

Abstract
De novo biosynthesis of amino acids uses intermediates provided by the TCA cycle that must be replenished by anaplerotic reactions to maintain the respiratory competency of the cell. Genome-wide expression analyses in Saccharomyces cerevisiae reveal that many of the genes involved in these reactions are repressed in the presence of the preferred nitrogen sources glutamine or glutamate. Expression of these genes in media containing urea or ammonia as a sole nitrogen source requires the heterodimeric bZip transcription factors Rtg1 and Rtg3 and correlates with a redistribution of the Rtg1p/Rtg3 complex from a predominantly cytoplasmic to a predominantly nuclear location. Nuclear import of the complex requires the cytoplasmic protein Rtg2, a previously identified upstream regulator of Rtg1 and Rtg3, whereas export requires the importin-β-family member Msn5. Remarkably, nuclear accumulation of Rtg1/Rtg3, as well as expression of their target genes, is induced by addition of rapamycin, a specific inhibitor of the target of rapamycin (TOR) kinases. We demonstrate further that Rtg3 is a phosphoprotein and that its phosphorylation state changes after rapamycin treatment. Taken together, these results demonstrate that target of rapamycin signaling regulates specific anaplerotic reactions by coupling nitrogen quality to the activity and subcellular localization of distinct transcription factors.
Keywords: gene expression, metabolism, phosphorylation, rapamycin, signal transduction
Introduction
Normal cell growth requires that cells adjust their metabolic activity according to nutrient availability and other environmental cues. Specialized signal transduction mechanisms exist that enable cells to perceive and integrate these cues in order to establish and/or maintain appropriate patterns of gene expression. Understanding how these pathways function is thus important for understanding both normal cellular behavior and the underlying basis of many human diseases, including cancer. One important signaling pathway used by all eukaryotic cells is the target of rapamycin (TOR) kinase pathway. This pathway was discovered through the action of the antibiotic rapamycin, a potent inhibitor of T cell proliferation, which combines with the small immunophilin FKBP and targets the large, evolutionarily conserved TOR kinase (Thomas and Hall 1997; Dennis et al. 1999). Rapamycin inhibits the growth of a wide variety of cell types and organisms, including the budding yeast, Saccharomyces cerevisiae. Yeast contains two TOR genes, TOR1 and TOR2, and the products of both are inhibited by the rapamcyin/FKBP complex (Heitman et al. 1991; Helliwell et al. 1994; Zheng et al. 1995). Treating yeast cells with rapamycin has several distinctive effects that mimic nutrient starvation, including inhibition of protein synthesis, cell cycle arrest at the G1/S boundary, onset of autophagy, and inhibition of ribosomal biogenesis (Heitman et al. 1991; Helliwell et al. 1994; Zheng et al. 1995; Barbet et al. 1996; Noda and Ohsumi 1998; Zaragoza et al. 1998; Powers and Walter 1999).
Recent studies have shown that TOR kinase activity is essential for the transcription of ribosomal RNA and ribosomal protein genes, as well as for the modulation of r-protein gene expression in response to changes in nutrient sources (Zaragoza et al. 1998; Powers and Walter 1999). These results demonstrate that control of gene expression at the level of transcription represents an important branch of TOR signaling. This conclusion has been extended dramatically by results of recent microarray studies where the expression of several hundred genes has been shown to be affected when TOR function is inhibited by rapamycin (Cardenas et al. 1999; Hardwick et al. 1999). In addition to genes involved in protein biosynthesis, many of the affected genes are involved in the glycolytic pathway, the TCA cycle, and nitrogen metabolism. Thus, one important role of TOR appears to be the coordination of the transcription of genes involved in several distinct nutrient-responsive cellular pathways.
One of the most striking sets of genes affected by rapamycin treatment is composed of genes involved in the use of different sources of assimilable nitrogen (Cardenas et al. 1999; Hardwick et al. 1999; C. Kao, T. Powers, P. Walter, G. Crabtree, and P. Brown, unpublished results). For example, a sharp decrease is observed in the expression of genes involved in the uptake and metabolism of preferred nitrogen sources, including glutamine and asparagine. In contrast, a corresponding increase is observed in the expression of genes involved in the uptake and use of alternative nitrogen sources, including urea, proline, and allantoin (a product of purine catabolism). These results establish a link between TOR signaling and nitrogen metabolism and suggest that one crucial role of this pathway is to couple nitrogen availability to continued cell growth. Moreover, they are consistent with results from studies of mammalian cells that demonstrate that TOR signaling is influenced by amino acid availability (Iiboshi et al. 1999b). Taken together, these results are particularly relevant given that levels of preferred nitrogen sources in human cells (e.g., glutamine and asparagine) play an important role in the progression of several diseases, including childhood acute lymphoblastic leukemia and these levels may be regulated, at least in part, by TOR (Iiboshi et al. 1999a).
The molecular mechanism by which TOR controls the expression of several genes involved in nitrogen metabolism, including GLN1, MEP2, and GAP1, has been shown recently to involve regulated changes in the subcellular localization of the Gln3 transcription factor (Beck and Hall 1999). Thus, in the presence of preferred sources of nitrogen, Gln3 is sequestered in the cytoplasm by association with the Ure2 protein, a previously identified negative regulator of Gln3 function. TOR promotes formation of a Gln3-Ure2 complex by a mechanism that involves inhibition of the Sit4 phosphatase and that requires the TOR effector protein Tap42 (Di Como and Arndt 1996; Beck and Hall 1999; Jiang and Broach 1999). Treating yeast cells with rapamycin or, alternatively, introducing them into nitrogen-poor media, causes Gln3 to become dephosphorylated and to dissociate from Ure2, where it then moves into the nucleus to activate its target genes (Beck and Hall 1999). Two additional studies suggest that TOR-dependent changes in the phosphorylation of Ure2 are also important for regulating the stability of the Gln3-Ure2 complex (Cardenas et al. 1999; Hardwick et al. 1999). Rapamycin treatment stimulates nuclear accumulation of two other transcriptional activators, Msn2 and Msn4, both of which respond to different sources of cellular stress, including carbon source limitation (Beck and Hall 1999). Thus, it is likely that additional nutrient-responsive transcription factors will also turn out to be regulated by TOR.
In addition to permeases and degradative enzymes required for the use of specific nitrogen sources, distinct pathways involved in carbon metabolism are also responsive to nitrogen availability. For example, it has been observed that several genes encoding enzymes involved in the TCA and glyoxylate cycles are required for glutamate prototrophy (Ogur et al. 1964, Ogur et al. 1965; Kim et al. 1986; Gangloff et al. 1990). These genes include ACO1, which encodes mitochondrial aconitase, and CIT1 and CIT2, which encode mitochondrial and peroxisomal forms of citrate synthase, respectively (Ogur et al. 1964; Kim et al. 1986; Rosenkrantz et al. 1986; Gangloff et al. 1990). Moreover, recent studies demonstrate that expression of these genes is subject to repression by glutamate (Liu and Butow 1999). These results reflect the fact that in addition to providing reduced NADH and FADH2 for mitochondrial respiration, the TCA cycle is also the source of many biosynthetic intermediates, including α-ketoglutarate, the primary precursor to glutamate (Stryer 1995). Thus, in the absence of exogenously supplied glutamate (or glutamine), these intermediates must be replaced through additional anaplerotic reactions to keep the TCA cycle operational and to maintain the respiratory competency of the cell. Recent studies have demonstrated that glutamate-mediated inhibition of ACO1, CIT1, and CIT2 involves regulation of the heterodimeric bHLH/Zip transcription factors Rtg1 and Rtg3 (Liu and Butow 1999). The mechanism by which these transcription factors are regulated according to nitrogen availability, however, is not well understood.
Studies of both prokaryotic and eukaryotic cells emphasize glutamate and glutamine as important regulators of nitrogen metabolism (reviewed by Magasanik 1992; Merrick and Edwards 1995; Marzluf 1997). Both are used in the biosynthesis of other amino acids and, as discussed above, can be readily converted to α-ketoglutarate for use in the TCA cycle. Glutamine is also an immediate precursor for the biosynthesis of nucleotides and other nitrogen containing molecules, including NAD+, and thus represents a primary means by which nitrogen is assimilated into cellular material. Not surprisingly, cells have evolved elaborate mechanisms to sense the intracellular levels of these amino acids and to use this information to regulate their uptake and/or synthesis. Studies of enteric bacteria have revealed a complex signaling pathway involving a two-component regulatory system that couples intracellular levels of glutamine to changes in gene expression (reviewed by Merrick and Edwards 1995). Whether TOR signaling responds specifically to intracellular levels of glutamine and/or glutamate in eukaryotic cells is presently unknown.
We are interested in understanding further both the scope and the mechanisms by which gene expression is modulated according to nitrogen availability in yeast. Toward this end, we have explored a novel use of genome-wide expression analysis by identifying genes that are expressed differentially when yeast cells are grown in the presence of two defined nitrogen sources, the primary source glutamine versus an alternative source, urea. We find that a surprisingly small number of genes (<40) show significant differences in their levels of expression under these two conditions, where each identified gene is either induced or repressed by glutamine. In addition to Gln3-dependent target genes, one of the most concise sets of genes subject to glutamine-mediated repression includes metabolic genes regulated by the transcription factors Rtg1 and Rtg3. We demonstrate that, like Gln3, Rtg1 and Rtg3 are regulated by changes in their subcellular localization according to available nitrogen and, moreover, that the TOR kinase pathway plays an essential role in this regulation. Our data further suggest that glutamine-responsive transcriptional modulation defines a distinct branch of TOR signaling in yeast.
Materials and Methods
Strains, Media, and General Methods
All strains of S. cerevisiae used in this study are listed in Table . The following culture media was used: YPD (1% yeast extract, 2% peptone, 2% dextrose); minimal dextrose (MD) (0.8% yeast nitrogen base without amino acids and ammonium sulfate, pH 5.5, 2% dextrose); synthetic complete dextrose (SCD) (0.7% yeast nitrogen base without amino acids, pH 5.5, 2% dextrose). MD media contained in addition one or more of the following nitrogen sources: glutamine, glutamate, ammonia, or urea, as indicated in the text, each at 0.2% final concentration. To supplement the auxotrophic requirements of strains used for the fluorescence microscopy experiment presented in Fig. 4 (below), required amino acids, adenine, and uracil were added to MD media at concentrations described by Sherman 1991. SCD media was also supplemented with appropriate amino acids, adenine and uracil as described by Sherman 1991. Yeast cultures were grown at 30°C for all experiments. Yeast transformations were performed using a DMSO-enhanced lithium acetate procedure (Hill et al. 1991). Rapamycin (Sigma-Aldrich) was dissolved in DMSO and added to a final concentration of 0.2 μg/ml unless stated otherwise.
Table 1.
Strains of S. cerevisiae Used in this Study
| S288c | Matα gal2 mal | Botstein Lab strain collection* |
|---|---|---|
| DBY7286 | Mata ura3 | Botstein Lab strain collection* |
| PYL037 | same as DBY7286, except for rtg1Δ::URA3 | This work |
| PLY039 | same as DBY7286, except for rtg3Δ::URA3 | This work |
| DBY8943 | Mata his3Δ::hisG | Botstein Lab strain collection* |
| PLY047 | same as DBY8943, except for RTG1-3HA:HIS3MX6 | This work |
| PLY050 | same as DBY8943, except for RTG3-3HA:HIS3MX6 | This work |
| PLY089 | same as DBY8943, except for RTG2-3HA:HIS3MX6 | This work |
| JK9-3da | Mata leu2-3, 112 ura3-52 rme1 trp1 his4 | Barbet et al. 1996 |
| JH11-1c | same as JK9-3da, except for TOR1-1 | Barbet et al. 1996 |
| PLY079 | same as EY0733, except for TOR1-1, pRTG1-GFP | This work |
| PLY083 | same as EY0735, except for TOR1-1, pRTG3-GFP | This work |
| K699 | Matα ade2-1 trp1-1 can1-100 leu2-3,112 his3-11,15 ura3 GAL+ | Nasmyth et al. 1990 |
| EY0733 | same as K699, except for rtg1Δ::TRP1 | This work |
| EY0734 | same as K699, except for rtg2Δ::TRP1 | This work |
| EY0735 | same as K699, except for rtg3Δ::TRP1 | This work |
| EY0736 | same as K699, except for msn5Δ::HIS3 | This work |
| EY0744 | same as K699, except for rtg2Δ::TRP1 msn5Δ::HIS3 | This work |
*See Materials and Methods.
