

Abstract
We show that RecN protein is recruited to a defined DNA double strand break (DSB) in Bacillus subtilis cells at an early time point during repair. Because RecO and RecF are successively recruited to DSBs, it is now clear that dynamic DSB repair centers (RCs) exist in prokaryotes. RecA protein was also recruited to RCs and formed highly dynamic filamentous structures, which we term threads, across the nucleoids. Formation of RecA threads commenced ∼30 min after the induction of DSBs, after RecN recruitment to RCs, and disassembled after 2 h. Time-lapse microscopy showed that the threads rapidly changed in length, shape, and orientation within minutes and can extend at 1.02 μm/min. The formation of RecA threads was abolished in recJ addAB mutant cells but not in each of the single mutants, suggesting that RecA filaments can be initiated via two pathways. Contrary to proteins forming RCs, DNA polymerase I did not form foci but was present throughout the nucleoids (even after induction of DSBs or after UV irradiation), suggesting that it continuously scans the chromosome for DNA lesions.
Introduction
DNA double strand breaks (DSBs) can be repaired through two principally different processes: error-prone direct end joining and recombination with the intact DNA duplex of the sister chromosome (Haber, 2000). The repair of DSBs through homologous recombination plays the more important role and occurs in three general steps. Initially, during presynapsis, DNA ends are primed for loading of the central homologous recombination protein RecA (Rad51 in eukaryotes) by the MRX (Mre11–Rad50–Xrs2) complex in eukaryotic cells or by RecBCD, RecN, RecF, RecO, and RecR proteins in bacteria (Kowalczykowski et al., 1994; Arnold and Kowalczykowski, 2000; Bork et al., 2001; Hopfner et al., 2002; Morimatsu and Kowalczykowski, 2003). During synapsis, RecA sets up strand exchange by introducing a single DNA strand from the break site into the intact homologous sister chromosome and vice versa. Finally, during postsynapsis, RecA-mediated three-way junctions are converted into true crossovers (or Holiday junctions) through the action of proteins such as the RecG helicase, and branch migration and resolution of Holiday junctions are achieved through the action of the RuvABC complex in bacteria (Cromie et al., 2001). It has recently become clear that the repair of DNA DSBs involves the orchestrated recruitment of various repair proteins to the sites of DSBs in eukaryotic cells. The first visible event is recruitment of the MRX complex to a DSB, which mediates end processing as well as signaling and, in turn, recruits single-stranded (ss) DNA protecting proteins and additional signaling proteins to the break. At a later time point, repair proteins such as Rad52 and Rad51 appear within the DSB repair center (RC; Lisby et al., 2004).
In the prokaryote Bacillus subtilis, three proteins that are involved in DSB repair have also been shown to assemble into discrete centers on the nucleoids after induction of DSBs. Like the MRX component Rad50, RecN protein is a member of the structural maintenance of chromosomes protein family, which are key players in a variety of chromosome dynamics from chromosome condensation and cohesion to transcriptional repression of whole chromosomes and DNA repair (Hirano, 2002). RecN assembles into discrete subcellular structures on the nucleoids within 15 to 30 min after induction of DSBs, whereas RecO is recruited to these sites within 60 min, followed by RecF, which becomes visible after ∼2 h (Kidane et al., 2004). Together with RecR, RecO is thought to facilitate the loading of RecA onto ssDNA, which needs to be generated by the action of an endonuclease/helicase enzyme assembly (RecBCD in Escherichia coli, AddAB in B. subtilis, and possibly RecJ and one of several helicases; Kowalczykowski et al., 1994; Hegde et al., 1996; Chedin et al., 2000; Bork et al., 2001). Biochemical data suggest that RecF protein might limit the spreading of RecA on ssDNA (Bork et al., 2001). RecA forms long nucleoprotein filaments with ssDNA in vitro, which most likely present its active form in DNA strand invasion and strand exchange (for review see Wyman and Kanaar, 2004). RecNOF has been shown to form DSB-induced foci in the absence of RecA protein (Kidane et al., 2004), suggesting that these proteins come to DSBs independently of RecA, but it has not been shown how and when RecA works at DSBs in live cells.
The replication machinery in B. subtilis and E. coli cells is a stationary protein complex that is located at the cell center during most of the cell cycle (Lemon and Grossman, 2001). Thus, the chromosome moves through the central replisome during DNA synthesis, and the duplicated sequences are segregated into each cell half by an active, but unknown, mechanism. In contrast to eukaryotic cells, in which sister chromosomes are paired during S phase until separation during anaphase in mitosis or meiosis (Nasmyth et al., 2000), chromosome segregation occurs concomitantly with DNA replication in eubacteria, with moderate to no chromosome cohesion occurring. Early after the initiation of replication, origin regions of the chromosome are rapidly separated toward opposite cell poles in several bacterial species (Gordon et al., 1997; Niki and Hiraga, 1998; Sharpe and Errington, 1998; Webb et al., 1998). All other replicated regions ensue such that genes are replicated, segregated, and positioned according to their order on the chromosome and such that in B. subtilis, E. coli, and Caulobacter crescentus cells, the chromosomes have a preferred arrangement (Teleman et al., 1998; Niki et al., 2000; Viollier et al., 2004). During most of the cell cycle, origin regions are positioned in a bipolar manner (one close to each cell pole), whereas terminus regions are located toward the cell center, and sequences between these two positions on the chromosome are positioned between the cell pole and cell center. It follows that for the repair of a break within a replicated DNA sequence, the intact copy for DNA repair is usually present within the other cell half. How repair of DSBs is achieved within time and space in prokaryotes has been unclear.
Results
RecN assembles at defined DNA DSBs
We have used the homothallic (HO) endonuclease system from Saccharomyces cerevisiae (Haber, 2002) to investigate whether RecN is indeed recruited to actual sites of DSBs. The HO endonuclease gene was stably integrated into the B. subtilis chromosome under the control of the inducible xylose promoter, and the HO cut site was integrated at two different places on the chromosome: close to the region containing the origin of replication and close to the terminus region. These regions were decorated with an array of tandem lacO sequences, and the cells expressed LacI-CFP and YFP-RecN. During induction of HO endonuclease or after the addition of mitomycin C (MMC), origin regions were located close to the cell poles (Fig. 1, A–C, white arrowheads), whereas the terminus region was close to the middle of the cells (Fig. 1 D). Thus, global chromosome orientation is retained after induction of DSBs and during repair. After induction of HO endonuclease, 25–35% of the cells showed clear YFP-RecN foci. In 62% of the cells having YFP-RecN foci and containing the cut site close to the replication origins, LacI-CFP and YFP-RecN foci were coincident, and 23% of the foci were in close proximity (within <0.3 μm). Only 15% of the foci were further than 0.3 μm apart. The distance between the lacO cassette and the cut site was ∼5,000 bp, which corresponds to 1.5 μm if the DNA is fully stretched between the cut site and cassette. This may explain, at least in part, why YFP-RecN and LacI-CFP signals were in close proximity, but were not coincident, in the relatively large number (23%) of cells. Interestingly, although a cut site exists close to both polar origins, only 12–15% of the cells contained two RecN foci that were coincident with the bipolar LacI-CFP foci in 90% of these cells (Fig. 1, B and C). To test if HO indeed cuts at the desired site and to monitor the effectiveness of its activity, we performed Southern blot analysis by using a probe that was located adjacent (120 bp) to the cut site. Fig. 2 shows that a 450-bp fragment, which was caused by the DSB, is visible 90 min after induction of HO endonuclease. However, it is clear that HO endonuclease does not cause a cut in all cells because the original 50-kb fragment is still detectable 90 min after HO induction. No clear break was visible 20 min after induction of HO endonuclease, showing that, in addition to its moderate activity, HO requires an extended period to administer a DSB break. These experiments suggest that the 25–35% of cells showing YFP-RecN foci is a result of the low activity of HO endonuclease and verify the specificity of the break. As a further control, we induced HO endonuclease in cells that expressed YFP-RecN but lacked the cut site. None of the 220 cells that were monitored contained detectable fluorescent signals, showing that the induction of YFP-RecN foci was caused by a DSB introduced at the HO cut site.
