

Abstract
Amyloid precursor protein (APP), implicated in Alzheimer's disease, is a trans-membrane protein of undetermined function. APP is cleaved by γ-secretase that releases the APP intracellular domain (AICD) in the cytoplasm. In vitro studies have implicated AICD in cell signaling and transcriptional regulation, but its biologic relevance has been uncertain and its in vivo function has not been examined. To investigate its functional role, we generated AICD transgenic mice, and found that AICD causes significant biologic changes in vivo. AICD transgenic mice show activation of glycogen synthase kinase-3β (GSK-3β) and phosphorylation of CRMP2 protein, a GSK-3β substrate that plays a crucial role in Semaphorin3a-mediated axonal guidance. Our data suggest that AICD is biologically relevant, causes significant alterations in cell signaling, and may play a role in axonal elongation or pathfinding.
Introduction
Amyloid precursor protein (APP), a cell surface protein of unknown function, is implicated in the pathogenesis of Alzheimer's disease (AD) (Price et al., 1998; Annaert and De Strooper, 2002; Selkoe, 2005). APP topology resembles that of a membrane receptor protein; it has a large extracellular portion, a single transmembrane segment, and a cytoplasmic tail domain that interacts with several proteins, including Fe65. Although the function of APP is not understood completely, it has been implicated in a variety of processes, including signal transduction, cell migration, and axonal elongation (see De Strooper and Annaert, 2000). APP is cleaved initially by β- or α-secretase, which sheds the extracellular portion and generates membrane-associated COOH-terminal fragments (APP-CTFs) that are cleaved further by γ-secretase within the plane of the membrane. The γ-cleavage results in the extracellular secretion of P3 or 40/42 residue–long Aβ peptides (which accumulate in amyloid plaques in AD brains), and simultaneous release of the APP intracellular domain (AICD) within the cell. The function of AICD or the relevance of γ-secretase cleavage in APP biology is unknown.
The generation of AICD peptide follows the general steps of “regulated intramembrane proteolysis” which results in the release of a membrane-tethered transcriptional regulator in response to an external signal (Brown et al., 2000). We and other investigators have shown that cleaved AICD enters the nucleus and regulates gene expression in vitro (Cao and Sudhof, 2001; Gao and Pimplikar, 2001; Baek et al., 2002). The AICD target genes are not firmly known, and a majority of support for its transcriptional role comes from the use of an artificial reporter gene. Although additional in vitro studies showed that AICD also alters cell signaling (Leissring et al., 2002) and induces apoptosis (Passer et al., 2000; Kinoshita et al., 2002), the physiologic relevance of AICD has been uncertain because its steady-state levels are reported to be low (Cupers et al., 2001; Kimberly et al., 2001). To examine the in vivo role of AICD, we generated transgenic mice that express AICD and Fe65 in the forebrain and hippocampal regions of the postnatal brain. We report that the transgenic mice show two- to threefold higher levels of AICD than control mice, and display robust activation of glycogen synthase kinase-3β (GSK-3β) and increased phosphorylation of a downstream substrate, CRMP2, a key component of the axonal guidance signaling pathway. We also demonstrate the presence of endogenous AICD in the membrane fractions from control mice, which suggests that the steady-state levels of AICD are higher than previously believed. Together, our in vivo findings support a biologic role for AICD in regulating gene expression and cell signaling.
Results
AICD transgenic mice
To examine the in vivo effects of AICD, we generated double transgenic mice expressing AICD and Fe65. The steady-state levels of AICD are reported to be exceedingly low. Ectopically expressed AICD is turned over rapidly in tissue culture cells, but can be stabilized when coexpressed with Fe65 (Cupers et al., 2001; Kimberly et al., 2001). Fe65 is a cytoplasmic protein that binds the “Y682ENPTY” motif in APP cytoplasmic domain through its PTB2 domain (Borg et al., 1996). The binding of Fe65 to holo-APP at the plasma membrane was proposed to regulate cell migration and control the growth cone movement in the neurons (Sabo et al., 2001, 2003). Fe65 also is required for the transcriptional activity of AICD (Cao and Sudhof, 2001; Baek et al., 2002). Therefore, we reasoned that coexpression of Fe65 might be required to stabilize AICD and to observe its full effects in transgenic animals. The specificity of AICD effects can be determined by comparing the AICD+Fe65 double transgenic mice with Fe65 single transgenic animals. We used the CaMKIIα promoter to drive the transgene expression, because its activity is restricted to forebrain and hippocampal regions of the brain (Abel et al., 1997), the areas that are affected widely in AD. Moreover, CaMKIIα promoter becomes active only at the 2-wk postnatal stage, thus avoiding possible lethal side effects during embryonic development. We expressed the 59-residue long AICD peptide, which is a product of “γ-cleavage” of APP. APP-CTF also undergoes “ɛ-cleavage,” which generates a 50-residue long AICD (Gu et al., 2001; Yu et al., 2001). The present study focused on characterizing the in vivo activity of AICD59 (referred to here as AICD).
We cloned myc-tagged Fe65 or AICD in plasmid NN265 that contained intron and SV40 polyadenylation sequences (Abel et al., 1997). A fragment that contained the intron, the transgene open reading frame, and polyA signal was excised and cloned into MM403, downstream of the 8-kb CaMKIIα promoter (Fig. 1 A). We mixed AICD and Fe65 expressing plasmids in 1:1 proportion, and co-injected the linearized plasmids into oocytes of C57BL/6 mice. Injected oocytes were implanted in pseudopregnant C57BL/6 mice; by PCR on tail DNA, 9 out of 49 pups obtained were found to have incorporated both transgenes. All 9 founder mice were mated with C57BL/6 mice. Germline transmission was observed in five lines, of which four of the founder lines transmitted both transgenes to F1 pups (unpublished data). In the current study, we present data from two of these four independent lines (named FeCγ.12 and FeCγ.25). The fifth line, called Fe.27, did not transmit the AICD transgene to pups (Fig. 1 B), and thereby, fortuitously created a Fe65 single transgenic line. The expression levels of Fe65 transgene were determined by Western blot analysis. Total brain homogenates (40 μg protein each) from two animals from each transgenic line or two nontransgenic littermates was separated by SDS-PAGE on a 10% gel, transferred to a nitrocellulose membrane, and probed using anti-myc antibodies to detect the transgene or anti-Fe65 3H6 antibody to detect total Fe65 (endogenous + transgene). The myc-Fe65 signal was apparent in mice from all three transgenic lines, but was absent in nontransgenic littermates (Fig. 1 C, top panel). The total levels of Fe65 in the three transgenic lines (Fig. 1 D; top panel) were comparable, and were approximately twice as high as the nontransgenic control animals, when normalized for the levels of tubulin (Fig. 1 E). The Fe65 levels in Fe.27 mice were not significantly different from those in FeCγ.12 or FeCγ.25 mice (P = 0.04 by Bonferroni/Dunn test).
Figure 1.

