

Abstract
The unique mechanical performance of animal cells and tissues is attributed mostly to their internal biopolymer meshworks. Its perplexing universality and robustness against structural modifications by drugs and mutations is an enigma in cell biology and provides formidable challenges to materials science. Recent investigations could pinpoint highly universal patterns in the soft glassy rheology and nonlinear elasticity of cells and reconstituted networks. Here, we report observations of a glass transition in semidilute F-actin solutions, which could hold the key to a unified explanation of these phenomena. Combining suitable rheological protocols with high-precision dynamic light scattering, we can establish a remarkable rheological redundancy and trace it back to a highly universal exponential stretching of the single-polymer relaxation spectrum of a “glassy wormlike chain.” By exploiting the ensuing generalized time-temperature superposition principle, the time domain accessible to microrheometry can be extended by several orders of magnitude, thus opening promising new metrological opportunities.
Keywords: biopolymers, light scattering, nonlinear rheology, wormlike chain
A major strategy in the investigation of complex materials is to reassemble them step by step from simpler subunits to trace back their material properties on the macroscopic scale to those of their elementary constituents. This strategy has been successfully applied to the biopolymer networks that constitute the basic scaffolding structure of animal cells, known as the cytoskeleton (1). Filamentous actin (F-actin), the major load-bearing element of the cytoskeleton has received particular attention. The success of simple viscoelastic models in the quantitative analysis of the rheological behavior of in vitro reconstituted biopolymer networks (1–5) has rendered them an attractive paradigm for rationalizing the mechanics of live cells (6, 7). Indeed, there appear to be striking mechanical analogies between cells and prestressed reconstituted networks (7–9) and strong correlations of cell functions with the viscoelastic parameters, suggesting the latter as viable indices for clinical diagnosis (10). Recently, this widely shared view has been challenged by observations (11) that live cells obey the highly universal and comparatively featureless pattern of “soft glassy rheology” that is ubiquitous in soft condensed matter (12). These observations seem to imply that the pertinent linear mechanical properties of a cell might be composed in a single number, the so-called “noise temperature” (13) of the glass, and on this basis it has indeed been suggested that we are all built of glass (14). Subsequently, it has become a central task in cell biophysics to understand how the two conflicting paradigms might be integrated into a unified picture (15–17).
Here, we aim to resolve the apparent puzzle by demonstrating that highly purified in vitro polymerized semidilute F-actin solutions undergo a glass transition as a function of various physiologically relevant physical control parameters. It manifests itself in a very robust exponential stretching of the relaxation spectrum evidenced by the logarithmic tails of the dynamic structure factor measured by high-precision dynamic light scattering (DLS), whereas no signs of diverging structural length scales can be detected. Spectacular consequences of this anomalous stretching also emerge in the nonlinear mechanical properties of the actin solution, notably in the finite-time nonlinear rheological response, which we find to be highly reminiscent of previous results for living cells (7) and cross-linked actin networks (3, 9, 18). The presence of a mechanical signature usually regarded as characteristic of biological cross-linkers in pure actin solutions hints at a close intrinsic similarity of the mechanics of (glassy) actin solutions with the mechanics of cross-linked in vitro networks and of the cytoskeleton of living cells. It suggests that a detailed analysis of the former may provide a key to a better understanding of the latter.
We start with the exposition of our rheological results. The data displayed in Fig. 1 were obtained by a series of short stress pulses (see Methods) of increasing amplitude σ for a representative actin sample. As can be seen from Fig. 1 Inset, the time dependence of the compliance, Jσ(t) = γ(t)/σ, on the time scale of the pulse duration exhibits two distinct regimes: a rapid elastic response followed by a comparably broad viscoelastic creep regime, which is characteristic of the internal relaxation times of the sample. The time dependence of the response is very similar for different pulse strengths σ, as evidenced by a neat collapse of all compliances onto a single master curve (data not shown), with practically perfect strain recovery after each pulse. The inverse of the creep compliance, Jt−1, at a given time t of the order of some seconds is thus a characteristic measure of the stiffness of the physical polymer network. The main plot in Fig. 1 shows Jt−1(σ) as a function of the stress-pulse amplitude σ for different delay times t. Independent of t, three different regimes can be distinguished: at low stress, the creep compliance is approximately constant, corresponding to a linear stress–strain relation. Above a critical stress of ≈0.06 Pa, one can discern a shear stiffening regime, where the elasticity increases with the pulse amplitude σ, approximately in accord with J−1 ∝ σx, x ≈ 0.4. Finally, at shear stresses >1 Pa, the elasticity drops sharply and the response ceases to be reversible, apparently because of irreversible structural changes.
