

Abstract
Cell migration is modulated by regulatory molecules such as growth factors, oncogenes, and the tumor suppressor PTEN. We previously described inhibition of cell migration by PTEN and restoration of motility by focal adhesion kinase (FAK) and p130 Crk-associated substrate (p130Cas). We now report a novel pathway regulating random cell motility involving Shc and mitogen-activated protein (MAP) kinase, which is downmodulated by PTEN and additive to a FAK pathway regulating directional migration. Overexpression of Shc or constitutively activated MEK1 in PTEN- reconstituted U87-MG cells stimulated integrin- mediated MAP kinase activation and cell migration. Conversely, overexpression of dominant negative Shc inhibited cell migration; Akt appeared uninvolved. PTEN directly dephosphorylated Shc. The migration induced by FAK or p130Cas was directionally persistent and involved extensive organization of actin microfilaments and focal adhesions. In contrast, Shc or MEK1 induced a random type of motility associated with less actin cytoskeletal and focal adhesion organization. These results identify two distinct, additive pathways regulating cell migration that are downregulated by tumor suppressor PTEN: one involves Shc, a MAP kinase pathway, and random migration, whereas the other involves FAK, p130Cas, more extensive actin cytoskeletal organization, focal contacts, and directionally persistent cell motility. Integration of these pathways provides an intracellular mechanism for regulating the speed and the directionality of cell migration.
Keywords: Shc, focal adhesion kinase, integrin, cell migration, PTEN
Cell migration is important for embryonic development, wound repair, inflammation, and cancer invasion. Although an extensive literature has implicated a variety of molecules in cell motility, relatively little is known about how the process of cell migration is integrated intracellularly to control the directionality and the speed of migration. Cell motility can be regulated or modulated by growth factors and cytokines, small G proteins, oncogenes, and the newly discovered tumor suppressor protein PTEN (Hynes and Lander 1992; Stossel 1993; Oliver et al. 1994; Huttenlocher et al. 1995; Nobes and Hall 1995; Lauffenburger and Horwitz 1996; Sheetz et al. 1998; Tamura et al. 1998, Tamura et al. 1999a). PTEN is a tumor suppressor gene mutated in a wide variety of human cancers, including breast, prostate, and brain cancer (Li and Sun 1997; Li et al. 1997; Steck et al. 1997). PTEN expression can suppress migration, invasion, tumorigenicity, and growth of human tumor cells (Furnari et al. 1997; Cheney et al. 1998; Li and Sun 1998; Tamura et al. 1998; Whang et al. 1998).
PTEN encodes a protein tyrosine phosphatase motif. Substrates of PTEN identified to date, both in vitro and in living cells, include the lipid phosphatidylinositol 3,4,5-trisphosphate (PIP3)1 (Maehama and Dixon 1998; Stambolic et al. 1998) and the phosphoprotein focal adhesion kinase (FAK) (Tamura et al. 1998). Purified PTEN can remove a specific phosphate group from PIP3, thereby inhibiting protein kinase B (also known as Akt), which in turn participates in cell growth control and inhibits the apoptosis pathway. Recent experiments using PTEN knockout mice and tumor cell lines indicate that PTEN is essential for embryonic development and sensitivity to apoptotic stimuli; the latter process has been linked to the lipid phosphatase activity of PTEN (Davies et al. 1998; Di Cristofano et al. 1998; Haas-Kogan et al. 1998; Li et al. 1998; Myers et al. 1998; Stambolic et al. 1998; Suzuki et al. 1998; Wu et al. 1998).
Because PTEN also has an NH2-terminal domain with extensive homology to tensin, a protein that interacts with actin filaments at focal adhesions, we have focused on analyzing roles of PTEN in integrin-mediated cell migration and signal transduction. We previously found that the G129E mutant of PTEN, which lacks lipid phosphatase activity but has protein phosphatase activity, can still inhibit integrin-mediated cell migration, spreading, focal adhesions, and tumor cell invasion, whereas a phosphatase-dead mutant (C124A) of PTEN cannot (Tamura et al. 1998, Tamura et al. 1999a), demonstrating an important role of protein phosphatase activity for PTEN function. PTEN directly associates with FAK and can reduce its tyrosine phosphorylation as well as that of a potential downstream effector, p130 Crk-associated substrate (p130Cas). Furthermore, overexpression of FAK or p130Cas can antagonize the effects of PTEN on cell migration and invasion (Tamura et al. 1999a).
We also observed that PTEN inhibits integrin- and growth factor–mediated mitogen-activated protein (MAP) kinase signaling pathways. MAP kinase activation could partially antagonize PTEN function with partial rescue of cell spreading on fibronectin impaired by PTEN. This inhibition of PTEN was associated with effects on Shc phosphorylation (Gu et al. 1998). Shc is an SH2-phosphotyrosine–binding adapter protein that links tyrosine kinases to Ras signaling by recruiting the Grb2-Sos complex to the plasma membrane in a tyrosine phosphorylation–dependent manner (Rozakis-Adcock et al. 1992; Pronk et al. 1994; Pawson and Scott 1997; Wary et al. 1998). PTEN can inhibit tyrosine phosphorylation of both FAK and Shc. Both proteins are implicated in integrin signaling and either one can bind Grb2 and potentially activate the Ras–MAP kinase pathway (Schlaepfer et al. 1994; Wary et al. 1996, Wary et al. 1998; Lin et al. 1997; Schlaepfer and Hunter 1997; Gu et al. 1998; Schlaepfer et al. 1998; Tamura et al. 1998). FAK can also promote integrin-mediated cell migration through the activation of p130Cas (Cary et al. 1998; Sieg et al. 1998). A general role for Shc in activation of the Ras–MAP kinase pathway is well established. Shc has been reported to be activated by only certain integrins and to regulate cell cycle progression in response to specific extracellular matrix proteins (Wary et al. 1996; Mainiero et al. 1997). Because the functions of Shc, FAK, and PTEN appear intertwined, we explored potential additional cell biological functions for Shc in regulating cell migration and the cytoskeleton.
In the present study, we examined the regulation of rates of cell motility versus directionality by the integrated effects of PTEN, Shc, and FAK. We tested for roles of Shc in cell migration and compared its effects and mechanisms with those of FAK. Shc was found to regulate integrin-mediated cell motility. Furthermore, Shc and constitutively activated MEK1 stimulated random cell migration. In contrast, FAK and p130Cas activated directional (persistent) cell migration in PTEN-reconstituted cells. These differences in types of migration patterns correlated with differences in the extent and organization of actin cytoskeleton. These findings indicate for the first time that a Shc pathway can selectively regulate integrin-mediated random cell motility and that PTEN can suppress cell motility by distinct pathways that diverge at the level of Shc and FAK. Integration of these three countervailing regulatory systems provides an intracellular mechanism for regulating the speed and the directionality of cell migration.
Materials and Methods
Expression Plasmids
Green fluorescent protein (GFP) expression plasmids based on pGZ21δxZ that contained no insert, full-length wild-type PTEN, or hemagglutinin (HA)-tagged FAK were constructed as described (Tamura et al. 1998). The dominant negative truncation sequence of FAK (FRNK) was PCR amplified from HA-FAK using the following forward and reverse primers: 5′-AGATCTAGATCTCGGATGAGGATGGAATCCAGAAG-3′ and 5′-GCGGCCGCTCAGTGTGGCCGTGTCTGCCCTAGCATTTT-3′. The PCR products were digested with BamHI and XbaI and cloned into pGZ21δxZ. Vesicular stomatis virus epitope–tagged FRNK was constructed by inserting the vesicular stomatis virus epitope at the 5′ end of FRNK and the full sequence was verified by DNA sequencing. The point mutations D92A and C124A were introduced into PTEN by site-directed mutagenesis as described (Tamura et al. 1998). pSSRa-Cas-Flag and pSSRa-ΔSD-Cas-Flag were constructed by inserting the epitope tag Flag (Eastman Kodak Co.) at the 3′ end of the p130Cas and ΔSD-p130Cas (dominant negative p130Cas) coding sequence in the expression vectors pSSRa-Cas and pSSRa-ΔSD-Cas, which were provided by Dr. Hisamaru Hirai (University of Tokyo, Tokyo, Japan) (Nakamoto et al. 1997). The plasmid ΔSD-p130Cas functions as a dominant interfering (dominant negative) inhibitor of p130Cas because it lacks the substrate domain, which contains 15 potential tyrosine phosphorylation sites for binding of molecules such as Crk and other proteins (Nakamoto et al. 1997).
A pcDNA/Flag-Shc construct was generated by inserting the epitope tag Flag at the 5′ end of the p52 Shc coding sequence in the expression vector pcDNA3.1(+). The L-p66-SN Shc cDNA that was used as a template for PCR was provided by Drs. E. Migliaccio and P.G. Pelicci (European Institute of Oncology, Milan, Italy). The point mutations Y239F or Y317F were introduced into the 52-kD isoform of Shc by site-directed mutagenesis or both were introduced to produce the double point mutant Y239/317F. Mutation of these two tyrosine phosphorylation sites generates a dominant negative inhibitor of Shc signaling that is initiated by integrins and growth factors (Wary et al. 1998). A plasmid containing pMCL⊕HA-tagged MEK1 (constitutively activated form) was provided by Dr. N.G. Ahn (Department of Chemistry and Biochemistry, University of Colorado) (Mansour et al. 1994). The puromycin resistance plasmid pHA262pur was provided by Dr. Hein te Riele (Netherlands Cancer Institute, Amsterdam, The Netherlands) (Lacalle et al. 1989). Wild-type Akt and dominant inhibitory Akt (Akt-K179A) in the pCIS2 expression vector were provided by Dr. Michael J. Quon (Hypertension-Endocrine Branch, National Institute of Diabetes and Digestive and Kidney Diseases, NIH) (Cong et al. 1997). COOH-terminal Src kinase (Csk) in the pME18SNeo expression vector was provided by Dr. Masato Okada (Institute for Protein Research, Osaka University, Osaka, Japan) (Nada et al. 1991).
Reagents and Antibodies
The mAb 2A7 (Upstate Biotechnology Inc.) directed against FAK was used for immunoprecipitation and a second mAb against FAK (Transduction Laboratories) was used for immunoblotting. Monoclonal anti-Shc as well as polyclonal anti-p44/42 MAP kinase antibodies were purchased from Santa Cruz Biotechnology. mAbs for p130Cas, paxillin, Csk, and phosphotyrosine (RC20) were obtained from Transduction Laboratories. Monoclonal anti–phospho-p44/42 MAP kinase antibody was from New England Biolabs, Inc. mAb against Flag (M2) was from Eastman Kodak Co., mAb against HA was purchased from BAbCO, and mAb against GFP was from CLONTECH Laboratories. Cy3-conjugated goat antibody to mouse immunoglobulin G (Jackson ImmunoResearch Laboratories, Inc.) was used at 1:500 dilution. Rhodamine-labeled phalloidin was from Molecular Probes. Culture medium and FBS were obtained from GIBCO BRL and Life Technologies, Inc.
Cell Culture, Transfection, and Selection
The PTEN-mutated glioblastoma cell line U-87MG was obtained from American Type Culture Collection. Cells were maintained in DME supplemented with 10% FBS, 100 U/ml penicillin, and 100 μg/ml streptomycin and cultured in 10% CO2 at 37°C. Transfections were performed by electroporation (LaFlamme et al. 1994). In brief, pGZ21δxZ (10 μg; cotransfection with 10 μg Flag-Shc, 10 μg HA-FAK, 10 μg Flag-Cas, or 3 μg constitutively activated HA-MEK1) containing either no insert or PTEN was transfected into 1.5 × 106 U-87MG cells by electroporation together with 3 μg pHA262pur. For cotransfections with FRNK or dominant negative Cas or Csk, we used 10 μg of each plasmid in this study. To increase the expression of transfected genes, 5 mM sodium butyrate was included in culture media. Cells were subcultured at a 1:3 dilution 24 h after transfection and were maintained for 2 d in 1 μg/ml puromycin-containing medium. The cells were cultured overnight in the absence of puromycin before use. This selection for transient transfectants resulted in ∼90% positive cells expressing GFP or GFP-PTEN as determined by fluorescence microscopy. For coimmunoprecipitation experiments, U-87MG cells were cotransfected with GFP tag only, or GFP-tagged wild-type PTEN, trapping mutant D92A, or inactive phosphatase mutant C124A (20 μg each) with pHA262pur, and then selected with puromycin as described above.
