

Abstract
Although the interaction of matrix proteins with integrins is known to initiate signaling pathways that are essential for cell survival, a role for tumor suppressors in the regulation of these pathways has not been established. We demonstrate here that p53 can inhibit the survival function of integrins by inducing the caspase-dependent cleavage and inactivation of the serine/threonine kinase AKT/PKB. Specifically, we show that the α6β4 integrin promotes the survival of p53-deficient carcinoma cells by activating AKT/PKB. In contrast, this integrin does not activate AKT/PKB in carcinoma cells that express wild-type p53 and it actually stimulates their apoptosis, in agreement with our previous findings (Bachelder, R.E., A. Marchetti, R. Falcioni, S. Soddu, and A.M. Mercurio. 1999. J. Biol. Chem. 274:20733–20737). Interestingly, we observed reduced levels of AKT/PKB protein after antibody clustering of α6β4 in carcinoma cells that express wild-type p53. In contrast, α6β4 clustering did not reduce the level of AKT/PKB in carcinoma cells that lack functional p53. The involvement of caspase 3 in AKT/PKB regulation was indicated by the ability of Z-DEVD-FMK, a caspase 3 inhibitor, to block the α6β4-associated reduction in AKT/PKB levels in vivo, and by the ability of recombinant caspase 3 to promote the cleavage of AKT/PKB in vitro. In addition, the ability of α6β4 to activate AKT/PKB could be restored in p53 wild-type carcinoma cells by inhibiting caspase 3 activity. These studies demonstrate that the p53 tumor suppressor can inhibit integrin-associated survival signaling pathways.
Keywords: p53, integrin, AKT/PKB, survival, caspase
Primary epithelial (Frisch and Francis 1994) and endothelial (Meredith et al. 1993) cells are prone to anoikis, a form of programmed cell death, when grown in the absence of growth factors and extracellular matrix proteins. This default apoptotic pathway is thought to be important in preventing cell growth at inappropriate anatomical sites. Survival signaling pathways associated with both growth factor receptors and cell adhesion molecules are important in protecting cells from anoikis. For example, growth factors such as EGF, PDGF, and insulin can promote the survival of serum-starved epithelial cells (Merlo et al. 1995; Rampalli and Zelenka 1995; Rodeck et al. 1997). Similarly, the binding of integrins such as αvβ3 (Stromblad et al. 1996), α5β1 (Zhang et al. 1995), and α6β1 (Howlett et al. 1995; Wewer et al. 1997; Farrelly et al. 1999) to the appropriate extracellular matrix protein can inhibit anoikis. These survival signals have been attributed to the ability of integrins to activate numerous molecules including focal adhesion kinase (Frisch et al. 1996), integrin-linked kinase (Radeva et al. 1997), AKT/PKB (Khwaja et al. 1997), and bcl-2 (Zhang et al. 1995; Stromblad et al. 1996). In addition, integrin survival functions have been associated with their ability to inhibit the activity of p53 (Stromblad et al. 1996; Ilic et al. 1998) and Rb (Day et al. 1997) tumor suppressors. Tumor cells acquire a partial resistance to anoikis as a result of their transformation, which is thought to activate select survival signaling pathways in these cells constitutively (Frisch and Francis 1994). For this reason, the identification of molecules that can inhibit survival signaling is crucial for developing strategies aimed at blocking tumor cell growth.
The α6β4 integrin, a receptor for the laminin family of extracellular matrix proteins, plays an important role in diverse cellular activities. In addition to serving an important structural role in the assembly of hemidesmosomes in epithelial cells (Borradori and Sonnenberg 1996; Green and Jones 1996), α6β4 promotes carcinoma cell migration and invasion (Tozeren et al. 1994; Chao et al. 1996; Shaw et al. 1997; O'Connor et al. 1998) in a phosphoinositide 3-OH kinase–dependent manner (Shaw et al. 1997). The β4 subunit of this integrin, which contains a cytoplasmic tail of ∼1,000 amino acids (Hemler et al. 1989; Kajiji et al. 1989; Kennel et al. 1989), has been shown to be crucial in the ability of this integrin to activate numerous signaling molecules, including phosphoinositide 3-OH kinase (Shaw et al. 1997), Shc (Mainiero et al. 1997), Ras (Mainiero et al. 1997), Jnk (Mainiero et al. 1997), p21WAF1/CIP1 (Clarke et al. 1995), and p53 (Bachelder et al. 1999). The diverse activities of this integrin are exemplified by its ability to promote both the survival of keratinocytes (Dowling et al. 1996) as well as the apoptosis of a number of carcinoma cell lines (Clarke et al. 1995; Kim et al. 1997; Sun et al. 1998; Bachelder et al. 1999). These apparently contradictory functions likely reflect the activation of distinct signaling pathways by this integrin in different cell types as well as the influence of other signaling pathways on α6β4 function.
In the present study, we define opposing signaling pathways that are activated by the α6β4 integrin that promote either carcinoma cell survival or apoptosis, depending on whether these cells express wild-type or functionally inactive mutants of p53. Specifically, we show that α6β4 can promote the AKT/PKB–dependent survival of p53-deficient carcinoma cells. However, this activity contrasts with the ability of α6β4 to stimulate the caspase-dependent cleavage and inactivation of AKT/PKB in p53 wild-type carcinoma cells. The ability of wild-type p53 to inhibit α6β4-associated survival signals suggests that the p53 status of an α6β4-expressing carcinoma cell influences its growth potential.
Materials and Methods
Cells
The RKO colon carcinoma cell line was obtained from M. Brattain (University of Texas, San Antonio, TX), and MDA-MB-435 breast carcinoma cells were obtained from the Lombardi Breast Cancer Depository (Georgetown University).
The cloning of the human β4 cDNA, the construction of the β4 cytoplasmic domain deletion mutant (β4-Δcyt), and their insertions into the pRc/CMV (β4) and pcDNA3 (β4-Δcyt) eukaryotic expression vectors, respectively, have been described (Clarke et al. 1995). RKO/β4Δcyt clone 3E1, RKO/β4 clone D4 (RKO/β4 clone 1), RKO/β4 clone A7 (RKO/β4 clone 2), MDA-MB-435/β4-Δcyt clone 3C12, MDA-MB-435/β4 clone 5B3 (MDA-MB-435/β4 clone 1), and MDA-MB-435/β4 clone 3A7 (MDA/β4 clone 2) were selected for analysis based on their expression of similar surface levels of α6β4 and α6β4-Δcyt, as we have previously demonstrated (Clarke et al. 1995; Shaw et al. 1997; Bachelder et al. 1999).
Dominant negative p53-expressing RKO/β4-Δcyt and RKO/β4 subclones were obtained by cotransfecting RKO/β4-Δcyt clone 3E1 and RKO/β4 clone D4 with plasmids expressing the puromycin resistance gene (Morgenstern and Land 1990) and a dominant negative p53 (dnp53) construct (provided by M. Oren, Weizmann Institute for Science, Israel) that encodes for a carboxy-terminal domain of p53 that can heterodimerize with endogenous p53 and inhibit its transcriptional activity. Dnp53-expressing subclones were obtained and those subclones expressing high levels of dnp53 were selected by FACS using the Pab122 mAb (Boehringer Mannhein), which recognizes a conserved, denaturation stable epitope in dnp53. In addition, RKO/β4 and RKO/β4-Δcyt cells were transfected with the puromycin resistance gene plasmid alone to obtain puromycin-resistant mock transfectants. All assays were performed using cell maintained below passage 10.
Stable transfectants of MDA/β4 clone 3A7 that expressed temperature-sensitive p53 were obtained by cotransfecting this cell line with plasmids expressing the puromycin resistance gene (1 mg) (Morgenstern and Land 1990) and a plasmid expressing a temperature-sensitive mutant of human p53 (tsp53; 4 μg) that assumes a functional conformation at 32°C, but not at 37°C (Zhang et al. 1994) using the Lipofectamine reagent (GIBCO BRL). After growing these transfectants in complete medium for 2 d, stable transfectants were selected by culturing these cells in puromycin-containing medium (2 μg/ml) for an additional 18 d. These bulk transfectants were expanded and tsp53 expression was confirmed by showing increased p53 levels in tsp53 transfectants relative to mock transfectants by immunoblotting with a goat anti-human p53, followed by HRP-conjugated donkey anti–goat IgG. All assays were performed on cells maintained below passage 5.
Dominant negative AKT (dnAKT)/PKB–expressing MDA-MB-435/mock and MDA-MB-435/β4 transient transfectants were generated by cotransfecting these cell lines using the Lipofectamine reagent (GIBCO BRL) with a plasmid encoding for green fluorescent protein (pEGFP-1; CLONTECH Laboratories; 1 μg) and a dnAKT/PKB construct that contains inactivating mutations in the catalytic domain of AKT/PKB (4 μg) (Dudek et al. 1997; Skorski et al. 1997; Eves et al. 1998).
Antibodies
The following antibodies were used: 439-9B, a rat mAb specific for the β4 integrin subunit (Falcioni et al. 1998), control rat IgG (Sigma Chemical Co.); Pab122, a polyclonal rabbit serum specific for p53 (Boehringer Mannheim); goat anti-human p53; rabbit polyclonal anti-AKT/PKB raised against a peptide corresponding to mouse AKT/PKB residues 466–479 (New England Biolabs); rabbit polyclonal anti-AKT/PKB phosphoserine 473 (New England Biolabs); rabbit anti-actin (Sigma Chemical Co.); and mouse anti-hemagglutinin (Boehringer Mannheim). Goat anti–mouse IgG and goat anti–rat IgG secondary antibodies, as well as HRP conjugates of these antibodies, were obtained from Jackson ImmunoResearch Laboratories, Inc. HRP-conjugated donkey anti–goat IgG was obtained from BioSource International.
Apoptosis Assays
To induce apoptosis in the RKO and MDA-MB-435 transfectants, the cells were plated in complete medium for 8 h in tissue culture wells (12-well plate; 2.5 × 105 cells/well) that had been coated overnight at 4°C with poly-l-lysine (Sigma Chemical Co.; 2 ml of 25 μg/ml stock) and blocked with 1% BSA. After 8 h, this medium was replaced with serum-free culture medium containing 1% BSA. After 15 h at 37°C, adherent and suspension cells were harvested, combined, and the level of apoptosis in these cells was assessed as described below.