Figure 4.

Rtg1 and Rtg3 are localized within the nucleus under glutamine-limiting conditions. rtg1Δ (EY0733) or rtg3Δ (EY0735) cells were transformed with pRtg1-GFP or pRtg3-GFP, respectively, and were grown to 0.5 OD600/ml in MD-glutamine or MD-urea and examined by fluorescence microscopy. Punctate nuclear fluorescence was observed for both Rtg1-GFP and Rtg3-GFP in MD-urea. The nuclear disposition of this GFP-based fluorescence was confirmed by its colocalization with DAPI-stained nuclear DNA (data not shown).
Gene Expression Analysis Using cDNA Microarrays
Strain S288c was grown with vigorous shaking to 0.5 OD600/ml in 1 liter of MD media containing appropriate nitrogen sources, as indicated in the text. Cells were immediately harvested by centrifugation, flash frozen in liquid nitrogen, and stored at −80°C. Relative mRNA levels were determined by hybridizing fluorescently labeled cDNAs to microarrays containing cDNAs representing virtually every yeast open reading frame (DeRisi et al. 1997; Lashkari et al. 1997). Arrays were produced under the auspices of J. Derisi at UCSF by a consortium of laboratories affiliated with the Department of Biochemistry and Biophysics. Primers for amplification of the yeast genome were purchased from Research Genetics and were provided by the laboratory of P. Walter. Arrays were scanned using a GenePix 4000a Microarray Scanner (Axon Instruments, Inc.) and analyzed using software provided by the manufacturer. Data were also analyzed using software available through the web site affiliated with the laboratories of P. Brown and D. Botstein at Stanford University (Stanford, CA; http://rana.Stanford.EDU/software/). Scatterplots shown in Fig. 2 (below) were constructed using Excel software (Microsoft Corp.). The complete data set for the nitrogen source experiments presented here will be made available upon request.
Figure 2.

Glutamine is both a global activator and repressor of gene expression. Scatter plots show pairwise comparisons of gene expression profiles of S288c cells grown in the presence of glutamine, urea, or glutamine + urea. (A) MD-glutamine versus MD-urea. (B) MD-urea versus MD-glutamine + urea. (C) MD-glutamine versus MD-glutamine + urea. (D) Control experiment comparing MD-glutamine with itself. For each plot, the x axis depicts cDNA samples labeled with Cy5 dye and the y axis depicts samples labeled with Cy3 dye.
Northern Blots
Northern-blot analysis was performed as described previously (Powers and Walter 1999). DNA probes were generated by PCR using genomic DNA from strain S288c as template and specific primers (purchased from Research Genetics) for individual genes (open reading frame, ORF, names are listed in Table ). Quantitation of blots was performed using a STORM 860 imaging system (Molecular Dynamics) and analyzed using software provided by the manufacturer.
Table 2.
Summary of Nitrogen-regulated Genes Identified by Microarray Analysis
| Functional class | ORF | Gene | Description | Expression ratio* | |
|---|---|---|---|---|---|
| Genes preferentially expressed in glutamine | |||||
| Transporters | YDR046C | BAP3 | Amino acid permease | 12.5 | |
| YBR068C | BAP2 | Amino acid permease | 17.4 | ||
| YCL025C | AGP1 | Amino acid permease | 12.3 | ||
| YBR069C | VAP1 | Amino acid permease | 3.5 | ||
| YOL020W | TAT2 | Tryptophan permease | 3.4 | ||
| YDR508C | GNP1 | Glutamine permease | 3.1 | ||
| YGR055W | MUP1 | Methionine permease | 10.6 | ||
| YER064C | Unknown | Putative small molecule transporter | 3.3 | ||
| YNL065W | Unknown | Putative small molecule transporter | 3.2 | ||
| Miscellaneous | YNL160W | YGP1 | Stationary phase secreted glycoprotein | 9.4 | |
| YCR021C | HSP30 | Plasma membrane heat shock protein | 4.6 | ||
| YFL014W | HSP12 | Glucose and lipid heat shock protein | 3.8 | ||
| Genes preferentially expressed in urea | |||||
| DAL cluster group | YIR027C | DAL1 | Allantoinase | 14.2 | |
| YIR029W | DAL2 | Allantoinase | 12.7 | ||
| YIR032C | DAL3 | Ureidoglycolate hydrolase | 5.3 | ||
| YIR028W | DAL4 | Allantoin permease | 18.8 | ||
| YJR152W | DAL5 | Allantoate and ureidosuccinate permease | 17.8 | ||
| YIR031C | DAL7 | Glyoxysomal malate synthase | 34.4 | ||
| YKR034W | DAL80 | Repressor of GLN3 function | 4.3 | ||
| Transporters | YKR039W | GAP1 | General amino acid permease | 5.3 | |
| YNL142W | MEP2 | Ammonia permease | 17.3 | ||
| YBR294W | SUL1 | Sulfate transport protein | 4.2 | ||
| YHL016C | DUR3 | Urea permease | 35.9 | ||
| YLR348C | DIC1 | Mitochondrial dicarboxylate carrier | 3.3 | ||
| YPL265W | DIP5 | Dicaroboxylic amino acid permease | 7.5 | ||
| Metabolism/Enzymes | YBR208C | DUR1,2 | Urea amidolyase | 5.4 | |
| YLR142W | PUT1 | Proline oxidase | 10.6 | ||
| YPR035W | GLN1 | Glutamine synthetase | 6.9 | ||
| YNL117W | MLS1 | Malate synthase 1 | 4.8 | ||
| YGL062W | PYC1 | Pyruvate carboxylase 1 | 7.5 | ||
| YLR304C | ACO1 | Aconitate hydratase | 9.5 | ||
| YNL037C | IDH1 | Isocitrate dehydrogenase 1 | 7.7 | ||
| YOR136W | IDH2 | Isocitrate dehydrogenase 2 | 8.4 | ||
| YCR005C | CIT2 | Peroxisomal citrate synthase | 10.5 | ||
| YEL071W | DLD3 | d-Lactate dehydrogenase 3 | 9.1 | ||
Prototrophic strain S288c was grown to 0.5 OD600/ml in 1 liter minimal dextrose media containing 0.2% glutamine or 0.2% urea as sole nitrogen sources. PolyA mRNA was isolated and used to prepare cDNAs that were fluorescently labeled with either Cy3 or Cy5 fluorescent dyes. Samples were mixed and applied to DNA microarrays containing .6,200 individual yeast genes and analyzed using a microarray laser reader. Shown are the results of three independent experiments. In two of these experiments, each sample was labeled with Cy3 as well as Cy5, generating a total of five independent microarrays used for analysis. Listed genes displayed a mean difference ratio of 3.0 or greater in each of the five microarrays. Several genes known from previous studies to be regulated by glutamine did not meet these strict criteria, including PUT4, ARG3, CAR1, and ASP3,4. Thus, this list is likely to represent an underestimate of the total number of glutamine-regulated genes. Gene names and descriptions were obtained from the SGD (http://genome-www.stanford.edu/Saccharomyces) and YPD (http://www.proteome.com/databases/index) databases. *Genes preferentially expressed in glutamine (glutamine vs. urea), genes preferentially expressed in urea (urea versus glutamine).
Plasmid Construction
Green fluorescent protein (GFP)–tagged plasmids were constructed by PCR amplification of the promoter and ORF of RTG1, RTG2, and RTG3. Primers were designed such that 500 base pairs of upstream promoter region and the entire ORF of each gene were amplified. Each 5′ upstream primer contained an XhoI restriction endonuclease site and each 3′ downstream primer contained an EcoRI restriction endonuclease site immediately following the stop codon. After digestion with XhoI and EcoRI, each fragment was introduced in the XhoI and EcoRI sites of pRS316-GFP, which contains GFPS65T (Kaffman et al. 1998b). The resulting plasmids, pRtg1-GFP, pRtg2-GFP, and pRtg3-GFP produced versions of Rtg1-Rtg3 that contained GFPS65T fused to their COOH termini. Plasmid pRtg1-GFP3, which contains RTG1 fused in frame with three tandem copies of GFPS65T, was constructed by replacing the promoter and coding region of pPHO4-GFP3 (Kaffman et al. 1998b) with the corresponding promoter and coding region of RTG1. pRtg3-zz was constructed by replacing the GFP in pRtg3-GFP with two Protein A z domains (zz) as described (Kaffman et al. 1998b). All plasmid-expressed, tagged genes were tested for their ability to complement the null phenotype of the appropriate deletion strains.
Construction of Yeast Strains
Strains derived from DBY7286 that were deleted for RTG1 or RTG3 were constructed using standard gene replacement techniques (Rothstein 1991). The entire coding regions of both genes were replaced with the URA3 gene from pRS306 (Sikorski and Heiter 1989). These strains were used for the experiments presented in Fig. 3 and Fig. 6 (below).
Figure 3.
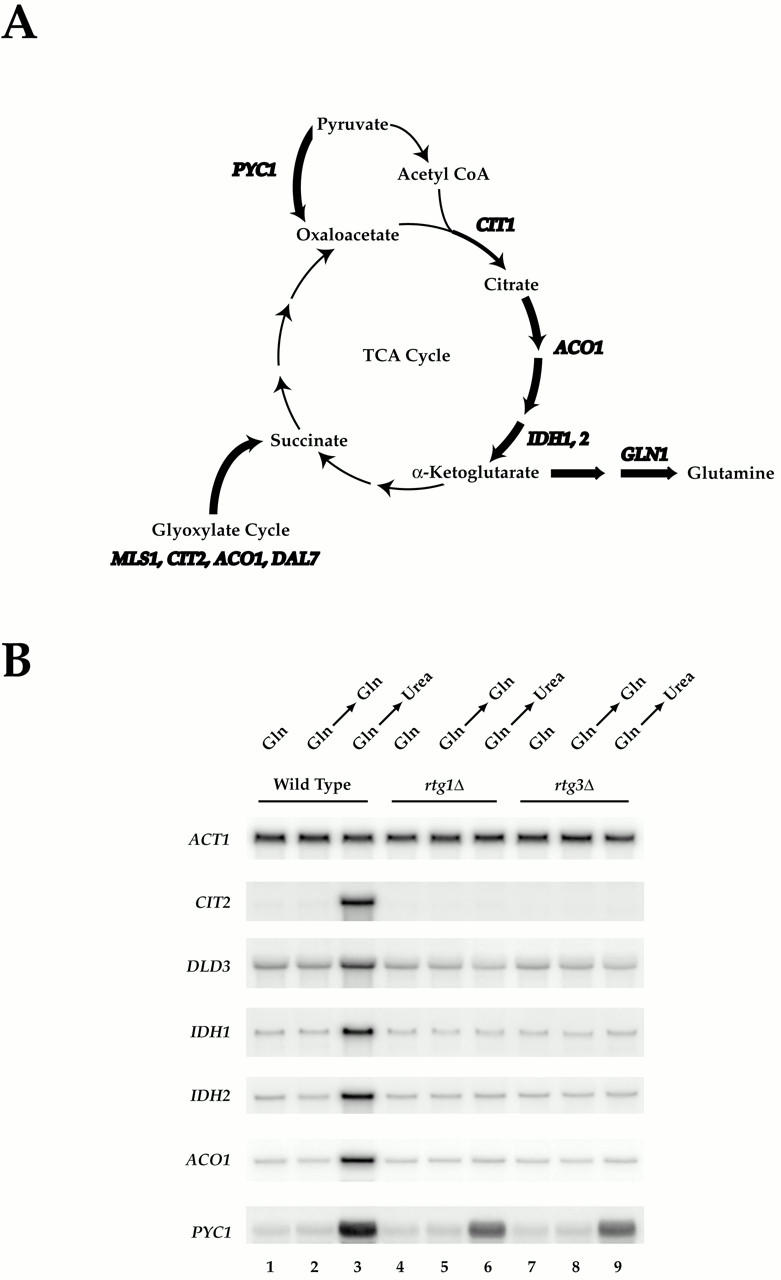
Rtg1 and Rtg3 are required for expression of distinct metabolic genes in MD-urea. (A) Summary of metabolic genes (bold) subject to glutamine-mediated transcriptional repression (see Table II; note that CIT1 is not listed in Table as its MD-glutamine/MD-urea expression ratio of ∼2.0 fell below the cut off value of 3.0 required for listing). Genes depicted were similarly repressed in MD-glutamine and MD-glutamate, except for GLN1 (see Fig. 1). (B) Nitrogen source shift experiment. Wild-type (S288c), rtg1Δ (PLY037), and rtg3Δ (PLY039) cells were grown in MD-glutamine until 0.5 OD600/ml and were either harvested (lanes 1, 4, and 7) or transferred to MD-glutamine (lanes 2, 5, and 8) or MD-urea (lanes 3, 6, and 9) media for 30 min before harvesting. RNA was prepared and analyzed by Northern blotting and probed for the specified mRNAs.
