex has also been demonstrated to play a role in apoptosis through caspase activation (Nauta et al., 2002). As a result, suppression of complement pathways could lead to anti-apoptotic effects, which are considered to be an important mode of action in peroxisome proliferator-induced carcinogenesis.
Figure 4. Suppression of immune response and induction of cell cycle in mouse liver by WY-14,643 is dependent on PPARα, not NADPH oxidase.
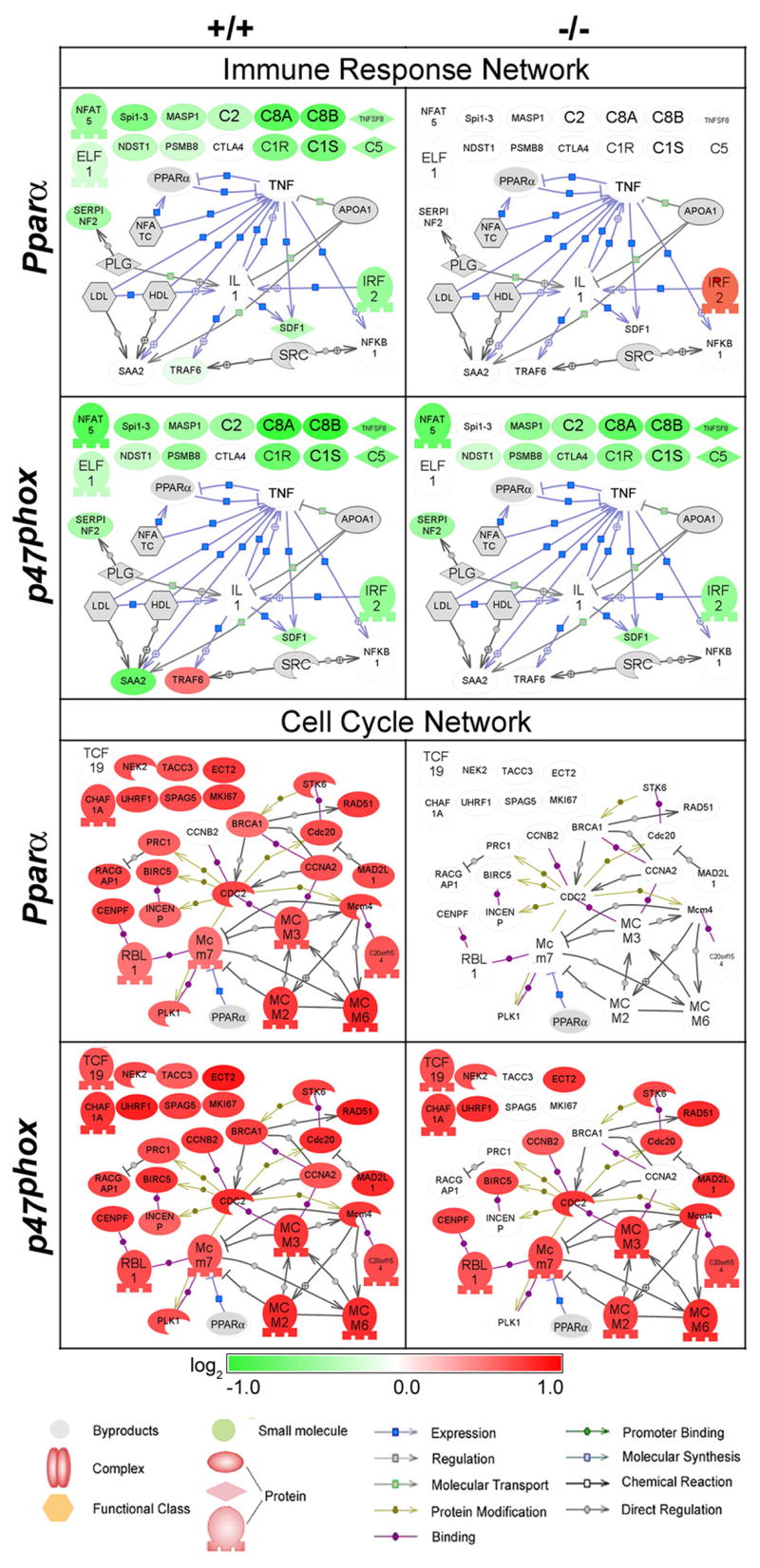
Expression of genes contributing to the Immune Response (top) and Cell Cycle (bottom) GO processes at 4 wks of treatment (see Figures 3 and 4) was visualized as networks using Pathway Studio software. Interactions among gene products include regulation (gray line) and induction of expression (blue line). Nodes shaded green are down-regulated and red nodes are up-regulated genes as compared to time/strain-matched control, and gray nodes are molecular mediators that were either not measured on the array or did not contribute to this GO analysis. No information regarding gene interactions was available using Pathway Studio for nodes that are not connected with arrows.
