

Abstract
Presenilin 1 (PS1) is the causative gene for an autosomal dominant familial Alzheimer's disease (AD) mapped to chromosome 14. Here we show that QM/Jun-interacting factor (Jif)-1, a negative regulator of c-Jun, is a candidate to mediate the function of PS1 in the cell. We screened for proteins that bind to PS1 from a human embryonic brain cDNA library using the two-hybrid method and isolated one clone encoding the QM/Jif-1 gene. The binding of QM/Jif-1 to full-length PS1 was confirmed in vitro by pull-down assay, and in vivo by immunoprecipitation assays with human samples, including AD brains. Immunoelectronmicroscopic analysis showed that QM/Jif-1 and PS1 are colocalized at the endoplasmic reticulum, and the nuclear matrix in human brain neurons. Chloramphenicol acetyltransferase assays in F9 cells showed that PS1 suppresses transactivation by c-Jun/c-Jun but not by c-Jun/c-Fos heterodimers, consistent with the reported function of QM/Jif-1. By monitoring fluorescent recombinant protein and by gel mobility shift assays, PS1 was shown to accelerate the translocation of QM from the cytoplasm to the nucleus and to thereby suppress the binding of c-Jun homodimer to 12-O-tetradecanoylphorbol-13- acetate (TPA)-responsive element (TRE). PS1 suppressed c-jun–associated apoptosis by retinoic acid in F9 embryonic carcinoma cells, whereas this suppression of apoptosis is attenuated by mutation in PS1. Collectively, the novel function of PS1 via QM/Jif-1 influences c-jun–mediated transcription and apoptosis.
Keywords: Alzheimer's disease, presenilin-1, c-jun, cell death, QM/Jif-1
Mutations in the presenilin 1 (PS1)1 gene have been shown to influence the intracellular metabolism of β-amyloid (Aβ) and increase the ratio of Aβ1-42 peptides, which appears to be more fibrillogenic than Aβ1-40 in patient plasma as well as in brains of transgenic mice (for reviews see Hardy 1997; Selkoe 1997; Masters and Beyreuther 1998). Furthermore, amyloid deposits are actually formed earlier in doubly transgenic mice coexpressing mutant PS1 and mutant amyloid precursor protein (APP) than in age-matched mice (Borchelt et al. 1997; Holcomb et al. 1997), consistent with human Alzheimer's disease (AD) pathology.
In addition to the gained function of mutant genes, PS1 is physiologically indispensable for embryonic morphogenesis. PS1−/− mice exhibited abnormal patterning of the axial skeleton and spinal ganglia and developed cerebral cavitation (Shen et al. 1997; Wong et al. 1997). PS1 is a multipass transmembrane protein homologous to SEL-12, a Caenorhabditis elegans protein that mediates the signaling from Notch/Lin-12 family cell surface receptors (Levitan and Greenwald 1995), and has been localized to endoplasmic reticulum (Kovacs et al. 1996), Golgi apparatus (Kovacs et al. 1996), nuclear membrane (Li et al. 1997), and plasma membrane (Dewji and Singer 1997). PS1 is proteolytically cleaved into two fragments (Thinakaran et al. 1996), then presumably degraded by proteasome (Kim et al. 1997). During development, mutant PS1 molecules appear to function normally, because before the onset of disease, neither morphological nor functional abnormalities are observed in patients associated with the PS1 mutations.
Functional analyses of PS1 still remain controversial. PS1 was shown to participate directly in the cleavage of APP (Wolfe et al. 1999). Meanwhile, a number of molecules, including calsenilin (Buxbaum et al. 1998), β-catenin (Zhang, Z., et al., 1998), filamin (Zhang, W., et al., 1998), and tau (Takashima et al. 1998), were reported to bind to PS1, some of which were assumed to affect cell death signaling. Furthermore, although amyloid plaque formation has been thought to be a central event in the pathogenesis of AD, it was reported that neuronal death enhanced by mutant PS1 transgene precedes extracellular amyloid deposition in aged transgenic mice (Chui et al. 1999).
Therefore, it is necessary to further characterize molecules interacting with PS1 in order to understand the mechanism underlying this complex and wide spectrum of cellular functions for the PS1 molecule. In this study, we found that QM/Jun-interacting factor (Jif)-1 (Dowdy et al. 1991; Monteclaro and Vogt 1993), a transcription factor that interacts with c-jun and inhibits its transcriptional activation, binds to full-length PS1. We also found that QM/Jif-1 and PS1 are colocalized in neurons of the human brain, including those affected by PS-1–linked familial AD, and that full-length PS1 mediates the effects of QM/Jif-1 on c-Jun–mediated function. These results suggest that QM/Jif-1 is another molecule mediating the function of PS1.
Materials and Methods
Two-Hybrid cDNA Cloning
cDNA encoding full-length PS1 was amplified from human hippocampal mRNA (Clontech) by reverse transcriptase PCR using primers F, 5′-AAAGAATTCATGACAGAGTTACCTGCACCGT-3′ (sequence data available from EMBL/GenBank/DDBJ under accession no. L42110; nucleotides [nt] 249–270) and R, 5′-AAACTCGAGCCATGGGATT- CTAACCGC-3′ (nt 1677–1660), and subcloned between EcoRI and XhoI sites of pEG202–LexA fusion plasmid. After confirming that cotransfection of the pEGPS1 and pJG4-5 plasmids does not show nonspecific binding, ∼5 × 105 clones were screened from a human embryonic brain cDNA library (provided by Dr. Roger Brent, Harvard Medical School, Boston, MA). A second screening was performed with both SG-HWUX-gal and SG-HWUL plates. Positive clones were subcloned into pBluescript KS+ (Stratagene) and sequenced by using an automatic sequencer (Applied Biosystems, Inc.).
Immunoprecipitation
100 mg of each cerebral cortex tissue was suspended in 1 ml of lysis buffer (10 mM Tris-HCl, pH 7.8, 1% NP-40, 150 mM NaCl, 1 mM EDTA, 1 mM PMSF, 10 μg/ml leupeptin, 10 μg/ml aprotinin), incubated at 4°C for 10 min, and disrupted by repeated aspiration through a 21-gauge needle. Cellular debris was removed by centrifugation at 10,000 g for 10 min. Aliquots of cell lysates were incubated with various antibodies for 1 h at 4°C, then precipitated with protein G–agarose (Oncogene Science). Anti–NH2-terminal antibody (αN) and anti-loop antibody (αL) were used finally at 1:200 dilution. Anti-QM (C-17) polyclonal antibody (Santa Cruz Biotechnology) was used at 1:1,000 dilution. αN antibody is specific for amino acids 21–80 of human PS1 (Lah et al. 1997). αL is a polyclonal antiserum that reacts with epitopes in the hydrophilic loop domain (amino acids 263–407) of human and mouse PS1 (Thinakaran et al. 1996). The beads were washed extensively with Radio-Immuno-Protein Assay buffer, then separated by 12% SDS-PAGE, blotted to Hybond-ECL membrane (Amersham Pharmacia Biotech), incubated with anti-QM polyclonal antibody (Santa Cruz Biotechnology), then detected with ECL Western blotting analysis system (Amersham Pharmacia Biotech).
Pull Down Assay
Expression vectors of various forms of PS1 as well as glutathione S-transferase (GST)-QM fusion proteins were constructed as follows. Full-length PS1 cDNA was amplified with primers F182 (5′-AAACTCGAGTCTATACAGTTGCTCCAATGAC, nt 232–254) and R182 (5′-AAATCTAGACCATGGGATTCTAACCGCA, nt 1677–1660) from human hippocampal mRNA, and subcloned between XhoI and XbaI sites of pCIneo mammalian expression vector (Promega). The structure and sequence were confirmed by DNA sequencing and restriction site analyses. For pCImPS1 carrying a mutation Met→Leu at codon 146 associated with early-onset familial AD, site-directed mutagenesis was performed with Transformer Site-directed Mutagenesis kit (Clontech) according to the commercial protocol.
For vectors expressing GST fusion proteins in Escherichia coli, QM cDNA was subcloned between BamHI and EcoRI pGEX3X (Amersham Pharmacia Biotech) by using PCR with synthetic primers to adjust the reading frame. Fusion proteins linked to deleted QM (see Fig. 2 c) was constructed similarly by using PCR. PS1 protein was synthesized and radiolabeled with [35S]methionine (NEN) by in vitro transcription and translation with TNT T7/T3–coupled reticulocyte lysate system (Promega). Interaction between PS1 and GST-QM proteins was performed according to the reported method (Chen et al. 1997).
Figure 2.
