

Abstract
In spite of advances in understanding the role of the cellular prion protein (PrP) in neural cell interactions, the mechanisms of PrP function remain poorly characterized. We show that PrP interacts directly with the neural cell adhesion molecule (NCAM) and associates with NCAM at the neuronal cell surface. Both cis and trans interactions between NCAM at the neuronal surface and PrP promote recruitment of NCAM to lipid rafts and thereby regulate activation of fyn kinase, an enzyme involved in NCAM-mediated signaling. Cis and trans interactions between NCAM and PrP promote neurite outgrowth. When these interactions are disrupted in NCAM-deficient and PrP-deficient neurons or by PrP antibodies, NCAM/PrP-dependent neurite outgrowth is arrested, indicating that PrP is involved in nervous system development cooperating with NCAM as a signaling receptor.
Introduction
The cellular prion protein (PrP) is a ubiquitous glycoprotein, prominently expressed in the brain and localized at the cell surface via a glycosylphosphatidylinositol (GPI) anchor. In prion diseases, PrP is converted to a conformationally altered form that accumulates in the brain (Prusiner, 1998; Weissmann and Flechsig, 2003). Mutations in the PrP gene have been linked to the Gerstmann-Sträussler-Scheinker syndrome, familial fatal insomnia, sporadic Creutzfeld-Jakob disease, and certain forms of dementia with cerebellar disorder and myopathy (Hsiao et al., 1989; Collinge, 1997).
Attempts to identify the functions of PrP are consistent with PrP functioning as a recognition molecule. PrP interacts or associates with the 67-kD laminin receptor, the 37-kD laminin receptor precursor protein, or the ECM glycoprotein laminin (Rieger et al., 1997; Graner et al., 2000; Gauczynski et al., 2001). PrP has been identified in a complex with the neural cell adhesion molecule (NCAM) by chemical cross-linking (Schmitt-Ulms et al., 2001). However, whether PrP binds directly to NCAM has remained unclear. In addition to its protective role in models of neurodegeneration due to oxidative stress, being probably linked to its metal ion binding ability (Milhavet and Lehmann, 2002), PrP has been implicated in neurite outgrowth and neuronal survival as a trans-interacting partner, that is, an interaction between the cell surface of one cell and a molecule from the ECM or from the cell surface of an adjacent cell. The binding partner for PrP at the neuronal cell surface has, however, remained elusive (Chen et al., 2003). Similar to the molecules associated with PrP, such as laminin and NCAM, PrP has been implicated in the physiology of neurons, affecting synaptic function (Collinge et al., 1994), neurite outgrowth, and neuronal survival (Chen et al., 2003). Because both PrP and NCAM have been implicated in signaling cascades involving the p59fyn nonreceptor tyrosine kinase (fyn) (Beggs et al., 1997; Mouillet-Richard et al., 2000) and because fyn is involved in NCAM-induced neurite outgrowth (Beggs et al., 1994), we investigated whether the two molecules may functionally cooperate with each other by engaging in cis and/or trans interactions. Furthermore, it seemed important to characterize the involvement of lipid-enriched microdomains, the so-called lipid rafts, at the cell surface as a signaling platform for PrP, which localizes to lipid rafts because of its GPI anchor (Gorodinsky and Harris, 1995; Walmsley et al., 2003) and for NCAM, which can be sequestered to lipid rafts due to palmitoylation, which is essential for promotion of neurite outgrowth (Niethammer et al., 2002). Here, we show that PrP interacts directly with NCAM, and in a heterophilic cis and trans configuration recruits to and stabilizes NCAM in lipid rafts, thereby activating fyn to induce NCAM-dependent neuritogenesis.
Results
PrP directly interacts with NCAM
To obtain insights into the function of PrP in the developing brain we first analyzed the association between PrP and NCAM in cultured hippocampal neurons. PrP partially colocalized with NCAM along neurites and in growth cones (Fig. 1 A). As a GPI-anchored protein, PrP mostly localizes to lipid rafts (Gorodinsky and Harris, 1995; Walmsley et al., 2003). We therefore analyzed whether NCAM colocalizes with PrP in lipid rafts by extracting neurons with cold 1% Triton X-100, a procedure used to isolate cytoskeleton-bound and raft-associated proteins (Ledesma, et al., 1998; Niethammer et al., 2002; Leshchyns'ka et al., 2003). In extracted neurons, PrP showed a patchy distribution along neurites (Fig. 1 B) (Madore et al., 1999), showing that PrP accumulates in subdomains at neuronal plasma membranes. Similar to PrP, NCAM showed a patchy distribution in clusters along neurites (Fig. 1 B). Clusters of NCAM overlapped with PrP accumulations (mean correlation between distributions of two proteins, r = 0.7 ± 0.01; Fig. 1 D). To verify whether this overlap was specific for NCAM, we analyzed the distribution of L1, another recognition molecule of the immunoglobulin superfamily and also present in lipid rafts of neurites (Nakai and Kamiguchi, 2002). In contrast to NCAM and PrP, L1 showed a more uniform distribution along extracted neurites (Fig. 1 C). The overall pattern of L1 and PrP localization was different from that between NCAM and PrP (mean correlation between distributions of L1 and PrP, r = 0.27 ± 0.02; Fig. 1, E and F).
Figure 1.
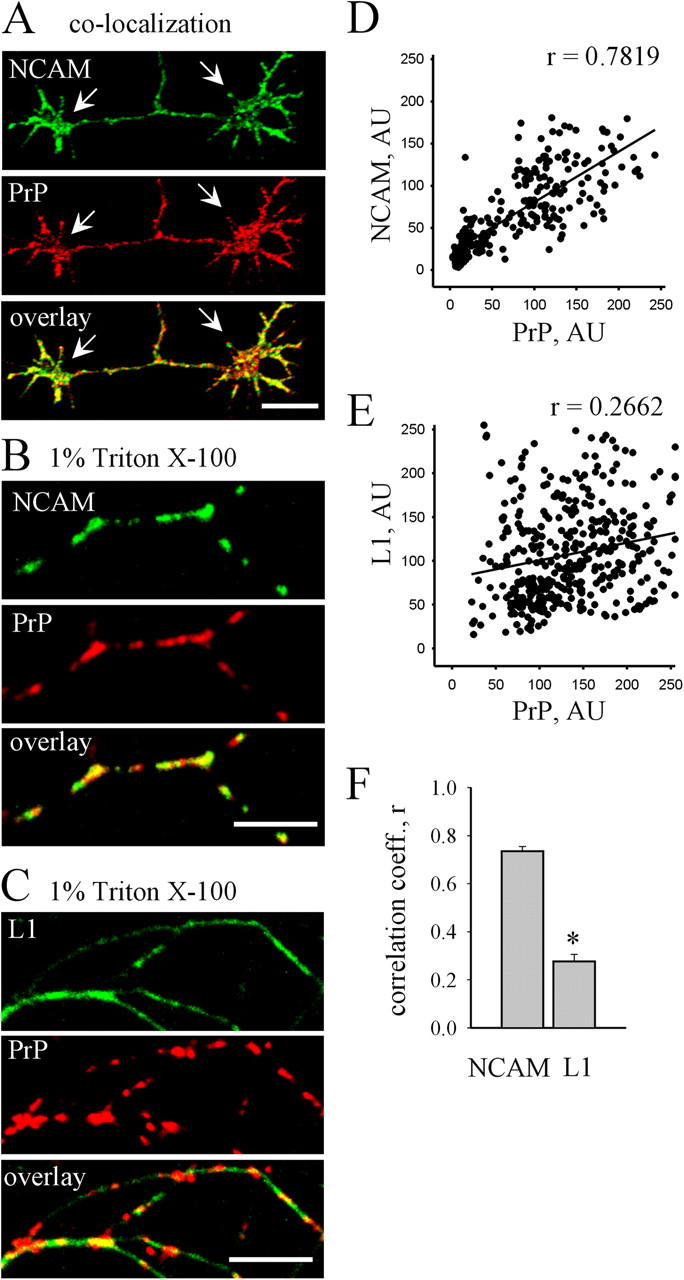
NCAM colocalizes with PrP in lipid rafts. (A) Neurite with two growth cones (arrows) double-labeled with NCAM and PrP antibodies. PrP and NCAM partially colocalize along neurites and in growth cones. (B and C) Neurons were extracted in cold 1% Triton X-100 and labeled with PrP and NCAM antibodies (B) or PrP and L1 antibodies (C). Detergent-insoluble clusters of PrP overlap with accumulations of NCAM. L1 shows a distribution distinct from PrP. Bars, 10 μm. (D and E) Examples of linear regression graphs comparing distributions of PrP and NCAM (D) or PrP and L1 (E) are shown. Corresponding correlation coefficients (r) are presented. (F) The diagram shows mean correlation coefficients (r) comparing distributions of PrP and NCAM or PrP and L1. The correlation between localization of NCAM and PrP is significantly higher than between localization of L1 and PrP. Mean values ± SEM (n > 30) are shown. *, P < 0.05, t test.
The similar localization of NCAM and PrP suggested that both proteins form a complex in lipid rafts. We thus cross-linked NCAM at the neuronal surface with NCAM antibodies applied to live neurons. NCAM clustering induced partial redistribution of PrP to NCAM-containing clusters (Fig. 2 A). We also noticed nonoverlapping clusters of NCAM and PrP (Fig. 2 A) in accordance with our observation that only a small portion of the neuronal NCAM140 and NCAM180 isoforms is in lipid rafts (Niethammer et al., 2002). To further investigate this phenomenon, we analyzed by an ELISA binding assay whether PrP and NCAM directly interact using recombinant PrP-Fc, which contains the extracellular domain of mouse PrP fused to the Fc portion of IgG (Chen et al., 2003), and NCAM purified from mouse brain. NCAM bound to PrP-Fc in a concentration-dependent manner, but not to BSA (Fig. 2 B). L1 also did not bind to PrP (Fig. 2 B), in accordance with results with extracted neurons (Fig. 1). NCAM is the carrier of polysialic acid (PSA), which may influence its binding to PrP. To verify whether PSA influences binding to PrP, we analyzed by ELISA the interaction between PrP and nonpolysialylated NCAM-Fc produced in CHO cells using recombinant PrP-AP, which contains the extracellular domain of PrP fused to alkaline phosphatase (Chen et al., 2003). We found that nonpolysialylated NCAM-Fc also bound to PrP-AP in a concentration-dependent manner, but not to BSA (Fig. 2 C).
Figure 2.