Immunoprecipitation and Western Blotting
Puromycin-selected U-87MG cells expressing the various constructs were detached by treating with 0.05% trypsin-EDTA, and then washed with medium without FBS. 3 × 105 cells were allowed to spread for the times indicated on 10-cm plastic tissue culture dishes coated with 10 μg/ml fibronectin. The cells were washed with ice-cold PBS and solubilized in 1% Triton X-100 lysis buffer (20 mM Tris, pH 7.5, 150 mM NaCl, 1 mM EDTA, 1 mM EGTA, 2.5 mM sodium pyrophosphate, 1 mM β-glycerophosphate, 1 mM sodium vanadate, 10 μg/ml leupeptin, 10 μg/ml aprotinin, 1 mM PMSF) for analysis of protein tyrosine phosphorylation.
For MAP kinase assays, puromycin-selected cells were serum-restricted overnight in media containing 0.2% FBS. The cells were washed with PBS and detached by treating with 0.05% trypsin-EDTA. Trypsin was inactivated with 1 mg/ml soybean trypsin inhibitor. The suspended cells were washed two times in DME with 1% BSA. Cell suspensions were incubated in the same medium at 37°C for 30 min on a rotator. Thereafter, cells were counted and allowed to spread for 10 min on fibronectin-coated dishes, and then solubilized as described above.
For coimmunoprecipitation experiments, cells were stimulated with EGF for 5 min, and then solubilized in modified CSK buffer (100 mM NaCl, 0.5% Triton X-100, 300 mM sucrose, 3 mM MgCl2, 10 mM Pipes, pH 6.8) containing 2 mM PMSF, 50 mM sodium fluoride, 1 mM sodium orthovanadate, and protease inhibitor mixture (Boehringer Mannheim). The homogenates were clarified by centrifugation at 20,000 g for 15 min at 4°C. Immunoprecipitates were suspended in reducing or nonreducing sample buffer, heated to 100°C for 5 min, resolved in 8 or 10% SDS–polyacrylamide gels (Novex), and electrophoretically transferred to nitrocellulose membrane (Novex) for 1.5 h at 150 mA. The filters were incubated with blocking buffer (5% nonfat dry milk; alternatively, 5% BSA for antiphosphotyrosine antibody in T-TBS[150 mM NaCl, 50 mM Tris-HCl, 0.1% Tween 20, pH 7.4]) for 1 h. Immunoblots for phosphotyrosine, activated ERK2, GFP, Shc, or other epitopes were visualized by the ECL system and Hyperfilm X-ray film (Amersham).
Protein Phosphatase Assays
PTEN dephosphorylation of Shc and FAK were examined using an in blot phosphatase assay as described (Tamura et al. 1998). In brief, histidine-tagged PTEN (His6-PTEN) was generated by inserting full-length PTEN cDNA into the pQE30 vector (Qiagen). The expressed recombinant protein was purified using Ni-NTA beads (Qiagen) under denaturing conditions, and then renatured by sequential dilution and concentration in renaturation buffer (PBS, pH 7.0, containing 2 mM MgCl2, 0.5 mM PMSF, 0.005% Tween 20, 10 mM DTT, protease inhibitor cocktail). Purity (>90%) was confirmed by SDS-PAGE and Coomassie blue staining. Phosphorylated FAK was obtained from immunoprecipitates using anti-FAK antibody from cell lysates of U-87MG cells that had spread on fibronectin for 1 h. Phosphorylated Shc and activated ERK2 were isolated as immunocomplexes from cell lysates of EGF-stimulated (10 ng/ml for 5 min) U-87MG cells transfected with Flag-Shc and HA-ERK2, and then immunoprecipitated using either anti-Flag or anti-HA antibodies, respectively. Immunoprecipitated FAK and Shc were mixed and subjected to 8% SDS-PAGE. Immunoprecipitates of ERK2 using anti-HA were subjected to 10% SDS-PAGE, and then electrotransferred to nitrocellulose. Blots were incubated with 20 μg/ml recombinant His6-PTEN in 100 mM Tris buffer, pH 7.0, containing 10 mM MgCl2, and 10 mM DTT at 30°C for 30 min. Phosphorylation of Shc and FAK was detected with RC20 antiphosphotyrosine antibody and activated ERK2 was detected by anti–phospho-ERK2 antibody.
PTEN phosphatase activity against all three isoforms of endogenous Shc was also examined under nondenaturing conditions in vitro using immunoprecipitated Shc before SDS-PAGE. Endogenous Shc was isolated from EGF-stimulated, nontransfected U-87MG cells homogenized in lysis buffer as described above by immunoprecipitation using anti-Shc mAb (4 μg/ml) and GammaBind G–Sepharose beads (Amersham Pharmacia Biotech) for 3 h at 4°C. The immunocomplexes were incubated with 0.5 μg recombinant PTEN in 30 μl of 50 mM Tris buffer, pH 7.0, containing 50 mM NaCl and 10 mM DTT at 30°C for 30 min. Controls were incubated without PTEN or with PTEN plus 2 mM sodium vanadate. The reaction was terminated by adding nonreducing SDS sample buffer and heating at 100°C for 5 min. After SDS-PAGE, immunoblotting was carried out using RC20 antiphosphotyrosine mAb.
Cell Motility
After puromycin selection, cells expressing various constructs were replated on 50-mm glass microwell dishes (Mattek Corp.) coated with 10 μg/ml fibronectin and cultured overnight in DME containing 10% FBS. Cell movements were monitored using a Zeiss inverted microscope. Video images were collected with a CCD camera (model 2400; Hamamatsu Photonics) at 20-min intervals, digitized, and stored as image stacks using MetaMorph Group 3.5 software (Universal Imaging Corp.). Image stacks were converted to QuickTime movies, the positions of nuclei were tracked to quantify cell motility, and their velocities were calculated in micrometers at 20-min points using the same software. Similar results with nonselected cells were obtained in preliminary experiments using GFP-tagged FAK or GFP-Shc and tracking of cell migration using time-lapse fluorescence microscopy. For testing the effects of PD98059 (a specific MEK1 inhibitor) and wortmannin (a phosphatidylinositol 3′-kinase inhibitor) on cell migration, we cultured the cells in 20 μM PD98059 or 30 nM wortmannin for 2 h, and then examined cell motility for three more hours with each inhibitor.
Immunofluorescence Microscopy
Glass coverslips (12 mm; Carolina Biological Supply Company) were incubated with 10 μg/ml fibronectin in PBS overnight at 4°C. The coverslips were blocked with 10 mg/ml BSA for an additional 1 h at 37°C. After puromycin selection, cells expressing various constructs were replated on the coverslips and cultured overnight in DME containing 10% FBS. Thereafter, the cells were fixed with 4% paraformaldehyde in PBS for 20 min, and then permeabilized with 0.5% Triton X-100 in PBS for 5 min. Focal adhesions were visualized by incubating first with mouse antipaxillin mAb, and then with Cy3-conjugated goat antibody to mouse immunoglobulin G. Actin filaments were stained with rhodamine-phalloidin. For semi-quantitative documentation of cytoskeletal organization, a square equivalent to 15 × 15 μm was overlaid randomly over each of the four quadrants of each cell. Rhodamine-phalloidin–stained actin microfilaments in each square were scored as appearing random or oriented in parallel. In this assay, the highest index for a cell occurs when all four test fields show oriented actin microfilaments, resulting in a maximal index score of 4.0.
Results
We examined for roles of Shc in regulating cell migration, because it is implicated in integrin signaling and is a prominent target of the tumor suppressor phosphatase PTEN, which is a newly identified regulator of cell migration and invasion. We find that Shc can enhance cell migration inhibited by PTEN and that Shc is a direct target for PTEN phosphatase activity. We compare this novel pathway regulating cell migration both mechanistically and biologically with the previously described FAK-p130Cas pathway (Cary et al. 1996, Cary et al. 1998; Tamura et al. 1998) including roles in regulating speed and the directionality of cell migration.
Shc Induces Cell Migration Inhibited by PTEN
To test for a role of Shc in cell migration modulated by PTEN, we cotransfected PTEN and puromycin resistance plasmids with Shc (or FAK as a positive control), and selected transfectants for 2 d using puromycin. This puromycin selection procedure routinely yielded ∼90% pure populations of transfectants according to fluorescence analyses using GFP markers. The surviving selected cells were replated on glass microwell dishes coated with 10 μg/ml fibronectin and cultured in DME containing 10% FBS overnight. To analyze cell motility, phase-contrast video images were recorded at 20-min intervals using a CCD camera and were analyzed for velocities of cell migration using MetaMorph image processing software. As shown in Fig. 1, reconstitution of PTEN in these cells lacking PTEN to protein levels similar to those in primary fibroblasts (1–2× according to immunoblotting) substantially inhibited cell movement. Migration was reduced to 39% of rates in controls without PTEN. Interestingly, coexpression of Shc with PTEN significantly rescued rates of cell motility on fibronectin, raising them from 39% of control migration rates with PTEN alone to 78% of controls after Shc coexpression with PTEN. These differences were significant at the P < 0.001 level.
Figure 1.

Shc accelerates cell movement inhibited by PTEN. U-87MG cells were transfected with various plasmids as indicated and transfectants were selected by puromycin as described in Materials and Methods. Cell movements were monitored by time-lapse video microscopy and motility was calculated as velocity (μm/3 h) using image processing software as described in Materials and Methods. Data from at least 10 cells selected by puromycin were collected and calculated in each experiment, and data were pooled from three independent experiments (each with similar results). Error bars indicate SD for at least 30 cells per condition. One asterisk, P < 0.001 versus controls transfected with GFP-PTEN only. Two asterisks, P < 0.01 versus each of the other conditions (except for control transfected with GFP-PTEN only or without plasmid).
Because PTEN can downmodulate the ERK type of MAP kinase signaling, we tested whether constitutively activated MEK1, a potential downstream effector of Shc, could also activate cell movement downmodulated by PTEN (Fig. 1). MEK1 coexpression was highly effective in reversing PTEN inhibition of migration (significant at the P < 0.001 level). Consistent with previous observations that FAK and p130Cas overexpression could rescue PTEN inhibition of cell migration measured by in vitro wound-healing assays (Tamura et al. 1999a), FAK and p130Cas also effectively rescued single cell movement in this system (Fig. 1). To test whether Shc and FAK stimulated cell motility via different or overlapping pathways, we performed a triple transfection experiment combining PTEN with both Shc and FAK. Cell movement was fully restored to 95% of controls by this triple transfection, as compared with 78% of controls for Shc plus PTEN double transfection and 82% for FAK plus PTEN. This simple additivity of migration suggests the existence of two parallel biological pathways originating from Shc and FAK affecting cell migration modulated by PTEN. In contrast, the Y397F mutant of FAK lacking the Src and phosphatidylinositol 3′-kinase binding site did not affect migration rates (data not shown).
Dominant Negative Shc Expression Inhibits Cell Migration
As a direct test of the role of Shc in cell migration (independent of PTEN), we examined whether expression of a dominant negative mutant of Shc to block endogenous Shc function could mimic the effects of PTEN. Transfection with dominant negative Shc (double point mutant Y239/317F) substantially reduced cell migration to 58% of controls (Fig. 2). A putative integrin-specific mutant of Shc in which only tyrosine 317 was mutated (Wary et al. 1998) produced less inhibition, suggesting a roughly 40:60% ratio of contributions of integrins versus serum growth factors to Shc stimulation of migration. Specifically, there was 16% inhibition with Y317F versus 42% inhibition with the double mutant.
Figure 2.

Dominant negative Shc inhibits cell motility. U-87MG cells were cotransfected with pHA262pur and GFP (−) with or without various plasmids and transfectants were selected as described in Materials and Methods. Shc (Dn) indicates the dominant negative Y239/317F Shc plasmid, Shc (Wt) indicates wild-type Shc, FRNK is a dominant negative FAK truncation, and P130Cas (Dn) indicates the ΔSD-p130Cas dominant negative mutant of p130Cas lacking the substrate domain. Cell motility was examined as described in Fig. 1. Error bars indicate SD for at least 30 cells per condition. Asterisk, P < 0.001 versus controls transfected with GFP (−) only.
