

Abstract
A screen for mutants of Saccharomyces cerevisiae secretory pathway components previously yielded sec34, a mutant that accumulates numerous vesicles and fails to transport proteins from the ER to the Golgi complex at the restrictive temperature (Wuestehube, L.J., R. Duden, A. Eun, S. Hamamoto, P. Korn, R. Ram, and R. Schekman. 1996. Genetics. 142:393–406). We find that SEC34 encodes a novel protein of 93-kD, peripherally associated with membranes. The temperature-sensitive phenotype of sec34-2 is suppressed by the rab GTPase Ypt1p that functions early in the secretory pathway, or by the dominant form of the ER to Golgi complex target-SNARE (soluble N-ethylmaleimide sensitive fusion protein attachment protein receptor)–associated protein Sly1p, Sly1-20p. Weaker suppression is evident upon overexpression of genes encoding the vesicle tethering factor Uso1p or the vesicle-SNAREs Sec22p, Bet1p, or Ykt6p. This genetic suppression profile is similar to that of sec35-1, a mutant allele of a gene encoding an ER to Golgi vesicle tethering factor and, like Sec35p, Sec34p is required in vitro for vesicle tethering. sec34-2 and sec35-1 display a synthetic lethal interaction, a genetic result explained by the finding that Sec34p and Sec35p can interact by two-hybrid analysis. Fractionation of yeast cytosol indicates that Sec34p and Sec35p exist in an ∼750-kD protein complex. Finally, we describe RUD3, a novel gene identified through a genetic screen for multicopy suppressors of a mutation in USO1, which suppresses the sec34-2 mutation as well.
Keywords: Sec34p, Sec35p, Rud3p, vesicle tethering, secretory pathway
The flow of material through the secretory pathway is mediated, at least in part, by membrane-bound vesicles or larger membrane-delimited structures ( Palade 1975). These transport intermediates, which are generated from numerous intracellular compartments, dock and fuse specifically with the next membrane compartment on their itinerary. Therefore, there must be a mechanism that allows the vesicle and target membranes to specifically recognize one another.
For some time, members of the rab GTPase family were suspected to be the principle determinants of this targeting specificity because distinct family members display unique organellar localizations that correlate with their site of action (for review see Simons and Zerial 1993). However, it has been demonstrated that a single chimeric rab protein can function at two transport steps ( Brennwald and Novick 1993; Dunn et al. 1993), indicating that rabs cannot be the sole targeting determinants. In a similar fashion, SNAREs (soluble N-ethylmaleimide sensitive fusion protein attachment protein receptor) proteins, which are integral membrane proteins found predominantly on vesicles (v-SNAREs) or target membranes (t-SNAREs), were proposed to encode specificity ( Söllner et al. 1993). Once more, however, evidence suggests that SNAREs cannot be the only targeting components: some SNAREs can function at several transport steps in vivo ( Fischer von Mollard et al. 1997; Fischer von Mollard and Stevens 1999), SNAREs that faithfully function at different transport steps in vivo can interact promiscuously in vitro ( Fasshauer et al. 1999; Yang et al. 1999), and some SNAREs have been found in multiple SNARE complexes by coimmunoprecipitation ( Lupashin et al. 1997; Holthuis et al. 1998).
Given that neither rabs nor SNAREs are the sole governors of targeting specificity, other components are likely to play important roles. Additional candidate players include the so-called tethering factors ( Lowe et al. 1998), which are proteins that bind membranes together before SNARE interactions ( Cao et al. 1998). Tethering factors are a diverse group of proteins whose only common feature is their tendency to form elongated, coiled-coil structures and/or to assemble into large, multimeric complexes (for reviews see Pfeffer 1999; Waters and Pfeffer 1999). However, because they are peripheral membrane proteins, and therefore must interact with integral membrane components with distinct localizations, once again, they cannot be the sole determinants of targeting specificity. Taken together, these results suggest that specificity in vesicle traffic might be generated by the interaction of several factors in such a way that no individual component plays the dominant role.
ER to Golgi complex traffic in the yeast Saccharomyces cerevisiae is one of the most intensively studied steps in membrane trafficking. The components involved in the consumption of ER-derived vesicles at the Golgi complex are related to those of many other steps. These include: the v-SNAREs Bet1p, Bos1p, Sec22p, and perhaps Ykt6p ( Novick et al. 1980; Newman and Ferro-Novick 1987; Dascher et al. 1991; Shim et al. 1991; McNew et al. 1997); the t-SNARE Sed5p ( Hardwick and Pelham 1992); the t-SNARE–associated protein Sly1p ( Dascher et al. 1991); and the rab GTPase Ypt1p ( Schmitt et al. 1986). In addition to these evolutionarily conserved components, several accessory factors, which do not appear to have sequence homologs employed at other steps within the cell, are also required.
Two of the accessory proteins required for ER to Golgi complex vesicle transport in yeast are Uso1p ( Nakajima et al. 1991), a homodimeric molecule with two heads and an extraordinarily long (∼150 nm) coiled-coil tail ( Yamakawa et al. 1996), and Sec35p ( Wuestehube et al. 1996), a novel 32-kD protein that is present in both cytosolic and membrane-associated pools ( VanRheenen et al. 1998). Genetic studies have shown that both Uso1p ( Sapperstein et al. 1996) and Sec35p ( VanRheenen et al. 1998) act upstream of the rab Ypt1p, the SNAREs, and Sly1p. These genetic data were explained mechanistically through the use of an in vitro system that recapitulates transport between the ER and the Golgi complex ( Barlowe 1997). In this system, both Uso1p ( Barlowe 1997) and Sec35p ( VanRheenen et al. 1998) are required for the stable interaction, or tethering, of ER-derived vesicles with the Golgi complex. This Uso1p- and Sec35p-dependent tethering step precedes, and is independent of, the essential function of the SNAREs and Sly1p ( Cao et al. 1998), and suggests that tethering might occur without trans-SNARE complex assembly.
In addition to Uso1p and Sec35p, another component that may function in the tethering of ER-derived vesicles is an ∼800-kD protein complex with ten subunits, termed TRAPP ( Sacher et al. 1998). TRAPP is predominantly localized to the cis-Golgi complex and it is required for the consumption of ER-derived vesicles there, which suggests that it is involved in either tethering, SNARE-mediated docking, or membrane fusion. Since the genetic interactions of two TRAPP genes (BET3 and BET5) with other genes involved in ER to Golgi complex traffic are very similar to those exhibited by USO1 and SEC35 ( Rossi et al. 1995; Jiang et al. 1998; Sacher et al. 1998), it seems likely that TRAPP functions in the tethering process as well.
In an effort to further elucidate the mechanism of vesicle tethering, we have studied SEC34. sec34 mutants were identified in a novel screen for secretion mutants in the early secretory pathway of S. cerevisiae and were shown to block ER to Golgi complex traffic concomitant with an accumulation of transport vesicles ( Wuestehube et al. 1996). In this report, we describe the cloning of SEC34 and analysis of its genetic interactions with other secretory genes. Genetic interaction between SEC34 and other genes encoding proteins involved in ER to Golgi complex tethering suggested that Sec34p may function in this process. Indeed, vesicle tethering was defective in an in vitro system generated from a sec34 mutant. Interestingly, we find that Sec34p is present in a large protein complex that contains Sec35p. These findings indicate that the Sec34p/Sec35p complex is a novel component required for tethering ER-derived vesicles to the yeast Golgi complex and, as such, may help to impart targeting specificity to this transport step. Lastly, we describe a novel gene, RUD3, which was originally identified as a high copy suppressor of a uso1 tethering mutant ( Sapperstein 1997), and which we now find acts as a multicopy suppressor of sec34 as well. This genetic result implicates Rud3p as functioning in, or downstream of, ER to Golgi vesicle tethering.
Materials and Methods
Media and Microbial Techniques
Bacterial media was prepared by standard protocols ( Miller 1972). Yeast strains were maintained on rich media (YPD) containing 1% Bacto-yeast extract, 2% Bacto-peptone, and 2% glucose, or on synthetic complete media (SC) containing 0.67% yeast nitrogen base without amino acids, 2% glucose, and the appropriate supplements ( Rose et al. 1990). SC media lacking histidine, leucine, and tryptophan used in the two-hybrid assay contained 2.5 mM 3-aminotriazole. Diploid strains were sporulated at room temperature in liquid media consisting of 1% potassium acetate and 0.02% glucose. Escherichia coli transformations were performed by the method of Hanahan 1983 and yeast transformations were performed by the method of Elble 1992, except for the yeast genomic library transformation, which was by the method of Schiestl and Gietz 1989.
Plasmid and Strain Construction
Plasmids used in this work are described in Table . Plasmid construction was as follows. To generate pSV22, the genomic library plasmid pB4 was digested with PvuII and HindIII, and the resulting 2.7-kb fragment containing YER157w/SEC34 was ligated into pRS416 that had been digested with SmaI and HindIII. To create pSV24 and pSV25, SEC34 was liberated from the polylinker of pSV22 with either HindIII and BamHI or XhoI and SpeI double-digests and ligated into pRS305 (digested with HindIII and BamHI) or pRS426 (digested with XhoI and SpeI), respectively. The insert for bacterial expression plasmids encoding glutathione S-transferase (GST)-Sec34p and His6-Sec34p fusion proteins was generated by PCR including a BamHI site adjacent to the codon for the first amino acid of Sec34p and a SmaI site downstream of the stop codon (5′ primer, 5′ gcc-gga-tcc-atg-gcg-aga-agt-aga-aag 3′; 3′ primer, 5′ tcc-ccc-ggg-gtt-tat-ttc-gtt-atg-gta-tc 3′). The PCR product was digested with BamHI and SmaI, and ligated into similarly digested pGEX4T-1 (Pharmacia Biotech, Inc.) and pQE30 (QIAGEN, Inc.), generating pSV28 and pSV30, respectively. To create the constructs expressing the Gal4p DNA binding domain (Gal4p-BD) or transcriptional activation domain (Gal4p-AD) fused to Sec34p, the SEC34 open reading frame (ORF) was amplified by PCR, placing a BamHI site upstream of the codon for the second amino acid residue of the protein and a PstI site downstream of the stop codon (5′ primer, 5′ cgc-gga-tcc-tgg-cga-gaa-gta-gaa-ag 3′; 3′ primer, 5′ cgc-gct-gca-gtt-tat-ttc-gtt-atg-gta-tc 3′). The resulting product was cleaved with BamHI and PstI, and ligated into a similarly digested pAS2 or pGAD424 (Clonetech, Inc.), yielding COOH-terminal fusions to Gal4p-BD (pSV37) and Gal4p-AD (pSV35), respectively. The constructs expressing the Gal4p-BD or Gal4p-AD fused to Sec35p were constructed in an identical manner, also placing a BamHI site upstream of the codon for the second amino acid residue of the protein and a PstI site downstream of the stop codon (5′ primer, 5′ cgc-gga-tcc-tgg-tca-aca-gtc-ata-g 3′; 3′ primer, 5′ cgc-gct-gca-ggt-ttt-ctc-cca-act-atg 3′), creating COOH-terminal fusions to Gal4p-BD (pSV34) and Gal4p-AD (pSV36), respectively. To delete one copy of SEC34 in a diploid strain by the γ-method ( Rose 1995), the sec34Δ plasmid, pSV27, was constructed in two stages. In the first stage, the region 5′ to SEC34 was excised from plasmid pB4 as a PstI/PvuII fragment (the location of restriction enzymes sites are shown in Fig. 1 a) and ligated into PstI/SmaI-digested pRS305. In the second stage, a HindIII fragment containing the region 3′ to the locus was released from plasmid pB4 and ligated into the plasmid generated from step one, which had been linearized with HindIII; the correct orientation of the HindIII fragment was confirmed by restriction digest. In the resulting construct, the inserts in the polylinker of pRS305 were placed such that the region directly 3′ to the ORF was placed upstream of the region directly 5′ of the ORF, with a unique restriction site, PstI, between the two sequences; digestion of this plasmid with PstI and transformation of the linearized plasmid into a diploid strain results in the replacement of the coding sequences of one allele of SEC34 with the sequences of the integrating vector. To generate pSK81, ORF YOR216c/RUD3 was isolated from the library plasmid pSOU7 ( Sapperstein 1997) as a 2.2-kb BamHI/SnaBI fragment and ligated into pRS426 that had been digested with BamHI and SmaI.