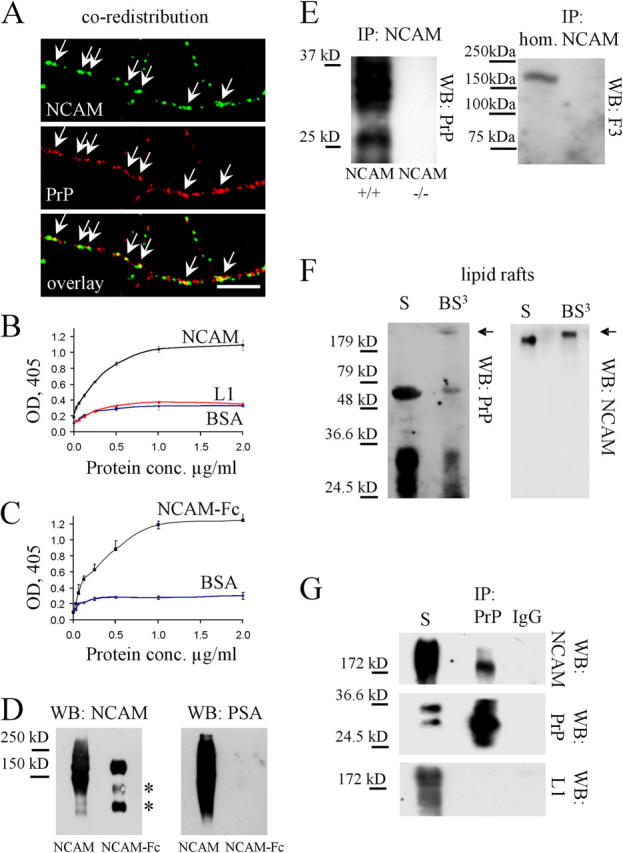
NCAM directly interacts with PrP. (A) Neurons incubated live with antibodies against NCAM to induce clustering of NCAM at the cell surface were fixed and labeled with antibodies against PrP. PrP partially redistributed to NCAM clusters (arrows). Bar, 10 μm. (B) Increasing concentrations of NCAM and L1, purified from mouse brain, were assayed by ELISA for their binding to plastic-bound PrP-Fc (5 μg/ml). Binding of NCAM to BSA (3 mg/ml) served as a control. Mean values (OD405) ± SEM (n = 6) are shown. (C) Increasing concentrations of NCAM-Fc were assayed by ELISA for their binding to plastic-bound PrP-AP (5 μg/ml). Binding of NCAM-Fc to BSA (3 mg/ml) served as a control. Mean values (OD405) ± SEM (n = 6) are shown. (D) NCAM from mouse brain and NCAM-Fc were immunoblotted (WB) with antibodies against NCAM and polysialic acid (PSA). Note that PSA immunoreactivity is found only on NCAM purified from mouse brain. The two bottom bands represent degradation products of NCAM-Fc (asterisk). (E) NCAM immunoprecipitates from the NCAM+/+ mouse brain were immunoblotted (WB) with PrP or F3 antibodies. NCAM−/− brains served as a control for PrP immunoprecipitation. Brain homogenate (hom.) was also probed for F3. PrP, but not F3, coimmunoprecipitated with NCAM. (F) Lipid rafts (S) or lipid rafts treated with BS3 (BS3) were immunoblotted with PrP antibodies (WB: PrP). Then, the membrane was stripped and labeled with NCAM antibodies (WB: NCAM). Note a PrP-immunoreactive band above 200 kD in the cross-linked material that overlaps with a shifted NCAM immunoreactive band (arrows). (G) Total membranes (S) or PrP immunoprecipitates from total membranes treated with BS3 (IP: PrP) were immunoblotted (WB) with antibodies to NCAM, PrP, and L1. Immunoprecipitation performed with nonimmune IgG served as a control (IP: IgG). Note that NCAM, but not L1, coimmunoprecipitated with PrP.
To investigate whether NCAM and PrP interact in brain tissue, we immunoprecipitated NCAM from brain homogenates and analyzed immunoprecipitates with antibodies against PrP. PrP coimmunoprecipitated with NCAM (Fig. 2 E), indicating that the two proteins are associated in brain. Another GPI-anchored immunoglobulin superfamily recognition molecule, F3/contactin, did not coimmunoprecipitate with NCAM, underscoring the specificity of NCAM interaction with PrP (Fig. 2 E).
Next, we examined whether NCAM and PrP exist in a complex in the same plasma membrane microenvironment by inducing covalent binding between primary amino groups of adjacent proteins in the lipid raft fraction from total brain homogenates using the homobifunctional BS3 chemical cross-linker with a spacer arm of 11.4 Å. It has been shown repeatedly that BS3 does not trigger nonspecific protein clustering (Bhamidipati et al., 2000; Culligan et al., 2001; Kaykas et al., 2004). In Western blots of cross-linked probes, PrP was observed in high molecular weight complexes as a PrP-positive band above 200 kD (Fig. 2 F). NCAM was also shifted to a higher molecular weight when compared with its molecular weight before cross-linking (Fig. 2 F). The high molecular weight PrP and NCAM immunoreactive bands overlapped (Fig. 2 F), suggesting PrP cross-linking with NCAM. Similar results were observed when isolated membrane fractions were used for cross-linking (unpublished data). To estimate the amount of NCAM complexing with PrP in brain membranes, we immunoprecipitated PrP from the cross-linked membranes. NCAM was coimmunoprecipitated with PrP (Fig. 2 G). Approximately 10% of all NCAM immunoreactivity was observed in this complex, corresponding to NCAM levels in lipid rafts (Niethammer et al., 2002). We conclude that NCAM interacts with PrP and that both molecules form a complex in neuronal cell surfaces.
PrP is involved in stabilization of NCAM in lipid rafts
NCAM140 and NCAM180 localize mostly to raft-free areas and redistribute to lipid rafts through palmitoylation of NCAM after NCAM activation (Niethammer et al., 2002; Leshchyns'ka et al., 2003). Because PrP localizes to lipid rafts, complex formation between NCAM and PrP should occur in lipid rafts. We thus hypothesized that interaction of NCAM with PrP may recruit NCAM to and stabilize it in lipid rafts. Indeed, absence of PrP in PrP−/− brains reduced the amount of NCAM140 and NCAM180 in lipid rafts (76.4 ± 6.7% in PrP−/− lipid rafts with PrP+/+ values set to 100%; Fig. 3 A) and growth cones (66.13 ± 10% in PrP−/− lipid rafts with PrP+/+ values set to 100%; Fig. 3 A). The GPI-anchored and lipid raft–localized NCAM120 was present in similar amounts in lipid rafts isolated from PrP+/+ and PrP−/− brains, indicating that lipid rafts were isolated with the same efficacy. Levels of L1 were similar in lipid rafts isolated from brains of PrP−/− and PrP+/+ mice (Fig. 3 B), confirming that PrP deficiency does not affect the overall raft composition. In contrast, overall levels of NCAM were increased in PrP−/− brains (Fig. 3 A), excluding the possibility that reduced levels of NCAM140 and NCAM180 in lipid rafts are due to reduced expression of NCAM in PrP−/− brains. We conclude that PrP is involved in stabilization of NCAM140 and NCAM180 in lipid rafts.
Figure 3.

Targeting of NCAM to lipid rafts is reduced in PrP −/− mice. (A) Brain homogenates or lipid rafts from brain homogenates and growth cones of PrP+/+ and PrP−/− mice (0–4 d old) were immunoblotted (WB) with antibodies against NCAM. The band at and above 180 kD represents polysialylated NCAM140 and NCAM180 (PSA-NCAM; Niethammer et al., 2002). In lipid rafts, a band representing nonpolysialylated NCAM120 is also observed. Diagrams show quantitation of PSA-NCAM and NCAM120. Note that the amount of PSA-NCAM is increased in brain homogenates and decreased in rafts isolated from brain homogenates and growth cones of PrP−/− mice. (B) Lipid rafts isolated from brain homogenates of PrP+/+ and PrP−/− mice (0–4 d old) were immunoblotted with L1 antibodies. Diagrams show quantitation of L1. Note that the amount of L1 is similar in rafts isolated from PrP−/− and PrP+/+ mice. Mean values ± SEM (n = 6) are shown. AU = arbitrary units. *, P < 0.05, paired t test (for A and B).
Cis and trans interactions between PrP and NCAM are important for NCAM stabilization in lipid rafts
Two distinct types of interaction between PrP and NCAM could account for the observed phenomena. PrP could stabilize NCAM in lipid rafts by cis interaction, i.e., both molecules associate in the plasma membrane of the same neuron. In accordance with this idea, NCAM and PrP were cross-linked with antibodies at live neuronal cell surfaces (Fig. 2 A). Chemical cross-linking of NCAM and PrP was also performed in low density cultures allowing only cis interactions between NCAM and PrP (unpublished data). To analyze the cis interaction in lipid rafts, we estimated the amount of detergent-insoluble NCAM in PrP+/+ and PrP−/− cultured hippocampal neurons. Only neurons without contacts were evaluated to assure that only cis interactions were analyzed. PrP−/− neurons extracted with cold Triton X-100 showed lower NCAM labeling intensity in NCAM clusters and along neurites when compared with PrP+/+ neurons, indicating a reduction of NCAM in lipid rafts (Fig. 4, C and D). This reduction was not due to a decrease in expression of NCAM in PrP−/− neurons because the mean labeling intensity of NCAM along neurites of nonextracted neurons was increased in PrP−/− neurons (Fig. 4, A and B), in accordance with our biochemical data (Fig. 3 A). We conclude that cis interactions between NCAM and PrP are important for stabilization of NCAM in lipid rafts.
Figure 4.