In addition, transfection with FRNK or dominant negative p130Cas also substantially reduced cell migration to 55 or 54% of controls, respectively (Fig. 2). In contrast, expression of GFP (−), Shc, FAK, constitutively activated MEK1, or p130Cas alone in the absence of PTEN had little or no effect on cell migration of U-87MG cells in this system (Fig. 2). Because it had been reported previously that FAK overexpression significantly increases CHO cell migration (Cary et al. 1996), we also compared FAK overexpression in CHO cells using our cell migration assay. We found that FAK overexpression did enhance cell migration of CHO cells in this system to 165% of controls transfected with GFP (−) alone (data not shown).
Although phosphatidylinositol 3′-kinase, an upstream regulator of PKB/Akt, has been implicated in cell movement (Keely et al. 1997; Shaw et al. 1997; Sander et al. 1998), to our knowledge there are no studies on the roles of Akt in cell movement. Because many studies indicate that PTEN inhibits cell growth and leads to apoptosis through inhibition of the PKB/Akt pathway, an obvious question is whether PTEN-mediated inhibition of Akt affects cell migration. U-87MG cells were cotransfected with the puromycin resistance plasmid pHA262pur and either wild-type Akt or dominant negative Akt, and transfected cells were selected as described above. We could not detect any significant differences in rates of cell migration between control cells and either type of transfectant affecting Akt (data not shown). Effects of PTEN on Akt are, therefore, not likely to play a role in PTEN regulation of cell migration.
Shc Interacts Physically with PTEN
Since Shc and PTEN appeared to be involved in an early step of a specific signaling pathway regulating cell migration, they might be expected to interact physically. Shc has three isoforms of 66, 52, and 46 kD, which are derived from alternative splicing and differential translation initiation at three ATG sites (Migliaccio et al. 1997). PTEN preferentially decreases tyrosine phosphorylation of the 52-kD isoform of Shc and thereby inhibits interaction with the adapter protein Grb2, resulting in decreased activation of the Ras/Raf/MEK/ERK pathway (Gu et al. 1998). Because phosphatases bind, but rapidly cleave and dissociate from substrates, we tested for physical interactions of PTEN with Shc in living cells using a trapping mutant D92A of PTEN. The latter mutant has inactivated phosphatase activity but retains its ability to bind and even to protect a substrate (Flint et al. 1997). Cells were cotransfected with control or PTEN plasmids and a puromycin resistance plasmid, and then selected with puromycin to enrich transfected cells. The surviving cells (90% positive) were cultured overnight without puromycin, and then stimulated with EGF for 5 min. Homogenates were immunoprecipitated with anti-Shc or anti-GFP followed by immunoblotting with the opposite or the same antibody.
As shown in Fig. 3 A, immunoprecipitated Shc retained substantial amounts of bound PTEN D92A trapping mutant. It also retained, to a lesser extent, the direct active site PTEN mutant C124A. But in cells transfected with control plasmid GFP (−) or wild-type GFP-PTEN, such associations with Shc could not be detected. Conversely, immunoprecipitation of the GFP-PTEN mutant D92A with anti-GFP antibody also retained Shc bound in higher quantities as compared with cells expressing the C124A mutant or wild-type PTEN (Fig. 3 B). These results using a substrate-trapping mutant strongly suggest that PTEN directly interacts with Shc. PTEN associated selectively with the 52-kD isoform of Shc (Fig. 3 B), which is consistent with the higher tyrosine phosphorylation of this 52-kD isoform. Similar but much weaker binding of the D92A PTEN trapping mutant to Shc was observed even in the absence of stimulation by EGF with minimal binding of wild-type or C124A mutant PTEN (data not shown).
Figure 3.
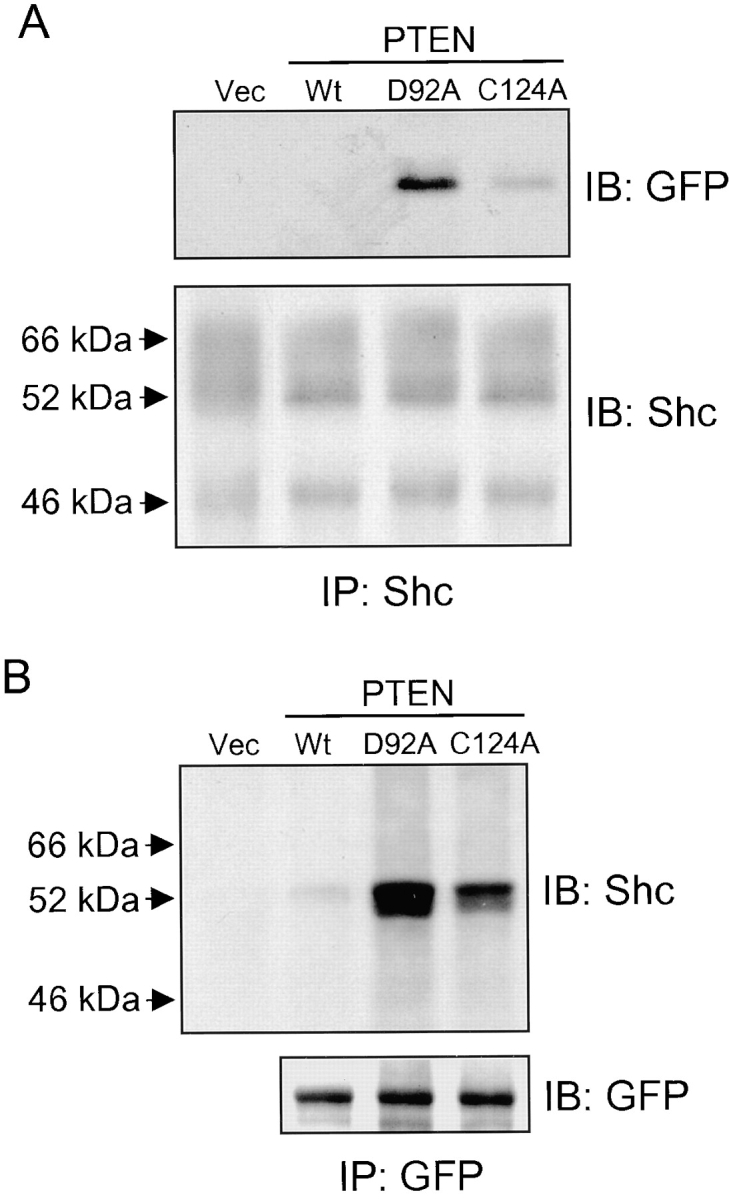
Physical interaction of PTEN with Shc. U-87MG cells were cotransfected with plasmids containing pHA262pur and GFP tag without PTEN (Vec), GFP-tagged wild-type PTEN (Wt), PTEN trapping mutant D92A, or inactivated phosphatase PTEN mutant C124A (20 μg each), and then selected with puromycin as described in Materials and Methods. After selection, cells were stimulated with EGF for 5 min, and then solubilized in modified CSK buffer as described in Materials and Methods. The cell lysates were immunoprecipitated with either anti-Shc (IP: Shc) or anti-GFP (IP: GFP) antibody and immunoblotted for anti-GFP (IB: GFP) or anti-Shc (IB: Shc) antibody. (A) Immunoprecipitates containing endogenous Shc immunoblotted with either anti-GFP (top panel) or anti-Shc antibody (bottom panel). (B) Immunoprecipitated GFP-tagged proteins immunoblotted with either anti-Shc antibody (top panel) or anti-GFP antibody to confirm equivalent expression of PTEN mutant proteins (bottom panel). Expression levels of GFP (−) lacking PTEN (Vec) were two to threefold higher than that of GFP-PTEN (data not shown).
PTEN Directly Dephosphorylates Shc
Next, we tested whether PTEN could directly dephosphorylate Shc using two types of in vitro phosphatase assays. An in blot phosphatase assay was used to examine the tyrosine-phosphorylated 52-kD isoform of Shc as a direct substrate of PTEN. FAK was used as a positive control (Tamura et al. 1998) and activated ERK2 as a negative control (Myers et al. 1997). Renatured recombinant PTEN reduced the tyrosine phosphorylation of the electroblotted 52-kD Shc by 67% (Fig. 4 A, lane 2, top) compared with controls to which we added 2 mM sodium vanadate, a general inhibitor of phosphatase activity (lane 1). This level of dephosphorylation of Shc was similar to the 70% reduction in tyrosine phosphorylation of FAK. In contrast, PTEN could not dephosphorylate activated ERK2 in vitro (Fig. 4 A, lane 2, bottom); the latter negative result was consistent with a previous report using a different assay system (Myers et al. 1997).
Figure 4.

Direct tyrosine dephosphorylation of Shc by PTEN. The capacity of PTEN to dephosphorylate Shc was evaluated either by an in blot tyrosine phosphatase assay using the transfected 52-kD isoform of PTEN or its mutants (A and C) or by an in vitro phosphatase assay using immunoprecipitated (IP), native Shc isoforms of 46, 52, and 66 kD followed by immunoblotting (IB) for phosphotyrosine (B). For A and C, U-87MG cells were transiently transfected with Flag-tagged 52-kD isoform of wild-type Shc (Wt), Y239F mutant, Y317F mutant, or HA-ERK2. 24 h after transfection, cells were stimulated by EGF for 5 min, and then immunoprecipitated (IP) with anti-Flag or anti-HA. For B, endogenous Shc was immunoprecipitated with anti-Shc mAb after nontransfected cells were stimulated by EGF for 5 min. Endogenous FAK was immunoprecipitated after stimulation by adhesion to fibronectin for 1 h as described in Materials and Methods. In A, immunoprecipitated FAK and Shc (wild-type) were mixed and subjected to 8% SDS-PAGE; immunoprecipitated ERK2 was analyzed using 10% SDS–polyacrylamide gels. After electroblotting, strips of blots were incubated with recombinant PTEN with (lane 1) or without (lane 2) 2 mM sodium vanadate at 30°C for 30 min. Shc and FAK phosphorylation were detected with antiphosphotyrosine antibody (A, top panels), and phospho-ERK2 was detected by anti–phospho-ERK1/2 antibody (A, bottom panels). Immunoprecipitates of endogenous Shc (B) and 52-kD isoform of wild-type or mutants (C) were directly incubated with 0.5 μg of renatured recombinant PTEN for 30 min at 30°C. The reaction mixtures were subjected to 8% SDS-PAGE for Western immunoblotting. Shc tyrosine phosphorylation was detected by an antiphosphotyrosine mAb. 0, no additions; 1, incubated with recombinant PTEN plus 2 mM sodium vanadate; and 2, recombinant PTEN without sodium vanadate. The same quantities of total protein were loaded in each lane.
We also tested whether PTEN could dephosphorylate native tyrosine–phosphorylated Shc in vitro. Incubation of recombinant PTEN with immunoprecipitated endogenous Shc showed that PTEN could dephosphorylate all three isoforms of Shc (Fig. 4 B). PTEN appeared to dephosphorylate both of the two major tyrosine phosphorylation sites because it equally effectively removed phosphotyrosine from mutant p52Shc molecules containing only one of the two sites after point mutations to phenylalanine in the Y239F or Y317F mutants (Fig. 4 C). The double point mutant Y239/317F Shc showed only very weak phosphorylation stimulated by EGF (data not shown), confirming that tyrosines 239 and 317 were the two major sites for phosphorylation.
Shc Overexpression Rescues Integrin-mediated MAP Kinase Activation Impaired by PTEN
The possible mechanisms of Shc regulation of PTEN-modulated adhesion were explored in more detail. We tested whether overexpression of Shc or FAK could attenuate the effects of PTEN on MAP kinase activation by fibronectin. We cotransfected GFP-PTEN and puromycin resistance plasmids with or without Shc or FAK, or with both Shc and FAK, and selected for transfectants using puromycin. The surviving selected cells were plated for 10 min on dishes coated with fibronectin, and then homogenized using lysis buffer. MAP kinase activation was assayed by direct examination of ERK1/2 phosphorylation by immunoblotting with anti–phospho-ERK1/2. MAP kinase activation was substantially suppressed in cells transfected with PTEN alone, as described previously (Gu et al. 1998) (Fig. 5 A, increases of only 1.2–1.3-fold after plating on fibronectin). However, co-overexpression of PTEN with Shc resulted in an increase of MAP kinase activation by 2.9-fold 10 min after plating on a fibronectin substrate compared with levels in cells maintained in suspension (Fig. 5 A). In comparison, control cells transfected with GFP (−) alone (no GFP-PTEN) showed a similar 2.5-fold increase in ERK activation after fibronectin stimulation (data not shown).
Figure 5.

