

Abstract
Background
Aside from muscle, brain is also a major expression site for dystrophin, the protein whose abnormal expression is responsible for Duchenne muscular dystrophy. Cognitive impairments are frequently associated with this genetic disease, we therefore studied the fate of brain and skeletal muscle dystrophins and dystroglycans in dystrophic animal models.
Results
All dystrophin-associated glycoproteins investigated were reduced in dystrophic muscle fibres. In Dp427-deficient mdx brain and Dp71-deficient mdx-3cv brain, the expression of α-dystroglycan and laminin was reduced, utrophin isoforms were up-regulated and β-dystroglycan was not affected. Immunofluorescence localization of β-dystroglycan in comparison with glial, endothelial and neuronal cell markers revealed co-localization of von Willebrand factor with β-dystroglycan. Its expression at the endothelial-glial interface was preserved in dystrophin isoform-deficient brain from mdx and mdx-3cv mice. In addition, chemical crosslinking revealed that the Dp71 isoform exists in mdx brain predominantly as a monomer.
Conclusions
This suggests an association of β-dystroglycan with membranes at the vascular-glial interface in the forebrain. In contrast to dystrophic skeletal muscle fibres, dystrophin deficiency does not trigger a reduction of all dystroglycans in the brain, and utrophins may partially compensate for the lack of brain dystrophins. Abnormal oligomerization of the dystrophin isoform Dp71 might be involved in the pathophysiological mechanisms underlying abnormal brain functions.
Background
The main hypotheses of how deficiency in dystrophin triggers muscular dystrophy suggest that the lack of this membrane cytoskeletal component weakens the sarcolemmal integrity, causes abnormal Ca2+-homeostasis and/or impairs proper clustering of ion channel complexes [1, 2]. Extensive biochemical and cell biological studies have demonstrated that one of the major functions of muscle dystrophin is to act as an actin-binding protein which mediates a link between the extracellular matrix component laminin and the sub-sarcolemmal membrane cytoskeleton [3,4]. Integral or surface-associated proteins that are relatively tightly connected with dystrophin are represented by α-,β-, γ-, and δ-sarcoglycan [5], α- and β-dystroglycan [6], sarcospan [7], α-, β1-, and β2-syntrophin [8], α- and β-dystrobrevin [9], laminin-2 [10] and cortical actin [11]. The backbone of this sarcolemma-spanning protein assembly is formed by the dystroglycans [6]. The extreme carboxy-terminus of 43 kDa β-dystroglycan contains a binding site for the second half of the hinge-4 region and the cysteine-rich domain of Dp427 [12], thereby indirectly connecti ng the actin membrane cytoskeleton via the amino-terminus of the dystrophin molecule to the surface membrane [13]. Since β-dystroglycan is also tightly associated with the peripheral merosin-binding protein α-dystroglycan, this complex provides a stable linkage to the laminin α2-chain in the basal lamina [10].
Deficiency in dystrophin triggers the disintegration of complexes normally formed by the above listed sarcolemmal components and thereby renders muscle fibres from patients afflicted with Duchenne muscular dystrophy (DMD) more susceptible to necrosis [1, 3]. In analogy to the pathobiochemical findings in DMD [3, 14], the dystrophic animal model mdx mouse also exhibits a drastic reduction in all dystrophin-associated glycoproteins in bulk skeletal muscle [15, 16]. This might explain at least partially the decreased osmotic stability [17] and higher vulnerability of stretch-induced injury [18] in dystrophin-deficient muscle fibres. An abnormal increase in cytosolic Ca2+- levels might trigger a drastic inc rease in net protein degradation and might be one of the initial steps in the molecular pathogenesis of inherited muscular dystrophy [19,20,21]. That the other members of the dystrophin -glycoprotein complex, besides dystrophin, play a role in the DMD pathology, is demonstrated by the fact that primary abnormalities in sarcoglycans and laminin are responsible for certain forms of limb-girdle muscular dystrophy and congenital muscular dystrophy, respectively [5, 22]. In contrast to muscle, much less is known about the molecular mechanisms underlying brain abnormalities in the most frequent neuromuscular disease in humans [23, 24]. One factor which probably makes pathophysiological studies of the dystrophic central nervous system more difficult is the greater complexity of dystrophin and utrophin isoforms present in the brain.
Seven promoters drive the tissue-specific expression of various dystrophin protein (Dp) isoforms from the human DMD gene [25], i.e. Dp427-M in skeletal and cardiac muscle, Dp427-B in brain, Dp427-P in Purkinje neurons, Dp-260 R in retina, Dp -140 - B/K in brain and kidney, Dp -116-S in Schwann cells, Dp-71-B/U in brain and many non-muscle tissues [13]. In addition, dystrophin-related proteins are represented by brain DRP-2 [26] and the autosomally-encoded dystrophin homologue utrophin, which forms a full-length 395 kDa isoform (Up395) [27] and two truncated molecular species named Up116 and Up71, also referred to as G-and U-utrophin [28]. Besides full-length brain Dp427 and a relatively low-abundance, carboxy-terminal isoform termed brain Dp140, in the central nervous system the major dystrophin isoform is represented by Dp71 [23]. While Dp427 was shown to be present in cortical neurons, hippocampal neurons and cerebellar Purkinje cells [29], probably mostly associated in these cell types with the postsynaptic density [30], the two smaller dystrophin brain isoforms were described to be associated with microvasular glial cells [31]. A developmental study suggests that dystrophin expression in perivascular astrocytes coincides with the formation of the blood-brain barrier [32]. Dystroglycans are also present in brain [33, 34] and a subpopulation localizes to the glial-vascular interface [31]. Recently, Blake et al. [35] showed that different dystrobrevin isoforms are present in neuronal versus glial dystrophin complexes. With respect to dystrophin-related proteins, full-length utrophin is more widely distributed in the central nervous system [36] and is possibly involved in the maintenance of regional specialization of the brain [37]. To complement these neurobiological studies and in order to determine the fate of dystroglycans in dystrophin-deficient forebrain, we employed two established genetic animal models. The mdx mouse is missing Dp427 due to a point mutation in exon 23 [38], while a mutation in exon 65 in the mdx-3cv mouse affects the splicing of both the 4.8 and 14 kb dystrophin mRNAs resulting in the additional loss of the Dp71 isoform [39]. Neurobehavioral studies have shown that the dystrophic animal models used in this study exhibit moderate alterations in associative learning and deficits in long-term consolidation memory [24, 40,41,42]. Our analysis of these mutant strains indicates that β-dystroglycan appears to be located at the endothelial-glial interface in the forebrain and that not all dystroglycans are reduced in dystrophic brain, making it different from dystrophic muscle fibres. Possibly an impaired oligomerization of the major brain Dp71 isoform plays a role in the molecular pathogenesis in the dystrophic central nervous system.
Results
In contrast to muscle tissues, relatively little is known about the function of brain dystrophins and their associated glycoproteins. To provide the necessary background for the rationale behind this study, the complexity of the dystrophin-glycoprotein complex, the structure and isoform expression pattern of dystrophins and the suitability of dystrophic animal models is summarized in Fig. 1. Dystrophin was previously shown to exist as a large multimeric complex at the cell periphery. As illustrated in the diagrammatic representation of Fig. 1a, the dystroglycan sub-complex provides the backbone structure of this plasmalemma-spanning complex thereby providing a linkage between the extracellular matrix and the membrane cytoskeleton [3, 22]. While the Dp427(-M) isoform exists in skeletal muscle, the central nervous system contains additional shorter dystrophin molecules [13, 25]. Three dystrophin isoforms exist in brain: Dp427(-B), Dp140 and Dp71 (Fig. 1b). Since they all share carboxy-terminal domains, antibodies against this region recognize all three isoforms [23]. In order to determine the effect of the absence of dystrophin on the dystrophin-associated glycoproteins α- and β-dystroglycan, we have used the established animal models mdx and mdx-3cv. These genetic models are due to a point mutation [38] and a genetic rearrangement [39], respectively (Fig. 1c).
Figure 1.
