

Abstract
Chromosomal translocations identified in hematopoietic and solid tumors result in deregulated expression of protooncogenes or creation of chimeric proteins with tumorigenic potential. In the pediatric solid tumor alveolar rhabdomyosarcoma, a consistent t(2;13)(q35;q14) or variant t(1;13)(p36;q14) translocation generates PAX3-FKHR or PAX7-FKHR fusion proteins, respectively. In this report, we demonstrate that in addition to functional alterations these translocations are associated with fusion product overexpression. Furthermore, PAX3-FKHR and PAX7-FKHR overexpression occurs by distinct mechanisms. Transcription of PAX3-FKHR is increased relative to wild-type PAX3 by a copy number-independent process. In contrast, PAX7-FKHR overexpression results from fusion gene amplification. Thus, gene-specific mechanisms were selected to overexpress PAX3-FKHR and PAX7-FKHR in alveolar rhabdomyosarcoma, presumably due to differences in regulation between the wild-type loci. We postulate that these overexpression mechanisms ensure a critical level of gene product for the oncogenic effects of these fusions.
Cytogenetics has demonstrated the presence of consistent chromosomal translocations in hematopoietic and solid malignancies (1). Molecular analysis of the consequences of these events has revealed two models for oncogene activation (1). Either protooncogene expression is deregulated by juxtaposition with strong regulatory elements or novel fusion proteins are created with altered function and tumorigenic potential.
Alveolar rhabdomyosarcoma (ARMS) is a pediatric soft tissue tumor that is associated with either a t(2;13)(q35;q14) or variant t(1;13)(p36;q14) translocation (2, 3). These translocations fuse either PAX3 or PAX7 with FKHR to generate chimeric genes that express PAX3-FKHR or PAX7-FKHR fusion products, respectively (4–7). These fusion proteins consist of the N-terminal DNA-binding domains of PAX3 or PAX7 fused to the C-terminal transcriptional activation domain of FKHR. Transient transfection experiments indicate that PAX3-FKHR functions as a transcription factor (8, 9). Moreover, PAX3-FKHR exhibits increased transcriptional potency relative to PAX3 due to swapping of PAX3 and FKHR C-terminal transactivation domains. Cell culture experiments also have demonstrated that PAX3-FKHR can induce phenotypic changes, including cellular transformation (10) and inhibition of myogenic differentiation (11).
While these studies of the ARMS translocations are consistent with the fusion protein model of oncogene activation, additional evidence indicates that altered expression of PAX3-FKHR and PAX7-FKHR is another fundamental characteristic of ARMS. Previous Northern blot analysis of a few ARMS cell lines demonstrated that while PAX3-FKHR RNA was readily detectable, PAX3 RNA was expressed at low or undetectable levels (4). Furthermore, amplification of the PAX3-FKHR and PAX7-FKHR fusion genes has been detected in tumor specimens (12). Finally, antisense oligonucleotide treatment of ARMS cells has shown that transient down-regulation of PAX3-FKHR expression is accompanied by induction of apoptosis (13).
In this study, we present RNase protection and immunoprecipitation analyses demonstrating that PAX3-FKHR and PAX7-FKHR overexpression is a consistent feature of ARMS. These findings indicate a fundamental role for fusion protein overexpression in the pathogenesis of ARMS. Furthermore, by analyzing gene copy number, RNA stability, and transcription levels for the wild-type and fusion genes, we demonstrate that PAX3-FKHR and PAX7-FKHR overexpression results from distinct gene-specific mechanisms.
MATERIALS AND METHODS
Riboprobe Plasmids.
