

Abstract
Severe bacterial infections leading to sepsis or septic shock can be induced by bacteria that utilize different factors to drive pathogenicity and/or virulence, leading to disease in the host. One major factor expressed by all clinical isolates of gram-negative bacteria is lipopolysaccharide (LPS); a second factor expressed by some Escherichia coli strains is a K1 polysaccharide capsule. To determine the role of the CD14 LPS receptor in the pathogenic effects of naturally occurring E. coli, the responses of CD14−/− and CD14+/+ mice to three different isolates of E. coli obtained from sepsis patients were compared; two isolates express both smooth LPS and the K1 antigen, while the third isolate expresses only LPS and is negative for K1. An additional K1-positive isolate obtained from a newborn with meningitis and a K1-negative isogenic mutant of this strain were also used for these studies. CD14−/− mice were resistant to the lethal effects of the K1-negative isolates. This resistance was accompanied by significantly lower levels of systemic tumor necrosis factor alpha (TNF-α) and interleukin-6 (IL-6) in these mice than in CD14+/+ mice, enhanced clearance of the bacteria, and significantly fewer additional gross symptoms. In contrast, CD14−/− mice were as sensitive as CD14+/+ mice to the lethal effects of the K1-positive isolates, even though they had significantly lower levels of TNF-α and IL-6 than CD14+/+ mice. These studies show that different bacterial isolates can use distinctly different mechanisms to cause disease and suggest that new, nonantibiotic therapeutics need to be directed against multiple targets.
Sepsis, characterized by a systemic bacterial infection and systemic inflammatory response syndrome, can lead to life-threatening conditions, including severe sepsis (sepsis with multiorgan failure) and septic shock (sepsis with persistent arterial hypotension) (1, 4, 25). Although the gross symptoms of sepsis and septic shock appear to be similar and independent of the specific type of the invading bacterium, the bacteria isolated from these patients can be highly varied in terms of the properties that promote their pathogenicity and/or virulence. Two widely studied bacterial factors that can influence pathogenicity are lipopolysaccharide (LPS) (or endotoxin) (20, 29, 33, 34) and some of the capsular polysaccharide K antigens (3, 7, 47).
LPS resides in the outer membrane of all gram-negative bacteria and can play a major role in pathophysiological responses by triggering production of large amounts of proinflammatory mediators, such as tumor necrosis factor alpha (TNF-α) and interleukin-6 (IL-6). This induction occurs through the interaction of LPS with its receptor, CD14 (13), and its signaling complex, Toll-like receptor 4 (TLR4)/MD2 (for a review, see reference 11). CD14 is expressed strongly on the surface of monocytes/macrophages (15) and weakly on granulocytes (18) and some dendritic cells (27, 48). Previous studies have shown that mice deficient in CD14 (CD14−/−) produce little or no proinflammatory cytokines (TNF-α, IL-1, IL-6) in response to LPS and Escherichia coli O111 and are highly resistant to the lethal effects of both of these factors (19). These observations indicate that CD14 can play a major role in the pathophysiological responses caused by LPS and/or live bacteria expressing LPS.
In addition to LPS, the surface of most E. coli strains is also covered with a layer of polysaccharides that can be distinguished serologically as K antigens (28, 47). K antigens vary in structure, but only a few of the more than 80 different serotypes described play a role in enhancing the virulence of a particular gram-negative bacterium. One example of a K antigen that promotes virulence is the K1 antigen, most commonly expressed by E. coli isolated from neonates suffering from septic shock and/or meningitis (39, 42). The K1-polysaccharide capsule masks surface structures of the bacterium, including LPS, as well as sites for the binding of opsonins. Masking of the latter enhances virulence in part by inhibiting the ability of the host to phagocytose K1-positive E. coli (37). Although LPS is also masked, it nevertheless has been suggested to play a role in the pathogenicity of K1-positive E. coli (5); however, the mechanism and exact role of LPS in the pathogenicity of K1-positive E. coli have not been clearly documented. It is conceivable that as the K1-positive bacteria divide, LPS and other membrane components are released, making them available to activate the innate immune system via CD14. In addition, since it has been proposed that CD14 acts as a pattern recognition receptor, direct interaction of the K1-polysaccharide capsule with CD14 may also trigger host responses.
The studies described here were designed to determine the role of CD14 in the pathogenic effects of three different isolates of E. coli obtained from sepsis patients that differ in several characteristics, including their O (LPS) antigen serotypes and the presence of a K1 capsule; two isolates express both smooth LPS and the K1 antigen, while the third isolate expresses only LPS and is negative for K1. An additional K1-positive E. coli strain isolated from a neonate with meningitis and a K1-negative isogenic mutant of this strain were also analyzed. Using CD14−/− and normal (CD14+/+) mice, the role of CD14 in the host's response to these five isolates was examined with respect to TNF-α and IL-6 production, bacterial clearance, and survival.
MATERIALS AND METHODS
Animals.
CD14−/− mice (10th backcross on BALB/c) (13, 19) and age- and weight-matched 6- to 8-week-old CD14+/+ wild-type BALB/c mice (Harlan, Indianapolis, IN) were housed in the animal facility of the City College of New York, New York, NY, or the Feinstein Institute of Medical Research, Manhasset, NY, and were provided with nonsterile laboratory chow (Harlan Teklad, Madison, WI) and water ad libitum. All animals were maintained and studied in accordance with recommendations of the Institute of Laboratory Animal Resources (National Academy of Sciences, Washington, DC) and the Institutional Animal Care and Use Committees of the City College of New York, New York, NY, and the Feinstein Institute for Medical Research, Manhasset, NY.
Bacteria.
Three clinical E. coli isolates utilized in these studies (E. coli isolates 59, 69, and 61) were obtained from patients and were characterized to determine various virulence factors as described by Picard et al. (31). The characteristics are summarized in Table 1. These bacteria were shown to belong to the same phylogenetic group (group 5), defined as nonhemolytic, carboxylase type B2 (Mf 62), and mannose-resistant hemagglutinin-positive organisms. These bacteria also expressed pap and aer operons; they were negative for the sfa/foc, afa, and hly operons and ibe-10 gene expression (31). In addition, E. coli isolates 69, 59, and 61 were biotyped by the Gastroenteric Disease Center (Pennsylvania State University, University Park) to determine specific O and H serotypes, fimbrial antigens, and toxin production (Table 1). An additional K1-positive isolate, RS218 (O18:K1:H7), obtained from a newborn with meningitis, and a K1-negative isogenic mutant of this isolate (O18:H7), have been described previously (22).
TABLE 1.
Characteristics of E. coli strains studied
| Characteristic |
E. coli isolate
|
||
|---|---|---|---|
| 59 | 61 | 69 | |
| Smooth LPS | +a | + | + |
| O-antigen typeb | 2 | 1 | 2 |
| H (flagella)b | Mc | 7 | M |
| K1 capsule | + | + | − |
| Serum (80%) sensitivity | − | − | − |
| Heat toxin (a and b)b | − | − | − |
| Shiga-like toxin types 1 and 2b | − | − | − |
| EAE (intimin)b | − | − | − |
| Cytotoxic necrotizing factors 1 and 2b | − | − | − |
| Fimbrial antigens (K88, K99, 987P, CS31A, F1845, F107)b | − | − | − |
| Bfp pili | − | − | − |
| aer operon | + | + | + |
| Alpha-hemolysin | − | − | − |
| pap operon | + | + | + |
| sfa/foc operon | − | − | − |
| afa operon | − | − | − |
| hly operon | − | − | − |
| ibe-10 | − | − | − |
| Mannose-resistant hemagglutinin antigen | + | + | + |
| Carboxylase B2 polymorphism type | 62 | 62 | 62 |
+, positive; −, negative.
Typing was performed by Pennsylvania State University (see Materials and Methods).
M, multiple (bacteria cross-reacted with H4, H23, H37, and H44 H antigens).
Serum sensitivity.
