

Abstract
The integral endoplasmic reticulum (ER) membrane protein Shr3p is required for proper plasma membrane localization of amino acid permeases (AAPs) in yeast. In the absence of Shr3p AAPs are uniquely retained in the ER with each of their twelve membrane-spanning segments correctly inserted in the membrane. Here, we show that the membrane domain of Shr3p specifically prevents AAPs from aggregating, and thus, plays a critical role in assisting AAPs to fold and correctly attain tertiary structures required for ER exit. Also, we show that the integral ER proteins, Gsf2p, Pho86p, and Chs7p, function similarly to Shr3p. In cells individually lacking one of these components only their cognate substrates, hexose transporters, phosphate transporters, and chitin synthase-III, respectively, aggregate and consequently fail to exit the ER membrane. These findings indicate that polytopic membrane proteins depend on specialized membrane-localized chaperones to prevent inappropriate interactions between membrane-spanning segments as they insert and fold in the lipid bilayer of the ER membrane.
Introduction
The mechanisms enabling polytopic membrane proteins, comprised of multiple transmembrane segments (TMS), to properly integrate and fold in the membrane of the ER in eukaryotes or in the cell membrane of prokaryotes are not well defined. Early in the secretory pathway of eukaryotic cells, integral plasma membrane (PM) proteins are inserted into the lipid bilayer of the ER (Lecomte et al., 2003; Turner, 2003; Alder and Johnson, 2004; Rapoport et al., 2004). Polytopic proteins fold concomitantly with insertion to attain native conformations that enable them to be recognized as cargo, and are incorporated into ER derived COPII-coated vesicles that target to the Golgi (Antonny and Schekman, 2001; Barlowe, 2003; Bickford et al., 2004). The hydrophobic amino acids within NH2-terminally localized TMS are sufficient to target membrane proteins to the ER where they interact with and enter a protein-conducting channel, or translocon, with a hydrophilic core. One of the most striking properties of the translocon is its ability to facilitate the insertion of TMS into the lipid bilayer of the ER membrane, a process that occurs laterally through an opening in the channel wall (Heinrich et al., 2000).
A highly conserved heterotrimeric complex of membrane proteins, i.e., the Sec61 complex in eukaryotes and SecY complex in bacteria forms the protein-conducting channel. The crystal structure of the Methanococcus jannaschii SecY translocon has recently been determined (van den Berg et al., 2004). In contrast to previous models of the translocon, the protein-conducting channel appears to be exclusively contained within SecY, the largest subunit of the heterotrimeric complex and the Sec61p homologue. According to this new information, the protein-conducting channel, although perhaps quite flexible, is too small to accommodate multiple TMS. Despite unresolved questions regarding the structure of an active Sec61/SecY translocon in vivo (Alder and Johnson, 2004; Rapoport et al., 2004), these new structural findings highlight persistent questions regarding how hydrophobic TMS of partially integrated polytopic proteins avoid inappropriate inter- and intramolecular interactions before the complete translocation and insertion of remaining TMS. Such inopportune interactions would likely hamper proper folding, and it has been suggested that membrane-localized chaperones exist to prevent such disadvantageous interactions (Lecomte et al., 2003; Alder and Johnson, 2004; Rapoport et al., 2004). In bacteria, the membrane protein YidC appears to play a chaperone-like role in the folding of lactose permease (LacY; Nagamori et al., 2004). To date, no such proteins have been identified in eukaryotes.
We use yeast amino acid permeases (AAPs) as models to address how polytopic membrane proteins insert into the ER membrane, fold, and subsequently enter ER-derived transport vesicles. The AAPs of Saccharomyces cerevisiae make up a conserved family of 18 proteins with 12 TMS (Gilstring and Ljungdahl, 2000). AAPs belong to the secondary transporter amino acid/polyamine/organocation protein family, members of which are found in bacteria, archaea, fungi, plants, and animals (Saier, 2000). All but one of the yeast AAPs function at the PM using the H+ gradient to drive the translocation of amino acids into the cell. One member of the AAP family with a large NH2-terminal extension, Ssy1p, functions as a core component of the SPS-sensor of extracellular amino acids (Forsberg and Ljungdahl, 2001).
AAPs are initially inserted into the ER membrane after which they are cotransported together with other secreted proteins from the ER to the Golgi via COPII-coated transport vesicles (Kuehn et al., 1996). The integral ER membrane component Shr3p is an accessory protein (Herrmann et al., 1999; Lee et al., 2004; Nyman et al., 2004) that is required for AAPs to exit the ER (Ljungdahl et al., 1992). The secretory block observed in cells carrying mutations in SHR3 is specific, other membrane-bound and secretory cargo exit the ER and are targeted to their correct intracellular locations (Ljungdahl et al., 1992; Kuehn et al., 1996). Shr3p itself does not exit the ER (Kuehn et al., 1996). Shr3p is a well-conserved protein in fungi, and homologues function similarly and interchangeably in Schizosaccharomyces pombe (Martínez and Ljungdahl, 2000) and Candida albicans (Martínez and Ljungdahl, 2004).
Shr3p is not required for insertion of AAPs; each of the TMS of the archetypal general AAP (Gap1p) integrates into the ER membrane in the correct orientation independently of Shr3p (Gilstring and Ljungdahl, 2000). The AAPs that accumulate in the membrane of the ER of shr3 null mutant cells do not induce the ER stress response pathway (Gilstring et al., 1999), indicating that the accumulated AAPs do not expose sequences that bind Kar2p, the homologue of mammalian BiP (Rutkowski and Kaufman, 2004). Despite having proper membrane topologies and not inducing ER stress, the AAPs in cells lacking Shr3p fail to be included in prebudding complexes and COPII transport vesicles (Kuehn et al., 1996, 1998).
Consistent with its specialized role in promoting the exit of AAPs from the ER, Shr3p physically associates with Gap1p, but not with other polytopic membrane proteins, such as Sec61p, Gal2p or Pma1p, in a transient complex that can be purified from detergent solubilized membranes (Gilstring et al., 1999). In addition, the COPII coatomer components Sec13p, Sec23p, Sec24p, and Sec31p copurify with the hydrophilic COOH-terminal domain of Shr3p (Gilstring et al., 1999). Based on the complete repertoire of interactions, it has been proposed that Shr3p functions to initiate the formation of transport vesicles in the vicinity of AAPs (Gilstring et al., 1999), perhaps by facilitating the presentation of ER exit sequences present in the COOH-terminal portion of AAPs (Malkus et al., 2002; Miller et al., 2003). Alternatively, it has been suggested that Shr3p acts as a “mismatch” chaperone (Levine et al., 2000). Mismatch chaperones are hypothetical entities that have been postulated to prevent inappropriate interactions between PM proteins during their residence in the ER, a membrane that is thought to be thinner than the average length of the hydrophobic TMS of PM proteins.
Here, we show that the primary function of Shr3p is to prevent the aggregation of AAPs thereby enabling them to fold correctly within the ER membrane. We also report that three additional ER components, Gsf2p, Pho86p, and Chs7p, act similarly to Shr3p, and their presence specifically prevents the aggregation of defined sets of polytopic membrane proteins, their cognate substrates. Strikingly, in cells lacking one of these ER proteins, we observe all-or-nothing effects, i.e., nearly quantitative aggregation and cross-linking that is limited to their cognate substrates. Our findings suggest that these ER proteins act as highly specialized membrane-localized chaperones that facilitate the folding of polytopic membrane proteins into their native tertiary membrane conformations, a requisite for ER exit.