Figure 6.

Induction of RTG-dependent target genes by rapamycin. Wild-type (S288c), rtg1Δ (PLY037), and rtg3Δ (PLY039) cells were grown in MD-glutamine to 0.5 OD600/ml and were treated either with drug vehicle (lanes 1, 5, and 9) or with rapamycin for 15 (lanes 2, 6, and 10), 30 (lanes 3, 6, and 9), or 60 (lanes 4, 8, and 12) min. Cells were then harvested and RNA was prepared and analyzed by Northern blotting, probing for the specified mRNAs.
Strains derived from DBY8943 that produced versions of Rtg1-Rtg3 tagged at their COOH termini with three copies of the hemagglutinin (HA) epitope were constructed using the PCR-based method described by Brachmann et al. 1998 and Longtine et al. 1998. As template for PCR, we used the plasmid pFA6a-3HA-HIS3MX6 that contained the Schizosaccharomyces pombe HIS3 homologue (Longtine et al. 1998). The tagged genes were determined to be functional based on the normal growth of each resulting strain, PLY047, PLY050, and PLY089, on MD–ammonia and –urea agar plates. These strains were used for the experiment presented in Fig. 9 A (below).
Figure 9.
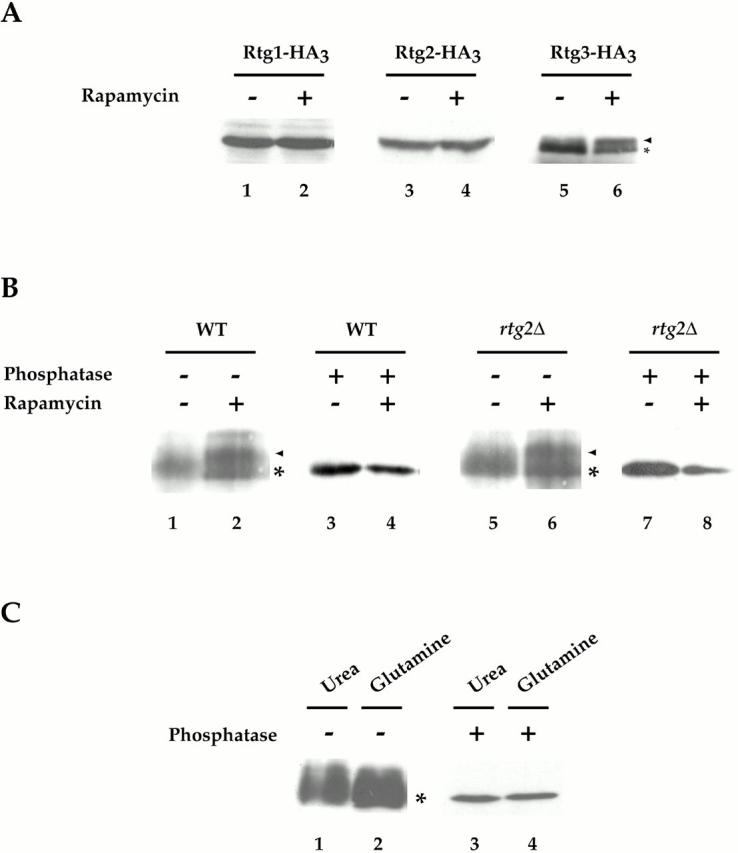
Rtg3 is a phosphoprotein and is differentially phosphorylated after rapamycin treatment. (A) Cells expressing Rtg1-HA3 (PLY047), Rtg2-HA3 (PLY089), and Rtg3-HA3 (PLY050) were grown to 0.5 OD600/ml in YPD and were treated either with drug vehicle alone or with rapamycin for 15 min. Extracts were prepared and Western blot analysis was performed using anti–HA monoclonal antibodies to detect each protein. No change in the abundance or relative mobility of Rtg1 or Rtg2 could be detected after rapamycin treatment. In contrast, a portion of Rtg3 showed an increased mobility (arrowhead) after rapamycin treatment, compared with its mobility in the absence of rapamycin (*). (B) Wild-type (K699) and rtg2Δ (EY0734) cells transformed with pRtg3-zz and were grown to 0.5 OD600/ml in SCD media lacking uracil. Cells were then treated with drug vehicle or with rapamycin for 15 min. Extracts were prepared and Rtg3-zz was immunoprecipitated with IgG-Sepharose and either mock-treated or treated with phosphatase before Western blot analysis, as indicated. Increased mobility of a portion of Rtg3-zz after rapamcyin treatment is indicated (arrowhead). (C) Wild-type (K699) cells carrying pRtg3-zz were grown to 0.5 OD600/ml in MD-glutamine or MD-urea and processed as in B. For each experiment in A–C, all samples were from the same gel. Identical results were obtained in three separate experiments.
Strains derived from K699 that were deleted for RTG1, RTG2, or RTG3 were made using the same PCR-based gene disruption technique described above, using the TRP1 gene of Candida glabrata as a selectable marker (Kitada et al. 1995). Primers used for PCR possessed 40 bases of homology that corresponded to the 5′ and 3′ ends of the open reading frame of each target gene. These strains, EY0733, EY0734, and EY0735, were transformed with an appropriate GFP fusion plasmid, described above, and used for the fluorescence microscopy experiments presented in Fig. 4, Fig. 5 A, and 7 (below).
Figure 5.
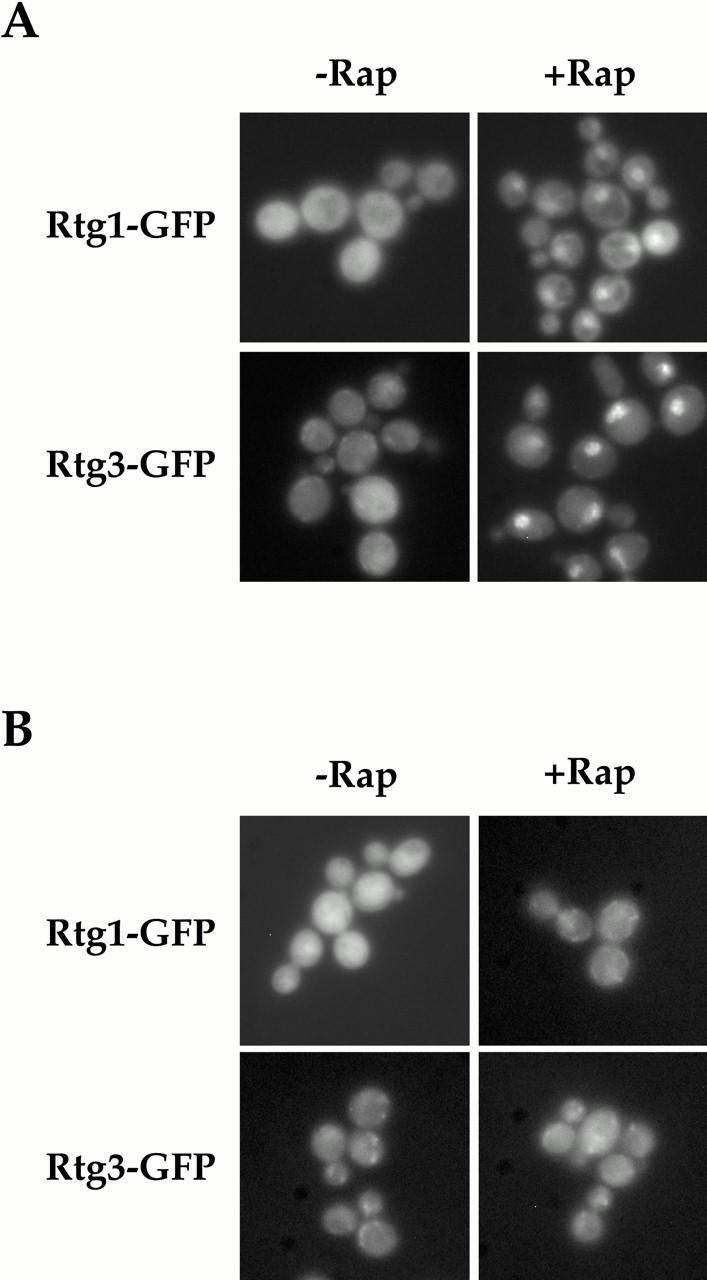
Rtg1 and Rtg3 are localized within the nucleus after rapamycin treatment in a TOR1-dependent manner. (A) rtg1Δ (EY0733) or rtg3Δ (EY0735) cells expressing Rtg1-GFP or Rtg3-GFP, respectively, were treated with drug vehicle alone (left) or with 1 μg/ml of rapamycin (right) for 5 min, followed by examination by fluorescence microscopy. Pronounced nuclear accumulation of both Rtg1-GFP and Rtg3-GFP was observed in cells treated with rapamycin. (B) The experiment in A was repeated using cells that carried the dominant rapamycin resistant TOR1-1 allele.
MSN5 was deleted from K699 using a HIS3-marked disruption vector, as described previously (Kaffman et al. 1998a). The rtg2Δ msn5Δ strain was made by mating the rtg2Δ and msn5Δ strains described above and selecting TRP+ HIS+ segregants after sporulation and tetrad dissection. The resulting msn5Δ and rtg2Δ msn5Δ strains, EY0736 and EY0744, respectively, were used for experiments presented in Fig. 8 (below).
Figure 8.
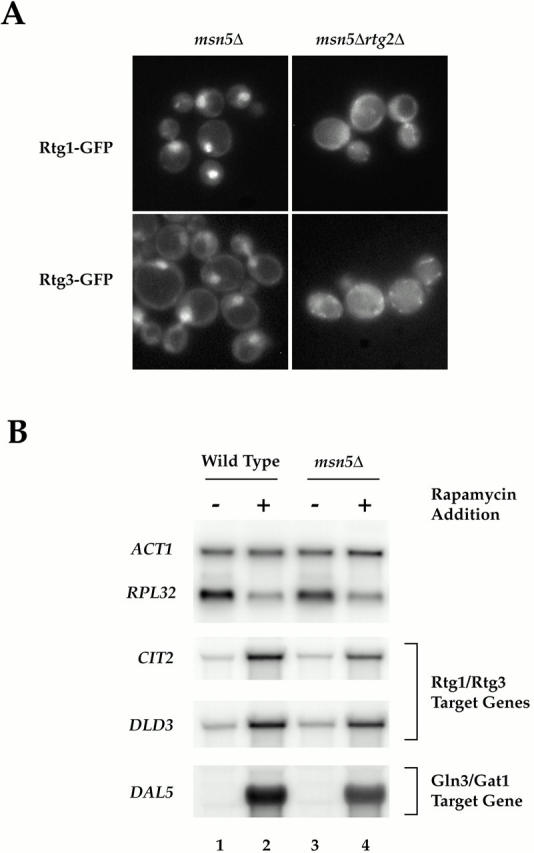
(A) Msn5 is required for export of Rtg1 and Rtg3 from the nucleus. (A) msn5Δ (EY0736) and msn5Δ rtg2Δ (EY0744) cells were transformed with pRtg1-GFP (top) or pRtg3-GFP (bottom) and grown to 0.5 OD600/ml in SCD media lacking uracil and were examined by fluorescence microscopy. Rtg1-GFP and Rtg3-GFP were localized constitutively within the nucleus in msn5Δ cells but not in msn5Δ rtg2Δ cells. (B) RTG-dependent target genes remain responsive to TOR signaling in msn5Δ cells. Wild-type (K699) and msn5Δ (EY0736) cells were grown in YPD until 0.5 OD600/ml and were treated either with drug vehicle (lanes 1 and 3) or with rapamycin (lanes 2 and 4) for 30 min (this time of incubation corresponded to the peak induction of RTG-dependent gene expression observed in the rapamycin time course in Fig. 6). Cells were then harvested and RNA was prepared and analyzed by Northern blotting, probing for the specified mRNAs.
