

Abstract
Francisella tularensis, the etiologic agent of tularemia in humans, is a potential biological threat due to its low infectious dose and multiple routes of entry. F. tularensis replicates within several cell types, eventually causing cell death by inducing apoptosis. In this study, a modified Himar1 transposon (HimarFT) was used to mutagenize F. tularensis LVS. Approximately 7,000 Kmr clones were screened using J774A.1 macrophages for reduction in cytopathogenicity based on retention of the cell monolayer. A total of 441 candidates with significant host cell retention compared to the parent were identified following screening in a high-throughput format. Retesting at a defined multiplicity of infection followed by in vitro growth analyses resulted in identification of approximately 70 candidates representing 26 unique loci involved in macrophage replication and/or cytotoxicity. Mutants carrying insertions in seven hypothetical genes were screened in a mouse model of infection, and all strains tested appeared to be attenuated, which validated the initial in vitro results obtained with cultured macrophages. Complementation and reverse transcription-PCR experiments suggested that the expression of genes adjacent to the HimarFT insertion may be affected depending on the orientation of the constitutive groEL promoter region used to ensure transcription of the selective marker in the transposon. A hypothetical gene, FTL_0706, postulated to be important for lipopolysaccharide biosynthesis, was confirmed to be a gene involved in O-antigen expression in F. tularensis LVS and Schu S4. These and other studies demonstrate that therapeutic targets, vaccine candidates, or virulence-related genes may be discovered utilizing classical genetic approaches in Francisella.
Francisella tularensis is a gram-negative intracellular pathogen and the etiologic agent of human tularemia. The CDC has classified F. tularensis as a category A select agent due to its highly infectious nature and ease of dissemination. Four subspecies of F. tularensis have been recognized, including (i) the virulent type A F. tularensis subsp. tularensis, (ii) the less virulent type B F. tularensis subsp. holarctica, (iii) F. tularensis subsp. mediasiatica, and (iv) F. tularensis subsp. novicida. The F. tularensis LVS (live vaccine strain) is derived from F. tularensis subsp. holarctica and is used as a model system to identify Francisella virulence factors since it is attenuated in humans but virulent in mice (8, 21). The limited genetic variation (2 to 4%) between the subspecies of Francisella suggests that there is potential overlap among genes related to pathogenesis (7, 54, 59). In fact, F. tularensis LVS and Schu S4 vary in genomic sequence by less than 1% (59). Regardless of the high sequence similarity at the genomic level, genome rearrangement and variation at the functional or regulatory level among the subspecies clearly result in phenotypes that impact virulence and pathogenesis (12, 15, 29, 54, 73, 74).
The life cycle of F. tularensis inside the macrophage begins with surface receptor binding and entry into a vacuole involving pseudopod loops, subsequent escape into the cytoplasm, where replication occurs, and release from the macrophage through apoptosis (for reviews, see references 44, 51, and 65). A role for autophagy after cytoplasmic replication has been described recently (11). Francisella can replicate intracellularly in macrophages, hepatocytes, endothelial cells, epithelial cells, fibroblasts, chicken embryos, and amoebae (for reviews, see references 19, 51, and 71). The fact that Francisella can thrive in such diverse environments suggests that a variety of virulence genes are required to facilitate its survival.
An initial barrier to using gene modification approaches to identify potential virulence factors in Francisella was the lack of genetic tools. Recent advances, however, have overcome this limitation with the development of shuttle plasmids (6, 40, 41, 57), transposons (23, 33, 42), and allelic replacement strategies (27, 36). The genes predicted to foster intracellular growth and virulence include the genes in operons encoding iglABCD and pdpABCD proteins present on the Francisella pathogenicity island (FPI), as well as genes involved in oxidative stress or protein turnover, capsule or lipopolysaccharide (LPS) biosynthesis, type IV pilin assembly, iron uptake, outer membrane channels, purine biosynthesis, and regulation through mglAB or pmrA (44, 46, 50, 51, 53, 56, 65, 66). Although information on the Francisella life cycle, Francisella intracellular growth, and several target virulence factors has been obtained, little is known about the mechanisms involved in pathogenesis in different subspecies and environmental niches. Genomic annotations for Francisella indicate that up to 30% of the open reading frames encode hypothetical proteins (23, 35, 54) which have the potential to function as novel virulence factors.
In this paper, we report the results of Himar1-based transposon (HimarFT) mutagenesis of F. tularensis LVS obtained utilizing a screen for isolates defective for cytopathogenicity in cultured J774A.1 macrophages. Genes with predicted functions that include a variety of transport processes, metabolic functions, capsule and LPS biosynthesis, posttranslational modification, and protein turnover, as well as hypothetical genes with unknown functions, were identified. Insertions in seven hypothetical genes resulted in strains that were attenuated for growth to various extents in macrophages and within mice in vivo. None of these mutants were defective for macrophage entry. One open reading frame for a predicted hypothetical membrane protein was identified as a gene involved in LPS biosynthesis in F. tularensis LVS and Schu S4.
MATERIALS AND METHODS
Bacterial strains, plasmids, and growth conditions.
All bacterial strains and plasmids used in this study are listed in Table 1. Studies involving type A F. tularensis strain Schu S4 were carried out under biosafety level 3 conditions according to federal and institutional select agent regulations. F. tularensis subspecies were routinely grown at 37°C in 2.1% Mueller-Hinton (MH) medium supplemented with glucose, iron, and IsoVitaleX as described previously (41). MH broth contained 1.24 mM CaCl2 and 1.03 mM MgCl2, and MH agar contained 0.5% NaCl, 1.6% agar, 1% tryptone (MHT) or protease peptone (MHP), and 2.5% calf or fetal bovine serum (Invitrogen, Carlsbad, CA). For in vivo infections, inocula were prepared in MHP broth. In some experiments, Chamberlain's chemically defined medium (CDM) was used (10). When required, medium was supplemented with kanamycin (10 μg ml−1) or nourseothricin (5 μg ml−1). Selection for nourseothricin resistance was performed at 30°C. Escherichia coli was grown aerobically at 37°C in Luria-Bertani medium supplemented with kanamycin (50 μg ml−1) or nourseothricin (50 μg ml−1) when required. Kanamycin was purchased from United States Biochemical Corporation (Cleveland, OH), and nourseothricin was purchased from WERNER BioAgents (Jena, Germany).
TABLE 1.
Bacterial strains, plasmids and primers used in this study
| Strain, plasmid, or primer | Description | Reference(s) and/or source |
|---|---|---|
| F. tularensis strains | ||
| LVS | F. tularensis subsp. holartica live vaccine strain; type B | 18; K. L. Elkins |
| U112 | F. tularensis subsp. novicida type B strain (isolated from water in Utah, 1950) | 34 |
| Schu S4 | F. tularensis subsp. tularensis type A strain (isolated from a human ulcer in Ohio, 1941) | 17; BEI Resources |
| TCZ1013 | LVS ΔpurMCD::groE-aph | 53 |
| LVS FTL_0741::HimarFT | LVS HimarFT mutant strain with an insertion in the hypothetical gene FTL_0741 used as a HimarFT-containing control strain displaying a wild-type phenotype in our in vitro and in vivo analyses | This study |
| LVS FTL_0706-1::HimarFT | LVS HimarFT mutant with an insertion in FTL_0706 with the groEL promoter (pgro) from HimarFT in the same orientation as FTL_0706 | This study |
| LVS FTL_0706-2::HimarFT | LVS HimarFT mutant with an insertion in FTL_0706 with the groEL promoter (pgro) from HimarFT in the orientation opposite that of FTL_0706 | This study |
| LVS wbtA1::HimarFT | LVS HimarFT mutant with an insertion in wbtA with the groEL promoter (pgro) from HimarFT in the orientation opposite that of wbtA | This study |
| LVS wbtA2::HimarFT | LVS HimarFT mutant with an insertion in wbtA with the groEL promoter (pgro) from HimarFT in the same orientation as wbtA | This study |
| LVS wbtM::HimarFT | LVS HimarFT mutant with an insertion in wbtM with the groEL promoter (pgro) from HimarFT in the same orientation as wbtM | This study |
| LVS wbtC::HimarFT | LVS HimarFT mutant with an insertion in wbtC with the groEL promoter (pgro) from HimarFT in the same orientation as wbtC | This study |
| Schu S4 FTT1238c::HimarFT (meriodipoid) | Schu S4 FTT1238c::HimarFT resulting from homologous recombination with SpeI circularized genomic DNA from LVS producing a single crossover in the Schu S4 genome | This study |
| Schu S4 FTT1238c::HimarFT | Schu S4 FTT1238c::HimarFT resulting from homologous recombination with SpeI circularized genomic DNA from LVS producing a double crossover in the Schu S4 genome | This study |
| E. coli strains | ||
| DH5α | F− φ80lacZΔM15 endA1 recA1 hsdR17 supE44 thi-1 gyrA96 relA1 Δ(lacZYA-argF)U169 | Invitrogen |
| DH5αλpir | As DH5α, lysogenized with λpir phage; used as host for HimarFT rescue | A. Camilli |
| TOP10 | F−mcrA Δ(mrr-hsdRMS-mcrBC) φ80lacZΔM15 ΔlacX74 deoR recA1 araD139 Δ(ara-leu)7697 galU galK rpsL(Smr) endA1 nupG; used as host for plasmids derived from pCR2.1-TOPO | Invitrogen |
| Plasmidsa | ||
| pCR2.1-TOPO | 3.9-kb plasmid for cloning PCR products; Kmr Apr | Invitrogen |
| pCR-Blunt II-TOPO | 3.5-kb plasmid for cloning PCR products; Kmr | Invitrogen |
| pAG36 | 6.1-kb plasmid used as the source of the nourseothricin resistance marker; Apr Ntr | Euroscarf |
| pFNLTP7 | 6.9-kb pFNLTP1 derivative with XhoI, StuI, PacI, SpeI, HpaI, and NdeI restriction enzyme sites (MCS3) cloned between the KpnI and BamHI sites; Kmr Apr | 41 |
| pFNLTP16 H3 | 10-kb derivative of pFNLTP16 containing HimarFT (Himar1 modified with the F. tularensis LVS groEL promoter in the BclI site upstream of aphA-2); Kmr Apr | 42 |
| pHimar H3 | 4.3-kb plasmid containing HimarFT and a minimal suicide vehicle containing the essential tnp for transposition; Kmr | This study |
| pFTNAT | 6.3-kb derivative of pFNLTP7 lacking kanamycin and ampicillin resistance and containing nourseothricin acetyltransferase flanked with NotI; Ntr | This study |
| pFTNAT-wbtALVS | 8.7-kb plasmid containing wbtA and 472-bp upstream sequence used for complementation; Ntr | This study |
| pFTNAT-wbtMLVS | 8.0-kb plasmid containing wbtM and 481-bp upstream sequence used for complementation; Ntr | This study |
| pFTNAT-FTL_0706 | 8.0-kb plasmid containing FTL_0706 and 175-bp upstream sequence used for complementation; Ntr | This study |
| Primersb | ||
| C1 | 5′-TGAGTAGCTATATTGGTAACTC-3′; forward primer for wbtC for RT-PCR of 366-bp fragment | |
| C2 | 5′-TCAGTTTGAAGCTTACTATCTC-3′; reverse primer for wbtC for RT-PCR of 366-bp fragment | |
| D1 | 5′-TGAATTGTTAGAAGCCTTTGC-3′; forward primer for wbtD for RT-PCR of 410-bp fragment | |
| D2 | 5′-TTAGCTCTAGCTTTATAGCTC-3′; reverse primer for wbtD for RT-PCR of 410-bp fragment | |
| H1 | 5′-CTAAAGTAGTTTTACTAGCAGC-3′; forward primer for 423-bp N-terminal FTL_0706 probe | |
| H2 | 5′-TCACTTATTATAAAATAATCTCCC-3′; reverse primer for 423-bp N-terminal FTL_0706 probe | |
| H3 | 5′-CGGGATCCAAAATGTAGAGTAGGAATGGGT; forward primer for 2-kb FTL_0706 fragment for complementation | |
| H4 | 5′-CCGCTCGAGGGATAATGAGTTGTTAGATGCT-3′; reverse primer for 2-kb FTL_0706 fragment for complementation | |
| IP1 | 5′-ATAAGAATGCGGCCGCTCCGCTTCCTTTAGCAGCC-3′; forward primer for inverse PCR of pFNLTP7 | |
| IP2 | 5′-ATAAGAATGCGGCCGCTTAAGCATTGGTAACTGTCAGA-3′; reverse primer for inverse PCR of pFNLTP7 | |
| K1 | 5′-GCTATTCGGCTATGACTG-3′; forward primer for 634-bp kanamycin marker (aphA2) probe | |
| K2 | 5′-CAGCAATATCACGGGTAG-3′; reverse primer for 634-bp kanamycin marker (aphA2) probe | |
| K3 | 5′-GCTTCCTCGTGCTTTACGG-3′; aphA-2 primer for HimarFT insertion sequencing | |
| R1 | 5′-TGCCACCTAAATTGTAAGCG-3′; R6K primer for HimarFT insertion sequencing | |
| T1 | 5′-CGTCGATATGTAACAATGG-3′; forward primer for 578-bp transposase (tnp) probe | |
| T2 | 5′-CCTTCAAGAGCGATACCAC-3′; reverse primer for 578-bp transposase (tnp) probe | |
| WA1 | 5′-CCGCTCGAGAATCCATGCTATGACTGATGC-3′; forward primer for 2.4-kb wbtA fragment for complementation | |
| WA2 | 5′-CCGCTCGAGGCTTATCTTTACCATATCGC-3′; reverse primer for 2.4-kb wbtA fragment for complementation | |
| WM1 | 5′-CCGCTCGAGATCTGCGCAGTAATGACAGG-3′; forward primer for 1.7-kb wbtM fragment for complementation | |
| WM2 | 5′-CCGCTCGAGAAATCTTGGCTATATGATGGCA-3′; reverse primer for 1.7-kb wbtM fragment for complementation |
Kmr, Apr, and Ntr indicate resistance to kanamycin, ampicillin, and nourseothricin, respectively.