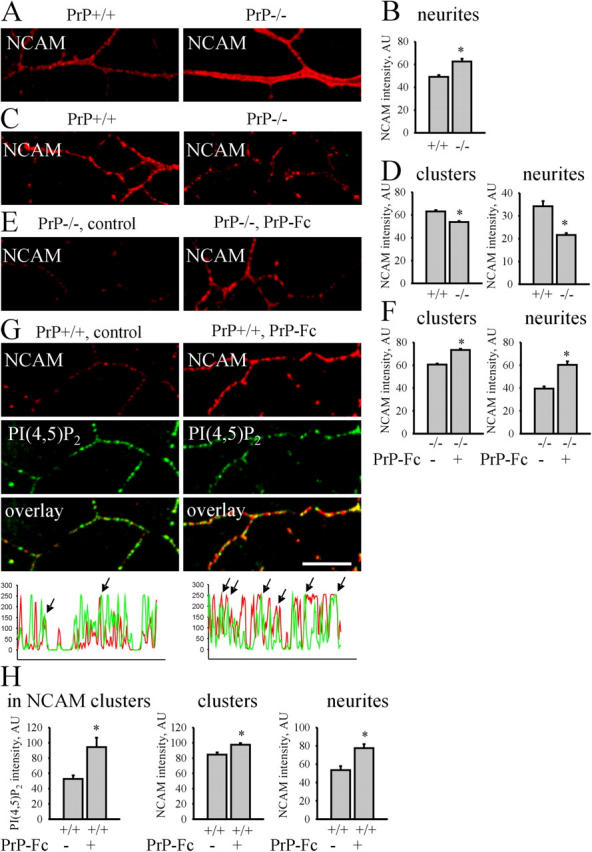
Cis and trans interactions between NCAM and PrP distribute NCAM to lipid rafts. (A and B) PrP+/+ and PrP−/− neurons were labeled with NCAM antibodies. Note enhanced levels of NCAM on PrP−/− neurites. (B) Diagram shows mean labeling intensity of NCAM along neurites. (C and D) PrP+/+ and PrP−/− neurons extracted in cold 1% Triton X-100 were labeled with NCAM antibodies. Note reduced levels of detergent-insoluble NCAM on PrP−/− neurites. (D) Diagrams show mean NCAM labeling intensity in NCAM clusters and along total neurites of PrP+/+ and PrP−/− neurons. (E and F) PrP−/− neurons were incubated with PrP-Fc, extracted in cold 1% Triton X-100, and labeled with NCAM antibodies. Note increased levels of detergent insoluble NCAM on neurites exposed to PrP-Fc when compared with the nonexposed group (control). (F) Diagrams show mean NCAM labeling intensity in NCAM clusters and along neurites of untreated and PrP-Fc–treated PrP−/− neurons. (G and H) NCAM+/+ neurons were incubated with PrP-Fc, extracted in cold 1% Triton X-100, and labeled with antibodies against NCAM and PI(4,5)P2. Examples of distributions of NCAM and PI(4,5)P2 labeling intensity along neurites of control and PrP-Fc–incubated groups are shown. Arrows show overlapping peaks. Note increased overlap of detergent-insoluble NCAM and PI(4,5)P2 after incubation with PrP-Fc. (H) Diagrams show mean labeling intensity of PI(4,5)P2 in NCAM clusters in control and PrP-Fc–treated groups and mean NCAM labeling intensity in NCAM clusters and along neurites. Mean values ± SEM are shown in arbitrary units (AU) (n > 50 neurites). *, P < 0.05, t test. Bar, 10 μm (for A–G).
PrP could also induce redistribution of NCAM to lipid rafts by trans interaction, i.e., NCAM binds to PrP on adjacent cells. To analyze the role of a trans interaction between NCAM and PrP, we applied soluble PrP-Fc to neurons from PrP−/− mice to evaluate the redistribution of NCAM to lipid rafts in absence of NCAM-to-PrP cis interaction. Covalent chemical cross-linking of PrP-Fc with NCAM in brain homogenates indicated that PrP-Fc indeed bound to endogenous NCAM in a trans fashion (unpublished data). Application of PrP-Fc increased the detergent-insoluble NCAM fraction in NCAM clusters and along neurites of PrP−/− neurons (Fig. 4, E and F). Because increase of NCAM levels in lipid rafts after application of PrP-Fc suggested a PrP-Fc–induced redistribution of NCAM to lipid rafts, we measured the association of NCAM with lipid rafts in response to PrP-Fc application in PrP+/+ neurons using the PI(4,5)P2 raft marker (Laux et al., 2000; Niethammer et al., 2002). Amounts of PI(4,5)P2 in detergent-insoluble NCAM clusters were increased after PrP-Fc application, indicating that PrP-Fc redistributed NCAM to lipid rafts (Fig. 4, G and H). The same result was obtained when endogenous PrP or ganglioside GM1 (Leshchyns'ka et al., 2003) were taken as raft markers (unpublished data). As for PrP−/− neurons, application of PrP-Fc to PrP+/+ neurons increased levels of detergent insoluble NCAM in NCAM clusters and along neurites (Fig. 4, G and H). We conclude that trans interaction of PrP with NCAM induces redistribution of NCAM to lipid rafts.
NCAM activation with NCAM-Fc or NCAM antibodies induces redistribution of NCAM to lipid rafts being necessary for NCAM-mediated neurite outgrowth (Niethammer et al., 2002; Leshchyns'ka et al., 2003). Indeed, NCAM-Fc or NCAM antibodies increased PrP levels in NCAM clusters in PrP+/+ neurons (Fig. 5, A and C), indicating that NCAM partially redistributes to lipid rafts as previously observed (Leshchyns'ka et al., 2003), and suggesting that association between NCAM and PrP is enhanced after NCAM activation. In spite of overall higher levels of NCAM expression in PrP−/− neurons (Fig. 4, A and B), NCAM levels in lipid rafts were reduced in stimulated PrP−/− neurons (Fig. 5, B and D), indicating that cis interactions between NCAM and PrP are important for redistribution of NCAM to lipid rafts in response to NCAM activation.
Figure 5.
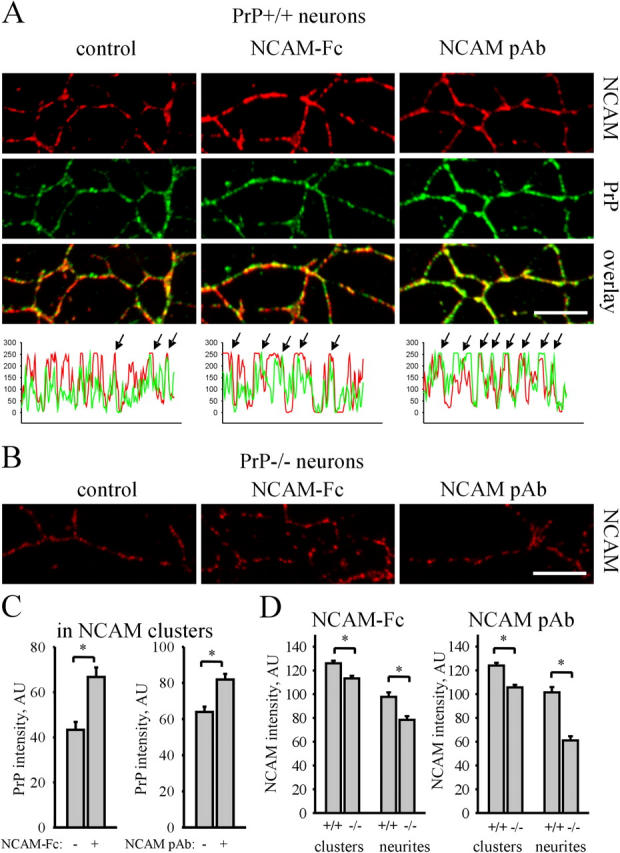
PrP is required for redistribution of NCAM to lipid rafts in response to NCAM activation. (A) PrP+/+ neurons were incubated with NCAM-Fc or polyclonal NCAM antibodies, extracted with 1% Triton X-100, and labeled with NCAM and PrP antibodies. Incubation with NCAM-Fc and NCAM pAb increased the overlap between NCAM and PrP, indicating that NCAM redistributed to lipid rafts. Examples of NCAM and PrP labeling intensities along neurites are shown below. Arrows show overlapping peaks. (B) In parallel with PrP+/+ neurons (A), PrP−/− neurons were incubated with NCAM-Fc or polyclonal NCAM antibodies (NCAM pAb), extracted with 1% Triton X-100, and labeled with NCAM antibodies. Note lower levels of detergent-insoluble NCAM in untreated (control), NCAM-Fc–, and NCAM pAb–treated PrP−/− versus PrP+/+ neurites. Bars, 10 μm (for A and B). (C) Diagrams show mean labeling intensity of PrP in NCAM clusters in control and NCAM-Fc– or NCAM pAb–incubated PrP+/+ neurons. (D) Diagrams show mean NCAM labeling intensity in NCAM clusters and along neurites of NCAM-Fc– or NCAM pAb–treated PrP+/+ and PrP−/− neurons. Mean values ± SEM (n > 50 neurites) are shown (for C and D). *, P < 0.05, t test.
Activation of the fyn kinase is reduced in PrP− /− mice
Accumulation of NCAM in lipid rafts is necessary for NCAM-mediated neurite outgrowth and implies activation of the fyn kinase pathway (Niethammer et al., 2002). Total levels of fyn kinase immunoprecipitable from brain homogenates and lipid rafts of PrP−/− mice were increased when compared with PrP+/+ mice (Fig. 6). However, levels of activated fyn were reduced in PrP−/− brain homogenates (ratio of activated fyn to the total fyn protein was 59.4 ± 15.3% for PrP−/− brains with PrP+/+ set to 100%) and lipid rafts (ratio of activated fyn to the total fyn protein was 26.7 ± 9.8% for PrP−/− rafts with PrP+/+ set to 100%). Because fyn forms a complex with NCAM (Beggs et al., 1997) and NCAM redistribution to lipid rafts activates NCAM-mediated fyn signaling (Niethammer et al., 2002; Bodrikov et al., 2005), reduction of activated fyn in PrP−/− brains could be due to reduction of activated fyn associated with NCAM.
Figure 6.
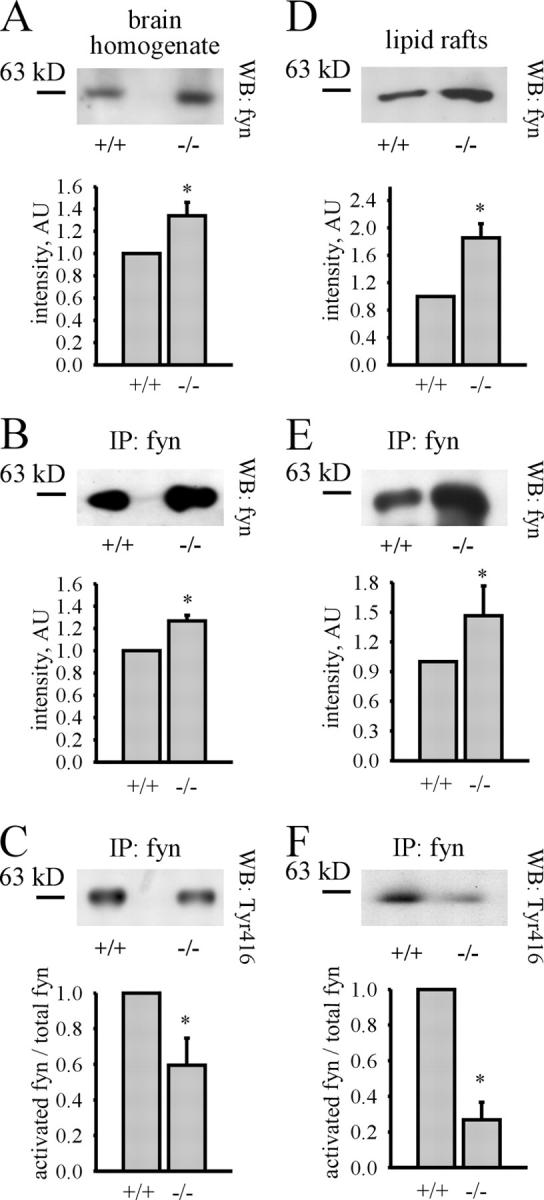
Activation of fyn kinase is reduced in PrP −/− mice. Fyn was immunoprecipitated from brain homogenates or lipid rafts of 0–4-d-old PrP+/+ and PrP−/− mice. Brain homogenates (A), lipid rafts (D), and immunoprecipitates from brain homogenates (B and C) or lipid rafts (E and F) were analyzed by immunoblotting (WB) with antibodies against fyn protein and activated Tyr416 phosphorylated fyn. Note that levels of fyn protein are increased in brain homogenates, lipid rafts, and immunoprecipitates from PrP−/− mice, whereas levels of activated fyn are decreased. Diagrams with quantitation of immunoblots are shown below. Mean values ± SEM (n = 6) are shown. AU = arbitrary units. *, P < 0.05, paired t test.