Table 1.
Plasmids Used in this Work
| Plasmid | Description | Source |
|---|---|---|
| pJG103 | 2μm SEC22 URA3 | S. Ferro-Novick (Yale University, New Haven, CT) |
| pSFN2d | 2μm BET1 URA3 | S. Ferro-Novick |
| pNB167 | 2μm YPT1 URA3 | S. Ferro-Novick |
| pGR3 | 2μm BET3 URA3 | S. Ferro-Novick |
| pSED5 | 2μm SED5 URA3 | H. Pelham (MRC, Cambridge, UK) |
| pNB142 | 2μm SEC4 URA3 | P. Novick (Yale University, New Haven, CT) |
| pSK47 | 2μm USO1 URA3 | This laboratory |
| pSK60 | 2μm YKT6 URA3 | This laboratory |
| pSK101 | 2μm BOS1 URA3 | This laboratory |
| pSV11 | CEN SLY1-20 URA3 | This laboratory |
| pSV17 | 2μm SEC35 URA3 | This laboratory |
| pVA3 | 2μm Gal4p-BD-p53 TRP1 | Clonetech, Inc. |
| pTD1 | 2μm Gal4p-AD-T-antigen LEU2 | Clonetech, Inc. |
| pB4 | CEN SEC34 URA3 (library plasmid) | This study |
| pA10 | CEN SEC34 URA3 (library plasmid) | This study |
| pSV22 | CEN SEC34 URA3 | This study |
| pSV24 | YIp SEC34 LEU2 | This study |
| pSV25 | 2μm SEC34 URA3 | This study |
| pSV27 | YIp sec34Δ-construct LEU2 | This study |
| pSV28 | GST-Sec34p | This study |
| pSV30 | His6-Sec34p | This study |
| pSV34 | 2μm Gal4p-BD-Sec35p TRP1 | This study |
| pSV35 | 2μm Gal4p-AD-Sec34p LEU2 | This study |
| pSV36 | 2μm Gal4p-AD-Sec35p LEU2 | This study |
| pSV37 | 2μm Gal4p-BD-Sec34p TRP1 | This study |
| pSOU7 | 2μm RUD3 URA3 (library plasmid) | This study |
| pSK81 | 2μm RUD3 URA3 | This study |
Figure 1.

The SEC34 genomic locus and the sequence of Sec34p. a, Diagrammatic representation of a 10-kb region of the right arm of chromosome V containing BEM2, YER156c, YER157w, and YER158c. Relevant restriction enzyme sites (see Materials and Methods) are indicated. b and c, Genomic fragments contained in library plasmids pB4 and pA10, respectively. d, The 2.6-kb PvuII/HindIII fragment that contains YER157w and restores temperature resistance to the sec34-2 strain. e, Representation of the SEC34 (YER157w) locus in the sec34Δ strain, in which the region between PvuII and HindIII is replaced by sequences in the LEU2 integrating vector. f, Predicted amino acid sequence of Sec34p. The nucleotide accession number for SEC34 is U18917 and the protein accession number is S50660.
Yeast strains used in this paper are described in Table . Strain construction was as follows. The sec34Δ::LEU2/SEC34 strain GWY127 was constructed by transforming GWY30 with PstI-digested pSV27. The presence of the SEC34 deletion in the Leu+ transformants was confirmed by PCR amplification of the novel junctions at the deletion locus. The diploid strain heterozygous for both the sec34-2 and sec35-1 alleles was created by mating GWY93 to GWY95 for 6 h on YPD. The diploid was identified by the distinct morphology of the zygote, and was isolated by micromanipulation.
Table 2.
Strains Used in this Work
| Strain | Genotype | Source |
|---|---|---|
| RSY255 | MATa ura3-52 leu2-3,-112 | R. Schekman (University of California at |
| Berkeley, Berkeley, CA) | ||
| RSY942 | MATa sec22-3 ura3-52 lys2-801 | R. Schekman |
| RSY944 | MATa bet1-1 ura3-52 lys2-801 | R. Schekman |
| RSY976 | MATa ypt1-3 ura3-52 | R. Schekman |
| RSY1074 | MATα sly1ts ura3-1 leu2-3,-112 trp1 ade2-1 | R. Schekman |
| RSY1157 | MATa pep4::HIS3 prb::HisG prc::HisG ura3-1 leu2-3,112 trp1-1 ade2-1 can1-100 | |
| SFNY314 | MATa bet3-1 ura3-52 leu2-3,-112 | S. Ferro-Novick (Yale University, |
| New Haven, CT) | ||
| PJ69-4A | MATa ura3-52 leu2-3,112 trp1-901 his3-200 gal4Δ gal80Δ LYS2::GAL1-HIS3 | P. James (University of |
| GAL2-ADE2 met2::GAL7-lacZ | Wisconsin, Madison, WI) | |
| GWY30 | MATa/α ura3-52/ura3-52 leu2Δ1/leu2Δ1 trp1Δ63/trp1Δ63 | This laboratory |
| GWY67 | MATα uso1-1 ura3-52 leu2-3,-112 | This laboratory |
| GWY93 | MATα sec35-1 ura3-52 leu2-3,-112 | This laboratory |
| GWY95 | MATa sec34-2 ura3-52 leu2-3,-112 lys2-801 | This laboratory |
| GWY127 | MATa/α sec34Δ::LEU2/SEC34 ura3-52/ura3-52 leu2Δ1/leu2Δ1 trp1Δ63/trp1Δ63 | This study |
| GWY132 | MATα sec34Δ::LEU2 ura3-52 leu2Δ1 trp1Δ63 +pSV11 (pSLY1-20 CEN URA3) | This study |
| GWY138 | MATa sec32-1 (bos1ts) ura3-52 his4-612 lys2-801 | This laboratory |
Cloning of SEC34
To clone SEC34, the sec34-2 strain GWY95 ( Wuestehube et al. 1996; VanRheenen et al. 1998) was transformed with a URA3 YCp50-based library ( Rose et al. 1987), and temperature-resistant colonies were selected at 38.5°C. Over 100,000 transformants were screened and ∼90 temperature-resistant colonies were isolated. Library plasmids from nine colonies were isolated, amplified in bacteria, and tested for the ability to confer temperature-resistance when retransformed into the sec34-2 strain. Four plasmids restored growth at 38.5°C to wild-type levels, three yielded partial suppression, and the remaining two did not display plasmid-linked suppression. The ends of the inserts of the seven constructs showing plasmid-linked suppression were sequenced with primers YEp24-F and YEp24-R ( Sapperstein et al. 1996) and the resulting sequences were used to search the Saccharomyces Genome Database. To mark the YER157w locus for purposes of integrative mapping, the sec34-2 strain GWY95 was transformed with pSV24 that had been linearized with BglII, an enzyme that cleaves internal to the ORF, such that integration is directed to the YER157w locus.
Antibody Production and Immunoblotting
GST-Sec34p and His6-Sec34p were expressed in strain XL1-Blue (Stratagene) from plasmids pSV28 and pSV30, respectively, and fusion proteins were purified according to the manufacturers' instructions (Pharmacia Biotech, Inc.; QIAGEN, Inc.). GST-Sec34p was used to immunize rabbits by standard procedures ( Harlow and Lane 1988) and the resulting serum was affinity-purified against His6-Sec34p that had been coupled to cyanogen bromide-activated Sepharose, as per the manufacturer's instruction (Pharmacia Biotech, Inc.). Crude yeast lysates used to characterize the affinity-purified antibody were made as described ( Ohashi et al. 1982), and the equivalent of 0.2 OD units of cells was analyzed for each strain. HRP-conjugated secondary antibodies were used at a dilution of 1:3,000. Incubations with both primary and secondary antibodies were for 1 h at room temperature and immunoblots were developed with a chemiluminescent detection kit (Amersham Corp.).
Extraction and Subcellular Fractionation
A 250-ml culture of wild-type yeast (RSY255) was grown in YPD at 30°C to midlogarithmic phase (OD595 = 1.4), washed in sterile water, and resuspended at ∼75 OD595/ml in Buffer 88 (25 mM Hepes, pH 7.0, 150 mM KOAc, 5 mM MgCl2, 1 mM DTT) containing protease inhibitors ( VanRheenen et al. 1998). 1/2 sample vol of acid-washed glass beads (425–600 μm; Sigma Chemical Co.) was added, and the material was vortexed eight times for 30 s, with 30 s on ice between each burst. The crude yeast lysate was centrifuged at 1,500 g (SA600 rotor, 3,300 rpm, 3 min, 4°C) to generate the S1 lysate. For extraction studies, yeast membranes were isolated on a step gradient in which 2 ml of S1 was layered over a step gradient consisting of 2 ml of 45% sucrose and 8 ml of 10% sucrose, both in Buffer 88. After centrifugation at 200,000 g (SW40 rotor, 40,000 rpm, 2 h, 4°C), membranes were collected from the 10%/45% sucrose interface by piercing the side of the tube with an 18 gauge needle and aspiration of the interface. The membranes (100 μl per reaction) were then mixed with an equal volume of Buffer 88 or a 2× extraction buffer (either 2% Triton X-100 in Buffer 88, 2 M NaCl in Buffer 88, or 0.2 M Na2CO3, pH 11.5, in water), diluted to 1 ml in Buffer 88 or 1× extraction buffer (either 1% Triton X-100 in Buffer 88, 1 M NaCl in Buffer 88, or 0.1 M Na2CO3, pH 11.5, in water), and incubated on ice for 45 min. The extraction mixtures (800 μl) were then layered over 200 μl 14% sucrose cushions made in the appropriate 1× extraction buffer and centrifuged at 175,000 g (TLA100.2 rotor, 70,000 rpm, 60 min, 4°C). 900 μl of the supernatant was removed from each sample, and 800 μL of Buffer 88 was added to the pellets (in the remaining 100 μl), which were resuspended with a Dounce homogenizer. Fractions were probed with antibodies against Sec34p, Sec35p ( VanRheenen et al. 1998), Sed5p ( Lupashin et al. 1997), or phosphoglycerate kinase (PGK; Molecular Probes). Subcellular fractionation was completed as previously described ( VanRheenen et al. 1998).