Effects of Shc or FAK overexpression on MAP kinase activation and Shc phosphorylation inhibited by PTEN. U-87MG cells were cotransfected with pHA262pur and GFP-PTEN with or without Flag-Shc or HA-FAK. After selection using puromycin, cells were serum-restricted overnight in 0.2% FBS. Cells were detached and incubated in serum-free medium for 30 min at 37°C as described under Materials and Methods. (A) For assaying MAP kinase activation, the cells were either maintained in suspension (−) or allowed to attach to fibronectin-coated dishes for 10 min (+), which is the time of maximal activation in these cells (Gu et al. 1998). Cell lysates were subjected to 10% SDS-PAGE. After electroblotting, blots were analyzed using anti–phospho-ERK1/2 antibody (top), and total quantities of ERK1/2 were confirmed using anti-ERK1/2 antibody (bottom). (B) After selection by puromycin, cells were either maintained in suspension (−) or allowed to attach to fibronectin-coated dishes for 30 min (+). Total Shc was immunoprecipitated with anti-Shc mAb, and then immunoblotted with antiphosphotyrosine (top) or total Shc (bottom). The level of Shc overexpression (52 kD) was increased 2.7-fold as compared with endogenous Shc (52 kD) as determined by densitometry. Ctr, control transfected with PTEN; and Vec, vector control transfectant without PTEN. The lanes labeled Shc, FAK, or Shc + FAK were cotransfected with PTEN plus the indicated expression plasmids.
The changes in MAP kinase activation accompanying overexpression of Shc in PTEN-reconstituted cells were associated with increases in Shc tyrosine phosphorylation as shown in Fig. 5 B. In contrast, overexpression of FAK produced little or no change in Shc tyrosine phosphorylation (Fig. 5 B), even though it substantially enhanced FAK phosphorylation (Fig. 6 A). These results in U-87MG cells differ from findings in 293 cells in which FAK overexpression produced elevated Shc phosphorylation and MAP kinase activation (Schlaepfer and Hunter 1997; Schlaepfer et al. 1998). We confirmed that transfection of FAK in 293 cells indeed produced elevated Shc phosphorylation (twofold) and MAP kinase activation; however, the levels of overexpression differed markedly in the two cell systems, with a two to threefold increase in total FAK levels in U-87MG cells and >10-fold increases in 293 cells, which may account for the differences (Fig. 5 and Fig. 6, and data not shown).
Figure 6.

Shc overexpression does not affect FAK and p130Cas phosphorylation. Cell lysates were prepared from cells cotransfected with PTEN and the indicated plasmids (labels at top) in suspension (−) or on fibronectin substrates (+) as described in Fig. 5. Proteins were immunoprecipitated from the lysates with either FAK or p130Cas antibodies, and then immunoblotted with antiphosphotyrosine. (A) Phospho-FAK (P-FAK, top) and total FAK (T-FAK, bottom); and (B) phospho-p130Cas (top) and total p130Cas (bottom). The level of FAK overexpression in FAK transfectants was increased 2.5-fold according to densitometry.
Shc Overexpression Does Not Affect FAK and p130Cas Phosphorylation
The activation of integrins by cell binding to extracellular matrix leads to increases in both Shc and FAK phosphorylation and enhances signaling pathways. We tested for possible overlaps between the Shc and FAK pathways by examining for effects of Shc overexpression on the FAK-p130Cas activation pathway by comparing FAK and p130Cas phosphorylation levels in U-87MG cells cotransfected with PTEN and Shc or FAK. As shown in Fig. 6 A, Shc overexpression did not increase FAK phosphorylation, which remained at levels similar to controls transfected with PTEN alone; in contrast, FAK overexpression clearly enhanced FAK phosphorylation as previously reported (Tamura et al. 1999a). Examining downstream p130Cas phosphorylation, FAK overexpression substantially enhanced p130Cas phosphorylation but Shc overexpression could not (Fig. 6 B). These results support the hypothesis that two separate pathways originating from Shc or from FAK are downregulated by PTEN.
Shc and FAK Regulate Cell Migration via Distinct Pathways
Cell migration was measured by time-lapse video microscopy and tracking of patterns of motility. Fig. 7 A, a, shows a representative set of motility records of U-87MG cells. PTEN inhibited movement of individual cells (Fig. 7 A, b), which is consistent with previous results (Tamura et al. 1999a). Unexpectedly, we found that Shc and FAK each regulated cell movement in a different manner. In cells cotransfected with Shc and PTEN, the cells moved more rapidly but in random directions with a relatively limited number of runs that persisted in the same direction (Fig. 7 A, c). In contrast, in cells cotransfected with FAK and PTEN, the cells tended to continue to migrate in a particular direction, i.e., persistent movement (Fig. 7 A, d).
Figure 7.


Shc and FAK reconstitute different aspects of PTEN-inhibited cell migration. (A) Representative examples of cell movements on fibronectin tracked at 20-min intervals over a span of 3 h. (a) Cells transfected with GFP (−) as a control; (b) cells transfected with GFP-PTEN only; (c) cells transfected with GFP-PTEN and Shc; and (d) cells transfected with GFP-PTEN and FAK. (B) Quantitation of persistence of migratory directionality. Relative D/T ratios represent the ratios of the direct distance from start point to end point (D) divided by the total track distance (T), expressed relative to the ratio after GFP-PTEN transfection alone (value set = 1.0). Error bars indicate SD of results pooled from three independent experiments (each with similar results and a total of at least 30 cells per condition). Asterisk, P < 0.0005 versus control transfected with GFP-PTEN alone. Two asterisks, P < 0.0005 versus samples cotransfected with GFP-PTEN plus Shc or activated MEK1 and also P < 0.001 versus samples cotransfected with GFP-PTEN and FAK or p130Cas. (C) Plot of mean square displacement against time. The net displacement (D) was measured every 40 min from start to end point. Error bars indicate SEM. At least 15 cells were measured for each point. Open circles represent the cells cotransfected with GFP-PTEN plus FAK and closed circles represent cells cotransfected with GFP-PTEN plus Shc. Note that the x-intercept for Shc transfectants is substantially closer to the origin than in the intercept for FAK transfectants.
To quantify these differences in migration patterns, we compared the ratios of the shortest direct distance from the starting point of each recording to the end point (D), to the total distance traversed by the cell (T). For ease of comparisons, the ratio D/T was normalized to a value of 1.0 for cells transfected with PTEN alone. As shown in Fig. 7 B, cotransfection of Shc with PTEN substantially reduced the ratio to 54% compared with controls transfected with PTEN alone. However, cotransfection by PTEN with FAK significantly increased the D/T value by 1.75-fold over controls transfected with PTEN alone. Interestingly, cotransfection of both Shc and FAK with PTEN resulted in an apparent reconstitution to a ratio characteristic of nontransfected cells (Fig. 7 B): the ratio was ∼1.25-fold higher than with PTEN alone, which represented restoration of the original ratio observed in control cells transfected with GFP (−) vector alone (i.e., no PTEN, Shc, or FAK transfection). The differences between the triple transfection (PTEN, Shc, and FAK) and double transfections (PTEN and Shc or PTEN and FAK) were significant at the P < 0.0005 and P < 0.001 levels, respectively. Furthermore, overexpression of constitutively activated MEK1 to enhance MAP kinase activation mimicked the actions of Shc and reduced the ratio as shown in Fig. 7 B. In contrast, overexpression of p130Cas produced effects similar to FAK and increased the ratio. Finally, even though transfection of cells with the dominant negative Shc construct reduced the speed of cell migration (Fig. 2), it resulted in a 1.5-fold increase in the D/T ratio (data not shown). These results strongly suggest that PTEN inhibits cell movement through at least two different pathways, i.e., Shc–MAP kinase and FAK-p130Cas.
To confirm random versus directional cell motility, we used a mean square displacement assay (Gail 1973). Net displacements (D) of cells from their location at time zero of video time-lapse microscopy was determined every 40 min and the mean square displacement (D2) was calculated and plotted against time as shown in Fig. 7 C. In pure random movement, the plot would be a straight line passing through the origin. The x-intercept for Shc was much closer to the origin than the intercept for FAK, indicating that Shc promotes relatively random movement, whereas FAK promotes considerably more directional migration.
Additional Evidence for Separate Shc and FAK Pathways
Overexpression of FAK in U-87MG cells did not significantly increase the level of tyrosine phosphorylation of Shc (Fig. 5 B) even though total phosphorylated FAK was considerably increased (Fig. 6 A). Dominant negative (dominant interfering) mutants of FAK, Cas, and Shc were used to test further the extent of separation of FAK and Shc pathways regulating migratory speed or directionality. U-87MG cells were cotransfected with PTEN to suppress migration, and then Shc or FAK cotransfectants were probed for specificity of each pathway using dominant negative FAK (the truncated version of FAK termed FRNK), dominant negative Cas (missing the substrate domain), or dominant negative Shc (Y239/317F). There were no significant effects of FRNK and dominant negative Cas on Shc-promoted cell motility: overexpression of Shc in PTEN-transfected cells plus FRNK or dominant interfering Cas cotransfection produced minimal effects on cell motility (Table ). Furthermore, D/T ratios were also only minimally affected compared with parallel transfectants without FRNK or dominant interfering Cas (Table ).
Table 1.
Minimal Cross-inhibition by Dominant Negative Inhibitors
| Transfectant | Additional plasmid | Migration rate ± SD | Directionality |
|---|---|---|---|
| μm/3 h | D/T ratio | ||
| PTEN + Shc | None | 110 ± 24 | 0.57 ± 0.20 |
| PTEN + Shc | FRNK | 101 ± 20 | 0.53 ± 0.25 |
| PTEN + Shc | Dn-Cas | 105 ± 18 | 0.50 ± 0.20 |
| PTEN + FAK | None | 117 ± 24 | 1.72 ± 0.14 |
| PTEN + FAK | Dn-Shc | 104 ± 21 | 1.81 ± 0.16 |
| PTEN + Cas | None | 118 ± 23 | 1.65 ± 0.17 |
| PTEN + Cas | Dn-Shc | 108 ± 18 | 1.82 ± 0.18 |
U-87MG cells were cotransfected with PTEN plus the indicated plasmids, and then analyzed by video time-lapse microscopy and image analysis for rates of cell migration and directionality as described in Materials and Methods. Dn-Cas, dominant negative Cas plasmid ΔSD-p130Cas lacking the substrate domain. Dn-Shc, dominant negative Shc plasmid Y239/317F with a double mutation in tyrosine phosphorylation sites. These dominant interfering plasmids substantially inhibited migration in cells in which the FAK and Shc pathways were not inhibited by PTEN (see Fig. 2).
Conversely, dominant negative Shc cotransfected with FAK or p130Cas also resulted in minimal effects on either rates of cell motility or the increase of D/T ratios dependent on the FAK pathway (Table ). These results reveal only minimal effects of Shc on the FAK-p130Cas pathway, whereas the same dominant negative Shc construct had substantial effects on migration when both putative pathways were active (Fig. 2).
Additional evidence for differences between the FAK and Shc pathways was provided by the use of MEK and phosphatidylinositol 3′-kinase inhibitors. The specific MEK inhibitor PD98059 abolished the increase in cell migration dependent on Shc (cells reconstituted with PTEN and cotransfected with Shc), producing a 96% reduction in cell motility compared with untreated controls (Table ). In clear contrast, there was no significant inhibition (10%) of cell migration by PD98059 in parallel cells cotransfected with FAK and PTEN (Table ). Furthermore, the phosphatidylinositol 3′-kinase inhibitor wortmannin substantially inhibited cell migration activated by FAK overexpression, producing a 65% reduction in rates of FAK-induced cell motility (Table ). A very recent report describes a similar inhibition by phosphatidylinositol 3′-kinase inhibitors of migration enhanced by FAK overexpression in CHO cells (80% inhibition; Reiske et al. 1999). In contrast, wortmannin had much less effect on our Shc-overexpressing cells, with a modest 23% decrease in the increased migration because of Shc.
Table 2.
Effects of Inhibitors on Shc- or FAK-induced Migration
| Transfectant | Inhibitor | Migration rate ± SD | Increase vs. PTEN | Percentage inhibition |
|---|---|---|---|---|
| μm/3 h | μm/3 h | |||
| PTEN | None | 54 ± 17 | — | — |
| PTEN + Shc | None | 110 ± 24 | 56 | 0% |
| PTEN + Shc | PD98059 | 56 ± 18 | 2 | 96% |
| PTEN + Shc | Wortmannin | 99 ± 18 | 43 | 23% |
| PTEN + FAK | None | 117 ± 24 | 63 | 0% |
| PTEN + FAK | PD98059 | 111 ± 15 | 57 | 10% |
| PTEN + FAK | Wortmannin | 76 ± 65 | 22 | 65% |
U-87MG cells, transiently transfected with the indicated expression plasmids, were treated with inhibitors or left untreated, and then were analyzed for rates of cell migration as described in Materials and Methods. The column entitled Increase shows net increases in the migration rate compared to the PTEN-reconstituted control.