To analyze this, we studied whether redistribution of NCAM to lipid rafts in response to stimulation with PrP-Fc would affect levels of activated fyn. Indeed, application of PrP-Fc to PrP+/+ neurons increased levels of activated fyn along neurites and in NCAM clusters (Fig. 7, A and B). When PrP-Fc was applied to PrP−/− neurons, levels of activated fyn were also significantly increased along neurites and in NCAM clusters (Fig. 7, A and B), indicating that trans interactions between NCAM and PrP induce fyn activation. However, the efficacy of fyn activation was lower in PrP−/− cells (200 and 140% along neurites of stimulated PrP+/+ and PrP−/− cells, respectively, with the level of activated fyn in control cells set to 100%), suggesting that cis interactions between NCAM and PrP are also important for fyn activation. To confirm this, we analyzed activation of fyn in response to NCAM-Fc in PrP−/− neurons, thereby excluding cis interactions between NCAM and PrP. Application of NCAM-Fc increased levels of activated fyn in NCAM clusters and along neurites of PrP+/+ neurons (150% along neurites of stimulated neurons, with the level of activated fyn in control cells set to 100%; Fig. 7 B). However, in PrP−/− neurons levels of activated fyn were not changed in response to NCAM-Fc, indicating that cis interactions between NCAM and PrP are required for NCAM-mediated fyn activation (Fig. 7 B). Levels of GM1 were similar in neurites of PrP+/+ and PrP−/− neurons (Fig. 7 C), excluding that reduced activation of fyn in PrP−/− neurons was due to reduced levels of lipid rafts in PrP−/− neurites. As control, nonspecific clustering of GM1 containing lipid rafts with cholera toxin (Harder et al., 1998) did not activate fyn (Fig. 7 D). To further confirm that NCAM is the major neuronal receptor required for PrP-mediated fyn activation, we analyzed activation of fyn in response to PrP-Fc application to NCAM−/− neurons: levels of activated fyn along neurites of NCAM−/− neurons were not changed (Fig. 7 E), indicating that NCAM is required for PrP-Fc induced fyn activation. In agreement, polyclonal PrP antibodies did not activate fyn, indicating that clustering of PrP alone is insufficient to activate fyn (Fig. 7 F). Interestingly, PrP antibodies completely inhibited NCAM-Fc–induced fyn activation (Fig. 7 F), probably by interfering with cis interaction between NCAM and PrP. Finally, levels of activated fyn coimmunoprecipitated with NCAM were also approximately two times lower in PrP−/− brains when compared with PrP+/+ brains (unpublished data), despite the overall increase of NCAM expression in PrP−/− brains. We conclude that NCAM is the receptor for PrP in trans and cooperates with PrP in cis to activate fyn.
Figure 7.
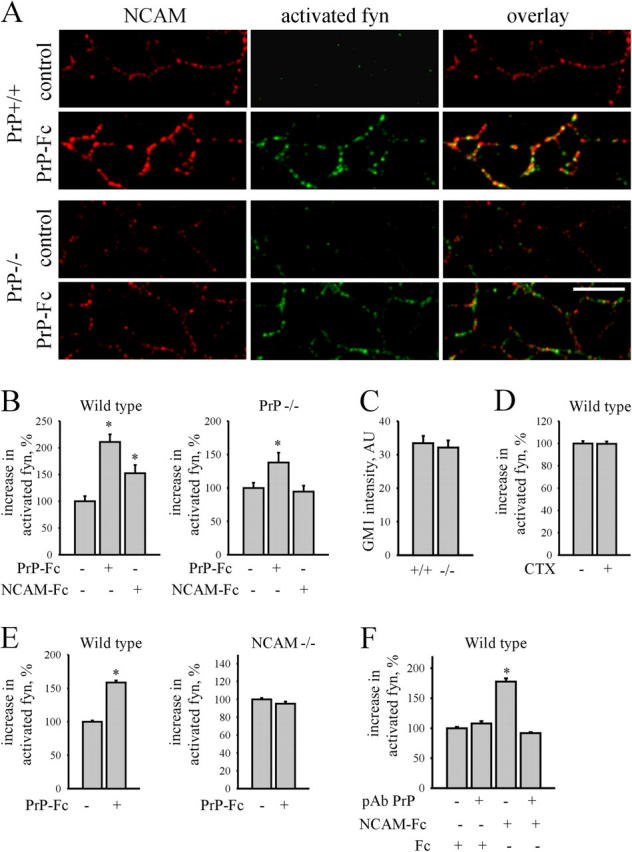
PrP-Fc activates fyn via its neuronal receptor NCAM. (A) PrP+/+ and PrP−/− neurons were incubated with PrP-Fc, extracted with 1% Triton X-100, and labeled with antibodies against NCAM and activated Tyr416 phosphorylated fyn. Note increased levels of activated fyn in NCAM clusters on PrP-Fc–treated PrP+/+ and PrP−/− neurites. Note lower levels of the activated fyn in PrP−/− neurons. Bar, 10 μm. (B) Application of PrP-Fc increased levels of activated fyn in PrP+/+ and less in PrP−/− neurons. Application of NCAM-Fc increased levels of activated fyn in PrP+/+ but not PrP−/− neurons. (C) Mean labeling intensity of GM1 along neurites of PrP+/+ and PrP−/− neurons extracted with 1% Triton X-100 is shown. (D) Aggregation of lipid rafts with cholera toxin (CTX) did not increase levels of activated fyn in PrP+/+ neurites. (E) Application of PrP-Fc increased levels of activated fyn in NCAM+/+ but not NCAM−/− neurites. (F) Application of polyclonal PrP antibodies before NCAM-Fc application blocked NCAM-Fc–induced fyn activation. (B, D–F) Diagrams show increase in mean labeling intensity of activated fyn along neurites. Mean labeling intensity of activated fyn in control neurites was taken as 100%. Mean values ± SEM (n > 50 neurites) are shown. *, P < 0.05, t test.
Coexpression of NCAM140 with PrP enhances targeting of NCAM140 to lipid rafts and fyn activation in CHO cells
To exclude that enzymes responsible for NCAM palmitoylation or fyn activation were nonspecifically affected by PrP ablation, we investigated whether PrP expression in PrP-negative CHO cells would affect NCAM140 targeting to lipid rafts and fyn activation. CHO cells were stably transfected with NCAM140 or PrP alone or cotransfected with NCAM140 and PrP. In low density CHO cell cultures and thus absence of trans interactions between NCAM and PrP, levels of NCAM140 were higher in lipid rafts from cells cotransfected with NCAM140 and PrP when compared with NCAM140-only transfected cells (Fig. 8 A), further confirming that cis interactions between NCAM140 and PrP target NCAM140 to lipid rafts. Application of PrP-Fc to NCAM140-transfected cells increased levels of NCAM140 in lipid rafts (Fig. 8 C), indicating that trans interactions between NCAM and PrP also target NCAM140 to lipid rafts. Furthermore, both types of interactions increased levels of activated fyn in the cells (Fig. 8, B and D).
Figure 8.
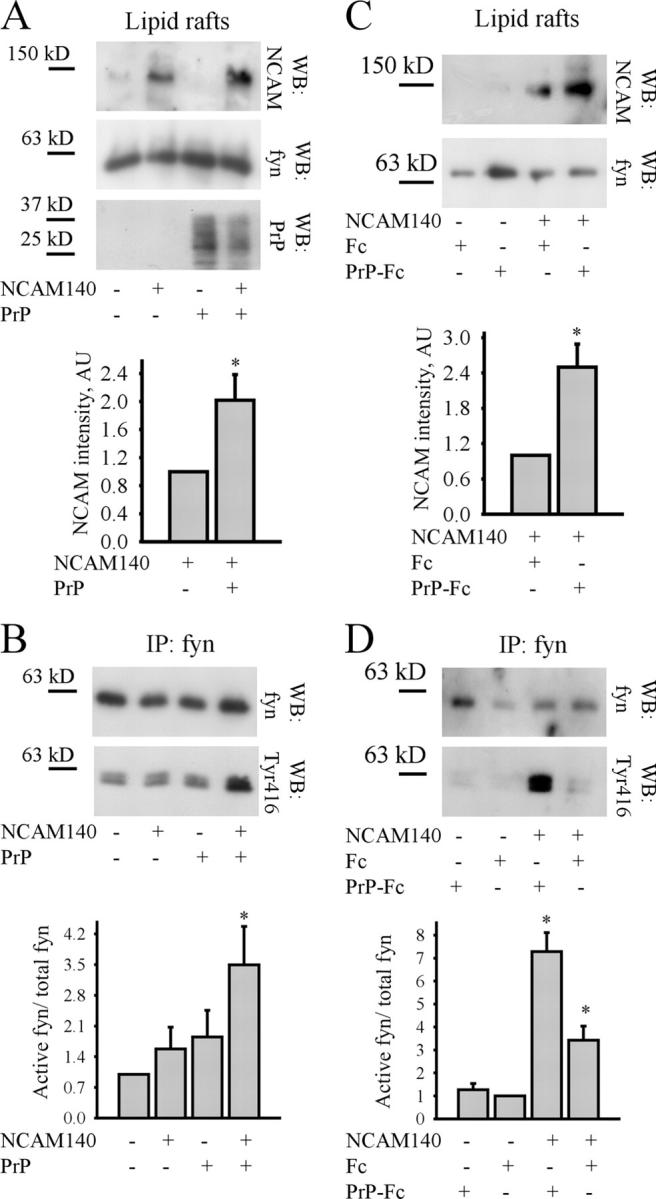
Cis and trans interactions between NCAM140 and PrP increase targeting of NCAM140 to lipid rafts and fyn activation in CHO cells. (A and B) Lipid rafts (A) or fyn immunoprecipitates from lipid rafts (B) from low density cultures of nontransfected CHO cells or CHO cells stably transfected with NCAM140, PrP, or cotransfected with NCAM140 and PrP were analyzed by immunoblotting (WB) with antibodies to NCAM, fyn protein, and Tyr416-phosphorylated fyn. Note that levels of NCAM140 and activated fyn are increased in lipid rafts from CHO cells cotransfected with NCAM140 and PrP, but not singly transfected cells. (C and D) Confluent cultures of nontransfected and NCAM140-transfected CHO cells were incubated with Fc or PrP-Fc. Lipid rafts (C) or fyn immunoprecipitates from lipid rafts (D) were analyzed by immunoblotting (WB) with antibodies to NCAM, fyn protein, and Tyr416-phosphorylated fyn. Note that levels of NCAM140 and activated fyn are increased after incubation with PrP-Fc in NCAM140-transfected cells. Slightly increased basal levels of activated fyn in control NCAM140 transfected cells compared with nontransfected cells are induced by trans interactions between NCAM molecules in these cultures (Niethammer et al., 2002). Immunolabeling for fyn (A and C) shows that lipid rafts were isolated with the same efficiency. Diagrams with quantitation of immunoblots (A–D) are shown below. Mean values ± SEM (n = 6) are shown. AU = arbitrary units. *, P < 0.05, paired t test.