For annexin V stains, cells were washed once with serum-containing medium, once with PBS, once with annexin V-FITC buffer (10 mM Hepes/NaOH, pH 7.4, 140 mM NaCl, 2.5 mM CaCl2), and incubated for 15 min at room temperature with annexin V-FITC (Bender MedSystems) at a final concentration of 2.5 μg/ml in annexin V buffer. After washing once with annexin V buffer, the samples were resuspended in the same buffer and analyzed by flow cytometry. Immediately before analysis, propidium iodide was added to a final concentration of 5 μg/ml to distinguish apoptotic from necrotic cells, and 5,000 cells were analyzed for each sample.
For ApopTag reactions, cells were harvested as described above, fixed in 1% paraformaldehyde for 15 min on ice, and washed twice with PBS. The samples were resuspended in 1 ml ice-cold 70% ethanol and stored at −20°C overnight. After centrifugation at 2,500 rpm for 15 min, cells were washed two times in PBS before performing ApopTag reactions (Oncor) according to the manufacturer's recommendations. These samples were analyzed by flow cytometry.
For in situ analysis of apoptosis in cells transfected transiently with the green fluorescent protein (GFP)–expressing vector pEGFP-1 (CLONTECH Laboratories) and dnAKT/PKB, the transfected cells were stained with annexin V-PE (PharMingen) according to the manufacturer's directions, and plated on coverslips. The percentage of GFP-positive cells that was annexin V-PE–positive was determined by fluorescence microscopy. A total of at least 80 GFP-positive cells from at least 10 microscopic fields were analyzed for each data point.
Analysis of AKT/PKB Expression and Activity
To assess the expression of endogenous AKT/PKB protein, cells were incubated with either rat Ig or 439-9B as described above in the presence of either DMSO (1:500), a caspase 3 inhibitor (Z-DEVD-FMK; Calbiochem-Novabiochem; 4 μg/ml), or a caspase 8 inhibitor (Z-IETD-FMK; Calbiochem-Novabiochem; 4 μg/ml). After washing with PBS, the cells were plated in serum-free medium containing 1% BSA in wells of a 12-well plate that had been coated with anti–rat Ig (13.5 μg/ml) and blocked for 1 h at 37°C with 1% BSA-containing medium. After a 1-h stimulation, adherent and suspension cells were harvested and extracted with AKT/PKB lysis buffer (20 mM Tris, pH 7.4, 0.14 M NaCl, 1% NP-40, 10% glycerol, 2 mM PMSF, 5 μg/ml aprotinin, 5 μg/ml pepstatin, 50 μg/ml leupeptin, 1 mM sodium orthovanadate). After removing cellular debris by centrifugation at 12,000 g for 10 min, equivalent amounts of total cell protein from these extracts were resolved by SDS-PAGE (8%) and transferred to nitrocellulose. The blots were probed with a rabbit anti-AKT/PKB antiserum, followed by HRP-conjugated goat anti–rabbit Ig, and the immunoreactive bands were visualized by enhanced chemiluminescence. These blots were also probed with a rabbit antiserum specific for actin to confirm the loading of equivalent amounts of protein. Relative AKT/PKB and actin expression levels were assessed by densitometry using IP Lab Spectrum software (Scanalytics).
To determine the level of serine 473–phosphorylated AKT/PKB, cells were transfected transiently using the Lipofectamine reagent (GIBCO BRL) with an HA-tagged AKT/PKB cDNA (provided by A. Toker, Boston Biomedical Research Institute, Boston, MA). 20 h after transfection, these cells were harvested by trypsinization and subjected to antibody-mediated integrin clustering. Specifically, cells were incubated on ice for 30 min with either control rat IgG or 439-9B at a concentration of 10 μg/ml. After washing with PBS, the cells were plated in serum-free medium containing 1% BSA onto wells of a 60-mM tissue culture dish that had been coated at 4°C with anti–rat Ig (13.5 μg/ml) and blocked for 1 h at 37°C in 1% BSA-containing medium. After 1 h, adherent and suspension cells were harvested and washed twice with PBS. Proteins from these cells were extracted with AKT/PKB lysis buffer (see above). After removing cellular debris by centrifugation at 12,000 g for 10 min at 4°C, equivalent amounts of total cellular protein were precleared with a 1:1 mixture of protein A and protein G–Sepharose for 1 h at 4°C. Immunoprecipitations were performed for 1 h on these precleared lysates using an HA-specific mAb (1 μg; Boehringer Mannheim) and protein A/protein G–Sepharose beads. Proteins from these immunoprecipitates were subjected to reducing SDS-PAGE (8%), transferred to nitrocellulose, and probed with an AKT/PKB phosphoserine 473–specific rabbit antiserum (New England Biolabs) followed by HRP-conjugated goat anti–rabbit IgG. Phospho-AKT/PKB was detected on these blots by chemiluminescence (Pierce Chemical Co.). These samples were also probed with rabbit anti-AKT/PKB. The relative intensity of phosphoserine AKT/PKB and AKT/PKB bands was assessed by densitometry, as described above.
Analysis of AKT/PKB Proteolysis
Baculovirus-expressed AKT/PKB (0.5 μg; provided by A. Toker) was incubated with either active recombinant caspase 8 (2 mg; Calbiochem-Novabiochem) or active recombinant caspase 3 (2 μg; Calbiochem-Novabiochem) at 37°C for 1 h in a final volume of 10 μl. Subsequently, the reaction mixtures were divided into two aliquots and resolved by SDS-PAGE (8%). The gels were silver stained using the GelCode SilverSNAP Stain Kit (Pierce Chemical Co.) or transferred to nitrocellulose and probed with a rabbit AKT/PKB antiserum as described above.
Results
The α6β4 Integrin Promotes the Survival of p53-deficient, but Not p53 Wild-type Carcinoma Cells
For our initial experiments, we used stable β4 transfectants of two α6β4-deficient carcinoma cell lines that differ in their p53 status: RKO colon carcinoma cells, which express wild-type p53 (Nagasawa et al. 1995); and MDA-MB-435 breast carcinoma cells, which express a mutant, inactive form of p53 (Lesoon-Wood et al. 1995). We also used RKO and MDA-MB-435 cells that express a cytoplasmic domain deletion mutant of α6β4 (RKO/β4-Δcyt; MDA/b4-Δcyt) that is signaling deficient. The characterization of these cells has been described previously (Clarke et al. 1995; Shaw et al. 1997).
To explore the potential influence of α6β4 expression on the survival of serum-starved carcinoma cells deprived of matrix attachment, the α6β4 and α6β4-Δcyt–expressing RKO and MDA-MB-435 subclones were plated on poly-l-lysine in serum-free medium. The level of apoptosis in these populations was determined either by staining with annexin V-FITC to detect cells in the early stages of apoptosis or by performing terminal deoxynucleotidyl transferase end labeling reactions (Apoptag) to detect DNA fragmentation (Fig. 1). In addition, we assessed the viability of these serum-deprived cells by measuring the cellular uptake of propidium iodide (Table ). The ability of α6β4 to promote the survival of these cells was determined by subtracting the percent apoptotic α6β4-expressing cells from the percent apoptotic α6β4-Δcyt–expressing cells. The expression of α6β4 in MDA-MB-435 cells significantly increased the survival of these cells relative to MDA-MB-435 cells expressing α6β4-Δcyt, as assessed by annexin V-FITC staining (Fig. 1), ApopTag staining (Fig. 1), and propidium iodide uptake (Table ). In contrast, the expression of α6β4 in RKO cells did not increase the survival of these cells relative to either the mock (Table ) or RKO/β4-Δcyt transfectants (Fig. 1). In fact, we observed a higher level of apoptosis and cell death in serum-starved RKO/β4 as compared with RKO/β4-Δcyt cells, in agreement with our previous demonstration that α6β4 can promote apoptosis in wild-type p53 carcinoma cells (Bachelder et al. 1999).
Figure 1.
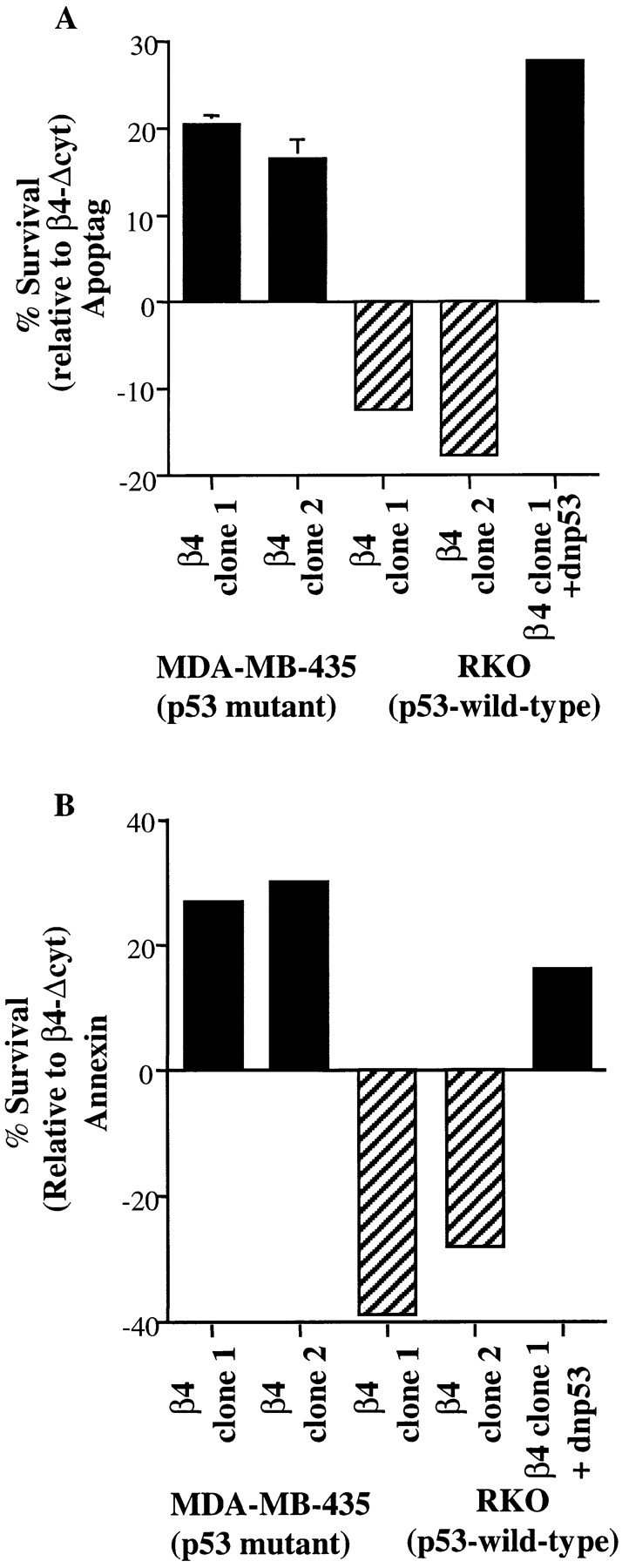
p53 inhibits α6β4-mediated survival. MDA-MB-435, RKO, and RKO + dnp53 cells that expressed either α6β4 (β4) or α6β4-Δcyt (β4-Δcyt) were plated on poly-l-lysine–coated tissue culture wells and cultured in the absence of serum. After 15 h, the cells were harvested, subjected to either ApopTag reactions (A) or annexin V-FITC staining (B), and analyzed by flow cytometry. A survival effect of α6β4 was quantified by subtracting the percentage of α6β4-expressing cells that were positive for either Apoptag (A) or annexin V-FITC (B) staining from the percentage of α6β4-Δcyt–expressing cells that were positive for these markers. This value was plotted on the bar graphs shown in A and B, with positive values indicating that the specified β4 clone exhibits increased survival relative to the relevant β4-Δcyt subclone, and negative values indicating an increased apoptosis of the indicated clone relative to the appropriate β4-Δcyt clone. The data in A represent the means (± SEM) from three independent experiments. Similar results to those shown in B were observed in three separate trials.