(a) The full-length PS1 was coprecipitated with QM by anti-QM antibody from cerebral cortex tissues of normal (samples 1 and 2), disease control (amytrophic lateral sclerosis) (sample 3), nonfamilial AD (samples 4 and 5), or PS1-linked AD brains (samples 6 and 7). Sample 6 was from a patient with a Met146Leu mutation, and sample 7 was from a patient with an Ala246Glu mutation. An approximately 50-kD band of the full-length PS1 was detected by αN and αL. Immunoprecipitate from a normal brain (sample 1) by nonspecific serum (NS) was similarly detected as a negative control. The same filters were reprobed by anti-QM antibody (lower panels) to confirm that equal amounts of the protein samples were loaded on the gels. (b) Western blot analysis of the same human brain samples with αN and αL. (c) QM was coprecipitated with PS1 by αN or αL antibodies from the cerebral cortex of normal human brain. Nonspecific serum (NS) was used for immunoprecipitation as a negative control.



Immunoelectronmicroscopy
5 mm3 of tissue was obtained from the cerebral cortex of a 37-yr-old woman 24 h after her death by loss of blood, and fixed with 4% paraformaldehyde. Sections for electronmicroscopic immunocytochemistry were postfixed in 1% osmium tetroxide, stained with 2% uranyl acetate, dehydrated using graded alcohol and propylene oxide, and embedded in Eponate 12 resin. 40-μm sections were stained by rat mAb against PS121–80 (Lah et al. 1997) at 1:100–500 dilution, and anti-QM polyclonal rabbit antibody (Santa Cruz Biotechnology) at 1:1,000 dilution, simultaneously. 10 nm gold-conjugated anti–rat Ig and 5 nm gold-conjugated anti–rabbit secondary antibodies were used to localize primary antibodies. Control experiments were performed without primary antibodies. For the analyses with diseased brains, sections of formaldehyde-fixed and paraffin-embedded cortex were deparaffinized and used for the incubation with antibodies. For immunohistochemistry of the mouse brain, 40-μm sections were stained by anti-QM polyclonal antibody (Santa Cruz Biotechnology) then visualized with 0.05% 3,3′-DAB tetrahydrochloride and 0.01% H2O2 in 50 mM Tris, pH 7.6, as described previously (Lah et al. 1997).
Chloramphenicol Acetyltransferase Assay
Transfection was performed as described previously (Okamoto et al. 1990; Okazawa et al. 1991). 10 μg of reporter plasmid and 10–20 μg of effector plasmids were used. 10–20 μg pBluescript KS+ (Stratagene) was added to equilibrate the total amount of plasmids for transfection. Construction of effector and reporter plasmids was reported previously (Angel et al. 1987). Efficiency of tranfection was verified by pCH110 (Amersham Pharmacia Biotech), a eukaryotic vector containing the simian virus early promoter and E. coli–β-galactosidase (LacZ) structural gene. Each experiment was repeated at least four times and variation of transfection efficiency was <20%.
Enhanced Green Fluorescent Protein Fusion Protein Expression Vectors
Expression vectors of fluorescent protein–QM fusion proteins were constructed by inserting various forms of QM cDNAs into pEGFPN1 (Clontech). Full-length QM cDNA was amplified by RT-PCR from human amygdala mRNA with the primers QMF (AACGAATTCCCATGGGCCGCCGCCCCGCCCGTT) and QMR (AATGGATCCGTGAGTGCAGGGCCCGCCA), and subcloned between EcoRI and BamHI sites of pEGFPN1.
c-Jun NH2-terminal Kinase Activity Assay
The effect of PS1 on c-Jun NH2-terminal kinase (JNK) activities was analyzed by transfecting 6 μg of T7-tagged JNK1 with 10 μg of pCIPS1 or pBS-KS as control into F9 cells. JNK1 protein was recovered by immunoprecipitation with mouse mAb against T7 epitope (Invitrogen). After the Sepharose resin was washed five times with lysis buffer containing 20 mM Tris-HCl, pH 8.0, 2 mM EDTA, 50 mM β-glycerophosphate, 0.1 mM Na3VO4, 1% Triton X-100, 0.5% deoxycholate, 0.1% SDS, 10% glycerol, 1 mM PMSF, 10 μg/ml leupeptin, and 10 μg/ml aprotinin, the proteins were recovered with SDS sample buffer and analyzed by Western blotting with anti–T7-Tag antibodies.
Retinoic Acid–induced Apoptosis
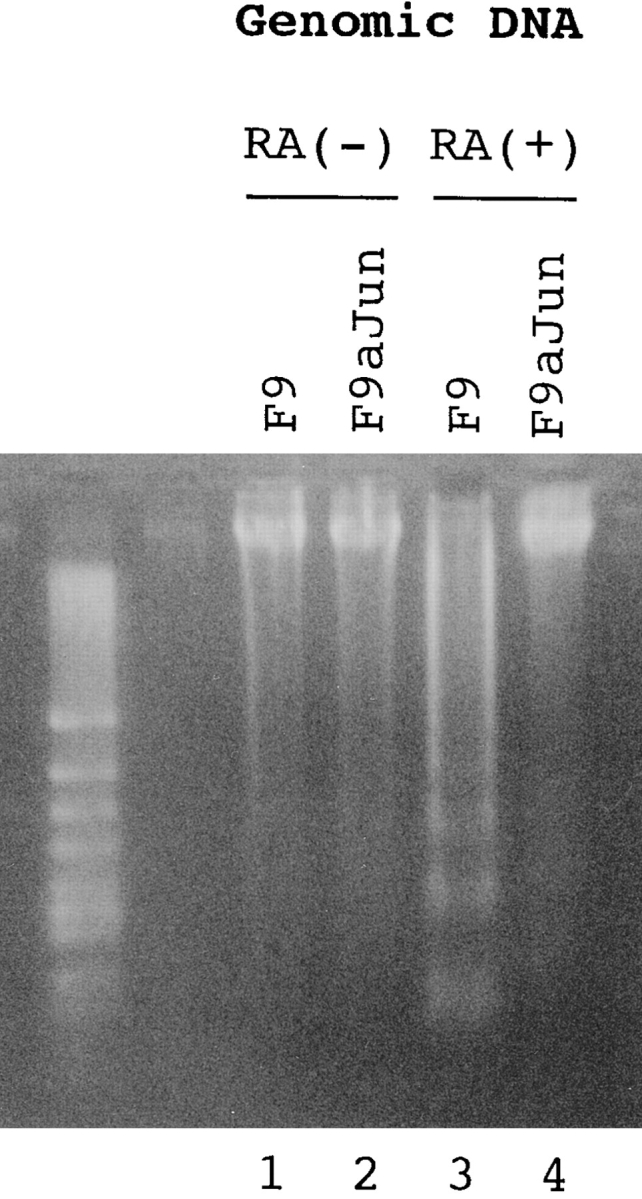
To make stable cells expressing antisense c-jun, pCIneo (Promega) containing the full-length c-jun cDNA at the reverse orientation was constructed and transfected into F9 cells. They were selected in α-medium (Sigma Chemical Co.) with 300 μg/ml G418 for 2 wk. Stable cells expressing normal or mutant PS1 were similarly made by transfecting pCIPS1 and pCImPS1. 1 × 105 F9 or stable cells were cultured in α-medium: 10% FBS with 1 μM all-trans retinoic acid (Sigma Chemical Co.) for 48 h. All the cells were collected and their genomic DNA were extracted and separated on 3% Nusieve-agarose gel (FMC BioProducts).
For analysis of the effect of deletion constructs of QM on apoptosis, 1 × 105 F9PS1 cells were transiently transfected with 20 μg of each expression vector described below using SuperFect (Qiagen) in a 10-cm tissue culture dish. 24 h after transfection, cells were treated with 1 μM all-trans retinoic acid (Sigma) for an additional 48 h. All the cells were collected and the percentage of cell death was estimated by trypan blue dye exclusion.
Deletion Constructs of QM/Jif-1
QM/Jif-1 cDNA fragments were amplified with RT-PCR from human hippocampal mRNA (Stratagene). The primers used were F1 (AAACTCGAGCCTGGTGTCGCCATG; sequence data available from EMBL/GenBank/DDBJ under accesion no. M64241; nt 30–44), R1 (AAAGAATTCAATTCGGGCAGCCTCCA, nt 251–235), F2 (AAAGAATTCCGCATCAACAAGATGT, nt 333–348), R2 (AAATCTAGAAGCCCTCATGAGTGCA, nt 691–676), and R3 (AAATCTAGAACATC-TTGTTGATGCG, nt 348–333). Different portions of QM cDNA were amplified with F1 and R3 for Δ1 or with F1 and R1 for Δ2, digested with XhoI and XbaI or with XhoI and EcoRI, and subcloned into corresponding sites of pCIneo vector (Promega), respectively. For Δ3, two cDNA fragments amplified with F1/R1 or F2/R2 were digested with XhoI/EcoRI or with EcoRI/XbaI, respectively, then subcloned between XhoI and XbaI sites of pCIneo.