NCAM interaction with PrP enhances NCAM-mediated neurite outgrowth
Redistribution of NCAM to lipid rafts in response to NCAM homophilic binding is required for NCAM-mediated neurite outgrowth (Niethammer et al., 2002; Leshchyns'ka et al., 2003). In this paradigm, substrate-coated or soluble NCAM interacts with and signals through NCAM at the neuronal cell surface. Because the binding of PrP-Fc to NCAM also redistributes NCAM lipid rafts (Fig. 4 G), we investigated whether PrP-Fc promotes NCAM-mediated neurite outgrowth. PrP-Fc was thus applied to cultured hippocampal neurons and neurite length was measured after 24 h. PrP-Fc increased neurite lengths when compared with the control group (Fig. 9 A) as previously observed (Chen et al., 2003), suggesting that PrP-Fc–induced redistribution of NCAM to lipid rafts promotes neurite outgrowth. Alternatively, clustering of GPI-anchored raft-associated proteins may also activate intracellular signaling cascades, leading to enhanced neurite outgrowth (Doherty et al., 1993). To exclude the possibility that PrP-Fc acts via clustering of PrP at cell surfaces and not via NCAM, we incubated neurons with different concentrations of polyclonal PrP antibodies, thereby clustering PrP at the cell surface. Unexpectedly, we found that PrP antibodies inhibited neurite outgrowth (Fig. 9 B), indicating that clustering of PrP is not sufficient to induce neurite outgrowth. Furthermore, it suggested that PrP antibodies inhibit cis interactions between PrP and a binding partner at the cell surface that was required for neurite outgrowth. To directly assess the role of NCAM in PrP-Fc–induced neurite outgrowth, we treated NCAM−/− neurons with PrP-Fc and found that, in contrast to NCAM+/+ neurons, NCAM−/− neurons did not respond to PrP-Fc (Fig. 9 C), confirming that NCAM is a major receptor for PrP in PrP-Fc–induced neurite outgrowth. To analyze the role of PrP-to-NCAM trans interaction in NCAM-mediated neurite outgrowth, we estimated neurite outgrowth in response to PrP-Fc in PrP−/− neurons, thereby abolishing cis interactions between NCAM and PrP. PrP-Fc enhanced neurite lengths (Fig. 9 D), indicating that trans interactions between NCAM and PrP are involved in promoting neurite outgrowth. To analyze the role of cis interactions between NCAM and PrP in NCAM-mediated neurite outgrowth, we compared neurite outgrowth in response to NCAM activation in PrP−/− neurons transfected with GFP alone or GFP together with PrP. Transfected PrP was delivered to the cell surface and partially colocalized with NCAM (unpublished data). In GFP-transfected neurons, treatment with NCAM-Fc enhanced neurite outgrowth when compared with Fc-treated cells (controls), indicating PrP-independent response to NCAM. However, PrP-transfected neurons treated with NCAM-Fc produced even longer neurites (Fig. 9 E). This increase in the NCAM-Fc–elicited response thus evolves from an NCAM-to-PrP cis interaction. Furthermore, PrP antibodies completely abolished the NCAM-Fc–induced response in wild-type neurons (Fig. 9 F), probably by interfering with cis interactions between NCAM and PrP at the cell surface and with fyn activation (Fig. 7). The combined observations indicate that NCAM is a major neuronal receptor for PrP presented in a trans fashion. We also conclude that cis interactions between NCAM and PrP at the neuronal cell surface enhance NCAM-induced neurite outgrowth when NCAM is presented to neurons in a trans fashion.
Figure 9.

NCAM-mediated neurite outgrowth depends on PrP. (A and B) Wild-type hippocampal neurons were incubated with PrP-Fc (A) or polyclonal PrP antibodies (B). Note enhanced neurite outgrowth after PrP-Fc application and reduced neurite outgrowth after incubation with polyclonal PrP antibodies. (C and D) NCAM−/− (C) or PrP−/− (D) neurons were incubated with PrP-Fc. PrP-Fc increased neurite lengths in PrP−/− but not NCAM−/− neurons. (E) Neurons from PrP−/− mice transfected with GFP alone or cotransfected with GFP and PrP were incubated with NCAM-Fc after transfection. NCAM-Fc increased neurite lengths of GFP-transfected neurons. NCAM-Fc–stimulated neurite outgrowth was further increased in neurons cotransfected with PrP. (F) PrP+/+ neurons were incubated with NCAM-Fc and NCAM-Fc together with polyclonal PrP antibodies. Note inhibition of NCAM-Fc–induced neurite outgrowth after incubation with PrP antibodies. Lengths of longest neurites were measured and mean values ± SEM are shown (n > 150 neurons, *, P < 0.05, t test, for A–F).
Discussion
In this study we have shown that PrP and NCAM are not only associated with each other at the surface of hippocampal neurons, but also directly interact with each other—features that have not been described previously. We also showed that the GPI-linked PrP that cannot interact directly with intracellular signaling pathways recruits to and stabilizes the transmembrane NCAM isoforms in lipid-rich microdomains to activate fyn and promote NCAM-mediated neurite outgrowth by cis and trans interactions.
Transmembrane recognition molecules are often segregated from their signaling cascades by colocalizing receptors and their downstream effectors to distinct membrane subdomains, such as lipid rafts. The mechanisms that collect receptors to their signaling platforms have only started to emerge. We show that NCAM uses its PrP-guided enrichment in lipid rafts to activate fyn, which becomes associated with lipid rafts rapidly after synthesis (van't Hof and Resh, 1997; Filipp et al., 2003). Interestingly, although the intracellular domain of NCAM does not contain sequences known to activate fyn, it directly associates with the receptor type protein phosphatase α (RPTPα), a fyn activator. At resting conditions, when not stimulated by cis or trans interactions, NCAM and RPTPα are segregated from fyn, which localizes to rafts. In response to NCAM activation NCAM binds RPTPα and both molecules are recruited to lipid rafts where RPTPα activates fyn (Bodrikov et al., 2005), a process that is regulated by PrP (Fig. 10). It is important in this respect that, in contrast to other GPI-anchored molecules such as Thy-1 (Doherty et al., 1993), clustering of PrP alone with PrP antibodies is not sufficient to induce neurite outgrowth but rather inhibits it by interfering with cis interactions between NCAM and PrP. Because PrP-Fc–induced fyn activation and neurite outgrowth are largely inhibited in NCAM−/− cells, the most plausible explanation is that NCAM is a major PrP receptor. These observations merge two previously separate venues of investigation, namely the requirement of NCAM to be localized to lipid rafts to induce neurite outgrowth via fyn activation together with FGF receptor activation (Niethammer et al., 2002) and the proposed activation of fyn kinase (Mouillet-Richard et al., 2000) by PrP, which binds to an unknown surface receptor (Chen et al., 2003). Whether NCAM cooperates with the laminin receptor, which is another surface receptor for PrP (Gauczynski et al., 2001), or whether PrP–laminin receptor interactions have distinct functions in neurite outgrowth remains to be investigated.
Figure 10.

Proposed model of NCAM-to-PrP interactions in NCAM/PrP-mediated neurite outgrowth. (A) In NCAM+/+ (wt) neurons, at resting conditions PrP accumulates in lipid rafts enriched in fyn. NCAM, which binds to RPTPα outside of lipid rafts (omitted for clarity) and activates fyn (Bodrikov et al., 2005), is largely excluded from lipid rafts. Application of bivalent NCAM-Fc or multivalent NCAM antibodies to neurons induces clustering of NCAM, resulting in its palmitoylation that redistributes NCAM to lipid rafts (Niethammer et al., 2002). In lipid rafts, cis interactions between NCAM and PrP further recruit and stabilize NCAM in lipid microdomains activating fyn via NCAM, and finally resulting in neurite outgrowth that is enhanced over neurite outgrowth in the absence of NCAM antibodies or NCAM-Fc. (B) Application of bivalent PrP-Fc to NCAM+/+ (wt) neurons to mimic trans interactions between NCAM at the cell surface and PrP on adjacent membranes and in the ECM of the brain also induces clustering of NCAM, favoring its redistribution to lipid rafts. Moreover, PrP-Fc may directly link NCAM to lipid-enriched microdomains due to the interactions between PrP-Fc and lipids (Walmsley et al., 2003). NCAM redistribution to lipid rafts results in fyn activation via NCAM and in enhanced neurite outgrowth. (C) In NCAM−/− neurons, application of PrP-Fc is ineffective because PrP-Fc cannot act through its neuronal receptor NCAM and thus the fyn-activating signal is lost, resulting in neurite outgrowth that is not different from neurite outgrowth in the absence of PrP. (D) Application of PrP-Fc to PrP−/− neurons induces clustering of NCAM and bridging it to lipid rafts favoring NCAM redistribution to lipid rafts, fyn activation, and neurite outgrowth (similarly to B). (E) Application of NCAM-Fc or NCAM antibodies to PrP−/− neurons induces clustering of NCAM and its redistribution to lipid rafts via palmitoylation of the NCAM intracellular domain. However, NCAM redistribution to lipid rafts in PrP−/− neurons is reduced because NCAM/PrP cis interactions stabilizing NCAM in lipid rafts are lost. This results in reduced levels of fyn activation and neurite outgrowth when compared with PrP+/+ neurons. (F) PrP antibodies applied to neurons induce clustering of PrP, but not fyn activation and neurite outgrowth induction because PrP does not have a fyn-activating signal. PrP antibodies block cis interactions between PrP and NCAM, thereby inhibiting NCAM-mediated neurite outgrowth.