In Vitro ER to Golgi Complex Transport Assay
Yeast semi-intact cells from either the wild-type (RSY255) or the sec34-2 strain (GWY95) were prepared from logarithmic phase cultures of strains grown at 23°C and were stored at −70°C ( Baker et al. 1988). Before assays, an aliquot of cells was quickly thawed and washed in Buffer 88 to remove cytosol, and 35S-pre-pro-α-factor was posttranslationally translocated into the ER of the semi-intact cells as previously described ( Baker et al. 1988). Vesicle tethering and transport assays were performed at either 23 or 29°C, as indicated ( Barlowe 1997; Cao et al. 1998). For tethering assays, vesicles that bud from the ER by the addition of COPII components are freely diffusible and remain in the supernatant fraction after centrifugation at 14,000 rpm; protease-protected glycosylated 35S-pro-α-factor (35S-gp-α-factor) contained in these vesicles was quantified after solubilization of membranes and precipitation with Concanavalin A–Sepharose. For transport assays, COPII proteins, in addition to purified Uso1p and LMA1, were added as indicated in the figure legends, and the amount of Golgi complex-modified 35S-gp-α-factor was measured by immunoprecipitation with anti-α1,6-mannose–specific antibodies. The data presented is the average of duplicate determinations and the error bars represent the range.
Partial Purification of the Sec34p/Sec35p Complex
The protease deficient RSY1157 strain was grown to late log phase (OD600 = 3.4) in 36 liters of YPD at 30°C, after which all manipulations were performed at 0–4°C. The cells were harvested by centrifugation and washed twice with water. The 334 g cell pellet was resuspended in 1 liter of 25 mM Tris-Cl, pH 8.0, 1 M KCl, 2 mM EGTA (lysis buffer) with protease inhibitors [0.5 mM 1:10 phenanthroline, 2 μM pepstatin A, 2 μg/ml aprotinin, 0.5 μg/ml leupeptin, 1 mM PMSF, 200 μM 4-(2-aminoethyl)-benzenesulfonyl fluoride (AEBSF)] and 1 mM DTT, and lysed in an EmulsiFlex-C5 (Avestin Inc.) at 18,000–20,000 psi. The lysate was centrifuged at 5,000 g (Sorvall SLA3000 rotor, 6,000 rpm, 10 min), and the supernatant (1.04 liter) was collected and centrifuged at 20,000 g (Sorvall SA600 rotor, 12,000 rpm, 20 min). The supernatant (960 ml) was removed, avoiding the loose pellet, and centrifuged at 175,000 g (Beckman 45Ti rotor, 44,000 rpm, 120 min). The supernatant (S175; 750 ml at 7.3 mg protein/ml) was removed, avoiding the pellets. The S175 was made 1 mM in EDTA, and (NH4)2SO4 was added to 35% saturation, dissolved, and the solution was stirred for 60 min. The (NH4)2SO4 precipitate was collected by centrifugation at 17,000 g (SLA3000 rotor, 10,000 rpm, 10 min), resuspended in enough 25 mM Tris-Cl, pH 8.0, 1 mM DTT to yield a conductivity equivalent to that of 25 mM Tris-Cl, pH 8.0, 100 mM KCl, 1 mM DTT (T8.0/100K/D). This material (1.55 g protein in 368 ml; 4.2 mg/ml) was loaded at 2 ml/min onto a 50 ml DEAE-Sepharose Fast Flow (Pharmacia Biotech, Inc.) column (2.5 cm i.d.) that had been equilibrated in T8.0/100K/D. The column was washed with 200 ml of T8.0/100K/D and eluted with a linear gradient (2 mM/ml) from T8.0/100K/D to T8.0/400K/D, collecting 10 ml fractions throughout. At this and each subsequent chromatographic step, fractions were analyzed for Sec34p and Sec35p content by immunoblotting. The fractions containing Sec34p and Sec35p, which eluted together at ∼160 mM KCl, were pooled (348 mg protein in 60 ml, 5.8 mg/ml). A 200 μl aliquot of this DEAE pool was chromatographed on a 24 ml Superose 6 (HR10/30 Pharmacia Biotech, Inc.) size exclusion column equilibrated in T8.0/150K/D at 0.3 ml/min, collecting 1 ml fractions. The remaining 60 ml of the DEAE pool was then concentrated to a volume of 17.8 ml (284 mg protein at 16 mg/ml) in an Amicon 8050 Ultrafiltration Cell (Millipore Inc.) with a YM100 (MWCO 100 kD) ultrafiltration membrane. The concentrated sample was loaded onto a 700 ml Sephacryl S-300 (Pharmacia Biotech, Inc.) column (2.5 cm i.d.) that had been equilibrated in 25 mM Tris-Cl, pH 7.6, 100 mM KCl, 1 mM DTT (T7.6/100K/D) and chromatographed at 1.5 ml/min in T7.6/100K/D, collecting 10 ml fractions. The fractions containing both Sec34p and Sec35p were pooled (66 mg protein in 47 ml, 1.4 mg/ml) and loaded onto an 8 ml MonoQ (HR 10/10, Pharmacia Biotech, Inc.) anion exchange column equilibrated in T7.6/100K/D at 1.5 ml/min. The column was washed with 36 ml of T7.6/100K/D and eluted with a linear gradient (2.5 mM/ml) from T7.6/100K/D to T7.6/500K/D, collecting 5 ml fractions. The fractions containing both Sec34p and Sec35p, which coeluted at ∼295 mM KCl, were pooled (16 mg protein in 18 ml, 0.89 mg/ml), dialyzed against 40 mM potassium phosphate, pH 6.8, 0 mM KCl, 1 mM DTT (KP/0K/D), and loaded onto a MonoS (HR 5/5, Pharmacia Biotech, Inc.) equilibrated in KP/0K/D at 0.5 ml/min. The column was washed with 4 ml of KP/0K/D and eluted with a linear KCl gradient (10 mM/ml) from KP/0K/D to KP/500K/D, collecting 2 ml fractions throughout. The fractions containing both Sec34p and Sec35p, which coeluted at ∼110 mM KCl, were pooled (1.5 mg protein in 8 ml, 0.19 mg/ml) and concentrated to a volume of 0.1 ml at 9.3 mg/ml with an Ultrafree BIOMAX centrifugal device (Millipore Inc.). The concentrated sample was loaded onto a 24 ml Superose 6 (HR10/30, Pharmacia Biotech, Inc.) size exclusion column that had been equilibrated in T7.6/200K/D, and chromatographed at 0.3 ml/min, collecting 0.5 ml fractions. After this point, protein concentration was no longer determined, to not decrease yield. The fractions containing Sec34p and Sec35p were pooled and loaded onto a Bio-Scale CHT2-I Hydroxyapatite column (Bio-Rad Laboratories). The column was washed with 8 ml 25 mM Tris-Cl, pH 8.0, 200 mM KCl, 0 mM potassium phosphate, 1 mM DTT (T7.6/200K/0Pi/D), and eluted with a linear potassium phosphate gradient (16.6 mM/ml) from T7.6/200K/0Pi/D to T7.6/200K/250Pi/D; Sec34p and Sec35p coeluted at ∼100 mM potassium phosphate.
Results
Cloning of SEC34
To clone SEC34, the sec34-2 strain was transformed with a low-copy (centromere, CEN) yeast genomic library and temperature-resistant colonies were selected at 38.5°C. Library plasmids were isolated from these colonies and retested for their ability to confer growth at 38.5°C. Four restored growth at the restrictive temperature to wild-type levels, and the remaining three yielded partial suppression. The ends of the inserts of the seven library plasmids were sequenced and found to contain overlapping regions of the right arm of chromosome V, a portion of which is shown in Fig. 1 a. The only complete ORF contained on each of the plasmids that conferred strong suppression of the temperature-sensitive phenotype (two of which are shown in Fig. 1b and Fig. c) was YER157w. Interestingly, the three plasmids that partially suppress the sec34-2 mutation contained identical inserts in which only the 5′ end of YER157w is present; the inability of these plasmids to fully suppress may be due to the absence of the COOH-terminal portion of the protein. The ORF YER157w was isolated from the genomic insert (as shown in Fig. 1 d) and transferred to a low-copy plasmid. This construct was demonstrated to suppress the temperature sensitivity of the sec34-2 strain, confirming that YER157w is responsible for the suppression conferred by each of the library plasmids. To address the possibility that YER157w was a suppressor of the sec34-2 mutation rather than the gene itself, integrative mapping was performed. A sec34-2 strain in which the YER157w locus was marked with LEU2 was constructed and subsequently mated to a wild-type strain. The resulting diploid strain was subjected to tetrad analysis and, of 38 tetrads examined, the temperature-sensitive phenotype did not segregate away from the marked locus. Thus, integrative mapping strongly suggests that YER157w is SEC34.
SEC34 is predicted to encode an 801-amino acid protein ( Fig. 1 f) with a molecular weight of 92.5 kD and a pI of 5.2. Sec34p lacks a signal sequence, as well as transmembrane domains or other motifs that could facilitate membrane attachment. Therefore, the protein is predicted to be either cytoplasmic or peripherally associated with membranes. Three putative orthologs of Sec34p have been detected. First, the C. elegans genome encodes a protein designated Y71F9A 290.A that is 25% identical and 35% similar to Sec34p. This 428-amino acid protein is ∼50% the size of Sec34p and is homologous to the NH2 terminus of Sec34p (spanning amino acid residues 111 to 459). Due to the disparity in size, it is unclear whether this protein is indeed an ortholog of Sec34p. Second, the genome of the fission yeast Schizosaccharomyces pombe contains a 735-amino acid protein (GenBank/EMBL/DDBJ accession number CAB51337, PID g5579050) that is 26% identical and 40% similar to Sec34p. Finally, several overlapping human expressed sequence tags (ESTs; GenBank/EMBL/DDBJ accession numbers AA280321, AA603511, AA429818, and z21241) have been isolated that show high similarity to Sec34p. Sequencing of clones containing the first two ESTs (provided by Genome Systems, Inc., St. Louis, MO) allowed us to analyze additional sequences previously unavailable in GenBank (data not shown). By combining our newly sequenced regions with the overlapping ESTs in the database, we obtained a 263-amino acid portion of the putative human protein. Comparison of this partial protein to Sec34p using the BLAST algorithm ( Altschul et al. 1990) revealed a 175-amino acid region of homology (encompassing amino acids 531 to 706 of Sec34p) between the human sequence and Sec34p. Within this region the two proteins are 25% identical and 50% similar. Since the 86-amino acid region 5′ to this homologous region does not display a high degree of similarity to Sec34p, the protein encoded by these ESTs may not be a true Sec34p homolog, but instead may contain a Sec34p-like domain.
The SEC34 Deletion Strain Displays a Severe Growth Defect
To evaluate the phenotype of a strain lacking SEC34, we constructed a diploid strain in which one allele of SEC34 had been deleted (as diagrammed in Fig. 1 e) and replaced with the gene LEU2. This strain (sec34Δ::LEU2/SEC34) was sporulated and dissected, and the resulting tetrads were incubated on rich media at 30°C. As shown in Fig. 2 (left), a clear 2+:2− segregation pattern was observed in which each tetrad contained two large and two very small colonies. The large colonies were without exception Leu− and thus contained the wild-type copy of the gene, while the small colonies were Leu+, indicating the presence of the sec34Δ locus. Therefore, although SEC34 is not an essential gene, haploid strains lacking SEC34 are at an extreme growth disadvantage. The growth defect of the sec34Δ strain is complemented by a plasmid bearing SEC34 since the presence of the construct in the diploid sec34Δ/SEC34 strain resulted in the restoration of a 4+:0− segregation pattern ( Fig. 2, right). In each tetrad, two segregants were Leu+ and Ura+, indicating the presence of both the deletion and the plasmid bearing SEC34, respectively.