Results
Identification of QM/Jif-1, a Negative Regulator of c-Jun, as a Binding Protein to PS1
We have investigated the function of PS1 by isolating the molecules that interact with PS1. We screened a human embryonic brain library using the yeast two-hybrid system. After double second screens using leucine-deficient plates and X-gal plates, we finally judged six clones showing strong interaction to be positive. In our experience with the two-hybrid system, this number of positive clones was small compared with other baits. Among them, we found a clone identical to QM/Jif-1. This molecule was isolated originally as a putative Wilms's tumor suppressor gene (Dowdy et al. 1991), then described as a transcription factor that interacts with c-Jun and inhibits its transcriptional activation (Monteclaro and Vogt 1993). Recently, it was shown to be identical to a ribosomal protein, L10 (Chan et al. 1996), which was therefore suggested to be a ribosomal protein possessing multiple extraribosomal functions (Wool 1996).
This clone (PS309-4) lacked an NH2-terminal portion of 43 amino acids and encoded a variant Ser202Asn (Fig. 1 a). QM/Jif-1 does not possess a leucine zipper but might compose a C2H2-type zinc finger (Fig. 1 a). Retransformation of EGY48 yeast cells by pEGPS1 and pJGPS309-4 plasmids showed high β-galactosidase activities in independent yeast colonies (Fig. 1 b).
Figure 1.
(a) Protein sequence of QM/PS309-4. Arrow indicates NH2-terminal end of the PS309-4 clone. Amino acid substitution in PS309-4 (Ser202Asn) is indicated with a bold letter. (b) Two-hybrid interaction between PS309-4 and PS1. pJGPS309-4 was retransfected into EGY48 yeast cells that had been transformed by pEGPS1 bait plasmid and pSH18-34 reporter plasmid. Two independent colonies were picked up from SD-HWU plate and tested on SG-HWUL and SG-HWU-Xgal plates. Negative (pEGPS1/pJG4-5) and positive (pEGShc/pJGShc) controls were equally tested. (c) Two-hybrid analysis on interaction between QM and various portions of the PS1 protein. Partial cDNAs corresponding to the number of amino acids of the deletion plasmids were amplified by PCR with synthetic primers and subcloned into pEG. EGY48 yeast cells were cotransfected with these pEG vectors, pJGPS309-4 and pSH18-34, and then incubated for 2 d on selection plates. Quantitative analysis of β-galactosidase activity was performed according to the standard protocol (Guarente 1983).



To determine the binding domain of PS1 molecule to QM/Jif-1, we performed deletion analysis of PS1 by two-hybrid assay. cDNAs corresponding to various regions of the PS1 molecule were subcloned into the pEG vector and cotransfected with pJG309-4 into yeast cells. Interestingly, any partial sequence of PS1 did not bind strongly to QM/Jif-1. Instead, full-length PS1 showed a strong interaction with QM/Jif-1 (Fig. 1 c). This finding corresponds well to the results of Western blot analysis using human brains described below.
In Vivo Interaction between QM/Jif-1 and PS1
To verify the interaction between QM and PS1 in vivo, we performed immunoprecipitation assay with human brains, including those of familial and nonfamilial AD patients. Approximately 50 kD full-length PS1 was detected in the precipitates by αN and by αL from normal, disease control (amyotrophic lateral sclerosis), nonfamilial AD, and PS1-linked AD brains (Fig. 2 a), whereas we could not observe clear bands corresponding to the cleaved PS1 fragments. This result in human brain, together with the results from two-hybrid deletion analyses (Fig. 1 c), suggest that multiple regions in the full-length structure of PS1 are necessary for tight interaction with QM/Jif-1. This idea might have some relationship to the recent finding that NH2- and COOH-terminal PS1 fragments reassociate and form a stable complex (Capell et al. 1998; Thinakaran et al. 1998).
Interestingly, the band was visible but very weak in lanes 6 and 7 loaded with samples from PS1-linked AD patients (Fig. 2 a). Compared with the PS1 bands in Western blot analysis using the same brain samples (Fig. 2 b), it is not due to difference of the PS1 protein amounts among brain samples. Instead, it could be due to the difference of residual neurons where QM and PS1 may interact, among samples, or due to the difference in affinity of QM to normal and mutant PS1. In the reverse immunoprecipitation assay, we detected QM in precipitates by αN as well as by αL (Fig. 2 c), reconfirming the interaction between PS1 and QM/Jif1 in vivo. We performed immunoprecipitation with nonimmune sera and with several nonspecific antisera using the same brain samples, but did not find QM or PS1 in the precipitates (data not shown).
PS1 and QM/Jif-1 Colocalize in Cortical Neurons
To observe the interaction between QM and PS1 in the brain morphologically, we performed immunohistochemical analyses. As QM expression has not been reported previously, we confirmed that the QM message is widely expressed in the brain by Northern blot analysis (Fig. 3). At the light microscopic level, anti-QM polyclonal antibody stained the cytoplasm of neurons in the mouse cerebral cortex (Fig. 4 a). To further characterize subcellular localization of the QM protein, we observed the mouse brain sample with electronmicroscopy and found that regions very close to tubular membrane structures, which possessed features of smooth endoplasmic reticulum, were predominantly stained (Fig. 4b and Fig. c). This subcellular localization of QM was exactly like that of PS1 reported to date (Lah et al. 1997).
Figure 3.

Northern blot analyses of the QM expression in various organs and various regions of the brain. PS309-4 cDNA was used as the probe. The final wash was 0.1× SSC/0.1% SDS at 60°C for 60 min. nucl, nucleus.
Figure 4.
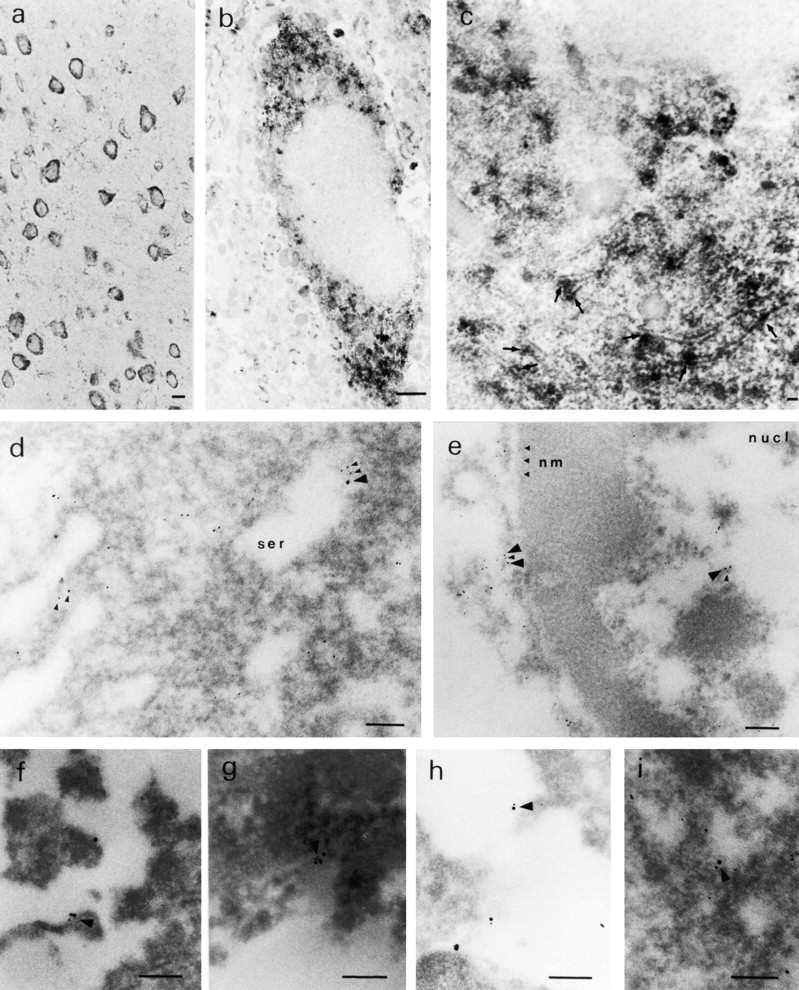
(a) Light microscopic immunohistochemistry of the mouse cerebral cortex. Cytoplasm of the cortical neurons are stained by anti-QM antibody. Bar, 5 μm. (b) Electron microscopic immunohistochemistry with DAB showing a single neuron in the mouse cerebral cortex. The cytoplasm is stained in a granular manner. Bar, 1 μm. (c) High power magnification of a neuron in the mouse cerebral cortex. DAB staining is located mainly at smooth tubular membrane structures (arrows). Bar, 100 nm. (d) Electron microscopic immunohistochemistry of a human cortical neuron with gold-conjugated secondary antibodies. QM (5-nm gold grains) and PS1 (10-nm gold grains) are located predominantly around smooth membrane structures. Bar, 100 nm. (e) Grains of PS1 and QM in the nucleus (nucl) of a human cortical neuron are fewer than those in the cytoplasm, but sometimes colocalized. Bar, 100 nm. (f–i) 5- and 10-nm grains exist close to each other (arrow heads) in normal human brain (f), nonfamilial AD human brain (g), PS1-linked AD human brain (h), and in amyotrophic lateral sclerosis human brain (i). Bar, 100 nm (f–i).