Results
The membrane domain of Shr3p is critical for function
Shr3p consists of two discrete domains, a membrane domain with four hydrophobic segments each predicted to span the ER membrane, and a hydrophilic COOH-terminal domain (Fig. 1 A). The membrane domain is important for binding AAPs and the hydrophilic COOH-terminal is required for interacting with cytoplasmically localized COPII coatomer (Gilstring et al., 1999). To gain a more precise understanding of the significance of these domains we determined the in vivo topology of Shr3p. A topological reporter construct (Gilstring and Ljungdahl, 2000) was inserted in-frame into the hydrophilic NH2- and COOH-terminal domains and each of the three hydrophilic loops (L1–L3) that separate the four hydrophobic segments of Shr3p (I–IV; Fig. 1 A). The resulting five gene-sandwich-fusions encode functional proteins that are capable of restoring amino acid uptake when introduced into shr3Δ cells. The glycosylation state of the fusion proteins was monitored in samples before and after incubation with endoglycosidase H. The results indicate that the topological reporter was efficiently glycosylated only when introduced into loops L1 and L3, indicating that these loops are oriented toward the ER lumen (Fig. 1 B). These results show that each of the hydrophobic segments of Shr3p span the membrane and that the NH2 and COOH termini are oriented toward the cytoplasm. The experimentally determined in vivo topology of Shr3p is consistent with its known biochemical activities (Kuehn et al., 1996; Gilstring et al., 1999).
Figure 1.

Structural and functional analysis of Shr3p. (A) Schematic illustration of the predicted membrane topology of Shr3p. The NH2 (NT) and COOH termini (CT) are predicted to be oriented toward the same side of the ER membrane. The four hydrophobic segments (I–IV) are separated by hydrophilic loops L1–L3. The suc2 topological reporter cassette (see Materials and methods) was inserted at the positions indicated; the numbers refer to the amino acid immediately preceding the individual cassette insertions. (B) The experimentally determined in vivo topology of Shr3p. Extracts from FGY212 (shr3Δ) cells carrying either plasmids (pJK40-pJK44) encoding the five fusion proteins or a control vector without insert (vector, pRS316) were treated with endoglycosidase H as indicated, and proteins were resolved by SDS-PAGE and immunoblotted with anti-Shr3p antibody. The orientation of each reporter cassette is indicated (cyt, cytosolic; lum, lumenal). (C) The hydrophobic membrane domain of Shr3p is sufficient for function. The wild-type SHR3 and the COOH-terminally truncated shr3ΔCT alleles are depicted with sequences encoding the four putative transmembrane domains (I–IV) are represented by the gray boxes. Serial dilutions (10-fold) of cell suspensions of strain FGY212 (shr3Δ) carrying a vector control (Δ, pRS316), or plasmids expressing either Shr3p (+, pPL210) or Shr3ΔCTp (ΔCT, pJK36) were applied to SD supplemented with lysine and the indicated toxic amino acid analogue. Plates were incubated at 30°C for 4 d and photographed. The abbreviations of amino acid analogues and concentrations used are as follows: Ethionine, d,l-Ethionine (60 μg ml−1); f-PA, p-fluoro-d,l-phenylalanine (400 μg ml−1); AzC, azetidine-2-carboxylate (500 μg ml−1).
The relative importance of the two domains of Shr3p was examined by constructing the shr3ΔCT allele encoding a truncated protein lacking the entire hydrophilic COOH terminus (Fig. 1 C). This allele was introduced into a shr3Δ null mutant and the ability of the expressed Shr3ΔCTp to restore amino acid uptake was initially assessed using growth-based assays. The shr3ΔCT allele restored sensitivity to toxic amino acid analogues including ethionine, fluoro-phenylalanine, and azetidine carboxylate (AzC; Fig. 1 C, compare dilution series ΔCT with Δ). To obtain a more quantitative measure of complementation, we assayed the rates of uptake of four representative amino acids (Table I). Uptake was determined at 4 μM substrate concentrations, a concentration that enables the activity of distinct high affinity AAPs to be monitored. The pleiotropic effect of the shr3 null mutation was clearly evident; the uptake rate of each of the amino acid was reduced to <4% of wild-type (SHR3). Consistent with the ability to restore sensitivity to toxic amino acid analogues, the strain carrying the shr3ΔCT allele exhibited remarkably robust rates of uptake that varied between 30 and 50% of wild-type activity.
Table I.
Amino acid uptake into SHR3 , shr3 ΔCT, and shr3 Δ null mutant strains
| Substratea
|
SHR3
|
shr3ΔCT
|
shr3Δ
|
shr3ΔCT/SHR3
|
|---|---|---|---|---|
|
|
units
b
|
units
|
units
|
ratio
|
| Glutamate | 0.230 | 0.075 | 0.008 | 0.33 |
| Proline | 0.075 | 0.043 | 0.001 | 0.57 |
| Leucinec | 1.40 | 0.784 | 0.052 | 0.56 |
| Phenylalaninec | 1.34 | 0.705 | 0.019 | 0.53 |
Initial rates of uptake were determined in SUD-grown strain JKY1 carrying either pPL210 (SHR3), pJK36 (shr3ΔCT), or pRS316 (shr3Δ) at a substrate concentration of 4 μM in buffer containing 2% glucose (Martínez and Ljungdahl, 2000). Uniformly, 14C-labeled l-amino acids were obtained from Amersham Biosciences. Subsamples were removed at 30, 60, and 180 s; the rates of glutamate and proline were linear throughout the subsampling period.
Unit = nmol min−1 mg−1 dry weight.
Approximate rate based on single point measurements at the 30-s time point.
We directly examined the intracellular location of a functional Gap1p-GFP fusion protein in SHR3 wild-type, shr3ΔCT, and shr3Δ null mutant strains (Fig. 2). The induced expression of the PGAL-GAP1-GFP construct (provided by B. André, Free University of Brussels, Brussels, Belgium) resulted in similar levels of Gap1p-GFP protein in each of the three strains analyzed (Fig. 2 B). In SHR3 cells the Gap1p-GFP fluorescence exhibited a marked PM rim-staining pattern (Fig. 2 A, top). In some of the wild-type cells a portion of Gap1p-GFP fluorescence was found associated with vacuoles (Fig. 2 A, compare GFP with the Nomarski images). In no instance did the GFP fluorescence colocalize with the fluorescence associated with DAPI-stained nuclei. In contrast, the Gap1p-GFP fluorescence in shr3Δ null mutants exhibited a pattern typical of ER localized proteins; the fluorescence was mainly restricted to internal membranes closely associated with DAPI-stained nuclei (Fig. 2 A, bottom). These observations confirm results from previous localization studies and substantiate that Shr3p is required for AAPs to exit the ER (Ljungdahl et al., 1992). The pattern of Gap1p-GFP fluorescence in cells expressing only the membrane domain of Shr3p (shr3ΔCT; Fig. 2 A, middle) exhibited a clear PM rim-staining pattern that was indistinguishable to that observed in SHR3 wild-type cells.
Figure 2.

Gap1p-GFP localizes to the PM in an Shr3p-dependent manner. Strain FGY135 (shr3Δ) carrying plasmid pJOD010 (PGAL-GAP1-GFP) was cotransformed with plasmids pJK64 (SHR3), pJK65 (shr3ΔCT), or pRS317 (shr3Δ, vector without insert). Cells were grown in allantoin media with 2% raffinose and 0.2% glucose to an OD600 of 2–3, and subsequently suspended in allantoin media with 4% galactose and 1 μg ml−1 DAPI for 5 h. (A) Cells immobilized in low melting point agar (0.4%) were viewed by Nomarski optics and the GFP and DAPI fluorescence was examined using an Axiophot microscope outfitted with a CCD camera. Bar, 5 μm. (B) Extracts from each of the transformants were prepared, proteins were resolved by SDS-PAGE, and immunoblotted with monoclonal anti-GFP antibody (Roche).