Strains containing the TOR1-1 allele combined with either rtg1Δ/pRTG1-GFP or rtg3Δ/pRTG3-GFP were constructed using the following approach. Strains EY0733 and EY0735 were transformed with pRTG1-GFP or pRTG3-GFP, respectively. The resulting transformants were mated to strain JH11-1c and diploids were selected by their ability to grow on SCD agar plates lacking both adenine and uracil. After sporulation, TRP+, URA+ segregants were isolated and tested for their ability to grow on plates containing 0.2 μg/ml rapamycin. The resulting selected strains, PLY079 and PLY083, were used for the experiment shown in Fig. 5 B (below).
All wild-type parental strains used for construction of the above strains were examined by Northern blot analysis to confirm that expression of the RTG-dependent target genes CIT2 and DLD3 was (a) repressed by preferred nitrogen sources, and (b) induced by rapamycin treatment.
Fluorescence Microscopy
For all microscopy experiments, cells were freshly transformed with plasmids that expressed appropriate GFP-fusion proteins. Cells were first grown overnight in SCD media that lacked uracil to select for plasmid maintenance. To lower the background auto fluorescence of the parent strain, additional adenine and tryptophan were added to a final concentration of 0.005%. Cells were then diluted to 0.005 OD600/ml in media appropriate for each experiment, as indicated in the text, and were examined directly by fluorescence microscopy when they reached 0.5 OD600/ml. Rapamycin was added to a final concentration of 1.0 μg/ml for microscopy experiments presented in the figures. Identical results were also obtained at the lower rapamycin concentration of 0.2 μg/ml. All images documenting GFP localization were collected on an microscope with a 100× objective (BX60; Olympus) and recorded with a CCD camera (Photometrics) using identical settings for each experiment and an average exposure time of 1.0–1.5 s.
Preparation of Cell Extracts and Western Blot Analysis
For detection of Rtg1-Rtg3 by immunoblotting, cells were grown in appropriate media, as described in the text, to 0.5 OD600/ml, treated with rapamycin where indicated, and harvested directly. Extracts were prepared as described previously (Kaffman et al. 1998b). As Rtg3 proved to be relatively unstable in cell extracts, even in the presence of protease inhibitors, fresh extracts were prepared for each experiment and were never frozen. To detect Rtg1-HA3, Rtg2-HA3, and Rtg3-HA3, 1–2 mg of extract was separated by SDS-PAGE as described (Kaffman et al. 1998b). Anti–HA Western blots were performed using affinity-purified 12CA5 antibodies (Babco). Rtg3-zz was immunoprecipitated using IgG-Sepharose beads as described (Amersham Pharmacia Biotech). For phosphatase experiments, immunoprecipitated Rtg3-zz was washed with IgG buffer and lambda phosphatase buffer (New England Biolabs, Inc.). Samples were split in half and beads were resuspended in 100 μl of lambda phosphatase buffer with 2 mM MnCl2. Approximately 800 U of lambda protein phosphatase were added to one of the two samples and the reactions were incubated for 30–45 min at 30°C. Samples were eluted by boiling the beads in 2× SDS sample buffer for 3 min and analyzed using anti–zz rabbit polyclonal antisera as described (Kaffman et al. 1998b).
Results
Identification of Nitrogen-regulated Genes
We used genome-wide gene expression analysis to compare mRNA levels following the growth of yeast cells in the presence of two distinct sources of assimilable nitrogen, glutamine and urea. Whereas glutamine is used directly in the biosynthesis of several nitrogen-containing compounds, urea must first be degraded, via a bifunctional enzyme encoded by the DUR1,2 gene, to ammonia and carbon dioxide before the ammonium ion can be incorporated into glutamate and glutamine (Sumrada and Cooper 1982). We chose urea in part because we observed that the prototrophic strain S288c grew with a similar doubling time of ∼110 min in minimal dextrose media containing either glutamine (MD-glutamine) or urea (MD-urea) as a sole source of nitrogen. This was in contrast to several other alternative nitrogen sources that we tested, including arginine and proline, which resulted in reduced growth rates (data not shown). Thus we reasoned that any observed differences in gene expression would be restricted to the use of glutamine and urea as nitrogen sources, rather than secondary effects due to differences in growth rate. Accordingly, S288c was grown to mid-log phase in MD-glutamine or MD-urea and PolyA mRNA was isolated. Fluorescently labeled cDNAs were then prepared and applied to DNA microarrays that contained nearly every yeast open reading frame (DeRisi et al. 1997; Lashkari et al. 1997).
Of the more than 6,200 genes examined, a surprisingly small number (<40) displayed differences in expression threefold or greater under these two nitrogen conditions; 12 were expressed preferentially in MD-glutamine and 24 were expressed preferentially in MD-urea (Table ). We confirmed these results for a representative number of genes directly by Northern blot analysis (Fig. 1). The majority of genes that were expressed better in MD-glutamine encoded permeases specific for amino acids associated with rich nutrient conditions, including GNP1 and TAT2, which encode high affinity permeases specific for glutamine and tryptophan, respectively (Zhu et al. 1996; Beck et al. 1999). In contrast, genes expressed preferentially in MD-urea could be grouped into one of three general classes: (a) transport permeases specific for poor nitrogen conditions, including DUR3, which encodes urea permease; (b) the majority of the DAL genes, which are involved in the uptake and catabolism of allantoin (Cooper et al. 1979; Cox et al. 1999); and (c) metabolic enzymes, including several associated with the citric acid and glyoxylate cycles (Table ). Given the relatively small number of genes identified in this experiment, these results suggest that a limited number of regulatory pathways are likely to be involved in the differential use of these two nitrogen sources.
Figure 1.

Examples of differences in gene expression during growth of yeast cells in the presence of different sources of assimilable nitrogen. Strain S288c was grown to mid-log phase in MD media containing the indicated nitrogen sources. Total mRNA was isolated and Northern blot analysis was performed, probing for the specified mRNAs. (A) Control transcripts showing no significant differences under the conditions tested. (B) Transcripts displaying similar levels of expression in MD-glutamine and MD-glutamate. (C) Transcripts displaying more complex patterns of expression.
To extend the above results, we determined the relative expression of a representative number of genes following the growth of cells in media containing one of two other preferred nitrogen sources, glutamate and ammonia. In general, the pattern of expression produced by cells in MD-glutamate was similar to that produced in MD-glutamine, particularly for the genes involved in the TCA and glyoxylate cycles (Fig. 1 B, compare lanes 1 and 2). Several differences were also observed, however. For example, GLN1, which encodes glutamine synthetase, was expressed in MD-glutamate but not in MD-glutamine (Fig. 1 C, compare lanes 1 and 2). This result demonstrates the extreme selectivity in gene expression that exists according to the precise nitrogen source provided (Magasanik 1992).
Very few differences in gene expression were observed when ammonia and urea were compared, one of the most notable being DUR3 (Fig. 1 C, compare lanes 3 and 4). Indeed, microarray analysis revealed that <10 genes were expressed differently when cells were grown in media containing each of these two nitrogen sources (T. Powers, unpublished results). These results were somewhat surprising given that ammonia is known to display many of the same regulatory properties as glutamine and glutamate (Magasanik 1992). One likely explanation for this result is that some strains of S. cerevisiae harbor mutations that affect ammonia-dependent regulation of certain genes (Courchesne and Magasanik 1983). The precise molecular basis for this observation has yet to be clarified.
Demonstration of Glutamine as a Global Regulator of Gene Activity
The results of the above microarray experiment revealed the scope of genes whose expression differed significantly in the presence of glutamine versus urea. This experiment could not distinguish, however, whether these differences resulted from the stimulation or repression of gene activity by either nitrogen source. We reasoned that we could address this issue using microarrays by pair-wise comparisons of cultures grown in the presence of one versus both nitrogen sources. The logic here was that a given gene should display the same level of expression (i.e., have a ratio ∼1.0) if the activating (or repressing) nitrogen source was present in the two samples being compared. Accordingly, we compared mRNA levels from cells grown in the following media: MD-glutamine, MD-urea, and MD-glutamine+urea. The results of this experiment are shown in the form of scatter plots in Fig. 2.
When mRNA levels from cells grown in MD-glutamine and MD-urea were compared, most genes appeared as points along a diagonal with a slope of ∼1.0 and corresponded to genes expressed similarly under the two conditions (Fig. 2 A). A characteristic number of points fell both above and below this diagonal and corresponded to genes (reported in Table ) that were preferentially expressed in glutamine or urea, respectively (Fig. 2 A). This pattern of expression was remarkably similar when cells grown in MD-urea and MD-glutamine+urea were compared, indicating that the presence of urea in both cultures did not significantly change the relative expression of any gene (Fig. 2, compare A and B; note that the plot in B appears as the reciprocal of the plot in A due to the arrangement of axes). In dramatic contrast, when MD-glutamine and MD-glutamine+urea samples were compared, essentially all points collapsed onto the diagonal, demonstrating the dramatic effect by glutamine on gene expression (Fig. 1 C). From these results, we conclude that essentially all differences in gene expression observed in these experiments result from glutamine acting as both an activator and a repressor of gene activity. This conclusion was confirmed for a number of representative genes by Northern blotting (Fig. 1, lanes 5–7) and is consistent with the demonstrated role of glutamine as an important regulator of nitrogen-dependent gene expression (Magasanik 1992).
RTG-dependent Gene Expression: An Interface between Carbon and Nitrogen Metabolism
Many of the differences in gene expression observed in the preceding experiments were likely to reflect altered metabolic needs as cells use distinct nitrogen sources. For example, de novo biosynthesis of amino acids requires intermediates provided by the TCA cycle, primarily oxaloacetate and α-ketoglutarate, that must be replaced through anaplerotic reactions to maintain the respiratory competency of the cell (Stryer 1995). These reactions include production of succinate via the glyoxylate cycle and formation of oxaloacetate directly from pyruvate, a reaction catalyzed by pyruvate carboxylase, encoded by the PYC1 gene in yeast (Stucka et al. 1991; Walker et al. 1991). Remarkably, it is precisely the genes encoding these enzymes, as well as several TCA cycle enzymes that catalyze steps leading to the formation of α-ketoglutarate, namely ACO1, IDH1, and IDH2, that we found to be repressed by glutamine (or glutamate) (Fig. 1 B and 3 A, and data not shown). Thus, one physiological response of cells growing in the absence of these nitrogen sources was increased expression of genes involved in these anaplerotic reactions. We decided to explore this regulation in greater detail.
Two distinct transcriptional regulatory complexes, namely HAP and RTG, have been demonstrated to regulate expression of ACO1, IDH1, and IDH2 (Forsburg and Guarente 1989; McNabb et al. 1995; Liu and Butow 1999). We reasoned that the RTG genes were most likely to be involved in nitrogen-regulated expression of these genes for the following reasons: (a) HAP gene control is subject to repression by glucose, which was used as a carbon source in all our experiments; (b) the RTG genes are responsible for regulating several other glutamine-repressed genes identified in our microarray experiments, including CIT2 and DLD3; (c) rtg mutants are reported to be glutamate auxotrophs, a phenotype that would be consistent with our observations (Liu and Butow 1999). The RTG family consists of three genes, RTG1–3, where RTG1 and RTG3 both encode members of the bZip family of transcription factors that form a heterodimeric complex (Jia et al. 1997; Rothermel et al. 1997). In agreement with results from a previous study (Liu and Butow 1999), strains containing single deletions in either RTG1 or RTG3 grew very poorly on plates containing urea or ammonia as sole nitrogen sources, yet grew normally on plates containing glutamine or glutamate (data not shown).