Next, we asked whether the QM and PS1 proteins are colocalized in the human brain neurons. Immunohistochemical analysis was performed with secondary antibodies conjugated to different sizes of gold particles and with the brain of a 37-yr-old woman who died due to loss of blood. We observed that the 5-nm grain of PS1 and 10-nm grain of QM were located very close to each other at the edge of smooth membrane structures in the cytoplasm of cortical neurons (Fig. 4 d). Most of those structures seemed to be smooth endoplasmic reticulum, whereas some of them possessed the features of Golgi apparatus (data not shown). Fewer grains were observed also in the nucleus (Fig. 4 e). Although the immunoreactivity was further reduced in the formaldehyde-fixed and paraffin-embedded tissues of nonfamilial or PS1-linked AD brains, we repeatedly found that these two grains were colocalized at membrane structure in the cytoplasm (Fig. 4, f–i).
PS1 Suppresses the Action of c-jun Homodimer
Next, we investigated the biological significance of the interaction between QM/Jif-1 and PS1. Jif-1 was isolated as a protein binding to c-Jun from a cDNA library screened with biotinylated c-Jun (Monteclaro and Vogt 1993). Work by Monteclaro and Vogt on the function of Jif-1 has shown that GST–QM/Jif-1 fusion proteins do not bind to 12-O-tetradecanoylphorbol-13-acetate (TPA)-responsive element (TRE) in gel mobility shift assays, but prevent the binding of a c-Jun homodimer to the TRE (Monteclaro and Vogt 1993). Moreover, the c-Jun/c-Fos heterodimer can override this inhibition by Jif-1. Through this binding inhibition, Jif-1 inhibits gene transactivation by c-Jun. QM/Jif-1 is extremely conserved during evolution from yeast to mammal (Farmer et al. 1994), suggesting that this factor is essential for basic cellular functions. From these observations, we supposed that expression of PS1 might affect the function of c-Jun through interaction with QM.
To test this hypothesis, we performed cotransfection chloramphenicol acetyltransferase (CAT) assays with c-jun or c-fos expression vectors as the first effector plasmid, normal or mutant PS1 expression vectors as the second effector plasmid (Fig. 5 a), and human collagenase (−517/−42) TKCAT vector containing TRE as reporter plasmid. We used F9 embryonic carcinoma cells in the assays because they possess almost no activated protein (AP)-1 activity (Angel et al. 1988), but express enough of the QM protein (data not shown). It was difficult to predict what kind of results would emerge with the addition of the PS1 expression vector, because the binding might keep QM in the endoplasmic reticulum where PS1 is most abundant (Kovacs et al. 1996), or it might assist the transport of QM to the nucleus where PS1 immunoreactivity is also detected (Li et al. 1997). It could be that cotransfection might not affect CAT activities due to a nonfunctional binding between the two molecules.
Figure 5.
(a) Constructions of PS1 expression vectors and 1× TRE CAT plasmids. PS1 cDNAs were subcloned into pCIneo directed by the cytomegalovirus immediate early enhancer/promoter. For 1× TRE CAT plasmids, synthetic oligonucleotides were inserted into tkCAT. (b) CAT assay showing the effect of normal or mutant (Met146Leu) PS1 on c-jun-, c-fos-, or c-jun/c-fos–mediated transactivation. Expression of normal as well as mutant PS1 repressed transactivation of the collagenase gene enhancer/promoter by c-jun but not by c-fos or c-jun/c-fos. As control, effect of GLUT1 on c-jun–induced transactivation was examined. pBluescript KS+ was added to equilibrate total amounts of plasmids. (c) CAT assay using various 1× TRE CAT reporter plasmids. Cotransfection of PS1 clearly suppressed transactivation by c-jun on 1× TRE vectors. (d) CAT assay using junB and junD expression vectors. PS1 expression suppressed junD-induced transactivation but not junB-induced transactivation. (e) Protein expression of c-Jun and of normal or mutant PS1 in transfected cells used for CAT assays. Proteins were prepared from the cells used for CAT assays, separated by SDS-PAGE, and detected by anti–c-Jun antibody (αJun) or by anti–PS1-loop antibody (αPS1L). The bands corresponding to c-Jun, full-length PS1, and COOH-terminal fragment of PS1 were indicated by arrowheads. Lanes 1–4 correspond to lanes 1–4 in b. (f) Histogram showing the mean fold transactivation of multiple assays.






Among these possibilities, we obtained the simplest and clearest outcome. c-jun or c-jun/c-fos transactivated the CAT gene expression (Fig. 5 b) as expected. The transactivation by c-Jun homodimer was suppressed by normal PS1 as well as mutant PS1, whereas the transactivation by c-Jun/c-Fos heterodimer was not affected by adding PS1 expression vectors (Fig. 5, a and b). This suppressive effect was considered to be specific, because overexpression of a multipass transmembrane protein, glucose transporter 1 (GLUT1), which is known to move from the endoplasmic reticulum to the Golgi apparatus, did not suppress CAT activity (Fig. 5 b). The weak transactivation by c-Fos, which might have been induced with a weak endogenous c-Jun activity, was not influenced by either normal or mutant PS1. These observations corresponded very well with the interacting behavior of QM to AP-1 molecules reported previously (Monteclaro and Vogt 1993). The suppression by mutant PS1 seemed weaker than that by normal PS1 (Fig. 5 b).
To examine whether the effect of PS1 is mediated by TRE, we changed the reporter plasmid to those that contained only TRE (Fig. 5 a). As expected, PS1 reproduced suppression of the c-Jun–mediated transactivation in collagenase 1× TRE CAT and metallothionein IIa 1× TRE CAT (Fig. 5 c). These results supported our hypothesis that PS1 affects gene regulation by c-jun through TRE. Interestingly, by using 1× TRE CAT plasmids, we observed more clearly that the suppression of c-Jun–induced transactivation was weaker in mutant PS1 than in normal PS1 (Fig. 5 c).
PS1 Represses Transactivation by junD but Not by junB
Second, we tested whether PS1 affects transcriptional regulation by the other c-jun family members forming an AP-1 complex. Expression of junB and junD enhanced transcription from collagenase 1× TRE CAT reporter plasmid (Fig. 5 d). Transactivation by junD was clearly suppressed by expression of normal and mutant PS1, whereas transactivation by junB was not affected (Fig. 5 d). In this case, suppression of junD-mediated transactivation by normal PS1 was not remarkably different from that by mutant PS1 (Fig. 5 d), in contrast to our observations with c-jun–mediated transactivation (Fig. 5b and Fig. c). In the CAT assays described above, we confirmed that expression of the c-Jun protein was not influenced by cotransfecting PS1, and that expression of normal and mutant PS1 proteins was equivalent (Fig. 5 e). We summarized results from all the CAT assays described above with a histogram showing mean fold transactivations (Fig. 5 f).
PS1 Promotes Translocation of QM/Jif-1 to the Nucleus
From the data described above, we hypothesized that PS1 somehow promotes translocation of the QM protein from the cytoplasm to the nucleus, inhibits the binding of c-Jun homodimer to TRE, and thereby suppresses transactivation by c-jun. We used a fluorescent protein (enhanced green fluorescent protein [EGFP]) fusion reporter plasmid to observe how intracellular transport of the QM protein is modulated by PS1, and observed that translocation of the fusion protein to the nucleus is actually accelerated by cotransfecting PS1 (Fig. 6 a). On the other hand, coexpression of mutant (Met146Leu) PS1 did not remarkably promote the nuclear translocation.
Figure 6.
(a) Translocation of EGFP-QM fusion protein to the nucleus was promoted by cotransfecting normal PS1. When pEGFQM was transfected alone into F9 cells, it was distributed predominantly in the cytoplasm. By coexpressing normal PS1, distribution of the EGFP-QM fusion protein was shifted to the nucleus. This nuclear translocation was not obvious when mutant (Met146Leu) PS1 was coexpressed. Expression of normal and mutant PS1 proteins was equivalent in Western blot analysis performed similarly to Fig. 5 e. Percentage of nuclear translocation was calculated by (number of cells with stronger fluorescence in the nucleus)/(total number of fluorescence-positive cells) × 100. About 100 fluorescence-positive cells were observed in each transfection. (b) Gel mobility shift assay showing the effect of normal or mutant PS1 on the DNA-protein binding of various AP-1 complexes. F9 cells were transfected by c-jun, c-jun/c-fos, junD, or junB expression vectors with pBluescript as control, normal PS1 (PS1), or mutant PS1 (mPS1). 10 μg of each plasmid was transfected by calcium phosphate method and nuclear extract was prepared 36 h after transfection. Oligonucleotide corresponding to metallothionein IIa TRE was radiolabeled with Klenow enzyme and used as the probe. C, DNA–protein complex. F, free probe. Cotransfection of normal PS1 remarkably suppressed the binding of c-Jun homodimer, whereas mutant PS1 suppressed weakly. The DNA–protein complex of the c-Jun/c-Fos heterodimer and JunB homodimer was not remarkably affected. The complex of the JunD homodimer was inhibited weakly.