Figure 2.

Deletion of SEC34 leads to a severe growth defect that can be suppressed by expression of YPT1 or SLY1-20. Left, The sec34Δ/SEC34 diploid strain (GWY127) was sporulated, dissected, and incubated on YPD at 30°C for six days. Ten representative tetrads are shown, each of which displays a 2+:2− segregation for size of colony. The small segregants in each tetrad are Leu+, indicating the presence of the SEC34 deletion. Right, The growth defect of the sec34Δ strain is complemented by SEC34 and suppressed by expression of YPT1 or SLY1-20. The sec34Δ/SEC34 diploid strain (GWY127) or containing plasmids pSEC34 (pSV22), pYPT1-2μm (pNB167), or pSLY1-20 (pSV11) was sporulated and subjected to tetrad analysis. Tetrads were incubated on YPD for six days at 30°C and two representative tetrads are shown for each.
Genetic Analysis of SEC34
Previous analysis of the sec34 mutant strain indicated a role for Sec34p in the docking or fusion of ER-derived vesicles with the cis-Golgi complex ( Wuestehube et al. 1996). By testing whether SEC34 displayed genetic interactions with other factors required at this stage, as well as at other stages of transport, we sought to confirm this assignment and to further explore the function of Sec34p. We thus tested whether the temperature-sensitive growth defect of the sec34-2 strain could be suppressed by overexpression of other genes required for transport. Two of the best suppressors were found to encode the rab protein Ypt1p and the dominant allele of the t-SNARE–associated protein, SLY1, termed SLY1-20 ( Fig. 3). Although neither gene could restore growth to the levels observed when the strain was complemented by SEC34 on a plasmid, both conferred a significant growth advantage to the mutant strain. A second gene, USO1, which encodes a tethering factor, also suppressed the temperature sensitivity, but to a lesser extent than either YPT1 or SLY1-20. Weak suppression was also observed upon overexpression of the v-SNAREs Sec22p, Bet1p, and Ykt6p, each of which has been implicated in the ER to Golgi complex stage of transport; no suppression was seen for another v-SNARE involved at this step, Bos1p ( Fig. 3).
Figure 3.

Suppression of the temperature-sensitive growth defect of the sec34-2 strain. Tenfold serial dilutions of a wild-type strain (RSY255) or the sec34-2 strain (GWY95) alone (---) or containing the indicated plasmids: pSEC34 (pSV22), pYPT1-2μm (pNB167), pSLY1-20 (pSV11), pRUD3-2μm (pSK81), pUSO1-2μm (pSK47), pSEC22-2μm (pJG103), pBET1-2μm (pSFN2), pYKT6-2μm (pSK60), pBOS1-2μm (pSK101), or vector (pRS426) were spotted onto YPD plates and incubated at 38.5°C for three days.
Several other genes were tested for their ability to suppress the sec34-2 mutation when overexpressed, but were found to have no effect. These genes encode the tethering factor Sec35p, the TRAPP complex component Bet3p, the cis-Golgi complex t-SNARE Sed5p, and the Golgi complex to plasma membrane rab protein Sec4p. The lack of suppression by the Golgi complex to plasma membrane rab indicates that suppression by the ER to Golgi complex rab is specific.
We also tested whether overexpression of SEC34 could suppress temperature-sensitive mutant alleles of several ER to Golgi complex docking factors. Interestingly, overexpression of SEC34 was capable of weakly suppressing the temperature-sensitive growth defect of the sec35-1 strain (data not shown). However, multicopy SEC34 was unable to suppress mutant alleles of all other secretory factors tested, including: the v-SNAREs Sec22p, Bos1p, and Bet1p; the t-SNARE Sed5p; the tethering factor Uso1p; the rab Ypt1p; and the TRAPP complex component Bet3p (data not shown). Therefore, overexpression of either SEC34 or SEC35 ( VanRheenen et al. 1998) is unable to suppress the temperature-sensitive growth defects of the majority of tethering and docking factors, yet multicopy SEC34 can improve the growth defect of a compromised allele of SEC35, and thus the two genes show a genetic interaction.
Since YPT1 and SLY1-20 were efficient suppressors of the temperature-sensitive growth defect of the sec34-2 strain, we explored whether they could also improve the severe growth defect of the sec34Δ strain. The sec34Δ/SEC34 diploid strain was therefore transformed with plasmids expressing either multicopy YPT1 or low-copy SLY1-20, and the resulting diploids were sporulated and subjected to tetrad analysis. Indeed, the presence of either plasmid significantly improved the growth of the sec34Δ strain ( Fig. 2, right). This result implies that Ypt1p and Sly1p most likely function downstream of, or in a parallel pathway with, Sec34p.
Isolation of RUD3 and its Genetic Interactions with SEC34 and USO1
Suppression of the sec34-2 temperature-sensitive growth defect was also observed upon overexpression of a gene designated RUD3 ( Fig. 3). RUD3 was isolated previously in our lab through a screen for genes that, when overexpressed, are able to suppress the temperature-sensitive growth defect of the uso1-1 strain ( Sapperstein et al. 1996; Sapperstein 1997). From a panel of multicopy suppressors, four library plasmids were isolated that contain overlapping regions of chromosome XV that include two hypothetical ORFs and one known gene. The suppressing activity was localized to the hypothetical ORF YOR216c, and this gene was named RUD3 because it relieves the uso1-1 transport defect. RUD3 is an efficient suppressor of the uso1-1 deficiency at 37.5°C, suppressing the growth defect ( Fig. 4 a), as well as the ER to Golgi complex transport block, as monitored through movement of the marker protein, carboxypeptidase Y (data not shown).
Figure 4.
Genetic interaction of USO1 and RUD3 and the predicted sequence of Rud3p. a, Suppression of the temperature-sensitive growth defect of the uso1-1 strain by overexpression of RUD3. The wild-type strain (RSY255) and the uso1-1 strain (GWY67), either alone (---) or containing the indicated plasmids: pUSO1-2μm (pSK47), pRUD3-2μm (pSK81), or vector (pRS426) were treated as described in Fig. 3, except that the strains were incubated at 37.5°C for two days. b, Predicted amino acid sequence of Rud3p with the two regions predicted to form a coiled-coil underlined. The nucleotide accession number for RUD3 is Z75124 and the protein accession number is S60943. c, Probability of a coiled-coil structure for Rud3p, as predicted by the PAIRCOIL program ( Berger et al. 1995).


RUD3 is predicted to encode a 484-amino acid protein with a molecular mass of 56.1 kD and a pI of 4.67 ( Fig. 4 b). Like Sec34p, this protein lacks any motifs to indicate a localization other than cytosolic. Both the PAIRCOIL and COILS programs ( Lupas et al. 1991; Berger et al. 1995) indicate a high probability that the protein assumes a coiled-coil secondary structure in a central, ∼215-amino acid stretch ( Fig. 4b and Fig. c). There is a region near the center of this coiled-coil domain, however, that has a decreased coiled-coil probability; this region could potentially be a hinge in the protein ( Lupas 1996). BLAST searches revealed a single putative homolog of Rud3p in the genome of S. pombe. This 401-amino acid protein (GenBank/EMBL/DDBJ accession number AL022117, PID g2959373) shares 28% identity and 41% similarity with Rud3p, and thus encodes a Rud3p ortholog that we designate spRud3p. Interestingly, spRud3p also contains a region predicted to form a coiled-coil secondary structure, although the homology between the proteins is not restricted to this motif. Indeed, the COOH termini of the proteins (ranging from amino acid 365 to 484 for Rud3p, and 280 to 401 for spRud3p) are most homologous, displaying 49% identity and 62% similarity.
To test whether Rud3p is encoded by an essential gene, a diploid strain was created in which one of the alleles of RUD3 was deleted and marked with LEU2. Sporulation and tetrad dissection of this strain yielded tetrads with four viable spores, two of which were Leu+. Upon incubation at 25, 30, and 37°C, the haploid rud3Δ strain did not display a significant growth defect as compared with a wild-type strain (data not shown), and thus RUD3 is not an essential gene.
Sec34p Is a Peripheral Membrane Protein
To analyze the Sec34 protein, we generated affinity-purified anti-Sec34p antibody. The antibody recognizes two proteins in crude yeast extracts, the larger of which corresponds to the predicted molecular weight of Sec34p ( Fig. 5 a). This protein is absent in a sec34Δ strain (which expressed SLY1-20 to enhance its propagation) and is overexpressed in a strain containing SEC34 on a multicopy plasmid, and thus represents Sec34p. The smaller protein, which is recognized despite the affinity purification, is not related to the Sec34p locus since it is present in the sec34Δ strain and its expression level is unaffected by overexpression of SEC34.
Figure 5.
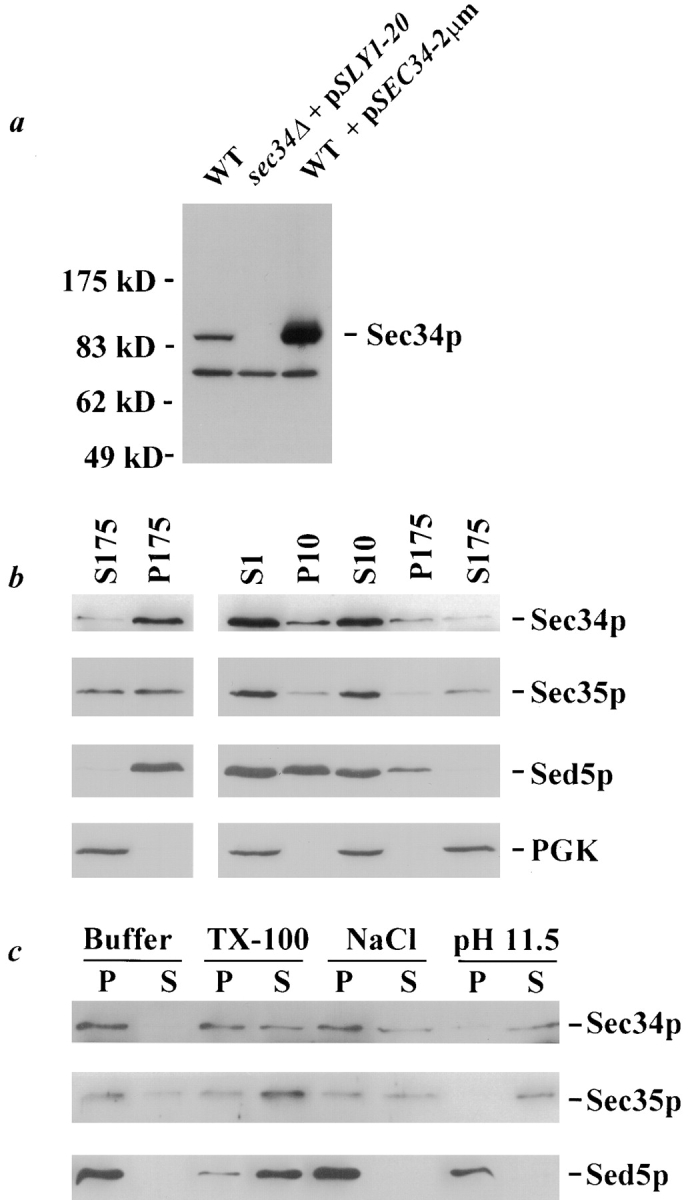
Biochemical characterization of Sec34p. a, Characterization of the anti-Sec34p antibody. Affinity-purified polyclonal antibodies against Sec34p were used to probe yeast lysate (derived from 0.2 OD units of cells per lane) from either a wild-type strain (RSY255), the sec34Δ strain expressing SLY1-20 to assist growth (GWY132), or a wild-type strain overexpressing SEC34 from a multicopy plasmid (RSY255 containing pSV25). Lysates were separated by SDS-7%PAGE and immunoblotted with affinity-purified antibody against Sec34p. The migration of molecular weight markers is indicated on the left. b, Subcellular fractionation of Sec34p. Left, Lysate (S1) from a wild-type strain (RSY255) was centrifuged at 175,000 g and separated into pellet (P) and supernatant (S) fractions. Right, S1 lysate was centrifuged at 10,000 g and separated into P10 and S10 fractions, and the S10 was further centrifuged at 175,000 g and separated into P175 and S175 fractions. c, Sec34p encodes a peripheral membrane protein. Isolated yeast membranes from a wild-type strain (RSY255) were incubated with buffer, 1% Triton X-100, 1 M NaCl, or 0.1 M Na2CO3, pH 11.5, as indicated, before centrifugation at 175,000 g and separation into pellet (P) and supernatant (S) fractions. b and c, Equivalent amounts (relative to the starting material) of each fraction were separated by SDS-12%PAGE and analyzed by immunoblotting with affinity-purified antibodies against Sec34p (only the upper, Sec34p-specific band is shown), Sec35p, Sed5p, and PGK, as indicated.