Fig. 1.

Nonlinear elasticity probed by the σ-pulse protocol. Inverse creep compliance, 1/J, as a function of applied stress pulse amplitude, σ, evaluated at delay times: t [s] = 1 (diamonds), 2 (circles), 3 (double triangles), 4 (triangles), 5 (squares) for actin concentration cA = 9.5 μM, temperature T = 21°C, and average polymer length L = 21 μm. (Inset) The time dependence of the compliance J(t) for different σ values.
The curves in Fig. 1 demonstrate that shear stiffening similar to that found for cross-linked networks is also possible for pure actin solutions. Indeed, the overall functional form of the curves in Fig. 1 resembles qualitatively results obtained for cross-linked (and prestressed) in vitro samples (3, 9, 18) or even for live cells (7). However, as seen from the data in Fig. 2, the stiffening is sensitively dependent on slight changes in temperature T, suggesting that the apparent power-law exponent x of the stress-stiffening relation might be less universal than thought (3, 4). The displayed inverse creep compliances measured at temperatures between 18°C and 27°C show that x decreases approximately linearly with increasing T, until the stiffening effect vanishes at T ≈ 25°C (compare Fig. 2 Inset). Curiously, neither in the linear regime of the curves in Fig. 2 nor in the frequency-dependent linear shear moduli in the range 0.01–10 Hz can any significant temperature dependence of the response be detected.
Fig. 2.

Temperature-induced transition from shear softening to shear stiffening (σ-pulse protocol). The inverse of the creep compliance, J, normalized by J(0.01 Pa), as a function of the applied stress σ for various temperatures: T [°C] = 18 (filled circles), 19 (filled squares), 21 (filled diamonds), 22 (filled triangles), 23 (open circles), 25 (open squares), 27 (open triangles) and cA = 9.5 μM, L = 21 μm. (Inset) Apparent power-law stiffening exponent x of 1/J(σ) ∝ σx vs. temperature T.
Completely analogous observations were made for other control parameters, notably ionic strength I, polymer length L, and actin concentration cA. For example, reducing the KCl concentration by a factor of 10 to 10 mM has no significant effect on the linear elastic response but strongly affects the nonlinear response in a way very similar to raising the temperature. Replacing 90 mM KCl by cosmotropic or chaotropic salts (NaCl, CsCl) did not affect the overall picture. Under both conditions the same mechanical response was observed, hinting at a purely electrostatic, ion-unspecific effect.
To obtain more comprehensive data, we turned to the more efficient γ̇-pulse protocol (see Methods and ref. 5). Taking the numerical derivative of the recorded stress-strain curves (see Fig. 3, for examples) yields the nonlinear differential modulus K, which may be parameterized either in terms of strain or stress. To facilitate comparison with Figs. 1 and 2, we have chosen the representation K(σ) in Fig. 4. On changing T, L, cA, and I, one finds again qualitatively the same characteristic nonlinear behavior as obtained in Fig. 2 for the σ-pulse method (see Fig. 4).
Fig. 3.

Stress–strain curves (γ̇-pulse protocol). The stress σ vs. the strain γ at 21°C (solid lines) and 25°C (dashed lines) for γ̇ [s−1] = 0.05, 0.1, 0.2, 0.4. For all curves L = 21 μm, cA = 9.5 μM. (Inset) Data collapse of 25 stress–strain curves for different T, L, cA, and γ̇ values onto a master relation upon rescaling each curve by its characteristic strain and stress values. The spread of the data at low-strain values is due to instrument limitations causing measurement uncertainties.
Fig. 4.

The transition as a function of polymer length and concentration (γ̇-pulse protocol). The differential modulus K (the numerical derivative of σ − γ curves) vs. the applied stress σ at γ̇ = 0.1 [s−1], both axes normalized by the linear modulus Klin (a) for various actin concentrations cA [μM] = 2.4, 4.8, 9.5, 19, and 38 at T = 21°C, L = 21 μm, and (b) for different filament lengths L [μm] = 5, 7, 10, and 21 at T = 21°C, cA = 9.5 μM.