Remarkably, not only binding of NCAM to PrP in the neuronal plasma membrane (cis interaction), but also binding of NCAM to PrP in trans interaction, redistributes NCAM to lipid rafts. It is thus interesting that besides its lipid domain–targeting GPI anchor, PrP may associate in trans interaction with lipid rafts via its ectodomain (Walmsley et al., 2003) and thereby may additionally recruit NCAM to lipid rafts. It is also conceivable that NCAM uses other signal transduction pathways independent of PrP and PrP's ability to recruit NCAM to lipid rafts for signal transduction. Because abnormalities in PrP−/− mice have been described as undetectable or mild, although closer scrutiny has identified abnormalities, it is conceivable that triggered palmitoylation of NCAM and/or recruitment of NCAM to other signaling platforms may compensate for some PrP functions.
Functional interactions of PrP with its binding partner(s) have been suggested previously (Telling et al., 1995; Shmerling et al., 1998): a cis- and/or trans-interacting PrP activates an unknown binding partner competing with the dominant-negative mutant of PrP truncated at the amino terminus, leading to ataxia and cerebellar lesions (Shmerling et al., 1998). We have now identified NCAM as binding partner for PrP that can cooperate with PrP in neurite outgrowth. It is likely that cooperation between these molecules also occurs in the adult. It is thus noteworthy that both molecules modify synaptic activity (Collinge et al., 1994; Cremer et al., 1994; Luthi et al., 1994; Mallucci et al., 2002). Interestingly, defects in NCAM- and PrP-dependent regulation of synaptic activity may not only be due to developmental abnormalities, but are seen in mice conditionally ablated for NCAM and PrP expression at a juvenile state (Mallucci et al., 2002; Bukalo et al., 2004). Furthermore, mutations in PrP in both humans and mice lead to abnormal sleep patterns, resulting in fatal familial insomnia in humans (Gambetti et al., 2003). Likewise, NCAM is involved in regulation of circadian body functions by entraining glutamatergic activity of the suprachiasmatic nuclei to the solar day in photic and nonphotic settings (Kleene and Schachner, 2004). These finding are remarkable in view of PrP and NCAM signaling through fyn, which is also implicated in synaptic functions (Grant et al., 1992; Kojima et al., 1997).
Finally, consequences of the trans and cis interactions between NCAM and PrP for transmissible and nontransmissible prion diseases, leading to infectious propagation of the mutation with its loss-of-function or gain-of-function consequences, should be viewed in the context of PrP interacting heterophilically with other molecules, such as NCAM. Interestingly, the incubation period of the scrapie conformer of prion protein (PrPSc) is not altered in NCAM−/− mice compared with NCAM+/+ mice, suggesting that NCAM does not affect PrPSc formation (Schmitt-Ulms et al., 2001). Also, it has been excluded that neurodegeneration occurs because of PrP deficiency (Mallucci et al., 2002). It is thus conceivable that interactions between PrP and NCAM are reduced by accumulation of PrPSc in the diseased nervous system. A reduced association between PrP and NCAM could also be caused by application of PrP antibodies that trigger rapid and extensive apoptosis in hippocampal and cerebellar neurons in vivo (Solforosi et al., 2004). Similarly, we found that application of PrP antibodies abrogates NCAM-induced neurite outgrowth. It is tempting to speculate that interference with NCAM-mediated signaling in the diseased brain may favor cell death and inhibit synaptic plasticity-related neuritogenesis. Because NCAM, on the one hand, and PrP, on the other, interact with different sets of cell surface and ECM molecules, the interplay of these interactions during development and in the adult brain will be the target of further investigations.
Materials and methods
Antibodies
Rabbit pAbs against the extracellular domain of NCAM (Niethammer et al., 2002) were used in immunoprecipitation, immunoblotting, and immunocytochemical experiments and rat mAbs H28 against mouse NCAM (Gennarini et al., 1984) were used in immunocytochemical experiments. Both antibodies react with the three major isoforms of NCAM. Hybridoma clone H28 was provided by Dr. Christo Goridis (CNRS UMR 8542, Paris, France). Mouse mAbs 8H4 and rabbit pAbs 340 against PrP were obtained from Dr. Man Sun Sy (Case Western Reserve University, Cleveland, OH); mouse mAbs against PI(4,5)P2 were from Dr. Kiyoko Fukami (University of Tokyo, Tokyo, Japan). Rat mAbs 555 against L1 were used in the ELISA binding assay and immunoblotting (Appel et al., 1993). The mAbs 735 against PSA have been described elsewhere (Frosch et al., 1985). Goat pAbs m20 against PrP and rabbit pAbs and mouse mAbs against fyn protein were from Santa Cruz Biotechnology, Inc. Rabbit pAbs against phosphorylated Tyr416 recognizing activated fyn were from Cell Signaling Technology. Cholera toxin B subunit labeled with fluorescein was purchased from Sigma-Aldrich. Secondary antibodies against rabbit, rat, and mouse Ig coupled to Cy2, Cy3, or Cy5 were from Dianova.
Animals
PrP−/− mice (Bueler et al., 1992; Weiss et al., 1997; Chen et al., 2003) were provided by Dr. Martin H. Groschup (Institute for Novel and Emerging Infectious Diseases, Greifswald, Germany) and Dr. Charles Weissmann (MRC Prion Unit, Institute of Neurology, London, UK). NCAM−/− mice were provided by Dr. Harold Cremer (Cremer et al., 1994). Both mutants were inbred for at least nine generations onto the C57BL/6J background. In the majority of experiments age-matched wild-type and mutant mice were used. Observed differences were also verified in experiments with wild-type and mutant littermates obtained from heterozygous breeding pairs that showed the same effects. In particular, levels of NCAM140 and NCAM180 in lipid rafts from PrP−/− littermates were 34.8 ± 2.4% with PrP+/+ values set to 100%. Ratio of activated fyn to total fyn protein in the brain homogenates was 63.6 ± 6.7% for PrP−/− littermates with PrP+/+ values set to 100%.
Culture and transfection of hippocampal neurons
Cultures of hippocampal neurons were prepared from 1–3-d-old C57BL/6J mice or from NCAM−/− or PrP−/− mice. For immunocytochemical experiments, neurons were grown in 10% horse serum on glass coverslips coated with 100 μg/ml poly-l-lysine in conjunction with 20 μg/ml laminin (Sytnyk et al., 2002). In experiments with stimulation of live neurons, 4 μg/ml NCAM-Fc or 2 μg/ml PrP-Fc was applied for 20 min. Control neurons were treated with 8 μg/ml human Fc. NCAM pAbs were applied to live neurons for 15 min followed by secondary antibodies applied for 5 min, all in a CO2 incubator. Control neurons were treated with 10 μg/ml nonimmune rabbit IgG. Neurons were transfected with GFP (CLONTECH Laboratories, Inc.) or mouse PrP (Lehmann and Harris, 1995) 6 h after plating using LipofectAMINE 2000 (Invitrogen) according to the manufacturer's instructions. Neurite outgrowth was assayed as described previously (Niethammer et al., 2002). In brief, neurons were incubated for 24 h with 2 μg/ml PrP-Fc or 4 μg/ml NCAM-Fc, with rabbit pAbs 340 against 10 μg/ml or 30 μg/ml PrP, or with goat pAbs m20 against 2 μg/ml PrP, and lengths of the longest neurites were measured (Chen et al., 1999). Control neurons were either treated with 8 μg/ml human Fc or 10 μg/ml nonimmune rabbit or goat IgG.
Detergent extraction of cultured neurons
Cells washed in PBS, pH 7.3, were incubated for 1 min in cold microtubule-stabilizing buffer (MSB) (2 mM MgCl2, 10 mM EGTA, and 60 mM Pipes, pH 7.0) and extracted 8 min on ice with 1% Triton X-100 in MSB as described previously (Ledesma et al., 1998). After washing with PBS, cells were fixed with cold 4% formaldehyde in PBS.
Immunofluorescence labeling
Indirect immunofluorescence staining was performed as described previously (Sytnyk et al., 2002; Leshchyns'ka et al., 2003). Primary antibodies were detected with corresponding secondary antibodies coupled to Cy2, Cy3, or Cy5 fluorochrome (Dianova). Clustering of NCAM was induced by incubating live cells for 15 min (5% CO2 at 37°C) with NCAM antibodies, and was visualized with secondary antibodies applied for 5 min (Leshchyns'ka et al., 2003). Coverslips were embedded in Aqua-Poly/Mount (Polysciences, Inc.). Images were acquired at RT using a confocal laser scanning microscope (LSM510; Carl Zeiss MicroImaging, Inc.), LSM510 software (version 3; Carl Zeiss MicroImaging, Inc.), and the oil Plan-Neofluar 40× objective (NA 1.3; Carl Zeiss MicroImaging, Inc.) at 3× digital zoom. Contrast and brightness of the images were further adjusted in Corel Photo-Paint 9 (Corel Corporation).
Immunofluorescence quantification
Colocalization analysis was performed as described previously (Leshchyns'ka et al., 2003). Images taken for quantification were acquired with the same settings and were not further manipulated. We defined an NCAM cluster as an accumulation of NCAM labeling with a mean intensity at least 30% higher than background. NCAM clusters were outlined using the threshold function of the Scion Image software (Scion Corporation). Within the outlined areas the mean intensities of NCAM and PrP, GM1, PI(4,5)P2, or activated fyn labeling associated with NCAM clusters were measured. The same threshold was used for all groups within one experiment. To determine the mean intensity of the labeling along neurites, neurites were manually outlined and the mean fluorescence intensity along the neurites was measured using Scion Image software. Colocalization profiles were analyzed using ImageJ software (National Institutes of Health, Bethesda, MD). Correlation coefficients were calculated using Excel software (Microsoft).
Protein ligand binding assay
PrP-Fc containing the extracellular domain of the mouse PrP in fusion with the Fc portion of human IgG or PrP-AP containing the extracellular domain of the mouse PrP in fusion with AP was produced as described previously (Chen et al., 2003). NCAM and L1 were purified from adult mouse brain homogenate as described previously (Rathjen and Schachner, 1984; Pollerberg et al., 1985). 5 μg/ml PrP-Fc or 5 μg/ml PrP-AP was immobilized overnight on 96-well polyvinyl chloride plates (Nunc) in PBS. Wells were then blocked for 1.5 h with PBS containing 3% BSA and incubated for 1.5 h at RT with NCAM, L1, or NCAM-Fc (0–2 μg/ml) diluted in PBS containing 0.05% Tween 20 (PBS-T) and 3% BSA. Plates were washed 3× with PBS-T and incubated for 1.5 h at RT with pAbs against NCAM, mAbs against L1, or pAbs against human Fc-tag (Jackson ImmunoResearch Laboratories) in PBS-T containing 3% BSA. After washing with PBS-T, wells were incubated with peroxidase-coupled secondary antibodies in PBS-T containing 3% BSA, washed 3×, and developed with 0.1% ABTS (Roche) in 100 mM acetate buffer, pH 5.0. The reaction was stopped with 100 mM sodium fluoride. OD was measured at 405 nm.