Table 1.
Influence of α6β4 Integrin on the Viability of RKO and MDA-MB-435 Cells
| Clone | Percent propidium iodide–positive cells |
|---|---|
| MDA/Mock | 21 |
| β4 Clone 1 | 13 |
| β4 Clone 2 | 9 |
| RKO/Mock | 32 |
| β4 Clone 1 | 49 |
| β4 Clone 2 | 47 |
Mock-transfected and β4-transfected MDA-MB-435 and RKO cells were plated on poly-l-lysine (25 μg/mL) in the absence of serum for 15 h, harvested, and incubated with propidium iodide (PI). The percentage of PI-positive cells was assessed by flow cytometry. Similar results were observed in four independent experiments.
Based on the fact that RKO and MDA-MB-435 cells differ in their p53 status, we reasoned that the ability of α6β4 to promote cell survival may be inhibited by p53. This hypothesis was examined by investigating the effect of α6β4 expression on the survival of RKO cells in which p53 activity had been inhibited by the expression of a dnp53 construct. Indeed, α6β4 expression promoted the survival of serum-starved, dnp53-expressing RKO cells as determined by ApopTag and annexin V-FITC staining (Fig. 1). These results demonstrate that p53 can suppress the survival signaling mediated by α6β4 in serum-starved carcinoma cells.
α6β4-Mediated Survival in p53-deficient Carcinoma Cells Is Inhibited by Dominant Negative AKT/PKB
Given the importance of the AKT/PKB kinase in numerous survival signaling pathways (Ahmed et al. 1997; Datta et al. 1997; Dudek et al. 1997; Songyang et al. 1997; Blume-Jensen et al. 1998; Crowder and Freeman 1998; Gerber et al. 1998), we investigated whether the survival function of α6β4 in serum-starved, p53-deficient carcinoma cells was AKT/PKB–dependent. The MDA-MB-435/β4–transfected clones, as well as the parental cells, were cotransfected with plasmids encoding for GFP and an HA-tagged, kinase-deficient AKT/PKB mutant that acts as a dominant negative construct (dnAKT/PKB) (Dudek et al. 1997; Skorski et al. 1997; Eves et al. 1998). Expression of this dnAKT/PKB construct was confirmed by immunoblotting extracts from these transfected cells with an HA-specific mAb (data not shown). After 15 h of serum starvation, the level of apoptosis in GFP-positive cells was assessed by annexin V-PE staining. As shown in Fig. 2, MDA-MB-435/β4 clones demonstrated significantly less apoptosis than parental MDA-MB-435 cells in agreement with the data shown in Table . Importantly, dnAKT/PKB expression inhibited this α6β4 survival function in each of the two MDA-MB-435/β4 clones examined, but it did not alter the level of apoptosis in parental MDA-MB-435 cells.
Figure 2.

Expression of a dominant negative AKT/PKB inhibits α6β4-mediated survival. Parental (neo) and α6β4-expressing (β4) MDA-MB-435 cells were transfected with either a GFP-expressing plasmid (mock) or both a GFP and a dnAKT/PKB–expressing construct (dnAKT/PKB), plated on poly-l-lysine, and cultured for 15 h in the absence of serum. Apoptosis in these cells was assessed by annexin V-PE staining. The data are reported as the percentage of GFP-positive cells that were stained by annexin V-PE. Similar results were observed in two additional experiments.
p53 Inhibits the Activation of AKT/PKB by α6β4
To understand the mechanism by which p53 inhibits α6β4-mediated survival, we investigated the possibility that p53 alters the ability of this integrin to activate AKT/PKB. Initially, we examined whether the antibody-mediated clustering of α6β4 in MDA-MB-435 cells resulted in the phosphorylation of AKT/PKB on serine 473, an event that has been shown to correlate with AKT/PKB activation (Alessi et al. 1996). MDA-MB-435/β4 subclones were transfected with an HA-tagged AKT/PKB construct. These cells were incubated with either a control rat IgG or the β4-specific antibody 439-9B and plated in the absence of serum on secondary antibody–coated tissue culture wells for 1 h. HA immunoprecipitations were performed on extracts from these cells, and the levels of serine-phosphorylated AKT/PKB were assessed by blotting these immunoprecipitates with an antiserum specific for AKT/PKB molecules phosphorylated on serine residue 473. As shown in Fig. 3 A, the antibody-mediated clustering of α6β4 stimulated an increase in the level of serine-phosphorylated AKT/PKB in each of the two MDA-MB-435/β4 subclones relative to control cells (2.1-fold increase, β4 clone 1; 5.5-fold increase, β4 clone 2). This α6β4-induced increase in AKT/PKB serine phosphorylation was dependent on α6β4 signaling based on the inability of α6β4-Δcyt clustering to increase the level of the serine 473–phosphorylated AKT/PKB in MDA-MB-435/β4-Δcyt subclones (data not shown).
Figure 3.
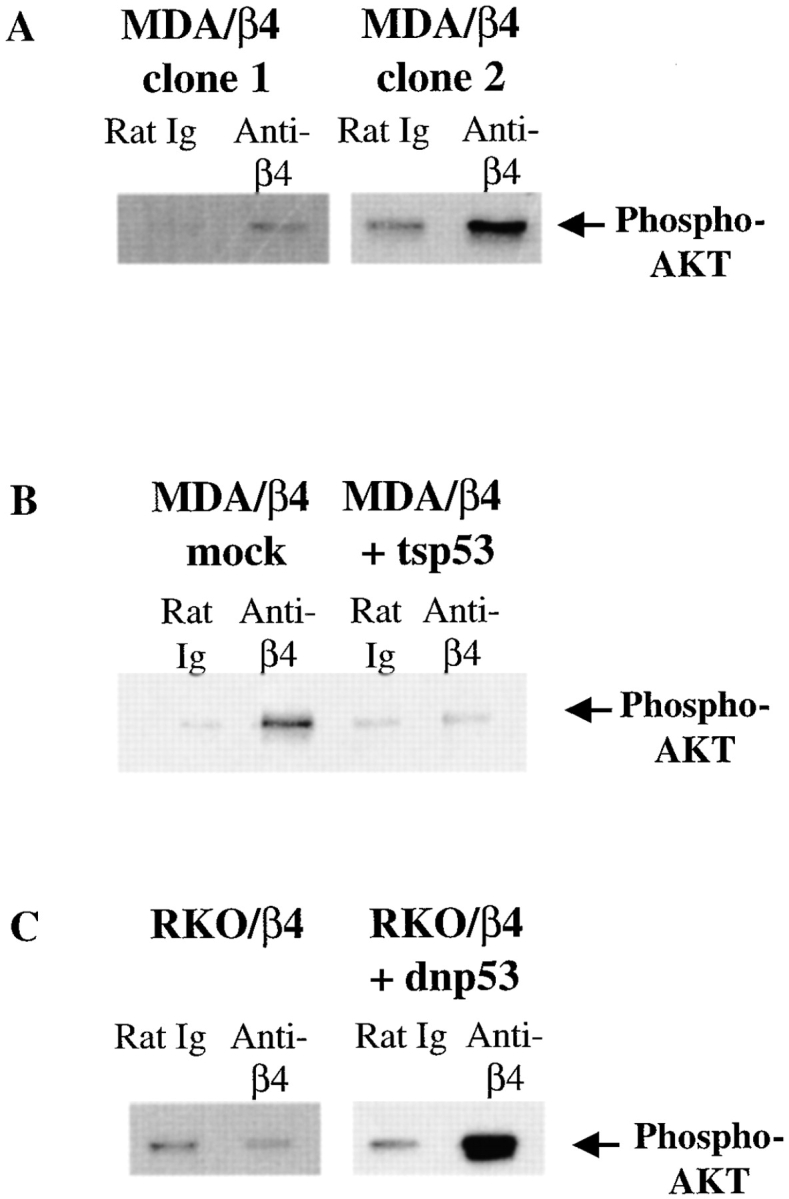
p53 inhibits the ability of α6β4 to induce AKT/PKB phosphorylation in carcinoma cells. MDA/β4, MDA/β4 + tsp53, RKO/β4, and RKO/β4 + dnp53 cells were transfected transiently with an HA-tagged AKT/PKB. These transfectants were incubated with the indicated primary antibodies, washed, and plated in the absence of serum on secondary antibody–coated tissue culture wells. HA-AKT/PKB–transfected MDA/β4 (A), RKO/β4 (C), and RKO/β4 + dnp53 (C) cells were stimulated for 1 h at 37°C. Alternatively, mock- and tsp53-transfected MDA/β4 cells (B) were stimulated for 1 h at 32°C to activate tsp53, followed by an additional hour at 37°C to activate AKT/PKB. Immunoprecipitations were performed with an HA-specific mAb on equal amounts of total extracted protein. The immunoprecipitates were resolved by SDS-PAGE (8%), transferred to nitrocellulose, and probed with a phosphoserine 473 AKT/PKB–specific rabbit antiserum (New England Biolabs), followed by HRP-conjugated goat anti–rabbit IgG. Phosphoserine-specific AKT/PKB bands were detected by chemiluminescence, and are noted by arrows.
