

Abstract
The development of needle-free vaccines is one of the recently defined “grand challenges in global health” (H. Varmus, R. Klausner, R. Klausner, R. Zerhouni, T. Acharya, A. S. Daar, and P. A. Singer, Science 302:398-399, 2003). To explore whether a natural pathway to the inductive site of the mucosa-associated lymphatic tissue could be exploited for atraumatic immunization purposes, replication-deficient viral vector vaccines were sprayed directly onto the tonsils of rhesus macaques. Tonsillar immunization with viral vector vaccines encoding simian immunodeficiency virus (SIV) antigens induced cellular and humoral immune responses. Viral RNA levels after a stringent SIV challenge were reduced, providing a level of protection similar to that observed after systemic immunization with the same vaccines. Thus, atraumatic oral spray immunization with replication-deficient vectors can overcome the epithelial barrier, deliver the vaccine antigen to the mucosa-associated lymphatic tissue, and avoid induction of tolerance, providing a novel approach to circumvent acceptability problems of syringe and needle vaccines for children and in developing countries.
The first immunization studies in humans with viral vectors encoding vaccine antigens have demonstrated induction of cellular and humoral immune responses (7, 20, 29, 30, 33). Several vector vaccines based on adenoviruses or poxviruses have shown promising preclinical results and are currently at different stages of clinical development for prophylactic or therapeutic use against infectious diseases or cancer.
In children, syringe and needle administration of vaccines faces acceptability problems, while in developing countries the inappropriate reuse of needles and syringes is associated with an increased risk of infection. A noninvasive oral vaccination strategy could greatly facilitate worldwide access to vaccines by, for example, enabling trained teachers to administer the vaccine. So far, only live attenuated vaccines have been used for oral vaccination in humans. The efficacy of these vaccines depends on subsequent replication and spread of the vaccines in the gastrointestinal tract. Although highly efficient, the spread of live attenuated vaccines to contact persons of the vaccinees or reversion of the vaccines to virulent forms limits the applicability of the live attenuated vaccine approach for many infectious diseases. Oral vaccination with replication-deficient viral vector vaccines might be able to substitute for the live attenuated vaccines, but it is questionable whether they can elicit substantial immune responses given that the excess amount of antigens taken up as food on a regular basis mostly leads to tolerance rather than immunity. In addition, if only the superficial layers of the mucosa are transduced by the viral vector vaccines, shedding of the transduced cells prior to expression of the vaccine antigen could be expected.
The epithelial barrier of the oral cavity to be passed by viral vector vaccines consists of a multilayer, nonkeratinized squamous epithelium. Below the epithelium, the oral cavity contains the Waldeyer's ring, an important member of the mucosa-associated lymphatic tissue (MALT), including the lingual and palatine tonsils. The crypts of these tonsils are lined by a lymphoepithelium with interspersed M cells that facilitate controlled entry of antigens through the epithelial barrier. Since the oral mucosa and especially the crypt epithelium of the tonsils are also rich in dendritic cells, delivery of the vaccine via the tonsillar crypts could be a promising vaccination approach.
Administering simian immunodeficiency virus (SIV) onto the tonsils of rhesus macaques led to efficient infection of exposed animals, providing a valuable atraumatic mucosal challenge model (36). Tonsillar delivery of live attenuated SIV vaccines also provided protection against subsequent challenge with homologous SIV and an SIV-human immunodeficiency virus hybrid virus (35, 38). Since protection induced by these live attenuated vaccines most likely has been due to systemic spread of the vaccine virus, we now analyzed the immunogenicity and efficacy of replication-deficient viral vector vaccines after immunization by the tonsillar route and compared them to those after systemic administration. Adenoviral vectors were selected for tonsillar immunizations, assuming that the natural infection pathway of adenoviruses would also allow efficient delivery of the vector-encoded vaccine antigens. For priming we also used a single-cycle immunodeficiency virus vaccine (SCIV). The SCIVs were produced by transient transfection of an SIV genome which was made replication deficient by mutations in the primer binding site and a deletion of vif (23). To allow a single round of replication, the primer binding site mutations were complemented in trans by a matched tRNA expression plasmid in vif-independent 293 producer cells. After administration to the vaccinees, the SCIVs can undergo only a single round of replication, leading to the production of noninfectious virus-like particles in vivo (12, 23). To increase in vivo expression levels, we pseudotyped the SCIVs with the G protein of vesicular stomatitis virus, which mediates efficient entry into a broad spectrum of cells (6), including dendritic cells (16). After repeated systemic immunizations with SCIVs, SIV-specific humoral and cellular immune responses were observed, peak viremia during primary infection with the SIVmac239 challenge virus was significantly reduced (13, 23), and in a long-term vaccination experiment persistent suppression of SIVmac239 viral load was achieved (24).
We now observed that adenoviral vectors delivered by a simple atraumatic spray procedure onto the tonsils led to expression of the encoded antigen in close proximity to the inductive site of the MALT. Oral immunization with the adenoviral vectors was sufficient to induce cellular and humoral immune responses to encoded SIV vaccine antigens, but the adenoviral vector vaccine alone did not reduce challenge virus load. By contrast, a tonsillar prime-boost regimen of SCIV and adenoviral vector vaccines induced higher levels of cellular immune responses and reduced viral RNA levels after challenge with neutralization-resistant SIVmac239.
MATERIALS AND METHODS
Preparation of the vaccines.