To detect PAX3, PAX7, PAX3-FKHR, PAX7-FKHR, and FKHR expression, we assembled riboprobe plasmids by cloning portions of each cDNA into pSP72 (Promega). PAX3 and PAX7 riboprobe plasmids contain PvuII fragments (365 bp and 353 bp, respectively) corresponding to exons 6–8. PAX3-FKHR and PAX7-FKHR riboprobe plasmids contain blunt-ended PvuII–NcoI fragments (396 bp and 384 bp, respectively) corresponding to exons 6–7 fused to FKHR exon 2. These blunt-end cDNA fragments were subcloned into the PvuII site of pSP72. The FKHR riboprobe plasmid was obtained by cloning a 412-bp BglII–ScaI fragment corresponding to exons 1–2 into BamHI–PvuII-digested pSP72. A glyceraldehyde-3-phosphate dehydrogenase (GAPDH) riboprobe plasmid (pTRI-GAPDH, Ambion) served as an internal control for the RNase protection experiments.
RNase Protection Analysis.
[32P]UTP-labeled antisense test and GAPDH riboprobes were synthesized from linearized riboprobe plasmids using SP6 RNA polymerase (MaxiScript, Ambion) and were used for RNase protection analysis (RPA II, Ambion). Total RNA (5 to 10 μg) was isolated using RNA STAT 60 extraction reagent (TEL TEST “B”) and was mixed with test and GAPDH riboprobes. The mixtures were denatured, hybridized at 42°C for at least 18 hr, and digested with RNases A and T1, and the resultant protected fragments were electrophoresed in a denaturing 6% polyacrylamide gel. Band intensities were quantified using a PhosphorImager (Molecular Dynamics). To correct for differences in specific activity among the riboprobes, the measured intensity of each protected fragment was divided by the number of uridines within the corresponding antisense riboprobe. To calculate a test RNA/GAPDH RNA ratio, normalized test values were divided by normalized GAPDH values. To standardize these experiments, all test/GAPDH ratios were normalized to PAX3 levels (arbitrarily set to 10 units) determined for the embryonal rhabdomyosarcoma cell line RD. Relative expression levels below two units are at the threshold of detection for this assay. Control experiments demonstrated that test/GAPDH ratios were not dependent upon the amount of input RNA and that the variation between experiments was at or below 20% (data not shown).
RNA Stability Analysis.
Subconfluent SJRH28 cultures were treated with 10 μg/ml actinomycin D (Sigma) at 37°C for various times and immediately lysed in RNA STAT 60, and total RNA was recovered for RNase protection analysis. An equivalent volume of ethanol carrier was added to mock-treated cells and did not significantly alter RNA expression (data not shown). The level of GAPDH expression did not change substantially after 240-min exposure to actinomycin D consistent with previous reports of its long half-life (14).
Immunoprecipitation of PAX3 and PAX3-FKHR Proteins.
Cells were metabolically labeled with 200 μCi of [35S]methionine/cysteine (NEN/DuPont) for 7 hr at 37°C and lysed in 1 ml RIPA/PEAL buffer as described (8). Lysates were precleared and incubated on ice for 2 hr with either 2 μg of anti-PAX3 IgG (5) or 2 μg of preimmune rabbit IgG. Immune complexes were collected by adsorption to Pansorbin (Calbiochem), washed with RIPA/PEAL, and eluted by boiling in 100 μl of 0.5% SDS. The eluate was subjected to another round of immunoprecipitation, except that 1 μg of anti-PAX3 or preimmune IgG was used. Immune complexes were eluted by boiling in Laemmli loading buffer and were fractionated by SDS/PAGE.
Nuclear Runoff Analysis.