The mechanism by which peroxisome proliferators elicit immunosuppression is not well understood. It has been suggested that immune response effects may be associated with lipid metabolism. One hypothesis is that activation of PPARα by leukotriene B4 (LTB4) and subsequent clearance of the pro-inflammatory agent through metabolism results in a negative-feedback loop that regulates inflammation (Devchand et al., 1996); however, LTB4 metabolism was not found to be increased as a result of PPARα activation. It is also likely that peroxisome proliferators may modulate immune response by altering serum lipids. WY-14,643 treatment causes a decrease in serum lipids, which may alter the proportion of energy from fatty acids to peripheral tissues (i.e., spleen, lymphatic tissue) that participate in the immune response (Yang et al., 2000). This response in NADPH oxidase-deficient mice may be further exaggerated due to immuno-compromised state of these animals and render them more susceptible to pro-proliferation effects of WY-14,643 in liver.
Considering that early activation of Kupffer cells and events mediated by these cells (i.e. cell proliferation, oxidative stress, etc.) are ephemeral, it is quite possible that they are coincidental and associative. However, sub-chronic PPARα-mediated immunosuppression likely effects the Kupffer cell defense mechanisms and cytokine production. Cytokines in the liver, which are mainly produced by Kupffer cells serve as important mediators of innate immune response, but also prime cells to proliferate or undergo apoptosis (Ramadori and Armbrust, 2001). There are conflicting reports with regards to the effects of peroxisome proliferators on cytokine production, and their importance to hepatocarcinogenesis. A number of studies suggested that cytokines may play a critical role in cell proliferation and apoptosis by peroxisome proliferators (Bojes et al., 1997; Hasmall et al., 2000a; Parzefall et al., 2001). At the same time, many reports questioned that cytokines are a requisite for altered cell turnover by peroxisome proliferators (Ledda-Columbano et al., 1998; Anderson et al., 2001). To address this controversy, this study looked at the temporal profile of expression changes related to cytokine signaling in liver. We found that transcription factors that are regulated by or involved in regulation of cytokine release, including E74-like factor 1 (Elf1), nuclear factor of activated T-cells 5 (Nfat5), and interferon regulator factor 2 (Irf2) were down-regulated by WY-14,643 treatment in a PPARα-mediated manner.
WY-14,643-induced PPARα-independent gene expression may be mediated by other PPARs
It is widely accepted that activation of PPARα is required for peroxisome proliferator-induced cell proliferation in vivo (Peters et al., 1997). However, the link between cell cycle regulatory genes and activated PPARα has not yet been clearly delineated. To gain additional insight into peroxisome proliferator-induced events associated with cell proliferation in mouse liver and their dependence on PPARα, gene signatures at 24 h and 4 wks were further assessed with particular focus on genes contributing to the “cell cycle” node (Figure 4, bottom; and Figure 5A). Interestingly, as early as 24 h (Figure 5A) we observed that differential expression of a number of genes largely involved in promoting mitosis was exclusive to Pparα-null mice; however, cell proliferation is not increased in these mice at this time point (data not shown). Furthermore, these effects were dramatically reversed by 4 weeks of treatment (Figure 4, bottom). It has been shown recently that mitogens are required for passage through the G2 phase of the cell cycle into M phase (Foijer and Te, 2006). Mitogen-independent progression to mitosis is possible, but not before a lengthy cell cycle arrest (~10hr). Given the suppressed state of the immune response in WY-14,643- responder strains (both wild types and p47phox-null mice), it is possible that reduced mitogen release can explain the less robust cell cycle signature at early time points.