The sensitivity of each bacterial isolate to serum (complement)-mediated killing was tested as described by Russo et al. (38), with some modifications. Briefly, fresh human serum was preadsorbed with log-phase E. coli to remove isoagglutinins, centrifuged at 14,000 × g for 5 min, filter sterilized, and stored in aliquots at −80°C. Each bacterial isolate was washed in Veronal buffer and resuspended (1 × 105 CFU/ml) in preadsorbed serum (with and without heat inactivation; final concentration, 80%) for 1 h at 37°C. Bacteria were then diluted in Veronal buffer and plated on tryptic soy agar, and the number of surviving bacteria was determined. All isolates were resistant to both normal and heat-inactivated adsorbed serum. Known serum-resistant (E. coli CP9) and serum-sensitive (E. coli CP923) strains (kind gifts of T. Russo [38]) were used as controls.
Electron microscopy of bacterial isolates.
The presence of a K1 capsule was confirmed by electron microscopy of ruthenium red (RR)-stained E. coli using the modified method of Luft (26). Briefly, fresh bacterial colonies from Trypticase soy agar plates (Difco, Detroit, MI) were suspended in 0.3 M sucrose, pelleted, fixed by resuspension in 8 ml of 2.5% glutaraldehyde in 0.1 M cacodylate buffer (pH 7.3) containing 0.15% (wt/vol) RR, and incubated overnight at 4°C. The fixed cells were rinsed in 0.1 M cacodylate buffer with RR, pelleted, and resuspended in 3 ml of buffered 2% osmium tetroxide containing 0.15% RR for 1 h. The cells were then rinsed two times in buffer containing RR and dehydrated in a graded ethanol series (25%, 50%, and finally 70% ethanol) containing RR for 15-min periods. The cells were then resuspended in a small volume (400 μl) and centrifuged with a Beckman 152 Microfuge. The intact pellet was dehydrated in the absence of RR with 95% ethanol followed by four changes of 100% ethanol and then infiltrated with effapoxy resin (2:1, 1:1, and 1:2 ethanol-effapoxy for 30-min periods and finally 100% effapoxy overnight at 4°C), and this was followed by embedding. Thin sections were stained with uranyl acetate and lead citrate and examined with a JEOL JEM100 CXII transmission electron microscope.
Bacterial culture.
Isolates 69, 59, and 61 were grown in Trypticase soy broth or agar (Difco, Detroit, MI). RS218 and an isogenic mutant of this strain were grown in tryptic soy broth or agar supplemented with 50 μg/ml streptomycin (for the K1-positive isolate) or 50 μg/ml streptomycin and 40 μg/ml chloramphenicol (for the K1-negative isolate). Individual isolates were grown in the appropriate broth media (5 ml) after inoculation of a single colony and incubated at 37°C overnight in an orbital shaker. The next day, 2.5 ml of each overnight culture was used to inoculate 47.5 ml of fresh broth and incubated for 2 h at 37°C with shaking. Bacterial growth was determined by measuring the optical density at 600 nm, and the number of bacteria (CFU/ml) was determined using a previously determined growth curve. The number and viability of bacteria were confirmed using a LIVE/DEAD Baclight kit (Molecular Probes, Eugene, OR) and by individual plating on agar following injection. For all experiments, bacteria in log phase were used. Pyrogen-free normal saline (Baxter Healthcare Corporation, Deerfield, IL) was used to prepare bacterial suspensions for injection.
Survival studies and determination of sublethal doses of bacteria.
Each isolate of E. coli was grown to log phase, concentrated by centrifugation, and diluted in saline, and various doses (0.2 ml) of each isolate were injected intraperitoneally (i.p.) into CD14+/+ BALB/c mice. The bacterial dose yielding close to 50% survival in this small-scale experiment (four mice per dose and four different doses for each bacterial isolate) was used for a larger-scale experiment to determine the sensitivity of CD14+/+ and CD14−/− mice to the different isolates of bacteria over a 7-day period. Following i.p. injection, the mice were observed for endotoxin shock-like symptoms, including ruffled fur, eye exudate, diarrhea, prostration, lack of reactivity, and death, for 7 days. The individual symptoms were each scored using a severity range of 0 to 3, the scores were combined to obtain a single score for each mouse ranging from 0 to 15, and the mean scores for all surviving mice were compared daily (Mann-Whitney test); the overall results shown by the two curves were compared by two-way analysis of variance (ANOVA). At the end of the observation period surviving mice were sacrificed with an inhalation overdose of CO2.
Preparation of tissue samples.
Mice were anesthetized with isofluorane, injected (i.p.) with an individual isolate (∼50% lethal dose), and at the appropriate time point euthanized with CO2 and exsanguinated. A sample (100 μl) of heparinized blood was taken for determination of the number of bacteria that it contained; the remainder of the blood was centrifuged to obtain plasma, which was sterile filtered, aliquoted, and stored at −80°C for cytokine analyses. The peritoneal lavage fluid was collected using 5 ml sterile normal saline; a portion (100 μl) was used for determination of the number of bacteria that it contained, and the remainder was sterile filtered, aliquoted, and stored at −80°C for cytokine analyses. A laparotomy was performed under aseptic conditions, and the lungs, liver, and spleen were collected in sterile saline, weighed, and homogenized using sterile tissue homogenizers (Tekmar, Cincinnati, OH).
Determination of the number of CFU.
Blood, peritoneal lavage fluid, and organ homogenates, all stored on ice, were serially diluted and plated on the appropriate agar plates. After overnight incubation at 37°C, bacterial colonies were counted and the number of live bacteria was normalized to the total volume of fluid or organ weight.
Activation of thioglycolate-elicited peritoneal macrophages with live bacteria.
Thioglycolate-elicited peritoneal macrophages were obtained and stimulated with live bacteria as previously described (14, 19). Briefly, CD14−/− or CD14+/+ BALB/c mice were injected with 3 ml of 4% thioglycolate broth (Difco, Detroit, MI) i.p. After 3 days, the mice were sacrificed, and the peritoneal lavage fluid was collected using RPMI 1640 medium (Gibco BRL, Grand Island, NY). The lavage fluid was centrifuged at 200 × g for 20 min at room temperature, and the cell pellet was washed once and resuspended in RPMI medium containing 1% autologous serum. The cells were seeded into 24-well plates (Becton Dickinson, Franklin Lakes, NJ) at a density of 2 × 106 cells per well, and the plates were incubated at 37°C in 5% CO2 for 3 h to allow the macrophages to adhere; the nonadherent cells were removed by washing twice with RPMI 1640 medium. Log-phase live bacteria (3 × 105 CFU) were then added to each well, and the plates were incubated for 3 h at 37°C. The supernatants were then collected, centrifuged, and filter sterilized, and the cell-free supernatants were stored at −80°C for further analysis.
Cytokine assays.
The amount of TNF-α in plasma or cell-free supernatant was measured using a double antibody sandwich enzyme-linked immunosorbent assay (ELISA) (R&D Systems, Minneapolis, MN) according to the manufacturer's instructions. The optical density at 450 nm was read using a Thermomax ELISA plate reader (Molecular Devices, Menlo Park, CA). The lower limit of detection of TNF-α was 10 pg/ml. The assays were linear up to a concentration of 1,500 pg/ml for TNF-α. Samples with levels above the upper limit were diluted and assayed again; samples with levels below the lower limit were assayed again using a lower dilution. IL-6 expression was measured by an ELISA (Biosource, Cararillo, CA) performed according to the manufacturer's instructions.
Statistics.
Results of cytokine production and bacterial clearance experiments were compared using the Mann-Whitney test, performed using GraphPad Prism version 4.0b for Macintosh (GraphPad Software, San Diego, CA) (www.graphpad.com). A P value of <0.05 was considered significant, and P values of <0.1 were also noted. Percent survival was calculated using the product limit (Kaplan-Meier) method, and curves were compared using the log rank test; all analyses were performed using GraphPad Prism software. Comparisons of symptoms of infected CD14+/+ and CD14−/− mice over time and clearance of bacteria by the two strains over time were performed by two-way ANOVA (GraphPad Prism).
RESULTS
Characteristics of E. coli isolates.