It is interesting to note that the effects of PS1 on transcription and on protein transport were remarkable although the expression level of PS1 was not as high as that of PS1 (Fig. 5 e), or as the endogenous expression level of QM/Jif-1 (data not shown). Considered with the function of PS1 assisting the nuclear transport of QM, transfected PS1 molecule might be recycled efficiently in cells, leading to the remarkable effect on c-Jun function via QM protein translocated to and accumulated in the nucleus.
Next, we performed gel mobility shift assays by using nuclear extracts prepared from F9 cells expressing AP-1 transcription factors with or without PS1. We found that PS1 suppresses the binding of c-Jun homodimer to TRE and very weakly suppresses that of JunD homodimer but not that of c-Jun/c-Fos heterodimer or JunB homodimer (Fig. 6 b). Furthermore, normal PS1 suppressed the binding to TRE more efficiently than mutant PS1 (Fig. 6 b). These findings are consistent with the results in CAT assay (Fig. 5, a–f) and support the idea that the nuclear translocation of QM/Jif-1 is promoted by normal PS1 thereby inhibiting the binding of c-Jun homodimer to TRE.
We tested another possibility that PS1 inhibits JNK and thereby suppresses transactivation by c-jun (for review see Minden and Karin 1997). T7-Tag-JNK1 expression vector was cotransfected with PS1, immunoprecipitated, and the JNK activity was tested with c-Jun produced in bacteria as a substrate. In this assay, we observed no change of JNK activity (data not shown). This finding indicates that PS1 represses the function of c-jun predominantly through the transport of QM/Jif-1, but not through c-Jun phosphorylation by JNK, although further analyses are necessary to determine whether or not JNK is partially involved in the suppression by PS1.
Mutation Attenuates Inhibition of c-jun–Mediated Apoptosis by PS1
c-jun had been characterized as a protooncogene promoting cellular proliferation, whereas recent data indicate that c-jun is involved in some types of apoptosis. Expression of c-jun dominant negative mutants protects sympathetic neurons against cell death induced by NGF withdrawal, and the overexpression of c-jun itself triggers apoptosis in sympathetic neurons (Ham et al. 1995). Transfection of a recombinant fusion protein chimera of c-Jun and hormone binding domain of estrogen receptor induces apoptosis in NIH 3T3 cells with β-estradiol added to the culture medium (Bossy-Wetzel et al. 1997). Furthermore, induction of c-fos is noted as an early event of programmed cell death (Smeyne et al. 1993), and c-fos is implicated in light-induced retinal degeneration (Hafezi et al. 1997). Interestingly, the time course of the protein expression in neuronal apoptosis induced by withdrawal of trophic factor was different. c-jun is upregulated at first and c-fos follows it; expression of junD does not change (Ham et al. 1995). These findings indicated that AP-1 plays important roles in cell death, although each member of the AP-1 complex might play a different role.
Considering these previous data, we examined whether PS1 affects c-jun–mediated apoptosis. We used F9 cells for this analysis, since retinoic acid treatment induces apoptosis (Atencia et al. 1994) and upregulation of the c-jun expression (Yamaguchi-Iwai et al. 1990). Suppression of c-jun by the antisense transcript expression reduced the retinoic acid–induced apoptosis (Fig. 7 a), indicating that c-jun mediates this type of cell death. Thus, we made stable transformants expressing >10 times the amount of PS1 protein than parental F9 cells (Fig. 7 b), and observed the effect of PS1 on retinoic acid–induced apoptosis. Apoptotic cells were detected by nick end labeling assay with terminal transferase (Fig. 7 c) and by trypan blue dye exclusion assay (Fig. 7 d) then analyzed statistically (Fig. 7 d). Normal PS1 significantly suppressed the percentage of apoptotic cells, whereas mutant PS1 suppressed it rather weakly. These results were consistently observed in the stable cell lines with high expression (n = 3). In addition, we confirmed these results by DNA fragmentation in cellular nuclei (Fig. 7 e). The weak antiapoptotic effect of mutant PS1 corresponds well to its weak suppressive effect on c-jun–induced transactivation in CAT assays (Fig. 5b, Fig. c, and Fig. f).
Figure 7.
(a) Retinoic acid–induced apoptosis was suppressed in stable cells expressing antisense c-jun. SDS-PAGE and Western blot analysis show that expression of the c-Jun protein was induced by retinoic acid treatment in F9 cells but not in the stable cells expressing antisense c-jun. Genomic DNA analysis of these cells shows that the DNA ladder formation corresponding to apoptosis was suppressed in the antisense stable cells. Similar results were obtained in the other stable cell lines (n = 3). (b) Western blot analysis with anti-PS1 loop antibody showing PS1 protein expression level in stable cells. F9PS1, a stable cell line expressing normal PS1; F9mPS1, a stable cell line expressing mutant (Met146Leu) PS1; FL, full-length PS1; CTF, COOH-terminal fragment of PS1. (c) Nick end labeling assay showing that retinoic acid–induced apoptosis was reduced remarkably in F9PS1 but mildly in F9mPS1. Fluorescent labeling of apoptotic nuclei was performed with Apoptosis Detection System (Promega) according to the commercial protocol. (d) Effects of PS1 or mPS1 on retinoic acid–induced apoptosis were detected by nick end labeling assay (white bars) and by trypan blue dye exclusion analysis (shaded bars). Three independent stable cell lines were analyzed in each overexpression. Percentage of apoptosis in eight visual fields (mean ± SD) was significantly decreased (ANOVA, P < 0.01) in F9PS1 (cell lines 1–3) and F9mPS1 (cell lines 1 and 2) compared with F9. (e) Genomic DNA analysis of F9 cells and stable cells expressing normal or mutant PS1 in retinoic acid–induced apoptosis. Cells were cultured in the presence of 0.1 μM all-trans retinoic acid for 48 h. Both floating and adhering cells were collected together for DNA preparation. MM, molecular weight marker IV (Boehringer Mannheim).




The Putative Zinc Finger Domain Is Essential for Interaction with PS1
At the end of this study, we investigated the role of the putative zinc finger domain (zif) of QM/Jif-1 in interaction with PS1, in transcriptional regulation, and in apoptosis. First, we made various deletion constructs of QM/Jif-1 (Fig. 8 a) and tested their binding to PS1 by pull-down assay. Normal or mutant (Met146Leu) PS1 radiolabeled with [35S]methionine by in vitro transcription/translation were interacted with GST fusion proteins of QM in vitro and pulled down by glutathione Sepharose 4B. Full-length QM interacted with normal PS1, whereas deletion constructs lacking zif did not bind to PS1 (Fig. 8 b), indicating that this region is essential for interaction. Mutant PS1 binds to the GST-QM fusion proteins similarly. However, the ratio between the input and the pulled-down amounts was lower in mutant PS1 (60%) than in normal PS1 (95%).
Figure 8.
(a) Deletion construct of QM/Jif-1 subcloned into a GST fusion protein expression vector or into a eukaryotic expression vector. (b) GST fusion proteins lacking the putative zinc finger domain (zif) did not bind to the full-length PS1 in pull-down assay. IP, input. (c) Deletion of zif affects transcriptional regulation. Cotransfection of full-length QM into F9PS1 cells suppressed c-jun–induced transactivation, whereas Δ1 enhanced c-jun–induced transactivation. This effect was not found in the other constructs lacking zif. 10 or 20 μg of Δ1–3 was cotransfected with 5 μg of c-jun expression vector, QM expression vector, and/or collagenase 1× TRE CAT into 1 × 106 F9PS1 cells. Total amount of the transfected plasmids was controlled with pBluescript KS+. (d) QM-GFP fusion proteins containing the deletion constructs of QM were expressed in F9PS1 cells, and their subcellular localization was analyzed. 1 or 2 (Δ2) cells were shown in each case. QM-GFP and Δ1-GFP were localized predominantly in the nucleus in F9PS1 cells. (e) F9PS1 cells were transfected with the vectors expressing full-length or deleted QM. Transfection of Δ1 increased apoptoic cells in the treatment with retinoic acid, whereas the other constructs did not affect the c-jun–associated apoptosis. Statistical analysis confirmed increase of the percentage apoptosis in transfection of Δ1 (ANOVA, P < 0.01) from the value in transfection of QM.





Next, we tested whether deletion of zif affects transcriptional regulation. As QM/Jif-1 is abundantly expressed in all the cell lines, we designed a dominant negative experiment. We selected F9PS1 cells in which transfected QM is translocated to the nucleus. Eukaryotic expression vectors containing the deletion constructs of QM were cotransfected with c-jun, and QM expression vectors into F9PS1 cells. Transfection of full-length QM suppressed c-jun–induced transactivation (Fig. 8 c, lanes 2 and 3). Δ1 antagonized this suppression by QM. The CAT activities in cotransfection of Δ1 (Fig. 8 c, lanes 4 and 5) was higher than those in c-jun–induced transactivation (Fig. 8 c, lane 2), suggesting that Δ1 antagonized endogenous QM in addition to transfected QM. This dominant negative effect was not observed in the other constructs without zif that cannot interact with PS1 (Fig. 8 c, lanes 6–9). Therefore, Δ1 probably inhibits binding of QM to PS1 in a competitive manner and represses the function of QM, since Δ1 transported to the nucleus does not have a suppressive effect on c-Jun. Consistently, translocation to the nucleus of the GFP fusion protein was observed in Δ1 but not in the other deletion constructs lacking zif (Fig. 8 d). Without transfecting c-jun expression vector, transactivation by Δ1 itself was not observed in F9PS1 cells, which do not express c-jun (data not shown).