Since a FAK-independent Src family kinase pathway has been described for the tyrosine phosphorylation of Shc (Sieg et al. 1998; Wary et al. 1998), we examined for possible effects of Src-related kinase activity on migration in these cells. Inhibition of function of Src kinases by the use of Csk overexpression had minimal inhibitory effects on the FAK pathway (FAK-overexpressing cells), with migration rates of 117 ± 24 μm/3 h in controls compared with 105 ± 25 μm/3 h in Csk-overexpressing cells; there were no detectable effects of Csk overexpression on Shc-enhanced motility. In these experiments, Csk transfection resulted in a 7-fold increase in Csk protein by Western blotting and a 2.5-fold enhancement of Src tyrosine phosphorylation, but no evidence for significant roles of Src in regulating migration of these cells could be demonstrated.
FRNK and Dominant Negative p130Cas Inhibit the Directionally Persistent Component of Movement Remaining in the Absence of Serum
To evaluate the contribution of growth factor stimulation to the Shc pathway stimulated by integrin ligation (as in Fig. 5B), we measured cell movements in the absence of FBS. Cell migration rates were reduced to ∼64% of controls in the presence of serum and the directionality of migration of the cells became markedly persistent in serum-free medium, as shown in Fig. 8 A (Ctr). Consistent with the prediction that this residual directional component of migration would be FAK-dependent, transfection by the FAK dominant negative construct termed FRNK or by dominant negative Cas resulted in inhibition of migration (Fig. 8a and Fig. B, FRNK and Dn-Cas [Dn-p130Cas]). The differences between transfection with vector alone (Ctr) and transfection with FRNK or dominant negative Cas were significant at the P < 0.0001 level. Moreover, overexpression of FRNK or dominant negative Cas also substantially reduced the directionality of migration, as indicated by a decrease in D/T ratios, which was also significant at the P < 0.0001 level.
Figure 8.

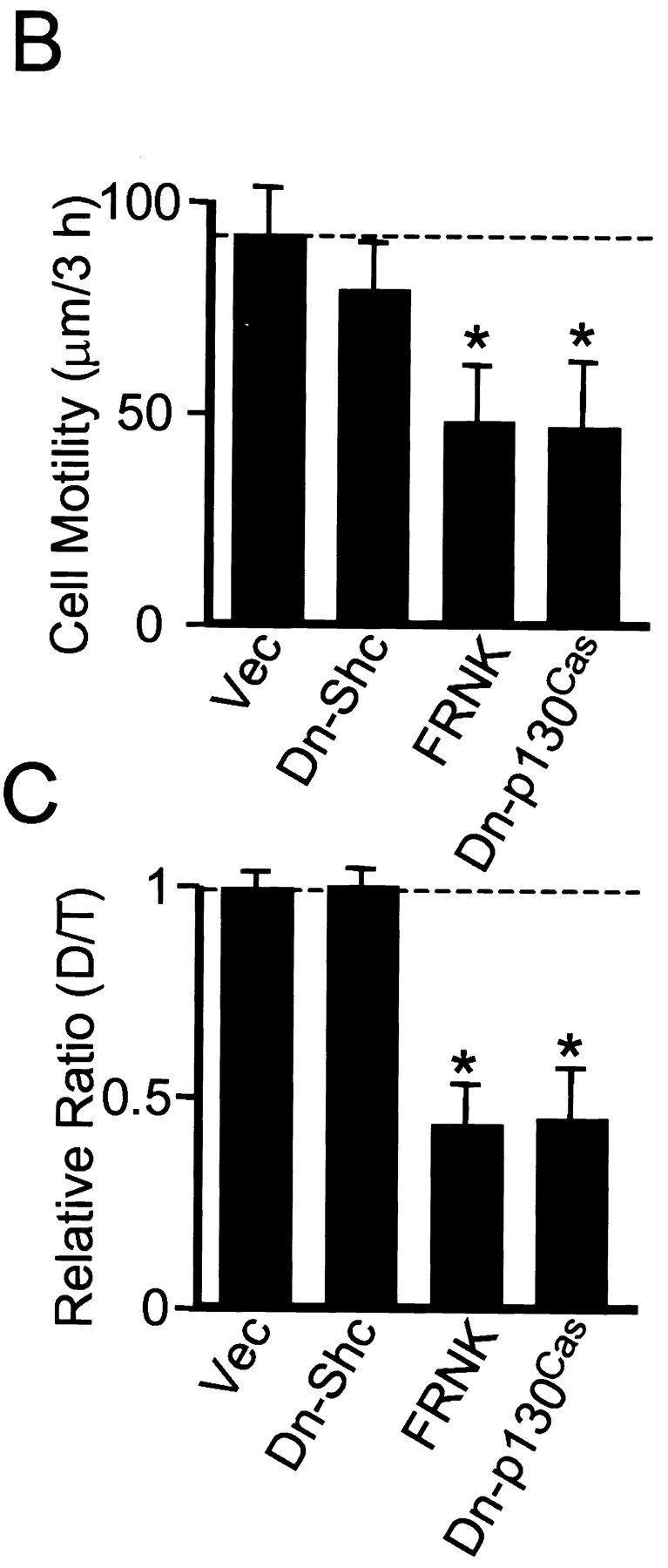
Dominant negative Shc and FRNK have different effects on cell migration in the absence of FBS. U-87MG cells were transfected with vector alone (Ctr) or dominant negative (Dn) plasmids of Shc, FAK, or p130Cas as indicated (Y239/317F Shc, the FRNK truncation of FAK, or ΔSD-p130Cas, respectively) and transfectants were selected by puromycin. After selection, cells were replated on fibronectin-coated dishes overnight without FBS. (A) Representative examples of cell movements on fibronectin tracked for 3 h without FBS. (B) Cell motility was calculated as velocity (μm/3 h). Data were collected from at least 20 cells from three independent experiments (each with similar results). Error bars indicate SD. (C) Quantitation of persistence of migratory directionality. The calculation of D/T ratios was performed as described for Fig. 7. The relative ratio value of vector (Vec) transfection alone was set as 1.0. Error bars indicate SD of results pooled from three independent experiments. Asterisk, P < 0.0001 versus control transfected with vector alone.
In contrast, transfection with dominant negative Shc had only minor effects on this FAK-dependent form of cell motility (Fig. 8, A–C). Taken together, these findings in Fig. 5 and Fig. 8 suggest that the Shc pathway in U-87MG cells involves both integrins and growth factors for Shc phosphorylation and its downstream effects, and that there are at least two distinct pathways regulating cell motility. These findings appear to be consistent with a previous report that Ras signaling (presumably including MAP kinase signaling) is involved in cell migration stimulated by PDGF (Kundra et al. 1994), yet cells expressing dominant negative Ras were still able to migrate on fibronectin (Kundra et al. 1995), which could have been due to involvement of the FAK-p130Cas pathway.
Shc and FAK Overexpression Have Different Effects on Actin Cytoskeleton and Focal Adhesions
Our previous studies had indicated that PTEN affects cell migration and invasion on fibronectin and had shown that FAK or p130Cas could rescue these functions (Tamura et al. 1999a). Moreover, transfection of constitutively activated MEK1 to induce MAP kinase activity could partially rescue cell spreading impaired by PTEN (Gu et al. 1998). In this study, Shc was found to enhance PTEN-downmodulated actin cytoskeletal organization (Fig. 9 A, P + Shc), but the actin microfilament bundles tended to be shorter than in control cells transfected with GFP tag only (Fig. 9 A, Ctr) with interrupted patterns of rhodamine-phalloidin staining. These Shc-transfected cells showed increased numbers of focal adhesions (Fig. 9 B, P + Shc), but not to the extent seen in control cells (Fig. 9 B, Ctr). Activated MEK1 produced similar patterns of partially enhanced actin microfilament organization (Fig. 9 A, P + Mek) that were not organized to the level of control cells. As reported above, both transfectants showed enhanced random motility.
Figure 9.
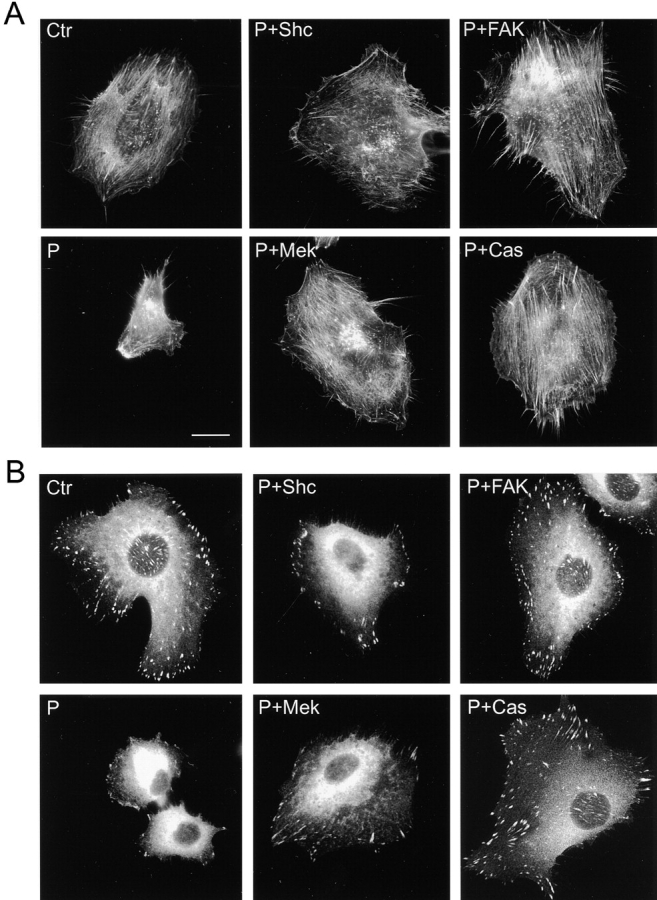
Differential effects of Shc and FAK pathways on enhancing actin cytoskeletal organization and focal contact formation downmodulated by PTEN. U-87MG cells were transfected with various constructs and selected as described above. They were allowed to spread on fibronectin-coated coverslips overnight in complete culture medium, and then stained with rhodamine-phalloidin to detect F-actin (A) and with antipaxillin antibody to detect focal contacts (B). Ctr, cells transfected with GFP (−) only; P, cells transfected with GFP-PTEN only; in the other frames, cells were cotransfected with GFP-PTEN plus each of the indicated plasmids; Mek, constitutively activated MEK1; and Cas, p130Cas. Bar, 20 μm.
In contrast, FAK-cotransfected cells showed relatively complete restoration of extensive and oriented patterns of actin microfilament bundles (Fig. 9 A, P + FAK) with extensive focal adhesions (Fig. 9 B, P + FAK) as detected by antipaxillin staining. Cells transfected with p130Cas, which also showed enhanced directional migration, also showed strongly organized actin cytoskeleton (Fig. 9 A, P + Cas) as well as well organized focal contacts (Fig. 9 B, P + Cas). These results establish distinct effects of Shc and FAK pathways on the actin cytoskeleton and focal adhesions, both of which are downmodulated by PTEN.
To quantify these apparent differences in morphological effects of signaling by FAK and Shc pathways, we applied a semi-quantitative morphometric measure for actin microfilament orientation that involves sampling and scoring a site within each quadrant of the cell for local actin filament orientation (see Materials and Methods). This actin orientation index confirmed the restoration of a striking degree of actin microfilament orientation in FAK- or p130Cas-overexpressing PTEN transfectant cells, as opposed to the relatively random organization of short actin filaments in Shc- and activated MEK1-overexpressing cells (Fig. 10, left). We also counted total numbers of focal adhesions in each cell and found that Shc and activated MEK1 had a lesser but significant ability to rescue focal adhesion formation downregulated by PTEN as compared with FAK and p130Cas (Fig. 10, right).
Figure 10.