The characteristics of three of the clinical isolates of E. coli used in these studies, designated strains 59, 61, and 69, are shown in Table 1. As previously described (31), these E. coli strains were isolated from sepsis patients and are identical in most characteristics. All three isolates express mannose-resistant hemagglutinin and carboxylase B (Mf 62), as well as the aer and pap operons; the aer operon encodes a molecule that competes with transferrin for iron uptake, and the pap operon encodes an extraintestinal attachment factor. Furthermore, all three isolates are negative for hemolysin, sfa/foc, afa, hly, and the ibe-10 operon and do not express toxins or fimbrial antigens (Table 1). However, the three isolates also differ from each other in at least one of three additional characteristics. E. coli isolates 59 and 61 differ in both O-antigen (LPS) and H-antigen (flagellum) serotypes; isolate 61 is O-antigen type 1 and H-antigen type 7, whereas isolate 59 is O-antigen type 2 and H-antigen type M. In addition, although both of these isolates express the capsular K1 antigen, as determined serologically using anti-Neisseria meningitidis group B antiserum (31), they differ from isolate 69, which does not express the K1 antigen. This characteristic, shown to be important in our subsequent studies, was confirmed by electron microscopy (Fig. 1). K1-positive E. coli isolates 59 and 61 show a heavy, electron-dense layer of acidic polysaccharides when they are stained with RR, which completely surrounds the bacterial cells on their outer wall surface. The K1-negative isolate 69 and our previously studied E. coli O111 isolate (also K1 negative) lack such staining (Fig. 1).
FIG. 1.

Transmission electron micrographs of K1-positive and K1-negative E. coli isolates. The arrows indicate the K1 capsular structures stained with RR.
Thus, of the 20 different characteristics compared, E. coli isolates 59 and 61 differ in their O- and H-antigen serotypes but are identical in all other respects, including expression of the K1 capsule. In contrast, E. coli isolate 69 lacks a K1 capsule but has the same O- and H-antigen types as isolate 59, which is K1 negative (Table 1).
CD14−/− mice show different levels of resistance to genetically distinct E. coli isolates.
We previously showed that CD14-deficient mice, in contrast to normal mice, are highly resistant to E. coli O111 (19); this strain types as O antigen 1 and is negative for the H (flagellum) antigen, as well as the K1 capsular antigen. In order to extend these observations to clinical isolates of E. coli, CD14−/− and CD14+/+ BALB/c mice were injected with isolate 59, 61, or 69 at a dose that was sublethal to CD14+/+ BALB/c mice (determined in a small-scale experiment as described in Materials and Methods). Whereas 50% of the CD14+/+ mice injected with a 50% lethal dose (2 × 106 CFU/g of body weight) of E. coli K1-negative isolate 69 died, all CD14−/− mice injected with isolate 69 survived (P < 0.005) (Fig. 2C). In contrast, both CD14+/+ and CD14−/− mice were sensitive to sublethal doses of K1-positive E. coli isolates 59 (8 × 104 CFU/g of body weight) and 61 (5 × 105 CFU/g of body weight) and displayed no significant differences in survival (Fig. 2A and B). Similar results (i.e., significant differences in survival for normal and CD14-deficient mice with isolate 69 but not with isolate 59 or 61) were obtained in three independent experiments (data not shown). Nevertheless, to confirm these differences between CD14−/− and CD14+/+ mice exposed to K1-negative isolate 69, a dose-response analysis was performed. CD14−/− mice showed a clear survival advantage over normal CD14+/+ mice (P = 0.0001, two-way ANOVA), all of which died at every bacterial dose tested (Table 2). It should be noted that this pattern of survival of CD14−/− mice but not normal CD14+/+ mice was previously observed for the other K1-negative strain, E. coli O111, that was previously studied (19).
FIG. 2.
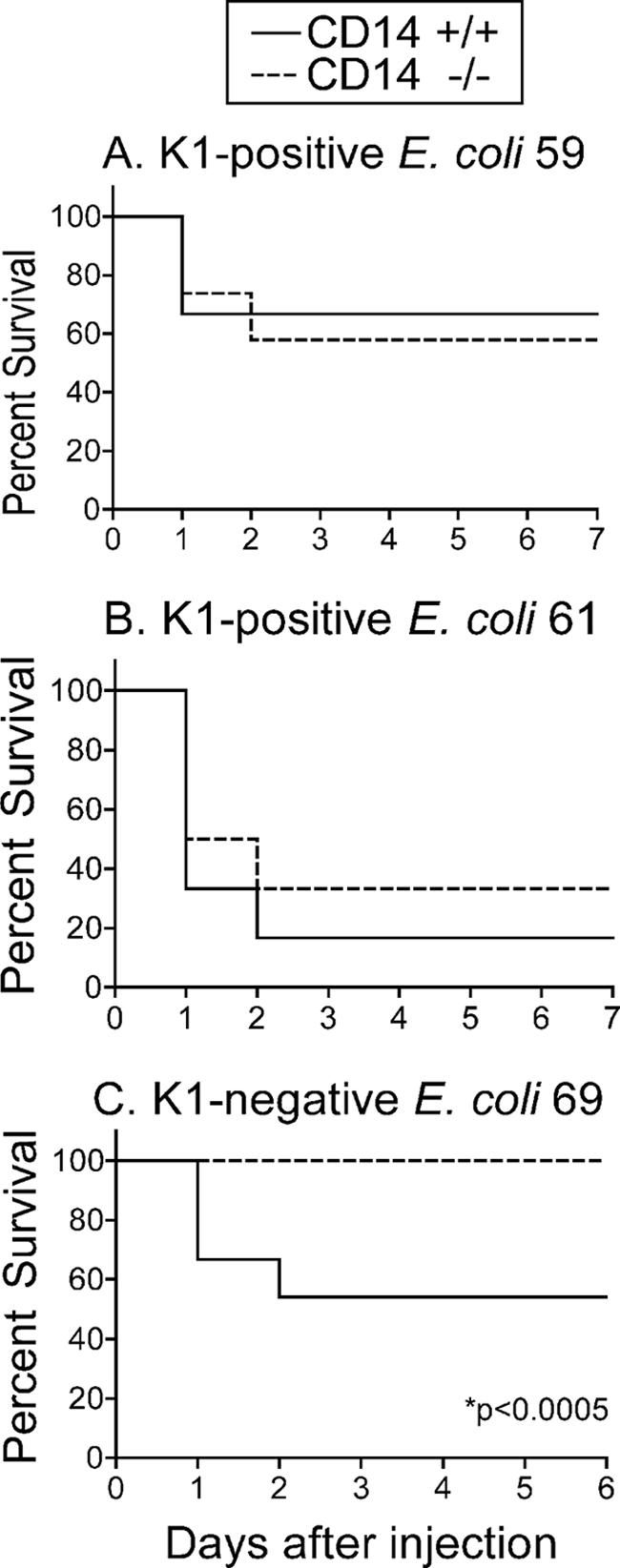
Survival of CD14+/+ and CD14−/− mice after i.p. injection of sublethal doses of K1-positive isolates 59 (A) and 61 (B) and K1-negative isolate 69 (C). The data in panels A and B are the means for 12 mice per group, and the data in panel C are the means of four pooled experiments (24 mice per group). The statistical significance of differences in survival curves was assessed by the log rank test using GraphPad Prism software.
TABLE 2.
Survival of mice injected with different amounts of K1-negative isolate 69a
| Amt injected (CFU/g of body wt) | No. alive/total no.
|
|||||
|---|---|---|---|---|---|---|
| Day 1
|
Day 2
|
Day 3
|
||||
| CD14+/+ mice | CD14−/− mice | CD14+/+ mice | CD14−/− mice | CD14+/+ mice | CD14−/− mice | |
| 3 × 106 | 2/4 | 4/4 | 0/4 | 4/4 | 0/4 | 4/4 |
| 4.5 × 106 | 2/4 | 2/4 | 0/4 | 2/4 | 0/4 | 2/4 |
| 6.75 × 106 | 0/4 | 3/4 | 0/4 | 2/4 | 0/4 | 2/4 |
| 10 × 106 | 0/4 | 0/4 | 0/4 | 0/4 | 0/4 | 0/4 |
Mice were injected i.p. with the doses of E. coli indicated and were observed for 7 days; no additional deaths occurred after 3 days. The P value was 0.0001 as determined by two-way ANOVA.