To determine directly whether the RTG transcription factors were required for regulated expression of the above metabolic genes under our experimental conditions, we performed the following nutrient-shift experiment. Wild type, rtg1Δ, or rtg3Δ cells were grown in MD-glutamine to early log phase, and were then transferred either to fresh MD-glutamine (as a control) or to MD-urea. Total RNA was isolated and mRNA levels of a representative number of these genes were analyzed by Northern blotting and normalized to actin mRNA levels (Fig. 3 B). As expected, each gene examined displayed increased expression, relative to actin, when wild-type cells were transferred to MD-urea but not to MD-glutamine (Fig. 3 B, compare lanes 1 and 2 with 3). In contrast, no increased expression of ACO1, IDH1, IDH2, CIT2, or DLD3 was observed upon transfer of either rtg1Δ or rtg3Δ cells to MD-urea (Fig. 3 B, lanes 6 and 9). Interestingly, PYC1 expression was increased in each mutant strain in MD-urea by about half the extent observed in wild-type cells (Fig. 3 B, compare lane 3 with 6 and 9). In addition, similar levels of expression were observed for MLS1 in both wild-type and each rtg mutant strains in MD-urea (data not shown). These latter results demonstrate that factors in addition to the RTG genes are likely to be involved in the regulated expression of these two metabolic genes under these conditions and is consistent with previous biochemical and molecular genetic analyses of RTG-dependent control of PYC1 expression (Small et al. 1995; Menendez and Gancedo 1998). Taken together, these results revealed a strong correlation between RTG-dependent expression for a number of metabolic genes and the ability of yeast to grow using urea as a nitrogen source.
Nitrogen-dependent Changes in the Subcellular Localization of Rtg1 and Rtg3
We wanted to understand the mechanism by which Rtg1 and Rtg3 activity is regulated. No significant differences were observed in the steady state levels of either RTG1 or RTG3 mRNAs, nor of Rtg1 or Rtg3 proteins in MD-glutamine versus MD-urea, suggesting that their activity was regulated post-translationally (data not shown; see below). Recently, a number of nutrient-responsive transcription factors have been shown to be regulated at the level of nuclear transport (reviewed in Beck and Hall 1999; Kaffman and O'Shea 1999). We therefore tested whether Rtg1 and/or Rtg3 might be similarly regulated in this manner. Toward this end, we fused the coding region of GFP in frame to the 3′ ends of both RTG1 and RTG3 to produce fusion proteins, termed Rtg1-GFP and Rtg3-GFP. Fluorescence microscopy was then used to examine the subcellular localization of Rtg1-GFP and Rtg3-GFP in cells grown under different nitrogen conditions. Control experiments demonstrated that each fusion protein complemented the glutamine auxotrophy of its respective rtg1 or rtg3 deletion strain (data not shown).
Both Rtg1-GFP and Rtg3-GFP appeared predominantly cytoplasmic when cells were grown in MD-glutamine (Fig. 4, left). Similar results were obtained when glutamate was used instead as a nitrogen source (data not shown). In contrast, both proteins were concentrated in the nucleus when cells were grown in MD-urea (Fig. 4, right). Also, in close agreement with the transcriptional responses described above, both Rtg1-GFP and Rtg3-GFP remained cytoplasmic when cells were grown in media that contained both glutamine and urea (data not shown). From these results, we conclude that RTG-dependent gene activation involves changes in the subcellular distribution of the Rtg1/Rtg3 complex.
Rtg1 and Rtg3 Activity and Subcellular Localization Is Regulated by the TOR Pathway
We wished to identify the signaling pathway(s) that linked nitrogen quality to the localization of the Rtg1/Rtg3 complex. Here a clue was provided by the fact that many RTG-dependent target genes become induced when cells are treated with rapamycin, a specific inhibitor of the TOR kinases (Hardwick et al. 1999; C. Kao, T. Powers, P. Walter, G. Crabtree, and P. Brown, unpublished observations). Accordingly, we localized Rtg1-GFP and Rtg3-GFP in cells grown in nitrogen-rich media in both the absence and presence of rapamycin. As expected, both proteins remained in the cytoplasm in the absence of rapamycin (Fig. 5 A, left). In striking contrast, strong nuclear accumulation of both Rtg1-GFP and Rtg3-GFP was observed within 5 min of rapamycin addition (Fig. 5 A, right). Nuclear accumulation of these proteins by rapamycin was due specifically to inhibition of the TOR pathway, since little or no accumulation was observed when this experiment was repeated using a strain that contained the dominant rapamycin-resistant TOR1-1 allele (Fig. 5 B, right). These results demonstrate that nuclear accumulation of Rtg1 and Rtg3 in rich nitrogen media is prevented by a functional TOR pathway.
A prediction of the above results was that inhibiting the TOR pathway might be sufficient to result in RTG-dependent gene activation. To test this directly, we performed a time course of rapamycin treatment of wild-type, rtg1Δ, and rtg3Δ cells grown in MD-glutamine, and then analyzed mRNA levels of several RTG-dependent targets by Northern blotting. In wild-type cells, each target gene examined showed increased expression within 15 min after addition of rapamycin (Fig. 6, compare lanes 1 and 2). The expression levels of these genes peaked at ∼30 min and were comparable with the levels observed in cells grown in MD-urea (Fig. 6, lane 3; compare with Fig. 3 B, lane 3).
In striking contrast, no induction was observed for any of these genes when rapamycin was added to rtg1Δ and rtg3Δ cells (Fig. 6 B, lanes 5–12). The sole exception was PYC1, which showed a level of induction in each mutant strain of about half that observed in wild-type cells. This latter result is thus reminiscent of the behavior of PYC1 in the nutrient shift experiment described above (Fig. 4 B) and indicates that an additional rapamycin-sensitive regulatory factor(s) is involved in the expression of this gene. As a control, we observed similar induction of two Gln3-dependent targets, DUR3 and DAL5, in both wild-type and the rtg deletion strains, demonstrating that the loss of induction in the rtg mutants is specific for RTG-dependent targets. The specificity of these results was also confirmed by an observed decrease in RPL32 mRNA levels in all strains after rapamycin treatment, as reported previously (Powers and Walter 1999). Taken together, these results directly link TOR activity to both the subcellular localization and the activity of the Rtg1/Rtg3 complex.
Interestingly, deletion of the genes encoding the GATA transcriptional regulators Gln3 and Gat1, whose nucleocytoplasmic transport is similarly regulated by TOR, confers weak resistance to rapamycin (Beck and Hall 1999). By contrast, we observed no change in the sensitivity of rtg1Δ or rtg3Δ cells to this drug, in comparison to wild-type cells (data not shown). Thus, we conclude that rapamycin-induced expression of RTG-dependent target genes does not contribute to the toxic effects of this drug on yeast cells.
Regulated Nucleocytoplasmic Transport of Rtg1 and Rtg3 Requires All Three RTG Genes
The third member of the RTG gene family, RTG2, encodes a cytoplasmic protein that contains an HSP70-like ATP binding domain and displays homology to certain bacterial polyphosphatases and phosphatases (Liao and Butow 1993; Koonin 1994). Genetic evidence indicates that this gene functions upstream of RTG1 and RTG3 and is essential for expression of RTG-dependent target genes (Rothermel et al. 1997). We asked whether Rtg2 was involved in TOR-regulated nuclear accumulation of Rtg1 and Rtg3 by monitoring the localization of Rtg1-GFP and Rtg3-GFP in rtg2Δ cells, in both the absence and presence of rapamycin. As in wild-type cells, both proteins were localized to the cytoplasm in the absence of drug (Fig. 7 A, left). In contrast, no nuclear accumulation of either protein was observed after addition of rapamycin (Fig. 7 A, right). Additional experiments demonstrated that Rtg2 was itself a cytoplasmic protein and that its localization did not change after rapamycin treatment (data not shown). Thus, these results demonstrate that Rtg2 is essential for rapamycin-induced nuclear accumulation of the Rtg1/Rtg3 complex and that it carries out this function in the cytoplasm.
Figure 7.
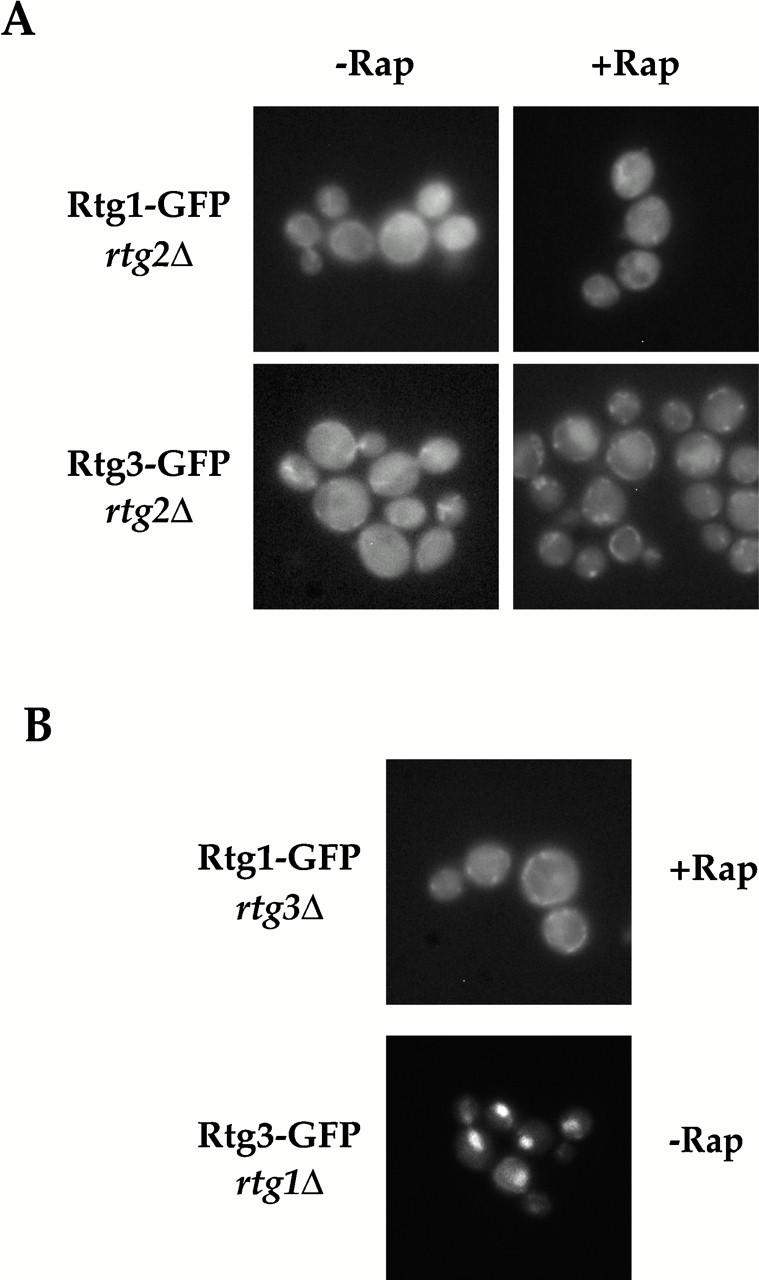
Regulated nucleocytoplasmic transport of Rtg1 and Rtg3 requires Rtg1, Rtg2, and Rtg3. (A) Rtg1-GFP and Rtg3-GFP were visualized in rtg2Δ (EY0734) cells in the absence (left) or presence (right) of rapamycin. In contrast to wild-type cells, neither Rtg1-GFP nor Rtg3-GFP relocate to the nucleus after rapamycin treatment (compare with Fig. 5 A). (B) Rtg1-GFP was visualized in rtg3Δ (EY0735) cells (top) and Rtg3-GFP was visualized in rtg1Δ (EY0733) cells (bottom). Note that Rtg1-GFP cannot accumulate in the nucleus upon rapamycin treatment in rtg3Δ cells. In contrast, Rtg3-GFP is localized constitutively to the nucleus in rtg1Δ cells, even in the absence of rapamycin addition.
We next determined whether both Rtg1 and Rtg3 were required for TOR-regulated nuclear transport of the Rtg1/Rtg3 complex by monitoring the localization of Rtg1-GFP in rtg3Δ cells or, alternatively, Rtg3-GFP in rtg1Δ cells, both before and after rapamycin treatment. We observed that Rtg1-GFP remained exclusively cytoplasmic in rtg3Δ cells, even after rapamycin addition (Fig. 7 B, top, and data not shown). Since Rtg1 is a relatively small protein of 177 amino acids, we wanted to exclude the possibility that the constitutive presence of Rtg1-GFP in the cytoplasm in rtg3Δ cells was not due simply to the diffusion of this protein out of the nucleus after rapamycin treatment. Toward this end, we fused three tandem copies of the coding region of GFP to the 3′ end of the RTG1 gene to create a much larger protein, termed Rtg1-GFP3. Control experiments confirmed that this fusion protein was functional (data not shown). We observed that Rtg1-GFP3 also remained in the cytoplasm in rapamycin-treated cells, indicating that Rtg1 cannot accumulate in the nucleus in the absence of Rtg3 (data not shown).