293T (293ts/A1609) (10), 293A (strongly adherent subclone of 293, Quantum Biotechnologies, Montreal, Canada), 293T-Rex (Invitrogen, Karlsruhe, Germany), and S-MAGI cells (8) were maintained in Dulbecco's modified Eagle's medium supplemented with 10% fetal calf serum, penicillin, streptomycin, and glutamine. The SCIVs were produced by transient transfection of 293T cells as previously described (23). Viral particles from the conditioned medium were thereafter concentrated by low-speed centrifugation (27). The preparation contained 235 μg/ml reverse transcriptase as measured by a commercial reverse transcriptase assay (Roche, Penzberg, Germany). Further characterization of the SCIV preparation revealed a similar ratio of Gag and Env bands in Western blot analyses as previously reported (23) and readily detectable levels of vesicular stomatitis virus G. For determination of the infectious titer, S-MAGI indicator cell lines, containing the beta-galactosidase gene downstream of the human immunodeficiency virus type 1 long terminal repeat, were seeded at a density of 2.5 × 104 cells per well of a 24-well plate. One day later, 200 μl of serial dilutions of the concentrated SCIV particles were incubated with the S-MAGI cells for 3 hours prior to addition of fresh medium. Two days later, S-MAGI cells were stained with 5-bromo-4-chloro-3-indolyl-β-d-galactopyranoside (X-Gal) essentially as described previously (8). The vector titer was calculated from the number of stained cells per well and was expressed as LacZ-forming units/ml. The two SCIV preparations had infectious titers of 3.5 × 109 and 2.3 × 108 infectious units/ml. The construction of the recombinant adenoviral vector expressing the codon-optimized SIV Gag-Pol (Ad-Sgpsyn) has been described previously (25). For the construction of the recombinant adenovirus expressing the SIV envelope (Ad-Senv-co), a codon-optimized version of the SIVmac239 envelope gene encoding a membrane-anchored Env with a truncated (amino acids 733 to 879) C terminus of the cytoplasmic domain (TM207) (41) was cloned into the HindIII-XbaI-digested pShuttle-TetO2 (25), generating pS-Senv-co. The leader peptide of SIV was replaced by the leader peptide of tissue plasminogen activator. The full-length adenoviral vector plasmid was generated by homologous recombination of the plasmids pS-Senv-co and pAdEasy1 (21) in BJ5183 cells (Stratagene, Amsterdam, The Netherlands) using standard procedures. Correctly recombined plasmids were transfected into 293T-Rex cells, and the recombinant viral vectors growing out from the transfected cells were analyzed for expression of the vaccine antigen by Western blot analyses. A second adenoviral vector (Ad-Srtenvco) encoding Rev and a secreted version of SIV Env was constructed in pShuttle-CMV by deleting the transmembrane domain of Env. The construct contains SIVmac239 nucleotides 6696 to 6859 (first exon of rev; numbering is according to GenBank entry M33262), the first 25 codons of the tissue plasminogen activator leader sequence, SIVmac239 env codons 23 to 683 in a codon-optimized version, and wild-type SIV nucleotides 9002 to 9499 (codons 715 to 879 of SIV env and second exon of rev). Full-length adenoviral vector constructs were generated by homologous recombination with pAdEasy1, and vector stocks were produced in 293 cells as described above. After purification of adenoviral vectors by CsCl gradient centrifugation, the particle concentration was measured by the optical density. The adenoviral vaccine preparations of Ad-Sgpsyn, Ad-Senvco, and Ad-Srtenvco had particle concentrations of 4.9 × 1012/ml, 4.8 × 1012/ml, and 2.4 × 1012/ml, respectively. The Ad-GFP vector was kindly provided by Kirsten Bender (Bochum) and had been constructed by homologous recombination using the pAdTrack-CMV plasmid and pAD-Easy1 (21). For the adenoviral neutralization assay, a second adenoviral vector, designated Ad-EGFP, was generated by cloning the enhanced green fluorescent protein (EGFP) gene of pEGFP-1 (Clontech, Mountain View, CA) into pShuttle-CMV (Qbiogene, Irvine, CA) and subsequent homologous recombination with pAD-Easy1.
Animal experiments.
Thirty-four purpose-bred young adult rhesus macaques were used in the present studies. Twenty-two of them were of Indian origin, and the 12 monkeys for the second immunization experiment had been imported from China. Animals were housed at the German Primate Centre, and animal care and use were in compliance with the German Animal Protection Law and relevant institutional guidelines.
Just prior to oral immunization, animals were treated intravenously with glycopyrroniumbromide at a dose of 40 μg. In the “Ad-oral” experiment, Ad-Sgpsyn and Ad-Senvco were each adjusted to a final concentration of 2 × 1011 particles/ml and administered in a total volume of 0.5 ml. In the prime-boost experiment, the adenoviral vectors Ad-Sgpsyn and Ad-Srtenvco were administered by oral spray immunization (0.5-ml total volume) or intramuscular immunization (final volume of 3 ml injected at five different sites) at doses of 0.5 × 1011 and 3 × 1011 particles per construct, respectively. For intravenous immunization with SCIV, 2 × 109 LacZ-forming units were injected in a final volume of 10 ml. Challenge virus exposure was performed with pathogenic SIVmac239 by the tonsillar route by touching the tonsils lightly with a cotton-wool swab soaked with culture medium containing approximately 6,000 median tissue culture infectious doses of cell-free SIVmac239 virus as described previously (24).
Immunohistochemistry and in situ hybridization. (i) Routine histology.
The tissues obtained by biopsy or autopsy were divided into two parts. One part was fixed overnight in 4% neutral buffered formalin and embedded in paraffin. The sections were stained with hematoxylin and eosin and Giemsa stain for routine histology. Portions of the fresh tissue were embedded in tissue freezing medium (Leica, Nussloch, Germany), snap frozen in liquid nitrogen, and stored at −70°C until use.
(ii) Immunohistochemistrty.
The staining for the detection of GFP was performed on frozen sections. The cryostat sections were fixed with acetone for 30 min and then incubated with rabbit polyclonal antibody GFP-Ch1P (1:300; Abcam, Cambridge, United Kingdom) at room temperature for 30 min. The antibody binding was visualized after incubation of the sections with a biotinylated goat anti-rabbit immunoglobulin (DakoCytomation, Copenhagen, Denmark) followed by streptavidin-APAAP (Dako). The color was developed with the alkaline phosphatase anti-alkaline phosphatase technique with New Fuchsin as red chromogen.
(iii) In situ hybridization.
SIV RNA in the axillary lymph nodes was detected on paraffin sections with a 35S-labeled single-stranded (antisense) RNA probe (Lofstrand Laboratories, Gaithersburg, MD) as described in detail before (39). From each biopsy, six to eight sections were hybridized. Spleen sections from naive infected monkeys were used as a positive control. As a negative control, sections from each lymph node specimen were hybridized with a sense-strand 35S-labeled probe. The slides were dipped in photo emulsion (NBT2; Kodak) and exposed in the dark at 4°C for 7 days. The slides were developed (D19; Kodak), fixed, counterstained with hematoxylin, and mounted. Examination of the sections was performed with a Zeiss Axiophot microscope (Carl Zeiss, Jena, Germany) equipped with transmission and epiluminescent illumination. Cells expressing SIV RNA were counted in all sections, and the mean value per section was calculated.
Immune monitoring and viral load measurement.
To determine SIV-specific T-cell responses, a gamma interferon (IFN-γ) enzyme-linked immunospot (ELISPOT) assay was employed essentially as described previously (37). The following antigenic stimuli were used: a 20-mer peptide pool of the SIVgag p27 region (EVA ARP714.1 to -22; 22 peptides), a pool of 15-mer peptides of SIVmac251/32H-Gag (EVA7066.1 to -16; 16 peptides), a pool of 15-mer peptides of SIVmac251/32H-Rev (EVA7068.1 to -8; 8 peptides), and four pools of 15-mer peptides of SIVmac239 Env (NIH AIDS Research & Reference Reagent Program, catalog no. 62049) comprising 55 or 56 peptides each (overall, 218 Env peptides). As an SIV-unrelated control stimulus, a pool of six 20-mer peptides derived from the gHCV NS3 gene (amino acids 1138 to 1157, 1198 to 1217, 1208 to 1227, 1458 to 1477, 1528 to 1547, and 1538 to 1557) was applied.
To measure humoral SIV-specific immune responses, a standard enzyme-linked immunosorbent assay for the detection of antibodies against the SIV polypeptides gp130 SU and p27 CA (36a) in a limiting-dilution format and a yield reduction assay for the determination of neutralizing antibodies (24) were employed. Recombinant SIVgp130 (EVA670, NIBSC) and SIVp27 (EVA643) for enzyme-linked immunosorbent assay were kindly provided by Programme EVA.
Adenovirus-neutralizing antibodies known to be induced in vaccination regimes employing adenoviral vectors were determined in a green fluorescent focus reduction assay as previously described (14) with slight modifications. Briefly, inactivated monkey sera were analyzed in serial fourfold dilutions starting at a 1:8 dilution. A 120-μl portion of diluted serum and 100 μl of a replication-deficient Ad5 vector expressing EGFP containing 2 × 105 PFU were incubated in triplicates at 4°C for 2 h. Then, 150 μl of this mixture was transferred onto 96-well flat-bottom plates which had been preseeded with 293 cells, and the plates were incubated at 37°C with 5% CO2 for 2 days. Thereafter the plates were examined under a fluorescence microscope. Sample dilutions exhibiting >50% reduction of green fluorescent foci compared to infected controls incubated with autologous monkey sera obtained before immunization were considered positive for Ad5 neutralizing antibodies. Viral RNA copy numbers in plasma were determined by real-time PCR as described previously (38).