Generation and characterization of double transgenic FeCγ and single Fe65 transgenic mice. (A) Construction of AICD and Fe65 transgenes. The horizontal lines with arrows shows the location of transgene specific primers. (B) A PCR reaction on tail DNA isolated from three pups from Fe.27 line from three different litters (lanes 1–3) using Fe65 (left) or AICD primers (right) was performed together with primers for mouse Xist gene. Lanes denoted “+” contained DNA from the founder mouse (Fe.27). Note that none of the pups carries the transgene for AICD. (C and D) Western blot analysis of brain homogenates from two animals from double transgenic lines (FeCγ.12 and FeCγ.25), single Fe65 transgenic line (Fe.27), and nontransgenic littermate controls. Blots were probed with anti-myc 9E10 (C; top panel) or anti Fe65 antibody 3H6 (D; top panel), and visualized by ECL. The blots were stripped and reprobed with anti-tubulin DM1A antibody as an internal control (bottom panels). (E) Quantitative analysis of total Fe65 levels as detected by 3H6 antibody. Protein levels were normalized to tubulin by reprobing the same blots after stripping. Quantification from three independent experiments. Values are the mean ± SEM; n = 6. Fe65 levels in FeCγ.12 and FeCγ.25 mice were significantly different from nontransgenic (nTg) animals (P < 0.0001), but not from Fe.27 mice (P = 0.04) by Bonferroni/Dunn test.
AICD transgene levels in transgenic mice
We next determined the AICD levels by Western blot following a protocol (see “Materials and Methods”) described by Pinnix et al. (2001), using antibody 0443, which is highly sensitive in detecting AICD. We fractionated total brain homogenates into cytosol (Fig. 2 A), and membrane or nuclear fractions (Fig. 2 B); 20 μg protein was separated on a NuPAGE Bis-Tris 4–12% gel and probed with antibody 0443 that was raised against the COOH-terminal 20 residues of APP. As a positive control, we loaded cell extract from COS-1 cells that were cotransfected with AICD and Fe65 (lane 1). We readily detected AICD59 in the cytosolic fraction from FeCγ.25 mice (Fig. 2 A, top panel: lanes 6 and 7). A longer exposure of the same blot also revealed the presence of AICD in FeCγ.12 mice (bottom panel: lanes 4, 5), but not in the cytosolic fractions from Fe.27 (lanes 8 and 9) or nontransgenic control animals (lanes 2 and 3). The expression levels of AICD transgene parallel those of Fe65 transgene because FeCγ.12 mice express lower levels of both transgenes compared with FeCγ.25 animals (Fig. 1 C).
Figure 2.
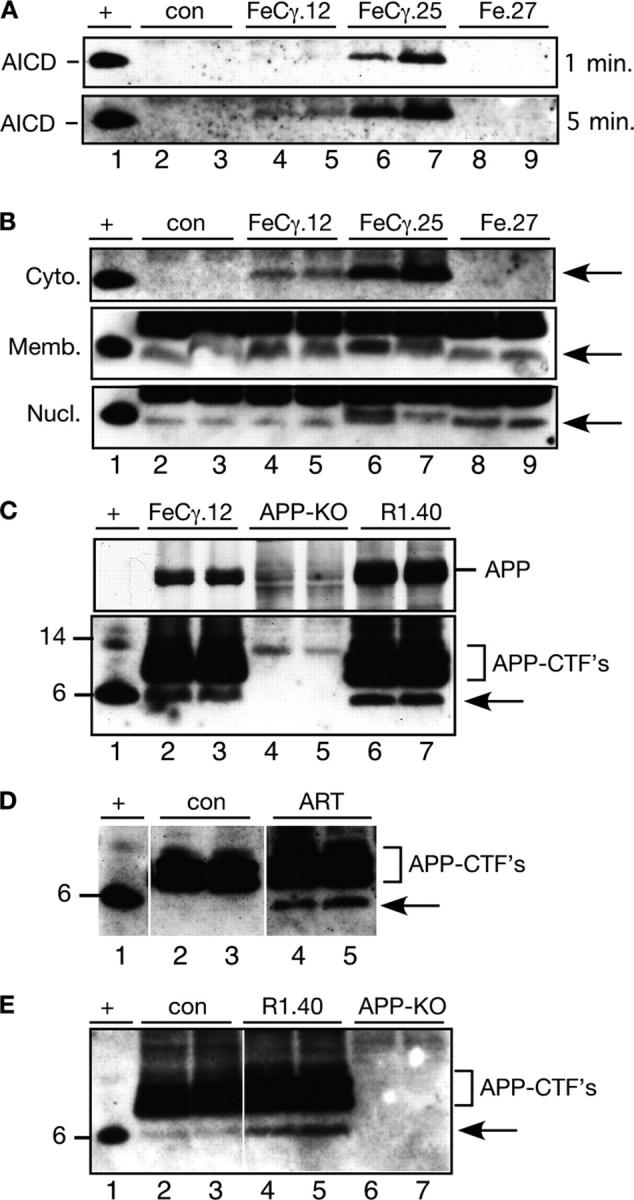
AICD transgenic mice show barely detectable levels of AICD in the membrane fraction. (A) Cytosolic proteins from control (con) or two transgenic mice from indicated lines were separated on a 4–12% Bis-Tris NuPAGE gel, and the blots were probed with antibody 0443 after antigen retrieval as described in “Methods and materials.” Cell lysate from COS-1 cells cotransfected with AICD and Fe65 was run as a positive control (lane +). AICD band is clearly visible in FeCγ.25 mice (top panel, lanes 6 and 7), whereas a longer exposure shows the AICD protein present in FeCγ.12 mice (bottom panel, lanes 4 and 5), but not in control (lanes 2 and 3) or Fe.27 mice (lanes 8 and 9). (B) Brain homogenates from two control (con) transgenic mice from indicated lines were separated into cytosolic (Cyto.), membrane (Memb.), or nuclear (Nucl.) fractions, and the blots were probed with antibody 0443 as indicated above. Cell lysate from COS-1 cells cotransfected with AICD and Fe65 was run as a positive control (lane +). Although AICD was detected in the cytosol of only FeCγ transgenic mice (arrow), the membrane and nuclear fractions of all animals showed the AICD co-migrating band (arrows). (C) The AICD co-migrating band is absent in APP-KO mice. Brain membranes from two FeCγ.12 mice (lanes 2 and 3), APP-KO mice (lanes 4 and 5), and R1.40 transgenic mice expressing human APP with “Swedish mutation” (lanes 6 and 7) were probed with antibody 0443, as described above, on a NuPAGE gel. Top panel shows that the APP band is present in FeCγ.12 and in higher amounts in R1.40 mice, but is absent in APP-KO mice. Similarly, the middle panel shows the absence of APP-CTFs in the APP-KO mice. Note that the AICD co-migrating band (arrow) also is absent in APP-KO mice. (D) The AICD peptide becomes detectable only after antigen retrieval treatment (ART). Equal amounts of brain membrane proteins from two R1.40 mice were separated in duplicate on a NuPAGE gel. After electrophoretic transfer, the membrane was cut lengthwise to give two identical gels. One was exposed to boiling PBS for 5 min (ART), whereas the other was kept in PBS at room temperature (con). Arrow shows the AICD band, which is detectable only in the ART samples. (E) AICD band (arrow) is present in increased amounts in R1.40 transgenic mice compared with the controls (con) and is absent in APP-KO mice. Membrane fractions from indicated mice were probed with antibody 0443.