Figure 5. PPARα-dependent and -independent effects of WY-14,643 in mouse liver.
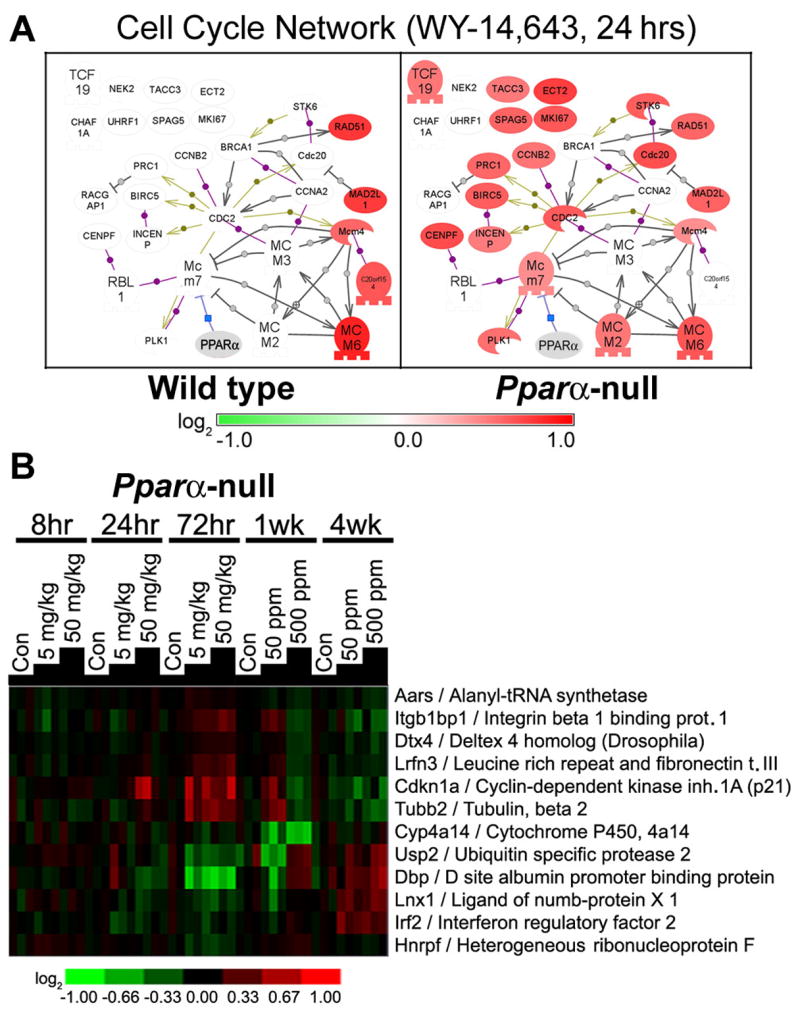
(A) Expression of genes contributing to the Cell Cycle GO process at 8 hrs after treatment (see Figures 3 and 4) was visualized as a network using Pathway Studio software. Please refer to Figure 5 for a symbol key. (B) Hierarchical clustering of liver genes in Pparα-null mice that respond to WY-14,643 treatment in temporal and dose-dependent manner. Twelve genes were found to be differentially expressed (FDR<0.05) using analysis detailed in Methods.