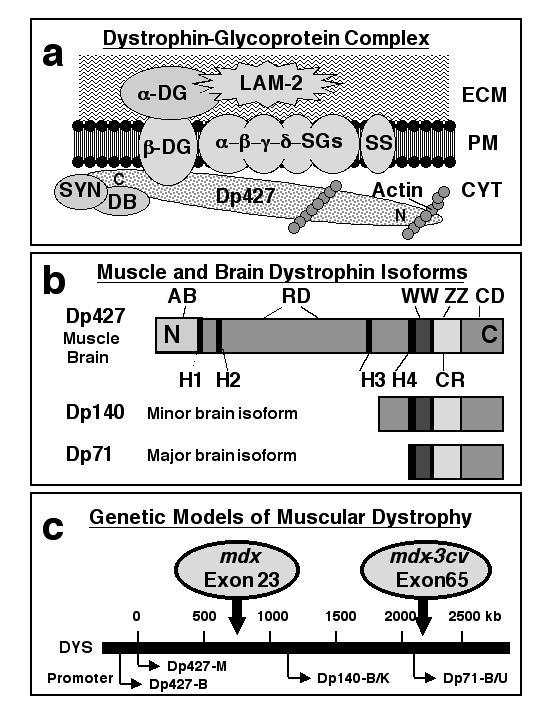
Diagrammatic representation of the dystrophin-glycoprotein complex, the structure of muscle and brain dystrophin isoforms and the genetic animal models of muscular dystrophy. In muscle, dystrophin forms a tightly associated complex with various surface components which provides a stabilizing linkage between the sub-sarcolemmal membrane cytoskeleton and the extracellular matrix. In panel (a) is shown the proposed spatial organization of this peripheral complex consisting of dystrophin (Dp427), α-, β-, γ-, and δ-sarcoglycan (SG), the sarcolemma-spanning backbone structure provided by α- and β-dystroglycan (DG), sarcospan (SS), various syntrophins (SYN) and dystrobrevins (DB), as well as laminin-2 (LAM-2) and cortical actin. Panel (b) outlines the various domains of dystrophin molecules with the N-terminal actin-binding domain (AB), hinge regions (H), the central spectrin-like rod domain (RD), as well as C-terminal binding domains such as the WW domain, the ZZ domain, the cysteine-rich region (CR) and the extreme carboxy-terminal domain (CD). While skeletal muscle fibres contain the Dp427 isoform of dystrophin, brain tissues express besides the full-length Dp427 molecule also two shorter isoforms termed Dp71 and Dp140 (b). Four of the seven promoters which drive the tissue-specific expression of dystrophins are shown in panel (c) illustrating that a point mutation in exon 23 or a mutation in exon 65 results in the absence of Dp427 in mdx mice and the absence of all brain isoforms of dystrophin in mdx-3cv mice.
For comparative purposes and in order to characterize the genetic animal models of muscular dystrophy used in this study, the fate of dystrophin-associated glycoproteins was evaluated in mdx and mdx-3cv skeletal muscle fibres and forebrain. Using indirect immunofluorescence microscopy, it was clearly shown that α- and β-dystroglycan are greatly reduced in muscle cells from both animal models. Antibodies to laminin, dystrophin, α-sarcoglycan and both members of the dystroglycan sub-complex almost exclusively immunolabeled the muscle cell periphery in normal mouse (Fig. 2a,d,g,j,s). Utrophin staining was restricted to the neuromuscular junction region (Fig. 2m) and overlapped with the visualization of the nicotinic acetylcholine receptor by α-bungarotoxin binding (Fig. 2n). In stark contrast, in cryosections taken from mdx and mdx-3cv skeletal muscle, which exhibited a complete absence of dystrophin (Fig. 2k,l), a greatly reduced immunofluorescence signal was detectable for α- and β-dystroglycan (Fig. 2e,f,h,i), as well as for α-sarcoglycan (Fig. 2t,u). Utrophin staining (Fig. 2o,q), α-bungarotoxin binding (Fig. 2p,r), and laminin labeling (2b, c) was not affected in dystrophic fibres. In control experiments, cryosections were labeled with antibodies to the membrane cytoskeletal protein spectrin, a component established not to be affected in muscular dystrophy [14]. As can be seen in Fig. 2v-x, both normal muscle and fibres from both dystrophic animal models exhibit almost exclusively peripheral staining for spectrin establishing the integrity of the cryosections analysed.
Figure 2.
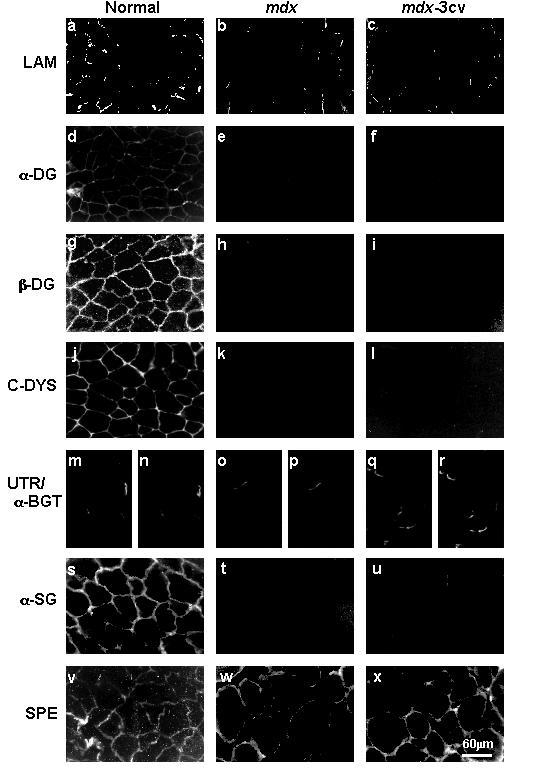
Immunofluorescence localization of β-dystroglycan and associated components in skeletal muscle fibres from dystrophic animal models. Shown are cryosections labeled with antibodies to laminin (LAM) (a-c), α-dystroglycan (α-DG) (d-f), β-dystroglycan (β-DG) (g-i), the carboxy-terminus of dystrophin (C-DYS) (j-l), utrophin (UTR) (m, o, q), α-sarcoglycan (α-SG) (s-u), and spectrin (SPE) (v-x). Panels (n), (p) and (r) represent labeling of tissue sections with α-bungarotoxin (α-BGT). Skeletal muscle specimens were taken from normal mice (a, d, g, j, m, n, s, v), mdx mice (b, e, h, k, o, p, t, w) and mdx-3cv mice (c, f, i, l, q, r, u, x). Bar = 60 μm.
Since initial immunofluorescence labeling experiments with antibodies to β-dystroglycan resulted in a distinct staining pattern in forebrain tissue, we performed more detailed co-localization experiments. Double-staining with antibodies to various common brain cell type markers revealed that this surface glycoprotein is highly enriched at the endothelial-glial interface in the forebrain (Fig. 3). As labels for distinct markers of glial, neuronal and endothelial cells we employed monoclonal antibody NR4 against the neurofilament of apparent 68 kDa, polyclonal antibody GA5 to the glial fibrillary acidic protein and a polyclonal antibody to von Willebrand factor, respectively [43, 44]. The neuronal marker strongly labeled this cell type but did not exhibit an overlap with the staining pattern of β-dystroglycan (Fig. 3a). In contrast, contact zones of overlapping staining were evident between β-dystroglycan and the glial marker (Fig. 3b), probably representing glial endfeet structures (Fig. 3c). A high degree of overlapping immunolabeling was clearly evident between von Willebrand factor and β-dystroglycan (Fig. 3d). Since antibodies to von Willebrand factor specifically label endothelial cells, this suggests high levels of β-dystroglycan at the endothelial-glial interface. To document the specificity of the antibody used for labeling von Willebrand factor, the restricted staining of the endothelial layer in rat aorta is shown in Fig. 3e,f.
Figure 3.
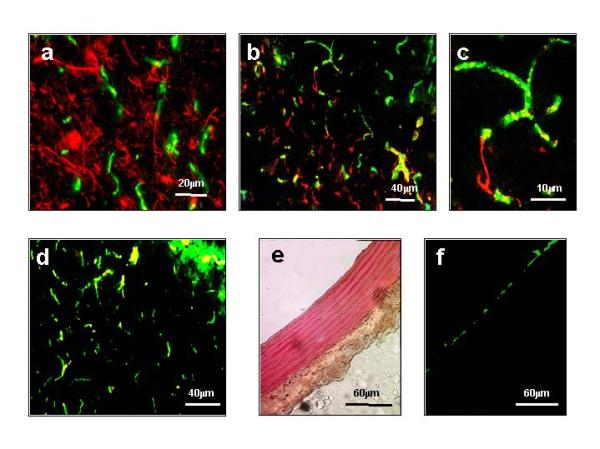
Colocalization of β-dystroglycan and von Willebrand factor in normal mouse forebrain. Shown are cryosections indirectly labeled with rhodamine-conjugated antibodies to the neurofilament of apparent 68 kDa (a), the glial fibrillary acidic protein (b, c) and von Willebrand factor (d). Sections (a) to (d) were indirectly double-labeled with a fluorescein-conjugated antibody against β-dystroglycan. To demonstrate the specificity of the antibody to von Willebrand factor, rat aorta sections are shown in (e) (Haematoxylin & Eosin staining) and (f) (immunofluorescence labeled). In (a), bar = 20 μm; in (b) and (d), bar = 40 μm; in (c), bar = 10 μm; and in (e) and (f), bar = 60 μm.