Figure 1.
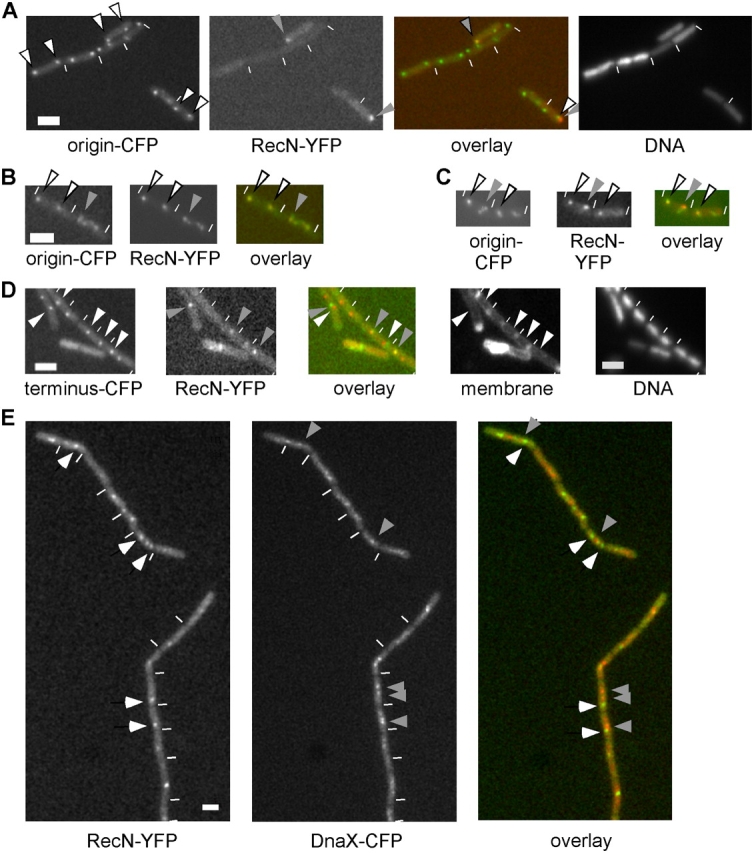
Fluorescence microscopy of B. subtilis cells in which DSBs were induced by different means. (A–C) Cells carrying the origin region tagged with LacI-CFP (origin-CFP, green in the overlay) and the HO cut site integrated next to the CFP-tagged site 1 h after induction of HO endonuclease. (A) White arrowheads indicate positions of origin regions; gray arrowheads indicate positions of RecN foci (red in the overlay). (B and C) White arrowheads indicate origins and RecN foci; gray arrowheads indicate abnormal origin signals. (D) Cells carrying LacI-CFP and the HO cut site close to the terminus region (terminus-CFP) 1 h after induction of HO endonuclease. White arrowheads indicate terminus regions (green in overlay); gray arrowheads indicate RecN foci (red in overlay) or colocalization in the overlay. Note that all cells contain DNA. Nucleoids are decondensed after DSBs, and staining is quite heterogeneous. (E) Strain AV2 (recN-yfp and dnaX-cfp) 60 min after the addition of 100 ng/ml MMC. White arrowheads indicate YFP-RecN foci (green in the overlay); gray arrows indicate DnaX-CFP foci (red in the overlay). (A–E) White lines indicate the ends of cells. Bars, 2 μm.
Figure 2.

Southern blot analysis of the efficiency and specificity of HO endonuclease. The first two lanes show no cut expression of HO endonuclease, and the remaining lanes show induction of HO endonuclease after indicated times. Sizes of marker fragments are indicated on the right.
When the cut site was moved to the terminus region, YFP-RecN foci colocalized with the centrally located LacI-CFP foci in 60% of the cells (Fig. 1 D) or were closely located in 21% of the cells, whereas both signals were clearly separate in only 19% of the cells (with 35–38% of all cells having YFP-RecN foci). Therefore, RecN is recruited to sites of DSBs, establishing that RecNOF foci constitute actual DSB RCs that are targeted to breaks in the chromosome. Furthermore, RCs can even form close to the terminus regions, although these have been replicated in only a minority of the cells because their duplication occurs late in the cell cycle. Thus, there is a sister chromosome for homologous recombination only infrequently. It is also possible that RCs form at sites close to the terminus only when these have been replicated but not in cells with a nonreplicated terminus. It will be highly interesting to test this point; however, we have not been able to address this as a result of technical difficulties.
RCs are generally separate from the replication machinery
To further characterize DSB RCs, we simultaneously localized repair foci and DNA polymerase (Pol) III after induction of DSBs by using MMC. Although YFP-RecN foci were often present close to the middle of the cells, YFP-RecN rarely colocalized with DnaX-CFP (τ subunit of DNA Pol III fused to CFP; Fig. 1 E). Rather, YFP-RecN foci were generally well separated from the central Pol. Although 5% of foci were coincident with DnaX-CFP, 35% of the foci were close to, but distinct from, DNA Pol III, and 60% of the foci were well separated from DNA Pol III (>150 cells analyzed). Similar results were obtained by using YFP-RecO as a marker for RCs (unpublished data). Thus, RCs are independent structures that appear to form at many positions on the nucleoids. We have recently shown that B. subtilis cells can survive in medium containing 100 but not 200 ng/ml MMC (Kidane et al., 2004). Interestingly, DNA Pol III was still visible as one or two central foci after the addition of 50 ng/ml MMC (Fig. 3, left), showing that the DNA replication machinery does not disintegrate after a sublethal amount of DSBs. However, after treatment with a lethal dose of 250 ng/ml MMC, DnaX-CFP foci were no longer apparent in most cells (Fig. 3, right), showing that a lethal dose of MMC interferes with maintenance of the replication machinery.
Figure 3.