Altogether, our data demonstrate that the characteristics of the transition from stiffening to softening are largely insensitive to the choice of the control parameter. To understand the origin of this remarkable rheological redundancy, consider again the stress–strain relations displayed in Fig. 3, which were obtained with the γ̇-pulse protocol at various fixed shear rates γ̇ and temperatures T. A comparison of the curves suggests that a decrease in temperature can be compensated by an appropriate decrease of the shear rate. Indeed, as demonstrated in Fig. 3 Inset with 25 curves for different T, L, cA, and γ̇, the stress–strain curves neatly collapse onto a single master curve on rescaling with their characteristic strain and stress values. This explains why rheological protocols imposing a fixed external time scale (19), like our pulse protocols, are a crucial prerequisite for a systematic study of the nonlinear mechanical consequences of the glass transition in F-actin solutions. By minimizing viscous creep as incurred during ordinary creep and oscillatory measurements in the nonlinear regime, they can detect the distinctive nonlinear elastic features characteristic of the particular (fixed) finite time scale that are otherwise “wiped out” in the measurement process.
The data collapse of the stress–strain relations in Fig. 3 moreover establishes a time–temperature superposition principle: the effect of a temperature change on the nonlinear rheological behavior amounts essentially to a rescaling of time. Moreover, with regard to the observed rheological redundancy, it suggests that changes not only of temperature but also of polymer length, concentration, ionic strength (and possibly further parameters) all affect the rheological response chiefly by a stretching of the relaxation spectrum. To support this far-reaching superposition principle, it would be desirable to measure in greater detail the equilibrium long-time dynamics of the samples on microscopic and macroscopic scales. Because of the technical difficulty of low-frequency linear rheometry for soft samples, we concentrate on microrheological measurements for that purpose. The linear microrheology is reliably and noninvasively probed over several orders of magnitude in time by high-precision DLS. Throughout the measurable time domain, the dynamic structure factor is uniquely determined by the transverse dynamic mean-square displacement of the polymers (20). By the fluctuation dissipation theorem, it may thus be translated into the mechanical susceptibility probed by an idealized weak periodic transverse point force applied to a test polymer, which is commonly reported in the form of microrheological moduli. Our DLS results presented in Fig. 5 show that the scattering function acquires a pronounced logarithmic tail at low T and large L, in stark contrast to more fluid samples at higher temperatures (21). The slopes of these logarithmic tails in the (semilogarithmic) plot determine the apparent power-law exponent characterizing the corresponding frequency-dependent microrheological moduli (11) as detailed in the supporting information (SI) Text and SI Fig. 10. As demonstrated in Fig. 5 Inset, the tails of the individual scattering curves measured for different actin concentrations (other conditions and scattering vector q = 8.04 μm−1 fixed) collapse on rescaling the time axis. The resulting master curve extends over >10 orders of magnitude in time of which ≈7 exhibit the logarithmic decay, providing an impressive illustration of the metrological potential of the superposition principle (here, for the parameter cA). Beyond an unambiguous demonstration of the stretching of the relaxation spectrum in form of the logarithmic decay, the DLS data thus represent strong independent evidence for the rheological redundancy inferred above from our nonlinear macroscopic rheological measurements.
Fig. 5.

Dynamic structure factor from DLS. Scattering functions for native actin (no gelsolin, cA = 17 μM, T = 15°C) at various scattering vectors q [μm−1] = 6.44, 8.04, 9.62, 11.71, 13.1, 14.47, 17.13, 19.64, 22.01, 24.22, and 29.66 (from top to bottom, the data outside the confidence time interval are truncated) fitted by the model proposed in the main text. The failure of the fits for low q indicates the breakdown of the single-polymer scattering theory. Quantitative evaluation is meaningful for q >10 μm−1 and yields 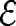 = 37 ± 6, Λ = 2.15 ± 0.13 μm (see SI Text and SI Figs. 7–9 for further details). (Inset) The logarithmic tails of the scattering curves at q = 8.04 μm−1 for various actin concentrations cA [μM] = 7.2, 12, 17, and 24 collapse on rescaling the time axis. Theoretical fits are dotted.