Furthermore, we examined the microarray data from Pparα-null mice to determine what genes do respond to WY-14,643 in time-and dose-dependent manner (Figure 5B). While only 12 genes were found to be significantly changed it is notable that even some putative PPARα-target genes, such as cytochrome P450 4a14 (Cyp4a14), are regulated in PPARα-independent manner, which has been previously demonstrated (Corton et al., 2004). WY-14,643 treatment caused a decrease in integrin beta 1 binding protein (Itgb1bp1) expression and an increase in Irf2, which inhibits transcriptional activation of interferon, suggesting that a PPARα-independent mechanism of immunosuppression by peroxisome proliferators may be involved. Cyclin-dependent kinase inhibitor 1A (Cdkn1a), also known as p21, which associates with p53 to impede cell cycle progression, is significantly up-regulated in Pparα-null mice with sub-acute WY-14,643 treatment and is suppressed at later time points. Tubulin 2 (Tubb2), which is also important to cell cycle regulation, particularly mitosis, exhibits a similar pattern of expression as p21. Tubb2 possesses a response element for both PPARγ and PPARδ (White et al., 2003), thus these observations may suggest that the ability of WY-14,643 to act via other PPARs is a small but detectable phenotype in Pparα-null mice.
Conclusions
This study provides an extensive time-course dataset of the effects of a model peroxisome proliferator WY-14,643 in liver of wild type and Pparα- and p47phox-null mice, animals that have been used to address the role of PPARα-dependent and -independent signaling events associated with these agents in rodent liver. This study provides several novel insights into the mechanisms of action of peroxisome proliferators in rodent liver. First, our data demonstrates that Kupffer cells are involved in immediate early responses to these agents, but may not play a role in chronic effects of peroxisome proliferators. To the contrary, PPARα-mediated molecular changes are prevalent with longer exposure to peroxisome proliferators. Second, we show that peroxisome proliferators cause a strong immunosuppressive response in mouse liver and suggest that this response may contribute to the anti-apoptotic effects of peroxisome proliferators. These effects may be linked to lipid metabolism in liver and controlled by PPARα as well as other PPARs.
Supplementary Material
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Anderson SP, Dunn C, Laughter A, Yoon L, Swanson C, Stulnig TM, Steffensen KR, Chandraratna RA, Gustafsson JA, Corton JC. Overlapping transcriptional programs regulated by the nuclear receptors peroxisome proliferator-activated receptor alpha, retinoid X receptor, and liver X receptor in mouse liver. Mol Pharmacol. 2004a;66:1440–1452. doi: 10.1124/mol.104.005496. [DOI] [PubMed] [Google Scholar]
- Anderson SP, Dunn CS, Cattley RC, Corton JC. Hepatocellular proliferation in response to a peroxisome proliferator does not require TNFalpha signaling. Carcinogenesis. 2001;22:1843–1851. doi: 10.1093/carcin/22.11.1843. [DOI] [PubMed] [Google Scholar]
- Anderson SP, Howroyd P, Liu J, Qian X, Bahnemann R, Swanson C, Kwak MK, Kensler TW, Corton JC. The transcriptional response to a peroxisome proliferator-activated receptor alpha agonist includes increased expression of proteome maintenance genes. J Biol Chem. 2004b;279:52390–52398. doi: 10.1074/jbc.M409347200. [DOI] [PubMed] [Google Scholar]