Quantitative analysis of effects of Shc and FAK signaling pathways on actin cytoskeletal organization and focal adhesions downregulated by PTEN. Cells were transfected with various plasmids and stained as described in Fig. 9. (Left) Actin orientation was analyzed by F-actin staining and quantified using four fields per cell as described in Materials and Methods. The actin orientation index represents the mean number of sampling boxes that were positive for actin orientation per cell. (Right) Focal adhesion plaques were stained by antipaxillin antibody and the number of focal adhesions was counted for each cell. Error bars indicate SD of results from at least 15 cells. Asterisk, P < 0.0001 versus transfection by GFP-PTEN alone or GFP (−) alone.
Discussion
Cell migration is a complex process that can be regulated by multiple mechanisms, including by the newly discovered tumor suppressor protein PTEN (Tamura et al. 1998, Tamura et al. 1999a). This phosphatase has both phosphoinositol lipid and phosphoprotein substrates (Maehama and Dixon 1998; Myers et al. 1998; Tamura et al. 1998). In this study, we have explored the integration of the regulation of cell migration by PTEN, Shc, and FAK pathways. We examined the intriguing possibility that the effects of PTEN on Shc phosphorylation levels and on cell migration might be causally related, e.g., through a previously undescribed Shc-initiated pathway for regulation of the speed or directionality components of cell migration.
Using transfection reconstitution, dominant negative, and biochemical approaches, we have found the following. (a) We have established a mechanism for our previous observation that PTEN transfection reduces the tyrosine phosphorylation of Shc and inhibits MAP kinase activation by demonstrating that PTEN can interact with Shc and can directly dephosphorylate it in vitro; we also show here that Shc overexpression can rescue PTEN-inhibited MAP kinase activation in U-87MG cells. (b) We have found that Shc overexpression can stimulate integrin-mediated cell migration and spreading downregulated by PTEN. (c) Conversely, we have demonstrated that cell migration is inhibited by a dominant negative mutant of Shc partially mimicking the action of PTEN. We also established that PTEN, Shc, and FAK regulate cell movement through two different mechanisms: one is a pathway from Shc through the MAP kinase pathway leading to the stimulation of random cell motility, and the other is from FAK through p130Cas leading to stimulation of directionally persistent cell migration. (e) We also have demonstrated that inhibition of the Shc component of migration results in slower but more directionally persistent migration because of retention of the FAK component of migration. (f) We have established that the increased random motility accompanying Shc and activated MEK1 action is associated with only partial cytoskeletal and focal contact enhancement, whereas the directional migration induced by FAK and p130Cas correlates with more extensive, oriented actin microfilament bundle (stress fiber) organization and focal contact formation. (g) Finally, we have demonstrated that the Shc/MEK1 pathway can enhance MAP kinase activation without affecting FAK/p130Cas phosphorylation, whereas moderate overexpression of FAK restores levels of tyrosine-phosphorylated FAK and p130Cas and stimulates migration with minimal effects on MAP kinase activation. These studies define two distinct pathways for regulating speed and directionality of cell migration that counterbalance and interdigitate with actions of the PTEN tumor suppressor protein.
The adapter protein Shc has been linked to specific integrin-dependent signaling pathways (Wary et al. 1996, Wary et al. 1998). Overexpression of Shc also reportedly enhances cell migration and growth in response to hepatocyte growth factor (Pelicci et al. 1995). Our studies provide, to our knowledge, the first report that Shc upregulates random cell migration mediated by integrins and serum factors in a process that opposes its downregulation by PTEN. Supporting this concept, overexpression of a dominant negative form of Shc, doubly mutated by changing tyrosines 239 and 317 to phenylalanine, substantially inhibits the random component of cell motility on fibronectin. A putative integrin-specific mutant in which only tyrosine 317 was mutated suggested that the ratio of integrin versus growth factor contribution to migration was roughly 40:60%.
We previously reported that PTEN inhibits integrin-mediated MAP kinase activation in this glioma cell line and find in this study that overexpression of Shc can rescue integrin-stimulated MAP kinase activation. Moreover, we find that transfection of constitutively activated MEK1 to activate MAP kinase can mimic the effects of Shc on random cell movement on fibronectin. In addition, the MEK inhibitor PD98059 substantially inhibits Shc-stimulated migration but does not inhibit FAK-stimulated migration. These findings suggest that Shc and PTEN can regulate cell motility by activation or suppression of the MAP kinase pathway. In fact, MAP kinase activation can accelerate integrin-mediated cell motility in some cells (Leavesley et al. 1993; Yenush et al. 1994; Klemke et al. 1997; Rigot et al. 1998), though not in others (Bornfeldt et al. 1994; Coffer et al. 1998). A recent study has shown that activated MAP kinase can directly phosphorylate and activate myosin light chain kinase, leading to phosphorylation of myosin light chains and promoting the cytoskeletal contraction necessary for cell movement (Klemke et al. 1997). Interestingly, EGF has been reported recently to stimulate random cell migration (Ware et al. 1998), which we speculate may also be related to its well-known enhancement of MAP kinase activation.
FAK also appears to have important roles in integrin signaling and cell migration. In CHO cells, FAK promotes integrin-mediated cell migration through the activation of p130Cas (Cary et al. 1998). Overexpression of FAK or p130Cas can also effectively rescue cell migration inhibited by PTEN (Tamura et al. 1999a and in this study). For comparing the roles of Shc versus FAK on cell motility, we used time-lapse video microscopy to examine rates and paths of cell motility, rather than only evaluating final outcomes using the Boyden chamber or in vitro scratch wound-healing assays. Our studies establish that Shc and downstream-activated MAP kinase (ERK) upregulate random cell motility. In clear contrast, FAK or downstream p130Cas upregulates directional motility. FAK may regulate migration using a pathway dependent on phosphatidylinositol 3′-kinase, e.g., as suggested by experiments using Wortmannin (this paper and Reiske et al. 1999). However, determining the mechanisms of phosphatidylinositol 3′-kinase involvement will require extensive future analysis. As summarized above, biochemical analyses in this cell line of the specificity (a) of Shc versus FAK for activation of MAP kinase, (b) of FAK but not Shc specificity for stimulating phosphorylation of FAK and p130Cas, and (c) of Shc but not FAK specificity for stimulation of Shc phosphorylation, also underscore the existence of distinct mechanisms. Taken together, our transfection and biochemical studies strongly suggest that there are at least two separate pathways for regulation of the velocity and directionality components of cell motility, and these pathways appear to be additive (Fig. 11).
Figure 11.

Model depicting proposed roles of PTEN, Shc, and FAK in integrin- and growth factor–mediated cell motility. Integrin and growth factors can collaboratively or separately stimulate the Shc-MAP kinase pathway. Integrin receptor engagement with fibronectin stimulates both FAK and Shc phosphorylation, and each initiates a distinct downstream signaling pathway activating either persistent movement or random migration; these pathways are additive. PTEN inhibits integrin-mediated FAK and Shc phosphorylation by direct dephosphorylation, thereby inhibiting cell migration and spreading. This model depicts at least two pathways for downstream regulation of cell migration in U-87MG cells: one involving Shc to a MAP kinase pathway producing random motility and the other a FAK to p130Cas pathway involving directional migration. For activation of the Shc random motility pathway, the contributions of integrins compared with serum growth factors were estimated to be roughly 40 versus 60% in these cells, as determined by a putative integrin-specific Shc mutant. It should also be noted, however, that higher levels of FAK overexpression can activate FAK and affect the Shc pathway in 293 cells as described (Schlaepfer and Hunter 1997, and this study; dashed lines). Summation of these regulatory processes controls the speed and the directionality of cell migration.
FAK provides one of several possible pathways for activation of the Ras–MAP kinase signaling pathway (Schlaepfer et al. 1994). However, in contrast to results with 293 and 3T3 cells, FAK does not appear to have strong effects on this pathway in U-87MG cells (data in this paper and Gu, J., unpublished results). In contrast to FAK, Shc is the more plausible effector for integrin- and growth factor–mediated MAP kinase activation in these cells since dominant negative Shc overexpression can effectively inhibit MAP kinase activation (data not shown). Cell migration can be viewed as a process regulated by counterbalanced signals that can control rates of motility by several mechanisms. Strength of cell adhesion is one mechanism, where suboptimal, optimal, or inhibitory degrees of cell adhesion can regulate speed of locomotion (Duband et al. 1991; Schmidt et al. 1993; Akasaka et al. 1995; Gilmore and Romer 1996; Huttenlocher et al. 1996; Palecek et al. 1997). In fact, extensive formation of focal adhesions has been linked to the slowing of cell migration (Couchman and Rees 1979; Duband et al. 1988; Dunlevy and Couchman 1993; Ilic et al. 1995). In addition, however, cytoskeletal systems are likely to play important roles in modulating rates and directionality of migration (Zigmond 1993; Oliver et al. 1994; Huttenlocher et al. 1995). The distinct pathways involving Shc–MAP kinase versus FAK-p130Cas defined in this paper produce distinct effects on the actin cytoskeleton and focal contact organization. Although both pathways produce cell spreading and increased organization of the actin-containing cytoskeleton downregulated by PTEN, Shc and MEK1 induced less actin organization compared with the more strongly organized and oriented actin bundles characteristic of FAK and p130Cas action. This enhanced orientation of the cytoskeleton is consistent with the maintenance of directional migration, although other mechanisms cannot be entirely excluded. It is noteworthy that this extent of focal contact formation and actin organization was obviously not sufficiently high to retard cell migration, which was accelerated. Taken together, these results suggest that an intermediate level of focal adhesion formation and actin microfilament organization are optimal for the highest velocity and directionality of migration of these cells and that speed and directionality of migration are separable.
Besides the phosphoproteins examined in this study, PTEN has a major lipid substrate that is important biologically. PTEN directly dephosphorylates PIP3, which is produced by phosphatidylinositol 3′-kinase and can activate the PKB/Akt signaling pathway. PTEN is thought to regulate cell growth and cell death by apoptosis and/or anoikis via this pathway (Davies et al. 1998; Haas-Kogan et al. 1998; Li et al. 1998; Myers et al. 1998; Stambolic et al. 1998; Suzuki et al. 1998; Tamura et al. 1999b; Wu et al. 1998). Nevertheless, the intertwined regulatory effects of Shc, PTEN, and FAK on migration that we describe do not appear to involve this PKB/Akt pathway. Even though phosphatidylinositol 3′-kinase is known to have regulatory effects on cell migration (Keely, et al. 1997; Shaw et al. 1997; Sander et al. 1998), recent studies indicate that phosphatidylinositol 3′-kinase induction of scattering acts through effectors other than PKB/Akt and requires at least basal MAP kinase function (Khwaja et al. 1998). In this study, inhibition of phosphatidylinositol 3′-kinase by Wortmannin also reduced the rate of cell migration of U-87MG cells, but it targeted the FAK pathway selectively. We have been unable to find any reports of PKB/Akt regulation of cell movement. In fact, we found that dominant negative Akt did not affect U-87MG cell movement on fibronectin, suggesting no role for PKB/Akt in regulating migration, at least in this cell system.
In conclusion, we propose that there are at least three signaling pathways regulated by PTEN: (1) a PIP3–PKB/Akt pathway affecting growth and apoptosis, (2) a Shc–MAP kinase pathway affecting random cell motility, and (3) a FAK-p130Cas pathway that contributes a directional motility component to cell migration. FAK and p130Cas have been related to effects of PTEN on regulating tumor cell invasiveness (Tamura et al. 1999a) and this study suggests that their effects were likely due to the directional component of migration. These different regulatory systems appear to be intertwined and provide countervailing influences on speed and directionality of integrin-mediated cell migration. Integration of their actions provides a mechanism for intracellular regulation of cell migration.
Acknowledgments
We thank Tim Springer (Harvard Medical School, Cambridge, MA) for suggesting application of the mean square displacement technique for characterizing random versus directional migration.
Erik Danen was supported by a fellowship from the Dutch Cancer Society and Takahisa Takino was supported by a fellowship from the Japan Society for the Promotion of Science.
Footnotes
1.used in this paper: Csk, COOH-terminal Src kinase; ERK, extracellular signal-related kinase; FAK, focal adhesion kinase; FRNK, dominant negative FAK truncation; GFP, green fluorescent protein; HA, hemagglutinin; MAP, mitogen-activated protein; MEK, MAP or ERK kinase; p130Cas, p130 Crk-associated substrate; PIP3, phosphatidylinositol 3,4,5-trisphosphate
The present address of J. Gu is Division of Protein Chemistry, Institute for Protein Research, Osaka University, 3-2 Yamadaoka, Suita, Osaka 565, Japan. The present address of M. Tamura is Second Department of Internal Medicine, University of Occupational and Environmental Health, 1-1 Iseigaoka, Yahata-nishi, Kitakyushu 807-8555, Japan. The present address of E.H.J. Danen is Division of Cell Biology, Netherlands Cancer Institute, Plesmanlaan 121, 1066 CX Amsterdam, The Netherlands.