The underlined regions are restriction enzyme sites for BamHI (primer H3), NotI (primers IP1 and IP2), and XhoI (primers H4, WA1, WA2, WM1, and WM2).
DNA manipulation and transformation.
Purification and manipulation of plasmid or genomic DNA, electroporation of F. tularensis, and chemical transformation of E. coli were performed as described previously (41). Custom oligonucleotide primers (Table 1) were synthesized by Operon (Huntsville, AL). DNA maps were constructed using MacPlasmap Pro 3.01 (CGC Scientific, Inc., Ballwin, MO). To determine insertion locations in F. tularensis LVS, genomic DNA from each strain was digested with SpeI and treated with T4 DNA ligase. HimarFT-containing fragments were recovered as plasmid DNA in E. coli DH5αλpir and subsequently used as a template for sequencing (9). Insertion locations for F. tularensis Schu S4 single- and double-crossover strains with mutations in FTT1238c were determined by sequencing directly from genomic DNA. Sequencing reactions and analysis were performed as described previously (41). Southern blotting of SpeI-digested genomic DNA was performed as described previously using either the Random Prime DNA labeling system (Invitrogen) and [α-32P]dCTP or a digoxigenin High Prime DNA labeling and detection starter kit (Roche Diagnostics Corporation, Indianapolis, IN). Probes constructed for identification of the aminoglycoside 3′-phosphotransferase (aphA-2) (64), the transposase (tnp), or the N terminus of FTL_0706 are described in Table 1.
Genome and bioinformatic analyses.
The HimarFT insertion site sequences were compared with the F. tularensis LVS genome (accession no. NC_007880) at http://bbrp.llnl.gov/bbrp/bin/f.tularensis_blast and the F. tularensis Schu S4 genome (NC_006570) available at http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?db=genome&cmd=Retrieve&dopt=Overview&list_uids=563. Additional F. tularensis subsp. genomic comparisons were performed using www.francisella.org.
Further analysis and annotation of candidate proteins were performed using the high-resolution tools PhyloBlocks (http://compbio.mcs.anl.gov/ulrich/phyloblock/) and Dragonfly (http://compbio.mcs.anl.gov/dragonfly/) available via the PUMA2 system (43). Candidate sequences were analyzed using BLAST, InterPro, SignalP, and TMHMM to identify global sequence alignments, local conserved domains, potential signal sequences, and potential transmembrane domains, respectively (1, 5, 47, 48). The results of these analyses were used to assign potential functions to the genes. Further identification of the conserved regions specific for predicted functions was done using PhyloBlocks. In brief, a set of orthologous sequences identified by BLAST were aligned using ClustalW and analyzed by Block Maker for prediction of the conserved motifs (31). Consensus sequences and HMM profiles were also generated for these sets of orthologs to be used for future analysis and classification.
Western blot analysis.
Whole-cell lysates (1× 109 cells in 100 μl) were boiled for 5 min in loading buffer, and 5 μl of each sample was loaded and separated on 10% polyacrylamide-sodium dodecyl sulfate gels. Western transfer, blocking, and antibody incubation were performed as described previously (41). Treatment with the mouse monoclonal anti-F. tularensis LPS primary antibody (1:8,000 dilution; Advanced ImmunoChemical Inc.) was followed by treatment with a horseradish peroxidase-conjugated goat anti-mouse immunoglobulin G secondary antibody (1:10,000 dilution; Roche).
F. tularensis LVS HimarFT library construction.
The pHimar H3 construct is a modified HimarFT suicide delivery vehicle constructed from temperature-sensitive pFNLTP16 H3 (42). Transposition using pHimar H3 was performed by electroporation of 100 ng of DNA into electrocompetent F. tularensis LVS. After outgrowth at 37°C on a shaker for 4 h, dilutions were plated onto MHT agar containing kanamycin and incubated at 37°C to select for HimarFT. In addition, dilutions were plated onto MHT agar without kanamycin to determine the total number of potential recipients. Colonies grown for 3 or 4 days at 37°C were picked, struck onto MHT agar containing kanamycin, and grown at 37°C to recover individual clones containing HimarFT insertions in the genome. For HimarFT library construction and storage, Kmr colonies were placed into 0.5 ml MH broth containing kanamycin in tissue culture plates with 96 deep wells. The last row in each plate was not inoculated as a control. Sterile lids and Parafilm were used to cover the plates for incubation and freezer storage. After 2 days of growth at 37°C and 200 rpm, 0.5 ml sterile 30% glycerol was added and mixed into each well to obtain a final glycerol concentration of 15%, and the plates were stored at −80°C. Recovery of the library was performed by using a 96-pin replicator and square agar plates (Nalge Nunc International Corp., Rochester, NY) containing MHT agar with kanamycin.
HimarFT library screen utilizing a macrophage infection model.
Stage 1 screening with J774A.1 macrophages (ATCC TIB-67) was performed using a 96-well format with a multiplicity of infection (MOI) of ∼50 to 200. Macrophages were seeded at a concentration of 6 × 104 cells per well and incubated overnight at 37°C in 5% CO2 before infection. Strains containing transposon insertions were grown on MHT agar with kanamycin for 3 days at 37°C and for 0 to 2 days at room temperature and transferred into a 96-well plate containing 100 μl of Dulbecco's modified Eagle's medium (DMEM) (Sigma-Aldrich, St. Louis, MO) supplemented with 10% fetal bovine serum and 4 mM l-glutamate (Invitrogen). The DMEM was aspirated from the macrophages, and 50-μl portions of test bacterial suspensions were placed onto macrophages. These steps were performed one row at a time to minimize drying of the monolayers. A 2-h incubation at 37°C in the presence of 5% CO2 was performed to allow bacterial attachment and entry. Each row of eight wells was then aspirated to remove the extracellular organisms, washed with 200 μl of Hanks' balanced salt solution, and aspirated again, and the contents were replaced with 200 μl of DMEM containing 5 μg ml−1 of gentamicin (Invitrogen) to kill extracellular bacteria. The plates were then incubated at 37°C in the presence of 5% CO2 for 2 days. Candidates, which retained the macrophage monolayer similar to uninoculated controls (groups A and B), were subjected to a second-stage 24-well analysis with 3.7 × 105 macrophages per well and defined inocula normalized using optical density to obtain an MOI of 100.
Crystal violet staining of J774A.1 macrophages.
The integrity of the macrophage monolayer was assessed by crystal violet staining. DMEM was aspirated from the macrophages, and the monolayers were washed with phosphate-buffered saline and then incubated with a 1% crystal violet solution for 5 min at room temperature. The crystal violet was removed, and the macrophage monolayers were washed with distilled water and allowed to dry before visual assessment of the stain intensity. Crystal violet was purchased from Hardy Diagnostics (Santa Maria, CA).
Quantitation of Francisella cytotoxicity with J774A.1 macrophages by LDH release.
The release of cytosolic lactate dehydrogenase (LDH) into the supernatant of infected cells was used as an indicator for lysis (CytoTox96; Promega Corporation, Madison, WI). Bacterial cell suspensions were prepared and normalized to an MOI of 100 based on the optical density at 550 nm (OD550) and subsequent plating to determine the actual number of CFU ml−1. The conditions used for J774A.1 macrophage infection were the same as those used for the 24-well stage 2 cytopathic screening. At 48 h postinfection, supernatants were harvested and the maximum lysis was determined (by addition of 100 μl of lysis buffer to an uninfected monolayer with 900 μl DMEM and incubation at 37°C for 45 min). Uninfected medium was used as a background control, and suitable dilutions of the other samples were included. The release of LDH was determined by measuring the OD490 using a plate reader and softmax pro software (Molecular Devices Corporation, Sunnyvale, CA).
Growth analysis of F. tularensis LVS mutants in bacteriological medium.
Growth in MH or CDM broth was assessed using a 96-well format with a plate reader and softmax pro software. One test colony was emulsified in 100 μl of CDM broth, 10 μl was placed into 190 μl of MH or CDM broth, and the microtiter plate was incubated at 37°C with shaking at 100 to 200 rpm. Turbidity changes in the cultures were monitored at OD550 for 24 h (data not shown). Some strains were grown on MH or CDM agar to confirm the observed phenotypes in broth.
In vivo analysis of HimarFT insertion mutants.
Six- to 8-week-old female BALB/c mice (Harlan Sprague Dawley, Inc., Indianapolis, IN) were infected by intraperitoneal (i.p.) injection of 0.1 ml (final volume) of phosphate-buffered saline containing the F. tularensis LVS wild type or mutant derivatives. The inocula were prepared from MHP broth cultures in the exponential phase of growth, and the concentrations were adjusted so that they represented approximately 1,000 50% lethal doses (21, 29). The actual number of bacteria delivered was determined by performing plate counts of the inoculum on MHP agar. Infected animals were closely monitored for 21 days, and the number of moribund animals during this time was determined. Animal infection experiments were approved by the Institutional Animal Care and Use Committee and the Institutional Biosafety Committee of the Medical College of Wisconsin.