These results unambiguously demonstrate that AAPs exit the ER at appreciable rates and correctly localize to the PM in cells expressing a truncated Shr3p lacking the COOH-terminal domain. Thus, the ability of Shr3p to interact with COPII coatomer components via its hydrophilic COOH-terminal domain (Gilstring et al., 1999) is not essential for promoting ER exit. Rather, the data indicate that interactions between the membrane domains of Shr3p and one or more of the 12 TMS of AAPs are important. This conclusion is consistent with an earlier study in which we found that the shr3-23 point mutation, resulting in an arginine residue replacing threonine 19 in the first TMS of Shr3p, encodes a nonfunctional protein that is unable to interact with AAPs (Gilstring et al., 1999). Together, the data suggest that the membrane domain of Shr3p possesses chaperone-like activity.
Shr3p influences the propensity of AAPs to be cross-linked
To address the possibility that Shr3p functions as a membrane-localized chaperone we probed the physical status of AAPs in vivo by examining the susceptibility of Gap1p to form cross-links in the presence of two membrane-permeable cross-linking reagents, i.e., thiol-cleavable Dithiobis[succinimidyl propionate] (DSP) or UV-activated N-succinimidyl 6-[4′-azido 2′-nitrophenylamino]hexanoate (SANPAH; Fig. 3 A). In contrast to Gap1p in membranes prepared from SHR3 wild-type cells (Fig. 3 A, lanes 1–5), Gap1p in membranes from shr3Δ mutant cells formed extensive cross-links as evidenced by the almost complete absence of detectable monomeric forms of Gap1p (Fig. 3 A, lanes 6–10). The propensity of Gap1p to be cross-linked in shr3Δ mutant cells was specific (Fig. 3 B). Two ER resident proteins, dolichol phosphate mannose synthase (Dpm1p) and Sec61p, and the PM hexose transporter Hxt1p did not become cross-linked; each of these proteins consistently migrated as monomers independent of the presence or absence of Shr3p. The ability to efficiently cross-link AAPs only in the absence of Shr3p is consistent with Shr3p having a critical role in enabling AAPs to attain native conformations. It should be noted that whereas the amounts of AAP present as monomers is reproducible from experiment to experiment, the amounts of AAPs present as high molecular weight smears is variable. This is a consequence of well established problems associated with blotting high molecular complexes.
Figure 3.

Gap1p specifically cross-links in the absence of Shr3p. (A) Extracts from isogenic SHR3 wild-type (CAY28) and shr3Δ null mutant (JKY1) strains grown in SAD at 23°C. Extracts were treated with the indicated concentration (mM) of DSP (left) or SANPAH (right). The DSP extracts were treated with DTT and extracts containing SANPAH were irradiated with UV as indicated. Proteins were resolved by SDS-PAGE, and immunoblotted with polyclonal anti-Gap1p antibody. (B) Immunoblot analysis of DSP treated extracts as in A with anti-Dpm1p and -Sec61p antibodies, and extracts from strains grown in SC with 4% glucose with anti-Hxt1p antibodies.
Shr3p functions to prevent AAP aggregation
AAPs insert into the ER membrane with the correct topology independently of Shr3p (Gilstring and Ljungdahl, 2000). Thus, the presence of Shr3p is not essential for the topogenic signals in AAPs to be properly interpreted by the Sec61 translocon. Although AAPs attain their correct topology, our cross-linking studies suggested that AAPs require Shr3p to fold correctly. We sought an independent experimental strategy to test this prediction and considered the possibility that AAPs may aggregate in the absence of Shr3p. The migration of Gap1p in polyacrylamide gels under nondenaturing conditions was examined; membrane preparations were partially solubilized using the nonionic detergent dodecyl-β-d-maltopyranoside (DM) and resolved by BN-PAGE (Schägger et al., 1994). Gap1p present in SHR3 wild-type membranes migrated as a diffuse band of monomers (Fig. 4 A, lanes 1 and 3). In contrast, the majority of Gap1p in membranes of shr3Δ mutant cells migrated as a high molecular weight smear (Fig. 4 A, lanes 2 and 4). Similar results were obtained when the migration of a second AAP, Agp1p (Iraqui et al., 1999), was examined (Fig. 4 B). These results, consistent with a chaperone-like function, indicate that in the absence of Shr3p, AAPs aggregate into high molecular weight complexes.
Figure 4.

Shr3p prevents AAP aggregation. (A) Extracts from isogenic SHR3 wild-type (CAY28) and shr3Δ null mutant (JKY1) strains grown in SAD at 23°C were prepared and solubilized with DM at the indicated concentrations (μg DM μg−1 protein). Solubilized proteins were separated by BN-PAGE and immunoblotted with anti-Gap1p antibody. (B) Agp1p in extracts from strains PLY127 (SHR3) and FGY212 (shr3Δ), carrying plasmid pJK60 (STP1Δ131) (Andréasson and Ljungdahl, 2002) to induce AGP1 expression, were solubilized and analyzed as in A using anti-Agp1p antibodies.
Next, we asked whether the membrane domain of Shr3p, which suffices to enable AAPs to exit the ER (Fig. 2), possessed the chaperone-like activity by examining the aggregation state of Gap1p in a strain carrying the shr3ΔCT allele. In membranes isolated from this strain a significant portion of the total Gap1p migrated as monomers (Fig. 5 A, lanes 2 and 5). The decreased efficiency to solubilize monomeric Gap1p in shr3ΔCT cells as compared with SHR3 wild-type cells (Fig. 5 A, compare lane 2 with 1, and lane 5 with 4) was not the consequence of diminished levels of Gap1p in shr3ΔCT cells. Each extract contained similar levels of Gap1p and the control ER marker protein Dpm1p (Fig. 5 B). These results are consistent with the ability of the COOH-terminal truncated protein to restore amino acid uptake (Fig. 1 C; Table I). This analysis demonstrates that the membrane domain of Shr3p is sufficient to enable AAPs to fold correctly and attain native structures compatible with ER exit.
Figure 5.
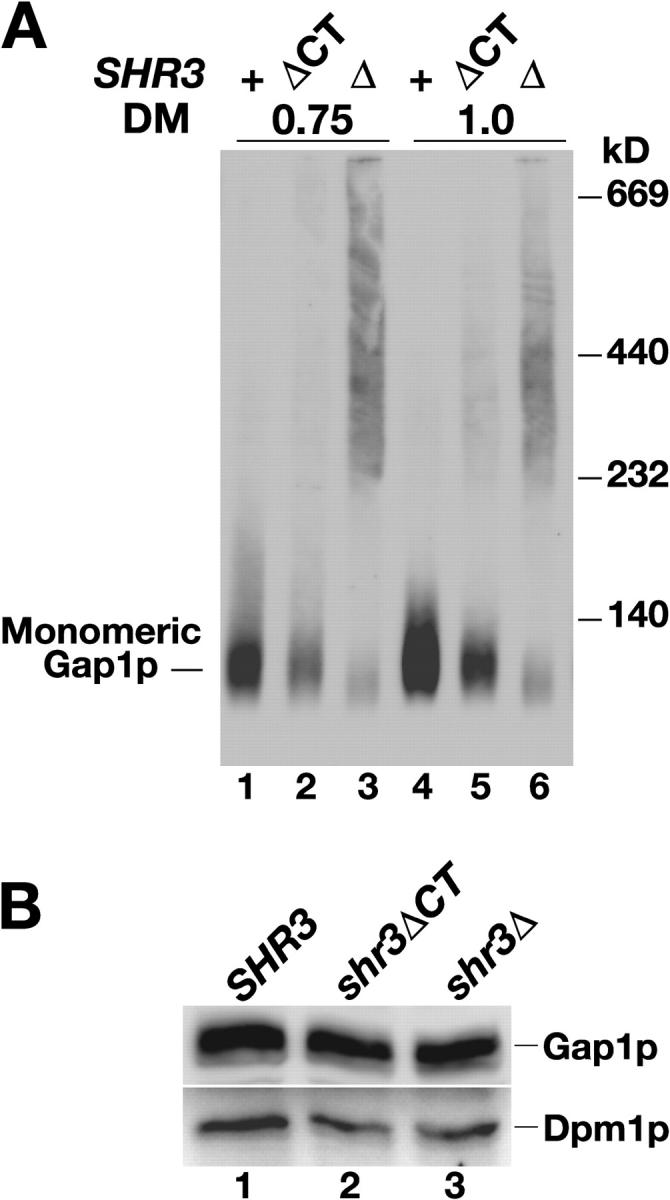
The membrane domain of Shr3p suffices to block AAP aggregation. Extracts prepared from strain JKY1 (shr3Δ) grown in SAD carrying a vector control (Δ), or plasmids expressing either Shr3p (+) or Shr3ΔCTp (ΔCT) were prepared. (A) Extracts were solubilized with DM at the indicated concentrations (μg DM μg−1 protein). Solubilized proteins were separated by BN-PAGE and immunoblotted with anti-Gap1p antibody. (B) Proteins within extracts were analyzed by SDS-PAGE and immunoblotted with anti-Gap1p and anti-Dpm1p antibodies as indicated.