Statistical analyses.
The mean number of spots per 106 peripheral blood mononuclear cells was determined from triplicate cultures for each peptide pool, and the mean number of spots in control cultures with an irrelevant hepatitis C virus peptide pool or lacking a peptide was subtracted. If data from several time points during the same treatment period (e.g., prior to immunization or after the first, second, or third immunization) were available, the mean ELISPOTs were calculated for this period for each peptide pool and each animal. The mean and standard deviation from each treatment period were determined for all animals and each peptide pool within the same group. A one-way analysis of variance (ANOVA) followed by pairwise multiple comparison using the Tukey test was performed for each peptide pool to test whether there is a significant difference in the ELISPOTs prior to immunization and after each immunization. Significantly enhanced ELISPOT responses during immunization within a treatment group were compared to the control group using the two-sided t test. The t test (two sided) was also used to analyze differences in the peak viral load, early set point RNA levels (mean from weeks 16, 20, and 24 for each animal), and late set point RNA levels (means from weeks 28 to 48). The Mann-Whitney rank sum test was used to evaluate differences in the median values of the log2 antibody titers.
RESULTS
Adenoviral vector immunization by the tonsillar route.
Given the tropism of adenoviruses of subgenus C for the upper respiratory tract and their persistence in submucosal lymphatic tissues, we first analyzed whether a replication-deficient adenoviral vector would allow expression of vaccine antigens in close proximity to the oral MALT. A replication-deficient adenoviral vector expressing GFP was therefore sprayed directly onto the tonsils of four rhesus monkeys. The adenoviral vector was administered at a concentration of 2 × 1011 particles/ml in total volume of 0.5 ml using a spray pump system (Pfeiffer, Radolfzell, Germany). Since ketamine anesthesia induced hypersalivation, monkeys were treated with glycopyrroniumbromide 10 to 15 min prior to the oral spray application, in order to enhance access of the vector preparations to the lymphoepithelium of the tonsils. Two days later, immunohistological staining revealed GFP-positive cells with epithelial cell-like morphology at the basal cell layer of the squamous epithelium of the tonsils in three of four animals. The close proximity to cells with lymphoid morphology (Fig. 1A and B) suggests that antigens could be delivered atraumatically to the induction site of the oral MALT by adenoviral vector immunization through the tonsillar route.
FIG. 1.

Detection of replication-deficient viral vectors in the lymphoid tissue. (A and B) GFP expression after tonsillar administration of an adenoviral vector (Ad-GFP) was detected by immunohistochemistry (red) in the crypt (A) and squamous epithelium (B) of the tonsil. The arrow (B) points to a large, nonlymphoid cell showing positivity for GFP in the lymphoid tissue. (C) In a control macaque, tonsils were not stained with the antibody to GFP. (D) As a positive control, 293T cells transfected with a GFP expression plasmid were also stained. (E to H) SCIV-producing cells in axillary lymph nodes 4 days after intravenous inoculation (E) or tonsillar application (F to H) of the vaccine. Cells expressing SIV RNA (greenish blue color with the combined reflected light and transillumination) are in the T-dependent zone (E), the germinal center (GC), and the efferent lymphatic vessel (G and H). This vessel harboring the SCIV-positive cell (G, arrow) is shown with higher magnification (H). The morphology of the positive cell (H, arrow) demonstrates that it is a lymphocyte (transillumination only; the silver grains are black). The number of hybridization signals appears low since the silver grains of the signal are located in a higher focal plane than the cells of the tissue section. Original magnifications: A, C, and E to G, ×100; B and H, ×160; D, ×50.
To explore whether oral immunization with replication-deficient vectors would indeed elicit immune responses, 10 rhesus monkeys received two doses of an adenoviral vector vaccine at weeks 0 and 4 by oral spray immunization (Fig. 2A). The SIV adenoviral vector vaccine consisted of two adenoviral vectors expressing gag-pol and env, respectively. Four weeks after the first immunization, low antibody titers against Env and Gag were detected in a minority of immunized monkeys (Fig. 2B). After the second immunization, significantly higher antibody titers to p27CA (Mann-Whitney rank sum test, P = 0.009) and gp130 (P < 0.001) than those prior to immunization were observed. Four of the 10 animals received a third oral immunization with the same adenoviral vector vaccine at week 16. After the third immunization, Env antibody titers increased significantly (P = 0.023), while the increase in the Gag antibody titers did not reach statistical significance.
FIG. 2.

Adenoviral vector immunization by the tonsillar route. (A) Experimental outline. The scale indicates the number of weeks after the first immunization. The type of vaccine or challenge virus is given above the time line. Four of the 10 monkeys of the Ad-oral group received a third oral immunization at week 16. (B) Mean titer and standard deviation of antibodies binding p27 CA or gp130 SU in 10 (weeks 0 to 12) and 4 (weeks 16 to 48) macaques of the Ad-oral group. The vertical dotted line in panel B indicates the time of challenge. (C) ELISPOT responses to the indicated peptide pools in the Ad-oral group were determined 4 and 0 weeks prior to immunization (Pre), and 2 and 4 weeks after the first, second, and third oral immunization with the adenoviral vector. ELISPOTs of unstimulated control cultures from each time point were subtracted, and the means for all available time points were used as single Pre, first, second, and third values for each animal. The means and standard deviations of the Pre, first, second, and third values for all animals after stimulation with the indicated peptide pools are shown. To determine whether there is a statistically significant difference between the means of the Pre, first, and second, ELISPOT values for each peptide, one-way ANOVA was used, followed by pairwise multiple comparison using the Tukey test. Numbers above the horizontal bars give the respective P values if a significant difference between two time points was obtained by ANOVA. Columns marked with an arrow indicate ELISPOT responses that are significantly higher (P < 0.05, t test) than the mock ELISPOT response to the respective peptide pool of the Ad-control group (D). For the 4 of the 10 animals that received a third oral immunization, a paired t test (#) was used to determine significant increases in ELISPOT responses between the second and third immunizations. (D) IFN-γ ELISPOT responses in six control animals were determined weeks −4, 0 (Pre), and 8 (mock) as described in for panel C. The t test was used to determine statistically significant differences between the Pre and mock ELISPOT responses for each peptide. *, due to failure of the equal variance test, ANOVA on ranks followed by pairwise comparison using Dunn's method was performed. (E) RNA load after challenge with SIVmac239. The mean and standard deviation of the viral RNA loads in the four animals receiving three oral adenoviral vector immunizations (Ad-oral) and in eight control monkeys infected in parallel are shown.