Following the immunolocalization of β-dystroglycan in normal forebrain, we analysed the relative expression levels of dystrophin and associated components by immunofluorescence microscopy in dystrophic forebrain. In contrast to dystrophin (Fig. 4j-l), dystroglycan labelling was not reduced in cryosections from mdx or mdx-3cv mouse forebrain. As illustrated in Fig. 4a-i, the intensity and pattern of immuno staining for laminin, α-dystroglycan and β-dystroglycan was not affected in the dystrophic specimens studied. Labeling with domain-specific antibodies to dystrophin revealed the presence of Dp71 and the absence of Dp427 in mdx forebrain, since the antibody to the carboxy terminus showed a distinct labeling pattern (Fig. 4k) while the probe to the rod domain did not stain any structures (not shown). All dystrophin isoforms which share the carboxy terminal domain were absent from mdx-3cv forebrain (Fig. 4l). Utrophin exhibited a similar localization pattern to dystrophin and was present in the forebrain from both dystrophic animal models (Fig. 4m-o).
Figure 4.
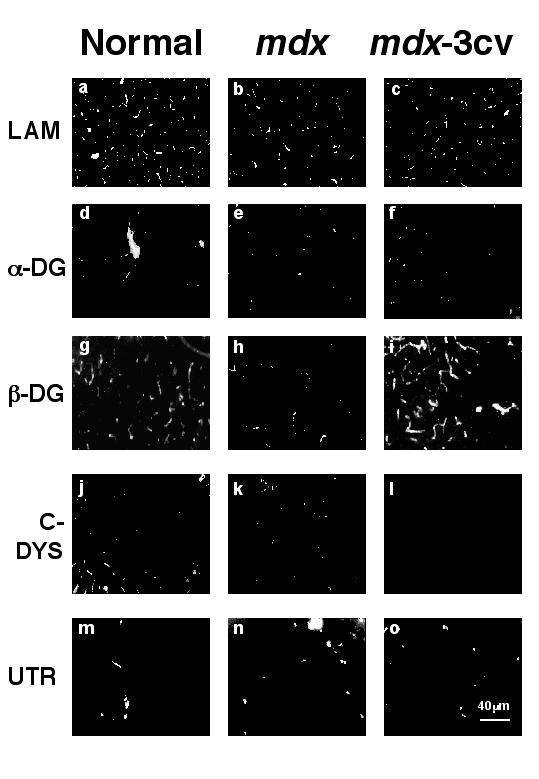
Immunofluorescence localization of β-dystroglycan and associated components in forebrain from dystrophic animal models. Shown are cryosections labeled with antibodies to laminin (LAM) (a-c), α-dystroglycan (α-DG) (d-f), β-dystroglycan (β-DG) (g-i), the carboxy-terminus of dystrophin (C-DYS) (j-l), and utrophin (UTR) (m-o). Forebrain specimens were taken from normal mice (a, d, g, j, m), mdx mice (b, e, h, k, n) and mdx-3cv mice (c, f, i, l, o). Bar = 40 μm.
To determine potential differences in the fate of dystrophin-associated surface components in dystrophic muscle and brain tissues, we also performed a comparative immunoblot analysis of components of the dystrophin-glycoprotein complex using the established animal models mdx and mdx-3cv. As illustrated in the immunoblot analysis shown in Fig. 5, the expression levels of laminin were not affected in the microsomal fraction isolated from dystrophic mdx muscle. On the other hand, this extracellular protein is clearly increased in its relative density in mdx-3cv membranes (Fig. 5a). Both, α- and β-dystroglycan, as well as α-sarcoglycan were found to be drastically reduced in their abundance in both dystrophic animal models (Fig. 5b-d). The dystrophin isoform Dp427 was demonstrated to be completely absent from mdx and mdx-3cv muscle microsomes (Fig. 5e). These findings agree with previous studies on the mdx mouse [15, 16, 22] and show that the same reduction in dystrophin-associated glycoproteins also occurs in the mdx-3cv genetic mouse model. Immunolabeling of full-length utrophin of apparent 395 kDa did not result in sufficient immuno-decoration for a proper comparison of its expression levels in normal versus dystrophic muscle membranes (not shown). For control purposes, an identical immunoblot as was used for the analysis of the dystrophin-glycoprotein complex, was immuno-decorated with an antibody to the α1-subunit of the dihydropyridine receptor. The relative abundance of this transverse-tubular membrane protein does not seem to be affected in microsomes isolated from dystrophic muscle fibres (Fig. 5f). Thus, the decrease in dystrophin-associated glycoproteins in skeletal muscle is a specific resu lt of the deficiency of dystrophin, and not a consequence of general muscle cell destruction in dystrophic fibres.
Figure 5.
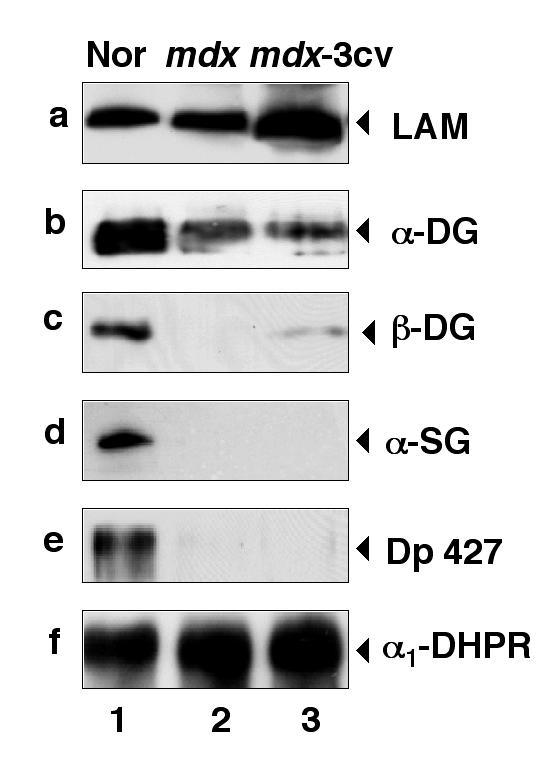
Immunoblot analysis of β-dystroglycan and associated components in normal and dystrophic skeletal muscle fibres. Shown are identical immunoblots labeled with antibodies to laminin (LAM) (a), α-dystroglycan (α-DG) (b), β-dystroglycan (β- DG) (c), α-sarcoglycan (α-SG) (d), full-length dystrophin of apparent 427 kDa (Dp427) (e), and the α1-subunit of the dihydropyridine receptor (α1-DHPR) (f). Lanes 1 to 3 represent microsomal membranes isolated from normal muscle fibres, mdx fibres, and mdx-3cv fibres, respectively. The position of immuno-decorated protein bands is indicated by arrow heads.
Following the analysis of microsomes from dystrophic muscle, we determined the relative expression levels of dystrophin and associated components by immunoblotting in total brain membranes. Prior to this comprehensive immunoblot analysis, membrane preparations from normal mice, mdx brain and mdx-3cv brain were compared by Coomassie staining and lectin overlay assays. Fig. 6 shows that the overall protein band pattern and lectin staining of distinct populations of glycoproteins was relatively comparable between the three different preparations. The only major difference between normal and dystrophic microsomes is the appearance of two protein bands of approximately 50 kDa in membranes isolated from the mdx and the mdx-3cv disease model. Staining with the Maclura pomifera lectin MPA and the Tritium vulgaris lectin WGA demonstrates that the deficiency in brain dystrophin isoforms does not trigger a general reduction in microsomal glycoproteins. In contrast, laminin and α-dystroglycan were clearly shown to be reduced in their relative expression in total brain microsomes from dystrophic animals, which is especially apparent in the mdx-3cv mouse (Fig. 7a,b). Interestingly, β-dystroglycan expression was not affected in both mdx and mdx-3cv total brain microsomes (Fig. 7c), which is a stark contrast to its drastic reduction in dystrophic skeletal muscle fibres (Fig. 5c). Since α-sarcoglycan does not exist in brain, the abundance of members of the sarcoglycan subcomplex were not studied. The immunoblot of Fig. 7d confirms the status of the mdx-3cv brain and demonstrates the absence of the Dp71 isoform in this animal model. In analogy to previous studies on dystrophic skeletal muscle [3], certain utrophin isoform levels were found to be elevated in Dp427-deficient and Dp71-deficient brain specimens. Both Up116 and Up71 were greatly increased in mdx-3cv brain microsomes (Fig. 7f,g), while full-length utrophin did not seem to be affected in dystrophic brain (Fig. 7e).
Figure 6.