We next determined the presence of AICD in the membrane and nuclear fractions (Fig. 2 B) from the brains of these animals. Whereas AICD is detected in the cytosolic fraction of only AICD transgenic mice (Fig. 2 B, upper panel), an AICD co-migrating band was observed in the membrane fractions of all animals (middle panel), and seemed to be present in slightly higher levels in the transgenic mice (lanes 4–7) compared with the control or Fe.27 mice. This finding is unexpected, because it is believed that the endogenously present AICD in brain only can be detected with a combined approach of immunoprecipitation and immunoblot. To rule out the possibility that this band was recognized nonspecifically by antibody 0443, we probed brain membrane fractions from APP knock-out (KO) mice and R1.40 transgenic mice that overexpress human APP with “Swedish mutation” by two- to threefold (Lamb et al., 1997). Antibody 0443 recognized the full-length APP in FeCγ.12 mice (Fig. 2 C, top panel), which was present in increased amounts in R1.40 mice (lanes 6 and 7) and absent in APP KO mice (lanes 4 and 5). Similarly, APP-CTF fragments were detected in FeCγ.12 and R1.40 mice, but not in the APP-KO mice (middle panel). The AICD59 (lane 1) co-migrating band was present only in FeCγ.12 and R1.40 mice, but not in APP-KO mice (arrow, middle panel). These data show that the co-migrating band is recognized specifically by antibody 0443, and must be an APP product because it is absent in APP-KO mice.
To understand why the membrane-associated AICD was not detected in previous studies, we separated membrane proteins from R1.40 brains in duplicate lanes on the same gel, and electrophoretically transferred them onto a nitrocellulose membrane. The membrane was cut lengthwise in two. One part received the antigen retrieval treatment (see “Materials and Methods”), whereas the other part was kept in PBS at room temperature. Both blots were blocked in 10% calf serum and processed identically. Fig. 2 D shows that although the AICD band is detected clearly upon antigen retrieval (arrow, lanes 4 and 5), it is not detected in the absence of the treatment, even though APP-CTFs are clearly visible (lanes 2 and 3). We verified these results further by comparing brain membranes from R1.40 mice with control C57BL/6 mice. R1.40 mice, which produce increased amounts of Aβ peptides (Lamb et al., 1997), also should give rise to increased amounts of AICD. AICD was present in greater amounts in R1.40 mice (Fig. 2 E, lanes 4 and 5) compared with controls (lanes 2 and 3), and was absent in APP-KO mice (lanes 6 and 7).
Together, these results show that the endogenous AICD can be detected in the membrane, but not the cytosolic fractions, of wild-type C57BL/6 animals (Fig. 2 B, also compare Fig. 2 E with Fig. 2 A). More importantly, the AICD transgenic mice express AICD at greater levels than do nontransgenic control mice, and similar to those levels observed in APP transgenic mice with familial Alzheimer's disease (FAD) mutation (Fig. 2 C). This lends strong support to the validity of our transgenic model.
Validation of AICD transgenic mice
Although no bona fide target genes of AICD have been identified genetically, a chromatin immunoprecipitation assay was used recently to show that AICD is recruited to KAI1 gene promoter and stimulates its transcription (Baek et al., 2002; Von Rotz et al., 2004). We sought to validate our animal model by examining the expression of KAI1 gene in the AICD transgenic mice. We probed the membrane fractions from nontransgenic littermate controls, two FeCγ transgenic lines, and Fe.27 animals with anti-KAI1 antibody (Fig. 3 A, top panel). A doublet of KAI1 protein band was visible in FeCγ.12 and FeCγ.25 mice (lanes 3–6), but not in nontransgenic controls (lanes 1 and 2) nor in Fe65 transgenic Fe.27 mice (lanes 7 and 8). When normalized for protein loading (Fig. 3 A, bottom panel), FeCγ.25 seem to express higher levels of KAI1 compared with FeCγ.12. These data confirm and extend in vivo the previous observations that AICD activates KAI1 gene expression (Baek et al., 2002; Von Rotz et al., 2004). These findings also validate the supposition that the γ-secretase cleaved, 59-residue long AICD exhibits biologic effects that are observed when APP is cleaved in vivo to generate AICD (Cao and Sudhof, 2001; Baek et al., 2002).
Figure 3.

Increased KAI1 levels in AICD transgenic mice. (A) The AICD transgenic mice, but not the control or Fe.27 mice, show expression of KAI1 gene. Membrane fractions from indicated mice were probed with anti-KAI1 antibody. FeCγ.25 mice showed higher expression of KAI1 protein compared with FeCγ.12 mice. (B) Membrane fractions from indicated mice were probed with anti-SERCA 2b antibody. No significant changes in SERCA 2b levels were reproducibly observed in FeCγ transgenic mice.
Although no gene, other than KAI1, has been shown conclusively to be an AICD target, a study implicated AICD in regulating genes that are involved in Ca2+ signaling (Leissring et al., 2002). Therefore, we examined the levels of SERCA 2b, a ubiquitously expressed Ca-ATPase that maintains the ER Ca2+ levels, in AICD transgenic mice. Membrane fractions from animals were analyzed by Western blotting using an antibody that specifically detects mouse SERCA 2b in nonmuscle tissue. We detected no consistent, significant changes in SERCA 2b levels in FeCγ.12 or FeCγ.25 mice compared with control animals (Fig. 3 B), when normalized for protein loading. SERCA 2b levels in Fe.27 mice were variable. Although it is possible that a small increase in SERCA 2b levels was not detected in the whole brain membrane fraction, a recent study (Mueller et al., 2004) also found no differences in SERCA 2b levels in AICD expressing cells by DNA microarray (Muller, T., and R. Egensperger, personal communication).
Elevated levels of active glycogen synthase kinase–3 in transgenic mice expressing AICD
Glycogen synthase kinase (GSK)–3β is a proline-directed Ser/Thr kinase that is implicated in AD pathogenesis (Jope and Johnson, 2004). In vitro observations suggest that ectopic expression of AICD results in a significant increase in the mRNA and protein levels of GSK-3β (Kim et al., 2003; Von Rotz et al., 2004). To examine whether AICD activates GSK-3β in vivo, we determined the status of GSK-3 by using antibodies that recognize the activated or inhibited forms of the enzyme. Phosphorylation of GSK-3β at Y216 stimulates its kinase activity (active form), whereas phosphorylation at S9 potently represses (inactive form) the kinase activity (Jope and Johnson, 2004). Brain cytosolic fractions were immunoblotted with antibody anti-pY216/279 that recognizes the phospho-Y216 on GSK-3β and the equivalent Y279 residue on GSK-3α. Fig. 4 A shows that activated forms of GSK-3α and -3β were elevated in FeCγ.12 and FeCγ.25 mice (top panel, lanes 3–6) when compared with nontransgenic controls (lanes 1 and 2) or Fe.27 mice (lanes 7 and 8). Densitometric analysis revealed (Fig. 4 B) an ∼1.7-fold increase in pGSK-3β levels in FeCγ animals (GSK-3α showed similar changes). We also analyzed the total GSK-3β levels by stripping the blots and reprobing with antibodies that recognize GSK-3β and observed no significant changes in the protein levels in transgenic mice compared with the control animals (Fig. 4 A, bottom panel).
Figure 4.