In striking contrast, we observed that Rtg3-GFP was localized exclusively in the nucleus in rtg1Δ cells, in both the presence and absence of rapamycin (Fig. 7 B, bottom, and data not shown). Thus, regulated transport of the Rtg1/Rtg3 complex requires that both proteins be present together. One potential explanation to account for the constitutive localization of these two proteins in different subcellular compartments is that Rtg1 contains a nuclear export signal, whereas Rtg3 contains the nuclear import signal (NLS) for the heterodimer. Consistent with this interpretation, a recent study has confirmed that the basic domain of the bHLH motif of Rtg3 contains a functional NLS (Sekito et al. 2000). Interestingly, however, we observed that Rtg3 was also localized constitutively to the nucleus when cells were deleted for both RTG1 and RTG2 (data not shown). Given that Rtg3 is localized exclusively in the cytoplasm when cells are singly deleted for RTG2 (Fig. 7 A), together these results suggest that Rtg1 may play a more antagonistic role in regulating Rtg3 nuclear import. A similar conclusion has also been reached recently by Sekito et al. 2000.
Export of Rtg1 and Rtg3 from the Nucleus Requires the β-Importin Homologue Msn5
Constitutive localization of Rtg3 in the nucleus in rtg1Δ cells suggested that export of the Rtg1/Rtg3 complex from the nucleus might play a role in the regulation of these transcription factors. Previous studies of another nutrient-responsive transcription factor, Pho4, has demonstrated that its export from the nucleus depends on the activity of Msn5, a member of the β-importin family of nuclear receptors (Kaffman et al. 1998a). We therefore tested whether export of Rtg1 and Rtg3 might similarly be regulated by this protein. Indeed, we observed that both Rtg1-GFP and Rtg3-GFP were constitutively nuclear in msn5Δ cells, demonstrating that this factor is required, either directly or indirectly, for export of the Rtg1/Rtg3 complex from the nucleus (Fig. 8 A, left). Interestingly, when we examined the localization of Rtg1-GFP and Rtg3-GFP in msn5Δ rtg2Δ cells, both proteins remained in the cytoplasm, consistent with the above observation that Rtg2 is absolutely required for nuclear entry of the Rtg1/Rtg3 complex (Fig. 8 A, right). Additional control experiments demonstrated that Rtg2-GFP remained in the cytoplasm in msn5Δ cells (data not shown).
If regulated access to the nucleus represents the primary mechanism by which the activity of the Rtg1/Rtg3 complex is controlled, then we expected to observe constitutive activation of their target genes in msn5Δ cells in the absence of rapamycin. However, no such increased expression of RTG-target genes was observed in msn5Δ cells compared with wild type (Fig. 8 B, compare lanes 1 and 3). This observation is consistent with studies of other regulated transcription factors; namely, that constitutive nuclear localization does not necessarily result in gene activation and that other regulatory mechanisms are involved (Komeili and O'Shea 1999). Remarkably, however, we observed rapid induction of RTG-dependent target genes in msn5Δ cells after addition of rapamycin (Fig. 8 B, compare lanes 3 and 4). This latter result demonstrates that despite its steady state nuclear localization in msn5Δ cells, the Rtg1/Rtg3 complex remains responsive to changes in TOR signaling.
TOR-dependent Changes in the Phosphorylation State of Rtg3
Previous studies have demonstrated that changes in phosphorylation of a transcription factor can be important for regulating its activity as well as concentration in the nucleus (Beck and Hall 1999; Komeili and O'Shea 1999). We therefore wished to determine whether rapamycin-induced nuclear accumulation of Rtg1 and Rtg3 also correlated with changes in the phosphorylation state of either of these proteins. Toward this end, we used immunoblot analysis to monitor the electrophoretic mobility of these proteins, as well as Rtg2, from extracts in which each protein was genomically tagged at its COOH terminus with the HA3 epitope (see Materials and Methods). Extracts were prepared from cells grown in rich nitrogen media that had been incubated with drug vehicle alone or with rapamycin, and then subsequently prepared in either the presence or absence of phosphatase inhibitors. No change in the relative abundance or mobility of Rtg1-HA3 or Rtg2-HA3 was observed in untreated versus rapamycin-treated cells (Fig. 9 A, lanes 1–4). In contrast, a slower migrating form of Rtg3-HA3 appeared after addition of rapamycin, which was observed only when extracts were prepared in the presence of phosphatase inhibitors (Fig. 9 A, compare lanes 5 and 6, data not shown). These results suggested that inhibition of TOR signaling resulted in a change in the phosphorylation state of Rtg3.
We wanted to confirm that Rtg3 was a phosphoprotein by treating purified Rtg3 with phosphatases in vitro; however, the HA epitope-tagged form of this protein was not efficiently immunoprecipitated from cell extracts. We therefore used a form of Rtg3 that was fused at its COOH terminus to two z domains from Protein A, termed Rtg3-zz, which could be immunoprecipitated quantitatively from extracts using immobilized-IgG (data not shown). The results confirmed that rapamycin treatment resulted in the conversion of a portion of Rtg3-zz to a more slowly migrating form (Fig. 9 B, compare lanes 1 and 2). Moreover, this slower form was abolished after treatment with phosphatase, confirming that the rapamycin-induced change in electrophoretic mobility of Rtg3-zz is due to changes in phosphorylation (Fig. 9 B, compare lanes 2 and 4; arrowhead, slower migrating form). Interestingly, the rapamycin-induced shift in Rtg3 mobility was also observed in rtg2Δ cells, suggesting that TOR influences Rtg3 phosphorylation independently from Rtg2 function (Fig. 9 B, compare lanes 5 and 6).
While the above results demonstrate that TOR influences the phosphorylation state of Rtg3, several other results indicate that phosphorylation-dependent regulation of this protein is likely to be complex. First, we observed that Rtg3-zz was phosphorylated in the absence of rapamycin treatment, a conclusion based on its increased mobility after phosphatase treatment, suggesting that Rtg3 is likely to be phosphorylated on multiple sites (Fig. 9 B, compare lanes 1 and 3). The existence of multiple sites of phosphorylation could account for the relatively poor resolution of Rtg3 on SDS-PAGE gels in the absence of phosphatase treatment. Second, we were unable to detect a difference in the mobility of Rtg3-zz in cells grown in MD-glutamine versus MD-urea (Fig. 9 C, lanes 1 and 2). This latter result suggests either that rapamycin-induced changes are transient or that additional changes in the phosphorylation state of this protein occur during steady state growth of cells in media lacking glutamine. Finally, multiple levels of regulation, possibly mediated by distinct changes in the phosphorylation state of Rtg3, would be consistent with our above observation that the Rtg1/Rtg3 complex is concentrated in the nucleus in msn5Δ cells, yet their target genes remain uninduced. Identifying the residue(s) of Rtg3 that are phosphorylated under these different conditions will be required to resolve these issues.
Discussion
Genome-wide expression analysis represents a powerful approach for exploring the scope of transcriptional regulation associated with different biological processes. The appeal of this method is most often attributed to the enormous amount of information that it can yield, as exemplified by studies in yeast of the diauxic shift, sporulation, and the cell cycle, where each of these processes involves sequential changes in the expression of groups of coregulated genes that number well into the hundreds (DeRisi et al. 1997; Chu et al. 1998; Spellman et al. 1998). By contrast, our results presented here demonstrate that by studying the effects of subtle changes in growth conditions (e.g., by varying the source of assimilable nitrogen), microarray analysis can help bring a specific biological problem into sharper focus by identifying a concise set of coregulated genes.
Specifically, we have demonstrated that a surprisingly small number of genes are differentially expressed when yeast cells use glutamine versus urea as their sole source of nitrogen (Table ). All of these genes are either induced or repressed by the presence of glutamine, a fact that is consistent with its established importance as a major regulator of nitrogen metabolism (Magasanik 1992; Marzluf 1997). One prominent group of glutamine-repressed genes encodes proteins involved in the uptake and metabolism of alternative sources of nitrogen, including urea and allantoin, and whose expression is controlled by the Gln3 and Ure2 regulatory proteins (Coffman et al. 1997; Cox et al. 1999). Recent studies have demonstrated that these genes are negatively regulated by the TOR signaling pathway via a mechanism that involves sequestration of the transcription factor Gln3 in the cytoplasm (Beck and Hall 1999).
A second prominent group of glutamine-repressed genes encodes enzymes involved in the TCA and glyoxylate cycles and whose expression in urea-containing media requires the heterodimeric transcription factors Rtg1 and Rtg3. Differential expression of these metabolic genes is consistent with the proposal by Liu and Butow 1999 that one important role of RTG-dependent gene expression is to provide adequate levels of α-ketoglutarate for use in the de novo biosynthesis of glutamate, which in turn is required for the synthesis of glutamine. In addition, our results extend involvement of RTG-dependent regulation to include PYC1, which provides an alternative route for the synthesis of oxaloacetate for use in both the TCA cycle and in amino acid biosynthesis (Fig. 3). These results thus highlight the intimate relationship that exists between nitrogen and carbon metabolism as well as its importance to normal cell growth.
Remarkably, we find that TOR signaling also regulates RTG-dependent gene activity. Specifically, inhibition of the TOR kinases by rapamycin results in both rapid nuclear accumulation of the Rtg1/Rtg3 complex as well as induction of their target genes. In addition, rapamycin treatment correlates with changes in the phosphorylation state of Rtg3, indicating that nucleocytoplasmic transport of the complex is likely to be regulated by differential phosphorylation of Rtg3. These results thus contribute to a growing body of evidence demonstrating that one important role of TOR is to control the activity of specific nutrient-responsive transcription factors. Moreover, our findings suggest that TOR signaling may provide an important mechanism by which carbon and nitrogen use are linked. Whether Rtg3 phosphorylation is influenced by the activity of the Tap42-Sit4 phosphatase complex, as has been shown recently for Gln3 (Beck and Hall 1999), remains to be determined.
Our results also shed light on the functional role of Rtg2 in regulating RTG-dependent gene expression, a previously identified upstream positive regulator of Rtg1 and Rtg3 (Liao and Butow 1993; Rothermel et al. 1997). Specifically, we find that Rtg2 is required for rapamycin-induced entry of the Rtg1/Rtg3 complex into the nucleus (Fig. 10). How Rtg2 acts mechanistically is unclear at present; however, we believe that Rtg2 is likely to be required for a step subsequent to the action of TOR, based upon the observation that rapamycin-induced changes in Rtg3 phosphorylation are observed in rtg2Δ mutant cells (Fig. 8 B). Furthermore, our examination of Rtg3 localization in rtg1Δ, rtg2Δ, and rtg1Δ rtg2Δ mutant cells suggests that Rtg2 may interact primarily with Rtg1 to regulate nuclear entry of the Rtg1/Rtg3 complex (Fig. 7, and data not shown).
Figure 10.
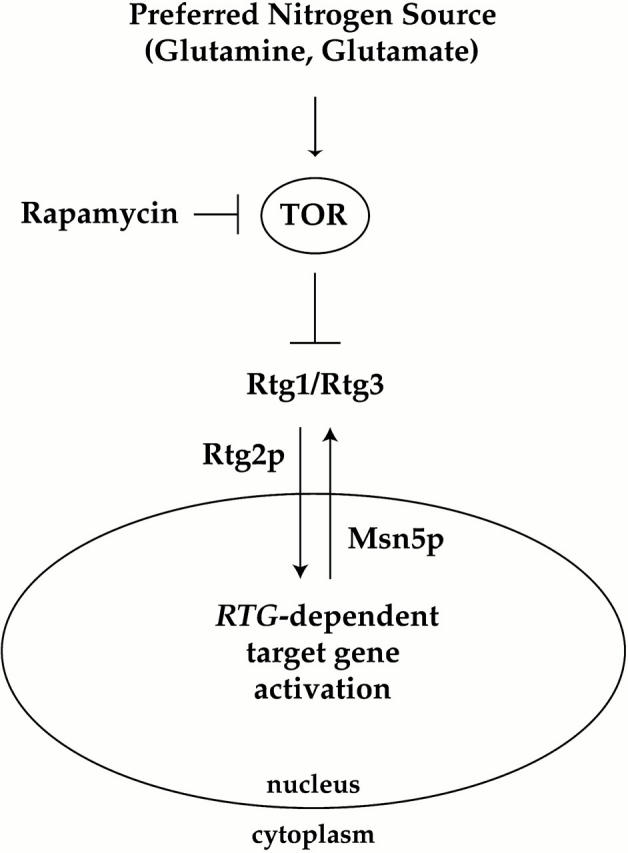
Model for involvement of the TOR pathway in nitrogen-dependent regulation of the Rtg1 and Rtg3 transcription factors. See text for details.