Culture and transfection of CHO cells
CHO cells were maintained in Glasgow modified Eagle's medium containing 10% FCS. Cells were stably transfected using LipofectAMINE Plus reagent (Invitrogen), following the manufacturer's instructions. NCAM140 expression vector was provided by Dr. Patricia Maness (University of North Carolina, Chapel Hill, NC). 24 h after transfection, 1 mg/ml G418 was added to the culture medium, which was replaced with fresh medium containing 1 mg/ml G418 every 3 d. After 3 wk in culture, single colonies were selected for subsequent screening. In experiments with stimulation, 2 μg/ml PrP-Fc or 8 μg/ml human Fc were applied to live CHO cells for 20 min in a CO2 incubator.
Subcellular fractionation and isolation of lipid-enriched microdomains
Rafts were prepared from crude membrane and growth cone fractions from 0–4-d-old mice as described previously (Leshchyns'ka et al., 2003). The same protocol was used to isolate lipid rafts from CHO cells.
Chemical cross-linking
Crude membrane fractions or lipid rafts from 0–4-d-old mice were incubated with 0.2 mM BS3 cross-linker (Pierce Chemical Co.) in 20 mM sodium phosphate buffer (pH 7.6) for 30 min at RT. The cross-linking reaction was stopped by the addition of 1 M Tris-HCl (pH 7.6) to a final concentration of 100 mM for 10 min. The cross-linked fractions were either separated by gel electrophoresis and then immunoblotted or were used for coimmunoprecipitation. Chemical cross-linking in brain homogenates with PrP-Fc as bait was performed using Sulfo-SBED biotin label transfer reagent (Pierce Chemical Co.), following the manufacturer's instructions.
Coimmunoprecipitation
Homogenates were prepared from the brains of 0–4-d-old mice in 50 mM Tris-HCl buffer (pH 7.5), containing 0.32 M sucrose, 1 mM CaCl2, 1 mM MgCl2, and 1 mM NaHCO3. Samples containing 1 mg of protein were lysed for 30 min in 50 mM Tris-HCl buffer (pH 7.5), containing 150 mM NaCl, 0.5% Triton X-100, 1% β-octyl-d-glucopyranoside, 1 mM sodium fluoride, 2 mM NaVO4, 0.1 mM PMSF, and EDTA-free protease inhibitor cocktail (Roche). The lysis buffer containing this combination of detergents completely solubilizes lipid rafts and has been used in a number of studies involving coimmunoprecipitation of lipid raft components (Trupp et al., 1999; Paratcha et al., 2001). Samples were then centrifuged for 15 min at 20,000 g and 4°C. Supernatants were cleared with protein A/G-agarose beads (Santa Cruz Biotechnology, Inc.) (for 3 h at 4°C) and incubated with pAbs against NCAM, p59fyn, or nonimmune rabbit Ig (overnight at 4°C), followed by precipitation with protein A/G-agarose beads (for 3 h at 4°C). The beads were washed 3× with RIPA buffer, once with PBS, and analyzed by immunoblotting (Leshchyns'ka et al., 2003). The approximate percentage of NCAM molecules bound to PrP in the total brain membranes (P) was quantified as: P = (NCAMIP/NCAMstart) × (PrPstart/PrPIP) × 100%, where NCAMstart and NCAMIP are labeling intensities of NCAM in the total brain membranes and PrP immunoprecipitates, respectively, and PrPstart and PrPIP are labeling intensities of PrP in the total brain membranes and PrP immunoprecipitates, respectively.
Gel electrophoresis and immunoblotting
Proteins were separated by 8% SDS-PAGE and electroblotted onto nitrocellulose transfer membrane (PROTRAN; Schleicher & Schuell) overnight at 5 mA. Immunoblots were incubated with appropriate primary antibodies followed by incubation with peroxidase-labeled secondary antibodies and were visualized using SuperSignal West Pico reagents (Pierce Chemical Co.) on BIOMAX film (Sigma-Aldrich). Molecular weight markers were prestained protein standards from Bio-Rad Laboratories. Chemiluminescence was quantified using TINA 2.09 software (University of Manchester, Manchester, UK). To allow quantitative comparisons of chemiluminescence between the lanes, the same amounts of total protein or equal amounts of immunoprecipitates were loaded in each lane and the intensity observed for the wild type was used for normalization. All preparations (brain homogenates, immunoprecipitations, or lipid rafts) were performed three times and at least two Western blots were performed with an individual sample (n ≥ 6). In each experiment, when PrP+/+ and PrP−/− brains were compared, PrP+/+ values were set to 1, and intensities for PrP−/− brains were normalized to PrP+/+ values. Values of all experiments were used to calculate mean values and SEM.
Acknowledgments
We thank Charles Weissmann and Martin H. Groschup for PrP−/− mice and Harold Cremer for NCAM-deficient mice; Man Sun Sy for antibodies against PrP; Kiyoko Fukami for antibodies against PI(4,5)P2; Christo Goridis for the H28 hybridoma clone; and Sylvain Lehmann and Patricia Maness for PrP and NCAM140 expression vectors. We are grateful to Achim Dahlmann and Eva Kronberg for genotyping and animal care, and to Suzhen Chen, Gabriele Loers, and Vsevolod Bodrikov for production of PrP-Fc, L1-Fc, and NCAM-Fc.
This work was supported by a European community grant (QLRT-2000-02353, to M. Schachner) and by Bundesministerium für Bildung und Forschung (01 KO 0204 to M. Schachner), Deutsche Forschungsgemeinschaft (to I. Leshchyns'ka, V. Sytnyk, and M. Schachner), and Zonta International, Hamburg-Alster (to I. Leshchyns'ka).
A. Santuccione, V. Sytnyk, and I. Leshchyns'ka contributed equally to this paper.
Abbreviations used in this paper: GPI, glycosylphosphatidylinositol; NCAM, neural cell adhesion molecule; PrP, prion protein; PSA, polysialic acid; RPTPα, receptor type protein phosphatase α.
References
- Appel, F., J. Holm, J.F. Conscience, and M. Schachner. 1993. Several extracellular domains of the neural cell adhesion molecule L1 are involved in neurite outgrowth and cell body adhesion. J. Neurosci. 13:4764–4775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhamidipati, A., S.A. Lewis, and N.J. Cowan. 2000. 149:1087-96. ADP ribosylation factor-like protein 2 (Arl2) regulates the interaction of tubulin-folding cofactor D with native tubulin. J. Cell Biol. 149:1087–1096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beggs, H.E., P. Soriano, and P.F. Maness. 1994. NCAM-dependent neurite outgrowth is inhibited in neurons from Fyn-minus mice. J. Cell Biol. 127:825–833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beggs, H.E., S.C. Baragona, J.J. Hemperly, and P.F. Maness. 1997. NCAM140 interacts with the focal adhesion kinase p125(fak) and the SRC-related tyrosine kinase p59(fyn). J. Biol. Chem. 272:8310–8319. [DOI] [PubMed] [Google Scholar]
- Bodrikov, V., I. Leshchyns'ka, V. Sytnyk, J. Overvoorde, J. den Hertog, and M. Schachner. 2005. RPTPα is essential for NCAM-mediated p59fyn activation and neurite elongation. J. Cell Biol. 168:127–139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bueler, H., M. Fischer, Y. Lang, H. Bluethmann, H.P. Lipp, S.J. DeArmond, S.B. Prusiner, M. Aguet, and C. Weissmann. 1992. Normal development and behaviour of mice lacking the neuronal cell-surface PrP protein. Nature. 356:577–582. [DOI] [PubMed] [Google Scholar]
- Bukalo, O., N. Fentrop, A.Y. Lee, B. Salmen, J.W. Law, C.T. Wotjak, M. Schweizer, A. Dityatev, and M. Schachner. 2004. Conditional ablation of the neural cell adhesion molecule reduces precision of spatial learning, long-term potentiation, and depression in the CA1 subfield of mouse hippocampus. J. Neurosci. 24:1565–1577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen, S., N. Mantei, L. Dong, and M. Schachner. 1999. Prevention of neuronal cell death by neural adhesion molecules L1 and CHL1. J. Neurobiol. 38:428–439. [DOI] [PubMed] [Google Scholar]
- Chen, S., A. Mange, L. Dong, S. Lehmann, and M. Schachner. 2003. Prion protein as trans-interacting partner for neurons is involved in neurite outgrowth and neuronal survival. Mol. Cell. Neurosci. 22:227–233. [DOI] [PubMed] [Google Scholar]
- Collinge, J. 1997. Human prion diseases and bovine spongiform encephalopathy (BSE). Hum. Mol. Genet. 6:1699–1705. [DOI] [PubMed] [Google Scholar]
- Collinge, J., M.A. Whittington, K.C. Sidle, C.J. Smith, M.S. Palmer, A.R. Clarke, and J.G. Jefferys. 1994. Prion protein is necessary for normal synaptic function. Nature. 370:295–297. [DOI] [PubMed] [Google Scholar]
- Cremer, H., R. Lange, A. Christoph, M. Plomann, G. Vopper, J. Roes, R. Brown, S. Baldwin, P. Kraemer, S. Scheff, et al. 1994. Inactivation of the N-CAM gene in mice results in size reduction of the olfactory bulb and deficits in spatial learning. Nature. 367:455–459. [DOI] [PubMed] [Google Scholar]
- Culligan, K., L. Glover, P. Dowling, and K. Ohlendieck. 2001. Brain dystrophin-glycoprotein complex: persistent expression of β-dystroglycan, impaired oligomerization of Dp71 and up-regulation of utrophins in animal models of muscular dystrophy. BMC Cell Biol. 2:2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doherty, P., A. Singh, G. Rimon, S.R. Bolsover, and F.S. Walsh. 1993. Thy-1 antibody-triggered neurite outgrowth requires an influx of calcium into neurons via N- and L-type calcium channels. J. Cell Biol. 122:181–189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Filipp, D., J. Zhang, B.L. Leung, A. Shaw, S.D. Levin, A. Veillette, and M. Julius. 2003. Regulation of Fyn through translocation of activated Lck into lipid rafts. J. Exp. Med. 197:1221–1227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frosch, M., I. Gorgen, G.J. Boulnois, K.N. Timmis, and D. Bitter-Suermann. 1985. NZB mouse system for production of monoclonal antibodies to weak bacterial antigens: isolation of an IgG antibody to the polysaccharide capsule of Escherichia coli K1 and group B meningiococci. Proc. Natl. Acad. Sci. USA. 82:1194–1198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gambetti, P., Q. Kong, W. Zou, P. Parchi, and S.G. Chen. 2003. Sporadic and familial CJD: classification and characterisation. Br. Med. Bull. 66:213–239. [DOI] [PubMed] [Google Scholar]