References
- Akasaka T., van Leeuwen R.L., Yoshinaga I.G., Mihm M.C., Jr., Byers H.R. Focal adhesion kinase (p125FAK) expression correlates with motility of human melanoma cell lines. J. Invest. Dermatol. 1995;105:104–108. doi: 10.1111/1523-1747.ep12313396. [DOI] [PubMed] [Google Scholar]
- Bornfeldt K.E., Raines E.W., Nakano T., Graves L.M., Krebs E.G., Ross R. Insulin-like growth factor-I and platelet-derived growth factor-BB induce directed migration of human arterial smooth muscle cells via signaling pathways that are distinct from those of proliferation. J. Clin. Invest. 1994;93:1266–1274. doi: 10.1172/JCI117081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cary L.A., Chang J.F., Guan J.-L. Stimulation of cell migration by overexpression of focal adhesion kinase and its association with Src and Fyn. J. Cell Sci. 1996;109:1787–1794. doi: 10.1242/jcs.109.7.1787. [DOI] [PubMed] [Google Scholar]
- Cary L.A., Han D.C., Polte T.R., Hanks S.K., Guan J.L. Identification of p130Cas as a mediator of focal adhesion kinase–promoted cell migration. J. Cell Biol. 1998;140:211–221. doi: 10.1083/jcb.140.1.211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheney I.W., Johnson D.E., Vaillancourt M.T., Avanzini J., Morimoto A., Demers G.W., Wills K.N., Shabram P.W., Bolen J.B., Tavtigian S.V., Bookstein R. Suppression of tumorigenicity of glioblastoma cells by adenovirus-mediated MMAC1/PTEN gene transfer. Cancer Res. 1998;58:2331–2334. [PubMed] [Google Scholar]
- Coffer P.J., Geijsen N., M'Rabet L., Schweizer R.C., Maikoe T., Raaijmakers J.A., Lammers J.W., Koenderman L. Comparison of the roles of mitogen-activated protein kinase kinase and phosphatidylinositol 3-kinase signal transduction in neutrophil effector function. Biochem. J. 1998;329:121–130. doi: 10.1042/bj3290121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cong L.N., Chen H., Li Y., Zhou L., McGibbon M.A., Taylor S.I., Quon M.J. Physiological role of Akt in insulin-stimulated translocation of GLUT4 in transfected rat adipose cells. Mol. Endocrinol. 1997;11:1881–1890. doi: 10.1210/mend.11.13.0027. [DOI] [PubMed] [Google Scholar]
- Couchman J.R., Rees D.A. The behaviour of fibroblasts migrating from chick heart explantschanges in adhesion, locomotion and growth, and in the distribution of actomyosin and fibronectin. J. Cell Sci. 1979;39:149–165. doi: 10.1242/jcs.39.1.149. [DOI] [PubMed] [Google Scholar]
- Davies M.A., Lu Y., Sano T., Fang X., Tang P., LaPushin R., Koul D., Bookstein R., Stokoe D., Yung W.K., Mills G.B., Steck P.A. Adenoviral transgene expression of MMAC/PTEN in human glioma cells inhibits Akt activation and induces anoikis. Cancer Res. 1998;58:5285–5290. [PubMed] [Google Scholar]
- Di Cristofano A., Pesce B., Cordon-Cardo C., Pandolfi P.P. PTEN is essential for embryonic development and tumour suppression. Nat. Genet. 1998;19:348–355. doi: 10.1038/1235. [DOI] [PubMed] [Google Scholar]
- Duband J.L., Nuckolls G.H., Ishihara A., Hasegawa T., Yamada K.M., Thiery J.P., Jacobson K. Fibronectin receptor exhibits high lateral mobility in embryonic locomoting cells but is immobile in focal contacts and fibrillar streaks in stationary cells. J. Cell Biol. 1988;107:1385–1396. doi: 10.1083/jcb.107.4.1385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duband J.L., Dufour S., Yamada S.S., Yamada K.M., Thiery J.P. Neural crest cell locomotion induced by antibodies to β1 integrins. A tool for studying the roles of substratum molecular avidity and density in migration. J. Cell Sci. 1991;98:517–532. doi: 10.1242/jcs.98.4.517. [DOI] [PubMed] [Google Scholar]
- Dunlevy J.R., Couchman J.R. Controlled induction of focal adhesion disassembly and migration in primary fibroblasts. J. Cell Sci. 1993;105:489–500. doi: 10.1242/jcs.105.2.489. [DOI] [PubMed] [Google Scholar]
- Flint A.J., Tiganis T., Barford D., Tonks N.K. Development of substrate-trapping mutants to identify physiological substrates of protein tyrosine phosphatases. Proc. Natl. Acad. Sci. USA. 1997;94:1680–1685. doi: 10.1073/pnas.94.5.1680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furnari F.B., Lin H., Huang H.S., Cavenee W.K. Growth suppression of glioma cells by PTEN requires a functional phosphatase catalytic domain. Proc. Natl. Acad. Sci. USA. 1997;94:12479–12484. doi: 10.1073/pnas.94.23.12479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gail, M. 1973. Time lapse studies on the motility of fibroblasts in tissue culture. In Ciba Foundation Symposium 14: Locomotion of Tissue Cells. Elsevier Science Publishing Co., Inc., North Holland, Amsterdam. 287–302. [DOI] [PubMed]
- Gilmore A.P., Romer L.H. Inhibition of focal adhesion kinase (FAK) signaling in focal adhesions decreases cell motility and proliferation. Mol. Biol. Cell. 1996;7:1209–1224. doi: 10.1091/mbc.7.8.1209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu J., Tamura M., Yamada K.M. Tumor suppressor PTEN inhibits integrin- and growth factor–mediated mitogen-activated protein (MAP) kinase signaling pathways. J. Cell Biol. 1998;143:1375–1383. doi: 10.1083/jcb.143.5.1375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haas-Kogan D., Shalev N., Wong M., Mills G., Yount G., Stokoe D. Protein kinase B (PKB/Akt) activity is elevated in glioblastoma cells due to mutation of the tumor suppressor PTEN/MMAC. Curr. Biol. 1998;8:1195–1198. doi: 10.1016/s0960-9822(07)00493-9. [DOI] [PubMed] [Google Scholar]
- Huttenlocher A., Sandborg R.R., Horwitz A.F. Adhesion in cell migration. Curr. Opin. Cell Biol. 1995;7:697–706. doi: 10.1016/0955-0674(95)80112-x. [DOI] [PubMed] [Google Scholar]
- Huttenlocher A., Ginsberg M.H., Horwitz A.F. Modulation of cell migration by integrin-mediated cytoskeletal linkages and ligand-binding affinity. J. Cell Biol. 1996;134:1551–1562. doi: 10.1083/jcb.134.6.1551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hynes R.O., Lander A.D. Contact and adhesive specificities in the associations, migrations, and targeting of cells and axons. Cell. 1992;68:303–322. doi: 10.1016/0092-8674(92)90472-o. [DOI] [PubMed] [Google Scholar]
- Ilic D., Furuta Y., Kanazawa S., Takeda N., Sobue K., Nakatsuji N., Nomura S., Fujimoto J., Okada M., Yamamoto T. Reduced cell motility and enhanced focal adhesion contact formation in cells from FAK-deficient mice. Nature. 1995;377:539–544. doi: 10.1038/377539a0. [DOI] [PubMed] [Google Scholar]
- Keely P.J., Westwick J.K., Whitehead I.P., Der C.J., Parise L.V. Cdc42 and Rac1 induce integrin-mediated cell motility and invasiveness through PI(3)K. Nature. 1997;390:632–636. doi: 10.1038/37656. [DOI] [PubMed] [Google Scholar]
- Khwaja A., Lehmann K., Marte B.M., Downward J. Phosphoinositide 3-kinase induces scattering and tubulogenesis in epithelial cells through a novel pathway. J. Biol. Chem. 1998;273:18793–18801. doi: 10.1074/jbc.273.30.18793. [DOI] [PubMed] [Google Scholar]
- Klemke R.L., Cai S., Giannini A.L., Gallagher P.J., de Lanerolle P., Cheresh D.A. Regulation of cell motility by mitogen-activated protein kinase. J. Cell Biol. 1997;137:481–492. doi: 10.1083/jcb.137.2.481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kundra V., Escobedo J.A., Kazlauskas A., Kim H.K., Rhee S.G., Williams L.T., Zetter B.R. Regulation of chemotaxis by the platelet-derived growth factor receptor-beta. Nature. 1994;367:474–476. doi: 10.1038/367474a0. [DOI] [PubMed] [Google Scholar]
- Kundra V., Anand-Apte B., Feig L.A., Zetter B.R. The chemotactic response to PDGF-BBevidence of a role for Ras. J. Cell Biol. 1995;130:725–731. doi: 10.1083/jcb.130.3.725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lacalle R.A., Pulido D., Vara J., Zalacain M., Jimenez A. Molecular analysis of the pac gene encoding a puromycin N-acetyl transferase from Streptomyces alboniger . Gene. 1989;79:375–380. doi: 10.1016/0378-1119(89)90220-5. [DOI] [PubMed] [Google Scholar]
- LaFlamme S.E., Thomas L.A., Yamada S.S., Yamada K.M. Single subunit chimeric integrins as mimics and inhibitors of endogenous integrin functions in receptor localization, cell spreading and migration, and matrix assembly. J. Cell Biol. 1994;126:1287–1298. doi: 10.1083/jcb.126.5.1287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lauffenburger D.A., Horwitz A.F. Cell migrationa physically integrated molecular process. Cell. 1996;84:359–369. doi: 10.1016/s0092-8674(00)81280-5. [DOI] [PubMed] [Google Scholar]
- Leavesley D.I., Schwartz M.A., Rosenfeld M., Cheresh D.A. Integrin β1- and β3-mediated endothelial cell migration is triggered through distinct signaling mechanisms. J. Cell Biol. 1993;121:163–170. doi: 10.1083/jcb.121.1.163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li D.M., Sun H. TEP1, encoded by a candidate tumor suppressor locus, is a novel protein tyrosine phosphatase regulated by transforming growth factor beta. Cancer Res. 1997;57:2124–2129. [PubMed] [Google Scholar]
- Li D.M., Sun H. PTEN/MMAC1/TEP1 suppresses the tumorigenicity and induces G1 cell cycle arrest in human glioblastoma cells. Proc. Natl. Acad. Sci. USA. 1998;95:15406–15411. doi: 10.1073/pnas.95.26.15406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J., Yen C., Liaw D., Podsypanina K., Bose S., Wang S.I., Puc J., Miliaresis C., Rodgers L., McCombie R. PTEN, a putative protein tyrosine phosphatase gene mutated in human brain, breast, and prostate cancer. Science. 1997;275:1943–1947. doi: 10.1126/science.275.5308.1943. [DOI] [PubMed] [Google Scholar]
- Li J., Simpson L., Takahashi M., Miliaresis C., Myers M.P., Tonks N., Parsons R. The PTEN/MMAC1 tumor suppressor induces cell death that is rescued by the AKT/protein kinase B oncogene. Cancer Res. 1998;58:5667–5672. [PubMed] [Google Scholar]
- Lin T.H., Aplin A.E., Shen Y., Chen Q., Schaller M., Romer L., Aukhil I., Juliano R.L. Integrin-mediated activation of MAP kinase is independent of FAKevidence for dual integrin signaling pathways in fibroblasts. J. Cell Biol. 1997;136:1385–1395. doi: 10.1083/jcb.136.6.1385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maehama T., Dixon J.E. The tumor suppressor, PTEN/MMAC1, dephosphorylates the lipid second messenger, phosphatidylinositol 3,4,5-trisphosphate. J. Biol. Chem. 1998;273:13375–13378. doi: 10.1074/jbc.273.22.13375. [DOI] [PubMed] [Google Scholar]