Formation of the replisome is influenced by DNA DSBs. Fluorescence microscopy of strain AV2 (dnaX-cfp and recN-yfp) 60 min after the addition of 50 (left) or 250 ng/ml (right) MMC. White lines indicate ends of cells. Bars, 2 μm.
RecA forms transient filament-like structures during DSB repair
Next, we analyzed the localization of the major recombination repair protein RecA. Although COOH-terminal GFP fusions to RecA were nonfunctional, an NH2-terminal GFP fusion retained almost full activity. RecA mutant cells grow considerably slower than wild-type cells, have abnormal nucleoid morphology, and are highly sensitive to DSB-inducing agents (Zyskind et al., 1992; Sciochetti et al., 2001; Cox, 2003). When GFP-RecA was expressed from an ectopic site on the chromosome and the original recA gene was deleted, the cells grew indistinguishably from wild-type cells and were as sensitive to MMC as wild-type cells (Fig. S1, available at http://www.jcb.org/cgi/content/full/jcb.200412090/DC1). Because nucleoids were somewhat abnormal compared with wild-type cells in only a small percentage of the cells (2–3%), these experiments show that GFP-RecA retains almost 100% of the wild-type activity. In exponentially growing cells, GFP-RecA localized throughout the nucleoids (Fig. 4 A), whereas this pattern of localization changed dramatically upon induction of DSBs by MMC. 30 min after the addition of 50 ng/ml MMC, RecA concentrated into patches on the nucleoid (nucleoids fuse after induction of DSBs [Kidane et al., 2004]) in usually one half or, less frequently, in both halves of the cells (Fig. 4 B) and further concentrated into irregular structures or defined foci between 45 and 60 min (Fig. 4 C). The latter structures coincided with RecN foci (Fig. 4 H) in >90% of the cells, showing that RecA is recruited to RCs after the recruitment of RecN. Starting 45–60 min after induction of DSBs, some GFP-RecA structures had filamentous extensions (we use the term threads) on one side, which were usually directed toward the opposite cell half (Fig. 4, C and D, indicated by cartoons). GFP-RecA threads extended in an increasing number of cells (Fig. 4 E) until, after 1 h, 84% of the cells contained highly extended GFP-RecA threads (with 4% of the cells showing GFP-RecA patches and 12% containing a diffuse GFP-RecA signal). The formation of RecA threads occurred with a similar timing compared with the recruitment of RecO into RCs (Kidane et al., 2004). Interestingly, many threads showed a helical pattern (Fig. 4, C–E). GFP-RecA threads became shorter 2 h after induction of DSBs (Fig. 4 F) and became more undetectable after 3 h when GFP-RecA returned to localize throughout the nucleoids (with three- to fourfold higher signal intensity compared with noninduced cells; Fig. 4 G). These data show that RecA threads are transient and dynamic structures that generally form from one side of the nucleoid to the other and initially emanate from RecA patches that surround RCs. It should be noted that the addition of 50 ng/ml MMC, as used in the aforementioned experiments, led to the death of 46% of all cells (Fig. S1). This shows that most of the cells are successfully repairing and surviving the induced DSBs, and it rules out the possibility that RecA threads are caused by abnormal localization in dying cells. However, because the NH2 terminus of RecA is required for the formation of nucleofilaments, we cannot rule out that some of the observed filamentation might be aberrant.
Figure 4.
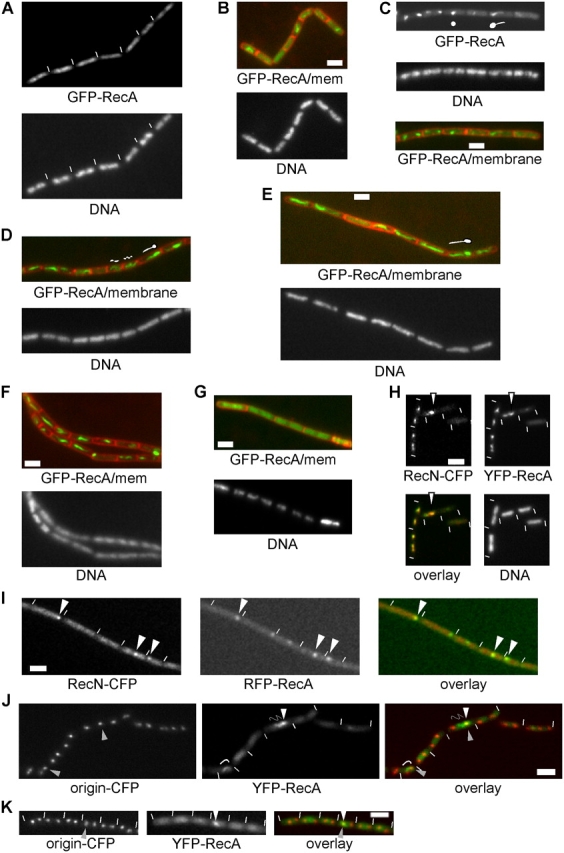
Fluorescence microscopy of B. subtilis cells expressing GFP-RecA. (A) Exponentially growing cells. (B–I) Cells at 30 (B), 45 (C), 60 (D), 90 (E), 120 (F), and 180 (G) min after induction of DSBs by MMC. Cartoons indicate the nature of the fluorescence in different panels. (H) Cells expressing CFP-RecN (red in overlay) and YFP-RecA (green in overlay) 45 min after induction of DSBs. Arrowheads indicate colocalization, with a GFP-RecA thread appearing to extend from the RC. (I) Cells expressing CFP-RecN (red in overlay) and RFP-RecA (green in overlay) 60 min after induction of DSBs. Arrowheads indicate RecA and RecN foci and their colocalization. (J and K) Cells containing origin regions that are decorated with LacI-CFP, a HO cut site close to the origin regions, and expressing YFP-RecA, in which HO endonuclease has been expressed 90 min before image acquisition. White arrowheads indicate RecA foci/patches; gray arrowheads indicate origin signals; curved lines indicate RecA filaments. White lines indicate the ends of cells. Bars, 2 μm.
To gain more insight into the nature of RecA threads, we generated a strain that expressed RecA-YFP, contained the HO endonuclease system with a cut site close to the origin regions, and had CFP-tagged origin regions. In the presence, but not in the absence, of xylose, 15% of cells showed YFP-RecA patches or foci and 7% contained YFP-RecA threads (with 120 cells analyzed; Fig. 4, J and K). These data show that the HO system is capable of inducing RecA patches and threads. However, either the HO system is less efficient in generating DSBs compared with the addition of MMC, or single DSBs may recruit less RecA such that its localization is below the limit of detection in many cells; most likely, both assumptions apply. In any event, 90% of the YFP-RecA patches or foci coincided with the signal of an origin region (Fig. 4 K), whereas in the other 10%, a YFP-RecA patch was present in the absence of an origin signal. These findings reinforce the concept that RecA is recruited to sites of DSBs. Fig. 4 J shows a YFP-RecA focus (white arrowheads) that coincides with an origin signal from which a thread (indicated by a snakelike line) extends toward the other origin that is located in the opposite cell half. In the cell on the left of the image, a thread extends from one origin to the other and appears to connect these chromosomal locations. Seven out of eight YFP-RecA threads showed such a pattern of localization, whereas one thread was localized between origin signals without clearly connecting with them. These data suggest that RecA threads extend away from a DSB toward the homologous region, which is located in the other cell half.