= 37 ± 6, Λ = 2.15 ± 0.13 μm (see SI Text and SI Figs. 7–9 for further details). (Inset) The logarithmic tails of the scattering curves at q = 8.04 μm−1 for various actin concentrations cA [μM] = 7.2, 12, 17, and 24 collapse on rescaling the time axis. Theoretical fits are dotted.
In the remainder, we briefly sketch the relevant features of the model that produces the fits in Fig. 5 and its relation to the nonlinear rheology reported above. A more comprehensive theoretical discussion of this “glassy wormlike chain” model is reported in ref. 22. Our starting point is the observation that for short times (or high frequencies) the dynamic structure factor and the micro- and macrorheological response can all be expressed in terms of single-polymer dynamics (20, 23–25). To account for the slowing down by interactions with the surrounding polymers at longer times, we modify the dynamics of an ordinary, weakly bending wormlike chain (see Methods) in the following way. The mode relaxation times τλ > τΛ of all eigenmodes of (half) wavelength, λ, longer than a characteristic interaction length, Λ, are multiplied by a factor exp(N) that grows exponentially in the number N ≡ λ/Λ − 1 of interactions per wavelength λ. The modification is reminiscent of the generic nonequilibrium trap models (12, 26) of soft glassy rheology (13), but concerns the equilibrium dynamics here. The stretching parameter controls the slopes of the logarithmic tails of the structure factor in a semilogarithmic plot and thus the nonuniversal apparent power-law exponents deduced from microrheological measurements. (Asymptotically for large ≫ 1, the “noise temperature” is given by 1 + 3/; see SI Text.) In view of the sensitivity of the rheology to temperature and ionic strength reported above, it seems natural to think of as the characteristic scale (in units of thermal energy) for energetic barriers. Whereas protein interactions remain poorly understood, many observations hint at temperature-sensitive unspecific (probably hydrophobic) adhesive contact interactions incompletely screened by electrostatic repulsion (27, 28), which would match well with this interpretation and with our observations. In the spirit of generic free-volume theories (29), the parameter might moreover accommodate free-energy contributions from caging and entanglement (22). In any case, an exponential scaling of the relaxation times in the wavelength λ seems plausible.
Crucially, appealing to the intuition of the stretching parameter as a (free) energy barrier, externally or internally generated stresses may arguably be expected to contribute additively to , which provides a tentative theoretical access to the observed stress softening/stiffening. Pertinent predictions suitable for a qualitative comparison with our nonlinear rheology data may be derived by subjecting the glassy wormlike chain to a prestress. To this end, we represent the effect of prestressing the sample by subjecting the chain to a tension f and by replacing the stretching parameter by − f/fT. Here, fT ≡ kBT/Δ represents some characteristic scale of the thermal equilibrium tension present in an unstressed sample, so that Δ may be interpreted as a characteristic (effective) width of the energetic traps and barriers. The complex frequency-dependent linear shear modulus of the prestressed system is then, within our simple scheme, equivalent to the much studied macroscopic frequency-dependent nonlinear differential modulus Kσ*(ω) (3, 7–9). In Fig. 6, its absolute value |Kσ*|, evaluated at a fixed frequency ω = 0.1τΛ−1 in the “slant-plateau” regime, is plotted as a function of the prestressing tension f, which is expressed as an equivalent prestress σ. The parameters, given in the legend to Fig. 6, were chosen to mimic our experimental conditions. Although the increase of the modulus is due to the stiffening of the polymers caused by the prestress, its sharp downturn signals the breakdown of the stretching of the relaxation spectrum when f ≈ fT and the sticky contacts yield to the stress. Qualitatively, the results compare favorably to our data in Figs. 2 and 4, thus providing further support for the proposed mechanism.
Fig. 6.

Nonlinear differential shear modulus of a glassy wormlike chain as function of prestress. Absolute value of Kσ*(ω) evaluated at ω = 0.1τΛ−1 for the glassy wormlike chain model discussed in the text. For the parameters pertaining to the free-energy wells we chose Δ = 0.25 μm, Λ = 1.5 μm, and = 4 … 40 from bottom to top. As in Fig. 4, both axes were normalized by the linear modulus |Kσ=0*(ω)|.