In addition to survival, mice were also examined daily for other gross symptoms of responsiveness often seen in endotoxemia, including ruffled fur, eye exudates, diarrhea, prostration, and lack of reactivity, and these symptoms were scored daily on the basis of severity from 0 to 3 (Fig. 3). In our previous studies (19), CD14-deficient mice showed few symptoms of shock, whereas normal mice quickly succumbed to E. coli O111. When the symptoms of CD14−/− mice infected with the K1-negative isolate, strain 69, were compared to those of normal CD14+/+ mice (Fig. 3C), it was observed that CD14−/− mice displayed significantly less severe symptoms than CD14+/+ mice (P = 0.0367), especially on day 1 (P < 0.03). In contrast, there were no differences in symptoms between CD14−/− and CD14+/+ mice infected with the K1-positive isolates, strains 59 and 61 (Fig. 3A and 3B), except on day 1, although the differences were less pronounced for mice injected with the K1-positive isolate 61 (Fig. 3B). These differences in severity of symptoms over an extended period of time correlated well with the differences in survival (Fig. 2 and 3). However, there was no correlation between symptoms on day 1 and survival; rather, the differences in symptoms observed at day 1 seemed to correlate well with levels of TNF-α expression (see below).
FIG. 3.

Symptoms of CD14+/+ and CD14−/− mice after injection of sublethal doses of K1-positive isolates 59 (A) and 61 (B) and K1-negative isolate 69 (C). The symptom index was calculated as described in Materials and Methods. Results at individual points were compared using the Mann-Whitney t test. The statistical significance of differences in symptoms over time was assessed by two-way ANOVA.
CD14−/− mice are resistant to a lethal dose of K1-negative E. coli but not to K1-positive E. coli.
To determine whether the differences in survival of CD14−/− mice infected with the nonisogenic K1-positive (isolates 59 and 61) and K1-negative (isolate 69) clinical isolates were due to the K1 capsule itself and not to one of the other factors that distinguish these isolates (Table 1), CD14−/− mice were injected with sublethal doses of one of two additional E. coli isolates, either RS218, a K1-positive E. coli obtained from a newborn with meningitis (O18:K1:H7) (22), or an isogenic mutant of RS218 that lacks the K1 capsule (O18:H7) (22). As was observed with the K1-negative nonisogenic isolate 69, CD14−/− survived a dose of the isogenic K1-negative mutant that was lethal for CD14+/+ mice (Fig. 4B), while both strains of mice were sensitive to the parental K1-positive E. coli RS218 strain (Fig. 4A). Thus, the presence of the K1 capsule alone was sufficient to eliminate the resistance of CD14−/− mice to E. coli.
FIG. 4.
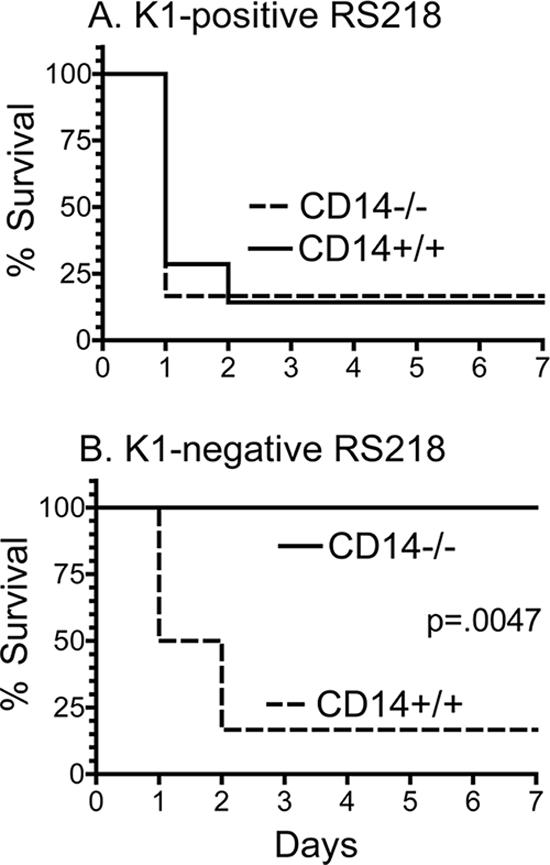
Survival of CD14+/+ and CD14−/− mice after i.p. injection of K1-positive RS218 E. coli (A) or its K1-negative isogenic mutant (B). The statistical significance of differences in survival curves was assessed by the log rank test using GraphPad Prism software.
CD14−/− mice produce low levels of TNF-α in response to both K1-positive and K1-negative E. coli.
To determine whether survival correlates with high or low levels of the proinflammatory cytokine TNF-α, the levels of in vivo production of TNF-α were compared in mice injected with the nonisogenic K1-positive and K1-negative E. coli strains. All three isolates induced comparable levels of circulating TNF-α in normal CD14+/+ mice, with values ranging from a mean of 1.877 ng/ml (K1-positive isolate 59) to 3.322 ng/ml (K1-negative isolate 69). However, the level of circulating TNF-α was significantly reduced in CD14−/− mice with all three isolates (Fig. 5). The decrease was most pronounced for the K1-negative isolate 69 (mean, 0.1409 ng/ml) and least pronounced for the K1-positive isolate 61 (mean, 0.7403 ng/ml). The difference in TNF-α induction between normal and CD14−/− mice was also observed with both the K-1 positive RS218 isolate and its K1-negative isogenic mutant and was maintained for up to 12 h (data not shown).
FIG. 5.
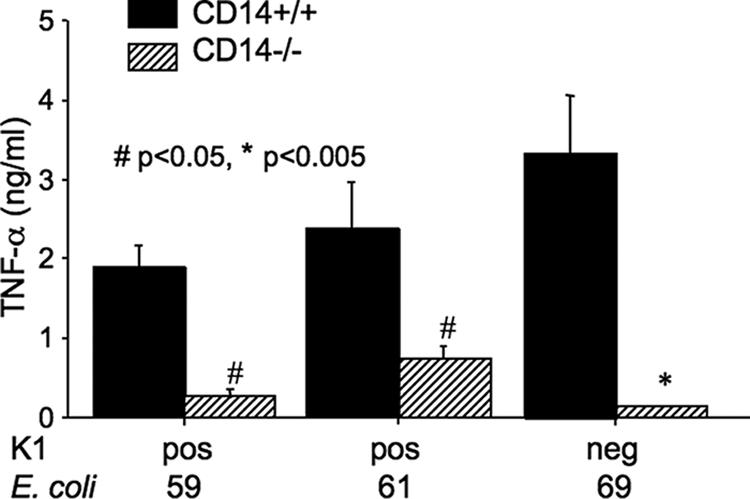
TNF-α levels in blood of CD14+/+ and CD14−/− mice 1.5 h following i.p. injection of K1-positive (isolates 59 and 61) or K1-negative (isolate 69) E. coli. The results are means ± standard errors of the means for six mice per group. All measurements were made in triplicate. Results were compared using the Mann-Whitney t test.
To examine whether the differences in TNF-α levels between CD14+/+ and CD14−/− mice in response to both K1-positive and K1-negative isolates reflected differences in the ability of CD14+/+ and CD14−/− macrophages to respond to these isolates, the abilities of all three nonisogenic isolates of E. coli to stimulate production of TNF-α from peritoneal macrophages isolated from both CD14+/+ and CD14−/− mice were determined. In these studies, a constant amount (3 × 105 CFU) of the isolates was used. All three isolates induced production of high levels of TNF-α from CD14+/+ macrophages, with values ranging from means of 6.649 ng/ml (K1-negative strain 69) to 13.91 ng/ml (K1-positive strain 61). However, the amount of TNF-α induced was significantly less for CD14−/− macrophages, irrespective of which isolate was used for stimulation (Fig. 6). Thus, for all three isolates, the ability to induce production of TNF-α was dependent on the presence of CD14. However, although the levels of TNF-α correlated with survival for mice injected with K1-negative isolate 69, there was no such correlation for mice injected with K1-positive isolates 59 and 61.
FIG. 6.