The AICD transgenic mice show activated GSK-3 levels. (A) Higher levels of activated form of GSK-3α and -3β in AICD transgenic mice. Immunoblot analysis of brain cytosol from animals from indicated lines was probed with anti-GSK antibody (pY279/216) that specifically recognizes the activated forms of GSK-3α and -3β enzymes (top panel). Note that mice from both AICD transgenic lines show higher levels of activated GSK-3α and -3β (lanes 3–6) as compared with control (con; lanes 1 and 2) or Fe.27 mice (lanes 7 and 8). Total GSK-3β protein levels are not changed (bottom panel). (B) Quantitative analysis of phospho–GSK-3β levels in transgenic and control mice. Quantification of GSK-3α levels gave similar results (not depicted). Protein levels were normalized to tubulin by reprobing the same blots after stripping. This experiment was repeated twice, and was performed on animals from an independent FeCγ.22 line. Values are the mean ± SEM; n = 6. *, P < 0.05 against nontransgenic (nTg) or Fe.27 mice by Fisher's PLSD test. (C and D) AICD transgenic mice show a dramatic reduction in the levels of inhibited form of GSK-3β, as detected by pS9–GSK-3β antibody (top panel). The total GSK-3β levels were not changed (bottom panel). Quantitative analysis of data in shown in (D). The protein levels were normalized to tubulin. This experiment was repeated twice and performed on animals from an independent FeCγ.22 line. Values are the mean ± SEM; n = 6. **, P < 0.001 against nontransgenic or Fe.27 mice. (E) Activated ERK1 and ERK2 levels are not altered significantly in transgenic mice as detected by pERK1/2 antibodies.
We corroborated these findings further by determining the levels of the inactive form of GSK-3β by using an antibody that specifically detects phospho-Ser9–GSK-3β. FeCγ double transgenic mice showed a dramatic reduction in the pS9–GSK-3β levels as compared with Fe.27 mice or nontransgenic littermates (Fig. 4 C, lanes 3–6, top panel), whereas the levels of total GSK-3β remained unaltered (bottom panel). A reduction in pS9–GSK-3β levels also was observed in a different double transgenic line (FeCγ.22; unpublished data). Quantification of these data shows that the levels of pS9–GSK-3β were reduced by 70% as compared with controls (Fig. 4 D). Together, these data show that low levels of AICD are able to activate GSK-3β in vivo.
In addition to the GSK-3 pathway, the extracellular signal-regulated kinase (ERK) pathway also is activated in AD brains (Zhu et al., 2003). To determine the selectivity of the AICD effect, we examined the levels of phosho-ERK1 and -ERK2 in the transgenic animals. Brain homogenates from FeCγ, Fe.27, and nontransgenic littermate animals were immunoblotted using pERK antibodies that recognize activated ERK1 and ERK2. We observed no significant differences in the levels of activated ERK1 or ERK2 (Fig. 4 E) in control or transgenic mice. Thus, these findings show that the ERK is not activated in 8–12-wk old FeCγ mice, and that AICD does not cause ERK activation. The lack of AICD on ERK activation is consistent with the reports the ERK activation occurs by way of Aβ peptides (Bell et al., 2004). However, the possibility cannot be ruled out that AICD modulates some other signaling pathways.
GSK-3β mRNA levels are not increased in transgenic mice
In contrast to the in vitro observations that AICD causes an increase in the GSK-3β protein levels in tissue culture cells, we did not observe an increase in the GSK-3β protein levels in the transgenic mice (Fig. 4, A and C). To examine this discrepancy in detail, we measured the mRNA levels in brain extracts by real-time PCR. Total RNA was extracted from brain tissue, subjected to reverse transcriptase reaction, and real-time PCR was performed. We used primers for GSK-3β and KAI1, and normalized the values by using β-actin. FeCγ.12 mice showed a 1.8-fold increase in KAI1 message when compared with nontransgenic or Fe.27 mice (Fig. 5 A). By contrast, we detected no change in GSK-3β transcripts when compared with nontransgenic or Fe.27 mice (Fig. 5 B). These observations suggest that GSK-3β activation in FeCγ transgenic mice is not due to increased transcription or translation of GSK-3β gene. Because GSK-3β kinase activity is regulated largely at the posttranslational level by upstream kinases and phosphatases (Jope and Johnson, 2004), it is possible that AICD activates GSK-3β by modulating upstream regulators. However, because the transgenes are expressed only in certain neuronal cells, we cannot rule out the possibility that a slight increase in GSK-3β mRNA or protein was undetected in whole brain extracts.
Figure 5.

Analysis of mRNA transcript levels by real-time PCR. Total RNA from at least three animals was isolated, subjected to reverse transcriptase, and real-time PCR was performed using BioRad iCycler and primers for KAI1 gene (A) or GSK-3β gene (B). Mouse β-actin primers were used as an internal control. Data are expressed as levels of KAI1 or GSK-3β transcripts relative to β-actin. Values are the mean ± SEM; n = 6. P = 0.08 for KAI1 transcript levels in FeCγ.12 against nontransgenic or Fe.27 mice by Fisher's PLSD test.
Phosphorylation of collapsin responsive mediator protein–2 in AICD transgenic mice
Many proteins are phosphorylated by GSK-3β on multiple Ser/Thr residues; the microtubule binding proteins, tau, microtubule-associated protein 1B, and collapsin responsive mediator protein–2 (CRMP2), are among the known GSK-3 substrates. These proteins bind and stimulate microtubule stability, but they fail to bind microtubules upon phosphorylation and cause microtubule depolymerization (Fukata et al., 2002; Jope and Johnson, 2004). We examined the status of CRMP2, a neuronal-specific protein that is involved in axonal repulsion and neuronal polarity, in our transgenic mice. CRMP2 mediates the repulsive action of Semaphorin3a (Sema3a), which binds the Plexin receptors in growth cones, activates GSK-3β, and phosphorylates CRMP2 on T509 and S522 (Uchida et al., 2005). An antibody, 3F4, which originally was raised against the neurofibrillary tangles (NFTs) from AD brains (Yoshida et al., 1998; Gu et al., 2000), specifically recognizes CRMP2 phosphorylated at T509 and S522 (Uchida et al., 2005). We examined the phosphorylation status of CRMP2 in our FeCγ transgenic mice by using the following antibodies: antibody 3F4 to detect CRMP2 phosphorylated at T509 and S522 (Uchida et al., 2005); antibody pT514 to recognize CRMP2 phosphorylated at T514 (Yoshimura et al., 2005); and antibody C4G that recognizes total CRMP2 (Gu et al., 2000). We immunoblotted brain cytosolic fractions from the indicated mice with antibody 3F4, and observed that the levels of phosphoCRMP2 were significantly higher in FeCγ.12 and FeCγ.25 animals compared with nontransgenic control or Fe.27 mice (Fig. 6 A). We stripped the blot and reprobed with antibody C4G that recognizes total CRMP2, and found that the total levels of CRMP2 were unaltered (Fig. 6 B). Quantification of the protein levels showed a two- to threefold increase in phosho-CRMP2 in the AICD transgenic mice compared with Fe.27 or normal mice (Fig. 6 C)
Figure 6.

Phosphorylation of CRMP2, a GSK-3β substrate, in transgenic mice. (A) Brain cytosol fractions were probed with antibody 3F4, which recognizes CRMP2 phosphorylated at T509 and S522. Note the elevated levels of phospho-CRMP2 in FeCγ transgenic mice compared with Fe.27 or nontransgenic control mice. (B). The blot used above was stripped and reprobed with antibody C4G, which recognizes total CRMP2. The levels of total CRMP2 are not changed. (C) Quantitative analysis of phospho-CRMP2 levels as detected by 3F4 antibody. Protein levels were normalized to tubulin by reprobing the same blots after stripping. Quantification from three independent experiments. Values are the mean ± SEM; n = 6. *, P < 0.05 against nontransgenic (nTg) or Fe.27 mice by Fisher's PLSD test. (D) Levels of CRMP2 phosphorylated at T514 were not changed in transgenic mice (top panel). The blot was stripped and reprobed with antitubulin antibodies as a loading control (bottom panel). (E) A hypothetical cascade of signaling events, similar to Sema3a signaling pathway, suggests a role for APP in axonal guidance. The cleavage of APP to release AICD activates GSK-3β and results in phosphorylation of CRMP-2 at S522 and T509–the same residues that mediate the repulsive action of Sema3a upon binding to Plexin/Neuropilin receptors. F-spondin, an extracellular signaling protein of floor plate and hippocampus which is involved in axonal pathfinding and neurite outgrowth, could be a candidate signaling protein (shown by “?”) because it binds APP and inhibits AICD production.