Complementation of selected HimarFT mutant strains using pFTNAT.
The complementation plasmid pFTNAT was constructed by inverse PCR using primers IP1 and IP2 (Table 1) and pFNLTP7 as the template DNA (41). Both the kanamycin and ampicillin resistance genes were removed, and the nourseothricin resistance marker was inserted at a NotI site (26). The pFTNAT plasmid retains the ability to replicate in both E. coli and F. tularensis. The complementation plasmids pFTNAT-FTL_0706, pFTNAT-wbtALVS, and pFTNAT-wbtMLVS were constructed by PCR amplification of genomic DNA with primer pairs H3/H4, WA1/WA2, and WM1/WM2 (Table 1), respectively, resulting in gene fragments with 175-, 472-, and 481-bp upstream sequences (putative promoter regions). All fragments were ligated into pCR-Blunt II-TOPO and subsequently subcloned into pFTNAT using EcoRI, XhoI, and XhoI, respectively.
RNA isolation and RT-PCR analysis of F. tularensis HimarFT strains with insertions in wbtA.
RNA was isolated using an RNeasy Protect mini kit (QIAGEN). Isolated RNA was treated with DNase I (Fermentas Inc., Hanover, MD) to remove contaminating genomic DNA for 1 h at 37°C and subsequently purified with the RNeasy Protect mini kit. RNA quality was verified by agarose gel analysis. Reverse transcription (RT)-PCR was performed using the SuperScript III One-Step RT-PCR system with Platinum Taq DNA polymerase (Invitrogen). Control PCRs were performed with Platinum Taq High Fidelity DNA polymerase (Invitrogen). Primers C1, C2, D1, and D2 were used for mRNA detection of either a 366-bp fragment of wbtC or a 410-bp fragment of wbtD downstream of wbtA (Table 1). All reaction mixtures included 50 ng of template DNA or RNA and were incubated at 50°C for 30 min for RT, followed by PCR for 25 cycles.
Statistical analysis.
Statistical significance was determined using single-factor analysis of variance.
RESULTS
Transposon mutagenesis of F. tularensis LVS using HimarFT.
In previous studies, a temperature-sensitive plasmid and a modified Himar1 transposon (HimarFT) were developed to perform random mutagenesis in F. tularensis (41, 42). While this system resulted in random, single, stable insertions without orientation bias, a drawback was observed when large-scale experiments were performed to saturate the genome. We noted that plasmid sequences were retained at a variable frequency. The failure to lose plasmid sequences may have been due to the length of time required for the plates to achieve the restrictive temperature, the number of generations required to dilute nonreplicating DNA, or the reversion of the point mutation in the replication initiation protein encoded by repA. Sequencing of repA from five isolates retaining the plasmid sequence confirmed the maintenance of the temperature-sensitive mutation. Plasmid rearrangements were not observed (data not shown). We noted that plasmid sequences were lost if strains were passaged at the restrictive temperature two additional times (49).
To avoid the labor-intensive steps of passaging individual isolates, the delivery vehicle was modified by removing the NotI fragment encoding pFNLTP16 to construct pHimar H3 (Fig. 1A). Deletion of the replicon for Francisella converted pFNLTP16 H3 to a suicide delivery system for HimarFT. The efficiency of transposition with pHimar H3 was similar to that obtained for pFNLTP16 H3, with ∼2 × 10−5 insertion per cell. The stable retention of HimarFT and the loss of plasmid sequences were confirmed by examining 100 mutagenized strains by colony blot analysis with 32P-labeled probes hybridizing to the aminoglycoside 3′-phosphotransferase (aphA-2) responsible for kanamycin resistance and the transposase (tnp), respectively. Southern blot analysis with genomic DNA isolated from 10 mutants after transposition demonstrated that there were random, single insertions and loss of the plasmid vehicle (Fig. 1B and data not shown). For construction of the insertion library, F. tularensis LVS was mutagenized with pHimar H3 and approximately 7,000 kanamycin-resistant colonies were arrayed into microtiter plates with 96 deep wells and preserved at −80°C.
FIG. 1.
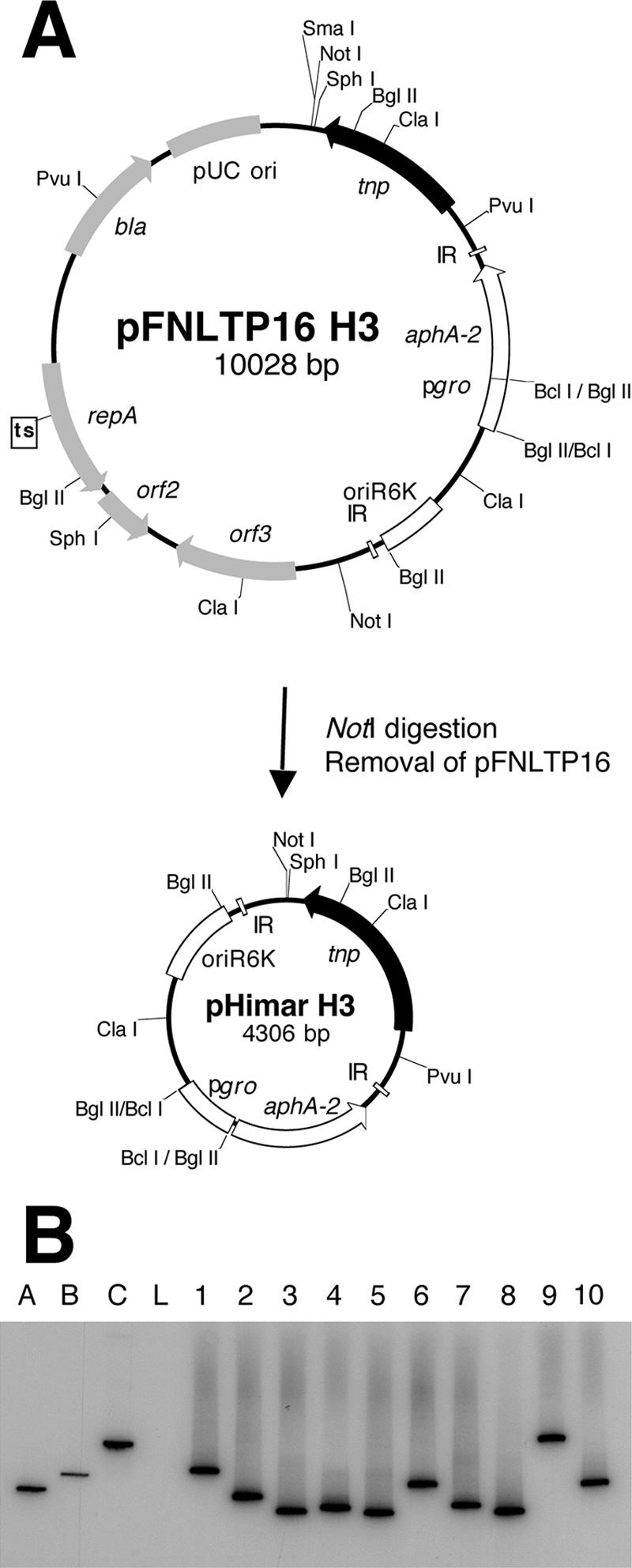
(A) Construction of a suicide delivery vehicle for HimarFT transposition. The original HimarFT transposon with the Francisella groEL promoter (pgro) facilitating expression of the aminoglycoside 3′-phosphotransferase (aphA-2) and transposase (tnp) genes on the temperature-sensitive pFNLTP16 vehicle is shown. pHimar H3 was constructed by removal of the NotI fragment containing pFNLTP16. (B) Verification of pHimar H3 insertion in F. tularensis LVS by Southern blot analysis of SpeI-digested genomic DNA. Hybridization with a 32P-labeled probe specific to aphA-2 was observed for 10 representative strains after pHimar H3 mutagenesis. Wild-type F. tularensis LVS genomic DNA (lane L) was included as a negative control. Three plasmids were included as markers (42): pMiniHimar (4 kb) (lane A), pFNLTP1 (6.9 kb) (lane B), and pFNLTP16 H3 (10 kb) (lane C). A probe specific to the transposase (tnp) gene on the plasmid did not hybridize with genomic DNA but produced a signal for markers A and C (data not shown).
Isolation of mutants with insertions in Francisella genes required for virulence in J774.A1 macrophages.
Francisella enters, replicates, and causes apoptosis-induced cytopathogenicity within macrophages (44, 51, 65). To screen for genes required for F. tularensis LVS virulence in J774.A1 macrophages, the integrity of macrophage monolayers infected with individual Kmr clones was assessed by staining with crystal violet. Preliminary experiments indicated that crystal violet staining at 2 days after infection at an MOI of 100 was optimal for the detection of cytopathogenicity (Fig. 2A). The initial screening (stage 1) was performed with an estimated MOI of 50 to 200 using a 96-well format. The retention of the monolayer and dark staining suggested that the clones were unable to enter, replicate, or cause apoptosis during the 2-day incubation. Noncytopathic candidates were retested in duplicate (stage 2) in 24-well plates at an MOI of 100 (Fig. 2B). Mutants were categorized into four groups based on staining intensity: no loss of the monolayer, as in the uninfected control well (group A), a small but detectable loss of monolayer (group B), detectable loss of cells (group C), and severe monolayer destruction, similar to that obtained with the parental strain, F. tularensis LVS (group D). To cull putative auxotrophs or other mutants defective in general metabolism, 140 candidates appearing to possess the most attenuated phenotypes (groups A and B) in macrophages were characterized for growth in complex and minimal bacteriological media (stage 3 screening) (data not shown). Duplicate cultures for each strain were arrayed in 96-well plates in MH or CDM broth, and growth was monitored over time by assessing the increase in optical density. Colony morphology was assessed using cultures transferred from microtiter broth cultures to MHT or CDM agar plates. Twenty-four strains failed to grow in or on CDM and are likely auxotrophic strains. Slower growth or inconsistent growth patterns were observed for another 24 candidates. In summary, the combined screens identified 89 strains with HimarFT insertions in genes potentially important for virulence in macrophages.
FIG. 2.
(A) Crystal violet staining of J774A.1 macrophages infected with F. tularensis LVS at various MOIs 2 days postinfection. The retention of the J774A.1 macrophage monolayer after infection is compared to the uninfected control (none). (B) F. tularensis LVS HimarFT mutant stage 2 macrophage screening using an MOI of 100. The uninfected control (well N) retains the monolayer and stains dark, while wild-type F. tularensis LVS (well L) is cytopathic, causing loss of the monolayer. Based on crystal violet retention, strains with HimarFT insertions were classified into four groups (A to D) represented by wells A to D, respectively: group A, similar to uninfected controls; group B, intermediate dye retention; group C, light dye retention; and group D, no dye retention, similar to infection by the parental strain. Phenotypes corresponding to groups A and B were characterized further.
Identification of the site of HimarFT insertion.