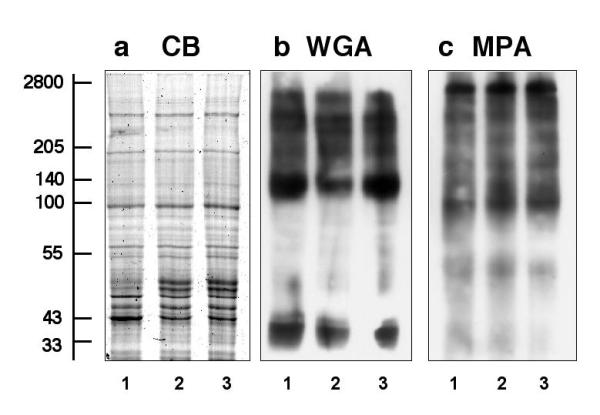
Analysis of microsomal preparations from normal, mdx and mdx-3cv brain. Shown is a Coomassie-stained gel (CB) (a) and identical blots labeled with the Tritium vulgaris lectin WGA (b) and the Maclura pomifera lectin MPA (c). Lanes 1 to 3 represent microsomal membranes isolated from normal brain, mdx brain, and mdx-3cv brain, respectively. The relative position of molecular mass standards (× 10-3) is indicated on the left.
Figure 7.
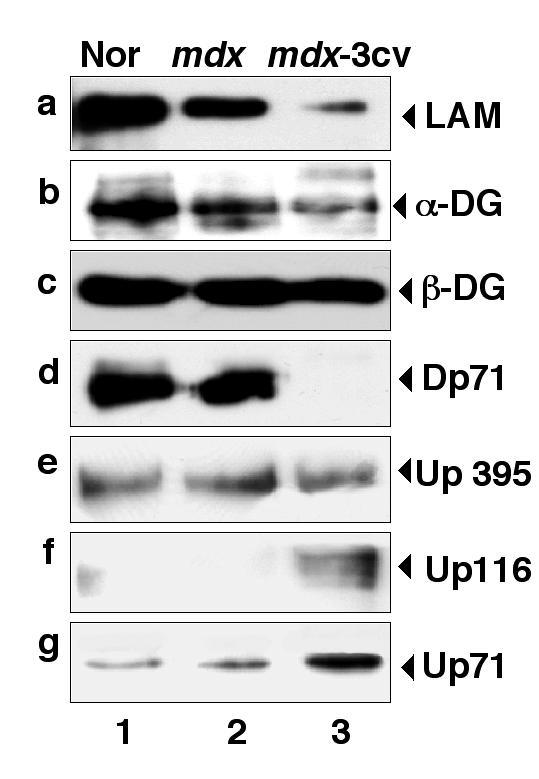
Immunoblot analysis of β dystroglycan and associated components in normal and dystrophic brain-membranes. Shown are identical immunoblots labeled with antibodies to laminin (LAM) (a), α-dystroglycan (α-DG) (b), β-dystroglycan (β-DG) (c), dystrophin of apparent 71 kDa (Dp71) (d), full-length utrophin of apparent 395 kDa (Up395) (e), the utrophin isoform of apparent 116 kDa (Up116) (f), and the utrophin isoform of apparent 71 kDa (Up71) (g). Lanes 1 to 3 represent microsomal membranes isolated from normal brain, mdx brain, and mdx-3cv brain, respectively. The position of immuno-decorated protein bands is indicated by arrow heads.
Since dystrophin does not exist in isolation at the cell surface but forms tightly associated multimeric complexes [3], it was of interest to determine the oligomeric status of the major brain isoform Dp71 in normal and dystrophic mice. Using previously optimized crosslinking conditions [45], we employed the hydrophilic 1.14 nm probe BS3 to stabilize high-molecular-mass complexes. The Coomassie-stained gel in Fig. 8a illustrates that incubation with the crosslinker did not trigger general protein clustering since the protein band pattern was relatively comparable between control and crosslinked membranes. On the other hand, a clear difference was observed for crosslinking-stabilized Dp71 complex formation between normal and mdx brain microsomes. While the crosslinker probe induced a shift to a high-molecular-mass complex in control samples, no decrease in electrophoretic mobility was detectable in dystrophic membranes (Fig. 8b). The major dystrophin isoform Dp71 was not detectable in mdx-3cv microsomes. Interestingly, crosslinking-induced complex formation of full-length utrophin was observed in normal brain, as well as in both dystrophic animal models studied (8c). Therefore, protein-protein int eractions between brain components and utrophin do not appear to be affected in dystrophic tissues. For control purposes, an identical immunoblot was labeled with an antibody to the α-subunit of the Na+/K+-ATPase. No shift to an extremely high-molecular-mass complex was observed for this brain surface protein following chemical crosslinking (Fig. 8d). This strongly suggests that the decrease in the relative electrophoretic mobility of Dp71 in normal brain microsomes is a specific result of crosslinker-induced stabilization of native membrane complexes.
Figure 8.
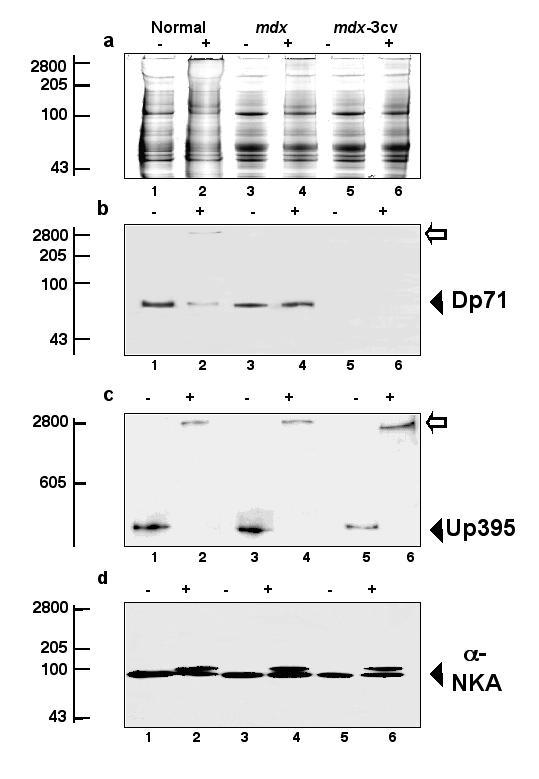
Chemical crosslinking analysis of brain dystrophin isoform Dp71 in normal and mdx mice. Shown is a Coomassie-stained gel (a) and identical immunoblots labeled with antibodies to the dystrophin isoform of apparent 71 kDa (Dp71) (b), full-length utrophin of apparent 395 kDa (Up395) (c), and the α-subunit of the Na+/K+-ATPase (α-NKA) (d). Lanes 1, 3 and 5 represent untreated control samples (-) and lanes 2, 4 and 6 are membranes treated with 200 μg crosslinker BS3 per mg protein (+). Lanes 1 and 2, 3 and 4, and 5 and 6 represent microsomal membranes isolated from normal brain, mdx brain, and mdx-3cv brain, respectively. The position of immuno-decorated monomers is indicated by closed arrow heads and crosslinking-stabilized high-molecular-mass complexes marked by open arrows. The relative position of molecular mass standards (x 10-3) is indicated on the left.
Discussion
Although the X-linked inherited disorder Duchenne muscular dystrophy (DMD) is primarily considered a muscle disease and most patients die of respiratory or cardiac failure [46], in a subpopulation of affected children non-progressive mental retardation preceeds degeneration of the muscular system [24]. These mental abnormalities do not correlate with the stage of the muscle disease [47] and can not be attributed to abnormal motor development [46]. Since all DMD patients experience a decrease in strength of limb and torso muscles, but only approximately one-third of dystrophic children suffer from cognitive impairments, it is believed that differences exist in the pathophysiolgical mechanisms between the central nervous system and muscle tissues [23, 24]. DMD children accomplish performance tasks at a normal level, but their verbal intelligence quotient is significantly lower as compared to age-matched normal boys [48]. Possibly cerebral or cerebellar hypermetabolism is involved in cognitive impairments in certain DMD patients [49], but no consistent abnormalities are detectable in dystrophic brain tissues [50].