To investigate the influence of p53 on the activation of AKT/PKB by α6β4, we explored whether α6β4 clustering induced the phosphorylation of AKT/PKB on serine residue 473 in MDA-MB-435/β4 that had been reconstituted with functional p53. Specifically, MDA-MB-435/β4 cells were transfected with a temperature-sensitive mutant of human p53 (tsp53) that assumes a functional conformation at 32°C but not at 37°C (Zhang et al. 1994). This construct has been used extensively to study the influence of p53 on signaling pathways involved in cell growth and apoptosis (Kobayashi et al. 1995; Owen-Schaub et al. 1995). Stable transfectants of these cells were selected, and tsp53 expression was confirmed by immunoblotting (data not shown). Tsp53 and mock-transfected cells were transfected transiently with HA-AKT/PKB. After incubating these cells with either rat IgG or 439-9B, they were plated on secondary antibody–coated wells and subjected to a 32°C incubation to stimulate p53 activity, followed by a 37°C incubation to activate AKT/PKB. HA immunoprecipitations were performed on extracts from these cells, and these immunoprecipitates were subjected to immunoblotting with phosphoserine 473 AKT/PKB–specific rabbit antiserum. As shown in Fig. 3 B, the clustering of α6β4 significantly increased the level of phosphoserine 473-AKT/PKB in mock-transfected MDA/β4 cells (7.9-fold increase), but not in tsp53-expressing MDA/β4 cells (1.2-fold increase). The importance of p53 in the inhibition of the α6β4-associated activation of AKT/PKB was indicated by the finding that α6β4 clustering increased the level of phosphoserine 473 AKT/PKB in MDA/β4 + tsp53 transfectants that had been incubated at 37°C, the nonpermissive temperature for this tsp53 construct (data not shown).
The ability of p53 to suppress the α6β4-mediated activation of AKT/PKB was explored further in RKO carcinoma cells, which express wild-type p53. In agreement with the results obtained in MDA/β4 cells that had been reconstituted with functional p53, the clustering of α6β4 in two independent RKO/β4 subclones did not result in increased amounts of serine phosphorylated AKT/PKB (Fig. 3 C and data not shown). Importantly, the expression of dnp53 in RKO/β4 cells restored the ability of α6β4 to activate AKT/PKB, as evidenced by an increase in phosphoserine 473-AKT/PKB immunoreactivity in RKO/β4 + dnp53 cells that had been subjected to antibody-mediated α6β4 clustering (8.6-fold increase), as described above (Fig. 3 C). The ability of α6β4 to stimulate AKT/PKB activity in RKO/β4 + dnp53 cells but not in RKO/β4 cells was confirmed by performing in vitro kinase assays using histone H2B as a substrate (data not shown). As a control for specificity, we also demonstrated that the clustering of α6β4 on dnp53-expressing RKO/β4-Δcyt cells did not stimulate AKT/PKB activity (data not shown).
α6β4 Stimulation Induces the Caspase 3–dependent Cleavage of AKT/PKB in a p53-dependent Manner
To define the mechanism by which p53 inhibits the ability of α6β4 to activate AKT/PKB, we investigated whether p53 alters AKT/PKB expression levels in response to α6β4 clustering. RKO/β4 and RKO/β4 + dnp53-expressing cells were incubated with either rat Ig or 439-9B and stimulated on secondary antibody–coated wells for 1 h. The amount of total AKT/PKB in equivalent amounts of total protein from these lysates was assessed by immunoblotting. Importantly, the antibody-mediated clustering of the α6β4 integrin on each of two RKO/β4 subclones resulted in a significant reduction in the total level of AKT/PKB in these cells (Fig. 4 A). In contrast, AKT/PKB levels were not reduced in dnp53-expressing RKO/β4 cells (Fig. 4 B) or in MDA-MB-435/β4 subclones (data not shown) after the antibody-mediated clustering of α6β4. We also observed decreased levels of HA-AKT/PKB protein in HA-AKT/PKB–transfected RKO/β4 cells, but not in HA-AKT/PKB–transfected RKO/β4 + dnp53 cells upon the antibody-mediated clustering of α6β4 (data not shown).
Figure 4.
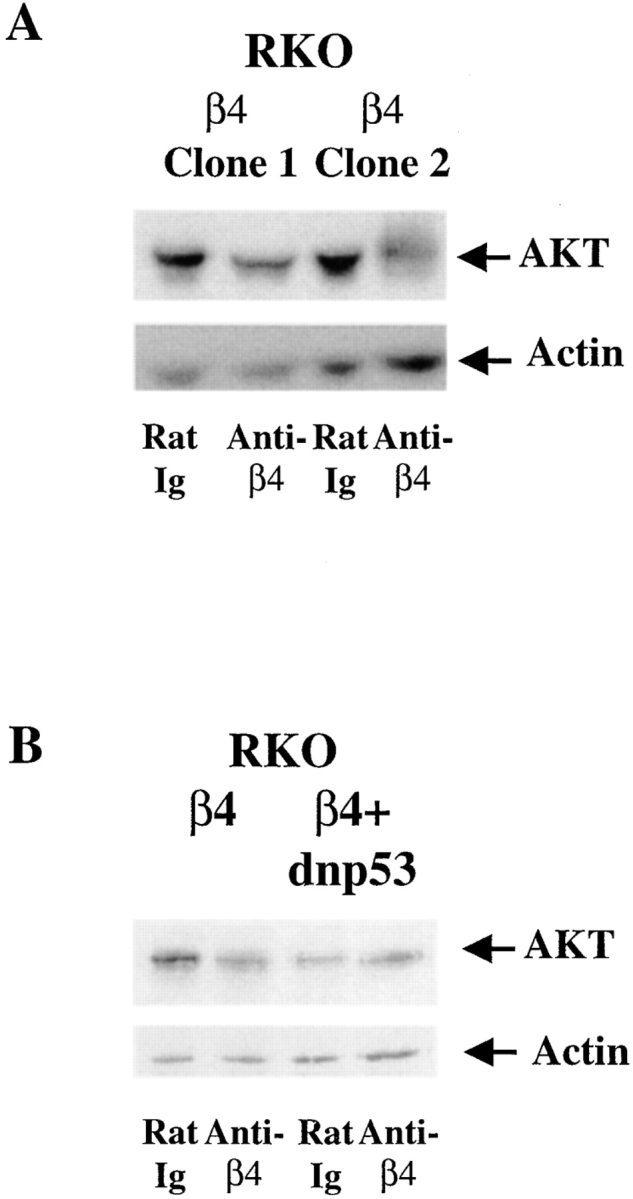
Clustering of the α6β4 integrin reduces AKT/PKB protein levels in p53-wild type but not in p53-deficient carcinoma cells. RKO/β4 (A and B) and RKO/β4 + dnp53 (B)–expressing cells were incubated with either rat Ig or 439-9B and plated on secondary antibody–coated wells for 1 h in the absence of serum. Equivalent amounts of total protein from lysates from these cells were resolved by SDS-PAGE (8%), transferred to nitrocellulose, and probed with an AKT/PKB–specific rabbit antiserum (New England Biolabs) followed by HRP-conjugated goat anti–rabbit IgG. These blots were also probed with an actin-specific rabbit antiserum (Sigma Chemical Co.) to confirm the loading of equivalent amounts of protein. The AKT/PKB and actin bands were detected by enhanced chemiluminescence, and are indicated by arrows. These bands were quantified by densitometry. α6β4 clustering decreased AKT/PKB levels in RKO/β4 subclones (1.7-fold decrease, β4 clone 1; 1.9-fold decrease, β4 clone 2), but not in RKO/β4 + dnp53 cells. Similar results were observed in four additional trials.
Based on the reported ability of caspases to cleave signaling molecules that promote cell survival (Cheng et al. 1997; Enari et al., 1998; Sakahira et al. 1998), we hypothesized that α6β4 may promote the caspase-dependent cleavage of AKT/PKB in wild-type p53-expressing carcinoma cells. Initially, we explored the importance of caspase 3 activity, which has been shown to play a crucial role in p53-dependent apoptotic pathways (Fuchs et al. 1997), in the α6β4-associated reduction of AKT/PKB expression levels. In agreement with the data shown in Fig. 4, the clustering of α6β4 in control RKO/β4 cells significantly reduced the level of AKT/PKB in these carcinoma cells (Fig. 5). However, RKO/β4 cells that had been pretreated with Z-DEVD-FMK, a cell permeable caspase 3 inhibitor, did not exhibit decreased levels of AKT/PKB in response to α6β4 clustering (Fig. 5). In contrast, we detected a decreased amount of AKT/PKB after the clustering of α6β4 in RKO/β4 cells that had been pretreated with Z-IETD-FMK, a cell permeable caspase 8 inhibitor (Fig. 5). Importantly, no effect of these inhibitors on AKT/PKB levels was observed upon the clustering of α6β4 on RKO/α6β4-Δcyt cells (data not shown).
Figure 5.

A caspase 3 inhibitor blocks α6β4-associated reductions in AKT/PKB protein levels. RKO/β4 cells were incubated with either rat Ig or 439-9B in the presence of DMSO (1:500), a caspase 3 inhibitor (Z-DEVD-FMK; 4 μg/ml), or a caspase 8 inhibitor (Z-IETD-FMK; 4 μg/ml). These cells were washed with PBS and plated onto secondary antibody–coated wells in the presence of the same drugs for 1 h in serum-free medium. Equivalent amounts of total protein were resolved by SDS-PAGE (8%), transferred to nitrocellulose, and probed with an AKT/PKB–specific rabbit antiserum (New England Biolabs) followed by HRP-conjugated goat anti–rabbit IgG. AKT/PKB was detected by enhanced chemiluminescence and quantified by densitometry. The antibody-mediated clustering of α6β4 decreased the level of AKT/PKB in DMSO-treated cells (2.0-fold decrease, β4 clone 1; 1.9-fold decrease, β4 clone 2), as well as in cells pretreated with a caspase 8 inhibitor (1.9-fold decrease). In contrast, the pretreatment of these cells with a caspase 3 inhibitor partially restored AKT/PKB levels in RKO/β4 cells subjected to α6β4 clustering (1.1-fold decrease, β4 clone 1; 1.1-fold decrease, β4 clone 2). By probing these blots with an actin-specific rabbit antiserum (Sigma Chemical Co.), we confirmed that equivalent amounts of actin were present in each lane (data not shown). Similar results were observed in three experiments.