CRMP2 also is phosphorylated at T514 by GSK-3β when primed by Cdk5; this phosphorylation event plays a crucial role in determining axonal versus dendritic fate of hippocampal neurites (Yoshimura et al., 2005). We used a pT514-specific antibody to test whether phosphorylation of CRMP2 at T514 also was stimulated in FeCγ mice. Fig. 6 D (top panel) shows that the levels of pT514-CRMP2 were not increased in our FeCγ double transgenic animals. We stripped the blot and reprobed with antitubulin antibody to show that equal amounts of proteins were loaded (Fig. 6 D, bottom panel). Together, these findings show that the phosphorylation of T509/S522, but not of T514, residue in CRMP2 protein is induced in FeCγ double transgenic mice.
Discussion
The biologic relevance of AICD to APP physiology or AD pathology has been proposed, but in vivo evidence that supports the hypothesis has been lacking. The current study was aimed at examining this hypothesis by expressing AICD postnatally and selectively in the forebrain and hippocampal regions of mouse brain. Here, we first show that AICD is present in brain membranes from normal control mice and can be detected by Western blot alone. We further demonstrate that the transgenic mice that were generated in this study express AICD at levels that are similar to those found in APP transgenic mice with FAD mutation, which are two- to threefold higher than in nontransgenic mice. These data strongly support the validity of our mouse model, which was verified further by observing increased expression of KAI1 gene in transgenic mice. Finally, we present evidence that the AICD transgenic mice display robust changes in GSK-3 signaling, which supports an in vivo role for AICD.
The finding that an AICD59–co-migrating peptide is present in the membrane fraction of the normal, wild-type C57BL/6 animals is novel and unanticipated because it was not detected in previous studies. We provide compelling data, by using various controls, that the membrane-associated, AICD59–co-migrating band is a product of APP. These include APP KO mice, which do not show the presence of AICD, and APP transgenic mice with FAD mutation, which show elevated AICD levels compared with control animals. The AICD peptide is not detected by Western blotting in the absence of antigen-retrieval treatment; this may explain why previous studies failed to detect AICD. Antigen-retrieval techniques have been used routinely in immunohistochemical detection of antigens in tissue sections, and our present findings indicate that such techniques also can be useful in Western blotting. The absence of AICD in the cytosol and its association with the membrane suggest that the AICD peptide is hydrophobic in nature. Studies that reported the “ɛ-cleavage” of APP-CTF found the resultant AICD peptide (AICD50) to be soluble and to fractionate in the cytosolic, but not in the membrane, fraction (Gu et al., 2001; Yu et al., 2001). These considerations make the AICD–co-migrating band that was identified in the present study more likely to be AICD59/57 than AICD50. The possibility that the endogenous band represents AICD57, rather than AICD59, cannot be ruled out. Future studies are required to establish unequivocally the length of the endogenous, membrane-associated AICD. In any case, our findings show that the steady-state levels of endogenously produced AICD peptides are higher than previously believed.
The FeCγ double transgenic mice that were generated in the current study showed robust stimulation of GSK-3β activity, without any significant changes in the mRNA or GSK-3β protein levels. These changes are caused primarily by AICD, because the Fe.27 mice express similar levels of Fe65 (Fig. 1 E) but do not exhibit altered GSK-3 signaling. However, it is possible that AICD may not be able to activate GSK-3 without Fe65. We do not know how AICD stimulates the GSK-3β activity in mice. Because the levels of mRNA transcripts and total protein of GSK-3β are not changed in the transgenic mice, our findings suggest a posttranscriptional signaling role for AICD in GSK-3 activation. Recently, Cao and Sudhof (2004) suggested that AICD acts catalytically in the context of membrane to “activate” Fe65, and bring about its biologic effects without necessarily being present in the nucleus. We detected AICD in the membrane and nuclear fractions in transgenic and control mice (Fig. 2 B). The presence of AICD in the membrane fraction is consistent with the above hypothesis. Nonetheless, the presence of AICD in the nuclear, but not in the cytosolic fraction of control mice shows that AICD accumulates in the nucleus and likely performs its function. Incidentally, our transgenic mice demonstrate the biologic relevance of the “γ-cleaved” AICD because they show that AICD59 peptide in vivo replicates the known activities of AICD (expression of KAI1 gene and activation of GSK-3β). Future studies on AICD50 transgenic mice will be required to determine the biologic effects of the “ɛ-cleaved” AICD peptide, which is not likely to remain associated with membranes.
The finding that AICD stimulates GSK-3 activity in vivo could be relevant to AD pathology. GSK-3β activity is elevated in AD brains (Pei et al., 1997; Kaytor and Orr, 2002; Bhat et al., 2004) and in the “Swedish mutation” mouse model, Tg2576, of AD (Tomidokoro et al., 2001). Higher GSK-3 activity in AD brains is responsible for phosphorylating the microtubule-binding protein, tau (Augustinack et al., 2002). Hyperphosphorylated tau aggregates into paired helical filaments and forms NFTs, a hallmark of AD pathology (Pei et al., 1999; Ishizawa et al., 2003). Significantly, the spatiotemporal distribution of active GSK-3β in AD brains coincides with the progression of NFTs and neurodegeneration, but not with the amyloid deposition (Pei et al., 1997; Pei et al., 1999). Thus, activated GSK-3β found in AD brains is one of the critical factors in the formation of NFTs. It is not clear what induces GSK-3β activation in AD brains, and our transgenic mice suggest that AICD could be a contributing factor. We observed elevated AICD levels in R1.40 transgenic mice that carry the “Swedish mutation” (Fig. 2), and our preliminary findings show that GSK-3 also is activated in these animals (unpublished data); however, R1.40 mice also show increased Aβ load. Because some in vitro studies suggest that Aβ can activate GSK-3β, it will be crucial to determine the contribution of these two APP-derived fragments toward GSK-3 activation in AD. We have initiated analysis of phospho-tau, a known substrate of GSK-3 in FeCγ transgenic mice. In the young animals that we examined so far (8–12 weeks old), we did not detect any significant changes in tau phosphorylation as detected by antibodies AT-8, AT-100, and TG-3 (not shown). It is not an uncommon observation that abnormal tau phosphorylation is detected in older animals, but not in young animals (Andorfer et al., 2003).