To recover DNA flanking the HimarFT insertion site, genomic DNA from each of the 89 candidates was digested with SpeI, ligated, and transformed into E. coli DH5αλpir. Of the 89 genomes, 70 (79%) were recovered as independently replicating plasmids containing HimarFT and adjacent genomic DNA, an efficiency similar to that reported elsewhere for transposons in Francisella (69). Recovered plasmids were used as templates for nucleic acid sequencing reactions with primers specific for HimarFT sequences corresponding to aphA-2 and the R6K origin (Table 1 and Fig. 1A). From this analysis, 26 unique genes were identified. Known or predicted functions for these genes included transport processes, metabolism of nucleotides, carbohydrates, amino acids, and coenzymes, capsule and LPS biosynthesis, and posttranslational modification/protein turnover (Table 2). The recovery of several HimarFT insertions in genes previously found to have a role in macrophage growth, including the caseinolytic protease gene clpB, the disulfide bond-related gene dsbB, the aspartate aminotransferase gene aspC2, and the putative capsular genes capBC and wbtA involved in O-antigen biosynthesis, validated the screening process used (28, 55, 58, 63, 67, 69, 70, 76). Several hypothetical proteins were also identified. Insertions appeared to be spread evenly throughout the genome (Fig. 3).
TABLE 2.
F. tularensis LVS HimarFT mutant strains attenuated for growth in macrophagesa
| Geneb | Gene locus
|
No. of insertions/ genec | Protein product | |
|---|---|---|---|---|
| LVS | Schu S4 | |||
| FTL_1806 | FTT0053 | 2 | Major facilitator superfamily transporter | |
| dsbB | FTL_1670 | FTT0107c | 1 | Disulfide bond formation protein, DsbB |
| glgB | FTL_0483 | FTT0413c | 5 | Glycogen branching enzyme, GlgB; polysaccharide metabolism |
| xasA | FTL_1583 | FTT0480c | 4 | Glutamate-aminobutyric acid antiporter, XasA; amino acid transport |
| FTL_0878 | FTT0610 | 1 | DNA/RNA endonuclease family | |
| FTL_0886 | FTT0618c | 1 | Conserved hypothetical protein YleA; possible tRNA-i (6)A37 methylthiotransferase | |
| capB | FTL_1416 | FTT0805 | 3 | Capsule biosynthesis protein CapB |
| capC | FTL_1415 | FTT0806 | 1 | Capsule biosynthesis protein CapC |
| FTL_1414 | FTT0807 | 2 | Hypothetical protein; possible capsule-related protein | |
| FTL_0439 | FTT0919 | 2 | Hypothetical outer membrane protein | |
| FTL_1262 | FTT0945 | 1 | Chorismate family binding protein; aromatic amino acid and folate biosynthesis | |
| FTL_1096 | FTT1103 | 1 | Hypothetical lipoprotein; ABC transporter and potential disulfide bond formation protein, DsbA | |
| metN | FTL_0838 | FTT1124 | 5 | d-Methionine transport protein (ABC transporter), MetN |
| metIQ | FTL_0837 | FTT1125 | 9 | d-Methionine transport protein (ABC transporter), MetIQ |
| aspC2 | FTL_0789 | FTT1165c | 3 | Aspartate aminotransferase; amino acid biosynthesis |
| ggt | FTL_0766 | FTT1181c | 7 | Gamma-glutamyltranspeptidase; amino acid, arachidonic acid, and glutathione metabolism |
| FTL_0706 | FTT1238c | 3 | Hypothetical membrane protein; LPS biosynthesis | |
| wbtM | FTL_0606 | FTT1450 | 1 | dTDP-glucose 4,6-dehydratase, WbtM, O-antigen polysaccharide biosynthesis |
| wbtC | FTL_0594 | FTT1462c | 1 | UDP-glucose-4-epimerase, WbtC, O-antigen polysaccharide biosynthesis |
| wbtA | FTL_0592 | FTT1464c | 5 | dTDP-glucose 4,6-dehydratase, WbtA, O-antigen polysaccharide biosynthesis |
| FTL_0304 | FTT1490 | 4 | Na+/H+ antiporter; regulation of pH | |
| FTL_0544 | FTT1564 | 1 | Hypothetical protein; polyphosphate kinase | |
| gplX | FTL_1701 | FTT1631c | 1 | Fructose-1,6-bisphosphatase, GplX, glucose metabolism |
| FTL_0050 | FTT1645 | 1 | Hypothetical protein; major facilitator superfamily | |
| FTL_0058 | FTT1688 | 3 | Aromatic amino acid transporter of HAAAP family | |
| clpB | FTL_0094 | FTT1769c | 2 | Caseinolytic protease, ClpB; response to unfolded protein |
Gene designations, loci, and protein designations correspond primarily to those in the Schu S4 genome annotation (http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?db=genome&cmd=Retrieve&dopt=Overview&list_uids=563). This gene list was compared with the F. tularensis LVS genome at www.francisella.org. Additional bioinformatics was performed by the Computational Biology Group at Argonne National Laboratory (indicated after the semicolon for some candidates). Hypothetical candidates are underlined.
The gene corresponds to the annotated gene designation if available.
The number of insertions per gene corresponds to the number of sequenced HimarFT insertions per open reading frame.
FIG. 3.

F. tularensis LVS genomic map of HimarFT insertion mutants recovered after stage 2 and 3 screening (designations above the F. tularensis LVS genome). Hypothetical HimarFT mutants are underlined. Several genes previously reported to be involved in growth inside macrophages were recovered, including clpB, FTL_0439, wbtA, capBC, aspC2, and dsbB (designated by asterisks; above the F. tularensis LVS genome). Regions and genes previously shown to be involved in macrophage growth but not recovered as strongly attenuated after the screening of our HimarFT library are indicated by the gray boxes corresponding to the Francisella pathogenicity islands (FPI#1 and FPI #2) and below the F. tularensis LVS genome, respectively.
In vivo analysis of mutants containing transposon insertions in genes encoding hypothetical proteins.
To focus on new gene discovery, strains with insertions in hypothetical genes were selected to test for attenuation in vivo. Groups of five BALB/c mice were given an intraperitoneal (i.p.) injection of mutant derivatives at doses ranging from 1,000 to 5,000 CFU or 3 logs above the reported 50% lethal dose (21, 29). Previous work indicated that BALB/c mice are moribund by day 4 to 8 when they are infected i.p. with 50 CFU or less (39, 53). Mice infected with strains bearing insertions in the hypothetical genes tested survived throughout the 21 days of monitoring (Table 3). We also challenged mice with F. tularensis FTL_0741::HimarFT, with a transposon insertion in a hypothetical locus that exhibited a parental phenotype in vitro. In vivo, LVS FTL_0741::HimarFT exhibited a parental phenotype, while LVS ΔpurMCD failed to induce active infection (53). These results suggest that a HimarFT insertion generates a specific phenotype related to its location in the chromosome.
TABLE 3.
BALB/c mice challenged with F. tularensis LVS hypothetical HimarFT mutants
| Strain | Mutant challenge (CFU)a | No. of mice surviving mutant challenge/total no. of miceb |
|---|---|---|
| LVS | 5,200 | 0/5 |
| LVS ΔpurMCD | 2,200 | 5/5 |
| LVS FTL_0886 | 1,010 | 5/5 |
| LVS FTL_1414 | 2,180 | 5/5 |
| LVS FTL_0439 | 5,100 | 5/5 |
| LVS FTL_1096 | 2,850 | 5/5 |
| LVS FTL_0741 | 1,990 | 0/5 |
| LVS FTL_0706 | 1,560 | 5/5 |
| LVS FTL_0544 | 2,640 | 5/5 |
| LVS FTL_0050 | 3,900 | 5/5 |
Mice were inoculated i.p. (0.1 ml) with 1,000 to 5,000 CFU of F. tularensis LVS HimarFT hypothetical (FTL) mutants. The mutant challenge was determined by counting CFU on MHP agar.
Survival was determined 21 days after infection. Mice challenged with F. tularensis LVS and LVS FTL_0741::HimarFT died 3 to 8 days after infection.
Characterization of transposon mutants for entry, initial replication, and cytotoxicity.
Attenuation in vivo for all mutants tested suggested that the in vitro cellular assay results correlated with pathogenicity. To determine if quantitative defects were associated with different stages of infection, mutants with insertions in hypothetical genes were tested with J774A.1 macrophages to determine their capacities to enter, replicate, and cause the release of LDH, an indicator of cell death (Fig. 4). An intracellular replication-deficient mutant used as a control, F. tularensis LVS ΔpurMCD, exhibited reduced replication and cytotoxicity, consistent with its attenuation in mice (53). Similarly, inactivation of FTL_0741 resulted in no change in replication and cytotoxicity, consistent with the wild-type phenotype in mice. There was no significant difference between LVS and the strains bearing HimarFT insertions in terms of the ability to enter J774A.1 macrophages (Fig. 4A). Statistically significant differences (P < 0.05) from the parental strain were observed when replication at 24 h and cytotoxicity at 48 h for mutants with insertions in FTL_0886, FTL_1414, FTL_0439, FTL_0706, and FTL_0050 were examined (Fig. 4B and C), consistent with the attenuation of these mutants in mice. Thus, defects in macrophage replication generally correlate with the inability to induce cytotoxicity. Interestingly, LVS FTL_0544::HimarFT displayed no significant changes in either cytotoxicity or replication, despite being attenuated in mice, compared to the wild type (Fig. 4B and C).
FIG. 4.
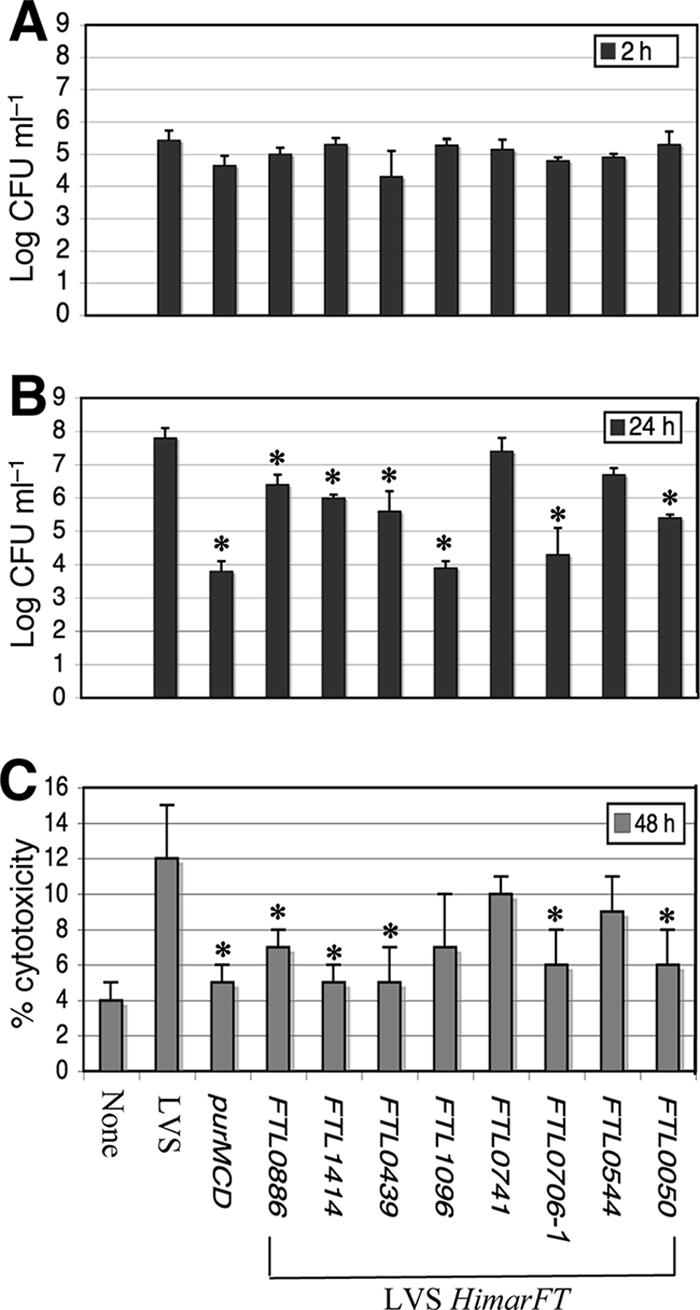
(A and B) Entry and replication in J774A.1 macrophages of the F. tularensis LVS wild type and ΔpurMCD, FTL_0741::HimarFT, and attenuated mutants with insertions in loci encoding hypothetical proteins. The experiments were performed using a 24-well format with an MOI of 100 in triplicate. The results are expressed as mean log CFU ml−1; the error bars indicate standard deviations. Mutants found to be significantly impaired in entry at 2 h or in replication at 24 h are indicated by asterisks (P < 0.05). (C) Quantitation of cytotoxicity by measuring LDH release into the supernatant compared to an uninfected monolayer (maximum lysis) at 48 h. LDH release was monitored alongside assessment for entry and replication defects as described above for panels A and B. Experiments were performed in triplicate, The bars indicate mean percentages of cytotoxicity, and the error bars indicate standard deviations. Mutants found to have a significant reduction in the percentage of cytotoxicity at 48 h are indicated by asterisks (P < 0.05).