Different overlapping peptide pools spanning Gag and Env of SIV were used in an IFN-γ ELISPOT assay to monitor cellular immune responses after immunization (Fig. 2C). Since Rev is not encoded by the vaccines used, a Rev peptide pool was also included as a negative control. To avoid arbitrary definitions of thresholds for ELISPOT positivity, which could bias statistical analyses, the ELISPOT values of nonstimulated control cultures of each monkey at each time point were subtracted from the values obtained after stimulation with different overlapping peptide pools. In nonimmunized animals and in animals prior to immunization, this led to low positive or negative values, depending on the peptide pools added, suggesting either weak unspecific stimulatory and inhibitory effects or random variation. For further statistical analyses, the mean of the ELISPOT response measured prior to immunization (weeks −4 and 0) was compared to the mean of the ELISPOT response at weeks 2 and 4 and at weeks 6 and 8 in order to determine the effect of the first and second adenoviral vector immunizations, respectively. Since the dynamics of the immune responses in individual animals were rather heterogeneous, with different animals showing the strongest ELISPOT responses at different time points after immunization, the average of the results from both time points analyzed after each immunization is given. Cellular immune responses could already be detected by the IFN-γ ELISPOT assay after a single oral adenoviral vector immunization, but in contrast to the antibody response, the second immunization did not boost the IFN-γ ELISPOT responses (Fig. 2C). In the four animals that received a third oral immunization, a booster effect on the IFN-γ ELISPOT response (mean of responses at weeks 14 and 16) was observed for the Gag peptide pool.
In the IFN-γ ELISPOT assay, seven different peptide pools deduced from SIV genes and consisting of 264 single peptides were used. To exclude the possibility that the increase in IFN-γ ELISPOTs is due to stimulation of T cells originally raised against non-SIV antigens by some of the peptides used, nonimmunized control animals were also analyzed in the IFN-γ ELISPOT assay. With the exception of the Env3 peptide pool, no significant differences between the preimmunization values and the values after mock immunization were detected in the control group (Fig. 2D). Importantly, the IFN-γ ELISPOT response to the Gag, p27, and Env2 peptide pool after the first, second, and/or third immunization was significantly higher in the orally immunized than in the nonimmunized group (Fig. 2C). Thus, oral spray immunization with adenoviral vectors induces cellular and humoral immune responses to the encoded vaccine antigen.
Challenge of monkeys immunized with adenoviral vectors by the oral route.
The four monkeys receiving three oral immunizations with the adenoviral vector and eight control monkeys were subsequently challenged orally with the SIVmac239 virus, a neutralization-resistant molecular clone homologous to the vaccine antigens. An oral challenge was performed, since this provides a reliable, truly atraumatic, mucosal infection route (36). All animals became infected, and peak viremia and set point RNA levels did not differ significantly between these two groups (Fig. 2E). Thus, the immune responses induced by oral immunization with the adenoviral vector did not protect against subsequent SIV challenge.
Oral prime-boost regimen.
To improve efficacy of the tonsillar immunization against SIV, we investigated whether a heterologous prime-boost regimen consisting of a SCIV prime and adenoviral vector boost would be beneficial. In addition, we compared oral spray with systemic vaccine application side by side. One group of four rhesus monkeys (systemic prime-boost group) was immunized by priming with SCIV at week 0 by the intravenous route and intramuscular boosting with the adenoviral vectors carrying gag-pol, env, and rev at week 8 (Fig. 3). A second group of four rhesus monkeys (oral prime-boost group) was immunized exclusively by spraying the vaccine preparations of SCIV and the adenoviral vectors directly onto the tonsils of the monkeys (Fig. 3). For the oral vaccination, each vaccine was given twice to compensate for the lower dose administered due to volume restrictions. To control for unspecific immunostimulatory effects of the adenoviral vector vaccines prior to challenge, two control monkeys each received an adenoviral vector encoding GFP by either the oral route (weeks 8 and 12) or the intramuscular route(week 8). All groups were challenged orally at week 20 with SIVmac239.
FIG. 3.
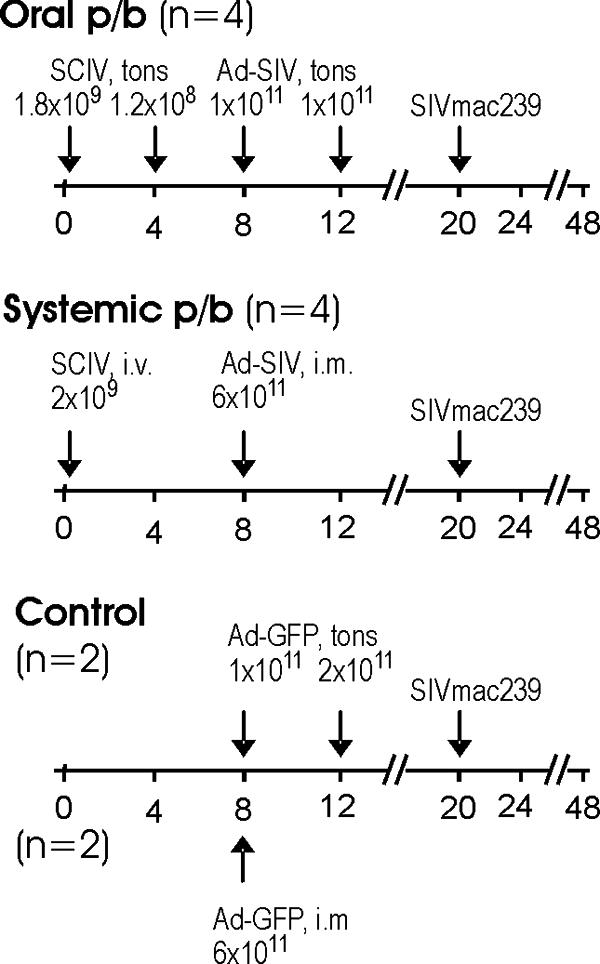
Prime-boost (p/b) immunization with viral vector vaccines. The different treatment groups and number of animals per group are given. The scale indicates the number of weeks after the first immunization. The type of vaccine or challenge virus, route of immunization, and dosages are given above the time line. tons, tonsillar; i.v., intravenous; i.m., intramuscular.
Systemic spread of SCIV-transduced cells after oral immunization.
We previously observed systemic infection after oral exposure to replication-competent SIV (36), indicating that the tonsils provide a port of entry for SIV particles. To explore whether systemic spread would also occur after oral immunization with VSV G-pseudotyped replication-deficient SCIVs, axillary lymph nodes were removed 4 days after immunization and a highly sensitive in situ hybridization technique was used to analyze expression of SCIV. A low number of SIV RNA-positive cells (1.87 RNA-positive cells per section) could be detected in the lymph nodes of three of the four immunized animals (Fig. 1E to H). The low number of positive cells did not allow further characterization of the vaccine-expressing cells by immunohistochemistry. However, cell morphology indicated that these cells were lymphocytes. The positive cells were present in the T-cell-dependent zone, the germinal centers and the sinuses. Importantly, we were able to detect such cells in the efferent lymphatics. These findings indicate that the vaccine reached all immunologically important regions of the node.
Humoral immune responses.
Priming with SCIV by the oral or intravenous route resulted in detectable antibodies to Gag in only one animal from each group, while Env antibodies were detectable at low levels in two animals after systemic immunization (Fig. 4). After the two oral boosts with the adenoviral vector, an increase in Gag antibody titers was observed in all four animals, and three of the four animals also developed Env antibodies. After boosting the systemic vaccination group with the adenoviral vector, a sharp increase in antibody titers was observed, which exceeded those seen after tonsillar immunization (Fig. 4). The anti-Env antibodies induced in the systemic vaccination group were able to neutralize the SIVmac251 strain, which had been used in the neutralization assay due to the neutralization-resistant phenotype of the SIVmac239 virus, while neutralizing antibodies in the oral prime-boost group remained undetectable (data not shown).