We also created a fusion of RecA to RFP with a shortened linker region (5 aa) compared with the GFP fusion (10 aa). The short linker probably interferes with the formation of RecA nucleofilaments because the NH2 terminus of RecA is important for filament formation. Interestingly, this fusion did not form any detectable threads upon induction of DSBs. Rather, RFP-RecA formed discrete foci that generally colocalized with GFP-RecN foci (Fig. 4 I), supporting the finding that RecA is recruited into RCs containing RecN. The expression of RFP-RecA as the sole source of RecA in the cells resulted in a recA-null phenotype; i.e., the cells were highly sensitive to MMC and grew as slowly as recA mutant cells. These findings show that the observed GFP-RecA threads are physiologically relevant, representing an intermediate step in recombination that is required for DSB repair. Some of the cells contained a RFP-RecA focus but no detectable CFP-RecN focus (Fig. 4 I). Because RecN assembles into foci considerably earlier than RecA, this observation suggests that RecN accumulation in RCs may be below the limit of detection in a low but considerable number of cells as a result of the much weaker fluorescence of CFP.
RecA threads assemble and disassemble dynamically in live cells
We performed time-lapse microscopy to further characterize the dynamics of RecA threads. Images taken at 1-min intervals showed that the filamentous RecA structures can rapidly grow and shrink and that they change dramatically in their structure and orientation (a total of 115 videos of cells with GFP-RecA threads were captured). Fig. 5 A shows a characteristic video with a GFP-RecA patch in the left part of the cell at 0 min, from which a filamentous structure appears to extend toward the right cell half starting at 1 min. The filamentous structure changes in shape between each 1-min interval, whereas the patch also assumed a threadlike morphology (Video 1, available at http://www.jcb.org/cgi/content/full/jcb.200412090/DC1). At 8 min, the extended threads are split into two filamentous structures, the left of which converts back into a more patchlike structure. A similar pattern is shown in Fig. 5 C, in which a GFP-RecA thread extends away from a GFP-RecA patch toward the opposite cell pole at 1 min. In this case, a second patch is formed at 3 min, which is converted back into a filamentous structure that moves toward the initial patch at the end of the series. Fig. 5 D shows an example of a GFP-RecA patch on the left and a thread on the right, which appear to connect with each other between 14 and 15 min and are clearly fused at 19 min. Again, the filamentous structure changes in shape between each interval, which can also be seen in Videos 2 and 3 (available at http://www.jcb.org/cgi/content/full/jcb.200412090/DC1). In Video 3 (taken ∼2 h after induction of DSBs), dynamic GFP-RecA threads move back and forth within the cells and finally convert back to diffuse localization on the nucleoids, which has been observed for RecA in the time course experiments at this time point (Fig. 4 G). These experiments suggest that RecA threads extend from DSBs toward the opposite cell half, possibly searching for sequence homology along the sister chromosome. Moreover, RecA threads appear to move between the DSB and the putative sister duplex within the other cell half until recombination is terminated. At this time point, RecA threads disassemble, and, most likely, RecA monomers are present throughout the nucleoids, as they are in exponentially growing cells. We were able to capture five videos in which the growth of GFP-RecA threads could be clearly followed over 5–6 min. Fig. 5 B shows an example of such an experiment (Video 4, available at http://www.jcb.org/cgi/content/full/jcb.200412090/DC1) in which the initial growth of a filament-like structure between 1 and 2 min is followed by a much more rapid extension of the thread (which could contain a bundle of RecA filaments) between 2 and 3 min and a decreasingly rapid extension for the last minutes. Three measurements of the extension of GFP-RecA threads are shown in Fig. 5 E, indicating that an early, slower extension period is followed by a maximal extension rate and a final cessation of extension. These findings agree with in vitro visualization of DNA-RecA filaments in which a slow nucleation period is ensued by a rapid polymerization phase (Sattin and Goh, 2004). We have measured the rate of extension of GFP-RecA threads (Fig. 5, B and C, arrowheads) in cells from 21 time-lapse experiments. The mean distance RecA threads extended between 1-min intervals was 1.02 μm (SD of 0.09 μm). GFP-RecA threads frequently appeared to take a large helical path within the cell (Fig. 5 A, minutes 1 and 2), which would complicate extension measurements. However, the relatively small deviation suggests that the deduced growth of RecA threads presents a reliable order of magnitude. In vitro, RecA filament growth was measured to reach a polymerization of 202 residues/min (Sattin and Goh, 2004), which translates into 320 nm/min given that six RecA monomers form a helix with a pitch of 9.5 nm. Thus, the extension of RecA filaments appears to be about threefold faster in vivo but 4–60-fold slower compared with the measured polymerization rate of actin (for review see Mogilner and Oster, 2003; Defeu Soufo and Graumann, 2004).
Figure 5.

Dynamic movement of RecA threads. (A–D) Time-lapse microscopy of cells expressing GFP-RecA 1 h after induction of DSBs. Numbers indicate time in minutes. White lines indicate the ends of cells; arrowheads indicate the position of growing RecA filaments. Bar, 2 μm (all images are equally scaled). (E) Rate of extension of GFP-RecA threads from three experiments, which are indicated by different symbols.
The formation of RecA threads depends on AddAB and RecJ activity
Several pathways have been proposed for the processing of DSBs and for loading of RecA onto ssDNA at the break, but it is still unclear which initial steps occur before the binding of RecA. To shed some light on this question and to further dissect DSB repair in live cells, we moved the GFP-RecA fusion into various mutant backgrounds and visualized RecA before and after induction of DSBs. The deletion of recO, recR, or recN still allowed for the formation of GFP-RecA threads after induction of DSBs (Table I and not depicted) but interfered with the localization of RecA in normally growing cells in that GFP-RecA localized throughout the cells, and patches were frequently found to accumulate at one position over or close to a nucleoid (in 25% of recO, in 14% of recN, or in 3% of recR mutant cells; Fig. 6 A and Table I). Because recA mutant cells display a segregation and growth defect in the absence of stress (Sciochetti et al., 2001), these findings indicate that the defect in growth and nucleoid morphology in recO mutant cells (Carrasco et al., 2004) and the formation of abnormal nucleoids in recN mutant cells (unpublished data) may be caused by an effect on the mislocalization of RecA. In recG or recU mutant cells, GFP-RecA threads formed normally after the addition of MMC (Table I); however, RecA threads were also detectable in 30 or 12% of mutant cells in the absence of externally induced DSBs, respectively (Fig. 6 B and Table I). These findings suggest that in exponentially growing cells, the loss of RecG or RecU leads to the formation of DSBs and RecA threads and, thus, to Holiday junctions, which may not be resolved because RecU is required for the cleaving of Holiday structures (Ayora et al., 2004). This would explain why recU and recG mutant cells show a defect in chromosome segregation and a slow growth phenotype (Pedersen and Setlow, 2000; Carrasco et al., 2004). Indeed, RecG has been shown to play an important function in the procession of stalled replication forks, which frequently arise during the cell cycle (McGlynn and Lloyd, 2001; Robu et al., 2004). In the absence of AddAB or RecJ, GFP-RecA localized normally in exponentially growing cells as well as in MMC-treated cells (not depicted; Table I; and Fig. 6, C and D). However, GFP-RecA threads were no longer detectable in addAB recJ mutant cells after induction of DSBs (Fig. 6 E). Instead, GFP-RecA formed a patch on the nucleoid, usually within one cell half, in 29% of the cells. Therefore, the function of either of the proteins or protein complexes is required for filament formation and, thus, for the proper function of RecA. Intriguingly, addAB recJ mutant cells are as sensitive to DSBs as recA mutant cells (unpublished data). This underlines the fact that the threads are the physiologically relevant form of RecA and suggests that at least two pathways exist for the presynaptic stage: one dependent on RecJ and an as yet unknown helicase and the other on AddAB.