In contrast to the role played by Rtg2, we find that Msn5, a member of the importin-β family of nuclear transport receptors, is required for nuclear export of the Rtg1/Rtg3 complex (Fig. 10). Thus, in msn5Δ cells, both Rtg1 and Rtg3 are constitutively localized to the nucleus. We do not know yet whether Msn5 interacts directly with Rtg1 and/or Rtg3 to facilitate their export from the nucleus, as has been demonstrated for Pho4 (Kaffman et al., 1998). It is possible that Msn5 is instead required for the proper localization of another protein(s) that is involved directly in the export of the Rtg1/Rtg3 complex.
While nucleocytoplasmic transport of the Rtg1/Rtg3 complex is essential for regulated RTG-dependent gene activation, our results indicate that additional control mechanisms are involved. This conclusion is based on our analyses of msn5Δ cells, where both Rtg1 and Rtg3 are concentrated in the nucleus yet their target genes remain uninduced (Fig. 9). As these genes can nevertheless be induced rapidly in msn5Δ cells following rapamycin treatment, these results raise the intriguing possibility that TOR regulates Rtg1/Rtg3 activity in both the cytoplasm as well as in the nucleus.
RTG1-RTG3 were originally identified as genes required for increased expression of CIT2 under conditions where mitochondrial respiratory function is impaired, as in rho0 petite mutants that lack mitochondrial DNA (Liao et al. 1991; Liao and Butow 1993). This process, termed retrograde regulation, is believed to involve intracellular signaling from the mitochondria to the nucleus (Liao and Butow 1993). Subsequently, the RTG genes have been shown to be involved in the expression of several genes that encode peroxisomal proteins in addition to glyoxylate cycle enzymes (Chelstowska and Butow 1995; Kos et al. 1995; McCammon 1996; Liu and Butow 1999). Indeed, the results of these studies suggest that both mitochondrial and peroxisomal functions are intimately linked via RTG-dependent gene expression. An important question prompted by our present results thus concerns the relationship between TOR signaling and retrograde regulation. While this manuscript was in preparation, Sekito et al. 2000 reported that Rtg1 and Rtg3 are localized constitutively to the nucleus in rho0 cells. Moreover, this localization depends on a functional Rtg2 protein. Given these similarities to what we present here, one possibility is that TOR may be integrated with retrograde signaling.
At odds with this conclusion, however, is the finding that nuclear accumulation of the Rtg1/Rtg3 complex in rho0 cells appears to correlate with a substantial decrease in phosphorylation of Rtg3 (Sekito et al. 2000). By contrast, our results suggest that nuclear import of this complex after rapamycin treatment may coincide with increased phosphorylation of Rtg3. Moreover, and very importantly, we were unable to detect a difference in the mobility of Rtg3 on SDS-PAGE following the growth of cells in glutamine- versus urea-containing media (Fig. 9 C). Nevertheless, the Rtg1/Rtg3 complex is localized in different subcellular compartments, cytoplasm versus nucleus, under these different nitrogen conditions (Fig. 4). Thus, the precise relationship between the phosphorylation state of Rtg3 and its nucleocytoplasmic disposition needs further clarification, which will require identification of residues in Rtg3 that are subject to phosphorylation under different cellular conditions. At present, it thus remains possible that retrograde and TOR signaling may represent independent routes by which Rtg1 and/or Rtg3 are regulated and that both pathways ultimately converge on the same nucleocytoplasmic transport step.
Inhibition of the TOR kinases by rapamycin affects the expression of a large number of genes, including those regulated by nitrogen and glucose-sensitive signaling pathways, glycolytic genes, genes expressed during the diauxic shift, and r-protein and rRNA genes (Beck and Hall 1999; Cardenas et al. 1999; Hardwick et al. 1999; Powers and Walter 1999; C. Kao, T. Powers, P. Walter, G. Crabtree, P. Brown, unpublished observations). By contrast, we find that only a small subset of these targets are expressed differentially in our nitrogen source experiments, in particular the GLN3- and RTG-dependent genes described above. These observations suggest that distinct branches of the TOR pathway may be regulated independently according to the precise nutritional state of the cell and, moreover, suggest that TOR activity may be modulated by multiple upstream nutritional signals. On the other hand, many of the glutamine-regulated genes identified in this study are affected to some extent by rapamycin treatment (T. Powers, unpublished results). Thus, we believe that TOR signaling is likely to represent an important mechanism by which the availability of glutamine and/or glutamate is coupled to changes in gene expression in eukaryotic cells. One challenge now at hand is to understand how TOR activity is regulated under different nitrogen conditions. Such studies will undoubtedly prove invaluable toward deciphering the role that this pathway plays in promoting normal cell growth.
Acknowledgments
We are grateful to J. Derisi for his expertise and leadership and to P. Walter and other members of the Department of Biophysics and Biochemistry for their help in establishing DNA microarray technology at UC San Francisco. We thank H. Bennett for her expert instruction in preparing fluorescently labeled cDNAs. We thank D. Botstein for strains and C. Kao for help with data analysis and for sharing her unpublished results. We thank K. Burtis for help with microarray development at UC Davis. We also thank the members of the Powers lab for their assistance during the course of this work as well as J. Derisi for valuable discussions. Finally, we thank S. Burgess, W. Heyer, M. Niwa, and M. Singer for comments on the manuscript.
This work was supported by laboratory start up funds as well as a faculty research grant from U.C. Davis (T. Powers) and by grant GM 59034 from the National Institutes of Health (E.K. O'Shea). A. Komeili is a Howard Hughes Medical Institute predoctoral fellow.
Footnotes
Abbreviations used in this paper: GFP, green fluorescent protein; HA, hemagglutinin; MD, minimal dextrose; ORF, open reading frame; SCD, synthetic complete dextrose; TOR, target of rapamycin.
References
- Barbet N.C., Schneider U., Helliwell S.B., Stansfield I., Tuite M.F., Hall M.N. TOR controls translation initiation and early G1 progression in yeast. Mol. Biol. Cell. 1996;7:25–42. doi: 10.1091/mbc.7.1.25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beck T., Hall M.N. The TOR signalling pathway controls nuclear localization of nutrient-regulated transcription factors. Nature. 1999;402:689–692. doi: 10.1038/45287. [DOI] [PubMed] [Google Scholar]
- Beck T., Schmidt A., Hall M.N. Starvation induces vacuolar targeting and degradation of the tryptophan permease in yeast. J. Cell Biol. 1999;146:1227–1238. doi: 10.1083/jcb.146.6.1227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brachmann C.B., Davies A., Cost G.J., Caputo E., Li J., Hieter P., Boeke J.D. Designer deletion strains derived from Saccharomyces cerevisiae S288Ca useful set of strains and plasmids for PCR-mediated gene disruption and other applications. Yeast. 1998;14:115–132. doi: 10.1002/(SICI)1097-0061(19980130)14:2<115::AID-YEA204>3.0.CO;2-2. [DOI] [PubMed] [Google Scholar]
- Cardenas M.E., Cutler N.S., Lorenz M.C., Di Como C.J., Heitman J. The TOR signaling cascade regulates gene expression in response to nutrients. Genes Dev. 1999;13:3271–3279. doi: 10.1101/gad.13.24.3271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chelstowska A., Butow R.A. RTG genes in yeast that function in communication between mitochondria and the nucleus are also required for expression of genes encoding peroxisomal proteins. J. Biol. Chem. 1995;270:18141–18146. doi: 10.1074/jbc.270.30.18141. [DOI] [PubMed] [Google Scholar]
- Chu S., DeRisi J.L., Eisen M., Mulholland J., Botstein D., Brown P.O., Herskowitz I. The transcriptional program of sporulation in budding yeast. Science. 1998;282:699–705. doi: 10.1126/science.282.5389.699. [DOI] [PubMed] [Google Scholar]
- Courchesne W.E., Magasanik B. Ammonia regulation of amino acid permeases in Saccharomyces cerevisiae . Mol. Cell. Biol. 1983;3:672–683. doi: 10.1128/mcb.3.4.672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coffman J.A., Rai R., Loprete D.M., Cunningham T., Svetlov V., Cooper T.G. Cross regulation of four GATA factors that control nitrogen catabolic gene expression in Saccharomyces cerevisiae . J. Bacteriol. 1997;179:3416–3429. doi: 10.1128/jb.179.11.3416-3429.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper T.G., Gorski M., Turoscy V. A cluster of three genes responsible for allantoin degradation in Saccharomyces cerevisiae . Genetics. 1979;92:383–396. doi: 10.1093/genetics/92.2.383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cox K.H., Pinchak A.B., Cooper T.G. Genome-wide transcriptional analysis in S. cerevisiae by mini-array membrane hybridization. Yeast. 1999;15:703–713. doi: 10.1002/(SICI)1097-0061(19990615)15:8<703::AID-YEA413>3.0.CO;2-Z. [DOI] [PubMed] [Google Scholar]
- Dennis P.B., Fumagalli S., Thomas G. Target of rapamycin (TOR)balancing the opposing forces of protein synthesis and degradation. Curr. Opin. Gen. Dev. 1999;9:49–54. doi: 10.1016/s0959-437x(99)80007-0. [DOI] [PubMed] [Google Scholar]
- DeRisi J.L., Vishwanath I.R., Brown P.O. Exploring the metabolic and genetic control of gene expression on a genomic scale. Science. 1997;278:680–686. doi: 10.1126/science.278.5338.680. [DOI] [PubMed] [Google Scholar]
- Di Como C.J., Arndt K.T. Nutrients, via the Tor proteins, stimulate the association of Tap42 with type 2A phosphatases. Genes Dev. 1996;10:1904–1916. doi: 10.1101/gad.10.15.1904. [DOI] [PubMed] [Google Scholar]
- Forsburg S.L., Guarente L. Communication between mitochondria and the nucleus in regulation of cytochrome genes in the yeast Saccharomyces cerevisiae . Annu. Rev. Cell Biol. 1989;5:153–180. doi: 10.1146/annurev.cb.05.110189.001101. [DOI] [PubMed] [Google Scholar]
- Gangloff S.P., Marguet D., Lauquin G.J.-M. Molecular cloning of the yeast mitochondrial aconitase gene (ACO1) and evidence of a synergistic regulation of expression by glucose plus glutamate. Mol. Cell. Biol. 1990;10:3551–3561. doi: 10.1128/mcb.10.7.3551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardwick J.S., Kuruvilla F.G., Tong J.K., Shamji A.F., Schreiber S.L. Rapamycin-modulated transcription defines the subset of nutrient-sensitive signaling pathways directly controlled by the Tor proteins. Proc. Natl. Acad. Sci. USA. 1999;96:14866–14870. doi: 10.1073/pnas.96.26.14866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heitman J., Movva N.R., Hall M.N. Targets for cell cycle arrest by the immunosuppressant rapamycin in yeast. Science. 1991;253:905–909. doi: 10.1126/science.1715094. [DOI] [PubMed] [Google Scholar]