With this antibody, we investigated whether Sec34p was capable of associating with membranes, as might be expected for a protein involved in secretion. A crude yeast extract (designated S1) was centrifuged at 175,000 g to separate the organelles of the secretory pathway, which are found in the pellet fraction, from cytosolic proteins, which are contained in the supernatant fraction. As shown in Fig. 5 b (left), the majority of Sec34p is found in the pellet fraction, along with the integral membrane protein Sed5p, while a small amount of Sec34p is contained in the supernatant, as is the cytosolic marker, PGK. This result is consistent with a peripheral membrane association for Sec34p. To explore the basis for the sedimentation of Sec34p, we attempted to extract the protein from an enriched membrane fraction using buffers containing Triton X-100, NaCl, or Na2CO3, pH 11.5. As expected for an integral membrane protein, Sed5p was released into the supernatant fraction after incubation with buffer containing Triton X-100, but not after treatment with salt or high pH, whereas the peripheral membrane protein Sec35p was released, at least partially, upon incubation with all three buffers. Sec34p was partially shifted into the supernatant fraction upon treatment with Triton X-100, salt, or high pH, and thus behaves as a peripheral membrane protein ( Fig. 5 c).
To further analyze the membrane association of Sec34p, differential centrifugation was employed. The S1 fraction was centrifuged at 10,000 g, separated into supernatant (S10) and pellet (P10) fractions, and the S10 fraction was further centrifuged at 175,000 g and separated into supernatant (S175) and pellet (P175) fractions. Under these conditions, the ER is contained primarily into the P10 fraction (data not shown), the Golgi complex partitions between the P10 and P175 fractions, as seen with the Golgi protein Sed5p, and cytosolic proteins remain in the supernatant fractions, as observed for PGK ( Fig. 5 b, right). The soluble portion of Sec34p is found in the S175 fraction, while the membrane-associated pool partitions between the P10 and P175, similar to the Golgi complex protein Sed5p. The fractionation pattern of Sec34p is quite similar to that observed for the peripheral membrane protein Sec35p, although Sec34p appears to have a greater proportion of the protein in the membrane fractions.
Sec34p Is Required for Tethering of ER-derived Vesicles to the cis-Golgi Complex
Because the mutant phenotype ( Wuestehube et al. 1996) and suppression profile of sec34-2 is similar to that of other factors required for the tethering stage of ER to Golgi complex transport, we investigated whether Sec34p was required for tethering as well. We thus employed two in vitro assays that, together, are able to distinguish the stages of budding, tethering, and fusion in ER to Golgi complex transport ( Barlowe 1997; Cao et al. 1998).
In the first assay ( Fig. 6 a), overall ER to Golgi complex transport is measured in semi-intact cells incubated with a mixture of purified protein components that drive all the stages of transport: vesicle formation from the ER is supported by the addition of COPII proteins, efficient vesicle tethering requires added Uso1p, and vesicle fusion requires a protein complex termed LMA1. Productive transport is monitored by following the addition of α1,6-mannose residues to gp-α-factor, an event that occurs in the cis-Golgi complex. Generation of this transport system from conditional mutants has shown that the system also requires the activities of several peripheral and integral membrane proteins including Ypt1p, Sec35p, and the SNAREs ( Cao et al. 1998; VanRheenen et al. 1998). Although there is no requirement for exogenous Sec34p in the system, Sec34p may be supplied to the assay by peripheral association with membranes of the semi-intact cells. Therefore, to test for a requirement for Sec34p, we generated the in vitro system from the sec34-2 strain. In wild-type or sec34-2 mutant cells at 23°C, movement of gp-α-factor from the ER to the Golgi complex in this in vitro system proceeds with a similar efficiency ( Fig. 6 a). In contrast, at 29°C in sec34-2 semi-intact cells, this process is very inefficient relative to wild-type, indicating that sec34-2 is defective for overall ER to Golgi transport in vitro.
Figure 6.

Sec34p is required for the tethering phase of ER to Golgi complex transport in vitro. a, Sec34p is required for in vitro transport. Semi-intact cells isolated from wild-type (left) and sec34-2 (right) strains were incubated at 23 or 29°C for 60 min. Reactions contained an ATP regeneration system alone (No addition, solid bars) or COPII, Uso1p, and LMA1 (Reconstituted, open bars). α1,6-mannose-modified forms of 35S-gp-α-factor were immunoprecipitated with α1,6-mannose-specific antibodies and the percent transport reflects the amount of total 35S-gp-α-factor (determined from precipitation with Concanavalin A) that obtained Golgi complex-specific outer chain modifications. b, Sec35p is required for vesicle tethering. Semi-intact cells prepared from wild-type (left) and sec34-2 (right) strains were incubated at 23 or 29°C for 30 min. Reactions contained an ATP regenerating system alone (No addition, solid bars), COPII (open bars), or COPII and Uso1p (hatched bars). Freely diffusible vesicles containing 35S-gp-α-factor were separated from semi-intact membranes by centrifugation at 18,000 g for 3 min. The percent diffusible vesicles reflects the fraction of protease-protected, Concanavalin A-precipitable 35S-gp-α-factor present in the supernatant.
The second assay we employed examined the functionality of the vesicle budding and vesicle tethering steps in the sec34-2 mutant ( Fig. 6 b). In this assay, release of vesicles from semi-intact cells is detected by the appearance of protease-protected gp-α-factor in a low-speed supernatant at the end of the reaction. Vesicles were efficiently generated upon addition of COPII components to wild-type or sec34-2 semi-intact cells at 23 or 29°C ( Fig. 6 b). These data indicate that Sec34p is not required for ER-derived vesicle budding. Because addition of Uso1p significantly reduced vesicle release, vesicle tethering was also functional in the semi-intact wild-type cells at 23 or 29°C, as well as in sec34-2 semi-intact cells at 23°C. In contrast, when Uso1p was added to the sec34-2-derived system at the restrictive temperature of 29°C, vesicle release was only slightly diminished ( Fig. 6 b). This result indicates that the sec34-2 mutant cannot efficiently tether ER-derived vesicles to the yeast Golgi complex.
sec34 and sec35 Display a Synthetic Lethal Interaction
The similar genetic interactions of SEC34 and SEC35 with genes involved in the docking stage of vesicular transport, taken together with their genetic interaction with one another, and with the finding that both proteins function in vesicle tethering, lead us to examine whether mutations in the two genes would display a synthetic lethal interaction. To do this, we generated a diploid strain heterozygous for both the sec34-2 and sec35-1 alleles and subjected it to tetrad analysis. Although both the sec34-2 and sec35-1 haploid strains are permissive for growth at both 21 and 30°C, tetrads from the diploid sec34-2/SEC34 SEC35/sec35-1 strain yielded numerous inviable colonies at either temperature ( Fig. 7 a). After incubation of the segregants for long periods of time, a small proportion of those previously characterized as inviable would form visible microcolonies. Since this phenotype was variable, we hypothesize that the microcolonies result from either the appearance of spontaneous suppressors of the inviability or from background mutations in the strain. Examination of the viable segregants in each tetrad for temperature sensitivity revealed a pattern in which the inviable segregants are predicted to be those containing both the sec34-2 and the sec35-1 alleles. To confirm this prediction, the diploid strain was transformed with low-copy plasmids bearing either SEC34 or SEC35 before tetrad dissection. The presence of either plasmid lead to a greater proportion of viable segregants than was observed for the untransformed strain, concurrent with the appearance of segregants that were sensitive to the drug 5-fluoro-orotic acid, which is toxic to cells that must maintain the plasmid to survive (data not shown). Therefore, the sec34-2 and sec35-1 alleles display a synthetic lethal phenotype, which can be complemented by the presence of either gene on a plasmid.
Figure 7.

Genetic and biochemical interactions between Sec34p and Sec35p. a, The sec34-2 and sec35-1 alleles display a synthetic lethal interaction. A diploid strain resulting from the mating of GWY95 (sec34-2) to GWY93 (sec35-1) was sporulated, dissected, and incubated on YPD at 30°C for three days. Ten representative tetrads are shown. b, Interaction of Sec34p and Sec35p by two-hybrid analysis. A two-hybrid strain (PJ69-4A) was transformed with plasmids expressing fusion proteins with the Gal4p-BD (either Gal4p-BD-Sec34p, pSV37; Gal4p-BD-Sec35p, pSV34; or Gal4p-BD-p53, pVA3) and the Gal4p-AD (either Gal4p-AD-Sec34p, pSV35; Gal4p-AD-Sec35p, pSV36; or Gal4p-AD-large T-antigen, pTD3) or with plasmids expressing the BD or AD alone (Gal4p-BD, pGAD424 and Gal4p-AD, pAS2, indicated by --), as shown on the left of the figure. Strains were grown to stationary phase in SC-Trp-Leu and analyzed as described in Fig. 3 with the following exceptions. The diluted samples were spotted onto either SC lacking adenine, leucine, and tryptophan (SC-Ade), or SC lacking histidine, leucine, and tryptophan, but containing 2.5 mM 3-aminotriazole (SC-His), and the plates were incubated at 30°C for three days.