FIG. 4.

Antibody titers to SIV p27 CA (left panels) and gp130 SU (right panels) after oral or systemic immunization and in control monkeys. The five-digit numbers are monkey designations, and arrows mark the time points of immunization. The vertical dotted lines indicate the time of challenge. p/b, prime-boost.
Cellular immune responses.
Cellular immune responses after immunization were monitored using the IFN-γ ELISPOT assay (Fig. 5). For statistical analyses, the mean of the ELISPOT response measured prior to immunization (weeks −4 and 0) for each group was compared to the mean of the ELISPOT response measured after priming (weeks 4 and 8) and boosting (weeks 10 and 12). After oral priming with SCIV, the ELISPOT response to the various peptide pools did not exceed the background response seen prior to immunization. However, after the first boost with the adenoviral vectors by the oral route, a significant increase in the IFN-γ ELISPOT responses after stimulation with the p27 and Env3 peptide pools was observed (Fig. 5A). Although considerably higher mean ELISPOT numbers after boosting were also seen by stimulation with the other peptide pools, this increase did not reach statistical significance. The systemic prime-boost regimen induced a broad IFN-γ ELISPOT response to Gag, Rev, and various Env peptide pools (Fig. 5B). Side by side with the vaccinated animals, ELISPOT responses were also measured for the control monkeys (Fig. 5C). With one exception, there were no significant differences between the boost values obtained for the control group and its prevaccination values, excluding the possibility that a systematic error due to variations in culture conditions is responsible for the positive ELISPOT responses seen after boosting in the vaccinated animals. A significant change in the ELISPOT response of the control group was observed only for the Env3 peptide pool after priming, suggesting fluctuation of T cells cross-reacting with a peptide of the Env3 peptide pool. Nevertheless, the increase in the Env3 IFN-γ ELISPOTs in the immunized groups seems to be vaccine induced, since the number of ELISPOTs after boosting was significantly lower in the control group than in the immunized groups. The ELISPOT response to the p27CA peptide pool after the oral booster immunization was also significantly higher than the ELISPOT response measured in parallel for the control group. Thus, the ELISPOT response to these peptide pools after the oral prime-boost immunization is significantly higher than the preimmunization values of the same group and the ELISPOT responses measured in parallel in the nonimmunized control group. Using the same criteria for the systemic SCIV prime-adenoviral vector boost immunization, a broader reactivity was observed (Fig. 5B), with only two peptide pools not showing a statistically significant ELISPOT response.
FIG. 5.

IFN-γ ELISPOT response after prime-boost immunization by the oral (A) or systemic (B) route. ELISPOT responses to the indicated peptide pools were determined 1 and 3 weeks prior to immunization (Pre), at weeks 4 and 8 (prime), and at weeks 10 and 12 (boost). ELISPOT responses are presented as described in the legend to Fig. 2. Numbers above the horizontal bars give the respective P values if a significant difference between two time points was obtained by ANOVA. Columns marked with an arrow indicate that the ELISPOT response is significantly higher (P < 0.05, t test) than the ELISPOT response to the respective peptide pool of the control group at the same time point (C). *, due to failure of the normality test, ANOVA on ranks followed by pairwise comparison using Dunn's method was performed. PBMC, peripheral blood mononuclear cell.
Viral load after challenge.
To investigate the efficacy of the oral prime-boost vaccination approach, monkeys were challenged with SIVmac239 by the tonsillar route, thus providing a mucosal challenge with a stringent form of SIV. All animals became infected (Fig. 6), but peak viral RNA loads (week 2), early-set-point RNA levels (mean viral RNA load at weeks 12 to 24), and late-set-point RNA levels (mean viral RNA load at weeks 28 to 48) were 83-, 35-, and 43-fold lower, respectively, in orally immunized monkeys than in the control animals. The differences in peak viral load between the two groups were statistically significant (P < 0.05). Of note, no differences in viral load levels were observed between orally and systemically immunized monkeys.
FIG. 6.

RNA load after challenge. (A) The means and standard deviations for the three groups are shown. (B to D) The viral RNA loads in monkeys immunized orally (B) or systemically (C) and in the nonimmunized control group (D) are shown for each of the macaques. The five-digit numbers are monkey designations. p/b, prime-boost.
Neutralizing antibodies against adenovirus.
Antibody titers to SIV after oral immunization with the adenoviral vector vaccine increased after the second administration (Fig. 2). This implies that oral immunization with adenoviral vectors does not prevent expression of the vaccine antigen after a second dose of the same adenoviral vector. Since it has been previously observed that intramuscular immunization with adenoviral vector vaccines limited the efficacy of booster immunization with homologous adenoviral vectors, we compared induction of neutralizing antibodies to the adenoviral vector after oral and intramuscular (systemic) immunization (Fig. 7). A single intramuscular injection of 6 × 1011 adenoviral vector particles induced antibody titers exceeding 1/100 in all monkeys (Fig. 7B). In contrast, a single oral immunization with 1 × 1011 or 2 × 1011 particles did not induces neutralizing antibodies, although high-titer neutralizing antibodies were detected after the oral booster immunization with the adenoviral vector (Fig. 7A and B). However, the neutralizing antibody titers declined rapidly. A third oral immunization with the adenoviral vectors 12 weeks after the second clearly boosted humoral immune responses to SIV antigens, indicating that neutralizing antibodies to the adenoviral vector did not prevent expression of the vaccine antigen after repeated oral administration.
FIG. 7.

Neutralizing antibodies to the adenoviral vector. (A) Neutralizing antibody titers to the adenoviral vector before the first (Pre) and third (pre 3rd) and 2 weeks after the first, second, and third adenoviral vector applications (dose, 2 × 1011 particles) by the tonsillar route (for the immunization regimen, see Fig. 2A). (B) Neutralizing antibody titers to the adenoviral vector were determined before (Pre) and 4 weeks after the first (prime) and second (boost) adenoviral vector applications by the tonsillar (oral; dose, 1 × 1011) or intramuscular (systemic; dose, 6 × 1011) route. The two control macaques receiving Ad-GFP either orally of systemically (for the immunization regimen, see Fig. 3A) were included in the analyses. The means and standard deviations for each group are shown.
DISCUSSION
Simple spraying of replication-deficient adenoviral vectors onto the tonsils either alone or after priming with SCIV induced cellular and humoral immune responses to the encoded vaccine antigens, indicating that the viral vectors can overcome the epithelial barrier, deliver the vaccine antigen to the inductive sites of the MALT, and avoid the induction of tolerance frequently observed after oral delivery of protein antigens. Viruses have evolved mechanisms to transit tight cell layers by a cellular transcytosis pathway without replicating in the cells they pass (1, 5, 22, 31, 34). Since even enveloped viruses are shuttled in this way through the cell and secreted at the basolateral site in an infectious form, transduction of basal cell layers by viral vector vaccines seems possible. In addition, the lymphoepithelium in the depth of the tonsillar crypts with its interspersed M cells should facilitate uptake of antigens. Following oral SCIV application, we could detect SIV RNA-positive cells in nondraining lymph nodes. The localization of the SIV RNA-positive cells in the sinuses of the lymph nodes and the efferent lymphatics suggests that SCIV-infected cells carry the viral vector vaccines through out the lymphatic tissues. Although we cannot formally exclude replication of SCIV in vivo, we consider this to be highly unlikely since (i) the primer complementation approach used to generate the SCIVs reduces infectivity in single-round replication assays at least 104-fold (17, 18); (ii) the SCIVs have an additional deletion of vif, which severely impairs replication in primary cells and rhesus macaques (9, 32), and (iii) we were never able to recover replication-competent virus from SCIV-injected animals despite repeated attempts (23).