Finally, we tested the role of zif in the c-jun–associated apoptosis. F9PS1 cells were transfected with the vectors expressing deleted QM/Jif-1. Transfection of Δ1 increased the percentage of cell death induced by retinoic acid treatment, whereas the other constructs did not affect the apoptosis (Fig. 8 e). This result again showed the dominant negative effect of Δ1 on apoptosis. Collectively, it was concluded that zif is essential for the interaction of QM with PS1 and for the effects of QM on c-Jun derived from the interaction.
Discussion
This study showed that PS1 binds to a negative cofactor of c-Jun, QM/Jif-1, and that PS1 regulates the functions of c-jun in transcription and cell death. Promotion of the nuclear translocation of QM/Jif-1 by PS1 seems to be the underlying mechanism that connects the first and second conclusions. A specific point in our results is that QM/Jif-1 binds to a full-length PS1. It was reported that most PS1 molecules are cleaved into two fragments which reassociate to form a heterodimer (Thinakaran et al. 1996, Thinakaran et al. 1998). However, Western blot analysis in this study (Fig. 2, a and b) and in the previous reports (Lah et al. 1997) shows that full-length PS1 also exists in the brain in vivo, although the function of this form has been unclear. Our findings suggest that the full-length form also possesses a functional role related to cell viability.
So far, more than five molecules, including APP (Waragai et al. 1997; Xia et al. 1997), calsenilin (Buxbaum et al. 1998), β-catenin (Zhang, Z., et al., 1998), filamin (Zhang, W., et al., 1998), and tau (Takashima et al. 1998), were isolated as binding proteins to PS1. The reason why many proteins bind to PS1 is not known. However, it might be explained by supposing that PS1 functions as a kind of intracellular transporter with a relatively low specificity. The multipass transmembrane structure of PS1, especially of the full-length form, does not contradict this hypothesis, and this kind of activity was actually shown in the case of amyloid protein transport (Naruse et al. 1998). Our observation on the nuclear translocation of QM/Jif-1 is also compatible with this idea.
Activation of c-jun, especially that mediated by JNK, has been suggested to participate in various types of cell death, including TNF- or Fas-induced apoptosis (Verheij et al. 1996). Numerous reports keep emerging on this apoptotic cascade. JNK was reported to enhance apoptosis induced by a breast cancer susceptibility gene, BRCA1, through activation of GADD45 (Harkin et al. 1999). The JNK-dependent apoptotic pathway has been implicated in the morphogenesis of Drosophila wing (Adachi-Yamada et al. 1999). Furthermore, kainic acid–induced apoptosis in the hippocampus was prevented in the mice lacking the Jnk3 gene (Yang et al. 1997), indicating that JNK3 is essential for this type of apoptosis. The role of JNK in kainic acid–induced apoptosis was reconfirmed in the knock-in mice carrying mutations at JNK-phosphorylation sites of the c-jun gene (Behrens et al. 1999). In the AD brain, the increase of c-Jun immunoreactivity was observed and strongly correlated with pathological changes (Anderson et al. 1996). Mutations in PS1 might influence such cascades at the final step of c-Jun and thereby affect apoptosis. Since our results showed that the inhibitory effect of mutant PS1 on c-jun–mediated apoptosis was weaker than that of normal PS1, mutation in PS1 could be a promoting factor in c-jun–mediated apoptosis.
However, there remain debates on the role of c-jun and JNK in the in vivo apoptosis. Although double gene disruption of Jnk1 and Jnk2 leads to severe dysregulation of apoptosis during development at specific regions in the brain, Jnk3 does not affect this apoptosis (Kuan et al. 1999). c-jun was reported to be dispensable in developmental cell death and axogenesis of the retina (Herzog et al. 1999). These discrepancies might suggest that the c-Jun/JNK cascade plays different roles in the developmental neuronal death and in the death of the mature neuron. Therefore, our results on the cascade from PS1 to c-Jun should be applied carefully to different types of cell death.
The functions of normal and mutant PS1 in cell death reported so far are still difficult to combine (for review see Barinaga 1998). Normal PS1 had been reported to promote cell death basically via affecting the Aβ metabolism. Meanwhile, accumulating data from independent laboratories indicated that PS1 also modifies various cell death cascades. It suppresses apoptosis associated with p53 and p21WAF1 activation (Roperch et al. 1998) and promotes the antiapoptotic function of β-catenin (Zhang, Z., et al., 1998). In these cases, normal PS1 suppresses cell death, whereas mutations in PS1 abort this function.
Suppression of c-Jun homodimer by PS1 might lead to a different outcome in signaling pathways other than the c-jun–mediated apoptosis examined in this study. For example, neurotrophins bind to tyrosine kinase–type membrane receptors and activate c-Jun through the mitogen-activated protein (MAP) kinase pathway. Many other trophic factors, including fibroblast (FGF), epidermal (EGF), platelet-derived (PDGF), and hepatocyte growth factors (HGF), induce similar signaling cascades and promote cell survival. In such a condition where the activation of c-Jun mainly contributes to survival, PS1 might promote apoptosis. Like neurotrophins which transduce survival and death signalings via the high affinity and low affinity receptors, respectively (for review see Dechant and Barde 1997), PS1 might transduce both types of signalings via different molecules binding to PS1.
In other words, our results might have proposed another type of explanation for why PS1 induces diverse effects on the cell fate. The PS1/QM/c-Jun cascade leads to the opposite outcome, survival or death, depending on the final effect of c-Jun, which is influenced by cellular conditions. Although it is not yet known which factors define the attitude of c-Jun in cells, other types of signaling cascades induced by PS1, including calcium release from endoplasmic reticulum (Guo et al. 1996) and G protein signaling (for review see Nishimoto 1998), might cross-talk and change the responding manner of cells to the c-Jun activation.
In the AD brains, which usually degenerate for more than several years, it is possible that both c-jun–associated and c-jun–nonassociated neuronal deaths occur in various situations. Although apoptosis itself is a rather rapid process in a single neuron, it occurs in numerous neurons of the brain at random and so the mass degeneration of the brain proceeds gradually. Therefore, we are speculating that c-jun–mediated apoptosis influenced by PS1 might be an additive factor to modify neuronal fate in the AD brain, and could function in parallel with the amyloid deposition promoted by PS mutations as well as the pathogenic mechanisms mediated by other binding proteins.
Acknowledgments
We thank Dr. Roger Brent for allowing us to use the two-hybrid system and human embryonic brain cDNA library, Dr. A.I. Levey (Emory University School of Medicine, Atlanta GA) for αN and αL PS1 mAbs, Drs. S.S. Sisodia and G. Thinakaran (University of Chicago, Chicago, IL and Johns Hopkins University, Baltimore, MD) for providing αL, Drs. T. Asano and K. Inukai (University of Tokyo, Tokyo, Japan) for pCAGGS-GLUT1, and Dr. K. Kamakura (National Defense Medical College, Saitama, Japan) for support in immunohistochemistry. We are grateful to Dr. Gary Lewin (Max-Delbruck Center for Molecular Medicine, Berlin, Germany) for critical reading of the manuscript and for his useful advice.
This work was supported by grants (07558233 and 10670574) from The Ministry of Education, Culture and Sports of Japan for H. Okazawa.
Footnotes
1.used in this paper: Aβ, β-amyloid; AD, Alzheimer's disease; αL, anti-loop antibody; αN, anti–NH2-terminal antibody; AP, activated protein; APP, amyloid precursor protein; CAT, chloramphenicol acetyltransferase; EGFP, enhanced green fluorescent protein; GLUT1, glucose transporter 1; GST, glutathione S-transferase; Jif, Jun-interacting factor; JNK, c-Jun NH2-terminal kinase; nt, nucleotide(s); PS1, presenilin 1; RT, reverse transcriptase; TRE, 12-O-tetradecanoylphorbol-13-acetate (TPA)-responsive element