- Bey EA, Cathcart MK. In vitro knockout of human p47phox blocks superoxide anion production and LDL oxidation by activated human monocytes. J Lipid Res. 2000;41:489–495. [PubMed] [Google Scholar]
- Bojes HK, Germolec DR, Simeonova P, Bruccoleri A, Schoonhoven R, Luster MI, Thurman RG. Antibodies to tumor necrosis factor alpha prevent increases in cell replication in liver due to the potent peroxisome proliferator, WY- 14,643. Carcinogenesis. 1997;18:669–674. doi: 10.1093/carcin/18.4.669. [DOI] [PubMed] [Google Scholar]
- Bojes HK, Thurman RG. Peroxisome proliferators activate Kupffer cells in vivo. Cancer Res. 1996;56:1–4. [PubMed] [Google Scholar]
- Cherkaoui-Malki M, Meyer K, Cao WQ, Latruffe N, Yeldandi AV, Rao MS, Bradfield CA, Reddy JK. Identification of novel peroxisome proliferator-activated receptor alpha (PPARalpha) target genes in mouse liver using cDNA microarray analysis. Gene Expr. 2001;9:291–304. doi: 10.3727/000000001783992533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corton JC, Apte U, Anderson SP, Limaye P, Yoon L, Latendresse J, Dunn C, Everitt JI, Voss KA, Swanson C, Kimbrough C, Wong JS, Gill SS, Chandraratna RA, Kwak MK, Kensler TW, Stulnig TM, Steffensen KR, Gustafsson JA, Mehendale HM. Mimetics of caloric restriction include agonists of lipid-activated nuclear receptors. J Biol Chem. 2004;279:46204–46212. doi: 10.1074/jbc.M406739200. [DOI] [PubMed] [Google Scholar]
- Currie RA, Bombail V, Oliver JD, Moore DJ, Lim FL, Gwilliam V, Kimber I, Chipman K, Moggs JG, Orphanides G. Gene ontology mapping as an unbiased method for identifying molecular pathways and processes affected by toxicant exposure: application to acute effects caused by the rodent non-genotoxic carcinogen diethylhexylphthalate. Toxicol Sci. 2005;86:453–469. doi: 10.1093/toxsci/kfi207. [DOI] [PubMed] [Google Scholar]
- Dennis G, Jr, Sherman BT, Hosack DA, Yang J, Gao W, Lane HC, Lempicki RA. DAVID: Database for Annotation, Visualization, and Integrated Discovery. Genome Biol. 2003;4:3. [PubMed] [Google Scholar]
- Devchand PR, Keller H, Peters JM, Vazquez M, Gonzalez FJ, Wahli W. The PPARalpha-leukotriene B4 pathway to inflammation control. Nature. 1996;384:39–43. doi: 10.1038/384039a0. [DOI] [PubMed] [Google Scholar]
- Eisen MB, Spellman PT, Brown PO, Botstein D. Cluster analysis and display of genome-wide expression patterns. Proc Natl Acad Sci U S A. 1998;95:14863–14868. doi: 10.1073/pnas.95.25.14863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fishelson Z, Attali G, Mevorach D. Complement and apoptosis. Mol Immunol. 2001;38:207–219. doi: 10.1016/s0161-5890(01)00055-4. [DOI] [PubMed] [Google Scholar]
- Flierman R, Daha MR. The clearance of apoptotic cells by complement. Immunobiology. 2007;212:363–370. doi: 10.1016/j.imbio.2006.11.005. [DOI] [PubMed] [Google Scholar]
- Foijer F, Te RH. Restriction beyond the restriction point: mitogen requirement for G2 passage. Cell Div. 2006;1:8. doi: 10.1186/1747-1028-1-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamadeh HK, Bushel PR, Jayadev S, Martin K, DiSorbo O, Sieber S, Bennett L, Tennant R, Stoll R, Barrett JC, Blanchard K, Paules RS, Afshari CA. Gene expression analysis reveals chemical-specific profiles. Toxicol Sci. 2002;67:219–231. doi: 10.1093/toxsci/67.2.219. [DOI] [PubMed] [Google Scholar]