The ability of the caspase 3 inhibitor to restore normal AKT/PKB levels suggested that AKT/PKB is cleaved by caspase 3 upon the clustering of α6β4 in carcinoma cells expressing wild-type p53. To establish the caspase 3–mediated cleavage of AKT/PKB more rigorously, we investigated whether a recombinant form of this cysteine protease could cleave baculovirus-expressed AKT/PKB in vitro. Proteins in these reactions were resolved by SDS-PAGE and detected by silver staining. The results obtained revealed that the incubation of baculovirus-expressed AKT/PKB (M r, 60 kD) with recombinant caspase 3 resulted in the formation of an AKT/PKB cleavage product (M r, 49 kD) (Fig. 6). In contrast, we did not detect an AKT/PKB cleavage product after the incubation of baculovirus AKT/PKB with recombinant caspase 8 (Fig. 6). The caspase 3–generated AKT/PKB cleavage product was also detected by immunoblotting with an antiserum specific for the carboxy terminus of AKT/PKB, suggesting that caspase 3 cleaves AKT/PKB at its amino terminus (data not shown).
Figure 6.

AKT/PKB is cleaved by recombinant caspase 3 in vitro. Baculovirus-expressed AKT/PKB (0.5 μg) was incubated either alone, with recombinant caspase 3 (2 μg) or with recombinant caspase 8 (2 μg) for 1 h at 37°C. Proteins in these reactions were resolved by SDS-PAGE (8%) and subjected to silver staining. AKT/PKB and its cleavage product are indicated by arrows. Similar results were observed in three trials.
Finally, to demonstrate that the caspase 3–dependent cleavage of AKT/PKB was responsible for the p53 inhibition of AKT/PKB activity in RKO/β4 cells, we explored the effects of a caspase 3 inhibitor on the ability of α6β4 to activate AKT/PKB. HA-AKT/PKB–transfected RKO/β4 cells were subjected to antibody-mediated α6β4 clustering in the presence of either DMSO or the caspase 3 inhibitor Z-DEVD-FMK. HA immunoprecipitates from extracts from these cells were subjected to immunoblotting with the phosphoserine 473 AKT/PKB–specific rabbit antiserum. As shown in Fig. 7, the pretreatment of RKO/β4 cells with Z-DEVD-FMK restored the ability of α6β4 to stimulate the phosphorylation of AKT/PKB in these cells. These results demonstrate that α6β4 stimulates the caspase 3–dependent cleavage and inactivation of AKT/PKB in p53 wild-type, but not in p53-deficient carcinoma cells.
Figure 7.

A caspase 3 inhibitor restores the ability of α6β4 to induce AKT/PKB phosphorylation. HA-AKT/PKB–transfected RKO/β4 cells were incubated with either rat Ig or 439-9B in the presence of DMSO (1:500) or a caspase 3 inhibitor (Z-DEVD-FMK; 4 μg/ml). After washing with PBS, these cells were plated on secondary antibody–coated wells in serum-free medium containing the indicated drugs for 1 h. HA immunoprecipitations were performed on equivalent amounts of total extracted protein from these samples. These immunoprecipitates were resolved by SDS-PAGE (8%), transferred to nitrocellulose, and probed with rabbit antiserum specific for phosphoserine 473-AKT/PKB, followed by HRP-conjugated goat anti–rabbit Ig. Phosphoserine 473-AKT/PKB was detected by enhanced chemiluminescence, and is indicated by an arrow. Total AKT/PKB levels were also assessed by stripping these membranes and probing with an AKT/PKB–specific rabbit antiserum (data not shown). Relative activity was assessed by determining the ratio of serine phosphorylated AKT/PKB to that of total AKT/PKB for each sample (relative AKT activity: lane 1 = 1.0; lane 2 = 1.3; lane 3 = 1.1; and lane 4 = 3.1). Similar results were observed in three experiments.
Discussion
The binding of extracellular matrix proteins to integrins initiates survival signals that inhibit anoikis, a form of apoptosis induced upon the detachment of cells from extracellular matrix (Meredith et al. 1993; Frisch and Francis 1994). In the current studies, we show that the α6β4 integrin suppresses anoikis exclusively in carcinoma cells that lack functional p53. Furthermore, we demonstrate that this α6β4-associated survival function depends on the ability of this integrin to activate the serine/threonine kinase AKT/PKB in p53-deficient cells. Finally, we provide evidence that p53 inhibits the α6β4-mediated activation of AKT/PKB by promoting the caspase 3–dependent cleavage of this kinase. Collectively, our findings establish that p53 can inhibit an integrin-associated survival function, a phenomenon that has important implications for tumor cell growth.
Our results suggest that the α6β4 integrin can enhance the survival of carcinoma cells in an AKT/PKB–dependent manner. Although previous studies have shown that cell attachment to matrix proteins promotes the survival of primary epithelial cells (Khwaja et al. 1997; Farrelly et al. 1999), α6β4 is the first specific integrin to be implicated in the delivery of AKT/PKB–dependent survival signals to carcinoma cells. The importance of AKT/PKB in α6β4 survival signaling was indicated in our studies by the ability of a dnAKT/PKB construct containing inactivating mutations in the catalytic domain to inhibit the survival effect of α6β4 in serum-starved MDA-MB-435 cells. Although this dnAKT/PKB has been used extensively to implicate AKT/PKB in survival pathways, it is possible that it associates with phosphoinositide-dependent kinases and inhibits their activity. However, our observation that the expression of a constitutively active AKT/PKB in MDA-MBA-435 enhances their survival (data not shown) strongly suggests that α6β4 expression promotes the survival of these cells by activating AKT/PKB.
Our demonstration that p53 can inhibit AKT/PKB kinase activity is of interest in light of the recent finding that the PTEN tumor suppressor can also inhibit cell growth by inhibiting AKT/PKB in a manner that is dependent on its lipid phosphatase activity (Myers et al. 1998; Stambolic et al. 1998; Davies et al. 1999; Ramaswamy et al. 1999; Sun et al. 1999). Together, our current findings on p53 and the previously described activities of PTEN highlight the impact of tumor suppressors on integrin-mediated functions. Moreover, our demonstration that p53 inhibits α6β4 survival signaling by promoting the caspase-dependent cleavage of AKT/PKB provides a mechanistic link between tumor suppressor function and the regulation of integrin signaling, similar to the phosphatase activities of PTEN. Although previous studies have demonstrated that caspases can be activated by p53 in both cell-free systems (Ding et al. 1998) as well as in response to DNA damage (Fuchs et al. 1997; Yu and Little 1998), our findings suggest that caspases can also be activated by an integrin in a p53-dependent manner. Indeed, it will be informative to determine if other activators of p53 such as DNA damage (Siegel et al. 1995; Komarova et al. 1997) can promote the caspase-dependent cleavage of AKT/PKB.
The finding that AKT/PKB activity can be regulated by caspase 3 substantiates the hypothesis that caspases play an important role in many forms of apoptosis based on their ability to cleave signaling molecules that influence cell survival. For example, caspases have been shown to cleave and inactivate an inhibitor of caspase-activated deoxyribonuclease (CAD). Importantly, the cleavage of this inhibitor results in the activation of CAD, which is the enzyme responsible for the DNA fragmentation that is characteristic of apoptosis (Enari et al., 1998; Sakahira et al. 1998). Caspase 3 has also been shown to cleave bcl-2, resulting in an inhibition of its anti-apoptotic function (Cheng et al. 1997). While AKT/PKB has been suggested to be a target of caspase activity based on the reduced levels of this kinase observed in T cells in response to fas stimulation (Widmann et al. 1998), our results extend this finding by establishing definitively that AKT/PKB is cleaved by caspase 3. More importantly, we provide evidence that this cleavage event results in the inhibition of AKT/PKB kinase activity, and implicate this event in the inhibition of α6β4 integrin survival function.
It is important to consider the mechanism by which the α6β4-induced, caspase-dependent cleavage of AKT/PKB inhibits its kinase activity. We detected an AKT/PKB fragment (M r, 49 kD) after the in vitro incubation of AKT/PKB with recombinant caspase 3. This fragment was recognized by a rabbit antiserum raised against a peptide corresponding to the extreme carboxy-terminal amino acids of the molecule, suggesting that caspase 3 cleaves AKT/PKB at its amino terminus. Interestingly, the pleckstrin homology domain, which resides in the amino terminus of AKT/PKB, is important in both the translocation of this kinase to the membrane and its subsequent activation (Franke et al. 1995; Andjelkovic et al. 1997). It is possible that the caspase 3–dependent cleavage of AKT/PKB prevents the membrane translocation of this kinase, thus, preventing its activation. However, we were unable to identify an AKT/PKB fragment in vivo after the clustering of α6β4, despite our detection of reduced AKT/PKB levels under these conditions. This result suggests that after the initial cleavage of AKT/PKB by caspase 3, this kinase is subjected to further cleavage by other caspases, as has been shown for ICAD (Tang and Kidd 1998). Moreover, our inability to detect AKT/PKB fragments in vivo after the clustering of α6β4 suggests that AKT/PKB cannot be detected by immunoblotting after its cleavage by multiple caspases. The ability of a caspase 3 inhibitor to restore both normal AKT/PKB levels as well as the α6β4-mediated activation of AKT/PKB suggests that the degradation of AKT/PKB observed in vivo is dependent on the initial cleavage of this kinase by caspase 3.
In contrast to our finding that p53-dependent, caspase 3 activity inhibits AKT/PKB, other studies have concluded that constitutively active AKT/PKB can delay p53-dependent apoptosis (Sabbatini and McCormick 1999), inhibit caspases (Cardone et al. 1998), and block caspase-dependent forms of apoptosis (Berra et al. 1998; Gibson et al. 1999). The demonstrated ability of AKT/PKB to inhibit p53 and caspase activity in these studies may relate to the kinetics of AKT/PKB activation. Specifically, the rapid stimulation of AKT/PKB may impede p53 or caspase activation. In contrast, the ability of α6β4 clustering to promote the caspase 3–dependent inactivation of AKT/PKB in p53 wild-type carcinoma cells may relate to the fact that α6β4 signaling stimulates caspase activity before AKT/PKB activity in these cells. Alternatively, it is possible that the ability of caspase 3 to cleave AKT/PKB was not observed in previous studies because insufficient amounts of endogenous caspase activity were present to inhibit the activity of exogenously introduced, active AKT/PKB. Nonetheless, these results suggest that an intimate crosstalk exists between AKT/PKB and caspases that contributes to the regulation of cell survival.