LPS phenotype of selected HimarFT mutant strains.
LPS biosynthesis has been previously shown to be important for intracellular replication (13, 58, 63, 70). Loss of a functional copy of wbtA results in loss of the O-antigen ladder (58, 63). Five F. tularensis LVS wbtA::HimarFT mutants from this study exhibited loss of O antigen, as predicted (Fig. 5A). Two additional mutations in the wbt locus, with HimarFT insertion in either wbtM or wbtC, resulted in complete O-antigen loss and loss of low-molecular-weight O antigen, respectively (Fig. 5A). These phenotypes have not been previously reported and likely correlate to the location in the pathway of LPS biosynthesis. Other mutations in the wbt locus, such as wbtDEF mutations, also result in a lack of O antigen (70). Strains with insertions in predicted capsule-related genes retained the O-antigen ladder, suggesting that mutation of these genes does not affect LPS (Fig. 5B). HimarFT mutations within hypothetical genes were analyzed by Western blotting to assess whether any of these genes represent unrecognized LPS biosynthetic genes. All of the insertions in hypothetical genes resulted in a parental pattern for the O-antigen side chain except for LVS FTL_0706::HimarFT (Fig. 5C). We concluded that the hypothetical protein encoded by FTL_0706 likely functions in the LPS biosynthetic pathway.
FIG. 5.

Western blot analysis of F. tularensis LVS or F. novicida U112 cell lysates with a mouse monoclonal anti-F. tularensis LPS antibody. The antibody recognizes LPS from F. tularensis LVS and Schu S4 but not LPS from F. novicida U112. Each HimarFT mutant strain is indicated by the gene in which HimarFT insertion was mapped, as shown in Table 2. Multiple strains with mutations in the same gene are indicated by the gene designation and a candidate number. The previously studied F. tularensis LVS ΔpurMCD mutant is indicated by MCD (53).
Complementation and HimarFT polarity.
HimarFT was engineered to contain a strong constitutive groEL promoter to transcribe the aphA-2 kanamycin resistance marker. Transcriptional stop sequences were not added, raising the possibility that the orientation of insertion may affect the transcription of adjacent genes. HimarFT mutants with insertions mapped to the beginning (wbtA) and the end (wbtM) of the wbt region were used to determine if the orientation of HimarFT influenced either the complementation pattern or transcription of downstream genes (Fig. 6A). Gene expression analyses were performed with F. tularensis LVS wbtA1::HimarFT and wbtA2::HimarFT, in which HimarFT had been mapped in the two orientations (Fig. 6A). To detect the presence of mRNA downstream of wbtA, RT-PCR was performed with primers designed to detect message containing wbtC or wbtD (Fig. 6A). Fragments that were the correct size were obtained in PCR and RT-PCR controls with F. tularensis LVS genomic DNA, verifying that there was primer-mediated amplification of the correct region (Fig. 6B). Products were not detectable when template DNA was not included or in PCRs utilizing DNase-treated RNA isolated from wild-type or mutant strains, indicating that the preparations of RNA were not contaminated with genomic DNA (Fig. 6B). RT-PCRs with RNA isolated from LVS, LVS wbtA1::HimarFT, and wbtA2::HimarFT each demonstrated that there was a message containing wbtD on a fragment whose size was similar to that of the genomic DNA control (Fig. 6B). Similarly, a product was detected for all three strains when wbtC was the target sequence (data not shown). The amounts of wbtC and wbtD cDNAs appeared to be reduced in strains containing an upstream HimarFT insertion. These data suggest that HimarFT insertion is partially polar.
FIG. 6.

Analysis of F. tularensis LVS HimarFT strains with mutations in the wbt locus by RT-PCR and complementation. (A) Map of the wbt loci (FTL_0592 to FTL_0595) in the F. tularensis LVS genome. The HimarFT insertion sites are indicated for F. tularensis LVS wbtA1::HimarFT, wbtA2, and wbtC mutant strains (indicated by the gene designation and candidate number). FTL_0606 (wbtM) is just downstream of the main wbt operon (FTL_0592 to FTL_0605). The genomic region used for complementation of the wbtA mutants is between the XhoI sites. Bars 1 and 2 correspond to the regions selected for detection of mRNA downstream of wbtA by RT-PCR. HimarFT insertion site maps for F. tularensis LVS wbtA1::HimarFT and wbtA2::HimarFT are shown, with LVS wbtA2::HimarFT containing the groEL promoter (pgro) in the same orientation as the gene. (B) Detection of mRNA containing wbtD downstream of wbtA in LVS wbtA1::HimarFT and wbtA2::HimarFT by RT-PCR. The reaction and template conditions used included RT-PCR with no template (lane 1), LVS genomic DNA (lane 2), and RNA isolated from either LVS wild-type (lane 3), wbtA1::HimarFT (lane 5), or wbtA2::HimarFT (lane 7). Control RT-PCRs without the reverse transcriptase were performed using RNA from LVS wild-type (lane 4), wbtA1::HimarFT (lane 6), and wbtA2::HimarFT (lane 8). (C) Plasmid pFTNAT containing the nourseothricin resistance gene used for complementation of HimarFT mutant strains. (D) Western blot analysis of cell lysates using a mouse monoclonal anti-F. tularensis LPS antibody. The strains analyzed include LVS with pFTNAT (lane 1); LVS wbtA1::HimarFT alone (lane 2), with pFTNAT (lane 3), or with pFTNAT-wbtALVS (lane 4); LVS wbtA2::HimarFT alone (lane 5), with pFTNAT (lane 6), or with pFTNAT-wbtALVS (lane 7); and LVS wbtM1::HimarFT alone (lane 8), with pFTNAT (lane 9), or with pFTNAT-wbtMLVS (lane 10).
To complement HimarFT mutants, we constructed a new shuttle vector containing a nourseothricin resistance marker, pFTNAT (Fig. 6C). Nourseothricin is an antibiotic that inhibits a broad spectrum of organisms and is postulated to inhibit protein synthesis by induction of miscoding events (26). It is an attractive marker for Francisella research as it is not used in human or veterinary medicine and has not been shown to display cross-resistance (42). Genomic fragments containing the genes and their predicted promoter regions for complementation were cloned into pFTNAT and transformed into the appropriate HimarFT mutant strains.
Complementation of mutants with insertions in either the wbt region or FTL_0706 was assessed both for O-antigen content and for cytopathogenicity in macrophages. O-antigen expression and cytopathogenicity were restored to F. tularensis LVS wbtM::HimarFT with pFTNAT-wbtMLVS in trans (Fig. 6D and data not shown). However, complementation of LVS wbtA1::HimarFT was not detected using a similar cloning strategy (pFTNAT-wbtALVS) (Fig. 6D). Partial restoration of O-antigen expression occurred when LVS wbtA2::HimarFT was transformed with pFTNAT-wbtALVS (Fig. 6D), but this strain still displayed reduced cytopathogenicity compared to LVS (data not shown). The presence of plasmid pFTNAT did not alter the strain phenotypes. The lack of complementation or partial complementation is consistent with results from RT-PCR analyses and suggests that the orientation of HimarFT influences the expression of adjacent genes. These data also support an operon organization for the wbt region.
F. tularensis LVS FTL_0706-1::HimarFT and FTL_0706-2::HimarFT mutants were complemented in trans using pFTNAT-FTL_0706 (Fig. 7A). FTL_0706 does not appear to be part of an operon based on the current annotation of the LVS genome. FTL_0706 provided in trans restored the O-antigen ladder in both FTL_0706::HimarFT mutant strains (Fig. 7B). Cytopathogenicity was restored in LVS FTL_0706-1::HimarFT and FTL_0706-2::HimarFT with pFTNAT-FTL_0706, resulting in loss of the macrophage monolayer, similar to the parental phenotype (Fig. 7C).
FIG. 7.

Complementation of F. tularensis LVS FTL_0706::HimarFT mutant strains in trans using pFTNAT-FTL_0706. (A) Two mutants containing HimarFT insertions within FTL_0706 in opposite orientations were assessed for complementation (locations are indicated by the gene designation and candidate number). LVS FTL_0706-1:: HimarFT contains the groEL promoter (pgro) in the same orientation as FTL_0706. F and R indicate the DNA fragment amplified from the genome, and the EcoRI sites indicate the fragment used for complementation. (B) Western blot analysis of cell lysates from LVS and LVS FTL_0706::HimarFT strains using a mouse monoclonal anti-F. tularensis LPS antibody. The strains analyzed include LVS without (lane 1) or with (lane 2) pFTNAT; LVS FTL_0706-1::HimarFT alone (lane 3), with pFTNAT (lane 4), and with pFTNAT-FTL_0706 (lane 5); and FTL_0706-2::HimarFT alone (lane 6), with pFTNAT (lane 7), and with pFTNAT-FTL_0706 (lane 8). (C) Cytopathic analysis with J774A.1 macrophages using crystal violet staining 2 days after infection at an MOI of 100. The retention of the monolayer after infection is compared to the uninfected control (well 1). Wells 2 and 3 contained wild-type LVS without and with pFTNAT. Wells 4 to 6 contained the F. tularensis LVS FTL_0706::HimarFT mutant candidates alone (wells 4), with pFTNAT (wells 5), and with pFTNAT-FTL_0706 (wells 6).
Mutagenesis of F. tularensis Schu S4.
FTL_0706 and FTT1238c are highly conserved hypothetical genes having variation in only two nucleotides in F. tularensis LVS and Schu S4, respectively (Fig. 8A). Therefore, we reasoned that introduction of the rescued plasmid carrying FTL_0706::HimarFT could be used to disrupt FTT1238c in Schu S4 (2, 3, 27, 36, 45). Electroporation of the nonreplicative plasmid into Schu S4 resulted in recovery of several Kmr clones, one of which was defective for the synthesis of O antigen like LVS FTL_0706-1::HimarFT. Other clones exhibited a parental O-antigen phenotype and were confirmed to be merodiploids by Southern blotting (Fig. 8B and C). Moreover, nucleotide sequence analysis of the Schu S4 mutants demonstrated that the first of two variable nucleotide differences in the locus was characteristic of LVS.
FIG. 8.