Aggregation reflects improper folding in the ER
AAPs in SHR3 wild-type cells are predominantly localized to the PM (Ljungdahl et al., 1992; Gilstring et al., 1999); we therefore investigated the possibility that the increased propensity of AAPs to cross-link and aggregate in shr3Δ mutant cells was merely a consequence of ER retention. We used the temperature-sensitive sec12-1 mutation to prevent AAPs from exiting the ER independently of Shr3p (Nakano et al., 1988). Subcellular fractionation experiments demonstrate the effectiveness of this approach; after Gap1p expression was induced and cells were shifted to the restrictive temperature (34°C), newly synthesized Gap1p cofractionated with the ER membrane marker protein Sec61p in both SHR3 sec12-1 and shr3Δ sec12-1 cells (Fig. 6 A, fractions 4–6). In contrast to the ER-retained Gap1p in SHR3 sec12-1 cells (Fig. 6 B, left, lanes 1–5), Gap1p in shr3Δ sec12-1 cells became cross-linked in the presence of DSP or SANPAH (Fig. 6 B, right, lanes 6–10). In addition, we examined the aggregation state of Gap1p in sec12-1 cells using BN-PAGE. Consistent with the cross-linking results, monomeric Gap1p was extracted from membranes derived from SHR3 sec12-1 cells (Fig. 6 C, lanes 1 and 3) but not from shr3Δ sec12-1 cells (Fig. 6 C, lanes 2 and 4). It is clear from these observations that the formation of AAP cross-links and AAP aggregation in shr3Δ mutants are not a consequence of an altered subcellular distribution.
Figure 6.

Shr3p-dependent cross-linking and aggregation occurs in the ER. (A) Gap1p is retained in the ER of sec12-1 mutant strains grown at nonpermissive temperatures. Strains JKY3 (sec12-1) and JKY4 (sec12-1 shr3Δ), grown at 22°C (permissive) in ammonium-based SC to repress GAP1 expression, were harvested, washed, and resuspended in allantoin containing medium (SAD) for 2 h at 34°C (nonpermissive) to induce GAP1 expression. Extracts were fractionated on 12–60% step sucrose gradients, and proteins within fractions 1–9 were separated by SDS-PAGE and analyzed by immunoblotting. Antibodies recognizing marker proteins Pma1p, Sec61p, and Kex2p were used to identify fractions containing PM, ER, and Golgi proteins, respectively. (B and C) Gap1p retained in the ER of sec12-1 strains cross-links and aggregates in an Shr3p-dependent manner. (B) Immunoblot analysis of DSP and SANPAH treated extracts as in A with anti-Gap1p antibody. (C) Extracts as in A were solubilized with DM at the indicated concentrations (μg DM μg−1 protein). Solubilized proteins were separated by BN-PAGE and immunoblotted with anti-Gap1p antibody.
Gsf2p, Pho86p, and Chs7p function similarly to Shr3p
In addition to AAPs, several polytopic PM proteins in yeast have been reported to require the assistance of accessory proteins to exit the ER. Three “Shr3p-like” accessory proteins have been described. These proteins, Gsf2p, Pho86p, and Chs7p, specifically facilitate the functional expression of the hexose transporters Hxt1p and Gal2p (with 12 TMS; Sherwood and Carlson, 1999), the phosphate transporter Pho84p (with 12 TMS; Lau et al., 2000), and the catalytic subunit of chitin synthase III Chs3p (with six TMS; Trilla et al., 1999), respectively. As observed with shr3 mutations, when the genes encoding these accessory proteins are mutated or deleted, only their cognate substrates fail to enter COPII transport vesicles and accumulate within the ER. Despite their apparent functional similarity, these accessory proteins do not share identifiable sequence homology with each other or with Shr3p.
Based on our novel understanding regarding the chaperone-like function of Shr3p, we examined the susceptibility of the cognate substrates of these accessory proteins to form cross-links. In contrast to Hxt1p in GSF2 wild-type cells (Fig. 7 A, lanes 1–5), Hxt1p in gsf2Δ mutant cells readily formed cross-links (Fig. 7 A, lanes 6–10). The propensity of Hxt1p to be cross-linked in gsf2Δ mutant cells was specific; Gap1p and Sec61p did not become cross-linked and migrated as monomers independent of the presence or absence of Gsf2p (Fig. 7 A, bottom two panels). Similarly, Pho84p exhibited Pho86p-dependent cross-linking (Fig. 7 B) and Chs3p exhibited Chs7p-dependent cross-linking (Fig. 7 C). Our data indicate that Gsf2p, Pho86p, and Chs7p function in a manner indistinguishable from Shr3p by acting to prevent inappropriate molecular interactions of their cognate substrates.
Figure 7.
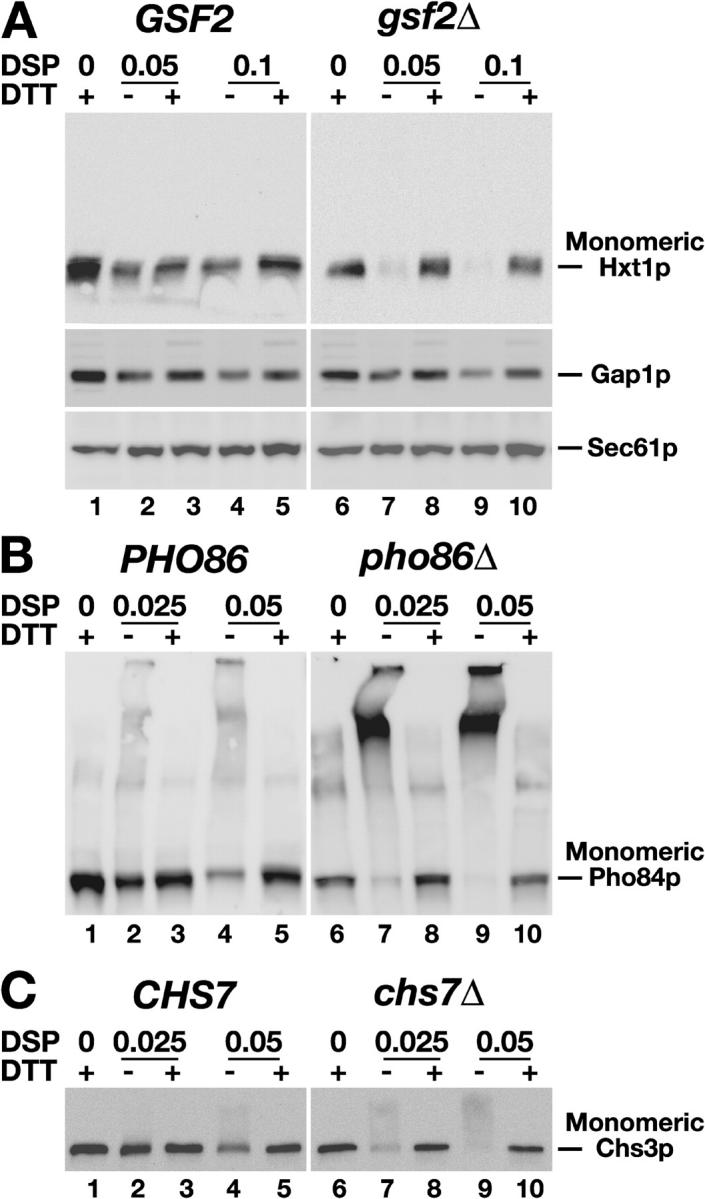
Specialized membrane-localized chaperones prevent the aggregation of specific sets of polytopic membrane proteins. (A) Whole cell extracts, prepared from isogenic GSF2 wild-type (CAY28) and gsf2Δ null mutant (JKY5) strains grown in SC (for the analysis of Hxt1p and Sec61p) or in SAD (for the analysis of Gap1p). The media were supplemented with 4% glucose to induce HXT1 expression. The extracts were treated with DSP at the concentrations (mM) and DTT as indicated. Proteins in the extracts were resolved by SDS-PAGE and immunoblotted with anti-Hxt1p, anti-Gap1p, and anti-Sec61 antibodies. (B) Pho84p expression was induced in isogenic PHO86 wild-type (CEN.PK113-5D/Pho84myc) and pho86Δ null mutant (JKY6) strains by growth in low phosphate medium. Extracts treated with DSP and DTT as in A, proteins were resolved by SDS-PAGE, and immunoblotted with anti-Myc antibody (9E10). (C) Analysis of DSP and DTT treated extracts from isogenic CHS7 wild-type (Y1306) and chs7Δ null mutant (JAY27) strains grown in SC. Proteins resolved by SDS-PAGE were immunoblotted with anti-HA antibody.