We have previously demonstrated that the α6β4 integrin activates p53 function (Bachelder et al. 1999). The current studies describe an important consequence of this α6β4 activity, namely the inhibition of AKT/PKB activity and its associated cell survival function. Similar to previous results from our laboratory (Clarke et al. 1995; Shaw et al. 1997; O'Connor et al. 1998) and others (Kim et al. 1997; Sun et al. 1998), the current studies demonstrate that the survival function of α6β4 is ligand-independent in β4-transfected, p53-deficient carcinoma cells. This ligand-independent survival function may be attributable to the ability of the β4 cytoplasmic domain to self-associate (Rezniczek et al. 1998).
In addition to demonstrating that p53 inhibits α6β4-mediated survival, we observed that α6β4 increases the level of apoptosis observed in serum-starved p53 wild-type carcinoma cells. This result suggests that the apoptotic signaling pathway activated by α6β4 can augment the apoptotic signaling initiated by serum deprivation. Although p53 has been implicated in the apoptosis induced in endothelial cells upon their detachment from matrix (Ilic et al. 1998), others have reported that epithelial cell anoikis is p53-independent (Boudreau et al. 1995). In agreement with the results of the latter study, we observed apoptosis in p53-deficient cells, including MDA-MB-435 cells and dnp53-expressing RKO cells, upon their detachment from matrix. These results indicate that carcinoma cells are subject to a p53-independent form of anoikis. In combination with our previous observation that α6β4 apoptotic signaling requires p53 activity (Bachelder et al. 1999), our findings suggest that the p53-independent apoptosis of carcinoma cells that occurs in response to matrix detachment can be enhanced by p53-dependent, α6β4 apoptotic signaling.
The current studies may explain why the α6β4 integrin has been implicated in the apoptosis of some cells and the survival of others. Specifically, α6β4 has been shown to induce growth arrest and apoptosis in several carcinoma cell lines (Clarke et al. 1995; Kim et al. 1997, Sun et al. 1998) as well as in endothelial cells (Miao et al. 1997). However, this integrin has also been shown to promote the proliferation (Mainiero et al. 1997; Murgia et al. 1998) and survival (Dowling et al. 1996) of keratinocytes. These apparently contradictory functions of α6β4 may relate to the fact that the functions of α6β4 are cell type–specific. The current studies establish that the p53 tumor suppressor is one critical signaling molecule that may influence α6β4 function in different cell types because this integrin promotes apoptosis only in wild-type p53-expressing cells and survival only in p53-deficient cells. Interestingly, the reported ability of α6β4 to promote keratinocyte survival (Dowling et al. 1996) may relate to the reported deficiency of p53 activity in these cells (Nigro et al. 1997).
One implication of our findings is that the α6β4 integrin is similar to a number of oncogenes that promote cell proliferation in some settings and cell death in others. The recent observation that oncogenes can deliver such death signals has led to their seemingly contradictory categorization as tumor suppressors in select environments. For example, although the stimulation of c-myc and E2F normally promotes cell proliferation, the activation of these oncogenes induces apoptosis in the presence of secondary stress signals such as p53 expression, serum starvation or hypoxia (Evan et al. 1992; Shi et al. 1992, Hermeking and Eick 1994; Qin et al. 1994; Wu and Levine 1994). The ability of these stress signals to stimulate oncogene-dependent apoptosis is thought to be important in eliminating tumor cells that escape normal proliferation checkpoints as a result of oncogene expression. Similarly, the α6β4 integrin, which promotes the survival of p53-deficient cells, could also be classified loosely as a tumor suppressor based on its apoptotic function in carcinoma cells that express wild-type p53. The current studies demonstrate that, similar to the activity of oncogenes, integrin function and signaling can be profoundly influenced by physiological stimuli that activate other signaling pathways in a cell.
In summary, we have described the ability of the α6β4 integrin to promote the survival of the p53 mutant, but not p53 wild-type carcinoma cells. This ability of p53 to influence integrin-mediated functions so markedly derives from its ability to activate the caspase 3–dependent cleavage of AKT/PKB. The fact that AKT/PKB overexpression has been suggested to contribute to the transformed phenotype of tumor cells (Bellacosa et al. 1995) suggests that the introduction of the α6β4 integrin into p53 wild-type tumors may inhibit their growth by inducing the cleavage of this transforming protein. The ability of α6β4 to induce the p53-dependent cleavage of AKT/PKB also suggests that the acquisition of inactivating mutations in either p53 or caspase 3 will provide a selective growth advantage for carcinoma cells by stimulating α6β4-mediated AKT/PKB–dependent survival signaling. Moreover, given our previous demonstration that α6β4 promotes carcinoma cell migration and invasion (Chao et al. 1996, Shaw et al. 1997; O'Connor et al. 1998), we suggest that carcinoma cells that express α6β4 and mutant forms of p53 or caspase 3 will have a distinct advantage in their ability to disseminate and survive as metastatic lesions.
Acknowledgments
We thank Moshe Oren, Alt Zantema, Alex Toker, and Phil Hinds (Harvard Medical School, Boston, MA) for reagents. We also thank Lewis Cantley, Alex Toker, Phil Hinds, Kathy O'Connor, and Leslie Shaw (Beth Israel Deaconess Medical Center, Boston, MA) for valuable discussions.
This work was supported by National Institutes of Health grants CA80789, AI39264 (both to A.M. Mercurio), and CA81697 (to R.E. Bachelder), as well as by the Italian Association for Cancer Research.
Footnotes
Abbreviations used in this paper: CAD, caspase-activated deoxyribonuclease; dnAKT, dominant negative AKT; dnp53, dominant negative p53; GFP, green fluorescent protein; HA, hemagglutinin; tsp53, temperature-sensitive p53.
References
- Ahmed N.N., Grimes H.L., Bellacosa A., Chan T.O., Tsichlis P.N. Transduction of interleukin-2 antiapoptotic and proliferative signals via Akt protein kinase. Proc. Natl. Acad. Sci. USA. 1997;94:3627–3632. doi: 10.1073/pnas.94.8.3627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alessi D.R., Andjelkovic M., Caudwell B., Cron P., Morrice N., Cohen P., Hemmings B.A. Mechanism of activation of protein kinase B by insulin and IGF-1. EMBO (Eur. Mol. Biol. Organ.) J. 1996;15:6541–6551. [PMC free article] [PubMed] [Google Scholar]
- Andjelkovic M., Alessi D.R., Meier R., Fernandez A., Lamb N.J., Frech M., Cron P., Cohen P., Lucocq J.M., Hemmings B.A. Role of translocation in the activation and function of protein kinase B. J. Biol. Chem. 1997;272:31515–31524. doi: 10.1074/jbc.272.50.31515. [DOI] [PubMed] [Google Scholar]
- Bachelder R.E., Marchetti A., Falcioni R., Soddu S., Mercurio A.M. Activation of p53 function in carcinoma cells by the α6β4 integrin. J. Biol. Chem. 1999;274:20733–20737. doi: 10.1074/jbc.274.29.20733. [DOI] [PubMed] [Google Scholar]
- Bellacosa A., de Feo D., Godwin A.K., Bell D.W., Cheng J.Q., Altomare D.A., Wan M., Dubeau L., Scambia G., Masciullo V. Molecular alterations of the AKT2 oncogene in ovarian and breast carcinomas. Intl. J. Cancer. 1995;64:280–285. doi: 10.1002/ijc.2910640412. [DOI] [PubMed] [Google Scholar]
- Berra E., Diaz-Meco M.T., Moscat J. The activation of p38 and apoptosis by the inhibition of Erk is antagonized by the phosphoinositide 3-kinase/Akt pathway. J. Biol. Chem. 1998;273:10792–10797. doi: 10.1074/jbc.273.17.10792. [DOI] [PubMed] [Google Scholar]
- Blume-Jensen P., Janknecht R., Hunter T. The kit receptor promotes cell survival via activation of PI 3-kinase and subsequent Akt-mediated phosphorylation of Bad on Ser136. Curr. Biol. 1998;8:779–782. doi: 10.1016/s0960-9822(98)70302-1. [DOI] [PubMed] [Google Scholar]
- Borradori L., Sonnenberg A. Hemidesmosomesroles in adhesion, signaling and human diseases. Curr. Opin. Cell Biol. 1996;8:647–656. doi: 10.1016/s0955-0674(96)80106-2. [DOI] [PubMed] [Google Scholar]
- Boudreau N., Sympson C.J., Werb Z., Bissell M.J. Suppression of ICE and apoptosis in mammary epithelial cells by extracellular matrix. Science. 1995;267:891–893. doi: 10.1126/science.7531366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cardone M.H., Roy N., Stennicke H.R., Salvesen G.S., Franke T.F., Stanbridge E., Frisch S., Reed J.C. Regulation of cell death protease caspase-9 by phosphorylation. Science. 1998;282:1318–1321. doi: 10.1126/science.282.5392.1318. [DOI] [PubMed] [Google Scholar]
- Chao C., Lotz M.M., Clarke A.C., Mercurio A.M. A function for the integrin alpha6 beta4 in the invasive properties of colorectal carcinoma cells. Cancer Res. 1996;56:4811–4819. [PubMed] [Google Scholar]