I. Imafuku and T. Masaki contributed equally to this work.
References
- Adachi-Yamada T., Fujimura-Kamada K., Nishida Y., Matsumoto K. Distortion of proximodistal information causes JNK-dependent apoptosis in Drosophila wing. Nature. 1999;400:166–169. doi: 10.1038/22112. [DOI] [PubMed] [Google Scholar]
- Anderson A.J., Su J.H., Cotmann C.W. DNA damage and apoptosis in Alzheimer's diseasecolocalization with c-Jun immunoreactivity, relationship to brain area, and effect of postmortem delay. J. Neurosci. 1996;16:1710–1719. doi: 10.1523/JNEUROSCI.16-05-01710.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Angel P., Imagawa M., Chiu R., Stein B., Imbra R.J., Rahmsdorf H.J., Jonat C., Herrlich P., Karin M. Phorbol ester-inducible genes contain a common cis element recognized by a TPA-modulated trans-acting factor. Cell. 1987;49:729–739. doi: 10.1016/0092-8674(87)90611-8. [DOI] [PubMed] [Google Scholar]
- Angel P., Allegretto E.A., Okino S.T., Hattori K., Boyle W.J., Hunter T., Karin M. Oncogene jun encodes a sequence-specific trans-activator similar to AP-1. Nature. 1988;322:166–171. doi: 10.1038/332166a0. [DOI] [PubMed] [Google Scholar]
- Atencia R., Garcia-Sanz M., Unda F., Arechaga J. Apoptosis during retinoic acid-induced F9 embryonal carcinoma cells. Exp. Cell. Res. 1994;214:663–667. doi: 10.1006/excr.1994.1304. [DOI] [PubMed] [Google Scholar]
- Barinaga M. Is apoptosis key in Alzheimer's disease? Science. 1998;281:1303–1304. doi: 10.1126/science.281.5381.1303. [DOI] [PubMed] [Google Scholar]
- Behrens A., Sibilia M., Wagner E.F. Amino-terminal phosphorylation of c-Jun regulates stress-induced apoptosis and cellular proliferation. Nat. Genet. 1999;21:326–329. doi: 10.1038/6854. [DOI] [PubMed] [Google Scholar]
- Borchelt D.R., Ratovitski T., van Lare J., Lee M.K., Gonzales V., Jenkins N.A., Copeland N.G., Price D.L., Sisodia S.S. Accelerated amyloid deposition in the brains of transgenic mice coexpressing mutant presenilin 1 and amyloid precursor proteins. Neuron. 1997;19:939–945. doi: 10.1016/s0896-6273(00)80974-5. [DOI] [PubMed] [Google Scholar]
- Bossy-Wetzel E., Bakiri L., Yaniv M. Induction of apoptosis by the transcription factor c-jun. EMBO (Eur. Mol. Biol. Organ.) J. 1997;7:1695–1709. doi: 10.1093/emboj/16.7.1695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buxbaum J.D., Choi E.K., Luo Y., Lilliehook C., Crowley A.C., Merriam D.E., Wasco W. Calsenilinsa calcium-binding protein that interacts with the presenilins and regulates the levels of a presenilin fragment. Nat. Med. 1998;4:1177–1181. doi: 10.1038/2673. [DOI] [PubMed] [Google Scholar]
- Capell A., Grunberg J., Pesold B., Diehlmann A., Citron M., Nixon R., Beyreuther K., Selkoe D.J., Haass C. The proteolytic fragments of the Alzheimer's disease–associated presenilin-1 form heterodimers and occur as a 100–150-kDa molecular mass complex. J. Biol. Chem. 1998;273:3205–3211. doi: 10.1074/jbc.273.6.3205. [DOI] [PubMed] [Google Scholar]
- Chan Y.-L., Diaz J.-J., Denoroy L., Madjar J.-J., Wool I.G. The primary structure of rat ribosomal protein L10relationship to a Jun-binding protein and to a putative Wilms' tumor suppressor. Biochem. Biophys. Res. Commun. 1996;225:952–956. doi: 10.1006/bbrc.1996.1277. [DOI] [PubMed] [Google Scholar]
- Chen H., Lin R.J., Schiltz R.L., Chakravarti D., Nash A., Nagy L., Privalsky M.L., Nakatani Y., Evans R.M. Nuclear receptor coactivator ACTR is a novel histone acetyltransferase and forms a multimeric activation complex with P/CAF and CBP/p300. Cell. 1997;90:569–580. doi: 10.1016/s0092-8674(00)80516-4. [DOI] [PubMed] [Google Scholar]
- Chui D.-H., Tanahashi H., Ozawa K., Ikeda S., Checler F., Ueda O., Suzuki H., Araki W., Inoue H., Shirotani K. Transgenic mice with Alzheimer presenilin 1 mutations show accelerated neurodegeneration without amyloid plaque formation. Nat. Med. 1999;5:560–564. doi: 10.1038/8438. [DOI] [PubMed] [Google Scholar]
- Dechant G., Barde Y.-A. Signalling through the neurotrophin receptor p75NTR. Curr. Opin. Neurobiol. 1997;7:413–418. doi: 10.1016/s0959-4388(97)80071-2. [DOI] [PubMed] [Google Scholar]
- Dewji N.N., Singer S.J. The seven-transmembrane spanning topography of the Alzheimer disease-related presenilin proteins in the plasma membranes of cultured cells. Proc. Natl. Acad. Sci. USA. 1997;94:14025–14030. doi: 10.1073/pnas.94.25.14025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dowdy S.F., Lai K.M., Weissman B.E., Matsui Y., Hogan B.L., Stanbridge E.J. The isolation and characterization of a novel cDNA demonstrating an altered mRNA level in nontumorigenic Wilms' microcell hybrid cells. Nucleic Acids Res. 1991;19:5763–5769. doi: 10.1093/nar/19.20.5763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farmer A.A., Loftus T.M., Mills A.A., Sato K.Y., Neill J.D., Tron T., Yang M., Trumpower B.L., Stanbridge E.J. Extreme evolutionary conservation of QM, a novel c-Jun associated transcription factor. Hum. Mol. Genet. 1994;3:723–728. doi: 10.1093/hmg/3.5.723. [DOI] [PubMed] [Google Scholar]
- Guarente L. Yeast promoters and lacZ fusions designed to study expression of cloned genes in yeast. Methods Enzymol. 1983;101:181–191. doi: 10.1016/0076-6879(83)01013-7. [DOI] [PubMed] [Google Scholar]
- Guo Q., Furukawa K., Sopher B.L., Pham D.G., Xie J., Robinson N., Martin G.M., Mattson M.P. Alzheimer's PS-1 mutation perturbs calcium homeostasis and sensitizes PC12 cells to death induced by amyloid beta-peptides. Neuroreport. 1996;8:379–383. doi: 10.1097/00001756-199612200-00074. [DOI] [PubMed] [Google Scholar]
- Hafezi F., Steinbach J.P., Marti A., Munz K., Wang Z.Q., Wagner E.F., Aguzzi A., Reme C.E. The absence of c-fos prevents light-induced apoptotic cell death of photoreceptors in retinal degeneration in vivo. Nat. Med. 1997;1:346–349. doi: 10.1038/nm0397-346. [DOI] [PubMed] [Google Scholar]
- Ham J., Babij C., Whitfield J., Pfarr C.M., Lallemand D., Yaniv M., Rubin L.L. A c-Jun dominant negative mutant protects sympathetic neurons against programmed cell death. Neuron. 1995;14:927–939. doi: 10.1016/0896-6273(95)90331-3. [DOI] [PubMed] [Google Scholar]
- Hardy J. Amyloid, the presenilins and Alzheimer's disease. Trends Neurosci. 1997;20:154–159. doi: 10.1016/s0166-2236(96)01030-2. [DOI] [PubMed] [Google Scholar]
- Harkin D.P., Bean J.M., Miklos D., Song Y.H., Truong V.B., Englert C., Christian F.C., Ellison L.W., Maheswaran S., Oliner J.D., Harber D.A. Induction of GADD45 and JNK/SAPK-dependent apoptosis following inducible expression of BRCA1. Cell. 1999;97:575–586. doi: 10.1016/s0092-8674(00)80769-2. [DOI] [PubMed] [Google Scholar]
- Herzog K.-H., Chen S.-C., Morgan J.I. c-jun is dispensable for developmental cell death and axogenesis in the retina. J. Neurosci. 1999;19:4349–4359. doi: 10.1523/JNEUROSCI.19-11-04349.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holcomb L., Gordon M.N., McGowan E., Yu X., Benkovic S., Jantzen P., Wright K., Saad I., Mueller R., Morgan D. Accelerated Alzheimer-type phenotype in transgenic mice carrying both mutant amyloid precursor protein and presenilin 1 transgenes. Nat. Med. 1997;4:97–100. doi: 10.1038/nm0198-097. [DOI] [PubMed] [Google Scholar]
- Kim T.-W., Pettingell W.H., Hallmark O.G., Moir R.D., Wasco W., Tanzi R.E. Endoproteolytic cleavage and proteasomal degradation of presenilin 2 in transfected cells. J. Biol. Chem. 1997;272:11006–11010. doi: 10.1074/jbc.272.17.11006. [DOI] [PubMed] [Google Scholar]