Discussion
We have examined the role of Shr3p in facilitating the AAP family of transport proteins in yeast to exit the ER, a requisite event for their correct localization to the PM. Our data indicate that AAPs specifically depend on the membrane domain of Shr3p to fold correctly within the membrane of the ER. This conclusion is based on several independent observations. First, in the absence of Shr3p, AAPs exhibit an enhanced propensity to form cross-links in the presence of membrane-soluble cross-linking agents (Fig. 3 A). Second, in cells lacking Shr3p, AAPs aggregate into large complexes that are resistant to detergent solubilization (Fig. 4). Third, the specificity of Shr3p in AAP folding, which was expected from previous studies (Ljungdahl et al., 1992; Kuehn et al., 1996, 1998; Gilstring et al., 1999; Malkus et al., 2002), was clearly evident; the absence of Shr3p did not increase the propensity of other ER resident proteins or membrane cargo to become cross-linked (Fig. 3 B). Finally, we found that AAPs are misfolded (Fig. 5) as a direct consequence of the lack of the four TMS of Shr3p (Fig. 1 B), and not due to secondary effects concomitant with an increased ER residence time (Fig. 6). The specific aggregation of AAPs in shr3Δ null mutant cells is consistent with their inability to exit the ER (Fig. 1 C and Fig. 2; Table I).
The observation that Gsf2p, Pho86p, and Chs7p function similarly to Shr3p (Fig. 7) suggests that polytopic membrane proteins require chaperone-like proteins to overcome common structural constraints associated with membrane insertion and folding. Strikingly, in cells lacking one of the four membrane-chaperones examined, cross-linking was found to be an almost all-or-nothing phenomenon. In wild-type cells, essentially none of the cognate substrates were cross-linked, whereas in isogenic null mutants lacking one of the membrane-chaperones, the propensity of cognate substrates to form cross-links was nearly complete. The quantitative nature of the requirement of Shr3p to prevent aggregation of AAPs was clearly observed using BN-PAGE. In SHR3 wild-type cells the AAPs were readily solubilized and migrated as monomers, whereas in shr3Δ null mutants the monomeric forms were barely detected (Figs. 4 and 5; Fig. 6 C). In no instance was dimers or trimers of AAPs observed in membranes isolated from wild-type cells, therefore in their native forms, these polytopic proteins are not likely to function as closely packed or covalently linked multimers.
Our data are reminiscent of recent results regarding the LacY in Escherichia coli (Nagamori et al., 2004). LacY depends on YidC, a member of a conserved family of membrane proteins (Abl3/Oxa1/YidC; Kuhn et al., 2003; Dalbey and Kuhn, 2004) to attain a native tertiary structure (Nagamori et al., 2004). Similar to Shr3p, YidC does not appear to be necessary for the insertion of the membrane spanning segments of LacY, but nonetheless, YidC must be present during translation of LacY for it to obtain a native conformation (Nagamori et al., 2004). Our findings of membrane-localized chaperones in yeast provide further evidence for the high degree of functional conservation of the process of membrane protein insertion in prokaryotic and eukaryotic cells.
Based on the all-or-nothing effect that we observe, it is likely that membrane-localized chaperones interact early with their cognate polytopic substrate proteins before the partitioning of all their TMS into the lipid phase of the ER membrane, and before the completely translated proteins are released from the Sec61 translocon. The crystal structures of bacterial PM transporters LacY and GlpT, each comprised of 12 TMS, have recently been elucidated (Abramson et al., 2003; Huang et al., 2003). The availability of these structures has clearly shown that interactions between TMS within the lipid phase of the membrane are not limited to immediately flanking TMS. Thus, it is possible that membrane-localized chaperones prevent TMS that do not normally interact in the mature protein from engaging in nonproductive interactions with flanking TMS as they sequentially partition into the membrane. Alternatively, the presence of membrane-localized chaperones may function to ensure the efficient insertion of discrete TMS into the membrane. It is known that exclusively hydrophobic TMS rapidly exit the protein-conducting channel of the translocon, and readily partition into the lipid phase of the membrane (Heinrich et al., 2000). In contrast, less hydrophobic TMS containing charged or polar residues partition into the membrane less readily, and are retained in close proximity to the translocon, as evidenced by their ability to cross-link to translocon associated proteins (Heinrich and Rapoport, 2003). Thus, in the case of AAPs, Shr3p may facilitate the partitioning of one or more of the TMS of AAPs that have charged residues (i.e., TMS II–VII; Gilstring and Ljungdahl, 2000). In the absence of Shr3p, the inability of these TMS to efficiently partition in the membrane could result in inappropriate interactions leading to aggregation.
The characterization of membrane-localized chaperones in yeast may provide a useful framework to better understand the expression of PM proteins controlling important biological processes in multicellular organisms. Several putative accessory proteins have been identified in cells of metazoan origin. For example, in mammalian cells, receptor-associated membrane proteins determine the intracellular transport and ligand specificity of G protein–coupled receptors (McLatchie et al., 1998; Bermak et al., 2001), and in Caenorhabditis elegans, a select subset of odorant receptors require the action of ODR-4 to be correctly localized to the PM (Dwyer et al., 1998; McClintock and Sammeta, 2003). In addition, the ER proteins BAP29/BAP31 have been postulated to affect the folding of a rather diverse set of membrane proteins and thereby influence their ability to exit the ER (Schamel et al., 2003). It is currently not known whether these proteins influence the membrane insertion and folding of their cognate substrates, however it would be interesting to apply the assays described here to examine the possibility that these metazoan accessory proteins function as membrane-localized chaperones.
Materials and methods
Strains and media
Yeast strains used are listed in Table II. Strain JKY1 is a meiotic segregant from a diploid obtained by crossing isogenic strains CAY29 and FGY212. Strain JKY5 was constructed by deleting the entire sequence of GSF2 in CAY28 with a PCR-amplified hphMX4 cassette (primers Fgsf2 and Rgsf2). Strains JKY3 and JKY4 are meiotic segregants from a cross between strains JKY1 and MAS26-1A (MAT a ura3-52 sec12-1) (Gilstring et al., 1999). Strain JKY6 was constructed as follows: the kanMX allele in CEN.PK113-5D/PHO84myc (obtained from B. Persson, Kalmar University, Kalmar, Sweden) was excised by the expression of the Cre-recombinase (pSH47), and the entire coding sequence of PHO86 was deleted using a PCR-amplified hphMX4 cassette (primers Fpho86 and Rpho86). Strains Y1306 and JAY27 were provided by C. Roncero (University of Salamanca, Salamanca, Spain).