- Cheng E.H., Kirsch D.G., Clem R.J., Ravi R., Kastan M.B., Bedi A., Ueno K., Hardwick J.M. Conversion of Bcl-2 to a Bax-like death effector by caspases. Science. 1997;278:1966–1968. doi: 10.1126/science.278.5345.1966. [DOI] [PubMed] [Google Scholar]
- Clarke A.S., Lotz M.M., Chao C.C., Mercurio A.M. Activation of the p21 pathway of growth arrest and apoptosis by the β4 integrin cytoplasmic domain. J. Biol. Chem. 1995;270:22673–22676. doi: 10.1074/jbc.270.39.22673. [DOI] [PubMed] [Google Scholar]
- Crowder R.J., Freeman R.S. Phosphatidylinositol 3-kinase and Akt protein kinase are necessary and sufficient for the survival of nerve growth factor-dependent sympathetic neurons. J. Neurosci. 1998;18:2933–2943. doi: 10.1523/JNEUROSCI.18-08-02933.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Datta S.R., Dudek H., Tao X., Masters S., Fu H., Gotoh Y., Greenberg M.E. Akt phosphorylation of BAD couples survival signals to the cell-intrinsic death machinery. Cell. 1997;91:231–241. doi: 10.1016/s0092-8674(00)80405-5. [DOI] [PubMed] [Google Scholar]
- Davies M.A., Koul D., Dhesi H., Berman R., McDonnell T.J., McConkey D., Yung W.K., Steck P.A. Regulation of Akt/PKB activity, cellular growth, and apoptosis in prostate carcinoma cells by MMAC/PTEN. Cancer Res. 1999;59:2551–2556. [PubMed] [Google Scholar]
- Day M.L., Foster R.G., Day K.C., Zhao X., Humphrey P., Swanson P., Postigo A.A., Zhang S.H., Dean D.C. Cell anchorage regulates apoptosis through the retinoblastoma tumor suppressor/E2F pathway. J. Biol. Chem. 1997;272:8125–8128. doi: 10.1074/jbc.272.13.8125. [DOI] [PubMed] [Google Scholar]
- Ding H.F., McGill G., Rowan S., Schmaltz C., Shimamura A., Fisher D.E. Oncogene-dependent regulation of caspase activation by p53 protein in a cell-free system. J. Biol. Chem. 1998;273:28378–28383. doi: 10.1074/jbc.273.43.28378. [DOI] [PubMed] [Google Scholar]
- Dowling J., Yu Q., Fuchs E. β4 integrin is required for hemidesmosome formation, cell adhesion and cell survival. J. Cell Biol. 1996;134:559–572. doi: 10.1083/jcb.134.2.559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dudek H., Datta S.R., Franke T.F., Birnbaum M.J., Yao R., Cooper G.M., Segal R.A., Kaplan D.R., Greenberg M.E. Regulation of neuronal survival by the serine-threonine protein kinase Akt. Science. 1997;275:661–666. doi: 10.1126/science.275.5300.661. [DOI] [PubMed] [Google Scholar]
- Enari M., Talanian R.V., Wong W.W., Nagata S. Sequential activation of ICE-like and CPP32-like proteases during Fas-mediated apoptosis. Nature. 1996;380:723–726. doi: 10.1038/380723a0. [DOI] [PubMed] [Google Scholar]
- Evan G.I., Wyllie A.H., Gilbert C.S., Littlewood T.D., Land H., Brooks M., Waters C.M., Penn L.Z., Hancock D.C. Induction of apoptosis in fibroblasts by c-myc protein. Cell. 1992;69:119–128. doi: 10.1016/0092-8674(92)90123-t. [DOI] [PubMed] [Google Scholar]
- Eves E.M., Xiong W., Bellacosa A., Kennedy S.G., Tsichlis P.N., Rosner M.R., Hay N. Akt, a target of phosphatidylinositol 3-kinase, inhibits apoptosis in a differentiating neuronal cell line. Mol. Cell. Biol. 1998;18:2143–2152. doi: 10.1128/mcb.18.4.2143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Falcioni R., Sacchi A., Resau J., Kennel S.J. Monoclonal antibody to human carcinoma protein complexquantitation in normal and tumor tissue. Cancer Res. 1998;48:816–821. [PubMed] [Google Scholar]
- Farrelly N., Lee Y.J., Oliver J., Dine C., Streuli C.H. Extracellular matrix regulates apoptosis in mammary epithelium through a control on insulin signaling. J. Cell Biol. 1999;144:1337–1347. doi: 10.1083/jcb.144.6.1337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franke T.F., Yang S.I., Chan T.O., Datta K., Kazlauskas A., Morrison D.K., Kaplan D.R., Tsichlis P.N. The protein kinase encoded by the Akt proto-oncogene is a target of the PDGF-activated phosphatidylinositol 3-kinase. Cell. 1995;81:727–736. doi: 10.1016/0092-8674(95)90534-0. [DOI] [PubMed] [Google Scholar]
- Frisch S.M., Francis H. Disruption of epithelial cell–matrix interactions induces apoptosis. J. Cell Biol. 1994;124:619–626. doi: 10.1083/jcb.124.4.619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frisch S.M., Vuori K., Ruoslahti E., Chan-Hui P.Y. Control of adhesion-dependent cell survival by focal adhesion kinase. J. Cell Biol. 1996;134:793–799. doi: 10.1083/jcb.134.3.793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuchs E.J., McKenna K.A., Bedi A. p53-dependent DNA damage-induced apoptosis requires Fas/APO-1-independent activation of CPP32beta. Cancer Res. 1997;57:2550–2554. [PubMed] [Google Scholar]
- Gerber H.P., McMurtrey A., Kowalski J., Yan M., Keyt B.A., Dixit V., Ferrara N. Vascular endothelial growth factor regulates endothelial cell survival through the phosphatidylinositol 3′-kinase/Akt signal transduction pathway. Requirement for Flk-1/KDR activation. J. Biol. Chem. 1998;273:30336–30343. doi: 10.1074/jbc.273.46.30336. [DOI] [PubMed] [Google Scholar]
- Gibson S., Tu S., Oyer R., Anderson S.M., Johnson G.L. Epidermal growth factor protects epithelial cells against Fas-induced apoptosis. Requirement for Akt activation. J. Biol. Chem. 1999;274:17612–17618. doi: 10.1074/jbc.274.25.17612. [DOI] [PubMed] [Google Scholar]
- Green K.J., Jones J.C. Desmosomes and hemidesmosomesstructure and function of molecular components. FASEB (Fed. Am. Soc. Exp. Biol.) J. 1996;10:871–881. doi: 10.1096/fasebj.10.8.8666164. [DOI] [PubMed] [Google Scholar]
- Hemler M.E., Crouse C., Sonnenberg A. Association of the VLA alpha 6 subunit with a novel protein. A possible alternative to the common VLA beta1 subunit on certain cell lines. J. Biol. Chem. 1989;264:6529–6535. [PubMed] [Google Scholar]
- Hermeking H., Eick D. Mediation of c-Myc-induced apoptosis by p53. Science. 1994;265:2091–2093. doi: 10.1126/science.8091232. [DOI] [PubMed] [Google Scholar]
- Howlett A.R., Bailey N., Damsky C., Petersen O.W., Bissell M.J. Cellular growth and survival are mediated by beta 1 integrins in normal human breast epithelium but not in breast carcinoma. J. Cell Sci. 1995;108:1945–1957. doi: 10.1242/jcs.108.5.1945. [DOI] [PubMed] [Google Scholar]
- Ilic D., Almeida E.A., Schlaepfer D.D., Dazin P., Aizawa S., Damsky C.H. Extracellular matrix survival signals transduced by focal adhesion kinase suppress p53-mediated apoptosis. J. Cell Biol. 1998;143:547–550. doi: 10.1083/jcb.143.2.547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kajiji S., Tamura R.N., Quaranta V. A novel integrin (alpha E beta 4) from human epithelial cells suggests a fourth family of integrin adhesion receptors. EMBO (Eur. Mol. Biol. Organ.) J. 1989;8:673–680. doi: 10.1002/j.1460-2075.1989.tb03425.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kennel S.J., Foote L.J., Falcioni R., Sonnenberg A., Stringer C.J., Crouse C., Hemler M.E. Analysis of the tumor-associated antigen TSP-180. Identity of alpha 6-beta 4 in the integrin superfamily. J. Biol. Chem. 1989;264:15515–15521. [PubMed] [Google Scholar]
- Khwaja A., Rodriguez-Viciana P., Wennstrom S., Warne P.H., Downward J. Matrix adhesion and Ras transformation both activate a phosphoinositide 3-OH kinase and protein kinase B/Akt cellular survival pathway. EMBO (Eur. Mol. Biol. Organ.) J. 1997;16:2783–2793. doi: 10.1093/emboj/16.10.2783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim S.Y., Bachman N.J., Nair T.S., Goldsmith S., Liebert M., Grossman H.B., Lomax M.I., Carey T.E. Beta 4 integrin transfection of UM-UC-2 (human bladder carcinoma) cellsstable expression of a spontaneous cytoplasmic truncation mutant with rapid loss of clones expressing intact beta 4. Cancer Res. 1997;57:38–42. [PubMed] [Google Scholar]
- Kobayashi T., Consoli U., Andreeff M., Shiku H., Deisseroth A.B., Zhang W. Activation of p21WAF1/Cip1 expression by a temperature-sensitive mutant of human p53 does not lead to apoptosis. Oncogene. 1995;11:2311–2316. [PubMed] [Google Scholar]
- Komarova E.A., Chernov M.V., Franks R., Wang K., Armin G., Zelnick C.R., Chin D.M., Bacus S.S., Stark G.R., Gudkov A.V. Transgenic mice with p53-responsive lacZp53 activity varies dramatically during normal development and determines radiation and drug sensitivity in vivo. EMBO (Eur. Mol. Biol. Organ.) J. 1997;16:1391–1400. doi: 10.1093/emboj/16.6.1391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lesoon-Wood L.A., Kim W.H., Kleinman H.K., Weintraub B.D., Mixson A.J. Systemic gene therapy with p53 reduces growth and metastases of a malignant human breast cancer in nude mice. Hum. Gene Ther. 1995;6:395–405. doi: 10.1089/hum.1995.6.4-395. [DOI] [PubMed] [Google Scholar]