Based on this lack of understanding of the exact neurobiology of DMD, we have performed here a comparative analysis of the expression of dystrophins and dystroglycans in brain and muscle tissues from animal models of muscular dystrophy. Forebrain β-dystroglycan was clearly shown to co-localize with the endothelial marker von Willebrand factor and it is not drastically affected in its relative abundance in brain lacking all neuronal dystrophin isoforms. The localization of this relatively abundant glycoprotein at the endothelial-glial interface agrees with previous immunolocalization studies on dystrophin-associated proteins [31, 32, 51,52,53,54]. Dystrophin isoforms of varying length, dystrobrevin and β-dystroglycan appear to be enriched around blood vessels in astrocytic endfeet in the cerebellum and at blood-ocular barrier sites in the retina [51,52,53,54]. Here we can show that the cellular localization of this integral membrane component at the endothelial-glial interface is neither changed in Dp427-deficient mdx forebrain or in Dp71-deficient mdx-3cv forebrain. Thus, in contrast to dystrophic mdx and DMD skeletal muscle fibres, which show a greatly reduced expression of sarcolemmal β-dystroglycan [3, 15, 16], this usually dystrophin-associated glycoprotein experiences a different fate during pathophysiological changes in the central nervous system of dystrophic mice. However, the relative expression of α-dystroglycan is reduced in dystrophic brain. This is unexpected, since both α- and β-dystroglycan are produced by post-translational cleavage of the product of a single transcript [10]. Although β-dystroglycan expression is preserved, this integral membrane protein might not be properly positioned in order to anchor extracellular α-dystroglycan to the outside of the membrane. Compensatory mechanisms to counteract the loss of dystrophin isoforms may induce conformational changes in β-dystroglycan units that interfere with stablising interactions within dystroglycan sub-complexes. Therefore, the preservation of β-dystroglycan does not seem to rescue the extracellular dystroglycan form.
Possibly up-regulation of utrophin isoforms partially compensates for the lack of brain dystrophins and thereby helps anchoring β-dystroglycans. This idea agrees with previous studies of extraocular muscle fibres from mdx and mdx-utrn-/- mice [55, 56]. In contrast to the neuromuscular junction-specific localization of utrophin in normal skeletal muscle [55], in dystrophin-deficient mdx extraocular muscle the full-length isoform of utrophin of apparent 395 kDa is up-regulated in its relative expression and also found in n on-junctional regions of the sarcolemma [57]. This replacement of dystrophin Dp427 by the large utrophin isoform seems to spare a large proportion of the extraocular muscle population from degeneration. However, mdx-utrn-/- mice lacking both dystrophin and utrophin exhibit severe dystrophic changes in these muscle groups strongly suggesting that the endogenous up-regulation of utrophin protects extraocular muscle in dystrophinopathies [58]. A similar protective mechanism might occur in dystrophic brain regions. We could previously show that most members of the dystrophin super-family of proteins, which share the carboxy-terminal binding domain for β-dystroglycan, exhibit very comparable biochemical properties [59, 60]. This was also confirmed for brain isoforms of dystrophin [61]. Since brain utrophins co-localize with the dystroglycan sub-complex in the forebrain, it seems likely that an up-regulation of utrophins anchors these components in Dp427- or Dp71-deficient membranes. Dp71 alone does not appear to properly oligomerize and anchor dystroglycans in mdx brain. Although Dp71 co-localizes with β-dystroglycan, the lack of full-length brain dystrophin seems to trigger a disturbed organization of the dystroglycan sub-complex resulting in a drastic reduction in the extracellular dystroglycan isoform. These findings show that we still have an incomplete understanding of the individual functions of dystrophin isoforms and of the interaction between short and long dystrophins in different tissues.
In contrast to established changes in the expression of dystrophins and utrophins in dystrophic brain [23,30,62], relatively little is known about the fate of dystrophin/utrophin-associated glycoproteins in human DMD brain. In contrast to mdx brain, DMD patient specimens appear to exhibit a reduction in β-dystroglycan levels [16, 63]. However, representative surveys of large patient populations with a varying degree of mental retardation have not yet been performed making it difficult to compare findings from genetic animal models with patient data. In this respect, the finding presented in this study that the major brain dystrophin isoform Dp71 does not appear to properly oligomerize in mdx brain might also be relevant for the human disease condition. The lack of crosslinker-induced complex stabilization indicates that Dp71 might trigger abnormal anchoring of dystroglycans, although it is present at normal concentrations. This in turn might destabilize certain brain structures and/or signal transduction pathways normally relying on the integrity of brain dystrophin-glycoprotein complexes. Since Ca2+-levels were found to be abnormal in dystrophic brain [64], similar pathophysiological changes, as suggested to be involved in muscular degeneration [19,20,21], could also render certain brain cells more susceptible to necrosis. An increased influx of Ca2+-ions might trigger cell destruction not only in Dp71-deficient cells but also in cellular structures with Dp71 molecules not capable of properly forming complexes with β-dystroglycan. In the dystrophic forebrain, abnormal anchoring of dystroglycans might therefore affect the proper establishment of the blood-brain barrier. However, since the cognitive impairment in DMD is non-progressive and exhibits great variations between individual patients, only a sub-population of brain cells may be affected by this pathophysiological mechanism. Deletions in the exon 45-52 region of the DMD gene have been reported to be associated with an increased incidence of cognitive abnormalities [24]. In these patients only the expression of the Dp 427 and Dp140 isoforms is impaired, but not the Dp71 protein [65]. Thus, probably a combination of different primary genetic defects in the DMD gene and variations in compensatory mechanisms result in the different degrees of mental insufficiencies in dystrophic children.
Conclusions
In conclusion, this report demonstrates that β-dystroglycan is not present at high concentrations in central neurons of the forebrain region, but seems to be mostly located at the interface between endothelial cells and glia. These structures possibly represent endfeet on astrocytes at the blood-brain barrier. In dystrophic forebrain, β-dystroglycan expression is not drastically affected, possibly due to the up-regulation of utrophin isoforms which partially compensate for the deficiency in brain dystrophins. Chemical crosslinking analysis showed that Dp71 exists in contrast to its normally oligomeric form in mdx brain as a monomeric protein. Thus, the lack in brain dystrophins does not necessarily lead to a loss in all associated glycoproteins and possibly abnormal oligomerization of the brain dystrophin might play a role in the molecular pathogenesis of abnormal brain functions in muscular dystrophy.
Materials and methods
Materials
Fluorescein-, rhodamine- or peroxidase-conjugated secondary antibodies were purchased from Boehringer Mannheim (Lewis, East Sussex, UK). Commercially available primary antibodies were from Novocastra Laboratories Ltd. (Newcastle upon Tyne, UK), Upstate Biotechnology (Lake Placid, NY, USA) and Sigma Chemical Company (Poole, Dorset, UK), and Texas Red-labeled α-bungarotoxin was purchased from Molecular Probes Europe BV (Leiden, The Netherlands). Superfrost Plus positively-charged microscope slides were from Menzel Glaesser (Braunschweig, Germany). Fuji Neopan 400ASA B/W photographic film was obtained from Fuji Photo Film Co. (Tokyo, Japan) and Kodacolor Gold 400ASA VR film from Eastman Kodak Company (Rochester, NY). Protease inhibitors and acrylamide were purchased from Boehringer Mannheim (Lewis, East Sussex, UK). Peroxidase-conjugated lectins were purchased from EY Labs (San Mateo, CA, USA). Western blotting chemiluminescence substrates and chemical crosslinkers were obtained from Pierce & Warriner (Chester, Cheshire, UK). Immobilon-P nitrocellulose was from Millipore Corporation (Bedford, MA, USA). All other chemicals were of analytical grade and purchased from Sigma Chemical Company (Poole, Dorset, UK).
Antibodies
Monoclonal and polyclonal antibodies employed in this study were characterized as previously described [66, 67]. Monoclonal antibodies NCL-43 against β-dystroglycan, NCL-a-SARC against α-sarcoglycan, DYS-1 to the Dp427 rod-domain, DYS-2 to the Dp427 carboxy-terminus, NCL-DRP1 to the carboxy -terminus of full-length utrophin and NCL-SPEC2 against spectrin were from Novocastra Laboratories Ltd. (Newcastle upon Tyne, UK). Monoclonal antibodies VIA41 to α-dystroglycan and c464.6 to the α-subunit of the Na+/K+-ATPase were purchased from Upstate Biotechnology (Lake Placid, NY, USA). Polyclonal antibodies to von Willebrand factor, laminin and the glial fibrillary acidic protein, as well as monoclonal antibody NR4 to the neurofilament of apparent 68 kDa were obtained from Sigma Chemical Company (Poole, Dorset, UK). A polyclonal antibody which recognizes the carboxy-terminal domain of the utrophin isoforms Up395, Up116 and Up71 [68] was a generous gift of Dr. Steve Winder (University of Glasgow). Monoclonal antibody IIID5 against the α1-subunit of the dihydropyridine receptor was a generous gift of Dr. Kevin P. Campbell (University of Iowa, Iowa City, IA). An antibody to the extreme carboxy-terminus of α-sarcoglycan was raised by 4 monthly injections of a peptide representing the last 15 residues of the carboxy-terminus [69] using a standard immunization protocol [70]. The peptide had been synthesized and coupled to KLH carrier by Research Genetics (Huntington, AL).