Table II. Yeast strains.
| Strain | Genotype | Source or reference |
|---|---|---|
| Isogenic derivatives of PLY127 | ||
| PLY127 | MATα ura3-52 lys2Δ201 | Kuehn et al., 1996 |
| FGY135a | MAT a ura3-52 lys2Δ201 leu2-3,112 shr3Δ6 gap1Δ2::LEU2 | Gilstring and Ljungdahl, 2000 |
| FGY212 | MATα ura3-52 lys2Δ201 shr3Δ6 | Gilstring and Ljungdahl, 2000 |
| CAY28 | MATα ura3-52 | Andréasson and Ljungdahl, 2002 |
| CAY29 | MAT a ura3-52 | Andréasson and Ljungdahl, 2002 |
| JKY1 | MATα ura3-52 shr3Δ6 | This work |
| JKY5 | MATα ura3-52 gsf2Δ::hphMX4 | This work |
| Isogenic derivatives of JKY3 | ||
| JKY3 | MATα ura3-52 sec12-1 | This work |
| JKY4 | MATα ura3-52 sec12-1 shr3Δ6 | This work |
| Isogenic derivatives of CEN.PK113-5D | ||
| CEN.PK113-5D/PHO84myc | MAT a ura3-52 PHO84::6His-2MYC-loxP-kanMX-loxP MAL2-8c SUC2 | Lagerstedt et al., 2002 |
| JKY6 | MAT a ura3-52 PHO84::6His-2MYC pho86Δ::hphMX4 MAL2-8c SUC2 | This work |
| Isogenic derivatives of Y1306 | ||
| Y1306 | MAT a ura3-52 lys2-801 ade2-101 trp1-901 his3Δ200 CHS3::3xHA | Trilla et al., 1999 |
| JAY27 | MAT a ura3-52 lys2-801 ade2-101 trp1-901 his3Δ200 CHS3::3xHA chs7Δ::HIS3 | Trilla et al., 1999 |
Standard media, YPD and SD supplemented as required, were prepared as described previously (Burke et al., 2000). Ammonium-based synthetic complex (SC; Andréasson and Ljungdahl, 2002) and minimal media containing proline as sole nitrogen source (SPD; Ljungdahl et al., 1992) have been described. Shr3p function was assessed using SPD supplemented with 30 mM l-histidine (Ljungdahl et al., 1992). SAD and SUD were prepared as SD, except that 1 g l−1 allantoin or urea replaced ammonium as the sole nitrogen source, respectively. Low phosphate medium was prepared according to Kaneko et al. (1982). Media were made solid with 2% (wt/vol) bacto Agar (Difco). Antibiotic selections were made on solid YPD, supplemented with 200 μg ml−1 G418 or 300 μg ml−1 hygromycin B.
DNA cloning
The sequences of oligonucleotide primers are available on request. The BamHI site in pPL210 (Ljungdahl et al., 1992) was destroyed by restriction and fill-in creating pJK5. Single-stranded pJK5 was used as a template to enable the introduction of novel BamHI sites into SHR3 by site-directed mutagenesis (Gilstring and Ljungdahl, 2000); oligonucleotides NT-BHI, L1-BHI, L2-BHI, L3-BHI, and CT-BHI were used to create plasmids pJK30–pJK34 that carry BamHI sites inserted immediately after sequences corresponding to amino acids 1, 44, 87, 125, and 210, respectively. A BamHI-flanked SUC2 cassette from plasmid pFG112 (Gilstring and Ljungdahl, 2000) was individually inserted into each BamHI site creating plasmids pJK40–pJK44, respectively. The sequences encoding the 50–amino acid hydrophilic COOH-terminal domain of SHR3 were deleted using oligonucleotide ΔCT and single-stranded pJK5 as a template. The resulting plasmid pJK36 encodes a truncated protein comprised of the first 160 aa of Shr3p (Shr3ΔCTp). Plasmid pJK60 was created by inserting a 4.3-kb SwaI–NotI fragment containing STP1Δ131 from pCA027 (Andréasson and Ljungdahl, 2002) into SwaI–NotI restricted pRS317. Plasmids pJK64 and pJK65 were created by inserting SalI–XbaI fragments from pPL210 (SHR3) and pJK36 (shr3ΔCT) into SalI–XbaI restricted pRS317, respectively.
Protein manipulations
The membrane topology of Shr3p was determined by assessing the glycosylation state of the topo-reporter cassette present in proteins expressed by plasmids pJK40 through pJK44 in strain FGY212 (shr3Δ) (Gilstring and Ljungdahl, 2000). The intracellular location of Gap1p in JKY3 (sec12-1) and JKY4 (sec12-1 shr3Δ6) was determined by fractionation on 12–60% sucrose gradients essentially as described previously (Egner et al., 1995). Cells were grown in SC at 22°C (permissive temperature) to an OD600 of 1.8, cells were harvested and washed twice in water. The washed cells were resuspended (OD600 of 1.8) in media containing allantoin (SAD) to induce Gap1p expression and incubated for 2 h at 34°C (nonpermissive temperature). Induced cells were collected by centrifugation, washed, and resuspended in 1 ml of lysis buffer (OD600 of 200). Cells were lysed by vigorous vortexing in the presence of glass beads (Gilstring et al., 1999). To remove unlysed cells and debris the lysates were centrifuged twice for 5 min at 2,000 g, and 1 ml of supernatant was layered onto gradients. A 50-μl aliquot of each fraction was mixed with equal amount of 3× SDS-PAGE sample buffer. Samples were heated for 10 min at 45°C, and proteins were resolved by SDS-PAGE and analyzed by immunoblotting.
Immunoblots were incubated as indicated with primary antibody in blocking buffer diluted as follows: rabbit α-Shr3p, 1:1,000; rabbit α-Gap1p, 1:20,000; rabbit α-Agp1p, 1:10,000; rat α-HA monoclonal (Roche), 1:1500; mouse α-Dpm1p monoclonal (Molecular Probes), 4 μg/ml; rabbit α-Sec61p, 1:5,000; rabbit α-Hxt1p, 1:1,000; mouse α-MYC monoclonal, 1:1,000; mouse α-Pma1p monoclonal, 1:10,000; rabbit α-Kex2p, 1:1,000; and mouse α-GFP monoclonal, 1:1,000 (Roche). Immunoreactive bands were visualized by chemiluminescence emanating from HRP-conjugated to a secondary antibody; α–rabbit Ig from donkey, α–mouse Ig from sheep or α–rat Ig from goat (Amersham Biosciences), using the LAS1000 system (Fuji Photo Film Co. Ltd.). The α-Gap1p and α-Agp1p antisera were provided by B. André. The α-Pma1p, α-Hxt1p, α-Kex2p, and α-Sec61p antibodies were obtained from J.P. Aris (University of Florida, Gainesville, FL), E. Boles (Goethe-Universitaet Frankfurt, Frankfurt, Germany), R. Fuller (University of Michigan Medical School, Ann Arbor, MI), and C. Stirling (University of Manchester, Manchester, UK), respectively.