The isolation of nuclei and nuclear transcription were performed as described (15) except that transcription was performed with 250 μCi of [α-32P]UTP (3,000 Ci/mmol, NEN/DuPont). The purification of 32P-labeled RNA was performed as described (15) except that unincorporated nucleotides were removed by purification over a NucTrap Probe Purification Column (Stratagene). To analyze the nuclear runoff products, 2.5 μg of linearized plasmid was denatured and immobilized onto Hybond N+ filters (Amersham) using a Milliblot-S slot blot manifold (Millipore). The 5′-PAX3 and 3′-PAX3 plasmids contain 3.4-kb HindIII and 4-kb HindIII-NotI fragments, respectively, from λ clone 21 (16). The plasmids pTRI-GAPDH (Ambion) and pBluescript II (Stratagene) were used as internal and negative hybridization controls, respectively. Individual filters were preincubated in hybridization solution [50% deionized formamide/5× SSPE (750 mM NaCl/50 mM NaH2PO4/5 mM EDTA, pH 7.7)/1% SDS/5× Denhardt’s solution (United States Biochemical)/100 μg/ml denatured herring sperm DNA and E. coli tRNA] at 42°C for 6–8 hr. 32P-labeled RNA was denatured, mixed with fresh hybridization solution, and allowed to hybridize to the filters at 42°C for 3 days. The filters were washed as described (17) and exposed to PhosphorImager plates or to film at −70°C. The quantified 5′- and 3′-PAX3 signals were divided by the number of uridines within the sense strands of the 3.4-kb HindIII and 4-kb HindIII-NotI fragments, respectively (16). Relative transcription levels were determined by dividing normalized 5′-or 3′-PAX3 signals by GAPDH signals.
Quantitative Southern Blot Analysis.
Genomic DNA (5 μg) was digested with restriction enzymes, electrophoresed in 0.75% agarose gels, and immobilized onto filters (Hybond N+, Amersham). DNA probes were isolated, labeled, and hybridized to filters as described previously (18). The 5′-PAX3 probes were isolated from intron 7-containing phage clones (16); 7I-ABg, 7I-XH, and 7I-BP correspond to a 0.8-kb ApaI-BglI fragment, a 0.9-kb XbaI-HindIII fragment, and a 0.5-kb BglII-PstI fragment, respectively. The 5′-PAX7 probes 187AE and 7.5 HB correspond to a 187-bp ApaLI-EcoRI cDNA fragment (7), and a 650-bp HindIII-BglII genomic fragment from the final intron of PAX7. Filters were exposed to PhosphorImager plates, the intensity of the wild-type and rearranged alleles was quantified, and an allele ratio was calculated. Only those restriction enzyme/probe combinations that resulted in fragment sizes below 23 kb were used in these calculations.
Cell Lines and Tumor Specimens.
ARMS cell lines containing the t(2;13) are SJRH5, SJRH18, SJRH28, SJRH30, and TTC487 (provided by T. Triche, Children’s Hospital, Los Angeles) and CW12 (5). RD is an embryonal rhabdomyosarcoma cell line (5). A673 is a peripheral primitive neuroectodermal tumor cell line (19). All cell lines were maintained by weekly passage in DMEM with high glucose (GIBCO), containing 10% fetal bovine serum (HyClone). Tumor specimens were collected and screened by reverse transcriptase–PCR for expression of the PAX3-FKHR and PAX7-FKHR fusion transcripts as described (20). Tumor specimens APMD101 and 102 were provided by W. Gerald (Memorial Sloan–Kettering, New York). Tumor specimen APMD105 was provided by P. H. B. Sorensen (British Columbia’s Children’s Hospital, Vancouver, Canada).
RESULTS
Wild-Type and Fusion Product Expression in t(2;13)-Containing Cell Lines.
Previous Northern blot analysis of a few t(2;13)-containing ARMS cell lines demonstrated readily detectable PAX3-FKHR mRNA, but low or undetectable wild-type PAX3 mRNA (4). These results suggest that PAX3-FKHR is overexpressed in ARMS. To refine this hypothesis, RNase protection was used to quantify wild-type and fusion transcripts in six t(2;13)-containing cell lines (Fig. 1 A and B). PAX3-FKHR transcripts were overexpressed relative to PAX3 in four lines; SJRH5, SJRH18, SJRH28, and TTC487 expressed PAX3-FKHR at high levels and PAX3 at low or undetectable levels. In contrast, SJRH30 and CW12 expressed PAX3-FKHR and PAX3 at equally low levels. FKHR expression was detectable in the four lines with high PAX3-FKHR expression and undetectable in the two lines with low PAX3-FKHR expression, suggesting coordinate regulation of these two genes. Expression levels of the reciprocal FKHR-PAX3 fusion were lower than PAX3 and FKHR levels or undetectable in all lines (data not shown).