- Gauczynski, S., J.M. Peyrin, S. Haik, C. Leucht, C. Hundt, R. Rieger, S. Krasemann, J.P. Deslys, D. Dormont, C.I. Lasmezas, and S. Weiss. 2001. The 37-kDa/67-kDa laminin receptor acts as the cell-surface receptor for the cellular prion protein. EMBO J. 20:5863–5875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gennarini, G., M. Hirn, H. Deagostini-Bazin, and C. Goridis. 1984. Studies on the transmembrane disposition of the neural cell adhesion molecule N-CAM. The use of liposome-inserted radioiodinated N-CAM to study its transbilayer orientation. Eur. J. Biochem. 142:65–73. [DOI] [PubMed] [Google Scholar]
- Gorodinsky, A., and D.A. Harris. 1995. Glycolipid-anchored proteins in neuroblastoma cells form detergent-resistant complexes without caveolin. J. Cell Biol. 129:619–627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graner, E., A.F. Mercadante, S.M. Zanata, O.V. Forlenza, A.L. Cabral, S.S. Veiga, M.A. Juliano, R. Roesler, R. Walz, A. Minetti, et al. 2000. Cellular prion protein binds laminin and mediates neuritogenesis. Brain Res. Mol. Brain Res. 76:85–92. [DOI] [PubMed] [Google Scholar]
- Grant, S.G., T.J. O'Dell, K.A. Karl, P.L. Stein, P. Soriano, and E.R. Kandel. 1992. Impaired long-term potentiation, spatial learning, and hippocampal development in fyn mutant mice. Science 258:1903–1910. [DOI] [PubMed] [Google Scholar]
- Harder, T., P. Scheiffele, P. Verkade, and K. Simons. 1998. Lipid domain structure of the plasma membrane revealed by patching of membrane components. J. Cell Biol. 141:929–942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsiao, K., H.F. Baker, T.J. Crow, M. Poulter, F. Owen, J.D. Terwilliger, D. Westaway, J. Ott, and S.B. Prusiner. 1989. Linkage of a prion protein missense variant to Gerstmann-Straussler syndrome. Nature. 338:342–345. [DOI] [PubMed] [Google Scholar]
- Kaykas, A., J. Yang-Snyder, M. Heroux, K.V. Shah, M. Bouvier, and R.T. Moon. 2004. Mutant Frizzled 4 associated with vitreoretinopathy traps wild-type Frizzled in the endoplasmic reticulum by oligomerization. Nat. Cell Biol. 6:52–58. [DOI] [PubMed] [Google Scholar]
- Kleene, R., and M. Schachner. 2004. Glycans and neural cell interactions. Nat. Rev. Neurosci. 5:195–208. [DOI] [PubMed] [Google Scholar]
- Kojima, N., J. Wang, I.M. Mansuy, S.G. Grant, M. Mayford, and E.R. Kandel. 1997. Rescuing impairment of long-term potentiation in fyn-deficient mice by introducing Fyn transgene. Proc. Natl. Acad. Sci. USA. 94:4761–4765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laux, T., K. Fukami, M. Thelen, T. Golub, D. Frey, and P. Caroni. 2000. GAP43, MARCKS, and CAP23 modulate PI(4,5)P2 at plasmalemmal rafts, and regulate cell cortex actin dynamics through a common mechanism. J. Cell Biol. 149:1455–1472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ledesma, M.D., K. Simons, and C.G. Dotti. 1998. Neuronal polarity: essential role of protein-lipid complexes in axonal sorting. Proc. Natl. Acad. Sci. USA. 95:3966–3971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehmann, S., and D.A. Harris. 1995. A mutant prion protein displays an aberrant membrane association when expressed in cultured cells. J. Biol. Chem. 270:24589–24597. [DOI] [PubMed] [Google Scholar]
- Leshchyns'ka, I., V. Sytnyk, J.S. Morrow, and M. Schachner. 2003. Neural cell adhesion molecule (NCAM) association with PKCβ2 via βI spectrin is implicated in NCAM-mediated neurite outgrowth. J. Cell Biol. 161:625–639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luthi, A., J.P. Laurent, A. Figurov, D. Muller, and M. Schachner. 1994. Hippocampal long-term potentiation and neural cell adhesion molecules L1 and NCAM. Nature. 372:777–779. [DOI] [PubMed] [Google Scholar]
- Madore, N., K.L. Smith, C.H. Graham, A. Jen, K. Brady, S. Hall, and R. Morris. 1999. Functionally different GPI proteins are organized in different domains on the neuronal surface. EMBO J. 18:6917–6926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mallucci, G.R., S. Ratte, E.A. Asante, J. Linehan, I. Gowland, J.G. Jefferys, and J. Collinge. 2002. Post-natal knockout of prion protein alters hippocampal CA1 properties, but does not result in neurodegeneration. EMBO J. 21:202–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milhavet, O., and S. Lehmann. 2002. Oxidative stress and the prion protein in transmissible spongiform encephalopathies. Brain Res. Brain Res. Rev. 38:328–339. [DOI] [PubMed] [Google Scholar]
- Mouillet-Richard, S., M. Ermonval, C. Chebassier, J.L. Laplanche, S. Lehmann, J.M. Launay, and O. Kellermann. 2000. Signal transduction through prion protein. Science. 289:1925–1928. [DOI] [PubMed] [Google Scholar]
- Nakai, Y., and H. Kamiguchi. 2002. Migration of nerve growth cones requires detergent-resistant membranes in a spatially defined and substrate-dependent manner. J. Cell Biol. 159:1097–1108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niethammer, P., M. Delling, V. Sytnyk, A. Dityatev, K. Fukami, and M. Schachner. 2002. Cosignaling of NCAM via lipid rafts and the FGF receptor is required for neuritogenesis. J. Cell Biol. 157:521–532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paratcha, G., F. Ledda, L. Baars, M. Coulpier, V. Besset, J. Anders, R. Scott, and C.F. Ibanez. 2001. Released GFRα1 potentiates downstream signaling, neuronal survival, and differentiation via a novel mechanism of recruitment of c-Ret to lipid rafts. Neuron. 29:171–184. [DOI] [PubMed] [Google Scholar]
- Pollerberg, E.G., R. Sadoul, C. Goridis, and M. Schachner. 1985. Selective expression of the 180-kD component of the neural cell adhesion molecule N-CAM during development. J. Cell Biol. 101:1921–1929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prusiner, S.B. 1998. Prions. Proc. Natl. Acad. Sci. USA. 95:13363–13383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rathjen, F.G., and M. Schachner. 1984. Immunocytological and biochemical characterization of a new neuronal cell surface component (L1 antigen) which is involved in cell adhesion. EMBO J. 3:1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rieger, R., F. Edenhofer, C.I. Lasmezas, and S. Weiss. 1997. The human 37-kDa laminin receptor precursor interacts with the prion protein in eukaryotic cells. Nat. Med. 3:1383–1388. [DOI] [PubMed] [Google Scholar]
- Schmitt-Ulms, G., G. Legname, M.A. Baldwin, H.L. Ball, N. Bradon, P.J. Bosque, K.L. Crossin, G.M. Edelman, S.J. DeArmond, F.E. Cohen, and S.B. Prusiner. 2001. Binding of neural cell adhesion molecules (N-CAMs) to the cellular prion protein. J. Mol. Biol. 314:1209–1225. [DOI] [PubMed] [Google Scholar]
- Shmerling, D., I. Hegyi, M. Fischer, T. Blattler, S. Brandner, J. Gotz, T. Rulicke, E. Flechsig, A. Cozzio, C. von Mering, et al. 1998. Expression of amino-terminally truncated PrP in the mouse leading to ataxia and specific cerebellar lesions. Cell. 93:203–214. [DOI] [PubMed] [Google Scholar]
- Solforosi, L., J.R. Criado, D.B. McGavern, S. Wirz, M. Sanchez-Alavez, S. Sugama, L.A. DeGiorgio, B.T. Volpe, E. Wiseman, G. Abalos, et al. 2004. Cross-linking cellular prion protein triggers neuronal apoptosis in vivo. Science 303:1514–1516. [DOI] [PubMed] [Google Scholar]
- Sytnyk, V., I. Leshchyns'ka, M. Delling, G. Dityateva, A. Dityatev, and M. Schachner. 2002. Neural cell adhesion molecule promotes accumulation of TGN organelles at sites of neuron-to-neuron contacts. J. Cell Biol. 159:649–661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Telling, G.C., M. Scott, J. Mastrianni, R. Gabizon, M. Torchia, F.E. Cohen, S.J. DeArmond, and S.B. Prusiner. 1995. Prion propagation in mice expressing human and chimeric PrP transgenes implicates the interaction of cellular PrP with another protein. Cell. 83:79–90. [DOI] [PubMed] [Google Scholar]
- Trupp, M., R. Scott, S.R. Whittemore, and C.F. Ibanez. 1999. Ret-dependent and -independent mechanisms of glial cell line-derived neurotrophic factor signaling in neuronal cells. J. Biol. Chem. 274:20885–20894. [DOI] [PubMed] [Google Scholar]
- van't Hof, W., and M.D. Resh. 1997. Rapid plasma membrane anchoring of newly synthesized p59fyn: selective requirement for NH2-terminal myristoylation and palmitoylation at cysteine-3. J. Cell Biol. 136:1023–1035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walmsley, A.R., F. Zeng, and N.M. Hooper. 2003. The N-terminal region of the prion protein ectodomain contains a lipid raft targeting determinant. J. Biol. Chem. 278:37241–37248. [DOI] [PubMed] [Google Scholar]
- Weissmann, C., and E. Flechsig. 2003. PrP knock-out and PrP transgenic mice in prion research. Br. Med. Bull. 66:43–60. [DOI] [PubMed] [Google Scholar]
- Weiss, S., D. Proske, M. Neumann, M.H. Groschup, H.A. Kretzschmar, M. Famulok, and E.L. Winnacker. 1997. RNA aptamers specifically interact with the prion protein PrP. J. Virol. 71:8790–8797. [DOI] [PMC free article] [PubMed] [Google Scholar]