- Mainiero F., Murgia C., Wary K.K., Curatola A.M., Pepe A., Blumemberg M., Westwick J.K., Der C.J., Giancotti F.G. The coupling of α6β4 integrin to Ras-MAP kinase pathways mediated by Shc controls keratinocyte proliferation. EMBO (Eur. Mol. Biol. Organ.) J. 1997;16:2365–2375. doi: 10.1093/emboj/16.9.2365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mansour S.J., Matten W.T., Hermann A.S., Candia J.M., Rong S., Fukasawa K., Vande Woude G.F., Ahn N.G. Transformation of mammalian cells by constitutively active MAP kinase kinase. Science. 1994;265:966–970. doi: 10.1126/science.8052857. [DOI] [PubMed] [Google Scholar]
- Migliaccio E., Mele S., Salcini A.E., Pelicci G., Lai K.M., Superti-Furga G., Pawson T., Di Fiore P.P., Lanfrancone L., Pelicci P.G. Opposite effects of the p52shc/p46shc and p66shc splicing isoforms on the EGF receptor-MAP kinase-fos signalling pathway. EMBO (Eur. Mol. Biol. Organ.) J. 1997;16:706–716. doi: 10.1093/emboj/16.4.706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Myers M.P., Stolarov J.P., Eng C., Li J., Wang S.I., Wigler M.H., Parsons R., Tonks N.K. P-TEN, the tumor suppressor from human chromosome 10q23, is a dual-specificity phosphatase. Proc. Natl. Acad. Sci. USA. 1997;94:9052–9057. doi: 10.1073/pnas.94.17.9052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Myers M.P., Pass I., Batty I.H., Van der Kaay J., Stolarov J.P., Hemmings B.A., Wigler M.H., Downes C.P., Tonks N.K. The lipid phosphatase activity of PTEN is critical for its tumor supressor function. Proc. Natl. Acad. Sci. USA. 1998;95:13513–13518. doi: 10.1073/pnas.95.23.13513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nada S., Okada M., MacAuley A., Cooper J.A., Nakagawa H. Cloning of a complementary DNA for a protein-tyrosine kinase that specifically phosphorylates a negative regulatory site of p60c-src. Nature. 1991;351:69–72. doi: 10.1038/351069a0. [DOI] [PubMed] [Google Scholar]
- Nakamoto T., Sakai R., Honda H., Ogawa S., Ueno H., Suzuki T., Aizawa S., Yazaki Y., Hirai H. Requirements for localization of p130cas to focal adhesions. Mol. Cell. Biol. 1997;17:3884–3897. doi: 10.1128/mcb.17.7.3884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nobes C.D., Hall A. Rho, rac, and cdc42 GTPases regulate the assembly of multimolecular focal complexes associated with actin stress fibers, lamellipodia, and filopodia. Cell. 1995;81:53–62. doi: 10.1016/0092-8674(95)90370-4. [DOI] [PubMed] [Google Scholar]
- Oliver T., Lee J., Jacobson K. Forces exerted by locomoting cells. Semin. Cell Biol. 1994;5:139–147. doi: 10.1006/scel.1994.1018. [DOI] [PubMed] [Google Scholar]
- Palecek S.P., Loftus J.C., Ginsberg M.H., Lauffenburger D.A., Horwitz A.F. Integrin-ligand binding properties govern cell migration speed through cell-substratum adhesiveness. Nature. 1997;385:537–540. doi: 10.1038/385537a0. [DOI] [PubMed] [Google Scholar]
- Pawson T., Scott J.D. Signaling through scaffold, anchoring, and adaptor proteins. Science. 1997;278:2075–2080. doi: 10.1126/science.278.5346.2075. [DOI] [PubMed] [Google Scholar]
- Pelicci G., Giordano S., Zhen Z., Salcini A.E., Lanfrancone L., Bardelli A., Panayotou G., Waterfield M.D., Ponzetto C., Pelicci P.G., Comoglio P.M. The motogenic and mitogenic responses to HGF are amplified by the Shc adaptor protein. Oncogene. 1995;10:1631–1638. [PubMed] [Google Scholar]
- Pronk G.J., de Vries-Smits A.M., Buday L., Downward J., Maassen J.A., Medema R.H., Bos J.L. Involvement of Shc in insulin- and epidermal growth factor-induced activation of p21ras. Mol. Cell. Biol. 1994;14:1575–1581. doi: 10.1128/mcb.14.3.1575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reiske H.R., Kao S.-C., Cary L.A., Guan J.-L., Lai J.-F., Chen H.C. Requirement of phosphatidylinositol 3-kinase in focal adhesion kinase-promoted cell migration. J. Biol. Chem. 1999;274:12361–12366. doi: 10.1074/jbc.274.18.12361. [DOI] [PubMed] [Google Scholar]
- Rigot V., Lehmann M., Andre F., Daemi N., Marvaldi J., Luis J. Integrin ligation and PKC activation are required for migration of colon carcinoma cells. J. Cell Sci. 1998;111:3119–3127. doi: 10.1242/jcs.111.20.3119. [DOI] [PubMed] [Google Scholar]
- Rozakis-Adcock M., McGlade J., Mbamalu G., Pelicci G., Daly R., Li W., Batzer A., Thomas S., Brugge J., Pelicci P.G. Association of the Shc and Grb2/Sem5 SH2-containing proteins is implicated in activation of the Ras pathway by tyrosine kinases. Nature. 1992;360:689–692. doi: 10.1038/360689a0. [DOI] [PubMed] [Google Scholar]
- Sander E.E., van Delft S., ten Klooster J.P., Reid T., van der Kammen R.A., Michiels F., Collard J.G. Matrix-dependent Tiam1/Rac signaling in epithelial cells promotes either cell–cell adhesion or cell migration and is regulated by phosphatidylinositol 3′-kinase. J. Cell Biol. 1998;143:1385–1398. doi: 10.1083/jcb.143.5.1385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlaepfer D.D., Hunter T. Focal adhesion kinase overexpression enhances ras-dependent integrin signaling to ERK2/mitogen-activated protein kinase through interactions with and activation of c-Src. J. Biol. Chem. 1997;272:13189–13195. doi: 10.1074/jbc.272.20.13189. [DOI] [PubMed] [Google Scholar]
- Schlaepfer D.D., Hanks S.K., Hunter T., van der Geer P. Integrin-mediated signal transduction linked to Ras pathway by GRB2 binding to focal adhesion kinase. Nature. 1994;372:786–791. doi: 10.1038/372786a0. [DOI] [PubMed] [Google Scholar]
- Schlaepfer D.D., Jones K.C., Hunter T. Multiple Grb2-mediated integrin-stimulated signaling pathways to ERK2/mitogen-activated protein kinasesummation of both c-Src- and focal adhesion kinase-initiated tyrosine phosphorylation events. Mol. Cell. Biol. 1998;18:2571–2585. doi: 10.1128/mcb.18.5.2571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt C.E., Horwitz A.F., Lauffenburger D.A., Sheetz M.P. Integrin–cytoskeletal interactions in migrating fibroblasts are dynamic, asymmetric, and regulated. J. Cell Biol. 1993;123:977–991. doi: 10.1083/jcb.123.4.977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaw L.M., Rabinovitz I., Wang H.H., Toker A., Mercurio A.M. Activation of phosphoinositide 3-OH kinase by the α6β4 integrin promotes carcinoma invasion. Cell. 1997;91:949–960. doi: 10.1016/s0092-8674(00)80486-9. [DOI] [PubMed] [Google Scholar]
- Sheetz M.P., Felsenfeld D.P., Galbraith C.G. Cell migrationregulation of force on extracellular-matrix-integrin complexes. Trends Cell Biol. 1998;8:51–54. doi: 10.1016/s0962-8924(98)80005-6. [DOI] [PubMed] [Google Scholar]
- Sieg D.J., Iiic D., Jones K.C., Damsky C.H., Hunter T., Schlaepfer D.D. Pyk2 and Src-family protein-tyrosine kinases compensate for the loss of FAK in fibronectin-stimulated signaling events but Pyk2 does not fully function to enhance FAK− cell migration. EMBO (Eur. Mol. Biol. Organ.) J. 1998;17:5933–5947. doi: 10.1093/emboj/17.20.5933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stambolic V., Suzuki A., de la Pompa J.L., Brothers G.M., Mirtsos C., Sasaki T., Ruland J., Penninger J.M., Siderovski D.P., Mak T.W. Negative regulation of PKB/Akt-dependent cell survival by the tumor suppressor PTEN. Cell. 1998;95:29–39. doi: 10.1016/s0092-8674(00)81780-8. [DOI] [PubMed] [Google Scholar]
- Steck P.A., Pershouse M.A., Jasser S.A., Yung W.K., Lin H., Ligon A.H., Langford L.A., Baumgard M.L., Hattier T., Davis T. Identification of a candidate tumour suppressor gene, MMAC1, at chromosome 10q23.3 that is mutated in multiple advanced cancers. Nat. Genet. 1997;15:356–362. doi: 10.1038/ng0497-356. [DOI] [PubMed] [Google Scholar]
- Stossel T.P. On the crawling of animal cells. Science. 1993;260:1086–1094. doi: 10.1126/science.8493552. [DOI] [PubMed] [Google Scholar]
- Suzuki A., de la Pompa J.L., Stambolic V., Elia A.J., Sasaki T., Barrantes I.B., Ho A., Wakeham A., Ltie A., Khoo W., Fukumoto M., Mak T.W. High cancer susceptibility and embryonic lethality associated with mutation of the PTEN tumor suppressor gene in mice. Curr. Biol. 1998;8:1169–1178. doi: 10.1016/s0960-9822(07)00488-5. [DOI] [PubMed] [Google Scholar]
- Tamura M., Gu J., Matsumoto K., Aota S., Parsons R., Yamada K.M. Inhibition of cell migration, spreading, and focal adhesions by tumor suppressor PTEN. Science. 1998;280:1614–1617. doi: 10.1126/science.280.5369.1614. [DOI] [PubMed] [Google Scholar]
- Tamura M., Gu J., Takino T., Yamada K.M. Tumor suppressor PTEN inhibition of cell invasion, migration, and growthdifferential involvement of focal adhesion kinase and p130cas Cancer Res. 59 1999. 442 449a [PubMed] [Google Scholar]
- Tamura M., Gu J., Danen E.H.J., Takino T., Miyamoto S., Yamada K.M. PTEN interactions with focal adhesion kinase and suppression of the extracellular matrix-dependent phosphatidylinositol 3-kinase/Akt cell survival pathway J. Biol. Chem. 274 1999. 20693 20703b [DOI] [PubMed] [Google Scholar]
- Ware M.F., Wells A., Lauffenburger D.A. Epidermal growth factor alters fibroblast migration speed and directional persistence reciprocally and in a matrix-dependent manner. J. Cell Sci. 1998;111:2423–2432. doi: 10.1242/jcs.111.16.2423. [DOI] [PubMed] [Google Scholar]
- Wary K.K., Mainiero F., Isakoff S.J., Marcantonio E.E., Giancotti F.G. The adaptor protein Shc couples a class of integrins to the control of cell cycle progression. Cell. 1996;87:733–743. doi: 10.1016/s0092-8674(00)81392-6. [DOI] [PubMed] [Google Scholar]
- Wary K.K., Mariotti A., Zurzolo C., Giancotti F.G. A requirement for caveolin-1 and associated kinase Fyn in integrin signaling and anchorage-dependent cell growth. Cell. 1998;94:625–634. doi: 10.1016/s0092-8674(00)81604-9. [DOI] [PubMed] [Google Scholar]
- Whang Y.E., Wu X., Suzuki H., Reiter R.E., Tran C., Vessella R.L., Said J.W., Isaacs W.B., Sawyers C.L. Inactivation of the tumor suppressor PTEN/MMAC1 in advanced human prostate cancer through loss of expression. Proc. Natl. Acad. Sci. USA. 1998;95:5246–5250. doi: 10.1073/pnas.95.9.5246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu X., Senechal K., Neshat M.S., Whang Y.E., Sawyers C.L. The PTEN/MMAC1 tumor suppressor phosphatase functions as a negative regulator of the phosphoinositide 3-kinase/Akt pathway. Proc. Natl. Acad. Sci. USA. 1998;95:15587–15591. doi: 10.1073/pnas.95.26.15587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yenush L., Kundra V., White M.F., Zetter B.R. Functional domains of the insulin receptor responsible for chemotactic signaling. J. Biol. Chem. 1994;269:100–104. [PubMed] [Google Scholar]
- Zigmond S.H. Recent quantitative studies of actin filament turnover during cell locomotion. Cell Motil. Cytoskelet. 1993;25:309–316. doi: 10.1002/cm.970250402. [DOI] [PubMed] [Google Scholar]