Notwithstanding the substantial success of the scheme above in accounting for the experimental observations, one should carefully consider the scope for other available theoretical explanations. Slow relaxation, as evidenced by our DLS data, also occurs in the vicinity of ordinary (e.g., percolation) critical points (30, 31). However, so far, there is neither direct evidence of fractal or percolating structures or other diverging length scales nor of a critical point at a finite temperature or at a semidilute concentration in actin solutions or in cells, but there is accumulating phenomenological evidence for (nonequilibrium) glassy behavior in cells (32–34). The anomalous stretching of the relaxation spectrum leading to generic apparent power-law fluid behavior with a parameter-dependent nonuniversal exponent is characteristic of soft glassy rheology and runs counter to an interpretation in terms of critical gelation. There are, moreover, good reasons why it might be physiologically advantageous for cells to behave as glassy rather than like a critical gel (33). Higher-order mode-coupling singularities might be considered as an alternative explanation, in analogy with what has been established for colloids (35) and suggested to extend to flexible polymer blends (36). However, given the very robust and generic nature of the effect in our F-actin solutions and the corresponding phenomenology in live cells, the fine-tuning of competing interactions required by that theory seems again somewhat unintuitive. In contrast, the characteristic patterns of soft glassy rheology arise very naturally from the glassy worm-like chain model introduced above and in ref. 22 (see also SI Text), which moreover offers considerable scope for pinpointing its microscopic origin and rationalizing additional features of nonlinear cell rheology (34) in future work.
It is an intriguing question as to what extent our results for pure F-actin solutions are representative also for the mechanics of cross-linked networks and live cells. First, we note that reversibly (and irreversibly) cross-linked networks are effectively contained (as the limiting case → ∞) in our theoretical discussion, which allows us to establish a very direct and well testable relation between the form of the linear and nonlinear viscoelastic moduli and the strength and concentration 3ξ−2Λ−1 (see Methods) of cross-links. Moreover, it is possible and would be worthwhile to develop the proposed scheme further to effectively account to some extent also for the diverse geometries and mechanical properties of various actin-binding proteins. Similar reduced descriptions are routinely applied with great success in colloidal science, where the physical effects of one species of particles in a binary mixture are completely absorbed into an effective interaction potential between the particles of the second species (37). Second, a comparison of our computed linear rheological moduli with published microrheological data for live cells (11, 32, 34) shows very satisfactory agreement. This might be interpreted as further evidence that cells and multicellular organisms do indeed live at the edge of a glass transition (14, 33), although a more comprehensive comparison with linear and nonlinear cell rheometry data clearly remains a major task for future studies. In particular, it would be interesting to analyze in greater detail to what extent the generalized time–temperature superposition principle and the striking rheological redundancy with respect to important physiological control parameters established above for actin solutions is also realized in living cells. Can it provide a unified explanation for the well known universality and robustness of the rheology of cells and reconstituted cytoskeletal networks against structural modifications by drugs and mutations (14, 38)? As we have demonstrated, the pronounced rheological redundancy observed in pure actin solutions finds a very natural explanation in the tight control of the dramatic stretching of the relaxation spectrum by a single characteristic free-energy scale , which generally has to be thought of as a complicated sum of a potentially large number of microscopic contributions to the free-energy barriers retarding the structural relaxation. Clearly, a complete microscopic derivation of the various anticipated contributions to is at the present stage elusive for actin solutions, let alone live cells. Also, the implications of the proposed scheme out of equilibrium remain to be explored. Yet, it certainly offers a promising new perspective on the widely reported observations of soft glassy rheology in cells and defines clear objectives for future theoretical work. Ultimately, it strongly supports expectations shared by many physicists that “simple polymer physics-based models may be able to explain the observed cell mechanical response and suggest mechanisms for mechano-sensing” (16).
In summary, we have found comprehensive evidence that small variations in the ambient conditions (temperature, salt) or the composition (polymer length, concentration) of F-actin solutions bring about dramatic changes in the long-time equilibrium dynamics and nonlinear rheology. We have proposed a uniform exponential stretching of the relaxation spectrum of the wormlike chain model as the universal origin of the glassy phenomenology and the corresponding rheological redundancy. This simple scheme yields predictions that compare favorably with experimental data and offers a unifying perspective on the relation between viscoelastic short-time and glassy long-time dynamics. Apart from its plausible physiological significance, the emerging superposition principle provides a powerful metrological tool for future investigations.
Methods
Actin Preparation.