- Hasmall SC, James NH, Macdonald N, Gonzalez FJ, Peters JM, Roberts RA. Suppression of mouse hepatocyte apoptosis by peroxisome proliferators: role of PPARalpha and TNFalpha. Mutat Res. 2000a;448:193–200. doi: 10.1016/s0027-5107(99)00236-5. [DOI] [PubMed] [Google Scholar]
- Hasmall SC, West DA, Olsen K, Roberts RA. Role of hepatic non-parenchymal cells in the response of rat hepatocytes to the peroxisome proliferator nafenopin in vitro. Carcinogenesis. 2000b;21:2159–2165. doi: 10.1093/carcin/21.12.2159. [DOI] [PubMed] [Google Scholar]
- Jackson SH, Gallin JI, Holland SM. The p47phox mouse knock-out model of chronic granulomatous disease. J Exp Med. 1995;182:751–758. doi: 10.1084/jem.182.3.751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kersten S, Mandard S, Escher P, Gonzalez FJ, Tafuri S, Desvergne B, Wahli W. The peroxisome proliferator-activated receptor alpha regulates amino acid metabolism. FASEB J. 2001;15:1971–1978. doi: 10.1096/fj.01-0147com. [DOI] [PubMed] [Google Scholar]
- Klaunig JE, Babich MA, Baetcke KP, Cook JC, Corton JC, David RM, Deluca JG, Lai DY, McKee RH, Peters JM, Roberts RA, Fenner-Crisp PA. PPARalpha agonist-induced rodent tumors: modes of action and human relevance. Crit Rev Toxicol. 2003;33:655–780. doi: 10.1080/713608372. [DOI] [PubMed] [Google Scholar]
- Lalwani ND, Reddy MK, Qureshi SA, Reddy JK. Development of hepatocellular carcinomas and increased peroxisomal fatty acid beta-oxidation in rats fed [4-chloro-6-(2,3-xylidino)-2-pyrimidinylthio] acetic acid (Wy-14,643) in the semipurified diet. Carcinogenesis. 1981;2:645–650. doi: 10.1093/carcin/2.7.645. [DOI] [PubMed] [Google Scholar]
- Lawrence JW, Wollenberg GK, Deluca JG. Tumor necrosis factor alpha is not required for WY14,643-induced cell proliferation. Carcinogenesis. 2001;22:381–386. doi: 10.1093/carcin/22.3.381. [DOI] [PubMed] [Google Scholar]
- Ledda-Columbano GM, Curto M, Piga R, Zedda AI, Menegazzi M, Sartori C, Shinozuka H, Bluethmann H, Poli V, Ciliberto G, Columbano A. In vivo hepatocyte proliferation is inducible through a TNF and IL-6-independent pathway. Oncogene. 1998;17:1039–1044. doi: 10.1038/sj.onc.1202018. [DOI] [PubMed] [Google Scholar]
- Lee SS, Pineau T, Drago J, Lee EJ, Owens JW, Kroetz DL, Fernandez-Salguero PM, Westphal H, Gonzalez FJ. Targeted disruption of the alpha isoform of the peroxisome proliferator- activated receptor gene in mice results in abolishment of the pleiotropic effects of peroxisome proliferators. Mol Cell Biol. 1995;15:3012–3022. doi: 10.1128/mcb.15.6.3012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leek JT, Monsen E, Dabney AR, Storey JD. EDGE: extraction and analysis of differential gene expression. Bioinformatics. 2006;22:507–508. doi: 10.1093/bioinformatics/btk005. [DOI] [PubMed] [Google Scholar]
- Li Z, Diehl AM. Innate immunity in the liver. Curr Opin Gastroenterol. 2003;19:565–571. doi: 10.1097/00001574-200311000-00009. [DOI] [PubMed] [Google Scholar]
- Nauta AJ, Daha MR, Tijsma O, van de WB, Tedesco F, Roos A. The membrane attack complex of complement induces caspase activation and apoptosis. Eur J Immunol. 2002;32:783–792. doi: 10.1002/1521-4141(200203)32:3<783::AID-IMMU783>3.0.CO;2-Q. [DOI] [PubMed] [Google Scholar]
- Nikitin A, Egorov S, Daraselia N, Mazo I. Pathway studio--the analysis and navigation of molecular networks. Bioinformatics. 2003;19:2155–2157. doi: 10.1093/bioinformatics/btg290. [DOI] [PubMed] [Google Scholar]