- Mainiero F., Murgia C., Wary K.K., Curatola A.M., Pepe A., Blumemberg M., Westwick J.K., Der C., Giancotti F.G. The coupling of α6β4 integrin to the ras-MAP kinase pathways mediated by shc controls keratinocyte proliferation. EMBO (Eur. Mol. Biol. Organ.) J. 1997;16:2365–2375. doi: 10.1093/emboj/16.9.2365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meredith J.E., Jr., Fazeli B., Schwartz M.A. The extracellular matrix as a cell survival factor. Mol. Biol. Cell. 1993;4:953–961. doi: 10.1091/mbc.4.9.953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merlo G.R., Basolo F., Fiore L., Duboc L., Hynes N.E. p53-Dependent and p53-independent activation of apoptosis in mammary epithelial cells reveals a survival function of EGF and insulin. J. Cell Biol. 1995;128:1185–1196. doi: 10.1083/jcb.128.6.1185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miao J., Araki S., Kaji K., Hayashi H. Integrin β4 is involved in apoptotic signal transduction in endothelial cells. Biochem. Biophys. Res. Comm. 1997;233:182–186. doi: 10.1006/bbrc.1997.6422. [DOI] [PubMed] [Google Scholar]
- Morgenstern J.P., Land H. Advanced mammalian gene transferhigh titre retroviral vectors with multiple drug selection markers and a complementary helper-free packaging cell line. Nucleic Acids Res. 1990;18:3587–3596. doi: 10.1093/nar/18.12.3587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murgia C., Blaikie P., Kim N., Dans M., Petrie H.T., Giancotti F.G. Cell cycle and adhesion defects in mice carrying a targeted deletion of the integrin beta4 cytoplasmic domain. EMBO (Eur. Mol. Biol. Organ.) J. 1998;17:3940–3951. doi: 10.1093/emboj/17.14.3940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Myers M.P., Pass I., Batty I.H., Van der Kaay J., Stolarov J.P., Hemmings B.A., Wigler M.H., Downes C.P., Tonks N.K. The lipid phosphatase activity of PTEN is critical for its tumor supressor function. Proc. Natl. Acad. Sci. USA. 1998;95:13513–13518. doi: 10.1073/pnas.95.23.13513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagasawa H., Li C.Y., Maki C.G., Imrich A.C., Little J.B. Relationship between radiation-induced G1 phase arrest and p53 function in human tumor cells. Cancer Res. 1995;55:1842–1846. [PubMed] [Google Scholar]
- Nigro J.M., Aldape K.D., Hess S.M., Tlsty T.D. Cellular adhesion regulates p53 protein levels in primary human keratinocytes. Cancer Res. 1997;57:3635–3639. [PubMed] [Google Scholar]
- O'Connor K.L., Shaw L.M., Mercurio A.M. Release of cAMP gating by the α6β4 integrin stimulates lamellae formation and the chemotactic migration of carcinoma cells. J. Cell Biol. 1998;143:1749–1760. doi: 10.1083/jcb.143.6.1749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Owen-Schaub L.B., Zhang W., Cusack J.C., Angelo L.S., Santee S.M., Fujiwara T., Roth J.A., Deisseroth A.B., Zhang W.W., Kruzel E. Wild-type human p53 and a temperature-sensitive mutant induce Fas/APO-1 expression. Mol. Cell. Biol. 1995;15:3032–3040. doi: 10.1128/mcb.15.6.3032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin X.Q., Livingston D.M., Kaelin W.G., Jr., Adams P.D. Deregulated transcription factor E2F-1 expression leads to S-phase entry and p53-mediated apoptosis. Proc. Natl. Acad. Sci. USA. 1994;91:10918–10922. doi: 10.1073/pnas.91.23.10918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Radeva G., Petrocelli T., Behrend E., Leung-Hagesteijn C., Filmus J., Slingerland J., Dedhar S. Overexpression of the integrin-linked kinase promotes anchorage-independent cell cycle progression. J. Biol. Chem. 1997;272:13937–13944. doi: 10.1074/jbc.272.21.13937. [DOI] [PubMed] [Google Scholar]
- Ramaswamy S., Nakamura N., Vazquez F., Batt D.B., Perera S., Roberts T.M., Sellers W.R. Regulation of G1 progression by the PTEN tumor suppressor protein is linked to inhibition of the phosphatidylinositol 3-kinase/Akt pathway. Proc. Natl. Acad. Sci. USA. 1999;96:2110–2115. doi: 10.1073/pnas.96.5.2110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rampalli A.M., Zelenka P.S. Insulin regulates expression of c-fos and c-jun and suppresses apoptosis of lens epithelial cells. Cell Growth Differ. 1995;6:945–953. [PubMed] [Google Scholar]
- Rezniczek G.A., de Pereda J.M., Relpert M., Aiche G. Linking integrin α6β4-based adhesion to the intermediate filament cytoskeletondirect interaction between the β4 subunit and plectin at multiple molecular sites. J. Cell Biol. 1998;141:2209–2225. doi: 10.1083/jcb.141.1.209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodeck U., Jost M., Kari C., Shih D.T., Lavker R.M., Ewert D.L., Jensen P.J. EGF-R dependent regulation of keratinocyte survival. J. Cell Sci. 1997;110:113–121. doi: 10.1242/jcs.110.2.113. [DOI] [PubMed] [Google Scholar]
- Sabbatini P., McCormick F. Phosphoinositide 3-OH kinase (PI3K) and PKB/AKT delay the onset of p53-mediated, transcriptionally dependent apoptosis. J. Biol. Chem. 1999;274:24263–24269. doi: 10.1074/jbc.274.34.24263. [DOI] [PubMed] [Google Scholar]
- Sakahira H., Enari M., Nagata S. Cleavage of CAD inhibitor in CAD activation and DNA degradation during apoptosis. Nature. 1998;391:96–99. doi: 10.1038/34214. [DOI] [PubMed] [Google Scholar]
- Shaw L.M., Rabinovitz I., Wang H.F., Toker A., Mercurio A.M. Activation of phosphoinositide 3-OH kinase by the α6β4 integrin promotes carcinoma invasion. Cell. 1997;91:949–960. doi: 10.1016/s0092-8674(00)80486-9. [DOI] [PubMed] [Google Scholar]
- Shi Y., Glynn J.M., Guilbert L.J., Cotter T.G., Bissonnette R.P., Green D.R. Role for c-myc in activation-induced apoptotic cell death in T cell hybridomas. Science. 1992;257:212–214. doi: 10.1126/science.1378649. [DOI] [PubMed] [Google Scholar]
- Siegel J., Fritsche M., Mai S., Brandner G., Hess R.D. Enhanced p53 activity and accumulation in response to DNA damage upon DNA transfection. Oncogene. 1995;11:1363–1370. [PubMed] [Google Scholar]
- Skorski T., Bellacosa A., Nieborowska-Skorska M., Majewski M., Martinez R., Choi J.K., Trotta R., Wlodarski P., Perrotti D., Chan T.O. Transformation of hematopoietic cells by BCR/ABL requires activation of a PI-3k/Akt-dependent pathway. EMBO (Eur. Mol. Biol. Organ.) J. 1997;16:6151–6161. doi: 10.1093/emboj/16.20.6151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Songyang Z., Baltimore D., Cantley L.C., Kaplan D.R., Franke T.F. Interleukin 3-dependent survival by the Akt protein kinase. Proc. Natl. Acad. Sci. USA. 1997;94:11345–11350. doi: 10.1073/pnas.94.21.11345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stambolic V., Suzuki A., de la Pompa J.L., Brothers G.M., Mirtsos C., Sasaki T., Ruland J., Penninger J.M., Siderovski D.P., Mak T.W. Negative regulation of PKB/Akt-dependent cell survival by the tumor suppressor PTEN. Cell. 1998;95:29–39. doi: 10.1016/s0092-8674(00)81780-8. [DOI] [PubMed] [Google Scholar]
- Stromblad S., Becker J.C., Yebra M., Brooks P.C., Cheresh D.A. Suppression of p53 activity and p21WAF1/CIP1 expression by vascular cell integrin alphavbeta3 during angiogenesis. J. Clin. Invest. 1996;98:426–433. doi: 10.1172/JCI118808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun H., Santoro S.A., Zutter M.M. Downstream events in mammary gland morphogenesis mediated by reexpression of the alpha2beta1 integrinthe role of the alpha6 and beta4 integrin subunits. Cancer Res. 1998;58:2224–2233. [PubMed] [Google Scholar]
- Sun H., Lesche R., Li D.M., Liliental J., Zhang H., Gao J., Gavrilova N., Mueller B., Liu X., Wu H. PTEN modulates cell cycle progression and cell survival by regulating phosphatidylinositol 3,4,5,-trisphosphate and Akt/protein kinase B signaling pathway. Proc. Natl. Acad. Sci. USA. 1999;96:6199–6204. doi: 10.1073/pnas.96.11.6199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang D., Kidd V.J. Cleavage of DFF-45/ICAD by multiple caspases is essential for its function during apoptosis. J. Biol. Chem. 1998;273:28549–28552. doi: 10.1074/jbc.273.44.28549. [DOI] [PubMed] [Google Scholar]
- Tozeren A., Kleinman H.K., Wu S., Mercurio A.M., Byers S.W. Integrin alpha 6 beta 4 mediates dynamic interactions with laminin. J. Cell Sci. 1994;107:3153–3163. doi: 10.1242/jcs.107.11.3153. [DOI] [PubMed] [Google Scholar]
- Wewer U.M., Shaw L.M., Albrechtsen R., Mercurio A.M. The integrin alpha 6 beta 1 promotes the survival of metastatic human breast carcinoma cells in mice. Am. J. Pathol. 1997;151:1191–1198. [PMC free article] [PubMed] [Google Scholar]
- Widmann C., Gibson S., Johnson G.L. Caspase-dependent cleavage of signaling proteins during apoptosis. A turn-off mechanism for anti-apoptotic signals. J. Biol. Chem. 1998;273:7141–7147. doi: 10.1074/jbc.273.12.7141. [DOI] [PubMed] [Google Scholar]
- Wu X., Levine A.J. p53 and E2F-1 cooperate to mediate apoptosis. Proc. Natl. Acad. Sci. USA. 1994;91:3602–3606. doi: 10.1073/pnas.91.9.3602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu Y., Little J.B. p53 is involved in but not required for ionizing radiation-induced caspase-3 activation and apoptosis in human lymphoblast cell lines. Cancer Res. 1998;58:4277–4281. [PubMed] [Google Scholar]
- Zhang W., Guo X.Y., Hu G.Y., Liu W.B., Shay J.W., Deisseroth A.B. A temperature-sensitive mutant of human p53. EMBO (Eur. Mol. Biol. Organ.) J. 1994;13:2535–2544. doi: 10.1002/j.1460-2075.1994.tb06543.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Z., Vuori K., Reed J.C., Ruoslahti E. The alpha 5 beta 1 integrin supports survival of cells on fibronectin and up-regulates Bcl-2 expression. Proc. Natl. Acad. Sci. USA. 1995;92:6161–6165. doi: 10.1073/pnas.92.13.6161. [DOI] [PMC free article] [PubMed] [Google Scholar]