Globular actin (G-actin) was prepared from rabbit skeletal muscle (39) and stored in lyophilized form at −20°C. For the measurements, the lyophilized actin was dissolved in deionized water and dialyzed against G-Buffer (2 mM Tris, 0.2 mM ATP, 0.2 mM CaCl2, 0.2 mM DTT, and 0.005% NaN3, pH 8) at 4°C. The G-actin solutions were stored at 4°C and used within 7 days of preparation. The average length L of filamentous actin (F-actin) was adjusted by adding gelsolin, which was isolated from bovine plasma serum (40). Throughout the text, we refer to the physically relevant average actin length L in place of the gelsolin concentration, exploiting the relation established in ref. 41. Samples were prepared by gently mixing deionized water with gelsolin and G-actin and buffered to 2 mM Tris, 2 mM MgCl2, 0.2 mM CaCl2, 0.2 mM DTT, 100 mM KCl, and 0.5 mM ATP.
Rheology.
Approximately 520 μl of sample volume were loaded within 1 min in a commercial stress-controlled rheometer (Physica MCR301, Anton Paar) with 50 mm of plate-plate geometry and 160 μm of plate separation. Our force protocols were designed to minimize viscous creep. In the “σ-pulse” protocol, short stress pulses are applied. The duration of the pulses (5 s) and the intermittent recovery periods (at least 45 s) were adjusted to allow for a 98% strain recovery of the sample between consecutive pulses. In the more efficient “γ̇-pulse” protocol, samples are sheared at a constant shear rate (0.01 s−1 ≤ γ̇ ≤ 0.4 s−1) and the differential nonlinear shear modulus, K(γ), as a function of the strain γ is obtained by taking the numerical derivative of the recorded stress–strain relations (5). We checked for certain parameter values throughout the whole parameter range that both methods yield qualitatively the same results.
DLS.
We used a light-scattering spectrometer SP/125 equipped with a single-photon detection unit SO-SIPD (ALV-Laser Vertriebsgesellschaft). The sample cuvettes were housed in a thermostated index-matching bath (ALV) filled with toluene. Measurement duration was up to 8·104 s. Therefore, instrument stability was a major concern and was checked by measuring the correlation function of toluene for 8·104 s. Values of g(2) − 1 did not exceed 0.003 with any statistical significance, so that we concluded that values exceeding 0.01 were measured reliably in our experiments. Such good stability of the instrument required a warm-up period of the Ar+ laser (Coherent) of 2 days, stability of the room temperature to within 3°C, and careful alignment of laser and goniometer. The intercept was very close to 2 (average value, 1.98; lowest value in all experiments that are presented here, 1.93). We measured each sample before and after all other light-scattering measurements at a scattering angle of 90°. Only experiments where these measurements yielded indistinguishable results were accepted.
Theory.
For convenience, we summarize a few basic formulae concerning the dynamics of the ordinary weakly bending wormlike chain, which is the basis of the glassy wormlike chain theory developed in the text. For more comprehensive background information on the wormlike chain, the reader is referred to ref. 42. For more specific details concerning the theory curves in Figs. 5 and 6, see SI Text and SI Figs. 7–9. The dynamics of a weakly bending wormlike chain of arc length s = 0 … L subject to an (optional) constant backbone tension, f, is described by the linear Langevin equation:
for its transverse excursions r⊥(s, t) from the straight ground state. We abbreviate partial time and arc length derivatives by dots and primes, respectively, and κ, ζ⊥, and ξ⊥(s, t) denote the bending rigidity, the solvent friction per length, and Gaussian white noise. (See SI Text for a subtle improvement concerning hydrodynamic interactions.) In three space dimensions, the persistence length is given by ℓp = κ/kBT. The equation of motion is solved by introducing eigenmodes. For the simple case of hinged-end boundary conditions, these are just sine functions with amplitudes rn(t) for discrete (half) wavelengths λn = L/n (n = 1, 2, 3, …)
The eigenmodes relax independently and exponentially. For the case of vanishing tension, f = 0,
with the characteristic mode relaxation time
referred to as τλ in the main text. By 〈…〉 we denote the averaging over the thermal noise ensemble. With the above formulae it is straightforward to calculate the transverse component 〈[r⊥(s, t) − r⊥(s, 0)]2〉 of the mean-square displacement (and similarly the longitudinal component) as a sum over the mode contributions (Eqs. 2 and 3), from which all other quantities follow by standard procedures. The case with finite tension f > 0 (interpreted as arising from a prestress σ = f/5ξ2 with ξ the mesh size) is treated along the same lines (22).