Analysis of HimarFT insertions with mutations in FTL_0706 and FTT1238c for F. tularensis LVS and Schu S4, respectively. (A) LVS and Schu S4 wild-type loci are indicated by gray and striped arrows, respectively. HimarFT insertion into FTL_0706 in LVS resulted in the loss of an SpeI site present in the parental chromosomal locus. The sequence for this locus in LVS and Schu S4 is highly conserved, with variation in only two nucleotides, as shown. Introduction of a circularized SpeI fragment containing the HimarFT insertion from LVS FTL_0706-1::HimarFT into Schu S4 promoted homologous recombination resulting in either a single- or double-crossover event as indicated. (B) Western blot analysis of the LVS and Schu S4 strains with a mouse monoclonal anti-F. tularensis LPS antibody demonstrated the presence or absence of the LPS ladder. The strains analyzed were LVS (lane 1), LVS FTL_0706-1::HimarFT (lane 2), Schu S4 (lane 3), Schu S4 FTT1238c::HimarFT merodiploid (lane 4), and Schu S4 FTT1238c::HimarFT (lane 5). (C) Southern blot analysis to assess the presence of both HimarFT and FTL_0706 in the represented strains. SpeI-digested genomic DNA was probed with digoxigenin-labeled fragments of aphA-2 (4-kb signal) or FTL_0706 (with and without HimarFT insertion; 4- and 1-kb signals, respectively). The lane contents are the same as those described above for panel B.
DISCUSSION
To gain a more comprehensive understanding of the gene products required for F. tularensis intracellular replication, a transposon-based mutagenesis strategy coupled with a simple high-throughput screen with macrophages was utilized. Several HimarFT mutants recovered after our screens contained insertions in previously identified genes important for macrophage growth, including clpB, dsbB, capBC, aspC2, and wbtA (28, 55, 58, 67, 69, 70, 76). However, some genes whose products are associated with virulence, including those located in the FPI, were not identified in our screen as strongly attenuated (2, 28, 44, 53, 55, 56, 69). The absence of some previously identified virulence factors in our pool could have been due to the growth and replication conditions used, the stringency of the screen, or the inability to rescue all of the clones with the highest potential of encoding critical functions for macrophage growth. Lastly, the results indicate that our library may not saturate the genome, although isolation of multiple HimarFT strains with mutations in the same genes suggests that good coverage was obtained.
The FPI has potential roles in secretion, phagosome biogenesis, and subsequent escape into the cytosol or as an effector or cofactor element involved in promoting an optimal intracellular niche (14, 35, 50, 61, 62). It is duplicated in F. tularensis LVS, making it unlikely that isolates with mutations within this locus would have a strongly attenuated phenotype. HimarFT mutants in group C, demonstrating light retention of the crystal violet stain after stage 2 screening, were also subjected to nucleotide sequence analysis (data not shown). This analysis yielded insertions in the FPI iglB, iglC, and iglD loci, suggesting that deletion of one copy of these genes may result in a partial loss of function in F. tularensis LVS. This is consistent with a slight reduction in the BALB/c mouse lung survival of an EZ::TN-derived iglC LVS mutant (67). Interestingly, allelic replacement of both iglC loci in LVS was required to inhibit growth in J774A.1 and mouse peritoneal macrophages (27). These data indicate that the copy number of the FPI members is important for virulence.
As expected, HimarFT mutagenesis in F. tularensis LVS yielded a variety of genes encoding functions predicted to be important for intracellular survival and replication, as well as a number of hypothetical open reading frames. The identification of virulence determinants can be influenced by growth conditions, the properties of the transposon element, the F. tularensis subspecies used, or the choice of screening criteria. Despite these differences, similar functional groups of genes have been identified utilizing a variety of strategies in Francisella (28, 55, 67, 69, 76). Replication, translation, and protein modification and/or turnover genes were represented, including genes encoding proteins with predicted functions in disulfide bond formation, glutathione metabolism, or proteolytic degradation. Genes involved in metabolism and transport were prevalent, consistent with intracellular persistence requiring a variety of substrates, including amino acids, iron, and extracellular proteins (4, 16, 30, 37, 53, 56, 68). We speculate that the differences in stresses induced during growth in broth or on plates compared with intracellular growth may account for the identification of this class of genes. One candidate with a potential role in protein folding/stability or transport (FTL_1096) is located downstream of a predicted macrophage infectivity potentiator. Protein processing, metabolic, and transport-deficient mutants have the potential to be the basis for a new vaccine since they can infect and transmit their surface antigens but are likely limited in promoting overt disease. Surprisingly, although purines are clearly essential for intracellular growth of Francisella (28, 53, 55, 56, 67, 69, 76), mutants with insertions in this pathway were not recovered from our library. This was likely due to culture conditions in which medium lacking protease peptone was used in the selection step for transposon insertions (53).
A variety of surface structures, including LPS (13, 58, 63, 70), capsule (60, 67), type IV pilin (20, 24, 30), and outer membrane channels or proteins (32, 52), are implicated in Francisella virulence through potential roles in adherence, uptake, or phagosomal escape. In this study, several insertions within genes encoding capsule, LPS, and transporters were identified. F. tularensis LPS displays low toxicity compared to other gram-negative bacteria, and the structures of the O antigen and its corresponding gene cluster (wbt locus) in F. tularensis LVS and Schu S4 are identical but distinctly different from that in F. tularensis subsp. novicida (44). This is consistent with variation in detection with anti-LPS antibodies and induced immunity and/or cross-protection (22, 70, 75). O-antigen expression is defective when HimarFT insertions map to wbtA, a result that is similar to results previously reported by other groups (13, 58, 63). HimarFT mutants with mutations in wbtM and wbtC also displayed an O-antigen-defective phenotype not previously reported. Although wbtA and wbtM both encode a predicted dTDP-glucose 4,6-dehydratase, the gene sequences and the protein domains are quite different, suggesting that there are distinct roles for each protein. Interestingly, mutation of the wbtA and wbtDEF loci in several subspecies results in O-antigen loss but is accompanied by variable effects on intracellular survival (13, 58, 63, 70). These differences were also observed in our study. LVS FTL_0706::HimarFT appears to be similar to mutant SC92 recovered for F. tularensis subsp. novicida (13), but the O-antigen and intracellular growth phenotypes were not congruent. The LVS FTL_0706::HimarFT strain had a complete loss of O antigen and a 1,000-fold reduction in intracellular replication. In contrast, a partial loss of O antigen and a 10-fold decrease in replication occurred in SC92. The variation in O antigen is due to either distinct roles in biosynthesis in different subspecies or variable recognition by the antibodies used. FTL_0706 is located near another locus predicted to be involved in LPS biosynthesis but does not appear to be part of an operon.
HimarFT mutants with insertions in hypothetical genes displayed several distinct phenotypes related to intracellular replication and cytotoxicity. There was no significant difference between F. tularensis LVS and the strains bearing HimarFT insertions in terms of the ability to enter J774A.1 macrophages. Five of the seven HimarFT mutants exhibited significantly reduced replication and cytotoxicity, consistent with their attenuation in mice. In general, the intracellular growth of these strains was variably hampered, and the strains exhibited reduced levels of cytotoxicity, similar to the pattern exhibited by LVS ΔpurMCD. The yield of viable bacteria from macrophages at 24 h for mutants with insertions in FTL_0886, FTL_1414, and FTL_0439 was significantly reduced compared to that of the parental strain, but these cultures contained approximately 100-fold more bacteria than cultures of the avirulent LVS ΔpurMCD mutant with a similar level of cytotoxicity. This suggests that cytotoxicity may be induced by specific bacterial activities perhaps encoded by FTL_0886, FTL_1414, or FTL_0439. Despite displaying attenuation in the mouse model of infection, mutation of FTL_1096 resulted in no significant change in cytotoxicity, and insertion in FTL_0544 resulted in no significant changes in either cytotoxicity or replication compared to the wild type. This phenomenon has been observed in several EZ::TN-derived transposon mutants of F. tularensis subsp. novicida (76). The use of several model systems and Francisella strains to dissect gene function should allow reconciliation between in vitro and in vivo phenotypes. For example, there were reported differences in attenuation among different isolates of LVS or Schu S4 (29, 74), when different in vitro models (diverse macrophage or hepatic cell lines) were assessed (55, 69), or when in vitro and in vivo results were compared (20, 25, 69).
Recent studies indicate that inactivation of FTT0918 in F. tularensis Schu S4 results in subsequent loss of a 58-kDa protein and attenuation in vivo (72). FTT0918/FTT0919 represents a new protein family with predicted signal peptides and/or coiled-coil domains commonly associated with membranes and/or secretion (32, 35, 72). Interestingly, the N-terminal domain of FTT0918 and the C-terminal domain of FTT0919 form a single hybrid protein in F. tularensis type A strain FSC043 and type B strain LVS (FTL_0439). It has been suggested that part of the observed attenuation of LVS could be due to the deletion and fusion events that altered the FTT0918/FTT0919 locus (54, 59, 72). In this study, inactivation of FTL_0439 in F. tularensis LVS resulted in attenuation in vitro and in vivo. Thus, despite the hybrid nature of FTL_0439, it appears to maintain an important role in the virulence of LVS and is not likely to be the cause of its attenuation.
Polarity is an important consideration in determining whether a specific transposon insertion results in the observed phenotype. To address whether HimarFT is polar, RT-PCR and complementation analyses were performed with strains containing insertions mapping to genes that are or are not postulated to be part of a larger operon structure. Semiquantitative RT-PCRs of the wbt operon demonstrated that when target regions downstream of HimarFT were assessed, there was a reduction in the amount of cDNA detected. The data indicate that HimarFT insertion affects distal gene expression. Previous complementation experiments have utilized a highly active promoter, such as groEL, to ensure gene expression in the absence of a native promoter. However, alteration in the level of expression of the gene in trans can be detrimental (14, 38, 61, 67). Since the native promoter was used for complementation in our studies, the partial restoration of O antigen in F. tularensis LVS wbtA2::HimarFT pFTNAT-wbtALVS was not due to overexpression of the gene in trans. In fact, a similar partial restoration of O antigen was observed in an LVS Himar1-based wbtA mutant when wbtA was driven by the groEL promoter in trans (63). Based on our RT-PCR and complementation patterns, we concluded that the groEL promoter from HimarFT transcribes into flanking gene regions. In trans complementation analysis verified that the noncytopathic phenotypes of mutants with insertions in both wbtM and FTL_0706 were due to the HimarFT insertion. Neither of these genes appears to be part of an operon.
In summary, after a HimarFT insertion library was screened for candidates deficient in cytopathogenicity in J774A.1 macrophages, 26 genes were identified as virulence determinants in Francisella. Several candidates have been reported in previous genetic analyses, validating our screening process, while others were recognized for the first time as potential virulence factors. Along with genes involved in transport, metabolism, protein stability/turnover, and surface components, seven mutants with insertions in hypothetical genes were recovered. All of these mutants were attenuated in a BALB/c mouse model of infection, suggesting that there is a direct correlation between in vitro and in vivo replication. Complementation analysis of selected strains confirmed that the phenotype observed was due to the gene in which HimarFT inserted. It also provided evidence for potential polar effects that can affect the expression of genes adjacent to HimarFT insertions. Additional studies of our candidate genes and the design of alternative screens should further define components important for the intracellular lifestyle of Francisella, provide insight into the mechanisms involved in infection, and facilitate the development of therapeutics and/or a defined vaccine effective for the identification and control of this invasive pathogen.
Acknowledgments
This work was sponsored by the NIH/NIAID Regional Center of Excellence for Biodefense and Emerging Infectious Diseases Research (RCE) Program, the Center for Biopreparedness and Infectious Disease at the Medical College of Wisconsin, and NIH funds (grant AI 063441 to D.W.F. and T.C.Z.). T.M.M. and C.W.D. were supported by Great Lakes Regional Center of Excellence career development fellowships CDP9 and CDP19.