For the adenoviral vector vaccine, we provide evidence of transduction of cells with epithelial cell-like morphology at the basal layer of the squamous epithelium. The close proximity of the transduced cells to lymphoid cells suggests that the tonsillar spray immunization indeed leads to atraumatic delivery of vaccine antigen to the oral MALT. The detection of adenoviral vector-transduced cells underneath intact superficial layers of the squamous epithelium provides the first in vivo evidence for transcytosis of adenoviral particles through these superficial layers.
Another atraumatic route used for immunization has been nasal application. Although the nasal MALT might be well suited for induction of immune responses, the anatomic link between nose and brain via the olfactory nerve suggests a note of caution particularly for the use of viral vector vaccines, since retrograde axonal transport of viral capsids has been well documented (15, 26).
Oral spray immunization with the adenoviral vectors alone also revealed induction of humoral and cellular immune responses. While the oral booster immunization with the adenoviral vectors 4 weeks after priming enhanced humoral immune responses, ELISPOT responses were not increased by this early boost. Since a third oral booster immunization with the adenoviral vector seemed to stimulate humoral and cellular immune responses, a longer time period between immunizations might be beneficial. However, the number of animals responding to the oral adenoviral vector immunization alone was lower than that after the prime-boost regimen. This suggests that the SCIV given orally primed for immune responses, although anti-SIV responses were mostly not detectable until after the adenoviral vector booster immunization. Since SIV-specific immune responses induced by adenoviral vector vaccines might inhibit the infectivity of the SCIV, the SCIVs, and not the adenoviral vectors, were used for priming.
Preexisting immunity to the vector could limit the use of adenoviral vectors in humans (40). In addition, neutralizing antibodies induced by priming with adenoviral vectors reduce the efficacy of booster immunizations with the same adenoviral vector serotype (3). We therefore compared induction of neutralizing antibodies to the adenoviral vector after oral and intramuscular immunizations. While a single intramuscular injection of the adenoviral vector vaccine induced neutralizing antibodies, substantial neutralizing antibodies to the adenoviral vector were observed only after two oral immunizations. Consistently, a strong booster effect of the second oral adenoviral vector immunization was evident, as humoral immune responses to the vector-encoded vaccine antigens were observed only after the second oral adenoviral vector immunization. Even a third oral adenoviral vector immunization given 12 weeks after the second boosted immune responses to the vaccine antigens in four out of four monkeys, suggesting that repeated administration of the same adenoviral vector is feasible for the oral immunization route. A rapid decline in neutralizing antibody titers to the adenoviral vector might explain the booster effects observed after the third oral adenoviral vector immunization. In addition to extended waiting periods between booster immunizations, recent advances with chimeric adenoviruses and/or the use of vectors based on adenovirus serotypes with low prevalence should also be able to overcome potential problems of oral adenoviral vector immunization with preexisting immunity in humans (reviewed in reference 2).
In addition to the immunogenicity of the oral SCIV prime-adenoviral boost immunization, we also observed substantial suppression of viral load after challenge with SIVmac239 in the absence of a sterilizing immunity. This indicates that oral immunization induced systemic immune responses inhibiting SIV replication. The mechanisms mediating this reduction in viral load remain to be defined. Neutralizing antibodies to the SIVmac239 challenge virus could not be detected, and there was no obvious inverse correlation between the magnitude of the IFN-γ ELISPOT responses and the viral load after challenge. A more detailed characterization of polyfunctional CD4 and CD8 T-cell responses (4, 19) might provide better correlates of protection. Whether the oral prime-boost immunization is indeed more efficient than oral immunization with adenoviral vectors alone cannot be concluded with certainty. While the adenoviral vector vaccine of the first study expressed a membrane-bound form of SIV Env, the adenoviral vector vaccine of the second study encoded Rev and a secreted form of SIV Env. Furthermore, rhesus monkeys of Indian origin were used in the first study, which are more susceptible to fatal consequences of SIV infection than the rhesus monkeys of Chinese origin used in the second study (28). Nevertheless, given the difficulties in inducing protective immune responses, particularly against the neutralization-resistant SIVmac239 virus, a 35- to 83-fold reduction in peak and set-point RNA levels after oral immunization is a striking observation, even in rhesus monkeys of Chinese origin.
Interestingly, subtopical delivery of modified vaccinia virus Ankara vaccines into the palatine tonsils of rhesus macaques via a needle-free injection device also induced substantial immune responses to the vector-encoded antigens and suppression of viral load after SHIV89.6P challenge (11). Thus, further studies on the efficacy of oral vaccination with viral vector vaccines against systemic and particularly respiratory tract infections seem warranted.
Acknowledgments
The recombinant SIV p27 CA was donated by I. Jones and the recombinant SIV gp130 SU by J. Raina (ImmunoDiagnostics). Both these proteins and the SIV Gag, SIV p27, and SIV Rev peptide pools were provided by the Centralised Facility for AIDS Reagents, NIBSC, United Kingdom, under the support of EU program EVA/MRC (contract QLKZ-CT-1999-00609) and the United Kingdom Medical Research Council. The SIVmac239 Env peptides were obtained through the NIH AIDS Research and Reference Reagent Program, Division of AIDS, NIAID, NIH.
This work was supported by grants from the EU (QLK2-CT-2002-00882, LSHP-CT-2004-012116, and LSHP-CT-2005-018685).
Footnotes
Published ahead of print on 26 September 2007.