Figure 1.

RNase protection (A-C) and immunoprecipitation (D) analyses of ARMS cell lines and tumor specimens. (A) Total RNA (5–10 μg) from the indicated cell lines was hybridized with the indicated [32P]UTP-labeled test and control riboprobes. Shown are equivalent exposures of the PAX3 and PAX3-FKHR protected fragments (Upper) and the corresponding GAPDH-protected fragments (Lower). (B) Relative expression levels of PAX3, PAX3-FKHR, and FKHR transcripts in six t(2;13)-containing cell lines. RNase protection analysis was performed on the indicated cell lines, the protected bands were quantified using a PhosphorImager and normalized for the number of uridines in each antisense riboprobe, and a test RNA/GAPDH RNA ratio was calculated. Results are shown as expression units relative to PAX3 levels in the embryonal rhabdomyosarcoma cell line RD (arbitrarily set to 10 units). Wild-type and fusion transcripts are indicated as: PAX3, black columns; PAX3-FKHR, stippled column; and FKHR, diagonal columns. (C) Relative expression levels of wild-type and fusion transcripts in PAX3-FKHR-positive (Left) or PAX7-FKHR-positive (Right) tumor specimens. Results are shown as in B. Differences in absolute expression levels among tumors may be partly attributed to variable numbers of nontumor cells within the specimens. Wild-type and fusion transcripts are indicated as: PAX3, black columns; PAX3-FKHR, stippled columns; PAX7, open columns; PAX7-FKHR, diagonal columns. (D) Immunoprecipitation analysis. Lysates from the indicated cell lines were incubated with either anti-PAX3 (α-PAX3) or pre-immune (pre) IgG. Immunoprecipitates were collected and resolved by SDS/PAGE. ARMS cell lines with the t(2;13) are SJRH5, SJRH28, and SJRH30. A673 is a peripheral primitive neuroectodermal tumor cell line lacking the t(2;13). Arrows indicate the PAX3 and PAX3-FKHR proteins. The sizes (in kDa) of protein markers are shown to the left.
We next performed immunoprecipitation analysis to examine expression at the protein level. In the peripheral primitive neuroectodermal tumor cell line A673, which lacks the t(2;13), PAX3 antiserum precipitated a 56-kDa protein corresponding to wild-type PAX3 (Fig. 1D). Wild-type PAX3 protein was detected at low levels in SJRH28 and was undetectable in SJRH5 and SJRH30 whereas higher amounts of the 97-kDa fusion protein (5) were detected in these ARMS lines. The relative amount of wild-type and fusion protein expressed in SJRH5 and SJRH28 parallels RNA expression. In contrast, the incongruity between protein and RNA expression in SJRH30 suggests a posttranscriptional mechanism favoring fusion protein expression in this line.
Wild-Type and Fusion Transcript Expression in t(2;13) and t(1;13) Tumor Specimens.
To investigate PAX3-FKHR expression in tumor specimens, we analyzed 19 cases by RNase protection. In 18 of 19 PAX3-FKHR-positive specimens, the fusion transcripts were expressed at 2- to 88-fold higher levels than PAX3 transcripts (Fig. 1C). Therefore, fusion gene overexpression is the predominant pattern in t(2;13)-containing ARMS. Only one specimen demonstrated comparable wild-type and fusion expression levels; the SJRH30 and CW12 lines may be representative of this small subset or may reflect culture-specific changes.
To extend this analysis to t(1;13)-containing ARMS, we assayed eight PAX7-FKHR-positive tumor specimens. This variant fusion was expressed at levels that were 12- to 76-fold higher than PAX7 levels in all eight specimens (Fig. 1C). Therefore, fusion gene overexpression is a common characteristic of both subtypes of ARMS.