We thank Marc A. Benson for his assistance with compiling the F. tularensis LVS HimarFT library. Travis McCarthy kindly assisted in generating the RT-PCR results.
Editor: J. B. Bliska
Footnotes
Published ahead of print on 6 August 2007.
REFERENCES
- 1.Altschul, S. F., W. Gish, W. Miller, E. W. Myers, and D. J. Lipman. 1990. Basic local alignment search tool. J. Mol. Biol. 215:403-410. [DOI] [PubMed] [Google Scholar]
- 2.Anthony, L. S., S. C. Cowley, K. E. Mdluli, and F. E. Nano. 1994. Isolation of a Francisella tularensis mutant that is sensitive to serum and oxidative killing and is avirulent in mice: correlation with the loss of MinD homologue expression. FEMS Microbiol. Lett. 124:157-165. [DOI] [PubMed] [Google Scholar]
- 3.Anthony, L. S. D., M. Z. Gu, S. C. Cowley, W. W. S. Leung, and F. E. Nano. 1991. Transformation and allelic replacement in Francisella spp. J. Gen. Microbiol. 137:2697-2703. [DOI] [PubMed] [Google Scholar]
- 4.Atkins, H. S., E. Dassa, N. J. Walker, K. F. Griffin, D. N. Harland, R. R. Taylor, M. L. Duffield, and R. W. Titball. 2006. The identification and evaluation of ATP binding cassette systems in the intracellular bacterium Francisella tularensis. Res. Microbiol. 157:593-604. [DOI] [PubMed] [Google Scholar]
- 5.Bendtsen, J. D., H. Nielsen, G. von Heijne, and S. Brunak. 2004. Improved prediction of signal peptides: SignalP 3.0. J. Mol. Biol. 340:783-795. [DOI] [PubMed] [Google Scholar]
- 6.Bina, X. R., C. Wang, M. A. Miller, and J. E. Bina. 2006. The Bla2 β-lactamase from the live-vaccine strain of Francisella tularensis encodes a functional protein that is only active against penicillin-class β-lactam antibiotics. Arch. Microbiol. 186:219-228. [DOI] [PubMed] [Google Scholar]
- 7.Broekhuijsen, M., P. Larsson, A. Johansson, M. Byström, U. Eriksson, E. Larsson, R. G. Prior, A. Sjöstedt, R. W. Titball, and M. Forsman. 2003. Genome-wide DNA microarray analysis of Francisella tularensis strains demonstrates genetic conservation within the species but identifies regions that are unique to the highly virulent F. tularensis subsp. tularensis. J. Clin. Microbiol. 41:2924-2931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Burke, D. S. 1977. Immunization against tularemia: analysis of the effectiveness of live Francisella tularensis vaccine in prevention of laboratory-acquired tularemia. J. Infect. Dis. 135:55-60. [DOI] [PubMed] [Google Scholar]
- 9.Camilli, A., D. A. Portnoy, and P. Youngman. 1990. Insertional mutagenesis of Listeria monocytogenes with a novel Tn917 derivative that allows direct cloning of DNA flanking transposon insertions. J. Bacteriol. 172:3738-3744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chamberlain, R. E. 1965. Evaluation of live tularemia vaccine prepared in a chemically defined medium. Appl. Microbiol. 13:232-235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Checroun, C., T. D. Wehrly, E. R. Fischer, S. F. Hayes, and J. Celli. 2006. Autophagy-mediated reentry of Francisella tularensis into the endocytic compartment after cytoplasmic replication. Proc. Natl. Acad. Sci. USA 103:14578-14583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chen, W., H. Shen, A. Webb, R. KuoLee, and J. W. Conlan. 2003. Tularemia in BALB/c and C57BL/6 mice vaccinated with Francisella tularensis LVS and challenged intradermally, or by aerosol with virulent isolates of the pathogen: protection varies depending on pathogen virulence, route of exposure, and host genetic background. Vaccine 21:3690-3700. [DOI] [PubMed] [Google Scholar]
- 13.Cowley, S. C., C. J. Gray, and F. E. Nano. 2000. Isolation and characterization of Francisella novicida mutants defective in lipopolysaccharide biosynthesis. FEMS Microbiol. Lett. 182:63-67. [DOI] [PubMed] [Google Scholar]
- 14.de Bruin, O. M., J. S. Ludu, and F. E. Nano. 2007. The Francisella pathogenicity island protein IglA localizes to the bacterial cytoplasm and is needed for intracellular growth. BMC Microbiol. 7:1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dempsey, M. P., J. Nietfeldt, J. Ravel, S. Hinrichs, R. Crawford, and A. K. Benson. 2006. Paired-end sequence mapping detects extensive genomic rearrangement and translocation divergence of Francisella tularensis subsp. tularensis and Fracisella tularensis subsp. holarctica populations. J. Bacteriol. 188:5904-5914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Deng, K., R. Blick, W. Liu, and E. J. Hansen. 2006. Identification of Francisella tularensis genes affected by iron limitation. Infect. Immun. 74:4224-4236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Eigelsbach, H. T., W. Braun, and R. D. Herring. 1951. Studies on the variation of Bacterium tularense. J. Bacteriol. 61:557-569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Eigelsbach, H. T., and C. M. Downs. 1961. Prophylactic effectiveness of live and killed tularemia vaccines. I. Production of vaccine and evaluation in the white mouse and guinea pig. J. Immunol. 87:415-425. [PubMed] [Google Scholar]
- 19.Ellis, J., P. C. Oyston, M. Green, and R. W. Titball. 2002. Tularemia. Clin. Microbiol. Rev. 15:631-646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Forslund, A. L., K. Kuoppa, K. Svensson, E. Salomonsson, A. Johansson, M. Bystrom, P. C. F. Oyston, S. L. Michell, R. W. Titball, L. Noppa, E. Frithz-Lindsten, M. Forsman, and A. Forsberg. 2006. Direct repeat-mediated deletion of a type IV pilin gene results in major virulence attenuation of Francisella tularensis. Mol. Microbiol. 59:1818-1830. [DOI] [PubMed] [Google Scholar]
- 21.Fortier, A. H., M. V. Slayter, R. Ziemba, M. S. Meltzer, and C. A. Nacy. 1991. Live vaccine strain of Francisella tularensis: infection and immunity in mice. Infect. Immun. 59:2922-2928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Fulop, M. J., T. Webber, R. J. Manchee, and D. C. Kelly. 1991. Production and characterization of monoclonal antibodies directed against the lipopolysaccharide of Francisella tularensis. J. Clin. Microbiol. 29:1407-1412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gallagher, L. A., E. Ramage, M. A. Jacobs, R. Kaul, M. Brittnacher, and C. Manoil. 2007. A comprehensive transposon mutant library of Francisella novicida, a bioweapon surrogate. Proc. Natl. Acad. Sci. USA 104:1009-1014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gil, H., J. L. Benach, and D. G. Thanassi. 2004. Presence of pili on the surface of Francisella tularensis. Infect. Immun. 72:3042-3047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gil, H., G. J. Platz, C. A. Forestal, M. Monfett, C. S. Bakshi, T. J. Sellati, M. B. Furie, J. L. Benach, and D. G. Thanassi. 2006. Deletion of TolC orthologs in Francisella tularensis identifies roles in multidrug resistance and virulence. Proc. Natl. Acad. Sci. USA 103:12897-12902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Goldstein, A. L., and J. H. McCusker. 1999. Three new dominant drug resistance cassettes for gene disruption in Saccharomyces cerevisiae. Yeast 15:1541-1553. [DOI] [PubMed] [Google Scholar]
- 27.Golovliov, I., A. Sjöstedt, A. N. Mokrievich, and V. M. Pavlov. 2003. A method for allelic replacement in Francisella tularensis. FEMS Microbiol. Lett. 222:273-280. [DOI] [PubMed] [Google Scholar]
- 28.Gray, C. G., S. C. Cowley, K. K. Cheung, and F. E. Nano. 2002. The identification of five genetic loci of Francisella novicida associated with intracellular growth. FEMS Microbiol. Lett. 215:53-56. [DOI] [PubMed] [Google Scholar]
- 29.Green, M., G. Choules, D. Rogers, and R. W. Titball. 2005. Efficacy of the live attenuated Francisella tularensis vaccine (LVS) in a murine model of disease. Vaccine 23:2680-2686. [DOI] [PubMed] [Google Scholar]
- 30.Hager, A. J., D. L. Bolton, M. R. Pelletier, M. J. Brittnacher, L. A. Gallagher, R. Kaul, S. J. Skerrett, S. I. Miller, and T. Guina. 2006. Type IV pili-mediated secretion modulates Francisella virulence. Mol. Microbiol. 62:227-237. [DOI] [PubMed] [Google Scholar]
- 31.Henikoff, S., J. G. Henikoff, W. J. Alford, and S. Pietrokovski. 1995. Automated construction and graphical presentation of protein blocks from unaligned sequences. Gene 163:GC17-GC26. [DOI] [PubMed] [Google Scholar]
- 32.Huntley, J. F., P. G. Conley, K. E. Hagman, and M. V. Norgard. 2007. Characterization of Francisella tularensis outer membrane proteins. J. Bacteriol. 189:561-574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kawula, T. H., J. D. Hall, J. R. Fuller, and R. R. Craven. 2004. Use of transposon-transposase complexes to create stable insertion mutant strains of Francisella tularensis LVS. Appl. Environ. Microbiol. 70:6901-6904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Larson, C. L., W. Wicht, and W. L. Jellison. 1955. A new organism resembling P. tularensis isolated from water. Public Health Rep. 70:253-258. [PMC free article] [PubMed] [Google Scholar]
- 35.Larsson, P., P. C. F. Oyston, P. Chain, M. C. Chu, M. Duffield, H.-H. Fuxelius, E. Garcia, G. Halltorp, D. Johansson, K. E. Isherwood, P. D. Karp, E. Larsson, Y. Liu, S. Michell, J. Prior, R. Prior, S. Malfatti, A. Sjostedt, K. Svensson, N. Thompson, L. Vergez, J. K. Wagg, B. W. Wren, L. Lindler, S. G. E. Andersson, M. Forsman, and R. W. Titball. 2005. The complete genome sequence of Francisella tularensis, the causative agent of tularemia. Nat. Genet. 37:153-159. [DOI] [PubMed] [Google Scholar]
- 36.Lauriano, C. M., J. R. Barker, F. E. Nano, B. P. Arulanandam, and K. E. Klose. 2003. Allelic exchange in Francisella tularensis using PCR products. FEMS Microbiol. Lett. 229:195-202. [DOI] [PubMed] [Google Scholar]
- 37.Lee, B.-Y., M. A. Horwitz, and D. L. Clemens. 2006. Identification, recombinant expression, immunolocalization in macrophages, and T-cell responsiveness of the major extracellular proteins of Francisella tularensis. Infect. Immun. 74:4002-4013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lindgren, H., I. Golovliov, V. Baranov, R. K. Ernst, M. Telepnev, and A. Sjöstedt. 2004. Factors affecting the escape of Francisella tularensis from the phagolysosome. J. Med. Microbiol. 53:953-958. [DOI] [PubMed] [Google Scholar]
- 39.Lopez, M. C., N. S. Duckett, S. D. Baron, and D. W. Metzger. 2004. Early activation of NK cells after lung infection with the intracellular bacterium, Francisella tularensis LVS. Cell. Immunol. 232:75-85. [DOI] [PubMed] [Google Scholar]
- 40.LoVullo, E. D., L. A. Sherrill, L. L. Perez, and M. S. J. Pavelka. 2006. Genetic tools for highly pathogenic Francisella tularensis subsp. tularensis. Microbiol. 152:3425-3435. [DOI] [PubMed] [Google Scholar]