REFERENCES
- 1.Banks, W. A., E. O. Freed, K. M. Wolf, S. M. Robinson, M. Franko, and V. B. Kumar. 2001. Transport of human immunodeficiency virus type 1 pseudoviruses across the blood-brain barrier: role of envelope proteins and adsorptive endocytosis. J. Virol. 75:4681-4691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Barouch, D. H. 2006. Rational design of gene-based vaccines. J. Pathol. 208:283-289. [DOI] [PubMed] [Google Scholar]
- 3.Barouch, D. H., M. G. Pau, J. H. Custers, W. Koudstaal, S. Kostense, M. J. Havenga, D. M. Truitt, S. M. Sumida, M. G. Kishko, J. C. Arthur, B. Korioth-Schmitz, M. H. Newberg, D. A. Gorgone, M. A. Lifton, D. L. Panicali, G. J. Nabel, N. L. Letvin, and J. Goudsmit. 2004. Immunogenicity of recombinant adenovirus serotype 35 vaccine in the presence of pre-existing anti-Ad5 immunity. J. Immunol. 172:6290-6297. [DOI] [PubMed] [Google Scholar]
- 4.Betts, M. R., M. C. Nason, S. M. West, S. C. De Rosa, S. A. Migueles, J. Abraham, M. M. Lederman, J. M. Benito, P. A. Goepfert, M. Connors, M. Roederer, and R. A. Koup. 2006. HIV nonprogressors preferentially maintain highly functional HIV-specific CD8+ T cells. Blood 107:4781-4789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bomsel, M. 1997. Transcytosis of infectious human immunodeficiency virus across a tight human epithelial cell line barrier. Nat. Med. 3:42-47. [DOI] [PubMed] [Google Scholar]
- 6.Burns, J. C., T. Friedmann, W. Driever, M. Burrascano, and J. K. Yee. 1993. Vesicular stomatitis virus G glycoprotein pseudotyped retroviral vectors: concentration to very high titer and efficient gene transfer into mammalian and nonmammalian cells. Proc. Natl. Acad. Sci. USA 90:8033-8037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Catanzaro, A. T., R. A. Koup, M. Roederer, R. T. Bailer, M. E. Enama, Z. Moodie, L. Gu, J. E. Martin, L. Novik, B. K. Chakrabarti, B. T. Butman, J. G. Gall, C. R. King, C. A. Andrews, R. Sheets, P. L. Gomez, J. R. Mascola, G. J. Nabel, and B. S. Graham. 2006. Phase 1 safety and immunogenicity evaluation of a multiclade HIV-1 candidate vaccine delivered by a replication-defective recombinant adenovirus vector. J. Infect. Dis. 194:1638-1649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chackerian, B., N. L. Haigwood, and J. Overbaugh. 1995. Characterization of a CD4-expressing macaque cell line that can detect virus after a single replication cycle and can be infected by diverse simian immunodeficiency virus isolates. Virology 213:386-394. [DOI] [PubMed] [Google Scholar]
- 9.Desrosiers, R. C., J. D. Lifson, J. S. Gibbs, S. C. Czajak, A. Y. Howe, L. O. Arthur, and R. P. Johnson. 1998. Identification of highly attenuated mutants of simian immunodeficiency virus. J. Virol. 72:1431-1437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.DuBridge, R. B., P. Tang, H. C. Hsia, P. M. Leong, J. H. Miller, and M. P. Calos. 1987. Analysis of mutation in human cells by using an Epstein-Barr virus shuttle system. Mol. Cell. Biol. 7:379-387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Earl, P. L., J. L. Americo, L. S. Wyatt, E. L. Anne, D. C. Montefiori, R. Byrum, M. Piatak, J. D. Lifson, A. R. Rao, H. L. Robinson, J. W. Huggins, and B. Moss. 2007. Recombinant modified vaccinia virus Ankara provides durable protection against disease caused by an immunodeficiency virus as well as long-term immunity to an orthopoxvirus in a non-human primate. Virology 366:84-97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Evans, D. T., J. E. Bricker, and R. C. Desrosiers. 2004. A novel approach for producing lentiviruses that are limited to a single cycle of infection. J. Virol. 78:11715-11725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Evans, D. T., J. E. Bricker, H. B. Sanford, S. Lang, A. Carville, B. A. Richardson, M. Piatak, Jr., J. D. Lifson, K. G. Mansfield, and R. C. Desrosiers. 2005. Immunization of macaques with single-cycle simian immunodeficiency virus (SIV) stimulates diverse virus-specific immune responses and reduces viral loads after challenge with SIVmac239. J. Virol. 79:7707-7720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Farina, S. F., G. P. Gao, Z. Q. Xiang, J. J. Rux, R. M. Burnett, M. R. Alvira, J. Marsh, H. C. Ertl, and J. M. Wilson. 2001. Replication-defective vector based on a chimpanzee adenovirus. J. Virol. 75:11603-11613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ghadge, G. D., R. P. Roos, U. J. Kang, R. Wollmann, P. S. Fishman, A. M. Kalynych, E. Barr, and J. M. Leiden. 1995. CNS gene delivery by retrograde transport of recombinant replication-defective adenoviruses. Gene Ther. 2:132-137. [PubMed] [Google Scholar]
- 16.Granelli-Piperno, A., L. Zhong, P. Haslett, J. Jacobson, and R. M. Steinman. 2000. Dendritic cells, infected with vesicular stomatitis virus-pseudotyped HIV-1, present viral antigens to CD4+ and CD8+ T cells from HIV-1-infected individuals. J. Immunol. 165:6620-6626. [DOI] [PubMed] [Google Scholar]
- 17.Grunwald, T., F. S. Pedersen, R. Wagner, and K. Überla. 2004. Reducing mobilization of simian immunodeficiency virus based vectors by primer complementation. J. Gene Med. 6:147-154. [DOI] [PubMed] [Google Scholar]
- 18.Hansen, A. C., T. Grunwald, A. H. Lund, A. Schmitz, M. Duch, K. Überla, and F. S. Pedersen. 2001. Transfer of primer binding site-mutated simian immunodeficiency virus vectors by genetically engineered artificial and hybrid tRNA-like primers. J. Virol. 75:4922-4928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Harari, A., V. Dutoit, C. Cellerai, P. A. Bart, R. A. Du Pasquier, and G. Pantaleo. 2006. Functional signatures of protective antiviral T-cell immunity in human virus infections. Immunol. Rev. 211:236-254. [DOI] [PubMed] [Google Scholar]
- 20.Harrer, E., M. Bauerle, B. Ferstl, P. Chaplin, B. Petzold, L. Mateo, A. Handley, M. Tzatzaris, J. Vollmar, S. Bergmann, M. Rittmaier, K. Eismann, S. Muller, J. R. Kalden, B. Spriewald, D. Willbold, and T. Harrer. 2005. Therapeutic vaccination of HIV-1-infected patients on HAART with a recombinant HIV-1 nef-expressing MVA: safety, immunogenicity and influence on viral load during treatment interruption. Antiviral Ther. 10:285-300. [PubMed] [Google Scholar]
- 21.He, T. C., S. Zhou, L. T. da Costa, J. Yu, K. W. Kinzler, and B. Vogelstein. 1998. A simplified system for generating recombinant adenoviruses. Proc. Natl. Acad. Sci. USA 95:2509-2514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hocini, H., and M. Bomsel. 1999. Infectious human immunodeficiency virus can rapidly penetrate a tight human epithelial barrier by transcytosis in a process impaired by mucosal immunoglobulins. J. Infect. Dis. 179(Suppl. 3):S448-S453. [DOI] [PubMed] [Google Scholar]
- 23.Kuate, S., C. Stahl-Hennig, P. T. Haaft, J. Heeney, and K. Überla. 2003. Single cycle immunodeficiency viruses provide strategies for uncoupling in vivo expression levels from viral replicative capacity and for mimicking live attenuated SIV vaccines. Virology 313:653-662. [DOI] [PubMed] [Google Scholar]