The FeCγ transgenic mice described here recapitulated the known in vitro effects of AICD, and revealed a new observation that AICD induces phosphorylation of CRMP2 on the residues that are crucially involved in growth cone collapse and axonal guidance. The phosphorylation of T509/S522 in CRMP2 plays a crucial role in mediating the repulsive action of Sema3a (Uchida et al., 2005). Sema3a is an extracellular matrix protein that binds Plexin/Neuropilin receptors in growth cones, activates GSK-3β, and phosphorylates CRMP2 on T509 and S522. These two residues are crucial in axonal guidance, because mutation of either residue abolishes the action of Sema3a and eliminates 3F4-reactivity (incidentally, 3F4-reactive phospho-CRMP2 is known to accumulate in the NFTs in AD brains [Yoshida et al., 1998; Gu et al., 2000; Cole et al., 2004]). Thus, increased phosphorylation of CRMP2 in the transgenic mice suggests an AICD-mediated signaling role for APP in axonal guidance or elongation (Fig. 6 E). The identity of an upstream ligand that binds APP and initiates the signaling cascade is unknown. F-spondin, a contact-repellent protein that is present in floor plate and hippocampus, may be a candidate because it binds APP and inhibits AICD production (Ho and Sudhof, 2004). The proposed role of AICD in mediating APP signaling and axonal elongation is consistent with the cortical dysplasia that is observed in mice lacking all three members of the APP family (Herms et al., 2004), or Fe65 and Fe65L1 proteins (Guenette et al., 2003).
Materials and methods
Transgenic animals
A cDNA encoding the 59 residue–long amyloid precursor protein–COOH-terminal fragment (APP-CTF) cleaved at the γ-secretase site was cloned into the vector NN265. A fragment containing an upstream intron, the APP intracellular domain (AICD)59 ORF, and a downstream SV40 polyadenylation signal was excised from this plasmid and cloned into MM403 plasmid containing the 5′ regulatory region of the CaMKIIα gene (both plasmids were the gift of T. Abel, University of Pennsylvania, Philadelphia, PA). Full-length cDNA encoding the entire ORF of rat Fe65 (gift of B. Margolis, University of Michigan, Ann Arbor, MI) was cloned as above. The transgenic constructs were linearized by digesting with BssHII (mixed in 1:1 proportion), microinjected into the male pronucleus of C57Bl/6, and implanted into pseudopregnant C57Bl/6 females. Founder mice were identified by PCR on tail DNA, and by using a forward primer (F1) in the intron region (GCGCTAAGATTGTCAGTTTCC) and reverse primer for AICD-R1 (TCTGCTGCATCTTGGACAGG) or Fe65-R1 (ACATTTCGGTTCTGGTCTCG). The sequence of primers that was used for the mouse endogenous Xist gene was as follows: forward: GGGACCTAACTGTTGGCTTTATCAG, reverse: GAAGTGAATTGAAGTTTTGGTCTAG. We also performed PCR using primers in the SV40 polyadenylation signal to ensure that the complete transgene was integrated.
Tissue preparation
The data presented here are from multiple 8–12-wk old, mixed gender animals from two double transgenic lines (FeCγ.12 and FeCγ.25) and a single Fe65 transgenic line (Fe.27). We used nontransgenic littermates as controls. Mice were killed by cervical dislocation and their brains were removed, divided sagittally (after removing cerebellum), and quick frozen. Tissues were homogenized in 10 volumes of Tris-buffered saline (50 mM Tris, pH 7.4, 150 mM NaCl, 1 mM EDTA) with freshly added 1 mM PMSF and protease and phosphatase inhibitor cocktail (Sigma-Aldrich). Total homogenates were centrifuged briefly to remove nuclei and unbroken tissue, and centrifuged at 150,000 g for 60 min to obtain cytosol (supernatant) and crude membrane (pellet) fractions. The nuclear extracts were prepared using the NE-PER kit (Pierce Chemical Co.). Protein determination was performed using a Bio-Rad Laboratories kit. Equal amounts of proteins (usually 10–25 μg per lane) were loaded on the gel.
Western blotting
Immunoblotting was performed as described before (Zheng et al., 1998; Gao and Pimplikar, 2001) using a 10% SDS-PAGE gel, except for AICD detection, which was performed as described by Pinnix et al. (2001), with some modifications. In brief, the proteins were separated using NuPAGE Novex Bris-Tris 4–12% gel (Invitrogen) and transferred on to nitrocellulose membrane. The membranes were incubated with boiling PBS for 5 min (antigen-retrieval treatment) before blocking in TBS containing 10% newborn calf serum for 2 h at room temperature. The blots were incubated with up to 1:10,000 dilution of antibody 0443 (gift of K. Sambamurti, Medical University of South Carolina, Charleston, SC) overnight at 4°C, and the blots were visualized using ECL. Other antibodies used are as follows: antibody 369 (gift of S. Gandy) to probe AICD; anti-myc antibody 9E10 (CLONTECH Laboratories, Inc.) to detect myc-Fe65 transgene; and antibody 3H6 (Upstate Biotechnology) to detect total Fe65 (this antibody detects mouse and rat Fe65). Anti-pS9–GSK-3β antibody to detect inhibited kinase was from Cell Signaling; anti-pY279/216 to detect activated GSK-3 (Cell Signaling) was a gift of M. Smith (Case Western Reserve University, Cleveland OH); and SERCA2 antibody, MA3-919, was from Affinity BioReagents, Inc. Antibody 3F4 recognizes CRMP2 phosphorylated at S522 and T509, and antibody C4G binds unphosphorylated CRMP2 (both antibodies were the gifts of Y. Ihara and Y. Morishima, University of Tokyo, Tokyo, Japan). Antibody that recognizes pT514 CRMP2 was a gift of K. Kaibuchi. Anti-pERK1/2 antibody was from Cell Signaling. Protein bands were visualized by ECL using Pierce SuperSignal system. Protein levels were quantified by scanning the films in Photoshop (Adobe), and measuring the band pixel densities in NIH Image J software (National Institutes of Health). The results were analyzed by ANOVA with Fisher's protected least significance difference (PLSD) test using StatView software (Abacus Concepts). The data presented here are from multiple animals from FeCγ.12 and FeCγ.25 lines and were repeated in the FeCγ.22 transgenic line.
Quantitative real-time PCR
Animals were killed by cervical dislocation, brains were dissected, cerebellum was discarded, and total RNA was isolated from the brain tissue with RNeasy kit (QIAGEN). First-strand cDNA synthesis was performed using 4 μg RNA with the Superscript First-Strand Synthesis system for RT-PCR (Invitrogen) according to the manufacturer's protocol. The cDNA mixture was diluted 1:20; 5 μl of the cDNA product was used for real-time PCR performed on the iCycler (Bio-Rad Laboratories), and detected by using iQ SYBR Green Supermix. The amplification cycle was 95°C for 10 s, 65°C for 15 s, and 72°C for 15 s. Primers were designed by using Primer 3 software to amplify the 3′ region of the ORF. Primer sequences were: β-actin (forward: TACAGCTTCACCACCACAGC; reverse: ATGCCACAGGATTCCATACC), KAI1 (forward: TCTGTGGGAGACAGGGTAGG; reverse: CTGCCAAGAAACACCAGTCC), and GSK-3β (forward: TCCATTCCTTTGGAATCTGC; reverse: CAATTCAGCCAACACACACAGC). Melting curve analysis confirmed that only one product was amplified. Specificity was confirmed by electrophoresis of PCR products through 1.5% agarose gels (stained with ethidium bromide). Only one product was observed with each primer set, and the product size matched that predicted from published cDNA sequences. Expression was normalized to β-actin.
Acknowledgments
We thank the two anonymous reviewers whose comments resulted in the vast improvement in this manuscript. We thank B. Lamb for R1.40 mice, his advice in generating the AICD transgenic mice, and for his comments on the manuscript.
This work was supported by a grant from the Alzheimer's Association to S.W. Pimplikar (IIRG-3894) and by a grant from the National Institute on Aging to UAC/CWRU ADRC (P50-AG08012).