- Helliwell S.B., Wagner P., Kunz J., Deuter-Reinhard M., Henriquez R., Hall M.N. TOR1 and TOR2 are structurally and functionally similar but not identical phosphatidylinositol kinase homologues in yeast. Mol. Biol. Cell. 1994;5:105–118. doi: 10.1091/mbc.5.1.105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hill J., Donald K.A., Griffiths D.E., Donald G. DMSO-enhanced whole cell yeast transformation. Nucleic Acids Res. 1991;19:5791. doi: 10.1093/nar/19.20.5791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iiboshi Y., Papst P.J., Hunger S.P., Terada N. l-Asparaginase inhibits the rapamycin-targeted signaling pathway Biochem. Biophys. Res. Commun. 260 1999. 534 539a [DOI] [PubMed] [Google Scholar]
- Iiboshi Y., Papst P.J., Kawasome H., Hosoi J., Abraham R.T., Houghton P.J., Terada N. Amino acid–dependent control of p70s6k. Involvement of tRNA aminoacylation in the regulation J. Biol. Chem. 274 1999. 1092 1099b [DOI] [PubMed] [Google Scholar]
- Jia Y., Rothermel B., Thornton J., Butow R.A. A basic helix-loop-helix-leucine zipper transcription complex in yeast functions in a signaling pathway from mitochondria to the nucleus. Mol. Cell. Biol. 1997;17:1110–1117. doi: 10.1128/mcb.17.3.1110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang Y., Broach J.R. Tor proteins and protein phosphatase 2A reciprocally regulate Tap42 in controlling cell growth in yeast. EMBO (Eur. Mol. Biol. Organ.) J. 1999;18:2782–2792. doi: 10.1093/emboj/18.10.2782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaffman A., O'Shea E.K. Regulation of nuclear localizationa key to a door. Annu. Rev. Cell Dev. Biol. 1999;15:291–339. doi: 10.1146/annurev.cellbio.15.1.291. [DOI] [PubMed] [Google Scholar]
- Kaffman A., Rank N.M., O'Neill E.M., Huang L.S., O'Shea E.K. The receptor Msn5 exports the phosphorylated transcription factor Pho4 out of the nucleus Nature. 396 1998. 482 486a [DOI] [PubMed] [Google Scholar]
- Kaffman A., Rank N.M., O'Shea E.K. Phosphorylation regulates association of the transcription factor Pho4 with its import receptor Pse1/Kap121 Genes Dev. 12 1998. 2673 2683b [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim K.-S., Rosenkrantz M.S., Guarente L. Saccharomyces cerevisiae contains two functional citrate synthase genes. Mol. Cell. Biol. 1986;6:1936–1942. doi: 10.1128/mcb.6.6.1936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitada K., Yamaguchi E., Arisawa M. Cloning of the Candida glabrata TRP1 and HIS3 genes, and construction of their disruptant strains by sequential integrative transformation. Gene (Amst.) 1995;165:203–206. doi: 10.1016/0378-1119(95)00552-h. [DOI] [PubMed] [Google Scholar]
- Komeili A., O'Shea E.K. Roles of phosphorylation sites in regulating activity of the transcription factor Pho4. Science. 1999;284:977–980. doi: 10.1126/science.284.5416.977. [DOI] [PubMed] [Google Scholar]
- Koonin E.V. Yeast protein controlling inter-organelle communication is related to bacterial phosphatases containing the Hsp70-type ATP-binding domain. Trends Biochem. Sci. 1994;19:156–157. doi: 10.1016/0968-0004(94)90275-5. [DOI] [PubMed] [Google Scholar]
- Kos W., Kal A.J., van Wilpe S., Tabak H.F. Expression of genes encoding peroxisomal proteins in Saccharomyces cerevisiae is regulated by different circuits of transcriptional control. Biochim. Biophys. Acta. 1995;1264:79–86. doi: 10.1016/0167-4781(95)00127-3. [DOI] [PubMed] [Google Scholar]
- Lashkari D.A., DeRisi J.L., McCusker J.H., Namath A.F., Gentile C., Hwang S.Y., Brown P.O. Yeast microarrays for genome wide parallel genetic and gene expression analysis. Proc. Natl. Acad. Sci. USA. 1997;94:13057–13062. doi: 10.1073/pnas.94.24.13057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao X., Butow R.A. RTG1 and RTG2two yeast genes required for a novel path of communication from mitochondria to the nucleus. Cell. 1993;72:61–71. doi: 10.1016/0092-8674(93)90050-z. [DOI] [PubMed] [Google Scholar]
- Liao X., Small W.C., Srere P.A., Butow R.A. Intramitochondrial functions regulate nonmitochondrial citrate synthase (CIT2) expression in Saccharomyces cerevisiae . Mol. Cell. Biol. 1991;11:38–46. doi: 10.1128/mcb.11.1.38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Z., Butow R.A. A transcriptional switch in the expression of yeast tricarboxylic acid cycle genes in response to a reduction or loss of respiratory function. Mol. Cell. Biol. 1999;19:6720–6728. doi: 10.1128/mcb.19.10.6720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Longtine M.S., McKenzie A., III, Demarini D.J., Shah N.G., Wach A., Brachat A., Philippsen P., Pringle J.R. Additional modules for versatile and economical PCR-based gene deletion and modification in Saccharomyces cerevisiae . Yeast. 1998;14:953–961. doi: 10.1002/(SICI)1097-0061(199807)14:10<953::AID-YEA293>3.0.CO;2-U. [DOI] [PubMed] [Google Scholar]
- Magasanik B. Regulation of nitrogen utilization. In: Jones E.W., Pringle J.R., Broach J.R., editors. The Molecular and Cellular Biology of the Yeast SaccharomycesGene Expression. Vol. 2. Cold Spring Harbor Laboratory Press; Cold Spring Harbor, NY: 1992. pp. 283–317. [Google Scholar]
- Marzluf G.A. Genetic regulation of nitrogen metabolism in the fungi. Microbiol. Mol. Biol. Rev. 1997;61:17–32. doi: 10.1128/mmbr.61.1.17-32.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCammon M.T. Mutants of Saccharomyces cerevisiae with defects in acetate metabolismisolation and characterization of Acn-mutants. Genetics. 1996;144:57–69. doi: 10.1093/genetics/144.1.57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McNabb D.S., Xing Y., Guarente L. Cloning of yeast HAP5a novel subunit of a heterotrimeric complex required for CCAAT binding. Genes Dev. 1995;9:47–58. doi: 10.1101/gad.9.1.47. [DOI] [PubMed] [Google Scholar]
- Menendez J., Gancedo C. Regulatory regions in the promoters of the Saccharomyces cerevisiae PYC1 and PYC2 genes encoding isozymes of pyruvate carboxylase. FEMS Microbiol. Lett. 1998;164:345–352. doi: 10.1111/j.1574-6968.1998.tb13108.x. [DOI] [PubMed] [Google Scholar]
- Merrick M.J., Edwards R.A. Nitrogen control in bacteria. Microbiol. Rev. 1995;59:604–622. doi: 10.1128/mr.59.4.604-622.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noda T., Ohsumi Y. Tor, a phosphatidylinositol kinase homologue, controls autophagy in yeast. J. Biol. Chem. 1998;273:3963–3966. doi: 10.1074/jbc.273.7.3963. [DOI] [PubMed] [Google Scholar]
- Nasmyth K., Adolf G., Lydall D., Seddon A. The identification of a second cell cycle control on the HO promoter in yeastcell cycle regulation of SW15 nuclear entry. Cell. 1990;62:631–647. doi: 10.1016/0092-8674(90)90110-z. [DOI] [PubMed] [Google Scholar]
- Ogur M., Coker L., Ogur S. Glutamate auxotrophs in Saccharomyces. I. The biochemical lesion in the glt-1 mutants. Biophys. Res. Commun. 1964;14:193–197. doi: 10.1016/0006-291x(64)90254-2. [DOI] [PubMed] [Google Scholar]
- Ogur M., Roshanmanesh A., Ogur S. Tricarboxylic acid cycle mutants in Saccharomyces cerevisiaecomparison of independently derived mutants. Science. 1965;147:1590. doi: 10.1126/science.147.3665.1590. [DOI] [PubMed] [Google Scholar]
- Powers T., Walter P. Regulation of ribosome biogenesis by the rapamycin-sensitive TOR-signaling pathway in Saccharomyces cerevisiae . Mol. Biol. Cell. 1999;10:987–1000. doi: 10.1091/mbc.10.4.987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenkrantz M.S., Alam T., Kim K., Clark B.J., Srere P.A. Mitochondrial and nonmitochondrial citrate synthase in Saccharomyces cerevisiae are encoded by distinct homologous genes. Mol. Cell. Biol. 1986;6:4509–4515. doi: 10.1128/mcb.6.12.4509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rothermel B.A., Thornton J.L., Butow R.A. Rtg3p, a basic helix-loop-helix/leucine zipper protein that functions in mitochondrial-induced changes in gene expression, contains independent activation domains. J. Biol. Chem. 1997;272:19801–19807. doi: 10.1074/jbc.272.32.19801. [DOI] [PubMed] [Google Scholar]
- Rothstein R. Targeting, disruption, replacement, and allele rescueintegrative DNA transformation in yeast. Methods Enzymol. 1991;194:281–301. doi: 10.1016/0076-6879(91)94022-5. [DOI] [PubMed] [Google Scholar]
- Sekito T., Thorton J., Butow R. Mitochondria-to-nuclear signaling is regulated by the subcellular localization of the transcription factors Rtg1p and Rtg3p. Mol. Biol. Cell. 2000;11:2103–2115. doi: 10.1091/mbc.11.6.2103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sherman F. Getting started with yeast. Methods Enzymol. 1991;194:3–21. doi: 10.1016/0076-6879(91)94004-v. [DOI] [PubMed] [Google Scholar]
- Sikorski R.S., Heiter P. A system of shuttle vectors and yeast host strains designed for efficient manipulation of DNA in Saccharomyces cerevisiae . Genetics. 1989;122:19–27. doi: 10.1093/genetics/122.1.19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Small W.C., Brodeur R.D., Sandor A., Fedorova N., Li G., Butow R.A., Srere P.A. Enzymatic and metabolic studies on retrograde regulation mutants of yeast. Biochemistry. 1995;34:5569–5576. doi: 10.1021/bi00016a031. [DOI] [PubMed] [Google Scholar]
- Spellman P.T., Sherlock G., Zhang M.Q., Iyer V.R., Anders K., Eisen M.B., Brown P.O., Botstein D., Futcher B. Comprehensive identification of cell cycle-regulated genes of the yeast Saccharomyces cerevisiae by microarray analysis. Mol. Biol. Cell. 1998;9:3273–3297. doi: 10.1091/mbc.9.12.3273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stryer L. Biochemistry. W.H. Freeman & Co; New York, NY: 1995. [Google Scholar]
- Stucka R., Dequin S., Salmon J.M., Gancedo C. DNA sequences in chromosomes II and VII code for pyruvate carboxylase isoenzymes in Saccharomyces cerevisiaeanalysis of pyruvate carboxylate deficient strains. Mol. Gen. Genet. 1991;229:307–315. doi: 10.1007/BF00272171. [DOI] [PubMed] [Google Scholar]
- Sumrada R.A., Cooper T.G. Urea carboxylase and allophanate hydrolase are components of a multifunctional protein in yeast. J. Biol. Chem. 1982;257:9119–9127. [PubMed] [Google Scholar]
- Thomas G., Hall M.N. TOR signalling and control of cell growth. Curr. Opin. Cell Biol. 1997;9:782–787. doi: 10.1016/s0955-0674(97)80078-6. [DOI] [PubMed] [Google Scholar]
- Walker M.E., Val D.L., Rohde M., Devenish R.J., Wallace J.C. Yeast pyruvate carboxylaseidentification of two genes encoding isoenzymes. Biochem. Biophys. Res. Commun. 1991;176:1210–1217. doi: 10.1016/0006-291x(91)90414-3. [DOI] [PubMed] [Google Scholar]
- Zaragoza D., Ghavidel A., Heitman J., Schultz M.C. Rapamycin induces the G0 program of transcriptional repression in yeast by interfering with the TOR signaling pathway. Mol. Cell. Biol. 1998;18:4463–4470. doi: 10.1128/mcb.18.8.4463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng X.-F., Fiorentino D., Chen J., Crabtree G.R., Schreiber S.L. TOR kinase domains are required for two distinct functions, only one of which is inhibited by rapamycin. Cell. 1995;82:121–130. doi: 10.1016/0092-8674(95)90058-6. [DOI] [PubMed] [Google Scholar]
- Zhu X., Garrett J., Schreve J., Michaeli T. GNP1, the high-affinity glutamine permease of S. cerevisiae . Curr. Genet. 1996;30:107–114. doi: 10.1007/s002940050108. [DOI] [PubMed] [Google Scholar]