Table I. Localization of RecA in different genetic backgrounds.
| Strain | Noninduced nucleoid localization |
Threads | Induced thread formationa |
|---|---|---|---|
| wild type | + | − | + |
| ΔrecN | − | − | + |
| ΔrecO | − | − | + |
| ΔrecF | + | − | + |
| ΔrecR | (+) | − | + |
| ΔaddAB | (+) | − | + |
| ΔrecJ | + | − | + |
| ΔrecJ, ΔaddAB | (+) | − | − |
| ΔrecG | (+) | + | + |
| ΔrecU | (+) | + | + |
(+) indicates a moderate defect.
Addition of 50 ng/ml MMC.
Figure 6.

Fluorescence microscopy of mutant B. subtilis cells expressing GFP-RecA. ΔrecO (A) and ΔrecG (B) cells growing exponentially. (A) Arrowheads indicate abnormal patches of RecA. (B) Arrowheads indicate RecA threads. (C–E) ΔaddAB (C), ΔrecJ (D), and ΔaddAB ΔrecJ (E) cells 1 h after induction of DSBs. White lines indicate the ends of cells. Bars, 2 μm.
DNA Pol I localizes throughout the nucleoids before and after induction of DSBs
DNA Pol I plays a crucial role in the repair of DNA damage and of DSBs. We wished to investigate the subcellular localization of DNA Pol I (encoded by polA) and, therefore, created a COOH-terminal YFP fusion that fully supported the growth of B. subtilis. Pol I–YFP was present on the nucleoids in growing cells (Fig. 7 A) without any apparent concentration at any specific site on the nucleoids. DNA Pol I retained its localization after the addition of MMC (Fig. 7 B); that is, it tracked with the fused nucleoids. In addition, fluorescence increased markedly, which is consistent with the view that the level of DNA Pol I increases after the occurrence of DNA DSBs. Likewise, after UV irradiation, DNA Pol I did not form fluorescent foci but remained localized throughout nucleoids that became slightly more decondensed (Fig. 7 C). We used doses between 10 and 60 Jm−2, which did not lead to a considerable change in localization. These results show that not all proteins required for DSB repair form foci upon DNA damage. However, the results also show that at least two different localization patterns exist for DSB repair proteins, constant association with the nucleoids or localization throughout the cells and specific recruitment into defined RCs after the occurrence of damage. Furthermore, our data suggest that even in the absence of DNA damage, DNA Pol I is constantly scanning the chromosome for lesions such that it is instantly present at sites of DNA damage.
Figure 7.

Localization of DNA Pol I in live cells. Fluorescence microscopy of strain DK6 (polA-yfp) growing exponentially (A), 1 h after induction of DSBs through MMC (B), or 1 h after UV irradiation with 60 Jm−2 (C). White lines indicate septa between cells. Bars, 2 μm.
Discussion
This study establishes that defined DSB RCs exit in prokaryotic cells, which is similar to eukaryotic cells (Haaf et al., 1995; Maser et al., 1997; Nelms et al., 1998; Lisby et al., 2004). RecN is the earliest protein found so far to assemble at defined breaks at different sites on the B. subtilis chromosome, whose arrangement is generally retained during DSB repair. The absence of RecN leads to a defect in the formation of RCs in that proteins that are later recruited to DSBs (RecO and RecF) frequently fail to assemble at DSBs (Kidane et al., 2004). Combined with this earlier finding, it is now clear that RecN plays an important role as an initiator of DSB repair. Additional evidence that RCs form at DSBs on any site on the chromosome came from our finding that RCs do not necessarily colocalize with the replication machinery but form at any site on the nucleoids (close to or far away from the replication fork) and, thus, are independent structures.
The fact that RecN, RecO, and RecF assemble at DSBs suggests that these breaks are repaired via homologous recombination with the nonbroken sister chromosome. An important question is how sister duplexes are paired in bacteria, because duplicated regions are separated into opposite cell halves during ongoing replication (Wu, 2004) and, in fact, soon after they have been replicated. Thus, a sister sequence is most likely located far away from a DSB (1–2 μm) within the other cell half. How bacteria achieve the task of finding the homologous sequence for a given DSB can partially be answered from our studies on the action of the major repair protein, RecA, which can be visualized in live cells in space and in time. After RecN assembly at DSBs, RecA also accumulates at these sites, forming foci or patches. Concomitant with the appearance of RecO at the RCs, RecA forms highly dynamic and transient filamentous structures that generally extend toward the opposite cell half, emanating from the RC. These RecA threads extend and retract within a minute or less and change their orientation and shape for ∼2 h until they dissipate, suggesting that the threads are composed of RecA nucleofilaments that are searching for the homologous duplex. We have observed cases in which a RecA thread extends from a patch on one side of the nucleoid toward the other cell half, where a new RecA patch is formed, possibly at the corresponding sister duplex of the DSB. We have also found examples in which RecA threads are exchanged between patches positioned in both cell halves, which can be interpreted as strand exchange processes between separated sister duplexes. Indeed, we observed that RecA threads extend away from a chromosomal site having a single, defined DSB and extend toward the homologous site on the sister chromosome. This strongly supports the notion that RecA threads communicate between a DSB and the nonbroken duplex. Interestingly, in exponentially growing cells and after the termination of DSB repair, RecA localizes throughout the nucleoids, showing that it is generally associated with the chromosome in spite of being an ssDNA-binding protein (Cox, 2003).