Supplementary Material
ACKNOWLEDGMENTS.
We thank M. Rusp for protein preparation. This work was supported by Volkswagen Stiftung Grant 77717 and by the Deutsche Forschungsgemeinschaft through the Nanosystems Initiative Munich, the Leipzig School of Natural Sciences–Building with Molecules and Nano-objects, and the TUM International Graduate School of Science and Engineering.
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
This article contains supporting information online at www.pnas.org/cgi/content/full/0705513104/DC1.
References
- 1.Bausch AR, Kroy K. Nat Phys. 2006;2:231–238. [Google Scholar]
- 2.Morse DC. Phys Rev E. 2001;63 031502. [Google Scholar]
- 3.Gardel ML, Shin JH, MacKintosh FC, Mahadevan L, Matsudaira P, Weitz DA. Science. 2004;304:1301–1305. doi: 10.1126/science.1095087. [DOI] [PubMed] [Google Scholar]
- 4.Storm C, Pastore JJ, MacKintosh FC, Lubensky TC, Janmey PA. Nature. 2005;435:191–194. doi: 10.1038/nature03521. [DOI] [PubMed] [Google Scholar]
- 5.Tharmann R, Claessens MM, Bausch AR. Phys Rev Lett. 2007;98 doi: 10.1103/PhysRevLett.98.088103. 088103. [DOI] [PubMed] [Google Scholar]
- 6.Wottawah F, Schinkinger S, Lincoln B, Ananthakrishnan R, Romeyke M, Guck J, Käs J. Phys Rev Lett. 2005;94 doi: 10.1103/PhysRevLett.94.098103. 098103. [DOI] [PubMed] [Google Scholar]
- 7.Fernandez P, Pullarkat PA, Ott A. Biophys J. 2006;90:3796–3805. doi: 10.1529/biophysj.105.072215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wang N, Tolić-Norrelykke IM, Chen J, Mijailovich SM, Butler JP, Fredberg JJ, Stamenovic D. Am J Physiol. 2002;282:C606–C616. doi: 10.1152/ajpcell.00269.2001. [DOI] [PubMed] [Google Scholar]
- 9.Gardel ML, Nakamura F, Hartwig JH, Crocker JC, Stossel TP, Weitz DA. Proc Natl Acad Sci USA. 2006;103:1762–1767. doi: 10.1073/pnas.0504777103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Guck J, Schinkinger S, Lincoln B, Wottawah F, Ebert S, Romeyke M, Lenz D, Erickson H M., Ananthakrishnan R, Mitchell D, et al. Biophys J. 2005;88:3689–3698. doi: 10.1529/biophysj.104.045476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Fabry B, Maksym G N., Butler JP, Glogauer M, Navajas D, Fredberg JJ. Phys Rev Lett. 2001;87:148102. doi: 10.1103/PhysRevLett.87.148102. [DOI] [PubMed] [Google Scholar]
- 12.Cipelletti L, Ramos L. J Phys: Condens Matter. 2005;17:R253–R285. [Google Scholar]
- 13.Sollich P, Lequeux F, Hébraud P, Cates ME. Phys Rev Lett. 1997;78:2020–2023. [Google Scholar]
- 14.Fabry B, Fredberg JJ. Respir Physiol Neurobiol. 2003;137:109–124. doi: 10.1016/s1569-9048(03)00141-1. [DOI] [PubMed] [Google Scholar]
- 15.Deng L, Trepat X, Butler J P., Millet E, Morgan KG, Weitz DA, Fredberg JJ. Nat Mater. 2006;5:636–640. doi: 10.1038/nmat1685. [DOI] [PubMed] [Google Scholar]
- 16.Hoffman B, Massiera G, Citters Van K, Crocker J. Proc Natl Acad Sci USA. 2006;103:10259–10264. doi: 10.1073/pnas.0510348103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rosenblatt N, Alencar AM, Majumdar A, Suki B, Stamenovic D. Phys Rev Lett. 2006;97:168101. doi: 10.1103/PhysRevLett.97.168101. [DOI] [PubMed] [Google Scholar]