Wild-Type and Fusion Gene Copy Number Analysis.
We previously found a high frequency of fusion gene amplification in PAX7-FKHR-positive, but not in PAX3-FKHR-positive tumors (12). In these previous experiments, we principally used a quantitative Southern blot assay that compares the relative copy number of FKHR sequences flanking the breakpoints. Probes from the 5′ and 3′ FKHR regions were simultaneously hybridized to Southern blots of tumor and control DNA digests, and amplification was detected as elevated 3′ to 5′ FKHR hybridization signal ratios. This method focuses on rearranged and wild-type FKHR alleles and actually calculates the ratio
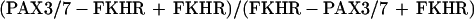 |
that is influenced by the number of FKHR and reciprocal FKHR-PAX3/7 gene copies.
To directly compare the copy numbers of wild-type and rearranged PAX3/7 alleles, we developed an alternative quantitative Southern blot strategy. Restriction fragments corresponding to wild-type and rearranged PAX3/7 alleles were separated by electrophoresis (Fig. 2A), hybridized to 5′ PAX3/7 probes, and quantified to calculate a rearranged to wild-type ratio (Fig. 2B). Analysis of t(2;13)-containing cell lines demonstrated ratios ranging from 0.5 to 3. In these pure tumor cell populations, the allele ratios do not correlate with expression differences. Analysis of tumor specimens indicated the presence of fusion gene amplification in 4 of 4 PAX7-FKHR-positive tumors and only 1 of 11 PAX3-FKHR-positive tumors (Fig. 2B). These findings demonstrate that fusion overexpression is directly related to amplification in PAX7-FKHR tumors, but is independent of copy number in PAX3-FKHR tumors.
Figure 2.

Quantitative Southern blot analysis of wild-type and rearranged PAX3 and PAX7 alleles. (A) Identification of t(2;13) and t(1;13) rearrangements in cell lines and tumor specimens. The indicated restriction enzymes (above each panel) and hybridization probes (below each panel) were selected to identify the wild-type PAX3 or PAX7 and rearranged PAX3-FKHR or PAX7-FKHR alleles (see Materials and Methods for a description of the probes). The wild-type alleles are indicated by arrowheads. (B) Wild-type and rearranged allele copy numbers. The intensities of the wild-type and rearranged alleles were quantified using a PhosphorImager, and a rearranged to wild-type allele ratio was calculated. The PAX3-FKHR-positive or PAX7-FKHR-positive cell lines and tumors are indicated by black or stippled columns, respectively. Please note that amplification of PAX7-FKHR in case CW15 was not detected in our previous analysis (12); this result can be explained by the presence of additional copies of wild-type FKHR or FKHR-PAX7, which will lower the 3′ to 5′ FKHR signal ratio but will not affect the PAX7-FKHR to PAX7 signal ratio in the current assay.
Transcriptional Activation of PAX3-FKHR Expression.
To determine the basis of PAX3-FKHR overexpression, we performed RNA stability analysis on SJRH28 (Fig. 3) and SJRH5 (data not shown). Cells were treated for various times with actinomycin D, and RNA was analyzed by RNase protection. The wild-type and fusion transcript levels decreased comparably after treatment, and thus there are no significant differences in stability between these transcripts.
Figure 3.

Stability analysis of PAX3 and PAX3-FKHR transcripts. SJRH28 cells were treated with 10 μg/ml actinomycin D for the indicated times. RNA was isolated, and the level of expression was determined by RNase protection in triplicate. Mock-treatment did not significantly alter RNA expression (data not shown). Results are shown as relative test (PAX3, open box; PAX3-FKHR, closed diamond) to GAPDH ratios.