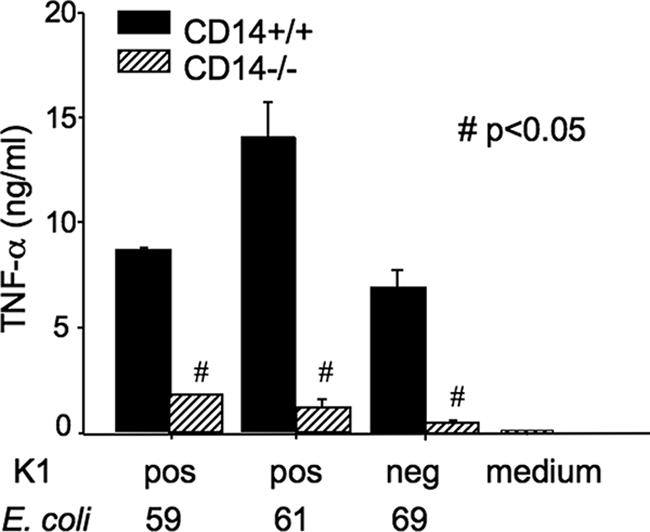
In vitro production of TNF-α by CD14+/+ and CD14−/− thioglycolate-elicited peritoneal macrophages after incubation with log-phase live K1-positive (isolates 59 and 61) or K1-negative (isolate 69) E. coli. The results are means ± standard errors of the means of two independent experiments. All measurements were made in triplicate. Where there are no error bars, they fall within the bar. Results were compared using the Mann-Whitney t test.
CD14−/− mice produce low levels of IL-6 in response to both K1-positive and K1-negative E. coli.
Studies by other workers have suggested that low levels of IL-6 at 6 h after infection correlate with survival in a polymicrobial model of infection (35, 44, 50). To determine whether survival after infection with K1-positive or K1-negative E. coli correlates with high or low levels of the proinflammatory cytokine IL-6, the levels of in vivo production of IL-6 at 6 h were compared in normal and CD14−/− mice injected with K1-positive and K1-negative bacteria. Both of the K1-positive isolates, isolates 59 and RS218, as well as the K1-negative isolates, isolate 69 and the isogenic mutant of RS218, induced production of significantly more IL-6 in normal mice than in CD14−/− mice (Fig. 7). However, in spite of the presence of low levels of IL-6, there was no survival advantage for CD14−/− mice infected with the K1-positive isolates as there was with CD14−/− mice infected with the K1-negative isolates.
FIG. 7.
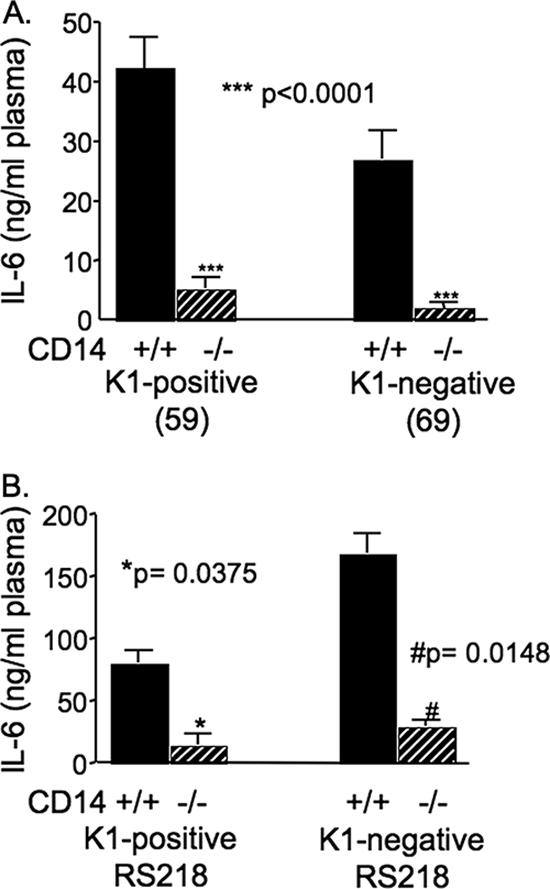
IL-6 levels in blood of CD14+/+ and CD14−/− mice 6 h following i.p. injection of (A) nonisogenic K1-positive isolate 59 and K1-negative isolate 69 and (B) K1-positive RS218 and its K1-negative isogenic mutant. Results were compared using the Mann-Whitney t test.
Survival correlates with enhanced clearance of bacteria.
To further examine the basis for the resistance of CD14−/− mice to K1-negative E. coli but not to K1-positive E. coli, the degrees of bacterial clearance of all three nonisogenic isolates were initially compared. At the time that the samples were analyzed (3 h after i.p. injection) all mice were bacteremic; however, the degree of bacteremia varied. The CD14−/− mice cleared the K1-negative E. coli isolate 69 much more efficiently than the two K1-positive isolates (Fig. 8). As shown in Fig. 8C, the bacterial counts of E. coli 69 in organs, the peritoneal cavity, and the blood were significantly reduced in CD14−/− mice compared to CD14+/+ mice. The bacterial counts in the blood and peritoneal lavage fluid of CD14−/− mice were 1/20 and 1/15 of the counts in their control counterparts, respectively. However, CD14−/− and CD14+/+ mice showed no difference in the bacterial counts in organs or fluids when they were injected with the two K1-positive E. coli isolates, strains 59 and 61 (Fig. 8A and B). Similarly, a pattern of enhanced bacterial clearance was observed at 3 h in CD14−/− mice compared to CD14+/+ mice when they were injected with the K1-negative isogenic mutant but not when they were injected with the parental K1-positive RS218 isolate (data not shown). Thus, the ability to clear bacteria, rather than TNF-α or IL-6 levels, correlated with survival.
FIG. 8.
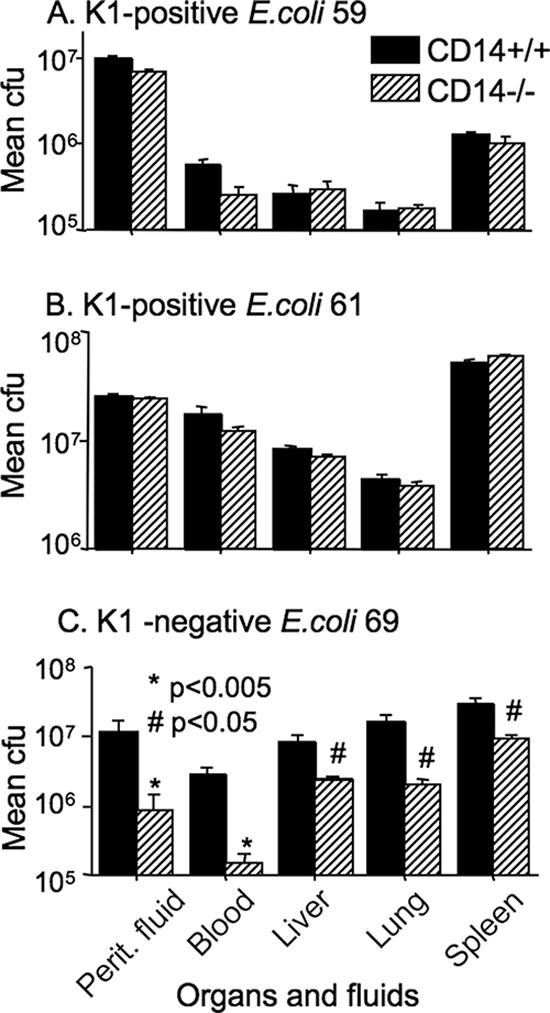
Bacterial counts (CFU/g of organ or CFU/ml of fluid) recovered 3 h following i.p. injection of K1-positive isolate 59 (A), K1-positive isolate 61 (B), or K1-negative isolate 69 (C). The data are expressed as means ± standard errors of the means on a log scale and include data for blood (CFU/ml), total peritoneal lavage fluid (Perit. fluid) (CFU) and the liver, lung, and spleen (CFU/g of organ). P values were determined by a Mann-Whitney t test.
To confirm that differences in bacterial clearance persisted over time, normal and CD14−/− mice were injected with a sublethal dose of either the K1-positive RS218 isolate or its K1-negative isogenic mutant and bacterial counts in peritoneal fluid and blood were determined at various times (1 to 12 h) after injection. As was observed with the nonisogenic isolates, the K1-negative isogenic mutant was rapidly cleared in CD14−/− mice but not in normal mice (Fig. 9B and D). In contrast, the number of K1-positive organisms rapidly increased over time in both normal and CD14−/− mice, although the increase was more pronounced in the blood than in the peritoneal cavity (Fig. 9A and C). A pattern of enhanced bacterial clearance similar to that shown in Fig. 8 was observed in the livers and spleens of CD14−/− mice injected with the K1-negative isogenic mutant but not in the livers and spleens of CD14−/− mice injected with the parental K1-positive RS218 isolate (data not shown).