Cross-linking
Cross-linking in the presence of either thiol cleavable DSP or UV-activated SANPAH cross-linkers (Pierce Chemical Co.) was performed as follows. Cells were grown to an OD600 of 0.8–1.0, harvested by centrifugation, washed twice in water, and resuspended at an OD600 of 150–200 in PBS, pH 7.4, containing protease inhibitors (Boehringer). Cells were lysed by vortexing in the presence of glass beads, and unlysed cells and debris were removed by centrifugation. DSP cross-linking: 40 μl reactions containing 10 μg protein in PBS and the indicated concentration of DSP were incubated for 30 min at 22°C. Free reactive groups were quenched in the presence of 30 mM Tris-HCl buffer, pH 7.5, for 30 min at 22°C. Samples were treated with 40 mM DTT for 30 min at 37°C as indicated, untreated samples were kept on ice. SANPAH cross-linking: 30 μl reactions containing 8 μg protein in PBS and the indicated concentration of SANPAH were incubated for 30 min at 22°C in the dark. Reactions were quenched as above for 15 min at 22°C. Photoactivation was performed by exposing samples to 366 nm UV light for 10 min.
Blue native–PAGE
Cells were grown to an OD600 of 0.8–1.0, harvested by centrifugation, washed twice in water, and resuspended at an OD600 of 150–200 in BN buffer (50 mM Tris-HCl, 150 mM NaCl, pH 7.5) containing protease inhibitors (Boehringer). Protein extracts were prepared as described for cross-linking. Proteins (1.8 mg ml−1 in BN buffer) were solubilized with DM (Roche) at 4°C for 30 min, and a 0.1 vol of BN-PAGE sample buffer (5% Serva blue G, 100 mM BisTris, 500 mM 6-aminocaproic acid, and 80% glycerol, pH 7.0) was added. Proteins were separated on 5–15% BN gradient gels as described previously (Schägger et al., 1994). High molecular weight markers (Amersham Biosciences) were used as standards.
Microscopy
Strain FGY135 carrying plasmid pJOD010 (PGAL-GAP1-GFP) was cotransformed with plasmids pJK64, pJK65, or pRS317 (vector without insert). Cells were grown in allantoin media with 2% raffinose and 0.2% glucose to an OD600 of 2–3, and subsequently suspended in allantoin media with 4% galactose and 1 μg ml−1 DAPI for 5 h. Cells were suspended in media containing 0.4% low melting point agar (37°C) and quickly mounted on slides for microscopic observation. Live cells were viewed at RT using an Axiophot microscope (Carl Zeiss MicroImaging, Inc.) with a Plan-Apochromat 63×/1.40 objective. Digital images of cells examined using Nomarski optics, and of the fluorescence associated with GFP and DAPI (standard filter sets), were captured using a C4742-95 CCD camera (Hamamatsu Photonics) and QED Imaging software (Media Cybernetics). Image files were incorporated into figures using Photoshop CS (Adobe Systems, Inc.).
Acknowledgments
We especially wish to thank C. Fredrik Gilstring for his extensive input during the early stages of this work and the preparation of polyclonal anti-Shr3p antibodies. The current members of Ljungdahl laboratory are acknowledged for constructive comments. We are indebted to Bruno André for his generous gifts of α-Gap1p and α-Agp1p antisera, and plasmid pJOD010 expressing Gap1-GFP. We thank John Aris, Eckhard Boles, Robert Fuller, and Colin Stirling for α-Pma1p, α-Hxt1p, α-Kex2p, and α-Sec61p antibodies, respectively. Finally, we thank Bengt Persson and Cesar Roncero for the strains carrying epitope-tagged PHO84 and CHS3 alleles, respectively.
This work was supported by the Ludwig Institute for Cancer Research and a grant from the EU (EFFEXPORT project; QLK3-CT-2001-00533).
Abbreviations used in this paper: AAP, amino acid permease; DM, dodecyl-β-d-maltopyranoside; Dpm1p, dolichol phosphate mannose synthase; DSP, Dithiobis[succinimidyl propionate]; LacY, lactose permease; PM, plasma membrane; SANPAH, N-succinimidyl 6-[4′-azido 2′-nitrophenylamino]hexanoate; SC, synthetic complex; TMS, transmembrane segments.
References
- Abramson, J., I. Smirnova, V. Kasho, G. Verner, H.R. Kaback, and S. Iwata. 2003. Structure and mechanism of the lactose permease of Escherichia coli. Science. 301:610–615. [DOI] [PubMed] [Google Scholar]
- Alder, N.N., and A.E. Johnson. 2004. Cotranslational membrane protein biogenesis at the endoplasmic reticulum. J. Biol. Chem. 279:22787–22790. [DOI] [PubMed] [Google Scholar]
- Andréasson, C., and P.O. Ljungdahl. 2002. Receptor-mediated endoproteolytic activation of two transcription factors in yeast. Genes Dev. 16:3158–3172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antonny, B., and R. Schekman. 2001. ER export: public transportation by the COPII coach. Curr. Opin. Cell Biol. 13:438–443. [DOI] [PubMed] [Google Scholar]
- Barlowe, C. 2003. Molecular recognition of cargo by the COPII complex: a most accommodating coat. Cell. 114:395–399. [DOI] [PubMed] [Google Scholar]
- Bermak, J.C., M. Li, C. Bullock, and Q.Y. Zhou. 2001. Regulation of transport of the dopamine D1 receptor by a new membrane-associated ER protein. Nat. Cell Biol. 3:492–498. [DOI] [PubMed] [Google Scholar]
- Bickford, L.C., E. Mossessova, and J. Goldberg. 2004. A structural view of the COPII vesicle coat. Curr. Opin. Struct. Biol. 14:147–153. [DOI] [PubMed] [Google Scholar]
- Burke, D., D. Dawson, and T. Stearns. 2000. Methods in Yeast Genetics: A Cold Spring Harbor Laboratory Course Manual. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY. 171–182.