Animal models
Muscle and brain samples from the mdx mouse, which lacks the Dp427 isoform of dystrophin due to a point mutation in exon 23 [38], and from the mdx-3cv mouse, which has a mutation in exon 65 that affects the splicing of both the 4.8 and 14 kb dystrophin mRNAs causing a loss of all dystrophin isoforms including the major brain dystrophin isoform Dp71 [39], were a generous gift from Dr. Harald Jockusch (Department of Developmental Biology, University of Bielefeld, Germany). For immunofluorescence microscopy, tissue specimens were taken from the tibialis anterior muscle and the forebrain region, quick-frozen in liquid nitrogen-cooled isopentane, transported on dry ice and stored at -70°C prior to cryosectioning. For immunoblot analysis, total brain and bulk skeletal muscle were dissected, quick-frozen in liquid nitrogen, transported in a container with dry ice and then stored at -70°C prior to homogenization.
Immunofluorescence microscopy
For immunolabeling of muscle and brain tissue sections, 12 μm cryosections were prepared using a standard cryostat (Microm, Heidelberg, Germany) and mounted on Superfrost Plus positively-charged microscope slides. Fixation, blocking, incubation with primary antibodies, washing steps, incubation with secondary antibodies, as well as photography was performed by established methodology [55]. Photographs were taken on Fuji Neopan 400ASA B/W photographic film or Kodak Gold Kodacolor 400ASA VR film. For double-staining procedures, a mixture of the appropriate primary antibodies were applied to tissue sections for 1 h at 37°C, cryosections washed, and then separately incubated for 30 min each with the appropriate secondary antibodies. In case of antibodies which had been generated in the same animal species, photographic images were obtained from concurrent areas in serial sections, and the labeling results overlayed.
Isolation of muscle and brain membranes
In order to compare the relative expression levels of members of the dystrophin-glycoprotein complex by immunoblotting, established protocols for the isolation of microsomal membranes from skeletal muscle [45] and brain [71] were employed. To minimize proteolytic degradation of membrane proteins, all buffers contained a protease inhibitor cocktail (0.2 mM pefabloc, 1.4 μM pepstatin, 0.15 μM aprotinin, 0.3 μM E-64, 1 μM leupeptin, 0.5 μM soybean trypsin inhibitor, and 1 mM EDTA) and all procedures were performed in a cold room at 0-4°C. Membrane pellets were resuspended at a protein concentration of 10 mg/ml and used immediately for gel electrophoretic analysis or quick-frozen in liquid nitrogen and then stored at -70°C prior to further usage. Protein concentration was determined by the method of Bradford [72] using bovine serum albumin as a standard.
Chemical crosslinking analysis
Chemical crosslinking was performed as previously described in detail [45, 66]. Microsomes (1 mg protein) were diluted to a final volume of 500 μl with 50 mM HEPES, pH 8.0 at 25°C. Using a stock solution of 5 mg/ml chemical crosslinker, bis-sulfosuccinimidyl-suberate (BS3) was added to the membrane suspension at a final concentration of 200 μg cross-linker per mg membrane protein. Since the cross -linker BS3 is water-soluble, it was dissolved in 50 mM citrate buffer, pH 5.0 in order to retard hydrolysis. Samples were incubated for 30 min with constant agitation at 25°C and then the crosslinking reactions terminated by the addition of 50 μl of 1 M ammonium acetate per ml reaction mixture. An equal volume of reducing sample buffer [73] was added and the solution incubated for 15 min at 37°C before being subjected to electrophoretic separation.
Gel electrophoresis, lectin staining and immunoblotting
Gel electrophoretic separation using 5% or 7% (w/v) resolving gels with a 5% (w/v) stacking gel in the presence of sodium dodecyl sulfate and dithiotreitol was performed for 200 Vh employing a Mini-MP3 electrophoresis system from Bio-Rad Laboratories (Hempel Hempstead, Herts., UK), whereby 25 μg protein was loaded per well [66, 73]. Chemically crosslinked samples were separated on gels lacking a stacking gel system. Nitrocellulose replica of polyacrylamide gels were produced as described by Towbin et al. [74]. Blot overlays with peroxidase-conjugated lectins (MPA, Maclura pomifera lectin; WGA, Tritium vulgaris lectin) were carried out as previously described [75]. For immunolabeling, nitrocellulose sheets were blocked and incubated with primary and secondary antibodies as previously described [45]. Immunodecoration was evaluated by the enhanced chemiluminescence technique [76]. Densitometric scanning of enhanced chemiluminescence blots was performed on a Molecular Dynamics 300S computing densitometer (Sunnyvale, CA) with ImageQuant V3.0 software.
Acknowledgments
Acknowledgements
Research was supported by project grants from the Irish Health Research Board (HRB-01/98) and Enterprise Ireland, Dublin (SC/2000/386), and a European travel grant from the Royal Society, London and the Royal Irish Academy, Dublin. The authors would like to thank Drs. H. Jockusch (University of Bielefeld, Germany), K.P. Campbell (University of Iowa, IA, USA) and S. Winder (University of Glasgow, Scotland) for providing our lab with animal models and antibodies.
Contributor Information
Kevin Culligan, Email: Kevin.Culligan@ucd.ie.
Louise Glover, Email: Louise.Glover@ucd.ie.
Paul Dowling, Email: Paul.Dowling@ucd.ie.
Kay Ohlendieck, Email: Kay.Ohlendieck@ucd.ie.
References
- Campbell KP. Three muscular dystrophies: loss of cytoskeleton-extracellular matrix linkage. Cell. 1995;80:675–679. doi: 10.1016/0092-8674(95)90344-5. [DOI] [PubMed] [Google Scholar]
- Carlson CG. The dystrophinopathies: an alternative to the structural hypothesis. Neurobiol Dis. 1998;5:3–15. doi: 10.1006/nbdi.1998.0188. [DOI] [PubMed] [Google Scholar]
- Ohlendieck K. Towards an understanding of the dystrophin-glycoprotein complex: linkage between the extracellular matrix and the subsarcolemmal membrane cytoskeleton. Eur J Cell Biol. 1996;69:1–10. [PubMed] [Google Scholar]
- Watkins SC, Cullen MJ, Hoffman EP, Billington L. Plasma membrane cytoskeleton of muscle: a fine structural analysis. Micros Res Tech. 2000;48:131–141. doi: 10.1002/(SICI)1097-0029(20000201/15)48:3/4<131::AID-JEMT2>3.0.CO;2-G. [DOI] [PubMed] [Google Scholar]
- Ozawa E, Noguchi S, Mizuno Y, Hagiwara Y, Yoshida M. From dystrophinopathy to sarcoglycanopathy: evolution of a concept of muscular dystrophy. Muscle Nerve. 1998;21:421–438. doi: 10.1002/(SICI)1097-4598(199804)21:4<421::AID-MUS1>3.0.CO;2-B. [DOI] [PubMed] [Google Scholar]
- Henry MD, Campbell KP. Dystroglycan inside and out. Curr Opin Cell Biol. 1999; 11:602–607. doi: 10.1016/S0955-0674(99)00024-1. [DOI] [PubMed] [Google Scholar]
- Crosbie RH, Heighway J, Venzke DP, Lee JC, Campbell KP. Sarcospan, the 25-kDa transmembrane component of the dystrophin-glycoprotein complex. J Biol Chem. 1997;272:31221–31224. doi: 10.1074/jbc.272.50.31221. [DOI] [PubMed] [Google Scholar]
- Adams ME, Butler MH, Dwyer TM, Peters MF, Murnae AA, Froehner SC. Two forms of mouse syntrophin, a 58 kD dystrophin-associated protein, differ in primary structure and tissue distribution. Neuron. 1993;11:531–540. doi: 10.1016/0896-6273(93)90157-m. [DOI] [PubMed] [Google Scholar]
- Peters MF, O'Brien KF, Sadoulet-Puccio HM, Kunkel LM, Adams ME, Froehner SC. β-Dystrobrevin, a new member of the dystrophin familiy. J Biol Chem. 1997;272:31561–31569. doi: 10.1074/jbc.272.50.31561. [DOI] [PubMed] [Google Scholar]
- Ibraghimov-Beskrovnaya O, Ervasti JM, Leveille CJ, Slaughter CA, Sernett SW, Campbell KP. Primary structure of dystrophin-associated glycoproteins linking dystrophin to the extracellular matrix. Nature. 1992;355:696–702. doi: 10.1038/355696a0. [DOI] [PubMed] [Google Scholar]