FIG. 9.

Time course of bacterial counts in the blood (A and B) and the peritoneal cavity (C and D) of CD14+/+ and CD14−/− mice following injection of K1-positive RS218 (top panels) or its K1-negative isogenic mutant (bottom panels). The statistical significance of differences in the number of live bacteria recovered over time was assessed by two-way ANOVA.
DISCUSSION
The present study was initiated to study the role of CD14 in the response of the host to different clinical isolates of E. coli obtained from sepsis patients. We used three extensively typed clinical isolates (designated strains 59, 61, and 69) that share the majority of genotypic and phenotypic characteristics but differ in at least one of three characteristics, the O-antigen (LPS) and/or H-antigen (flagellum) antigen serotype or the presence of a K1 capsule (Table 1). Both LPS and the K1 capsule are known to be important virulence factors for E. coli. LPS is a potent activator of innate immune responses, resulting in the release of pro- and anti-inflammatory mediators from hematopoetic and nonhematopoetic cells (16, 45). The smooth form of LPS, found in most strains of gram-negative bacteria, consists of a serologically defined O-antigen polysaccharide linked to core polysaccharides and a lipid A moiety; the lipid A moiety is embedded in the outer bacterial membrane. The primary receptor for LPS, CD14, is expressed on the surface of monocytes, macrophages, neutrophils, and some dendritic cells (15, 18, 27, 48). It functions in the activation of cellular responses, including release of proinflammatory cytokines and induction of endotoxemia (9, 10, 13, 16, 17, 19, 23, 24, 40, 41, 49), playing a major role in the symptoms leading to severe sepsis and/or septic shock (4, 20, 29, 33). In general, it is widely accepted that low-level expression of proinflammatory cytokines and other inflammatory mediators is beneficial to the host's ability to eliminate the infecting bacteria, while high levels of these mediators lead potentially to death.
In addition to LPS, the presence of a polysaccharide capsule loosely associated with the outer surface of gram-negative bacteria may be associated with increased virulence; the polysaccharides show much greater structural variability than LPS. Capsular polysaccharides (serologically typed as K antigens) represent a major surface antigen of E. coli, and more than 80 distinct K serotypes have been described (47). However, only a few K-antigen serotypes have been associated with virulence; these include K1, K5, K10, and K54 (47).
Structurally, the K1 capsule is a homopolymer of α2,8-linked N-acetylneuraminic acid. Unlike the toxic properties of LPS that cause activation of host cells, the virulence of bacteria expressing such a capsule appears to be associated with evading the host response. Several mechanisms by which the capsular polysaccharide aids the bacteria in eluding host defenses have been postulated. Such mechanisms include masking of LPS and thus preventing activation of the innate immune system, as well as masking sites for deposition of opsonins and thus inhibiting phagocytosis and sites required for complement-mediated killing (37). Thus, the available literature suggests that the K1 capsule may be capable of inhibiting some aspects of the early innate immune response that may normally be beneficial early in infection.
In the course of performing these studies we observed major differences in the responses of CD14-deficient and normal mice to three different E. coli isolates (isolates 59, 61, and 69) that seemed to correlate with the presence of a K1 capsule. Accordingly, we used a K1-positive isolate, RS218, obtained from a newborn with meningitis, and a K1-negative isogenic mutant of this isolate to confirm observations made with the nonisogenic isolates.
Our results demonstrate that CD14−/− mice, but not CD14+/+ mice, are resistant to K1-negative E. coli, similar to what we previously found using an E. coli O111 strain (19). Furthermore, as was previously observed with E. coli O111, the K1-negative organisms are rapidly cleared and induce very little TNF-α or IL-6 production in CD14−/− mice. In contrast, although the levels of TNF-α and IL-6 induced by the K1-positive isolates in CD14−/− mice are dramatically reduced compared to the levels in CD14+/+ mice, there is no increase in survival; both CD14+/+ and CD14−/− mice die within 48 h. Furthermore, there is no accelerated clearance of the K1-positive bacteria, as was observed for the two K1-negative isolates, in CD14−/− mice; thus, CD14+/+ and CD14−/− mice showed no difference in bacterial counts when they were injected with any of the K1-positive isolates.
In contrast to our observation that CD14−/− mice are resistant to a lethal dose of K1-negative E. coli, Cross et al. (6) found that C3H/HeJ mice that are deficient in a functional TLR4 molecule required for LPS-CD14 signaling (21, 32) are as sensitive as normal mice (C3H/HeN) to a lethal dose of K1-negative E. coli, as measured by survival and bacterial clearance (5, 6). Furthermore, although CD14−/− mice show sensitivity to K1-positive E. coli similar to that of CD14+/+ mice (Fig. 2), Cross et al. (6) found that C3H/HeJ mice lacking TLR4 were >1,000-fold more sensitive to K1-positive E. coli than their parental control. The differences in the resistance of CD14-deficient and TLR4-deficient mice to both K1-negative and K1-positive E. coli suggest that TLR4 functions independent of CD14 in response to some bacterial components.
Our studies also contrast with those of Bernheiden et al. (2), who showed that CD14−/− and TLR4−/− mice are more susceptible to Salmonella enterica serovar Typhimurium than controls. This difference between our observations and those of Bernheiden and colleagues may be due to the fact that S. enterica serovar Typhimurium is an intracellular pathogen, while the E. coli isolates used in these studies are extracellular pathogens.
The resistance of CD14−/− mice to the K1-negative isolates but not to the K1-positive isolates indicates that entirely different mechanisms are responsible for the symptoms and death induced by K1-positive E. coli and by K1-negative E. coli. Production of large amounts of proinflammatory cytokines like TNF-α is induced during gram-negative bacterial infections, which are thought to be critical for inducing mortality during septic shock (8, 43). For the K1-negative isolates, survival correlates with the decrease in TNF-α production and rapid clearance of the organism. In contrast, although there is a significant decrease in TNF-α production in CD14−/− mice injected with the K1-positive organisms, there is no enhanced survival, suggesting that TNF-α is not responsible for death caused by K1-positive E. coli. Furthermore, there is no beneficial effect from the low levels of TNF-α, in contrast to the studies of Cross et al. (5), who found that injection of a combination of TNF-α and IL-1 reduced the sensitivity of C3H/HeJ (TLR4-deficient) mice to K1-positive bacteria. It should be noted that the small amount of TNF-α induced by the K1-positive isolates, especially isolate 61, in CD14−/− mice indicates that there is a bacterial component capable of inducing TNF-α production via a non-CD14 mechanism.
The role of IL-6 in sepsis-induced morbidity and mortality has been controversial. In previous studies, individual variation in IL-6 expression was used to predict the survival of mice in the early (acute) phase of cecal ligation and puncture-induced sepsis (35, 44, 50). However, studies with IL-6-deficient mice (36), as well as antibody therapy targeted to IL-6 (46), clearly show that IL-6 is an indicator of the severity of disease rather than a cause of disease. As was the case with IL-6-deficient mice in a cecal ligation and puncture model of sepsis (36), the low levels of IL-6 expression in CD14-deficient mice compared to normal mice did not prevent mortality when mice were infected with the K1-positive E. coli isolates, even though CD14−/− mice displayed reduced symptoms of sepsis on day 1 compared to normal mice (Fig. 3).
Further evidence that the mechanisms resulting in death induced by the K1-negative and K1-positive isolates are distinct is based on the pattern of symptoms observed after injection. Initially (within 24 h), mice injected with the K1-positive organisms show significantly greater gross symptoms of a response than those injected with the K1-negative bacteria (Fig. 3). Furthermore, whereas these symptoms diminish significantly after day 1 in surviving mice injected with the K1-positive isolates, a similar significant decrease is not seen in mice injected with K1-negative bacteria.