Sec34p and Sec35p Interact in the Two-hybrid Assay
In many cases, synthetic lethality between alleles of two genes involved in secretion indicates that their gene products are involved in the same stage of secretion ( Kaiser and Schekman 1990). Furthermore, such genetic interactions can also be indicative of a physical interaction of the proteins encoded by those genes, as is the case for mutations in the α and β subunits of tubulin ( Huffaker et al. 1987). Therefore, we investigated whether Sec34p and Sec35p physically interact using the two-hybrid system ( Fields and Song 1989). A strain in which transcription of both the ADE2 and HIS3 genes is under the control of the Gal4p transcriptional activator ( James et al. 1996) was transformed with plasmids expressing either the Gal4p-BD or Gal4p-AD, either alone or fused to Sec34p or Sec35p. Activation of transcription of the ADE2 and HIS3 genes was assessed by the ability of the strain to grow on media lacking adenine or histidine, respectively. As an example of a positive two-hybrid interaction, coexpression of p53 fused to the Gal4p-BD and the large T-antigen fused to the Gal4p-AD was demonstrated to activate both reporter genes ( Fig. 7 b), as has been described previously ( Iwabuchi et al. 1993). Strains expressing either the Sec34p-Gal4p-BD or the Sec35p-Gal4p-AD fusion protein were unable to grow on either media. However, when Sec34p-Gal4p-BD was expressed along with the Sec35p-Gal4p-AD, the strain grew well on media lacking either adenine or histidine ( Fig. 7 b), indicating that the interaction of Sec34p with Sec35p was able to localize the Gal4p-AD to the promoters of these genes, activating transcription. When the converse experiment was completed, in which Sec35p was fused to the Gal4p-BD and Sec34p was fused to the Gal4p-AD, expression of the ADE2 and HIS3 genes was also observed only when both fusion proteins were expressed. Thus, Sec34p and Sec35p interact.
Sec34p and Sec35p Are Components of a Large Protein Complex
To characterize the interaction of Sec34p and Sec35p further, we employed immunoblotting to monitor the behavior of these proteins during fractionation of yeast cytosol. Sec34p and Sec35p coprecipitated in 35% saturated ammonium sulfate and cofractionated precisely by DEAE anion exchange chromatography (data not shown). An aliquot of the Sec34p/Sec35p anion exchange pool was then subjected to size exclusion chromatography on Superose 6 ( Fig. 8 a), and once again, Sec34p and Sec35p precisely cofractionate. Interestingly, they elute from the column slightly before thyroglobulin, a 669-kD globular protein. These results are consistent with Sec34p and Sec35p existing in a large protein complex with a mass of up to ∼750 kD. A small amount of monomeric Sec35p is also evident upon gel filtration, suggesting either that some Sec35p has dissociated from the complex or that a cytosolic pool of monomeric Sec35p exists. No such monomeric Sec34p has been detected in cytosolic fractions. To further purify the Sec34p/Sec35p complex, the remainder of the DEAE anion exchange pool was subjected to several sequential chromatographic steps (see Materials and Methods), including Sephacryl S-300 gel filtration (data not shown), MonoQ anion exchange ( Fig. 8 b), MonoS cation exchange ( Fig. 8 c), Superose 6 gel filtration (data not shown), and ceramic hydroxyapatite ( Fig. 8 d). Once again, Sec34p and Sec35p precisely comigrate through each step, strongly indicating that Sec34p and Sec35p are present in a large protein complex.
Figure 8.
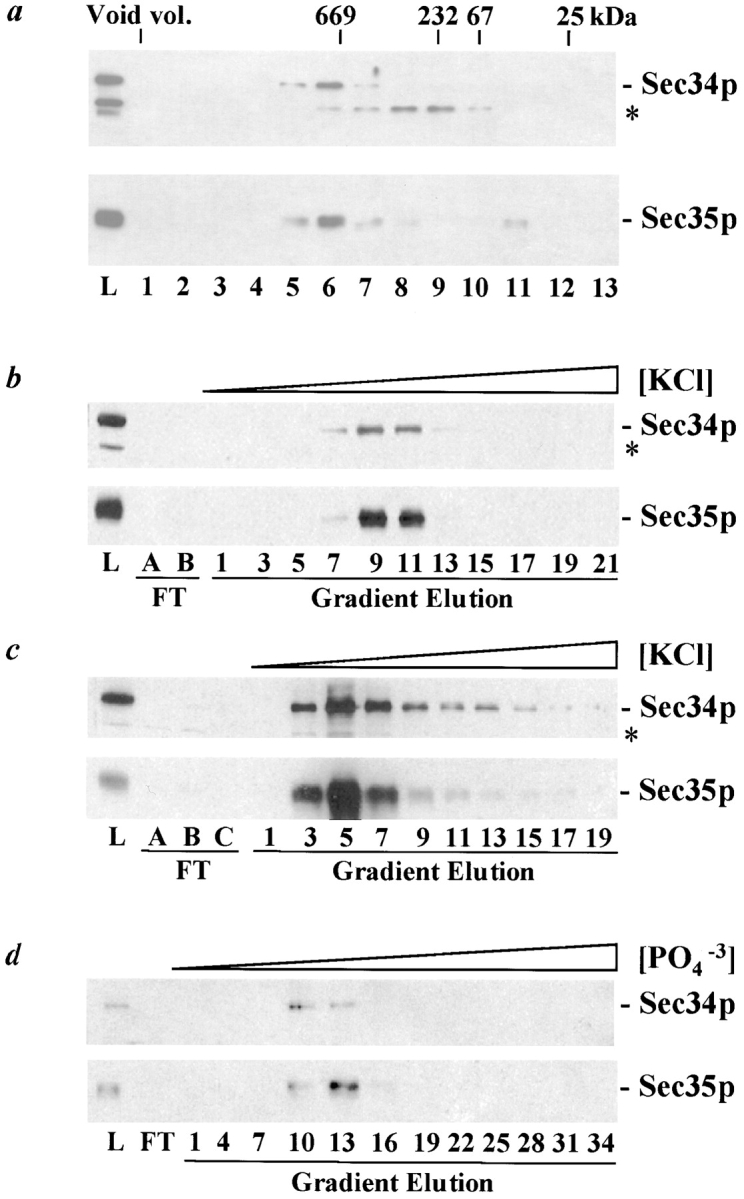
Sec34p and Sec35p are components of a large protein complex. a, Sec34p and Sec35p cofractionate by Superose 6 size-exclusion chromatography. Pooled fractions from a DEAE anion exchange column containing Sec34p and Sec35p were fractionated by size-exclusion chromatography. Proteins contained in the load (L), as well as the column, fractions were separated by SDS-11%PAGE and immunoblotted with antibodies against Sec34p or Sec35p, as indicated. The immunoreactive protein marked with an asterisk (*) is unrelated to Sec34p (see Fig. 5 a). The elution position of 25, 67, 232, and 669 kD protein standards, as well as the location of the void volume, is indicated at the top. b–d, Sec34p and Sec35p cofractionate through sequential chromatographic steps. b, Sec34p and Sec35p cofractionate by MonoQ anion exchange chromatography. The remainder of the DEAE anion exchange pool containing Sec34p and Sec35p was subjected to Sephacryl S-300 gel filtration chromatography, and the pooled Sec34p- and Sec35p-containing fractions were loaded onto a MonoQ anion exchange column and eluted with a linear KCl gradient. Proteins contained in the load (L), flow-through fractions (FT), and every second gradient elution fraction were analyzed as described in a. c, Sec34p and Sec35p cofractionate by MonoS cation exchange chromatography. Fractions 8–11 from the MonoQ step described in b were loaded onto a MonoS anion exchange column and eluted with a linear KCl gradient. Proteins contained in the load (L), flow-through fractions (FT), and every second gradient elution fraction were analyzed as described in a. d, Sec34p and Sec35p cofractionate on ceramic hydroxyapatite. Fractions 4–7 from the MonoS chromatographic step described in c were subjected to Superose 6 gel filtration chromatography and the Sec34p- and Sec35-containing fractions were pooled, loaded onto a ceramic hydroxyapatite column, and eluted with a linear potassium phosphate (PO4 −3) gradient. Proteins in the load (L), flow-through (FT), and every third gradient elution fraction were analyzed as described in a. The apparent difference in Sec34p and Sec35p abundance in fractions 10 and 13 is likely an artifact of chemiluminescent detection because reanalysis of every fraction in this region yields precise comigration and relative abundance (data not shown).
Discussion
Much effort has been extended towards gaining an understanding of the mechanism of transport vesicle docking in the secretory pathway. Several families of proteins are involved in this event, including the rab family of small GTP-binding proteins and the SNARE family of integral membrane proteins. Recently, another class of proteins has been described, the tethering factors. Although these proteins do not display homology with one another and thus do not define a family, they share a similar function in docking, that of connecting the vesicle to the target compartment before the interaction of v- and t-SNAREs (for reviews see Pfeffer 1999; Waters and Pfeffer 1999). The docking event can therefore be separated into two distinct substages, tethering and SNARE-dependent docking.
A recent genetic screen identified temperature-sensitive alleles of two genes, SEC34 and SEC35, that, when incubated at the restrictive temperature, are defective in ER to Golgi complex transport and accumulate large numbers of vesicles ( Wuestehube et al. 1996). Mutant alleles of these genes are also able to be suppressed by the dominant allele of SLY1, SLY1-20, a trait shared with all previously characterized ER to Golgi complex tethering factors ( Sapperstein et al. 1996; Cao et al. 1998; VanRheenen et al. 1998). Based on these data, we hypothesized that SEC34 and SEC35 might be involved in tethering, and this was demonstrated to be the case for SEC35 by the discovery that this gene both displayed a genetic interaction with genes involved in tethering and is required in this process as revealed by an in vitro assay ( VanRheenen et al. 1998). We therefore investigated whether Sec34p functions in tethering as well.
To begin our study of SEC34, we cloned the gene by complementation of the temperature-sensitive phenotype of a strain bearing the sec34-2 mutation. SEC34 was discovered to be a novel gene encoding a protein with a predicted molecular weight of 93 kD. Deletion of SEC34 in a haploid strain resulted in a severe growth defect, and thus SEC34 is essential for wild-type growth rates, although not for viability.
To investigate the genetic interactions of SEC34 we employed multicopy suppressor analysis. The best suppression of the sec34-2 temperature-sensitive growth defect was conferred by overexpression of Ypt1p, the rab required in ER to Golgi complex transport, or by expression of Sly1-20p, the dominant form of the t-SNARE–associated factor, Sly1p. Suppression of the SEC34 deletion strain allowed us to order the action of Sec34p with respect to Ypt1p and Sly1p. Since either YPT1 or SLY1-20 can suppress both mutations in, and a deletion of, SEC34, yet overexpression of SEC34 cannot suppress mutations in either YPT1 or SLY1, we hypothesize that Ypt1p and Sly1p function downstream of Sec34p.
Weaker suppression of the sec34-2 mutation was observed upon overexpression of the tethering factor Uso1p, or the v-SNAREs Sec22p, Bet1p, or Ykt6p. The suppression of sec34-2 by the v-SNAREs may be through mass action, in which vesicles containing supernumerary v-SNAREs are able to compensate for a deficiency in tethering, albeit with a very low efficiency. This phenomena has been observed previously for mutations in the tethering factors Uso1p ( Sapperstein et al. 1996) and Sec35p ( VanRheenen et al. 1998), as well as components of the putative tethering complex TRAPP ( Jiang et al. 1998). Interestingly, no suppression of the sec34-2 mutation was observed upon overexpression of either the v-SNARE Bos1p or the cis-Golgi complex t-SNARE Sed5p. Overexpression of Bos1p was also unable to suppress a temperature-sensitive growth defect of the sec35-1 strain ( VanRheenen et al. 1998) or the inviability of the uso1Δ strain ( Sapperstein et al. 1996). This lack of suppression could result from inefficient expression of the gene or may indicate a functional difference between Bos1p and the other v-SNAREs that mediate the ER to Golgi complex transport step. The lack of suppression of the sec34-2 mutant strain by high-copy expression of Sed5p was not unexpected, because it has been demonstrated that overexpression of this t-SNARE is toxic to cells ( Hardwick and Pelham 1992).