From our experiments, the following scenario can be deduced: RecN protein is the first or among the first proteins to assemble at a DSB, where it appears to form large protein and nucleoprotein complexes within 15 to 30 min (Kidane et al., 2004). RecJ and AddAB are present at the DSB concomitant with or soon after RecN because their activity is required for the formation of RecA filaments. RecJ has exonuclease activity similar to that of AddAB, which additionally has helicase activity (Lovett and Kolodner, 1989; Chedin et al., 2000). Both enzymatic properties are required to generate ssDNA overhangs as a substrate for RecA binding to ssDNA. RecA accumulates at and around DSBs and commences to form filamentous structures that are accompanied by the arrival of RecO at the RC (Kidane et al., 2004), which, together with RecR, appears to facilitate loading of RecA onto ssDNA (Bork et al., 2001; Morimatsu and Kowalczykowski, 2003). RecA threads are highly dynamic structures, which we propose contain ssDNA from one or both ends of the DSB that is used to scan the whole nucleoid for the homologous sister duplex. RecA threads extend toward the other end of the nucleoid within the other cell half, where the homologous duplex from the duplicated sister chromosome is located. We favor the idea that RecA threads extend and retract back and forth between the sister duplexes, exchanging strands and possibly bringing the D-loop from the sister duplex to the original DSB. This way, Holiday junctions could be set up between sister duplexes that are far apart, aided by the action of helicases such as RecG and the RuvAB–RecU complex, which should come into play at this time point. Concomitant with the disassembly of RecA filaments 2–3 h after induction of DSBs, RecF is recruited to RCs (Kidane et al., 2004), possibly inducing shrinkage of filaments or blocking their growth. It will be interesting to investigate whether the RecA filament capping factor RecX (Drees et al., 2004) is also recruited to RCs at this time point. However, not all proteins that are involved in DSB repair form discrete foci or filaments. DNA Pol I continues to be present throughout the nucleoids and is, therefore, assumed to be present in a sufficient amount to be present in RCs for the filling of gaps. Our finding that DNA Pol I is present throughout the cells, and even in exponentially growing cells, suggests that the enzyme is constantly scanning for gaps in DNA and is, therefore, instantly present for all types of DNA repair.
Future experiments should address the questions of whether RecN is a sensor for DSBs, how sensing occurs, and whether several DSBs are recruited into single RCs for repair like in some eukaryotic cells (Lisby et al., 2003; Aten et al., 2004), as seems to be the case based on earlier experiments on B. subtilis (Kidane et al., 2004).
Materials and methods
Growth conditions
E. coli XL1-blue (Stratagene) was grown in Luria Bertani–rich medium supplemented with 50 μg/ml ampicillin where appropriate. B. subtilis strains were grown in Luria Bertani–rich medium at 37°C. All of the strains in this study are listed in Table S1 (available at http://www.jcb.org/cgi/content/full/jcb.200412090/DC1). For microscopy, cells were grown in S750 medium (Mascarenhas et al., 2002). 50 ng/ml MMC was added for induction of DSBs, or 0.5% xylose was added for induction of HO endonuclease.
Construction of vectors and strains
To create an NH2-terminal fusion of RecA with GFP for double crossover integration into the chromosome, the entire recA ORF was amplified by PCR using primers RecAamyup315 and RecAamydw310 (Table S2, available at http://www.jcb.org/cgi/content/full/jcb.200412090/DC1) and was cloned into ApaI and EcoRI sites of pSG1729 (Feucht and Lewis, 2001). B. subtilis PY79 wild-type cells were transformed with the resulting plasmid (selecting for spectinomycin [spec]), which stably integrated into the amylase locus on the chromosome, establishing strain DK37 (Table S1). A YFP fusion of RecA was constructed from Gfpmut2 by site-directed mutagenesis using primers Gfpmut2up and Gfpmut2dw. For the construction of a red fluorescent variant of RecA (RFP-RecA), plasmid pSG1729 was modified by removing the gfp gene and substituting it with mrfp, which was amplified from pRSETB (gift from R. Tsien, University of California, San Diego, La Jolla, CA) using primers mRFPup and mRFPdw. For induction of GFP-RecA fusions, 0.01–0.5% xylose was used (Table S2), yielding similar results. To generate an inducible HO endonuclease system, the entire HO endonuclease ORF was amplified from plasmid adc3-OH (gift from H. Ulrich, London Research Institute, London, UK) using primers Hoendoup304 and Hoendodw305. Then, it was cloned into pSG1193 using restriction sites ApaI and EcoRI under the control of the xylose promoter, which was integrated into the amy locus by selecting for spec (DK52). 200 bp containing the HO cut site were PCR amplified from strain W3749-14C (gift from M. Lisby and R. Rothstein, Columbia University, New York, NY) using primers Hocutup318 and Hocutdw319 and were cloned into the EcoRI site of pSG1164 (Feucht and Lewis, 2001). To integrate the HO cut site at 359°C on the B. subtilis chromosome, the spo0J (841 bp) gene was amplified using primers Spo0Jup and Spo0Jdw and was cloned into the ApaI site on the plasmid containing the HO cut site. The resulting plasmid was introduced into PY79 cells by selecting for chloramphenicol (Cm) resistance (strain DK53). To change the resistance cassette in DK53, the strain was transformed with plasmid pCm/Nm (Bacillus Genetic Stock Center), exchanging Cm for kanamycin resistance via double crossover integration. This generated strain DK57. Strain PG26 (carrying the CFP origin tag) was transformed with chromosomal DNA from DK01 (recN-yfp), and the resulting strain, DK55, was transformed with chromosomal DNA from DK52 (HO endonuclease). The resulting strain, DK56, was transformed with chromosomal DNA from DK57 (cut site at the origin), establishing DK58. For integration at the terminus region on the chromosome, the cgeB gene (811 bp; 180°C) was amplified using primers CgeB180up and CgeB180dw and was cloned into the ApaI site next to the HO cut site. The resulting plasmid was used for single crossover integration of the HO cut site at 180°C on the chromosome, generating strain DK59. DK59 was transformed with chromosomal DNA from strain DK01 by selecting for tetracycline (tet) resistance. This was followed by transformation with DNA from strain PG25 (lacI-cfp at threonine locus; 25 μg/ml lincomycin/2 μg/ml erythromycin resistance; Mascarenhas et al., 2002) and, finally, with DNA from strain AT54 (lacO cassette at 180°C [cgeD]; Teleman et al., 1998) by selecting for Cm resistance. This established DK63.
Strain PG26 was transformed with plasmid pSG1729, which contained the yfp-recA gene that integrated at the amy locus, by selecting for spec and Cm resistance. The final screening for double crossover integration by using starch plates resulted in strain DK72. The HO endonuclease ORF was amplified from strain W3749-14C by using primers Hoendoup518 and Hoendodw519 and was cloned into NheI and SphI sites into the modified plasmid pJQ43 (containing the IPTG-inducible hyperspank promoter). The cotF ORF (839 bp) was amplified by PCR and was cloned into the resulting plasmid at the SphI site (adjacent to the HO endonuclease gene). The final plasmid was integrated into the B. subtilis chromosome via single crossover integration into the cotF locus by selecting for Cm, generating strain DK70. Afterwards, the Cm resistance cassette was changed into tet by using the plasmid pCm/tet (containing a cm gene disrupted with the tet gene; Bacillus Genetic Stock Center), which gave rise to strain DK71. Strain DK72 was transformed with chromosomal DNA from DK71 by selecting for tet and Cm resistance, which generated strain DK73. DK73 was transformed with chromosomal DNA from strain DK57 (carrying the HO cut site at the origin region), establishing strain DK74.