- 24.Kuate, S., C. Stahl-Hennig, H. Stoiber, G. Nchinda, A. Floto, M. Franz, U. Sauermann, S. Bredl, L. Deml, R. Ignatius, S. Norley, P. Racz, K. Tenner-Racz, R. M. Steinman, R. Wagner, and K. Uberla. 2006. Immunogenicity and efficacy of immunodeficiency virus-like particles pseudotyped with the G protein of vesicular stomatitis virus. Virology 351:133-144. [DOI] [PubMed] [Google Scholar]
- 25.Kuate, S., D. Stefanou, D. Hoffmann, O. Wildner, and K. Uberla. 2004. Production of lentiviral vectors by transient expression of minimal packaging genes from recombinant adenoviruses. J. Gene Med. 6:1197-1205. [DOI] [PubMed] [Google Scholar]
- 26.Kuo, H., D. K. Ingram, R. G. Crystal, and A. Mastrangeli. 1995. Retrograde transfer of replication deficient recombinant adenovirus vector in the central nervous system for tracing studies. Brain Res. 705:31-38. [DOI] [PubMed] [Google Scholar]
- 27.Leurs, C., M. Jansen, K. E. Pollok, M. Heinkelein, M. Schmidt, M. Wissler, D. Lindemann, C. von Kalle, A. Rethwilm, D. A. Williams, and H. Hanenberg. 2003. Comparison of three retroviral vector systems for transduction of nonobese diabetic/severe combined immunodeficiency mice repopulating human CD34(+) cord blood cells. Hum. Gene Ther. 14:509-519. [DOI] [PubMed] [Google Scholar]
- 28.Ling, B., R. S. Veazey, A. Luckay, C. Penedo, K. Xu, J. D. Lifson, and P. A. Marx. 2002. SIV(mac) pathogenesis in rhesus macaques of Chinese and Indian origin compared with primary HIV infections in humans. AIDS 16:1489-1496. [DOI] [PubMed] [Google Scholar]
- 29.Mwau, M., I. Cebere, J. Sutton, P. Chikoti, N. Winstone, E. G. Wee, T. Beattie, Y. H. Chen, L. Dorrell, H. McShane, C. Schmidt, M. Brooks, S. Patel, J. Roberts, C. Conlon, S. L. Rowland-Jones, J. J. Bwayo, A. J. McMichael, and T. Hanke. 2004. A human immunodeficiency virus 1 (HIV-1) clade A vaccine in clinical trials: stimulation of HIV-specific T-cell responses by DNA and recombinant modified vaccinia virus Ankara (MVA) vaccines in humans. J. Gen. Virol. 85:911-919. [DOI] [PubMed] [Google Scholar]
- 30.Ondondo, B. O., H. Yang, T. Dong, K. di Gleria, A. Suttill, C. Conlon, D. Brown, P. Williams, S. L. Rowland-Jones, T. Hanke, A. J. McMichael, and L. Dorrell. 2006. Immunisation with recombinant modified vaccinia virus Ankara expressing HIV-1 gag in HIV-1-infected subjects stimulates broad functional CD4+ T cell responses. Eur. J. Immunol. 36:2585-2594. [DOI] [PubMed] [Google Scholar]
- 31.Ouzilou, L., E. Caliot, I. Pelletier, M. C. Prevost, E. Pringault, and F. Colbere-Garapin. 2002. Poliovirus transcytosis through M-like cells. J. Gen. Virol. 83:2177-2182. [DOI] [PubMed] [Google Scholar]
- 32.Park, I. W., K. Myrick, and J. Sodroski. 1994. Effects of vif mutations on cell-free infectivity and replication of simian immunodeficiency virus. J. Acquir. Immune Defic. Syndr. 7:1228-1236. [PubMed] [Google Scholar]
- 33.Peters, B. S., W. Jaoko, E. Vardas, G. Panayotakopoulos, P. Fast, C. Schmidt, J. Gilmour, M. Bogoshi, G. Omosa-Manyonyi, L. Dally, L. Klavinskis, B. Farah, T. Tarragona, P. A. Bart, A. Robinson, C. Pieterse, W. Stevens, R. Thomas, B. Barin, A. J. McMichael, J. A. McIntyre, G. Pantaleo, T. Hanke, and J. Bwayo. 2007. Studies of a prophylactic HIV-1 vaccine candidate based on modified vaccinia virus Ankara (MVA) with and without DNA priming: effects of dosage and route on safety and immunogenicity. Vaccine 25:2120-2127. [DOI] [PubMed] [Google Scholar]
- 34.Rokos, K., H. Wang, J. Seeger, A. Schafer, and G. Pauli. 1998. Transport of viruses through fetal membranes: an in vitro model of perinatal transmission. J. Med. Virol. 54:313-319. [DOI] [PubMed] [Google Scholar]
- 35.Stahl-Hennig, C., R. M. Steinman, P. ten Haaft, K. Uberla, N. Stolte, S. Saeland, K. Tenner-Racz, and P. Racz. 2002. The simian immunodeficiency virus ΔNef vaccine, after application to the tonsils of rhesus macaques, replicates primarily within CD4+ T cells and elicits a local perforin-positive CD8+ T-cell response. J. Virol. 76:688-696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Stahl-Hennig, C., R. M. Steinman, K. Tenner-Racz, M. Pope, N. Stolte, K. Matz-Rensing, G. Grobschupff, B. Raschdorff, G. Hunsmann, and P. Racz. 1999. Rapid infection of oral mucosal-associated lymphoid tissue with simian immunodeficiency virus. Science 285:1261-1265. [DOI] [PubMed] [Google Scholar]
- 36a.Stolte-Leeb, N., U. Sauermann, S. Norley, Z. Fagrouch, J. Heeney, M. Franz, G. Hunsmann, and Ch. Stahl-Hennig. 2006. Sustained conservation of CD4+ T-cell in multiprotein triple modality immunized rhesus macaques after intrarectal challenge with simian immunodeficiency virus. Viral Immunol. 19:448-457. [DOI] [PubMed] [Google Scholar]
- 37.Suh, Y. S., K. S. Park, U. Sauermann, M. Franz, S. Norley, D. Wilfingseder, H. Stoiber, Z. Fagrouch, J. Heeney, G. Hunsmann, C. Stahl-Hennig, and Y. C. Sung. 2006. Reduction of viral loads by multigenic DNA priming and adenovirus boosting in the SIVmac-macaque model. Vaccine 24:1811-1820. [DOI] [PubMed] [Google Scholar]
- 38.Tenner-Racz, K., H. C. Stahl, K. Uberla, H. Stoiber, R. Ignatius, J. Heeney, R. M. Steinman, and P. Racz. 2004. Early protection against pathogenic virus infection at a mucosal challenge site after vaccination with attenuated simian immunodeficiency virus. Proc. Natl. Acad. Sci. USA 101:3017-3022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Tenner-Racz, K., H. J. Stellbrink, J. van Lunzen, C. Schneider, J. P. Jacobs, B. Raschdorff, G. Grosschupff, R. M. Steinman, and P. Racz. 1998. The unenlarged lymph nodes of HIV-1-infected, asymptomatic patients with high CD4 T cell counts are sites for virus replication and CD4 T cell proliferation. The impact of highly active antiretroviral therapy. J. Exp. Med. 187:949-959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Thorner, A. R., R. Vogels, J. Kaspers, G. J. Weverling, L. Holterman, A. A. Lemckert, A. Dilraj, L. M. McNally, P. M. Jeena, S. Jepsen, P. Abbink, A. Nanda, P. E. Swanson, A. T. Bates, K. L. O'Brien, M. J. Havenga, J. Goudsmit, and D. H. Barouch. 2006. Age dependence of adenovirus-specific neutralizing antibody titers in individuals from sub-Saharan Africa. J. Clin. Microbiol. 44:3781-3783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40a.Varmus, H., R. Klausner, R. Zerhouni, T. Acharya, A. S. Daar, and P. A. Singer. 2003. Grand challenges in global health. Science 302:398-399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.West, J. T., P. B. Johnston, S. R. Dubay, and E. Hunter. 2001. Mutations within the putative membrane-spanning domain of the simian immunodeficiency virus transmembrane glycoprotein define the minimal requirements for fusion, incorporation, and infectivity. J. Virol. 75:9601-9612. [DOI] [PMC free article] [PubMed] [Google Scholar]