Biochemical analysis of the Sec34 protein reveals that it is a peripheral membrane protein. Although a small amount of the protein is soluble, the remainder partitions between the P10 and P175 fractions, similar to the Golgi protein Sed5p; it is possible, therefore, that Sec34p is associated with the Golgi complex. Due to the association of Sec34p with membranes, we used semi-intact cells made from the sec34-2 strain to test the requirement for Sec34p in tethering through an assay that reconstitutes ER to Golgi complex transport. These semi-intact cells were demonstrated to be able to bud vesicles from the ER, but these vesicles failed to efficiently tether to the Golgi complex at the restrictive temperature, indicating that Sec34p is required for the tethering of ER-derived vesicles to the cis-Golgi complex. Since cytosolic proteins are removed from the sec34-2 semi-intact cells, the membrane-associated pool of Sec34-2p is most likely the source of the tethering defect. In addition, since the membranes involved in tethering are restricted to those of the vesicle and the cis-Golgi complex, Sec34p is most likely associating with one, or both, of these membranes.
SEC34 was found to display two interesting genetic interactions with the tethering factor gene SEC35. First, multicopy SEC34 weakly suppresses a temperature-sensitive allele of SEC35. Since overexpression of SEC34 cannot suppress the cold-sensitive lethality of the sec35Δ strain, Sec34p is able to assist a handicapped allele of SEC35, but cannot replace its function. Second, the sec34-2 and sec35-1 alleles display a synthetic lethal interaction. Although strains bearing either allele alone are permissive for growth at 23 and 30°C, a haploid strain containing both the sec34-2 and sec35-1 alleles is inviable at either temperature. This synthetic phenotype is more severe than the conditional synthetic lethality of the sec35-1 allele in combination with a mutant allele of either YPT1 or USO1, in which the double mutants are viable at 23°C, but not at 30°C ( VanRheenen et al. 1998). This finding suggests a close functional interaction of the Sec34 and Sec35 proteins.
Based on these results, we investigated whether Sec34p and Sec35p could physically interact through the two-hybrid assay. Indeed, Sec34p and Sec35p were found to interact. The interaction between the two proteins may explain the ability of multicopy SEC34 to suppress the sec35-1 allele, but not the sec35Δ allele: increased levels of Sec34p could stabilize a defective form of Sec35p but would be ineffectual in the absence of Sec35p, especially if the interaction of the two proteins is essential to their function in tethering. To further explore the interaction of Sec34p and Sec35p we examined the behavior of the soluble pool of these proteins through several chromatographic steps. The proteins cofractionated through ammonium sulfate precipitation and anion exchange, cation exchange, ceramic hydroxyapatite, and size exclusion chromatographic steps, providing strong evidence that the two proteins are in a complex with one another. Intriguingly, the Sec34p/Sec35p complex appears quite large, with an estimated molecular weight (if globular) of ∼750 kD. This size, which is larger than the combined molecular weights of the two proteins (124 kD), suggests several possibilities for the structure of the complex. First, the complex could be homodimeric, containing one molecule of each protein, but highly elongated such that it migrates rapidly through a size exclusion column. We consider this unlikely because the sequences of Sec34p and Sec35p lack motifs (such as coiled-coil domains) that would be indicative of an elongated structure. Second, the complex could contain two or more molecules of at least one protein, resulting in a more massive structure. Finally, the complex could be multimeric, containing heretofore unidentified component(s) in addition to Sec34p and Sec35p. We are currently purifying the Sec34p/Sec35p complex to address this issue and identify any additional components. It appears, however, that Uso1p is unlikely to be a component of the Sec34p/Sec35p complex since immunoblotting fractions from the purification with an antibody against this protein revealed that Uso1p did not comigrate with Sec34p and Sec35p (data not shown).
The 750-kD complex containing Sec34p and Sec35p is reminiscent of the TRAPP complex, which migrates at ∼800 kD by size exclusion chromatography ( Sacher et al. 1998). However, two pieces of data indicate that the TRAPP complex is distinct from the Sec34p/Sec35p complex. First, the identities of the low molecular weight members of the TRAPP complex have been elucidated, and none corresponds to Sec35p, whose mobility on SDS-PAGE was within the range of the proteins that have been sequenced ( Sacher et al. 1998). In addition, the known members of the TRAPP complex display genetic interactions with one another ( Jiang et al. 1998; Sacher et al. 1998), yet no interaction was discerned between the gene encoding the TRAPP component Bet3p and either SEC34 or SEC35 (this work and VanRheenen et al. 1998).
Since many secretory factors are evolutionarily conserved, we explored whether the components of the Sec34p/Sec35p complex were conserved in higher eukaryotes. The genome of the nematode C. elegans was discovered to contain a protein designated Y71F9A 290.A that is very similar to Sec34p. However, the C. elegans protein is ∼50% the size of Sec34p and therefore may not be a true ortholog. We also discovered a C. elegans protein with moderate homology to Sec35p (22% identical and 33% similar), designated C35A5.6. While the similarity is not high, the proteins are similar in size (C35A5.6 is comprised of 273 amino acid residues, whereas Sec35p is comprised of 275 amino acid residues), and thus, this C. elegans protein is a putative ortholog of Sec35p. Searches of GenBank for additional homologs of these proteins did not reveal additional Sec35p homologs, but several human ESTs were discovered with a high degree of similarity to Sec34p. Interestingly, the sequences contained on these ESTs were homologous to Sec34p over only a portion of the analyzed region of the putative human protein, and thus the protein may contain a Sec34p-like domain and may not be a true Sec34p ortholog. These data indicate that there may be orthologs of the Sec34p/Sec35p complex in higher organisms, but functional experiments will be required to unambiguously address this point. Finally, a putative ortholog of Sec34p was discovered in the genome of S. pombe. No paralogs of either Sec34p or Sec35p exist in S. cerevisiae, and thus these proteins do not define a family of related proteins.
Finally, we describe the identification and characterization of a gene designated RUD3 that displays a genetic interaction with SEC34. RUD3, which encodes a novel nonessential protein with a predicted molecular weight of 56 kD, was originally identified in a screen for multicopy suppressors of a temperature-sensitive allele of the tethering factor, USO1 ( Sapperstein et al. 1996; Sapperstein 1997), and is also able to suppress the temperature-sensitive growth defect of the sec34-2 strain. Interestingly, RUD3 is unable to suppress mutations in other ER to Golgi complex docking factors such as Sec35p, Ypt1p, Sec22p, Bet1p, or Bos1p, and thus the suppression is specific to mutant alleles of SEC34 and USO1. Overexpression of RUD3 can weakly suppress the inviability of the uso1Δ strain (data not shown). Taken together, these data suggest that Rud3p either acts at, or downstream of, the tethering stage of ER to Golgi complex transport. Rud3p does not appear to be a component of the Sec34p/Sec35p complex since the majority of the protein fractionates away from the complex during its purification (data not shown).
In summary, we describe the characterization of a novel secretory factor, Sec34p, and its role in tethering of ER-derived vesicles to the cis-Golgi complex. Unlike the SNAREs and rabs, the tethering factors described thus far at different intracellular transport steps are not members of a protein family. Nevertheless, they do share structural similarity, since they are either elongated or present in a large multimeric complex ( Pfeffer 1999; Waters and Pfeffer 1999). The large size may be related to the requirement for the tethering factors to span the distance between the vesicle and the target compartment, before trans-SNARE complex formation. Interestingly, three factors meet this criteria in the yeast ER to Golgi complex transport step: the extended homodimer Uso1p, the TRAPP complex, and the Sec34p/Sec35p complex. It will be very exciting to discover in the future how these large protein complexes function to secure a vesicle to its target membrane, and whether their function is more complex than simply connecting vesicle and target membranes.
Acknowledgments
We thank Randy Schekman for the original sec34 and sec35 strains. We are grateful to S. Ferro-Novick, P. James, P. Novick, H. Pelham, M. Rose, R. Schekman, and members of their laboratories for generously supplying reagents and strains, and D. Hasara for expert assistance in antibody production. We thank Bryan Kraynack, Barbara Reilly, and Misha Rosenbach for assistance with the two-hybrid assay.
This work was supported by grants from the American Cancer Society (RPG-98-050-01-CSM to M.G. Waters), the National Institutes of Health (GM52549 to C. Barlowe), the Pew Scholars Program in the Biomedical Science (C. Barlowe), and a fellowship from the Lucille P. Markey Charitable Trust (M.G. Waters). S.M. VanRheenen and E.C. Chiang were supported by a National Institutes of Health training grant (GM07312) and S.K. Sapperstein was supported by an American Heart Association predoctoral fellowship.
Footnotes
Stephanie K. Sapperstein's present address is Department of Neurobiology, Stanford University Medical Center, Stanford, CA 94305.
Vladimir V. Lupashin's present address is Department of Physiology and Biophysics, University of Arkansas for Medical Sciences, Little Rock, AR 72205.
Abbreviations used in this paper: CEN, centromere; Gal4p-AD, Gal4p transcriptional activation domain; Gal4p-BD, Gal4p DNA-binding domain; gp-α-factor, glycosylated pro-α-factor; GST, glutathione S-transferase; ORF, open reading frame; PGK, phosphoglycerate kinase; SNARE, soluble N-ethylmaleimide sensitive fusion protein attachment protein receptor; t-SNAREs, SNAREs found predominantly on target membranes; v-SNAREs, SNAREs found predominantly on vesicles.