We next used nuclear runoff analysis to determine if PAX3 and PAX3-FKHR are transcribed at different levels. For this assay we used unlabeled hybridization probes from genomic regions flanking the t(2;13) breakpoint in SJRH5 (Fig. 4A). Filters containing control probes and either 5′- or 3′-PAX3 probes were hybridized with 32P-labeled RNA isolated from either A673 or SJRH5 nuclei (Fig. 4B). In A673, which lacks the t(2;13), we detected equivalent signals from the 5′- and 3′-PAX3 regions. In contrast, we detected 5′-PAX3 signals that were 5.6-fold higher than 3′-PAX3 signals in SJRH5 (Fig. 4C). In this cell line, 5′-PAX3 probes hybridize to wild-type PAX3 and PAX3-FKHR transcripts, while 3′-PAX3 probes hybridize to wild-type PAX3 and FKHR-PAX3 transcripts. Therefore, PAX3-FKHR is transcribed at least 4.6-fold higher than PAX3. This difference is an underestimate depending upon the level of FKHR-PAX3 transcription. Thus, the difference in transcription levels can account for most of the difference between steady-state PAX3-FKHR and PAX3 RNA levels.
Figure 4.

Nuclear runoff analysis. Nuclei were isolated from A673 and SJRH5, and nuclear transcription was performed with [α-32P]UTP. 32P-labeled RNA was purified and hybridized to linearized plasmids immobilized onto slot blots. (A) PAX3 intron 7/exon 8 sequences used as 5′- and 3′-PAX3 hybridization probes. Exons 6–8 are represented by the black boxes. The 5′-and 3′-PAX3 hybridization probes are indicated by the brackets below the horizontal line. The t(2;13) breakpoint region in SJRH5 is indicated by the arrow. (B) 32P-labeled RNA from A673 lacking the t(2;13) (Left) and SJRH5 containing the t(2;13) (Right) was hybridized to filters containing either 5′-or 3′-PAX3 hybridization probes. Plasmids GAPDH and pBluescript served as internal and negative controls, respectively. (C) Relative transcription levels were determined by normalizing hybridization signals for the number of uridines in the sense strands of the 5′- or 3′-PAX3 hybridization probes and calculating a 5′- or 3′-PAX3 to GAPDH ratio. Averages and standard deviations were calculated from three experiments.
DISCUSSION
Our findings demonstrate that fusion products are overexpressed in both t(2;13) and t(1;13) subsets of ARMS and support the hypothesis that expression above a threshold level is an important oncogenic parameter. Consistent with this hypothesis, transient down-regulation of PAX3-FKHR expression by antisense oligonucleotide treatment is accompanied by a decrease in cell viability and induction of apoptosis, indicating that the level of fusion protein is important for cell survival (13). This threshold effect also may be an important factor for the function of the fusion proteins in inducing cellular transformation and inhibiting terminal differentiation (10, 11).
The overexpression of these fusion proteins is postulated to influence their function as transcription factors. Overexpression may reflect a requirement for PAX3-FKHR and PAX7-FKHR to deregulate target genes with low affinity binding sites or abrogate competitive interactions with other proteins. Because the transcriptional potency of PAX3 is 10- to 100-fold lower than PAX3-FKHR (9), PAX3 may counteract the influence of PAX3-FKHR on target gene regulation by competition for binding site occupancy.
The mechanism of PAX3-FKHR overexpression involves increased transcription and is copy number-independent, reminiscent of the model of protooncogene activation by juxtaposition with expression elements of other genes (1). The likely explanation for this increased transcription is recombination of cis-acting expression elements by the t(2;13) chromosomal translocation. Because expression of FKHR-PAX3 is lower than that of FKHR, the expression consequences of the balanced translocation appear to be reciprocal. These reciprocal effects could be achieved by swapping a negative 3′-PAX3 or a positive 3′-FKHR regulatory element. Our finding of similar patterns of PAX3-FKHR and FKHR expression in ARMS cell lines is consistent with the presence of a positive 3′-FKHR element.