- Rybakova IN, Amann KJ, Ervasti JM. A new model for the interaction of dystrophin with F-actin. J Cell Biol. 1996;135:661–672. doi: 10.1083/jcb.135.3.661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung D, Yang B, Meyer J, Chamberlain JS, Campbell KP. Identification and characterization of the dystrophin anchoring site of β-dystroglycan. J Biol Chem. 1995;270:27305–27310. doi: 10.1074/jbc.270.45.27305. [DOI] [PubMed] [Google Scholar]
- Winder SJ. The membrane-cytoskeleton interface: the role of dystrophin and utrophin. J Muscle Res Cell Motil. 1997;18:617–629. doi: 10.1023/A:1018627705273. [DOI] [PubMed] [Google Scholar]
- Ohlendieck K, Matsumura K, Ionasescu VV, Towbin JA, Bosch P, Weinstein SL, Sernett SW, Campbell KP. Duchenne muscular dystrophy: deficiency of dystrophin-associated proteins in the sarcolemma. Neurology. 1993;43:795–800. doi: 10.1212/wnl.43.4.795. [DOI] [PubMed] [Google Scholar]
- Ohlendieck K, Campbell KP. Dystrophin-associated proteins are greatly reduced in skeletal muscle from mdx mice. J Cell Biol. 1991;115:1685–1694. doi: 10.1083/jcb.115.6.1685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finn D, Culligan K, Ohlendieck K. Reduction of brain β-dystroglycan in Duchenne muscular dystrophy but not the mdx animal model. Biochem Biophys Res Comm. 1998;249:231–235. doi: 10.1006/bbrc.1998.9119. [DOI] [PubMed] [Google Scholar]
- Menke A, Jockusch H. Decreased osmotic stability of dystrophin-less muscle cells from mdx mouse. Nature. 1991;349:69–71. doi: 10.1038/349069a0. [DOI] [PubMed] [Google Scholar]
- Petrof BJ, Schrager JB, Stedman HH, Kelly AM, Sweeney HL. Dystrophin protects the sarcolemma from stresses developed during muscle contraction. Proc Natl Acad Sci USA. 1993;90:3710–3714. doi: 10.1073/pnas.90.8.3710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turner PR, Fong P, Denetclaw WF, Steinhardt RA. Increased calcium influx in dystrophic muscle. J Cell Biol. 1991;115:1701–1712. doi: 10.1083/jcb.115.6.1701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alderton JM, Steinhardt RA. Calcium influx through calcium leak channels is responsible for the elevated levels of calcium-dependent proteolysis in dystrophic myotubes. J Biol Chem. 2000;275:9452–9460. doi: 10.1074/jbc.275.13.9452. [DOI] [PubMed] [Google Scholar]
- Mallouk N, Jacquemond V, Allard B. Elevated subsarcolemmal Ca2+ in mdx mouse skeletal muscle fibres detected with Ca2+-activated K+ channels. Proc Natl Acad Sci USA. 2000;97:4950–4955. doi: 10.1073/pnas.97.9.4950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Culligan K, Mackey A, Finn D, Maguire PB, Ohlendieck K. Role of dystrophin isoforms and associated glycoproteins in muscular dystrophy (review). Int J Mol Med. 1998;2:639–648. doi: 10.3892/ijmm.2.6.639. [DOI] [PubMed] [Google Scholar]
- Blake DJ, Kröger S. The neurobiology of Duchenne muscular dystrophy: learning lessons from muscle? Trends Neurosci. 2000;23:92–99. doi: 10.1016/S0166-2236(99)01510-6. [DOI] [PubMed] [Google Scholar]
- Mehler MF. Brain dystrophin, neurogenetics and mental retardation. Brain Res Rev. 2000;32:277–307. doi: 10.1016/S0165-0173(99)00090-9. [DOI] [PubMed] [Google Scholar]
- Ahn AH, Kunkel LM. The structural and functional diversity of dystrophin. Nat Genet. 1993;3:283–291. doi: 10.1038/ng0493-283. [DOI] [PubMed] [Google Scholar]
- Roberts RG, Freeman TC, Kendall E, Vetrie DLP, Dixon AK, Shaw-Smith C, Bone Q, Bobrow M. Characterization of DRP2, a novel human dystrophin homologue. Nat Genet. 1996;13:223–226. doi: 10.1038/ng0696-223. [DOI] [PubMed] [Google Scholar]
- Tinsley JM, Blake DJ, Roche A, Fairbrother U, Riss J, Byth BC, Knight AE, Kendrick-Jones J, Suthers GK, Love DR, Edwards YH, Davies KE. Primary structure of dystrophin-related protein. Nature. 1992;360:591–593. doi: 10.1038/360591a0. [DOI] [PubMed] [Google Scholar]
- Nguyen TM, Helliwell TR, Simmons C, Winder SJ, Kendrick-Jones J, Davies KE, Morris C. Full-length and short forms of utrophin, the dystrophin-related protein. FEBS Lett. 1995;258:262–266. doi: 10.1016/0014-5793(94)01441-3. [DOI] [PubMed] [Google Scholar]
- Lidov HGW, Byers TJ, Watkins SC, Kunkel LM. Localization of dystrophin to postsynaptic regions of central nervous system cortical neurons. Nature. 1990;348:725–728. doi: 10.1038/348725a0. [DOI] [PubMed] [Google Scholar]
- Kim TW, Wu K, Xu JL, Black IB. Detection of dystrophin in the postsynaptic density of rat brain and deficiency in a mouse model of Duchenne muscular dystrophy. Proc Natl Acad Sci USA. 1992;89:11642–11644. doi: 10.1073/pnas.89.23.11642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian M, Jacobson C, Gee SH, Campbell KP, Jucker M. Dystroglycan in the cerebellum is a laminin alpha 2-chain binding protein in the glial -vascular interface and is expressed in Purkinje cells. Eur J Neurosci. 1996;8:2739–2747. doi: 10.1111/j.1460-9568.1996.tb01568.x. [DOI] [PubMed] [Google Scholar]
- Jancsik V, Hajos F. The demonstration of immunoreactive dystrophin and its developmental expression in perivascular astrocytes. Brain Res. 1999;831:200–205. doi: 10.1016/S0006-8993(99)01445-6. [DOI] [PubMed] [Google Scholar]
- Mummery R, Sessay A, Lai FA, Beesley PW. Beta-dystroglycan: subcellular localisation in rat brain and detection of a novel immunologically related, postsynaptic density-enriched protein. J Neurochem. 1996;66:2455–2459. doi: 10.1046/j.1471-4159.1996.66062455.x. [DOI] [PubMed] [Google Scholar]
- Cavaldesi M, Macchia G, Barca S, Defilippi P, Tarone G, Petrucci TC. Association of the dystroglycan complex isolated from bovine brain synaptosomes with proteins involved in signal transduction. J Neurochem. 1999;72:1648–1655. doi: 10.1046/j.1471-4159.1999.721648.x. [DOI] [PubMed] [Google Scholar]
- Blake DJ, Hawkes R, Benson MA, Beesley PW. Different dystrophin-like complexes are expressed in neurons and glia. J Cell Biol. 1999;147:645–657. doi: 10.1083/jcb.147.3.645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khurana TS, Watkins SC, Kunkel LM. The subcellular distribution of chromosome 6-encoded dystrophin-realted protein in the brain. J Cell Biol. 1992;119:357–366. doi: 10.1083/jcb.119.2.357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khurana TS, Kunkel LM, Frederickson AD, Carbonetto S, Watkins SC. Interaction of chromosome-6-encoded dystrophin related protein with the extracellular matrix. J Cell Sci. 1995;108:173–185. doi: 10.1242/jcs.108.1.173. [DOI] [PubMed] [Google Scholar]
- Sicinski P, Geng Y, Ryder-Cook AS, Barnard EA, Darlison MG, Barnard PG. The molecular basis of muscular dystrophy in the mdx mouse: a point mutation. Science. 1989;244:1578–1580. doi: 10.1126/science.2662404. [DOI] [PubMed] [Google Scholar]
- Cox GA, Phelps SF, Chapman VM, Chamberlain JS. New mdx mutation disrupts expression of muscle and nonmuscle isoforms of dystrophin. Nat Genet. 1993;4:87–93. doi: 10.1038/ng0593-87. [DOI] [PubMed] [Google Scholar]
- Muntoni F, Mattedu G, Serra G. Passive avoidance behaviour deficit in the mdx mouse. Neuromusc Disord. 1991;1:121–123. doi: 10.1016/0960-8966(91)90059-2. [DOI] [PubMed] [Google Scholar]
- Vaillend C, Rendon R, Misslin A, Ungerer A. Retention deficits at long delays in spontaneous alteration and bar-pressing tasks. Behav Genet. 1995;25:569–579. doi: 10.1007/BF02327580. [DOI] [PubMed] [Google Scholar]
- Vaillend C, Ungerer A. Behavioral characterization of mdx3cv mice deficient in C-terminal dystrophins. Neuromusc Disord. 1999;9:296–304. doi: 10.1016/S0960-8966(99)00029-2. [DOI] [PubMed] [Google Scholar]
- Franke FE, Schachenmayr W, Osborn M, Altmannsberger M. Unexpected immunoreactivities of intermediate filament antibodies in human brain-tumors. Am J Pathol. 1991;139:67–79. [PMC free article] [PubMed] [Google Scholar]
- Risau Differentiation of endothelium. FASEB J. 1995;9:926–933. [PubMed] [Google Scholar]
- Murray B, Ohlendieck K. Crosslinking analysis of the ryanodine receptor and α1 - dihydropyridine receptor in rabbit skeletal muscle triads. Biochem J. 1997;324:689–696. doi: 10.1042/bj3240689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engel AG, Yamamoto M, Fischbeck KH. Dystrophinopathies. In Myology (Engel, AG, Yamamoto, M, Fischbeck, KM, Eds) McGraw-Hill, Inc, New York, NY, 1994. pp. 1133–1187.