- Kovacs D.M., Fausett H.J., Page K.J., Kim T.W., Moir R.D., Merriam D.E., Hollister R.D., Hallmark O.G., Mancini R., Felsenstein K.M. Alzheimer-associated presenilins 1 and 2neuronal expression in brain and localization to intracellular membranes in mammalian cells. Nat. Med. 1996;2:224–229. doi: 10.1038/nm0296-224. [DOI] [PubMed] [Google Scholar]
- Kuan C.Y., Yang D.D., Samanta Roy D.R., Davis R.J., Rakic P., Flavell R.A. The Jnk1 and Jnk2 kinases are required for regional specific apoptosis during early brain development. Neuron. 1999;22:667–676. doi: 10.1016/s0896-6273(00)80727-8. [DOI] [PubMed] [Google Scholar]
- Lah J.J., Heilman C.J., Nash N.R., Rees H.D., Yi H., Counts S.E., Levey A.I. Light and electron microscopic localization of presenilin-1 in primate brain. J. Neurosci. 1997;17:1971–1980. doi: 10.1523/JNEUROSCI.17-06-01971.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levitan D., Greenwald I. Facilitation of lin-12-mediated signaling by sel-12, a Caenorhabditis elegans S182 Alzheimer's disease gene. Nature. 1995;377:351–354. doi: 10.1038/377351a0. [DOI] [PubMed] [Google Scholar]
- Li J., Xu M., Zhou H., Ma J., Potter H. Alzheimer presenilins in the nuclear membrane, interphase kinetochores, and centrosomes suggest a role in chromosome segregation. Cell. 1997;90:917–927. doi: 10.1016/s0092-8674(00)80356-6. [DOI] [PubMed] [Google Scholar]
- Masters C.L., Beyreuther K. Alzheimer's disease. BMJ. 1998;316:446–448. doi: 10.1136/bmj.316.7129.446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minden A., Karin M. Regulation and function of the JNK subgroup of MAP kinases. Biochim. Biophys. Acta. 1997;1333:F85–F104. doi: 10.1016/s0304-419x(97)00018-8. [DOI] [PubMed] [Google Scholar]
- Monteclaro F.S., Vogt P.K. A Jun-binding protein related to a putative tumor suppresser. Proc. Natl. Acad. Sci. USA. 1993;90:6726–6730. doi: 10.1073/pnas.90.14.6726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naruse S., Thinakaran G., Luo J.J., Kusiak J.W., Tomita T., Iwatsubo T., Qian X., Ginty D.D., Price D.L., Borchelt D.R. Effects of PS1 deficiency on membrane protein trafficking in neurons. Neuron. 1998;21:1213–1221. doi: 10.1016/s0896-6273(00)80637-6. [DOI] [PubMed] [Google Scholar]
- Nishimoto I. A new paradigm for neurotoxicity by FAD mutants of betaAPPa signaling abnormality. Neurobiol. Aging. 1998;19:S33–S38. doi: 10.1016/s0197-4580(98)00040-2. [DOI] [PubMed] [Google Scholar]
- Okamoto K., Okazawa H., Okuda A., Sakai M., Muramatsu M., Hamada H. A novel octamer binding transcription factor is differentially expressed in mouse embryonic cells. Cell. 1990;60:461–472. doi: 10.1016/0092-8674(90)90597-8. [DOI] [PubMed] [Google Scholar]
- Okazawa H., Okamoto K., Ishino F., Ishino-Kaneko T., Takeda S., Toyoda Y., Muramatsu M., Hamada H. The oct-3 gene, a gene for an embryonic transcription factor is controlled by a retinoic acid repressible enhancer. EMBO (Eur. Mol. Biol. Organ.) J. 1991;10:2997–3005. doi: 10.1002/j.1460-2075.1991.tb07850.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roperch J.P., Alvaro V., Prieur S., Tuynder M., Nemani M., Lethrosne F., Piouffre L., Gendron M.C., Israeli D., Dausset J. Inhibition of presenilin 1 expression is promoted by p53 and p21WAF-1 and results in apoptosis and tumor suppression. Nat. Med. 1998;4:835–838. doi: 10.1038/nm0798-835. [DOI] [PubMed] [Google Scholar]
- Selkoe D.J. Alzheimer's diseasegenotypes, phenotypes, and treatments. Science. 1997;275:630–631. doi: 10.1126/science.275.5300.630. [DOI] [PubMed] [Google Scholar]
- Shen J., Bronson R.T., Chen D.F., Xia W., Selkoe D.J., Tonegawa S. Skeletal and CNS defects in presenilin-1-deficient mice. Cell. 1997;89:629–639. doi: 10.1016/s0092-8674(00)80244-5. [DOI] [PubMed] [Google Scholar]
- Smeyne R.J., Vendrell M., Hayward M., Baker S.J., Miao G.G., Schilling K., Robertson L.M., Curran T., Morgan J.I. Continuous c-fos expression precedes programmed cell death in vivo. Nature. 1993;363:166–169. doi: 10.1038/363166a0. [DOI] [PubMed] [Google Scholar]
- Takashima A., Murayama M., Murayama O., Kohno T., Honda T., Yasutake K., Nihonmatsu N., Mercken M., Yamaguchi H., Sugihara S., Wolozin B. Presenilin 1 associates with glycogen synthetase kinase-3 beta and its substrate tau. Proc. Natl. Acad. Sci. USA. 1998;95:9637–9641. doi: 10.1073/pnas.95.16.9637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thinakaran G., Borchelt D.R., Lee M.K., Slunt H.H., Spitzer L., Kim G., Ratovitsky T., Davenport F., Nordstedt C., Seeger M. Endoproteolysis of presenilin 1 and accumulation of processed derivatives in vivo. Neuron. 1996;17:181–190. doi: 10.1016/s0896-6273(00)80291-3. [DOI] [PubMed] [Google Scholar]
- Thinakaran G., Regard J.B., Bouton C.M., Harris C.L., Price D.L., Borchelt D.R., Sisodia S.S. Stable association of presenilin derivatives and absence of presenilin interactions with APP. Neurobiol. Dis. 1998;4:438–453. doi: 10.1006/nbdi.1998.0171. [DOI] [PubMed] [Google Scholar]
- Verheij M., Bose R., Lin X.H., Yao B., Jarvis W.D., Grant S., Birrer M.J., Szabo E., Zon L.I., Kyriakis J.M. Requirement for ceramide-initiated SAPK/JNK signaling in stress-induced apoptosis. Nature. 1996;380:75–79. doi: 10.1038/380075a0. [DOI] [PubMed] [Google Scholar]
- Waragai M., Imafuku I., Takeuchi S., Kanazawa I., Oyama F., Udagawa Y., Kawabata M., Okazawa H. Presenilin 1 binds to amyloid precursor protein directly. Biochem. Biophys. Res. Commun. 1997;239:480–482. doi: 10.1006/bbrc.1997.7488. [DOI] [PubMed] [Google Scholar]
- Wolfe M.S., Xia W., Ostazewski B.L., Diehl T.S., Kimberly W.T., Selkoe D.J. Two transmembrane aspartates in presenilin-1 required for presenilin endoproteolysis and γ-secretase activity. Nature. 1999;398:513–516. doi: 10.1038/19077. [DOI] [PubMed] [Google Scholar]
- Wong P.C., Zheng H., Chen H., Becher M.W., Sirinathsinghji D.J., Trumbauer M.E., Chen H.Y., Price D.L., Van der Ploeg L.H., Sisodia S.S. Presenilin 1 is required for Notch 1 and Dll 1 expression in the paraaxial mesoderm. Nature. 1997;387:288–292. doi: 10.1038/387288a0. [DOI] [PubMed] [Google Scholar]
- Wool I.G. Extraribsomal functions of ribosomal proteins. Trends Biochem. 1996;Sci. 21:164–165. [PubMed] [Google Scholar]
- Xia W., Zhang J., Perez R., Koo E.H., Selkoe D.J. Interaction between amyloid precursor protein and presenilins in mammalian cellsimplication for the pathogenesis of Alzheimer disease. Proc. Natl. Acad. Sci. USA. 1997;94:8208–8213. doi: 10.1073/pnas.94.15.8208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamaguchi-Iwai Y., Satake M., Murakami Y., Sakai M., Muramatsu M., Ito Y. Differentiation of F9 embryonal carcinoma cells induced by the c-jun and activated c-Has-ras oncogenes. Proc. Natl. Acad. Sci. USA. 1990;87:8670–8674. doi: 10.1073/pnas.87.21.8670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang D.D., Kuan C.-Y., Whitmarsh A.J., Rincon M., Zheng T.S., Davis R.J., Rakic P., Flavell R.A. Absence of excitotoxicity-induced apoptosis in the hippocampus of mice lacking the Jnk3 gene. Nature. 1997;389:865–870. doi: 10.1038/39899. [DOI] [PubMed] [Google Scholar]
- Zhang W., Han S.W., McKeel D.W., Goate A., Wu J.Y. Interaction of presenilins with the filamin family of actin-binding proteins. J. Neurosci. 1998;18:914–922. doi: 10.1523/JNEUROSCI.18-03-00914.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Z., Hartmann H., Do V.M., Abramowski D., Sturchler-Pierrat C., Staufenbiel M., Sommer B., van de Wetering M., Clevers H., Saftig P. Destabilization of beta-catenin by mutations in presenilin-1 potentiates neuronal apoptosis. Nature. 1998;395:698–702. doi: 10.1038/27208. [DOI] [PubMed] [Google Scholar]