- Palmer CN, Hsu MH, Griffin KJ, Raucy JL, Johnson EF. Peroxisome proliferator activated receptor-alpha expression in human liver. Mol Pharmacol. 1998;53:14–22. [PubMed] [Google Scholar]
- Parzefall W, Berger W, Kainzbauer E, Teufelhofer O, Schulte-Hermann R, Thurman RG. Peroxisome proliferators do not increase DNA synthesis in purified rat hepatocytes. Carcinogenesis. 2001;22:519–523. doi: 10.1093/carcin/22.3.519. [DOI] [PubMed] [Google Scholar]
- Peters JM, Cattley RC, Gonzalez FJ. Role of PPAR alpha in the mechanism of action of the nongenotoxic carcinogen and peroxisome proliferator Wy-14,643. Carcinogenesis. 1997;18:2029–2033. doi: 10.1093/carcin/18.11.2029. [DOI] [PubMed] [Google Scholar]
- Peters JM, Cheung C, Gonzalez FJ. Peroxisome proliferator-activated receptor-alpha and liver cancer: where do we stand? J Mol Med. 2005;83:774–785. doi: 10.1007/s00109-005-0678-9. [DOI] [PubMed] [Google Scholar]
- Peters JM, Rusyn I, Rose ML, Gonzalez FJ, Thurman RG. Peroxisome proliferator-activated receptor alpha is restricted to hepatic parenchymal cells, not Kupffer cells: implications for the mechanism of action of peroxisome proliferators in hepatocarcinogenesis. Carcinogenesis. 2000;21:823–826. doi: 10.1093/carcin/21.4.823. [DOI] [PubMed] [Google Scholar]
- Ramadori G, Armbrust T. Cytokines in the liver. Eur J Gastroenterol Hepatol. 2001;13:777–784. doi: 10.1097/00042737-200107000-00004. [DOI] [PubMed] [Google Scholar]
- Roberts RA, James NH, Cosulich S, Hasmall SC, Orphanides G. Role of cytokines in non-genotoxic hepatocarcinogenesis: cause or effect? Toxicol Lett. 2001;120:301–306. doi: 10.1016/s0378-4274(01)00282-x. [DOI] [PubMed] [Google Scholar]
- Rose ML, Rusyn I, Bojes HK, Germolec DR, Luster M, Thurman RG. Role of Kupffer cells in peroxisome proliferator-induced hepatocyte proliferation. Drug Metab Rev. 1999;31:87–116. doi: 10.1081/dmr-100101909. [DOI] [PubMed] [Google Scholar]
- Rusyn I, Asakura S, Pachkowski B, Bradford BU, Denissenko MF, Peters JM, Holland SM, Reddy JK, Cunningham ML, Swenberg JA. Expression of base excision DNA repair genes is a sensitive biomarker for in vivo detection of chemical-induced chronic oxidative stress: Identification of the molecular source of radicals responsible for DNA damage by peroxisome proliferators. Cancer Res. 2004;64:1050–1057. doi: 10.1158/0008-5472.can-03-3027. [DOI] [PubMed] [Google Scholar]
- Rusyn I, Kadiiska MB, Dikalova A, Kono H, Yin M, Tsuchiya K, Mason RP, Peters JM, Gonzalez FJ, Segal BH, Holland SM, Thurman RG. Phthalates rapidly increase production of reactive oxygen species in vivo: role of Kupffer cells. Mol Pharmacol. 2001;59:744–750. doi: 10.1124/mol.59.4.744. [DOI] [PubMed] [Google Scholar]
- Rusyn I, Peters JM, Cunningham ML. Modes of action and species-specific effects of di-(2-ethylhexyl)phthalate in the liver. Crit Rev Toxicol. 2006;36:459–479. doi: 10.1080/10408440600779065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rusyn I, Yamashina S, Segal BH, Schoonhoven R, Holland SM, Cattley RC, Swenberg JA, Thurman RG. Oxidants from nicotinamide adenine dinucleotide phosphate oxidase are involved in triggering cell proliferation in the liver due to peroxisome proliferators. Cancer Res. 2000;60:4798–4803. [PubMed] [Google Scholar]