Abbreviations used in this paper: AD, Alzheimer's disease; AICD, APP intracellular domain; APP, amyloid precursor protein; CRMP2, collapsin responsive mediator protein–2; CTF, COOH-terminal fragment; ERK, extracellular signal-regulated kinase; FAD, familial Alzheimer's disease; GSK, glycogen synthase kinase; Sema3a, Semaphorin3a.
References
- Abel, T., P.V. Nguyen, M. Barad, T.A. Deuel, E.R. Kandel, and R. Bourtchouladze. 1997. Genetic demonstration of a role for PKA in the late phase of LTP and in hippocampus-based long-term memory. Cell. 88:615–626. [DOI] [PubMed] [Google Scholar]
- Andorfer, C., Y. Kress, M. Espinoza, R. de Silva, K.L. Tucker, Y.A. Barde, K. Duff, and P. Davies. 2003. Hyperphosphorylation and aggregation of tau in mice expressing normal human tau isoforms. J. Neurochem. 86:582–590. [DOI] [PubMed] [Google Scholar]
- Annaert, W., and B. De Strooper. 2002. A cell biological perspective on Alzheimer's disease. Annu. Rev. Cell Dev. Biol. 18:25–51. [DOI] [PubMed] [Google Scholar]
- Augustinack, J.C., A. Schneider, E.M. Mandelkow, and B.T. Hyman. 2002. Specific tau phosphorylation sites correlate with severity of neuronal cytopathology in Alzheimer's disease. Acta Neuropathol. (Berl.). 103:26–35. [DOI] [PubMed] [Google Scholar]
- Baek, S.H., K.A. Ohgi, D.W. Rose, E.H. Koo, C.K. Glass, and M.G. Rosenfeld. 2002. Exchange of N-CoR corepressor and Tip60 coactivator complexes links gene expression by NF-kappaB and beta-amyloid precursor protein. Cell. 110:55–67. [DOI] [PubMed] [Google Scholar]
- Bell, K.A., K.J. O'Riordan, J.D. Sweatt, and K.T. Dineley. 2004. MAPK recruitment by beta-amyloid in organotypic hippocampal slice cultures depends on physical state and exposure time. J. Neurochem. 91:349–361. [DOI] [PubMed] [Google Scholar]
- Bhat, R.V., S.L. Budd Haeberlein, and J. Avila. 2004. Glycogen synthase kinase 3: a drug target for CNS therapies. J. Neurochem. 89:1313–1317. [DOI] [PubMed] [Google Scholar]
- Borg, J.P., J. Ooi, E. Levy, and B. Margolis. 1996. The phosphotyrosine interaction domains of X11 and FE65 bind to distinct sites on the YENPTY motif of amyloid precursor protein. Mol. Cell. Biol. 16:6229–6241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown, M.S., J. Ye, R.B. Rawson, and J.L. Goldstein. 2000. Regulated intramembrane proteolysis: a control mechanism conserved from bacteria to humans. Cell. 100:391–398. [DOI] [PubMed] [Google Scholar]
- Cao, X., and T.C. Sudhof. 2001. A transcriptionally [correction of transcriptively] active complex of APP with Fe65 and histone acetyltransferase Tip60. Science. 293:115–120. [DOI] [PubMed] [Google Scholar]
- Cao, X., and T.C. Sudhof. 2004. Dissection of amyloid-beta precursor protein-dependent transcriptional transactivation. J. Biol. Chem. 279:24601–24611. [DOI] [PubMed] [Google Scholar]
- Cole, A.R., A. Knebel, N.A. Morrice, L.A. Robertson, A.J. Irving, C.N. Connolly, and C. Sutherland. 2004. GSK-3 phosphorylation of the Alzheimer epitope within collapsin response mediator proteins regulates axon elongation in primary neurons. J. Biol. Chem. 279:50176–50180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cupers, P., I. Orlans, K. Craessaerts, W. Annaert, and B. De Strooper. 2001. The amyloid precursor protein (APP)-cytoplasmic fragment generated by gamma-secretase is rapidly degraded but distributes partially in a nuclear fraction of neurones in culture. J. Neurochem. 78:1168–1178. [DOI] [PubMed] [Google Scholar]
- De Strooper, B., and W. Annaert. 2000. Proteolytic processing and cell biological functions of the amyloid precursor protein. J. Cell Sci. 113:1857–1870. [DOI] [PubMed] [Google Scholar]
- Fukata, Y., T.J. Itoh, T. Kimura, C. Menager, T. Nishimura, T. Shiromizu, H. Watanabe, N. Inagaki, A. Iwamatsu, H. Hotani, and K. Kaibuchi. 2002. CRMP-2 binds to tubulin heterodimers to promote microtubule assembly. Nat. Cell Biol. 4:583–591. [DOI] [PubMed] [Google Scholar]
- Gao, Y., and S.W. Pimplikar. 2001. The gamma-secretase–cleaved C-terminal fragment of amyloid precursor protein mediates signaling to the nucleus. Proc. Natl. Acad. Sci. USA. 98:14979–14984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu, Y., N. Hamajima, and Y. Ihara. 2000. Neurofibrillary tangle-associated collapsin response mediator protein-2 (CRMP-2) is highly phosphorylated on Thr-509, Ser-518, and Ser-522. Biochemistry. 39:4267–4275. [DOI] [PubMed] [Google Scholar]
- Gu, Y., H. Misonou, T. Sato, N. Dohmae, K. Takio, and Y. Ihara. 2001. Distinct intramembrane cleavage of the beta-amyloid precursor protein family resembling gamma-secretase–like cleavage of Notch. J. Biol. Chem. 276:35235–35238. [DOI] [PubMed] [Google Scholar]
- Guenette, S.Y., Y. Chang, T. Hiesberger, J.A. Richardson, and J. Herz. 2003. Mice deficient for the Fe65 and Fe65L1 proteins have neurological defects. Society for Neuroscience Abstract. 336:10. [Google Scholar]
- Herms, J., B. Anliker, S. Heber, S. Ring, M. Fuhrmann, H. Kretzschmar, S. Sisodia, and U. Muller. 2004. Cortical dysplasia resembling human type 2 lissencephaly in mice lacking all three APP family members. EMBO J. 23:4106–4115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ho, A., and T.C. Sudhof. 2004. Binding of F-spondin to amyloid-beta precursor protein: a candidate amyloid-beta precursor protein ligand that modulates amyloid-beta precursor protein cleavage. Proc. Natl. Acad. Sci. USA. 101:2548–2553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishizawa, T., N. Sahara, K. Ishiguro, J. Kersh, E. McGowan, J. Lewis, M. Hutton, D.W. Dickson, and S.H. Yen. 2003. Co-localization of glycogen synthase kinase-3 with neurofibrillary tangles and granulovacuolar degeneration in transgenic mice. Am. J. Pathol. 163:1057–1067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jope, R.S., and G.V. Johnson. 2004. The glamour and gloom of glycogen synthase kinase-3. Trends Biochem. Sci. 29:95–102. [DOI] [PubMed] [Google Scholar]
- Kaytor, M.D., and H.T. Orr. 2002. The GSK3 beta signaling cascade and neurodegenerative disease. Curr. Opin. Neurobiol. 12:275–278. [DOI] [PubMed] [Google Scholar]