- Dalbey, R.E., and A. Kuhn. 2004. YidC family members are involved in the membrane insertion, lateral integration, folding, and assembly of membrane proteins. J. Cell Biol. 166:769–774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dwyer, N.D., E.R. Troemel, P. Sengupta, and C.I. Bargmann. 1998. Odorant receptor localization to olfactory cilia is mediated by ODR-4, a novel membrane-associated protein. Cell. 93:455–466. [DOI] [PubMed] [Google Scholar]
- Egner, R., Y. Mahé, R. Pandjaitan, and K. Kuchler. 1995. Endocytosis and vacuolar degradation of the plasma membrane-localized Pdr5 ATP-binding cassette multidrug transporter in Saccharomyces cerevisiae. Mol. Cell. Biol. 15:5879–5887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forsberg, H., and P.O. Ljungdahl. 2001. Sensors of extracellular nutrients in Saccharomyces cerevisiae. Curr. Genet. 40:91–109. [DOI] [PubMed] [Google Scholar]
- Gilstring, C.F., M. Melin-Larsson, and P.O. Ljungdahl. 1999. Shr3p mediates specific COPII coatomer-cargo interactions required for the packaging of amino acid permeases into ER-derived transport vesicles. Mol. Biol. Cell. 10:3549–3565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilstring, C.F., and P.O. Ljungdahl. 2000. A method for determining the in vivo topology of yeast polytopic membrane proteins demonstrates that Gap1p fully integrates into the membrane independently of Shr3p. J. Biol. Chem. 275:31488–31495. [DOI] [PubMed] [Google Scholar]
- Heinrich, S.U., and T.A. Rapoport. 2003. Cooperation of transmembrane segments during the integration of a double-spanning protein into the ER membrane. EMBO J. 22:3654–3663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heinrich, S.U., W. Mothes, J. Brunner, and T.A. Rapoport. 2000. The Sec61 complex mediates the integration of a membrane protein by allowing lipid partitioning of the transmembrane domain. Cell. 102:233–244. [DOI] [PubMed] [Google Scholar]
- Herrmann, J.M., P. Malkus, and R. Schekman. 1999. Out of the ER–outfitters, escorts and guides. Trends Cell Biol. 9:5–7. [DOI] [PubMed] [Google Scholar]
- Huang, Y., M.J. Lemieux, J. Song, M. Auer, and D.N. Wang. 2003. Structure and mechanism of the glycerol-3-phosphate transporter from Escherichia coli. Science. 301:616–620. [DOI] [PubMed] [Google Scholar]
- Iraqui, I., S. Vissers, F. Bernard, J.O. de Craene, E. Boles, A. Urrestarazu, and B. André. 1999. Amino acid signaling in Saccharomyces cerevisiae: a permease-like sensor of external amino acids and F-Box protein Grr1p are required for transcriptional induction of the AGP1 gene, which encodes a broad-specificity amino acid permease. Mol. Cell. Biol. 19:989–1001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaneko, Y., A. Toh-e, and Y. Oshima. 1982. Identification of the genetic locus for the structural gene and a new regulatory gene for the synthesis of repressible alkaline phosphatase in Saccharomyces cerevisiae. Mol. Cell. Biol. 2:127–137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuehn, M.J., R. Schekman, and P.O. Ljungdahl. 1996. Amino acid permeases require COPII components and the ER resident membrane protein Shr3p for packaging into transport vesicles in vitro. J. Cell Biol. 135:585–595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuehn, M.J., J.M. Herrmann, and R. Schekman. 1998. COPII-cargo interactions direct protein sorting into ER-derived transport vesicles. Nature. 391:187–190. [DOI] [PubMed] [Google Scholar]
- Kuhn, A., R. Stuart, R. Henry, and R.E. Dalbey. 2003. The Alb3/Oxa1/YidC protein family: membrane-localized chaperones facilitating membrane protein insertion? Trends Cell Biol. 13:510–516. [DOI] [PubMed] [Google Scholar]
- Lagerstedt, J.O., R. Zvyagilskaya, J.R. Pratt, J. Pattison-Granberg, A.L. Kruckeberg, J.A. Berden, and B.L. Persson. 2002. Mutagenic and functional analysis of the C-terminus of Saccharomyces cerevisiae Pho84 phosphate transporter. FEBS Lett. 526:31–37. [DOI] [PubMed] [Google Scholar]
- Lau, W.T., R.W. Howson, P. Malkus, R. Schekman, and E.K. O'Shea. 2000. Pho86p, an endoplasmic reticulum (ER) resident protein in Saccharomyces cerevisiae, is required for ER exit of the high-affinity phosphate transporter Pho84p. Proc. Natl. Acad. Sci. USA. 97:1107–1112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lecomte, F.J., N. Ismail, and S. High. 2003. Making membrane proteins at the mammalian endoplasmic reticulum. Biochem. Soc. Trans. 31:1248–1252. [DOI] [PubMed] [Google Scholar]
- Lee, M.C., E.A. Miller, J. Goldberg, L. Orci, and R. Schekman. 2004. Bi-directional protein transport between the ER and Golgi. Annu. Rev. Cell Dev. Biol. 20:87–123. [DOI] [PubMed] [Google Scholar]
- Levine, T.P., C.A. Wiggins, and S. Munro. 2000. Inositol phosphorylceramide synthase is located in the Golgi apparatus of Saccharomyces cerevisiae. Mol. Biol. Cell. 11:2267–2281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ljungdahl, P.O., C.J. Gimeno, C.A. Styles, and G.R. Fink. 1992. SHR3: A novel component of the secretory pathway specifically required for the localization of amino acid permeases in yeast. Cell. 71:463–478. [DOI] [PubMed] [Google Scholar]
- Malkus, P., F. Jiang, and R. Schekman. 2002. Concentrative sorting of secretory cargo proteins into COPII-coated vesicles. J. Cell Biol. 159:915–921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martínez, P., and P.O. Ljungdahl. 2000. The SHR3 homologue from S. pombe demonstrates a conserved function of ER packaging chaperones. J. Cell Sci. 113:4351–4362. [DOI] [PubMed] [Google Scholar]
- Martínez, P., and P.O. Ljungdahl. 2004. An ER packaging chaperone determines the amino acid uptake capacity and virulence of Candida albicans. Mol. Microbiol. 51:371–384. [DOI] [PubMed] [Google Scholar]
- McClintock, T.S., and N. Sammeta. 2003. Trafficking prerogatives of olfactory receptors. Neuroreport. 14:1547–1552. [DOI] [PubMed] [Google Scholar]
- McLatchie, L.M., N.J. Fraser, M.J. Main, A. Wise, J. Brown, N. Thompson, R. Solari, M.G. Lee, and S.M. Foord. 1998. RAMPs regulate the transport and ligand specificity of the calcitonin-receptor-like receptor. Nature. 393:333–339. [DOI] [PubMed] [Google Scholar]
- Miller, E.A., T.H. Beilharz, P.N. Malkus, M.C. Lee, S. Hamamoto, L. Orci, and R. Schekman. 2003. Multiple cargo binding sites on the COPII subunit Sec24p ensure capture of diverse membrane proteins into transport vesicles. Cell. 114:497–509. [DOI] [PubMed] [Google Scholar]
- Nagamori, S., I.N. Smirnova, and H.R. Kaback. 2004. Role of YidC in folding of polytopic membrane proteins. J. Cell Biol. 165:53–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakano, A., D. Brada, and R. Schekman. 1988. A membrane glycoprotein, Sec12p, required for protein transport from the endoplasmic reticulum to the Golgi apparatus in yeast. J. Cell Biol. 107:851–863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nyman, T., J. Kota, and P.O. Ljungdahl. 2004. Ancillary proteins in membrane targeting of transporters. Topics in Current Genetics – Molecular Mechanisms Controlling Transmembrane Transport. E. Boles and R. Krämer, editors. Springer-Verlag, Heidelberg. 207–234.
- Rapoport, T.A., V. Goder, S.U. Heinrich, and K.E.S. Matlack. 2004. Membrane-protein integration and the role of the translocation channel. Trends Cell Biol. 14:568–575. [DOI] [PubMed] [Google Scholar]
- Rutkowski, D.T., and R.J. Kaufman. 2004. A trip to the ER: coping with stress. Trends Cell Biol. 14:20–28. [DOI] [PubMed] [Google Scholar]
- Saier, M.H., Jr. 2000. Families of transmembrane transporters selective for amino acids and their derivatives. Microbiol. 146:1775–1795. [DOI] [PubMed] [Google Scholar]
- Schägger, H., W.A. Cramer, and G. von Jagow. 1994. Analysis of molecular masses and oligomeric states of protein complexes by blue native electrophoresis and isolation of membrane protein complexes by two-dimensional native electrophoresis. Anal. Biochem. 217:220–230. [DOI] [PubMed] [Google Scholar]
- Schamel, W.W.A., S. Kuppig, B. Becker, K. Gimborn, H.P. Hauri, and M. Reth. 2003. A high-molecular-weight complex of membrane proteins BAP29/BAP31 is involved in the retention of membrane-bound IgD in the endoplasmic reticulum. Proc. Natl. Acad. Sci. USA. 100:9861–9866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sherwood, P.W., and M. Carlson. 1999. Efficient export of the glucose transporter Hxt1p from the endoplasmic reticulum requires Gsf2p. Proc. Natl. Acad. Sci. USA. 96:7415–7420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trilla, J.A., A. Duran, and C. Roncero. 1999. Chs7p, a new protein involved in the control of protein export from the endoplasmic reticulum that is specifically engaged in the regulation of chitin synthesis in Saccharomyces cerevisiae. J. Cell Biol. 145:1153–1163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turner, R.J. 2003. Understanding the biogenesis of polytopic integral membrane proteins. J. Membr. Biol. 192:149–157. [DOI] [PubMed] [Google Scholar]
- van den Berg, B., W.M. Clemons Jr., I. Collinson, Y. Modis, E. Hartmann, S.C. Harrison, and T.A. Rapoport. 2004. X-ray structure of a protein-conducting channel. Nature. 427:36–44. [DOI] [PubMed] [Google Scholar]