- Teissier E, Nohara A, Chinetti G, Paumelle R, Cariou B, Fruchart JC, Brandes RP, Shah A, Staels B. Peroxisome proliferator-activated receptor alpha induces NADPH oxidase activity in macrophages, leading to the generation of LDL with PPAR-alpha activation properties. Circ Res. 2004;95:1174–1182. doi: 10.1161/01.RES.0000150594.95988.45. [DOI] [PubMed] [Google Scholar]
- Troyanskaya O, Cantor M, Sherlock G, Brown P, Hastie T, Tibshirani R, Botstein D, Altman RB. Missing value estimation methods for DNA microarrays. Bioinformatics. 2001;17:520–525. doi: 10.1093/bioinformatics/17.6.520. [DOI] [PubMed] [Google Scholar]
- Tusher VG, Tibshirani R, Chu G. Significance analysis of microarrays applied to the ionizing radiation response. Proc Natl Acad Sci U S A. 2001;98:5116–5121. doi: 10.1073/pnas.091062498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wheeler MD, Smutney OM, Check JF, Rusyn I, Schulte-Hermann R, Thurman RG. Impaired Ras membrane association and activation in PPARalpha knockout mice after partial hepatectomy. Am J Physiol Gastrointest Liver Physiol. 2003;284:G302–G312. doi: 10.1152/ajpgi.00175.2002. [DOI] [PubMed] [Google Scholar]
- White IR, Man WJ, Bryant D, Bugelski P, Camilleri P, Cutler P, Hayes W, Holbrook JD, Kramer K, Lord PG, Wood J. Protein expression changes in the Sprague Dawley rat liver proteome following administration of peroxisome proliferator activated receptor alpha and gamma ligands. Proteomics. 2003;3:505–512. doi: 10.1002/pmic.200390064. [DOI] [PubMed] [Google Scholar]
- Wong JS, Gill SS. Gene expression changes induced in mouse liver by di(2-ethylhexyl) phthalate. Toxicol Appl Pharmacol. 2002;185:180–196. doi: 10.1006/taap.2002.9540. [DOI] [PubMed] [Google Scholar]
- Woods CG, Burns AM, Bradford BU, Ross PK, Kosyk O, Swenberg JA, Cunningham ML, Rusyn I. WY-14,643-induced cell proliferation and oxidative stress in mouse liver are independent of NADPH oxidase. Toxicol Sci. 2007a;98:366–374. doi: 10.1093/toxsci/kfm104. [DOI] [PubMed] [Google Scholar]
- Woods CG, Burns AM, Maki A, Bradford BU, Cunningham ML, Connor HD, Kadiiska MB, Mason RP, Peters JM, Rusyn I. Sustained formation of alpha-(4-pyridyl-1-oxide)-N-tert-butylnitrone radical adducts in mouse liver by peroxisome proliferators is dependent upon peroxisome proliferator-activated receptor-alpha, but not NADPH oxidase. Free Radic Biol Med. 2007b;42:335–342. doi: 10.1016/j.freeradbiomed.2006.10.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Q, Xie Y, DePierre JW. Effects of peroxisome proliferators on the thymus and spleen of mice. Clin Exp Immunol. 2000;122:219–226. doi: 10.1046/j.1365-2249.2000.01367.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeeberg BR, Feng W, Wang G, Wang MD, Fojo AT, Sunshine M, Narasimhan S, Kane DW, Reinhold WC, Lababidi S, Bussey KJ, Riss J, Barrett JC, Weinstein JN. GoMiner: a resource for biological interpretation of genomic and proteomic data. Genome Biol. 2003;4:R28. doi: 10.1186/gb-2003-4-4-r28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeeberg BR, Qin H, Narasimhan S, Sunshine M, Cao H, Kane DW, Reimers M, Stephens RM, Bryant D, Burt SK, Elnekave E, Hari DM, Wynn TA, Cunningham-Rundles C, Stewart DM, Nelson D, Weinstein JN. High-Throughput GoMiner, an ‘industrial-strength’ integrative gene ontology tool for interpretation of multiple-microarray experiments, with application to studies of Common Variable Immune Deficiency (CVID) BMC Bioinformatics. 2005;6:168. doi: 10.1186/1471-2105-6-168. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.