- 41.Maier, T. M., A. Havig, M. Casey, F. E. Nano, D. W. Frank, and T. C. Zahrt. 2004. Construction and characterization of a highly efficient Francisella shuttle plasmid. Appl. Environ. Microbiol. 70:7511-7519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Maier, T. M., R. Pechous, M. Casey, T. C. Zahrt, and D. W. Frank. 2006. In vivo Himar1-based transposon mutagenesis of Francisella tularensis. Appl. Environ. Microbiol. 72:1878-1885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Maltsev, N., E. Glass, D. Sulakhe, A. Rodriguez, M. Syed, T. Bompada, and Y. Zhang. 2006. PUMA2—grid-based high-throughput analysis of genomes and metabolic pathways. Nucleic Acids Res. 34:D369-D372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.McLendon, M. K., M. A. Apicella, and L.-A. H. Allen. 2006. Francisella tularensis: taxonomy, genetics, and immunopathogenesis of a potential agent of biowarfare. Annu. Rev. Microbiol. 60:167-185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Mdluli, K. E., L. S. Anthony, G. S. Baron, M. K. McDonald, S. V. Myltseva, and F. E. Nano. 1994. Serum-sensitive mutation of Francisella novicida: association with an ABC transporter gene. Microbiology 140:3309-3318. [DOI] [PubMed] [Google Scholar]
- 46.Mohapatra, N. P., S. Soni, B. L. Bell, R. Warren, R. K. Ernst, A. Muszynski, R. W. Carlson, and J. S. Gunn. 2007. Identification of an orphan response regulator required for Francisella virulence and transcription of pathogenicity island genes. Infect. Immun. 75:3305-3314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Moller, S., M. D. R. Croning, and R. Apweiler. 2001. Evaluation of methods for the prediction of membrane spanning regions. Bioinformatics 17:646-653. [DOI] [PubMed] [Google Scholar]
- 48.Mulder, N. J., R. Apweiler, T. K. Attwood, A. Bairoch, D. Barrell, A. Bateman, D. Binns, M. Biswas, P. Bradley, P. Bork, P. Bucher, R. R. Copley, E. Courcelle, U. Das, R. Durbin, L. Falquet, W. Fleischmann, S. Griffiths-Jones, D. Haft, N. Harte, N. Hulo, D. Kahn, A. Kanapin, M. Krestyaninova, R. Lopez, I. Letunic, D. Lonsdale, V. Silventoinen, S. E. Orchard, M. Pagni, D. Peyruc, C. P. Ponting, J. D. Selengut, F. Servant, C. J. Sigrist, R. Vaughan, and E. M. Zdobnov. 2003. The InterPro Database, 2003 brings increased coverage and new features. Nucleic Acids Res. 31:315-318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Nallapareddy, S., K. Singh, and B. Murray. 2006. Construction of improved temperature-sensitive and mobilizable vectors and their use for constructing mutations in the adhesion-encoding acm gene of poorly transformable clinical Enterococcus faecium strains. Appl. Environ. Microbiol. 72:334-345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Nano, F. E., N. Zhang, S. C. Cowley, K. E. Klose, K. K. M. Cheung, M. J. Roberts, J. S. Ludu, G. W. Letendre, A. I. Meierovics, G. Stephens, and K. L. Elkins. 2004. A Francisella tularensis pathogenicity island required for intramacrophage growth. J. Bacteriol. 186:6430-6436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Oyston, P. C. F., A. Sjostedt, and R. W. Titball. 2004. Tularaemia: bioterrorism defence renews interest in Francisella tularensis. Nat. Rev. 2:967-978. [DOI] [PubMed] [Google Scholar]
- 52.Pavkova, I., M. Hubálek, J. Zechovska, J. Lenco, and J. Stulik. 2005. Francisella tularensis live vaccine strain: proteomic analysis of membrane proteins enriched fraction. Proteomics 5:2460-2467. [DOI] [PubMed] [Google Scholar]
- 53.Pechous, R., J. Celli, R. Penoske, S. F. Hayes, D. W. Frank, and T. C. Zahrt. 2006. Construction and characterization of an attenuated purine auxotroph in a Francisella tularensis live vaccine strain. Infect. Immun. 74:4452-4461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Petrosino, J. F., Q. Xiang, S. E. Karpathy, H. Jiang, S. Yerrapragada, Y. Liu, J. Gioia, L. Hemphill, A. Gonzalez, T. M. Raghavan, A. Uzman, G. E. Fox, S. Highlander, M. Reichard, R. J. Morton, K. D. Clinkenbeard, and G. M. Weinstock. 2006. Chromosome rearrangement and diversification of Francisella tularensis revealed by the type B (OSU18) genome sequence. J. Bacteriol. 188:6977-6985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Qin, A., and B. J. Mann. 2006. Identification of transposon insertion mutants of Francisella tularensis tularensis strain Schu S4 deficient in intracellular replication in the hepatic cell line HepG2. BMC Microbiol. 6:69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Quarry, J. E., K. E. Isherwood, S. L. Michell, H. Diaper, R. W. Titball, and P. C. F. Oyston. 2007. A Francisella tularensis subspecies novicida purF mutant, but not a purA mutant, induces protective immunity to tularemia in mice. Vaccine 25:2011-2018. [DOI] [PubMed] [Google Scholar]
- 57.Rasko, D. A., C. D. Esteban, and V. Sperandio. 2007. Development of novel plasmid vectors and a promoter trap system in Francisella tularensis compatible with the pFNL10 based plasmids. Plasmid 58:159-166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Raynaud, C., K. L. Meiborn, M. A. Lety, I. Dubail, T. Candela, E. Frapy, and A. Charbit. 2007. Role of the wbtA locus of Fransicella tularensis in lipopolysaccharide O-antigen biogenesis and pathogenicity. Infect. Immun. 75:536-541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Rohmer, L., M. Brittnacher, K. Svensson, D. Buckley, E. Haugen, Y. Zhou, J. Chang, R. Levy, H. Hayden, M. Forsman, M. Olson, A. Johansson, R. Kaul, and S. I. Miller. 2006. Potential source of Francisella tularensis live vaccine strain attenuation determined by genome comparison. Infect. Immun. 74:6895-6906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Sandström, G., S. Löfgren, and A. Tärnvik. 1988. A capsule-deficient mutant of Francisella tularensis LVS exhibits enhanced sensitivity to killing by serum but diminished sensitivity to killing by polymorphonuclear leukocytes. Infect. Immun. 56:1194-1202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Santic, M., M. Molmeret, J. R. Barker, K. E. Klose, A. Dekanic, M. Doric, and Y. Abu Kwaik. 21 May 2007, posting date. A Francisella tularensis pathogenicity island protein essential for bacterial proliferation within the host cytosol. Cell. Microbiol. doi: 10.1111/j. 1462-5822.2007.00968.x. (Subsequently published, Cell. Microbiol. 9:2391-2403, 2007.) [DOI] [PubMed] [Google Scholar]
- 62.Santic, M., M. Molmeret, K. E. Klose, S. Jones, and Y. Abu Kwaik. 2005. The Francisella tularensis pathogenicity island protein IglC and its regulator MglA are essential for modulating phagosome biogenesis and subsequent bacterial escape into the cytoplasm. Cell. Microbiol. 7:969-979. [DOI] [PubMed] [Google Scholar]
- 63.Sebastian, S., S. T. Dillon, J. G. Lynch, L. T. Blalock, E. Balon, K. T. Lee, L. E. Comstock, J. W. Conlan, E. J. Rubin, A. O. Tzianabos, and D. L. Kasper. 2007. A defined O-antigen polysaccharide mutant of Francisella tularensis live vaccine strain has attenuated virulence while retaining its protective capacity. Infect. Immun. 75:2591-2602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Shaw, K. J., P. N. Rather, R. S. Hare, and G. H. Miller. 1993. Molecular genetics of aminoglycoside resistance genes and familial relationships of the aminoglycoside-modifying enzymes. Microbiol. Rev. 57:138-163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Sjostedt, A. 2006. Intracellular survival mechanisms of Francisella tularensis, a stealth pathogen. Microbes Infect. 8:561-567. [DOI] [PubMed] [Google Scholar]
- 66.Sjöstedt, A. 2003. Virulence determinants and protective antigens of Francisella tularensis. Curr. Opin. Microbiol. 6:66-71. [DOI] [PubMed] [Google Scholar]
- 67.Su, J., J. Yang, D. Zhao, T. H. Kawula, J. A. Banas, and J. R. Zhang. 2007. Genome-wide identification of Francisella tularensis virulence determinants. Infect. Immun. 75:3089-3101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Sullivan, J., E. Jeffery, J. Shannon, and G. Ramakrishnan. 2006. Characterization of the siderophore of Francisella tularensis and role of fslA in siderophore production. J. Bacteriol. 188:3785-3795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Tempel, R., X. H. Lai, L. Crosa, B. Kozlowicz, and F. Heffron. 2006. Attenuated Francisella novicida transposon mutants protect mice against wild-type challenge. Infect. Immun. 74:5095-5105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Thomas, R. M., R. W. Titball, P. C. Oyston, K. Griffin, E. Waters, P. G. Hitchen, S. L. Michell, I. D. Grice, J. C. Wilson, and J. L. Prior. 2007. The immunologically distinct O-antigens from Francisella tularensis subspecies tularensis and Francisella novicida are both virulence determinants and protective antigens. Infect. Immun. 75:371-378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Titball, R. W., A. Johansson, and M. Forsman. 2003. Will the enigma of Francisella tularensis virulence soon be solved? Trends Microbiol. 11:118-123. [DOI] [PubMed] [Google Scholar]
- 72.Twine, S., M. Bystrom, W. Chen, M. Forsman, I. Golovliov, A. Johansson, J. Kelly, H. Lindgren, K. Svensson, C. Zingmark, W. Conlan, and A. Sjostedt. 2005. A mutant of Francisella tularensis strain Schu S4 lacking the ability to express a 58-kilodalton protein is attenuated for virulence and is an effective live vaccine. Infect. Immun. 73:8345-8352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Twine, S. M., N. C. Mykytezuk, M. D. Petit, H. Shen, A. Sjostedt, W. J. Colan, and J. F. Kelly. 2006. In vivo proteomic analysis of the intracellular bacterial pathogen, Francisella tularensis isolated from mouse spleen. Biochem. Biophys. Res. Commun. 345:1621-1633. [DOI] [PubMed] [Google Scholar]
- 74.Twine, S. M., H. Shen, J. F. Kelly, W. Chen, A. Sjostedt, and J. W. Conlan. 2006. Virulence comparison in mice of distinct isolates of type A Franicsella tularensis. Microb. Pathog. 40:133-138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Vinogradov, E., W. J. Conlan, J. S. Gunn, and M. B. Perry. 2004. Characterization of the lipopolysaccharide O-antigen of Francisella novicida (U112). Carbohydr. Res. 339:649-654. [DOI] [PubMed] [Google Scholar]
- 76.Weiss, D. S., A. Brotcke, T. Henry, J. J. Margolis, K. Chan, and D. M. Monack. 2007. In vivo negative selection screen identifies genes required for Francisella virulence. Proc. Natl. Acad. Sci. USA 104:6037-6042. [DOI] [PMC free article] [PubMed] [Google Scholar]