A COOH-terminal fusion of polA with yfp was created by cloning the 3′ (600 bp) region of polA using primers polAup and polAdw and by cloning into KpnI and EcoRI sites of modified pSG1164 that contained a yfp gene (Kidane et al., 2004). This integrated at the polA locus by single crossover and by selecting for Cm, establishing strain DK6.
Southern blot analysis
To monitor DNA breakage, strains carrying the HO cut site (DK58) were grown until rich log phase and were induced with 0.5% xylose. DNA samples were extracted before and after induction in 20-, 30-, 90-, and 150-min time intervals. The extracted genomic DNA was digested with the BglII restriction enzyme, run on a 1% agarose gel under Tris-EDTA buffer, and blotted on nitrocellulose paper. Products were detected by using a 450-bp DNA probe that was amplified from strain DK39 with primers Gfpmut2-527 and Gfpmut2-528 and were labeled by random primed DNA labeling, incorporating digoxigenin-11-dUTP during PCR. Detection was performed by using colorimetric detection with nitro blue tetrazolium and BCIP based on a digoxigenin Nucleic Acid Labeling and Detection system (Boehringer).
Image acquisition
Fluorescence microscopy was performed on a microscope (model AX70; Olympus) using a 100× UplanAPO objective with an aperture of 1.35. Cells were mounted on agarose pads containing S750 medium on object slides at RT. Images were acquired with a digital CCD camera (MicroMax; Roper Scientific); signal intensities and cell length were measured using Metamorph 4.6 software (Universal Imaging Corp.). Images were processed and assembled using Canvas 7 software (Deneba). DNA was stained with 0.2 ng/ml DAPI, and membranes were stained with 1 nM FM4-64.
Online supplemental material
Online supplemental material includes four videos showing the dynamics of GFP-RecA filaments during DSB repair. Fig. S1 shows the survival rate of wild-type and GFP-RecA strains after treatment with MMC. Fig. S2 shows the formation of GFP-RecA filaments during DSB repair with lower induction levels of GFP-RecA and also provides the sequences of the fluorescent protein–linker regions that were used in this study. Tables S1 and S2 show the strains and primers, respectively, that were used in this study. Online supplemental material is available at http://www.jcb.org/cgi/content/full/jcb.200412090/DC1.
Acknowledgments
We would like to thank Helle Ulrich, M. Lisby, and R. Rothstein for the gift of valuable strains and Johannes Gescher for help with Southern blotting.
This work was supported by grants from the Deutsche Forschungsgemeinschaft and the Fonds der Chemischen Industrie.
D. Kidane's and P.L. Graumann's present address is Institut für Mikrobiologie, Albert-Ludwigs Universität Freiburg, 79104 Freiburg, Germany.
Abbreviations used in this paper: Cm, chloramphenicol; DSB, double strand break; HO, homothallic; MMC, mitomycin C; Pol, polymerase; RC, repair center; spec, spectinomycin; ss, single stranded; tet, tetracycline.
References
- Arnold, D.A., and S.C. Kowalczykowski. 2000. Facilitated loading of RecA protein is essential to recombination by RecBCD enzyme. J. Biol. Chem. 275:12261–12265. [DOI] [PubMed] [Google Scholar]
- Aten, J.A., J. Stap, P.M. Krawczyk, C.H. van Oven, R.A. Hoebe, J. Essers, and R. Kanaar. 2004. Dynamics of DNA double-strand breaks revealed by clustering of damaged chromosome domains. Science. 303:92–95. [DOI] [PubMed] [Google Scholar]
- Ayora, S., B. Carrasco, E. Doncel, R. Lurz, and J.C. Alonso. 2004. Bacillus subtilis RecU protein cleaves Holliday junctions and anneals single-stranded DNA. Proc. Natl. Acad. Sci. USA. 101:452–457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bork, J.M., M.M. Cox, and R.B. Inman. 2001. The RecOR proteins modulate RecA protein function at 5′ ends of single-stranded DNA. EMBO J. 20:7313–7322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carrasco, B., M.C. Cozar, R. Lurz, J.C. Alonso, and S. Ayora. 2004. Genetic recombination in Bacillus subtilis 168: contribution of Holliday junction processing functions in chromosome segregation. J. Bacteriol. 186:5557–5566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chedin, F., S.D. Ehrlich, and S.C. Kowalczykowski. 2000. The Bacillus subtilis AddAB helicase/nuclease is regulated by its cognate Chi sequence in vitro. J. Mol. Biol. 298:7–20. [DOI] [PubMed] [Google Scholar]
- Cox, M.M. 2003. The bacterial RecA protein as a motor protein. Annu. Rev. Microbiol. 57:551–577. [DOI] [PubMed] [Google Scholar]
- Cromie, G.A., J.C. Connelly, and D.R. Leach. 2001. Recombination at double-strand breaks and DNA ends: conserved mechanisms from phage to humans. Mol. Cell. 8:1163–1174. [DOI] [PubMed] [Google Scholar]
- Defeu Soufo, H.-J., and P.L. Graumann. 2004. Dynamic movement of actin-like proteins within bacterial cells. EMBO Rep. 5:789–794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drees, J.C., S.L. Lusetti, S. Chitteni-Pattu, R.B. Inman, and M.M. Cox. 2004. A RecA filament capping mechanism for RecX protein. Mol. Cell. 15:789–798. [DOI] [PubMed] [Google Scholar]
- Feucht, A., and P.J. Lewis. 2001. Improved plasmid vectors for the production of multiple fluorescent protein fusions in Bacillus subtilis. Gene. 264:289–297. [DOI] [PubMed] [Google Scholar]
- Gordon, G.S., D. Sitnikov, C.D. Webb, A. Teleman, R. Losick, A.W. Murray, and A. Wright. 1997. Chromosome and low copy plasmid segregation in E. coli: visual evidence for distinct mechanisms. Cell. 90:1113–1121. [DOI] [PubMed] [Google Scholar]
- Haaf, T., E.I. Golu, G. Reddy, C.M. Radding, and D.C. Ward. 1995. Nuclear foci of mammalian Rad51 recombination protein in somatic cells after DNA damage and its localization in synaptonemal complexes. Proc. Natl. Acad. Sci. USA. 92:2298–2302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haber, J.E. 2000. Partners and pathways repairing a double-strand break. Trends Genet. 16:259–264. [DOI] [PubMed] [Google Scholar]
- Haber, J.E. 2002. Uses and abuses of HO endonuclease. Methods Enzymol. 350:141–164. [DOI] [PubMed] [Google Scholar]