The lack of a major role for CD14 in the response to K1-positive E. coli, coupled with the host's inability to phagocytose and kill K1-positive E. coli, may explain the results of other workers studying the therapeutic effects of anti-CD14 monoclonal antibodies in models of septic shock or pneumonia induced by K1-positive E. coli. In these studies, anti-CD14 reduced symptoms of endotoxemia but had no effect on bacterial clearance (12, 30). The importance of bacterial clearance in ameliorating the effects of septic shock is further supported by studies in mouse models of polymicrobial infection which show that survival during the chronic phase of infection correlates best with the ability to control bacterial overgrowth and not with proinflammatory cytokine levels (50).
These studies suggest that there is significant diversity in the mechanisms and/or molecules associated with symptoms occurring in response to bacteria expressing different virulence factors. Indeed, although some bacteria may use specific cell receptors, such as CD14, to mediate their effects, other bacteria may use other receptors to induce severe sepsis or septic shock. Accordingly, it may be difficult to treat all patients with gram-negative septicemia with a single drug, as has been tried previously with anti-TNF antibodies. Only a thorough understanding of the various mechanisms employed by different organisms to induce sepsis/septic shock will make it possible to treat these syndromes effectively.
Acknowledgments
This work was supported in part by National Institutes of Health grant RO1 AI23859 (to S.M.G.) and by a grant from la Fondation pour la Recherche Médicale, Paris (to S.C.G.).
We thank Ana Pino (Laboratory of Innate Immunity, CUNY Medical School/Sophie Davis School of Biomedical Research) and Jeffrey Moyse (Electron Microscopy Laboratory, Manhasset, NY) for their excellent technical assistance.
Editor: J. B. Bliska
Footnotes
Published ahead of print on 20 August 2007.
REFERENCES
- 1.American College of Chest Physicians/Society of Critical Care Medicine Consensus Conference Committee. 1992. Definitions for sepsis and organ failure and guidelines for the use of innovative therapies in sepsis. Crit. Care Med. 20:864-874. [PubMed] [Google Scholar]
- 2.Bernheiden, M., J. M. Heinrich, G. Minigo, C. Schutt, F. Stelter, M. Freeman, D. Golenbock, and R. S. Jack. 2001. LBP, CD14, TLR4 and the murine innate immune response to a peritoneal Salmonella infection. J. Endotoxin Res. 7:447-450. [PubMed] [Google Scholar]
- 3.Bliss, J. M., and R. P. Silver. 1996. Coating the surface: a model for expression of capsular polysialic acid in Escherichia coli K1. Mol. Microbiol. 21:221-231. [DOI] [PubMed] [Google Scholar]
- 4.Bone, R. C., C. L. Sprung, and W. J. Sibbald. 1992. Definitions for sepsis and organ failure. Crit. Care Med. 20:724-726. [DOI] [PubMed] [Google Scholar]
- 5.Cross, A., L. Asher, M. Seguin, L. Yuan, N. Kelly, C. Hammack, J. Sadoff, and P. Gemski, Jr. 1995. The importance of a lipopolysaccharide-initiated, cytokine-mediated host defense mechanism in mice against extraintestinally invasive Escherichia coli. J. Clin. Investig. 96:676-686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cross, A. S., J. C. Sadoff, N. Kelly, E. Bernton, and P. Gemski. 1989. Pretreatment with recombinant murine tumor necrosis factor alpha/cachectin and murine interleukin 1 alpha protects mice from lethal bacterial infection. J. Exp. Med. 169:2021-2027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cross, A. S. 1990. The biologic significance of bacterial encapsulation. Curr. Top. Microbiol. Immunol. 150:87-95. [DOI] [PubMed] [Google Scholar]
- 8.Damas, P., A. Reuter, P. Geysen, J. Demonty, M. Lamy, and P. Franchimont. 1989. Tumor necrosis factor and interleukin-1 serum levels during severe sepsis in humans. Crit. Care Med. 17:975-978. [DOI] [PubMed] [Google Scholar]
- 9.Ferrero, E., C. L. Hsieh, U. Francke, and S. M. Goyert. 1990. CD14 is a member of the family of leucine-rich proteins and is encoded by a gene syntenic with multiple receptor genes. J. Immunol. 145:331-336. [PubMed] [Google Scholar]
- 10.Ferrero, E., D. Jiao, B. Z. Tsuberi, L. Tesio, G. W. Rong, A. Haziot, and S. M. Goyert. 1993. Transgenic mice expressing human CD14 are hypersensitive to lipopolysaccharide. Proc. Natl. Acad. Sci. USA 90:2380-2384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Fitzgerald, K. A., D. C. Rowe, and D. T. Golenbock. 2004. Endotoxin recognition and signal transduction by the TLR4/MD2-complex. Microbes Infect. 6:1361-1367. [DOI] [PubMed] [Google Scholar]
- 12.Frevert, C. W., G. Matute-Bello, S. J. Skerrett, R. B. Goodman, O. Kajikawa, C. Sittipunt, and T. R. Martin. 2000. Effect of CD14 blockade in rabbits with Escherichia coli pneumonia and sepsis. J. Immunol. 164:5439-5445. [DOI] [PubMed] [Google Scholar]
- 13.Gangloff, S. C., U. Zahringer, C. Blondin, M. Guenounou, J. Silver, and S. M. Goyert. 2005. Influence of CD14 on ligand interactions between lipopolysaccharide and its receptor complex. J. Immunol. 175:3940-3945. [DOI] [PubMed] [Google Scholar]
- 14.Gangloff, S. C., N. Hijiya, A. Haziot, and S. M. Goyert. 1999. Lipopolysaccharide structure influences the macrophage response via CD14-independent and CD14-dependent pathways. Clin. Infect. Dis. 28:491-496. [DOI] [PubMed] [Google Scholar]
- 15.Goyert, S. M., E. Ferrero, W. J. Rettig, A. K. Yenamandra, F. Obata, and M. M. Le Beau. 1988. The CD14 monocyte differentiation antigen maps to a region encoding growth factors and receptors. Science 239:497-500. [DOI] [PubMed] [Google Scholar]
- 16.Grone, A. 2002. Keratinocytes and cytokines. Vet. Immunol. Immunopathol. 88:1-12. [DOI] [PubMed] [Google Scholar]
- 17.Haziot, A., S. Chen, E. Ferrero, M. G. Low, R. Silber, and S. M. Goyert. 1988. The monocyte differentiation antigen, CD14, is anchored to the cell membrane by a phosphatidylinositol linkage. J. Immunol. 141:547-552. [PubMed] [Google Scholar]
- 18.Haziot, A., B.-Z. Tsuberi, and S. M. Goyert. 1993. Neutrophil CD14: biochemical properties and role in the secretion of tumor necrosis factor-α in response to lipopolysaccharide. J. Immunol. 150:5556-5565. [PubMed] [Google Scholar]
- 19.Haziot, A., E. Ferrero, F. Kontgen, N. Hijiya, S. Yamamoto, J. Silver, C. L. Stewart, and S. M. Goyert. 1996. Resistance to endotoxin shock and reduced dissemination of gram-negative bacteria in CD14-deficient mice. Immunity 4:407-414. [DOI] [PubMed] [Google Scholar]
- 20.Horn, D. L., D. C. Morrison, S. M. Opal, R. Silverstein, K. Visvanathan, and J. B. Zabriskie. 2000. What are the microbial components implicated in the pathogenesis of sepsis? Report on a symposium. Clin. Infect. Dis. 31:851-858. [DOI] [PubMed] [Google Scholar]
- 21.Hoshino, K., O. Takeuchi, T. Kawai, H. Sanjo, T. Ogawa, Y. Takeda, K. Takeda, and S. Akira. 1999. Cutting edge: Toll-like receptor 4 (TLR4)-deficient mice are hyporesponsive to lipopolysaccharide: evidence for TLR4 as the Lps gene product. J. Immunol. 162:3749-3752. [PubMed] [Google Scholar]