- Kim, H.S., E.M. Kim, J.P. Lee, C.H. Park, S. Kim, J.H. Seo, K.A. Chang, E. Yu, S.J. Jeong, Y.H. Chong, and Y.H. Suh. 2003. C-terminal fragments of amyloid precursor protein exert neurotoxicity by inducing glycogen synthase kinase-3beta expression. FASEB J. 17:1951–1953. [DOI] [PubMed] [Google Scholar]
- Kimberly, W.T., J.B. Zheng, S.Y. Guenette, and D.J. Selkoe. 2001. The intracellular domain of the beta-amyloid precursor protein is stabilized by Fe65 and translocates to the nucleus in a notch-like manner. J. Biol. Chem. 276:40288–40292. [DOI] [PubMed] [Google Scholar]
- Kinoshita, A., C.M. Whelan, O. Berezovska, and B.T. Hyman. 2002. The gamma secretase-generated carboxyl-terminal domain of the amyloid precursor protein induces apoptosis via Tip60 in H4 cells. J. Biol. Chem. 277:28530–28536. [DOI] [PubMed] [Google Scholar]
- Lamb, B.T., L.M. Call, H.H. Slunt, K.A. Bardel, A.M. Lawler, C.B. Eckman, S.G. Younkin, G. Holtz, S.L. Wagner, D.L. Price, et al. 1997. Altered metabolism of familial Alzheimer's disease-linked amyloid precursor protein variants in yeast artificial chromosome transgenic mice. Hum. Mol. Genet. 6:1535–1541. [DOI] [PubMed] [Google Scholar]
- Leissring, M.A., M.P. Murphy, T.R. Mead, Y. Akbari, M.C. Sugarman, M. Jannatipour, B. Anliker, U. Muller, P. Saftig, B. De Strooper, et al. 2002. A physiologic signaling role for the gamma-secretase–derived intracellular fragment of APP. Proc. Natl. Acad. Sci. USA. 99:4697–4702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mueller, T., D. Koegel, J.H.M. Prehn, and R. Egensperger. 2004. Role of the β-amyloid precursor protein intracellular domain (AICD) and Fe65 in regulation of gene expression and calcium dependent neuronal cell death. Society for Neuroscience Abstract. 335:9. [Google Scholar]
- Passer, B., L. Pellegrini, C. Russo, R.M. Siegel, M.J. Lenardo, G. Schettini, M. Bachmann, M. Tabaton, and L. D'Adamio. 2000. Generation of an apoptotic intracellular peptide by gamma-secretase cleavage of Alzheimer's amyloid beta protein precursor. J. Alzheimers Dis. 2:289–301. [DOI] [PubMed] [Google Scholar]
- Pei, J.J., E. Braak, H. Braak, I. Grundke-Iqbal, K. Iqbal, B. Winblad, and R.F. Cowburn. 1999. Distribution of active glycogen synthase kinase 3beta (GSK-3beta) in brains staged for Alzheimer disease neurofibrillary changes. J. Neuropathol. Exp. Neurol. 58:1010–1019. [DOI] [PubMed] [Google Scholar]
- Pei, J.J., T. Tanaka, Y.C. Tung, E. Braak, K. Iqbal, and I. Grundke-Iqbal. 1997. Distribution, levels, and activity of glycogen synthase kinase-3 in the Alzheimer disease brain. J. Neuropathol. Exp. Neurol. 56:70–78. [DOI] [PubMed] [Google Scholar]
- Pinnix, I., U. Musunuru, H. Tun, A. Sridharan, T. Golde, C. Eckman, C. Ziani-Cherif, L. Onstead, and K. Sambamurti. 2001. A novel gamma-secretase assay based on detection of the putative C-terminal fragment-gamma of amyloid beta protein precursor. J. Biol. Chem. 276:481–487. [DOI] [PubMed] [Google Scholar]
- Price, D.L., R.E. Tanzi, D.R. Borchelt, and S.S. Sisodia. 1998. Alzheimer's disease: genetic studies and transgenic models. Annu. Rev. Genet. 32:461–493. [DOI] [PubMed] [Google Scholar]
- Sabo, S.L., A.F. Ikin, J.D. Buxbaum, and P. Greengard. 2001. The Alzheimer amyloid precursor protein (APP) and FE65, an APP-binding protein, regulate cell movement. J. Cell Biol. 153:1403–1414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabo, S.L., A.F. Ikin, J.D. Buxbaum, and P. Greengard. 2003. The amyloid precursor protein and its regulatory protein, FE65, in growth cones and synapses in vitro and in vivo. J. Neurosci. 23:5407–5415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selkoe, D.J. 2005. Defining molecular targets to prevent Alzheimer disease. Arch. Neurol. 62:192–195. [DOI] [PubMed] [Google Scholar]
- Tomidokoro, Y., K. Ishiguro, Y. Harigaya, E. Matsubara, M. Ikeda, J.M. Park, K. Yasutake, T. Kawarabayashi, K. Okamoto, and M. Shoji. 2001. Abeta amyloidosis induces the initial stage of tau accumulation in APP(Sw) mice. Neurosci. Lett. 299:169–172. [DOI] [PubMed] [Google Scholar]
- Uchida, Y., T. Ohshima, Y. Sasaki, H. Suzuki, S. Yanai, N. Yamashita, F. Nakamura, K. Takei, Y. Ihara, K. Mikoshiba, et al. 2005. Semaphorin3A signalling is mediated via sequential Cdk5 and GSK3beta phosphorylation of CRMP2: implication of common phosphorylating mechanism underlying axon guidance and Alzheimer's disease. Genes Cells. 10:165–179. [DOI] [PubMed] [Google Scholar]
- Von Rotz, R.C., B.M. Kohli, J. Bosset, M. Meier, T. Suzuki, R.M. Nitsch, and U. Konietzko. 2004. The APP intracellular domain forms nuclear multiprotein complexes and regulates the transcription of its own precursor. J. Cell Sci. 117:4435–4448. [DOI] [PubMed] [Google Scholar]
- Yoshida, H., A. Watanabe, and Y. Ihara. 1998. Collapsin response mediator protein-2 is associated with neurofibrillary tangles in Alzheimer's disease. J. Biol. Chem. 273:9761–9768. [DOI] [PubMed] [Google Scholar]
- Yoshimura, T., Y. Kawano, N. Arimura, S. Kawabata, A. Kikuchi, and K. Kaibuchi. 2005. GSK-3beta regulates phosphorylation of CRMP-2 and neuronal polarity. Cell. 120:137–149. [DOI] [PubMed] [Google Scholar]
- Yu, C., S.H. Kim, T. Ikeuchi, H. Xu, L. Gasparini, R. Wang, and S.S. Sisodia. 2001. Characterization of a presenilin-mediated amyloid precursor protein carboxyl-terminal fragment gamma. Evidence for distinct mechanisms involved in gamma -secretase processing of the APP and Notch1 transmembrane domains. J. Biol. Chem. 276:43756–43760. [DOI] [PubMed] [Google Scholar]
- Zheng, P., J. Eastman, S. Vande Pol, and S.W. Pimplikar. 1998. PAT1, a microtubule-interacting protein, recognizes the basolateral sorting signal of amyloid precursor protein. Proc. Natl. Acad. Sci. USA. 95:14745–14750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu, X., Z. Sun, H.G. Lee, S.L. Siedlak, G. Perry, and M.A. Smith. 2003. Distribution, levels, and activation of MEK1 in Alzheimer's disease. J. Neurochem. 86:136–142. [DOI] [PubMed] [Google Scholar]