In contrast to activation of PAX3-FKHR transcription, PAX7-FKHR overexpression is a consequence of gene amplification. The difference in overexpression mechanisms suggests important underlying differences in the control of PAX3 and PAX7 gene expression. Evidence of these expression regulatory differences is derived from studies of murine embryogenesis that demonstrate overlapping, but distinct, patterns of Pax3 and Pax7 expression (21, 22). The finding of myogenic cell types in which Pax3 and Pax7 are divergently expressed indicates cis-acting elements that are differentially active in various myogenic cell environments. We postulate that in ARMS cells the PAX7 promoter may not be as strong or as responsive to 3′-FKHR elements as the PAX3 promoter; the fusion of PAX7 and FKHR genes therefore may not be sufficient for PAX7-FKHR overexpression in the ARMS cell environment.
Few studies to date have examined the control of fusion gene expression in tumors. After fusion of 5′-EWS with 3′-FLI1 or 3′-ERG in peripheral primitive neuroectodermal tumor and Ewing sarcoma, available data indicates that EWS-FLI1/ERG transcripts are expressed at similar levels as wild-type EWS transcripts whereas wild-type FLI1/ERG is not detectably expressed (23–25). As EWS is ubiquitously expressed, promoter swapping is likely a explanation for the EWS-FLI1/ERG expression pattern. A promoter swapping mechanism is not sufficient to explain the expression pattern in ARMS because PAX3-FKHR and PAX3 share the same 5′-region.
The tumorigenic significance of fusion gene overexpression will be clarified in future experiments by dissection of the functional differences between the wild-type and fusion proteins. In addition, the molecular basis of fusion gene overexpression in ARMS will be refined by characterization of the cis-acting regulatory elements and corresponding transcription factors that control PAX3-FKHR and PAX7-FKHR expression.
Acknowledgments
We thank L. Nauta, J. Ginsberg, and J. Hollows for technical assistance. This work was supported in part by National Institutes of Health Grants CA64202 and CA71838 and the Dr. Louis Sklarow Memorial Fund.
ABBREVIATIONS
- ARMS
alveolar rhabdomyosarcoma
- GAPDH
glyceraldehyde-3-phosphate dehydrogenase
References
- 1.Rabbitts T H. Nature (London) 1994;372:143–149. doi: 10.1038/372143a0. [DOI] [PubMed] [Google Scholar]
- 2.Whang-Peng J, Knutsen T, Theil K, Horowitz M E, Triche T. Genes Chromosomes Cancer. 1992;5:299–310. doi: 10.1002/gcc.2870050405. [DOI] [PubMed] [Google Scholar]
- 3.Biegel J A, Meek R S, Parmiter A H, Conard K, Emanuel B S. Genes Chromosomes Cancer. 1991;3:483–484. doi: 10.1002/gcc.2870030612. [DOI] [PubMed] [Google Scholar]
- 4.Barr F G, Galili N, Holick J, Biegel J A, Rovera G, Emanuel B S. Nat Genet. 1993;3:113–117. doi: 10.1038/ng0293-113. [DOI] [PubMed] [Google Scholar]
- 5.Galili N, Davis R J, Fredericks W J, Mukhopadhyay S, Rauscher F J, III, Emanuel B S, Rovera G, Barr F G. Nat Genet. 1993;5:230–235. doi: 10.1038/ng1193-230. [DOI] [PubMed] [Google Scholar]
- 6.Shapiro D N, Sublett J E, Li B, Downing J R, Naeve C W. Cancer Res. 1993;53:5108–5112. [PubMed] [Google Scholar]