- 22.Kim, K. J., S. J. Elliott, F. Di Cello, M. F. Stins, and K. S. Kim. 2003. The K1 capsule modulates trafficking of E. coli-containing vacuoles and enhances intracellular bacterial survival in human brain microvascular endothelial cells. Cell. Microbiol. 5:245-252. [DOI] [PubMed] [Google Scholar]
- 23.Knuefermann, P., S. Nemoto, A. Misra, N. Nozaki, G. Defreitas, S. M. Goyert, B. A. Carabello, D. L. Mann, and J. G. Vallejo. 2002. CD14-deficient mice are protected against lipopolysaccharide-induced cardiac inflammation and left ventricular dysfunction. Circulation 106:2608-2615. [DOI] [PubMed] [Google Scholar]
- 24.Leturcq, D. J., A. M. Moriarty, G. Talbott, R. K. Winn, T. R. Martin, and R. J. Ulevitch. 1996. Antibodies against CD14 protect primates from endotoxin-induced shock. J. Clin. Investig. 98:1533-1538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Levy, M. M., M. P. Fink, J. C. Marshall, E. Abraham, D. Angus, D. Cook, J. Cohen, S. M. Opal, J. L. Vincent, and G. Ramsay. 2003. 2001 SCCM/ESICM/ACCP/ATS/SIS International Sepsis Definitions Conference. Intensive Care Med. 29:530-538. [DOI] [PubMed] [Google Scholar]
- 26.Luft, J. H. 1971. Ruthenium red and violet. I. Chemistry, purification, methods of use for electron microscopy and mechanism of action. Anat. Rec. 171:347-368. [DOI] [PubMed] [Google Scholar]
- 27.Mahnke, K., E. Becher, P. Ricciardi-Castagnoli, T. A. Luger, T. Schwarz, and S. Grabbe. 1997. CD14 is expressed by subsets of murine dendritic cells and upregulated by lipopolysaccharide. Exp. Med. Biol. 417:145-159. [DOI] [PubMed] [Google Scholar]
- 28.McCabe, W. R., B. Kaijser, S. Olling, M. Uwaydah, and L. A. Hanson. 1978. Escherichia coli in bacteremia: K and O antigens and serum sensitivity of strains from adults and neonates. J. Infect. Dis. 138:33-41. [DOI] [PubMed] [Google Scholar]
- 29.Morrison, D. C., and J. L. Ryan. 1987. Endotoxin and disease mechanisms. Annu. Rev. Med. 38:417-432. [DOI] [PubMed] [Google Scholar]
- 30.Opal, S. M., J. E. Palardy, N. Parejo, and R. L. Jasman. 2003. Effect of anti CD14 monoclonal antibody on clearance of Escherichia coli bacteremia and endotoxemia. Crit. Care Med. 31:929-932. [DOI] [PubMed] [Google Scholar]
- 31.Picard, B., J. S. Garcia, S. Gouriou, P. Duriez, N. Brahimi, E. Bingen, J. Elion, and E. Denamur. 1999. The link between phylogeny and virulence in Escherichia coli extraintestinal infection. Infect. Immun. 67:546-553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Poltorak, A., X. He, I. Smirnova, M. Y. Liu, C. Van Huffel, X. Du, D. Birdwell, E. Alejos, M. Silva, C. Galanos, M. Freudenberg, P. Ricciardi-Castagnoli, B. Layton, and B. Beutler. 1998. Defective LPS signaling in C3H/HeJ and C57BL/10ScCr mice: mutations in Tlr4 gene. Science 282:2085-2088. [DOI] [PubMed] [Google Scholar]
- 33.Raetz, C. R., R. J. Ulevitch, S. D. Wright, C. H. Sibley, A. Ding, and C. F. Nathan. 1991. Gram-negative endotoxin: an extraordinary lipid with profound effects on eukaryotic signal transduction. FASEB J. 5:2652-2660. [DOI] [PubMed] [Google Scholar]
- 34.Raetz, C. R. 1990. Biochemistry of endotoxins. Annu. Rev. Biochem. 59:129-170. [DOI] [PubMed] [Google Scholar]
- 35.Remick, D. G., G. R. Bolgos, J. Siddiqui, J. Shin, and J. A. Nemzek. 2002. Six at six: interleukin-6 measured 6 h after the initiation of sepsis predicts mortality over 3 days. Shock 17:463-467. [DOI] [PubMed] [Google Scholar]
- 36.Remick, D. G., G. Bolgos, S. Copeland, and J. Siddiqui. 2005. Role of interleukin-6 in mortality from and physiologic response to sepsis. Infect. Immun. 73:2751-2757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Roberts, I. S. 1996. The biochemistry and genetics of capsular polysaccharide production in bacteria. Annu. Rev. Microbiol. 50:285-315. [DOI] [PubMed] [Google Scholar]
- 38.Russo, T. A., G. Sharma, C. R. Brown, and A. A. Campagnari. 1995. Loss of the O4 antigen moiety from the lipopolysaccharide of an extraintestinal isolate of Escherichia coli has only minor effects on serum sensitivity and virulence in vivo. Infect. Immun. 63:1263-1269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Schiffer, M. S., E. Oliveira, M. P. Glode, G. H. McCracken, Jr., L. M. Sarff, and J. B. Robbins. 1976. A review: relation between invasiveness and the K1 capsular polysaccharide of Escherichia coli. Pediatr. Res. 10:82-87. [DOI] [PubMed] [Google Scholar]
- 40.Schimke, J., J. Mathison, J. Morgiewicz, and R. J. Ulevitch. 1998. Anti CD14 mAb treatment provides therapeutic benefit after in vivo exposure to endotoxin. Proc. Natl. Acad. Sci. USA 95:13875-13880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Tasaka, S., A. Ishizaka, W. Yamada, M. Shimizu, H. Koh, N. Hasegawa, Y. Adachi, and K. Yamaguchi. 2003. Effect of CD14 blockade on endotoxin-induced acute lung injury in mice. Am. J. Respir. Cell Mol. Biol. 29:252-258. [DOI] [PubMed] [Google Scholar]
- 42.Timmis, K. N., G. J. Boulnois, D. Bitter-Suermann, and F. C. Cabello. 1985. Surface components of Escherichia coli that mediate resistance to the bactericidal activities of serum and phagocytes. Curr. Top. Microbiol. Immunol. 118:197-218. [DOI] [PubMed] [Google Scholar]
- 43.Tracey, K. J., B. Beutler, S. F. Lowry, J. Merryweather, S. Wolpe, I. W. Milsark, R. J. Hariri, T. J. Fahey III, A. Zentella, J. D. Albert, et al. 1986. Shock and tissue injury induced by recombinant human cachectin. Science 234:470-474. [DOI] [PubMed] [Google Scholar]
- 44.Turnbull, I. R., P. Javadi, T. G. Buchman, R. S. Hotchkiss, I. E. Karl, and C. M. Coopersmith. 2004. Antibiotics improve survival in sepsis independent of injury severity but do not change mortality in mice with markedly elevated interleukin 6 levels. Shock 21:121-125. [DOI] [PubMed] [Google Scholar]
- 45.Van Amersfoort, E. S., T. J. Van Berkel, and J. Kuiper. 2003. Receptors, mediators, and mechanisms involved in bacterial sepsis and septic shock. Clin. Microbiol. Rev. 16:379-414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Vyas, D., P. Javadi, P. J. Dipasco, T. G. Buchman, R. S. Hotchkiss, and C. M. Coopersmith. 2005. Early antibiotic administration but not antibody therapy directed against IL-6 improves survival in septic mice predicted to die on basis of high IL-6 levels. Am. J. Physiol. Regul. Integr. Comp. Physiol. 289:R1048-R1053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Whitfield, C., and I. S. Roberts. 1999. Structure, assembly and regulation of expression of capsules in Escherichia coli. Mol. Microbiol. 31:1307-1319. [DOI] [PubMed] [Google Scholar]
- 48.Woodhead, V. E., M. H. Binks, B. M. Chain, and D. R. Katz. 1998. From sentinel to messenger: an extended phenotypic analysis of the monocyte to dendritic cell transition. Immunology 94:552-559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wright, S. D., R. A. Ramos, P. S. Tobias, R. J. Ulevitch, and J. C. Mathison. 1990. CD14, a receptor for complexes of lipopolysaccharide (LPS) and LPS binding protein. Science 249:1431-1433. [DOI] [PubMed] [Google Scholar]
- 50.Xiao, H., J. Siddiqui, and D. G. Remick. 2006. Mechanisms of mortality in early and late sepsis. Infect. Immun. 74:5227-5235. [DOI] [PMC free article] [PubMed] [Google Scholar]