References
- Altschul S.F., Gish W., Miller W., Myers E.W., Lipman D.J. Basic local alignment search tool. J. Mol. Biol. 1990;215:403–410 . doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- Baker D., Hicke L., Rexach M., Schleyer M., Schekman R. Reconstitution of SEC gene product-dependent intercompartmental protein transport. Cell. 1988;54:335–344 . doi: 10.1016/0092-8674(88)90196-1. [DOI] [PubMed] [Google Scholar]
- Barlowe C. Coupled ER to Golgi transport reconstituted with purified cytosolic components. J. Cell Biol. 1997;139:1097–1108 . doi: 10.1083/jcb.139.5.1097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berger B., Wilson D.B., Wolf E., Tonchev T., Milla M., Kim P.S. Predicting coiled coils by use of pairwise residue correlations. Proc. Natl. Acad. Sci. USA. 1995;92:8259–8263 . doi: 10.1073/pnas.92.18.8259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brennwald P., Novick P. Interactions of three domains distinguishing the Ras-related GTP-binding proteins Ypt1 and Sec4. Nature. 1993;362:560–563 . doi: 10.1038/362560a0. [DOI] [PubMed] [Google Scholar]
- Cao X., Ballew N., Barlowe C. Initial docking of ER-derived vesicles requires Uso1p and Ypt1p but is independent of SNARE proteins. EMBO (Eur. Mol. Biol. Organ.) J. 1998;17:2156–2165 . doi: 10.1093/emboj/17.8.2156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dascher C., Ossig R., Gallwitz D., Schmitt H.D. Identification and structure of four yeast genes (SLY) that are able to suppress the functional loss of YPT1, a member of the RAS superfamily. Mol. Cell. Biol. 1991;11:872–885 . doi: 10.1128/mcb.11.2.872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunn B., Stearns T., Botstein D. Specificity domains distinguish the Ras-related GTPases Ypt1 and Sec4. Nature. 1993;362:563–565 . doi: 10.1038/362563a0. [DOI] [PubMed] [Google Scholar]
- Elble R. A simple and efficient procedure for transformation of yeasts. BioTechniques. 1992;13:18–20 . [PubMed] [Google Scholar]
- Fasshauer D., Antonin W., Margittai M., Pabst S., Jahn R. Mixed and non-cognate SNARE complexes. Characterization of assembly and biophysical properties. J. Biol. Chem. 1999;274:15440–15446 . doi: 10.1074/jbc.274.22.15440. [DOI] [PubMed] [Google Scholar]
- Fields S., Song O. A novel genetic system to detect protein–protein interactions. Nature. 1989;340:245–246 . doi: 10.1038/340245a0. [DOI] [PubMed] [Google Scholar]
- Fischer von Mollard G., Stevens T.H. The Saccharomyces cerevisiae v-SNARE Vti1p is required for multiple membrane transport pathways to the vacuole. Mol. Biol. Cell. 1999;10:1719–1732 . doi: 10.1091/mbc.10.6.1719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischer von Mollard G., Nothwehr S.F., Stevens T.H. The yeast v-SNARE Vti1p mediates two vesicle transport pathways through interactions with the t-SNAREs Sed5p and Pep12p. J. Cell Biol. 1997;137:1511–1524 . doi: 10.1083/jcb.137.7.1511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanahan D. Studies on transformation of Escherichia coli with plasmids. J. Mol. Biol. 1983;166:557–580 . doi: 10.1016/s0022-2836(83)80284-8. [DOI] [PubMed] [Google Scholar]
- Hardwick K.G., Pelham H.R. SED5 encodes a 39-kD integral membrane protein required for vesicular transport between the ER and the Golgi complex. J. Cell Biol. 1992;119:513–521 . doi: 10.1083/jcb.119.3.513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harlow E., Lane D. Antibodies. A Laboratory Manual. Cold Spring Harbor Laboratory Press; Cold Spring Harbor, NY : 1988. [Google Scholar]
- Holthuis J.C., Nichols B.J., Dhruvakumar S., Pelham H.R. Two syntaxin homologues in the TGN/endosomal system of yeast. EMBO (Eur. Mol. Biol. Organ.) J. 1998;17:113–126 . doi: 10.1093/emboj/17.1.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huffaker T.C., Hoyt M.A., Botstein D. Genetic analysis of the yeast cytoskeleton. Ann. Rev. Gen. 1987;21:259–284 . doi: 10.1146/annurev.ge.21.120187.001355. [DOI] [PubMed] [Google Scholar]
- Iwabuchi K., Li B., Bartel P., Fields S. Use of the two-hybrid system to identify the domain of p53 involved in oligomerization. Oncogene. 1993;8:1693–1696 . [PubMed] [Google Scholar]
- James P., Halladay J., Craig E.A. Genomic libraries and a host strain designed for highly efficient two-hybrid selection in yeast. Genetics. 1996;144:1425–1436 . doi: 10.1093/genetics/144.4.1425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang Y., Scarpa A., Zhang L., Stone S., Feliciano E., Ferro-Novick S. A high copy suppressor screen reveals genetic interactions between BET3 and a new gene. Evidence for a novel complex in ER-to-Golgi transport. Genetics. 1998;149:833–841 . doi: 10.1093/genetics/149.2.833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaiser C.A., Schekman R. Distinct sets of SEC genes govern transport vesicle formation and fusion early in the secretory pathway. Cell. 1990;61:723–733 . doi: 10.1016/0092-8674(90)90483-u. [DOI] [PubMed] [Google Scholar]
- Lowe M., Nakamura N., Warren G. Golgi division and membrane traffic. Trends Cell Biol. 1998;8:40–44 . doi: 10.1016/s0962-8924(97)01189-6. [DOI] [PubMed] [Google Scholar]
- Lupas A. Coiled coilsnew structures and new functions. Trends Biochem. Sci. 1996;21:375–382 . [PubMed] [Google Scholar]
- Lupas A., Van Dyke M., Stock J. Predicting coiled coils from protein sequences. Science. 1991;252:1162–1164 . doi: 10.1126/science.252.5009.1162. [DOI] [PubMed] [Google Scholar]
- Lupashin V.V., Pokrovskaya I.D., McNew J.A., Waters M.G. Characterization of a novel yeast SNARE protein implicated in Golgi retrograde traffic. Mol. Biol. Cell. 1997;8:2659–2676 . doi: 10.1091/mbc.8.12.2659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McNew J.A., Søgaard M., Lampen N.M., Machida S., Ye R.R., Lacomis L., Tempst P., Rothman J.E., Söllner T.H. Ykt6p, a prenylated SNARE essential for ER–Golgi transport. J. Biol. Chem. 1997;272:17776–17783 . doi: 10.1074/jbc.272.28.17776. [DOI] [PubMed] [Google Scholar]
- Miller J.H. Experiments in Molecular Genetics. Cold Spring Harbor Press; Cold Spring Harbor, NY : 1972. [Google Scholar]
- Nakajima H., Hirata A., Ogawa Y., Yonehara T., Yoda K., Yamasaki M. A cytoskeleton-related gene, uso1, is required for intracellular protein transport in Saccharomyces cerevisiae . J. Cell Biol. 1991;113:245–260 . doi: 10.1083/jcb.113.2.245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newman A.P., Ferro-Novick S. Characterization of new mutants in the early part of the yeast secretory pathway isolated by a [3H]mannose suicide selection. J. Cell Biol. 1987;105:1587–1594 . doi: 10.1083/jcb.105.4.1587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Novick P., Field C., Schekman R. Identification of 23 complementation groups required for post-translational events in the yeast secretory pathway. Cell. 1980;21:205–215 . doi: 10.1016/0092-8674(80)90128-2. [DOI] [PubMed] [Google Scholar]
- Ohashi A., Gibson J., Gregor I., Schatz G. Import of proteins into mitochondria. The precursor of cytochrome c1 is processed in two steps, one of them heme-dependent. J. Biol. Chem. 1982;257:13042–13047 . [PubMed] [Google Scholar]
- Palade G. Intracellular aspects of the process of protein secretion. Science. 1975;189:347–358 . doi: 10.1126/science.1096303. [DOI] [PubMed] [Google Scholar]
- Pfeffer S.R. Transport vesicle targetingtethers before SNAREs. Nature Cell Biol. 1999;1:E17–E22 . doi: 10.1038/8967. [DOI] [PubMed] [Google Scholar]
- Rose M., Winston F., Hieter P. Methods in Yeast Genetics. A Laboratory Course Manual. Cold Spring Harbor Laboratory Press; Cold Spring Harbor, NY : 1990. [Google Scholar]
- Rose M.D. Modern and post-modern genetics in Saccharomyces cerevisiae . Yeasts. 1995;6:69–120 . [Google Scholar]
- Rose M.D., Novick P., Thomas J.H., Botstein D., Fink G.R. A Saccharomyces cerevisiae genomic plasmid bank based on a centromere-containing shuttle vector. Gene. 1987;60:237–243 . doi: 10.1016/0378-1119(87)90232-0. [DOI] [PubMed] [Google Scholar]
- Rossi G., Kolstad K., Stone S., Palluault F., Ferro-Novick S. BET3 encodes a novel hydrophilic protein that acts in conjunction with yeast SNAREs. Mol. Biol. Cell. 1995;6:1769–1780 . doi: 10.1091/mbc.6.12.1769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sacher M., Jiang Y., Barrowman J., Scarpa A., Burston J., Zhang L., Schieltz D., Yates I.J., Abeliovich H., Ferro-Novick S. TRAPP, a highly conserved novel complex on the cis-Golgi that mediates vesicle docking and fusion. EMBO (Eur. Mol. Biol. Organ.) J. 1998;17:2494–2503 . doi: 10.1093/emboj/17.9.2494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sapperstein, S. 1997. Analysis of the intra-Golgi transport factor p115, and its Saccharomyces cerevisiae homolog Uso1p. In Department of Molecular Biology. Princeton University, Princeton, NJ. 195 pg.
- Sapperstein S.K., Lupashin V.V., Schmitt H.D., Waters M.G. Assembly of the ER to Golgi SNARE complex requires Uso1p. J. Cell Biol. 1996;132:755–767 . doi: 10.1083/jcb.132.5.755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schiestl R.H., Gietz R.D. High efficiency transformation of intact yeast cells using single stranded nucleic acid as a carrier. Curr. Genet. 1989;16:339–346 . doi: 10.1007/BF00340712. [DOI] [PubMed] [Google Scholar]
- Schmitt H.D., Wagner P., Pfaff E., Gallwitz D. The ras-related YPT1 gene product in yeasta GTP-binding protein that might be involved in microtubule organization. Cell. 1986;47:401–412 . doi: 10.1016/0092-8674(86)90597-0. [DOI] [PubMed] [Google Scholar]
- Shim J., Newman A.P., Ferro-Novick S. The BOS1 gene encodes an essential 27-kD putative membrane protein that is required for vesicular transport from the ER to the Golgi complex in yeast. J. Cell Biol. 1991;113:55–64 . doi: 10.1083/jcb.113.1.55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simons K., Zerial M. Rab proteins and the road maps for intracellular transport. Neuron. 1993;11:789–799 . doi: 10.1016/0896-6273(93)90109-5. [DOI] [PubMed] [Google Scholar]
- Söllner T., Whiteheart S.W., Brunner M., Erdjument-Bromage H., Geromanos S., Tempst P., Rothman J.E. SNAP receptors implicated in vesicle targeting and fusion. Nature. 1993;362:318–324 . doi: 10.1038/362318a0. [DOI] [PubMed] [Google Scholar]
- VanRheenen S.M., Cao X., Lupashin V.V., Barlowe C., Waters M.G. Sec35p, a novel peripheral membrane protein, is required for ER to Golgi vesicle docking. J. Cell Biol. 1998;141:1107–1119 . doi: 10.1083/jcb.141.5.1107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waters M.G., Pfeffer S.R. Membrane tethering in intracellular transport. Curr. Opin. Cell. Biol. 1999;11:453–459 . doi: 10.1016/s0955-0674(99)80065-9. [DOI] [PubMed] [Google Scholar]
- Wuestehube L.J., Duden R., Eun A., Hamamoto S., Korn P., Ram R., Schekman R. New mutants of Saccharomyces cerevisiae affected in the transport of proteins from the endoplasmic reticulum to the Golgi complex. Genetics. 1996;142:393–406 . doi: 10.1093/genetics/142.2.393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamakawa H., Seog D.-H., Yoda K., Yamasaki M., Wakabayashi T. Uso1 protein is a dimer with two globular heads and a long coiled-coil tail. J. Struct. Biol. 1996;116:356–365 . doi: 10.1006/jsbi.1996.0053. [DOI] [PubMed] [Google Scholar]
- Yang B., Gonzalez L., Jr., Prekeris R., Steegmaier M., Advani R.J., Scheller R.H. SNARE interactions are not selective. Implications for membrane fusion specificity. J. Biol. Chem. 1999;274:5649–5653. doi: 10.1074/jbc.274.9.5649. [DOI] [PubMed] [Google Scholar]