- 18.Lieleg O, Bausch AR. Phys Rev Lett. 2007;99:158105. doi: 10.1103/PhysRevLett.99.158105. [DOI] [PubMed] [Google Scholar]
- 19.Xu JY, Tseng Y, Wirtz D. J Biol Chem. 2000;275:35886–35892. doi: 10.1074/jbc.M002377200. [DOI] [PubMed] [Google Scholar]
- 20.Kroy K, Frey E. In: Scattering in Polymeric and Colloidal Systems. Brown W, Mortensen K, editors. London: Gordon and Breach; 2000. pp. 197–248. [Google Scholar]
- 21.Janmey P, Hvidt S, Käs J, Lerche D, Maggs A, Sackmann E, Schiliwa M, Stossel TP. J Biol Chem. 1994;269:32503–32513. [PubMed] [Google Scholar]
- 22.Kroy K, Glaser J. New J Phys. 9:416. [Google Scholar]
- 23.Hohenadl M, Storz T, Kirpal H, Kroy K, Merkel R. Biophys J. 1999;77:2199–2209. doi: 10.1016/S0006-3495(99)77060-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Liu J, Gardel ML, Kroy K, Frey E, Hoffman BD, Crocker JC, Bausch AR, Weitz DA. Phys Rev Lett. 2006;96:118104. doi: 10.1103/PhysRevLett.96.118104. [DOI] [PubMed] [Google Scholar]
- 25.Gittes F, MacKintosh FC. Phys Rev. E. 1998;58:R1241. [Google Scholar]
- 26.Monthus C, Bouchaud JP. J Phys A Math Gen. 1996;29:3847–3869. [Google Scholar]
- 27.Carpineti M, Piazza R. Phys Chem Chem Phys. 2004;6:1506–1511. [Google Scholar]
- 28.Piazza R. Curr Opin Coll Int Sci. 2004;8:515–522. [Google Scholar]
- 29.Stinchcombe R, Depken M. Phys Rev Lett. 2002;88:125701. doi: 10.1103/PhysRevLett.88.125701. [DOI] [PubMed] [Google Scholar]
- 30.Hess W, Vilgis TA, Winter HH. Macromolecules. 1988;21:2536–2542. [Google Scholar]
- 31.Höfling F, Franosch T, Frey E. Phys Rev Lett. 2006;96:165901. doi: 10.1103/PhysRevLett.96.165901. [DOI] [PubMed] [Google Scholar]
- 32.Bursac P, Lenormand G, Fabry B, Oliver M, Weitz DA, Viasnoff V, Butler JP, Fredberg JJ. Nat Mater. 2005;4:557–561. doi: 10.1038/nmat1404. [DOI] [PubMed] [Google Scholar]
- 33.Lenormand G, Fredberg JJ. Biorheology. 2006;43:1–30. [PubMed] [Google Scholar]
- 34.Trepat X, Deng L, An SS, Navajas D, Tschumperlin DJ, Gerthoffer WT, Butler JP, Fredberg JJ. Nature. 2007;447:592–596. doi: 10.1038/nature05824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Pham KN, Puertas AM, Bergenholtz J, Egelhaaf SU, Moussaid A, Pusey PN, Schofield AB, Cates ME, Fuchs M, Poon WCK. Science. 2002;296:104–106. doi: 10.1126/science.1068238. [DOI] [PubMed] [Google Scholar]
- 36.Moreno AJ, Colmenero J. J Chem Phys. 2006;124:184906. doi: 10.1063/1.2198202. [DOI] [PubMed] [Google Scholar]
- 37.Poon WCK. J Phys Condens Matter. 2002;14:R859–R880. [Google Scholar]
- 38.Sackmann E. In: Modern Optics, Electronics, and High Precision Techniques in Cell Biology. Isenberg G, editor. Heidelberg: Springer; 1997. pp. 213–259. [Google Scholar]
- 39.Spudich J, Watt S. J Biol Chem. 1971;246:4866–4871. [PubMed] [Google Scholar]
- 40.Cooper J, Bryan J, Schwab B, Frieden C, Loftus D, Elson E. J Cell Biol. 1987;104:491–501. doi: 10.1083/jcb.104.3.491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Janmey P, Peetermans J, Zanker K S., Stossel TP, Tanaka T. J Biol Chem. 1986;261:8357–8362. [PubMed] [Google Scholar]
- 42.Hallatschek O, Frey E, Kroy K. Phys Rev E. 2007;75 doi: 10.1103/PhysRevE.75.031905. 031905. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.