- 7.Davis R J, D’Cruz C M, Lovell M A, Biegel J A, Barr F G. Cancer Res. 1994;54:2869–2872. [PubMed] [Google Scholar]
- 8.Fredericks W J, Galili N, Mukhopadhyay S, Rovera G, Bennicelli J L, Barr F G, Rauscher F J., III Mol Cell Biol. 1995;15:1522–1535. doi: 10.1128/mcb.15.3.1522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bennicelli J L, Edwards R H, Barr F G. Proc Natl Acad Sci USA. 1996;93:5455–5459. doi: 10.1073/pnas.93.11.5455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Scheidler S, Fredericks W J, Rauscher F J, III, Barr F G, Vogt P K. Proc Natl Acad Sci USA. 1996;93:9805–9809. doi: 10.1073/pnas.93.18.9805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Epstein J A, Lam P, Jepeal L, Maas R L, Shapiro D N. J Biol Chem. 1995;270:11719–11722. doi: 10.1074/jbc.270.20.11719. [DOI] [PubMed] [Google Scholar]
- 12.Barr F G, Nauta L E, Davis R J, Schafer B W, Nycum L M, Biegel J A. Hum Mol Genet. 1996;5:15–21. doi: 10.1093/hmg/5.1.15. [DOI] [PubMed] [Google Scholar]
- 13.Bernasconi M, Remppis A, Fredericks W J, Rauscher F J, III, Schafer B W. Proc Natl Acad Sci USA. 1996;93:13164–13169. doi: 10.1073/pnas.93.23.13164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dani C, Piechaczyk M, Audigier Y, El Sabouty S, Cathala G, Marty L, Fort P, Blanchard J M, Jeanteur P. Euro J Biochem. 1984;145:299–304. doi: 10.1111/j.1432-1033.1984.tb08552.x. [DOI] [PubMed] [Google Scholar]
- 15.Groudine M, Peretz M, Weintraub H. Mol Cell Biol. 1981;1:281–288. doi: 10.1128/mcb.1.3.281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Macina R A, Barr F G, Galili N, Riethman H C. Genomics. 1995;26:1–8. doi: 10.1016/0888-7543(95)80076-x. [DOI] [PubMed] [Google Scholar]
- 17.Greenberg M E, Bender T P. In: Current Protocols in Molecular Biology. Ausubel F M, Brent R, Kingston R E, Moore D D, Seidman J G, Smith J A, Struhl K, editors. New York: Wiley; 1988. pp. 4.10.1–4.10.11. [Google Scholar]
- 18.Barr F G, Davis R J, Eichenfield L, Emanuel B S. Proc Natl Acad Sci USA. 1992;89:942–946. doi: 10.1073/pnas.89.3.942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sorensen P H B, Liu X F, Delattre O, Rowland J M, Biggs C A, Thomas G, Triche T J. Diagn Mol Pathol. 1993;2:147–157. [PubMed] [Google Scholar]
- 20.Barr F G, Chatten J, D’Cruz C M, Wilson A E, Nauta L E, Nycum L M, Biegel J A, Womer R B. J Am Med Assoc. 1995;27:553–557. [PubMed] [Google Scholar]
- 21.Goulding M D, Chalepakis G, Deutsch U, Erselius J R, Gruss P. EMBO J. 1991;10:1135–1147. doi: 10.1002/j.1460-2075.1991.tb08054.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jostes B, Walther C, Gruss P. Mech Devel. 1991;33:27–37. doi: 10.1016/0925-4773(90)90132-6. [DOI] [PubMed] [Google Scholar]
- 23.Sorensen P H B, Triche T J. Semin Cancer Biol. 1996;7:3–14. doi: 10.1006/scbi.1996.0002. [DOI] [PubMed] [Google Scholar]
- 24.Sorensen P H B, Lesnick S L, Lopez-Terrada D, Liu X F, Triche T J, Denny C T. Nat Genet. 1994;6:146–151. doi: 10.1038/ng0294-146. [DOI] [PubMed] [Google Scholar]
- 25.Zucman J, Delattre O, Desmaze C, Plougastel B, Joubert I, Melot T, Peter M, De Jong P, Rouleau G, Aurias A, Thomas G. Genes Chromosomes Cancer. 1992;5:271–277. doi: 10.1002/gcc.2870050402. [DOI] [PubMed] [Google Scholar]