- Hegde, S.P., M.H. Qin, X.H. Li, M.A. Atkinson, A.J. Clark, M. Rajagopalan, and M.V. Madiraju. 1996. Interactions of RecF protein with RecO, RecR, and single-stranded DNA binding proteins reveal roles for the RecF-RecO-RecR complex in DNA repair and recombination. Proc. Natl. Acad. Sci. USA. 93:14468–14473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirano, T. 2002. The ABCs of SMC proteins: two-armed ATPases for chromosome condensation, cohesion, and repair. Genes Dev. 16:399–414. [DOI] [PubMed] [Google Scholar]
- Hopfner, K.P., C.D. Putnam, and J.A. Tainer. 2002. DNA double-strand break repair from head to tail. Curr. Opin. Struct. Biol. 12:115–122. [DOI] [PubMed] [Google Scholar]
- Kidane, D., H. Sanchez, J.C. Alonso, and P.L. Graumann. 2004. Visualization of DNA double-strand break repair in live bacteria reveals dynamic recruitment of Bacillus subtilis RecF, RecO and RecN proteins to distinct sites on the nucleoids. Mol. Microbiol. 52:1627–1639. [DOI] [PubMed] [Google Scholar]
- Kowalczykowski, S.C., D.A. Dixon, A.K. Eggleston, S.D. Lauder, and W.M. Rehrauer. 1994. Biochemistry of homologous recombination in Escherichia coli. Microbiol. Rev. 58:401–465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemon, K.P., and A.D. Grossman. 2001. The extrusion-capture model for chromosome partitioning in bacteria. Genes Dev. 15:2031–2041. [DOI] [PubMed] [Google Scholar]
- Lisby, M., U.H. Mortensen, and R. Rothstein. 2003. Colocalization of multiple DNA double-strand breaks at single Rad52 repair centre. Nat. Cell Biol. 5:572–577. [DOI] [PubMed] [Google Scholar]
- Lisby, M., J.H. Barlow, R.C. Burgess, and R. Rothstein. 2004. Choreography of the DNA damage response: spatiotemporal relationships among checkpoint and repair proteins. Cell. 118:699–713. [DOI] [PubMed] [Google Scholar]
- Lovett, S.T., and R.D. Kolodner. 1989. Identification and purification of a single-stranded-DNA-specific exonuclease encoded by the recJ gene of Escherichia coli. Proc. Natl. Acad. Sci. USA. 86:2627–2631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mascarenhas, J., J. Soppa, A. Strunnikov, and P.L. Grauman. 2002. Cell cycle-dependent localization of two novel prokaryotic chromosome segregation and condensation proteins in Bacillus subtilis that interact with SMC protein. EMBO J. 21:3108–3118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maser, R.S., K.J. Monsen, B.E. Nelms, and J.H. Petrini. 1997. hMre11 and hRad50 nuclear foci are induced during the normal cellular response to DNA double-strand breaks. Mol. Cell. Biol. 17:6087–6096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGlynn, P., and R.G. Lloyd. 2001. Rescue of stalled replication forks by RecG: simultaneous translocation on the leading and lagging strand templates supports an active DNA unwinding model of fork reversal and Holliday junction formation. Proc. Natl. Acad. Sci. USA. 98:8227–8234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mogilner, A., and G. Oster. 2003. Polymer motors: pushing out the front and pulling up the back. Curr. Biol. 13:R721–R733. [DOI] [PubMed] [Google Scholar]
- Morimatsu, K., and S.C. Kowalczykowski. 2003. RecFOR proteins load RecA protein onto gapped DNA to accelerate DNA strand exchange. A universal step of recombinational repair. Mol. Cell. 11:1337–1347. [DOI] [PubMed] [Google Scholar]
- Nasmyth, K., J.M. Peters, and F. Uhlmann. 2000. Splitting the chromosome: cutting the ties that bind sister chromatids. Science. 288:1379–1385. [DOI] [PubMed] [Google Scholar]
- Nelms, B.E., R.S. Maser, J.F. MacKay, M.G. Lagally, and J.H. Petrini. 1998. In situ visualization of DNA double-strand break repair in human fibroblasts. Science. 280:590–592. [DOI] [PubMed] [Google Scholar]
- Niki, H., and S. Hiraga. 1998. Polar localization of the replication origin and terminus in Escherichia coli nucleoids during chromosome partitioning. Genes Dev. 12:1036–1045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niki, H., Y. Yamaichi, and S. Hiraga. 2000. Dynamic organization of chromosomal DNA in Escherichia coli. Genes Dev. 14:212–223. [PMC free article] [PubMed] [Google Scholar]
- Pedersen, L.B., and P. Setlow. 2000. Penicillin-binding protein-related factor A is required for proper chromosome segregation in Bacillus subtilis. J. Bacteriol. 182:1650–1658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robu, M.E., R.B. Inman, and M.M. Cox. 2004. Situational repair of replication forks: roles of RecG and RecA proteins. J. Biol. Chem. 279:10973–10981. [DOI] [PubMed] [Google Scholar]
- Sattin, B.D., and M.C. Goh. 2004. Direct observation of the assembly of RecA/DNA complexes by atomic force microscopy. Biophys. J. 87:3430–3436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sciochetti, S.A., G.W. Blakely, and P.J. Piggot. 2001. Growth phase variation in cell and nucleoid morphology in a Bacillus subtilis recA mutant. J. Bacteriol. 183:2963–2968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharpe, M.E., and J. Errington. 1998. A fixed distance for separation of newly duplicated copies of oriC in Bacillus subtilis: implications for co-ordination of chromosome segregation and cell division. Mol. Microbiol. 28:981–990. [DOI] [PubMed] [Google Scholar]
- Teleman, A.A., P.L. Graumann, D.C.H. Lin, A.D. Grossman, and R. Losick. 1998. Chromosome arrangement within a bacterium. Curr. Biol. 8:1102–1109. [DOI] [PubMed] [Google Scholar]
- Viollier, P.H., M. Thanbichler, P.T. McGrath, L. West, M. Meewan, H.H. McAdams, and L. Shapiro. 2004. Rapid and sequential movement of individual chromosomal loci to specific subcellular locations during bacterial DNA replication. Proc. Natl. Acad. Sci. USA. 101:9257–9262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Webb, C.D., P.L. Graumann, J. Kahana, A.A. Teleman, P. Silver, and R. Losick. 1998. Use of time-lapse microscopy to visualize rapid movement of the replication origin region of the chromosome during the cell cycle in Bacillus subtilis. Mol. Microbiol. 28:883–892. [DOI] [PubMed] [Google Scholar]
- Wu, L.J. 2004. Structure and segregation of the bacterial nucleoid. Curr. Opin. Genet. Dev. 14:126–132. [DOI] [PubMed] [Google Scholar]
- Wyman, C., and R. Kanaar. 2004. Homologous recombination: down to the wire. Curr. Biol. 14:R629–R631. [DOI] [PubMed] [Google Scholar]
- Zyskind, J.W., A.L. Svitil, W.B. Stine, M.C. Biery, and D.W. Smith. 1992. RecA protein of Escherichia coli and chromosome partitioning. Mol. Microbiol. 6:2525–2537. [DOI] [PubMed] [Google Scholar]