- Dubowitz V. Intellectual impairment in muscular dystrophy. Arch Dis Child. 1965;4:296–301. doi: 10.1136/adc.40.211.296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zellweger H, Handon JW. Psychometric studies in muscular dystrophy type IIIa (Duchenne). Develop Med Child Neurol. 1967;9:576–583. doi: 10.1111/j.1469-8749.1967.tb02327.x. [DOI] [PubMed] [Google Scholar]
- Bresolin N, Castelli E, Comi GP, Felisari G, Bardoni A, Perani D, Grassi F, Turconi A, Mazzuchelli F, Gallotti D, Moggio M, Prelle A, Ausenda C, Fazio G, Scarlato G. Cognitive impairment in Duchenne muscular dystrophy. Neuromusc Disord. 1994;4:359–369. doi: 10.1016/0960-8966(94)90072-8. [DOI] [PubMed] [Google Scholar]
- Jagadha J, Becker LE. Brain morphology in Duchenne muscular dystrophy. Pediatr Neurol. 1988;4:87–92. doi: 10.1016/0887-8994(88)90047-1. [DOI] [PubMed] [Google Scholar]
- Ueda H, Baba T, Terada N, Kato Y, Fujii Y, Takayamma I, Mei X, Ohno S. Immunolocalization of dystrobrevin in the astrocytic endfeet and endothelial cells in the rat cerebellum. Neurosci Lett. 2000;283:121–124. doi: 10.1016/S0304-3940(00)00925-3. [DOI] [PubMed] [Google Scholar]
- Ueda H, Gohdo T, Ohno S. Beta-dystroglycan localization in the photoreceptor and Muller cells in the rat retina revealed by immunoelectron microscopy. J Histochem Cytochem. 1998;46:185–191. doi: 10.1177/002215549804600207. [DOI] [PubMed] [Google Scholar]
- Ueda H, Baba T, Ohno S. Current knowledge of dystrophin and dystrophin-associated proteins in the retina. Histo Histopathol. 2000;15:753–760. doi: 10.14670/HH-15.753. [DOI] [PubMed] [Google Scholar]
- Ueda H, Baba T, Kashiwagi K, Iijima H, Ohno S. Dystrobrevin localization in photoreceptor axon terminals and at blood-ocular barrier sites. Invest Ophthalmol Vis Sci. 2000;41:3908–3914. [PubMed] [Google Scholar]
- Ohlendieck K, Ervasti JM, Matsumur K, Kahl SD, Leveille CJ, Campbell KP. Dystrophin-related protein is localized to neuromuscular junctions of adult skeletal muscle. Neuron. 1991;7:499–508. doi: 10.1016/0896-6273(91)90301-f. [DOI] [PubMed] [Google Scholar]
- Rafael JA, Tinsley JM, Potter AC, Deconinck AE, Davies KE. Skeletal muscle-specifi c expression of utrophin transgene rescues utrophin-dystrophin deficient mice. Nat Genet. 1998;19:79–82. doi: 10.1038/ng0598-79. [DOI] [PubMed] [Google Scholar]
- Matsumura K, Ervasti JM, Ohlendieck K, Kahl SD, Campbell KP. Association of dystrophin -related protein with dystrophin-associated proteins in mdx mouse. Nature. 1992;360:588–591. doi: 10.1038/360588a0. [DOI] [PubMed] [Google Scholar]
- Porter JD, Rafael JA, Ragusa RJ, Brueckner JK, Trickett JI, Davies KE. The sparing of extraocular muscle in dystrophinopathy is lost in mice lacking utrophin and dystrophin. J Cell Sci. 1998;111:1801–1811. doi: 10.1242/jcs.111.13.1801. [DOI] [PubMed] [Google Scholar]
- Ohlendieck K, Campbell KP. Dystrophin constitutes five percent of membrane cytoskeleton in skeletal muscle. FEBS Lett. 1991;283:230–234. doi: 10.1016/0014-5793(91)80595-T. [DOI] [PubMed] [Google Scholar]
- Ohlendieck K. Membrane cytoskeletal characterisation of utrophin in highly purified sarcolemma vesicles. Biochim Biophys Acta. 1996;1283:215–222. doi: 10.1016/0005-2736(96)00102-2. [DOI] [PubMed] [Google Scholar]
- Finn D, Ohlendieck K. Rabbit brain and muscle isoforms containing the carboxy-terminal domain of 427 kDa skeletal muscle dystrophin exhibit similar biochemical properties. Neurosci Lett. 1997;222:1–4. doi: 10.1016/S0304-3940(97)13326-2. [DOI] [PubMed] [Google Scholar]
- Kim TW, Wu K, Black IB. Deficiency of brain synaptic dystrophin in human Duchenne muscular dystrophy. Ann Neurol. 1995;38:446–449. doi: 10.1002/ana.410380315. [DOI] [PubMed] [Google Scholar]
- Uchino M, Hara A, Mizuno Y, Fujiki M, Nakamura T, Tokunaga M, Hirano T, Yamashita T, Uyama E, Ando Y, Mita S, Ando M. Distribution of dystrophin and dystrophin-associated protein 43DAG (β-dystroglycan) in the central nervous system of normal controls and patients with Duchenne muscular dystrophy. Intern Med. 1996;35:189–194. doi: 10.2169/internalmedicine.35.189. [DOI] [PubMed] [Google Scholar]
- Hopf FW, Steinhardt RA. Regulation of intracellular free calcium in normal and dystrophic mouse cerebellar neurons. Brain Res. 1992;578:49–54. doi: 10.1016/0006-8993(92)90228-2. [DOI] [PubMed] [Google Scholar]
- Lidov HG, Selig S, Kunkel LM. Dp140: a novel 140 kDa CNS transcript from the dystrophin locus. Hum Mol Genet. 1995;4:329–335. doi: 10.1093/hmg/4.3.329. [DOI] [PubMed] [Google Scholar]
- Finn D, Ohlendieck K. Oligomerization of β-dystroglycan in rabbit diaphragm and brain as revealed by chemical crosslinking. Biochim Biophys Acta. 1998;1370:325–336. doi: 10.1016/S0005-2736(97)00283-6. [DOI] [PubMed] [Google Scholar]
- Ohlendieck K, Ervasti JM, Snook JB, Campbell KP. Dystrophin glycoprotein complex is highly enriched in skeletal muscle sarcolemma. J Cell Biol. 1991;112:135–148. doi: 10.1083/jcb.112.1.135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- James M, Nutall A, Ilsey JL, Ottersbach K, Tinsley JM, Sudol M, Winder SJ. Adhesion-dependent tyrosine phosphorylation of (beta)-dystroglycan regulates its interaction with utrophin. J Cell Sci. 2000;113:1717–1726. doi: 10.1242/jcs.113.10.1717. [DOI] [PubMed] [Google Scholar]
- Roberds SL, Leturcq F, Allamand V, Piccolo F, Jeanpierre M, Anderson RD, Lim LE, Lee JC, Tome FMS, Romero NB, Fardeau M, Beckmann JS, Kaplan JC, Campbell KP. Missense mutations in the adhalin gene linked to autosomal recessive muscular dystrophy. Cell. 1994;78:625–633. doi: 10.1016/0092-8674(94)90527-4. [DOI] [PubMed] [Google Scholar]
- Harlow E, Lane D. Antibodies. A Laboratory Manual. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY, 1998. pp. 53–138.
- McPherson PS, Campbell KP. Solubilization and biochemical characterization of the high-affinity [3H] ryanodine receptor from rabbit brain membranes. J Biol Chem. 1990;265:18454–18460. [PubMed] [Google Scholar]
- Bradford MM. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem. 1976;72:248–254. doi: 10.1006/abio.1976.9999. [DOI] [PubMed] [Google Scholar]
- Laemmli UK. Cleavage of bacteriophage T7 early RNAs and proteins on slab gels. Nature. 1970;227:680–685. [Google Scholar]
- Towbin H, Staehelin T, Gordon J. Electrophoretic transfer of proteins from polyacrylamide gels to nitrocellulose sheets: procedure and some applications. Proc Natl Acad Sci USA. 1979;76:4350–4354. doi: 10.1073/pnas.76.9.4350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohlendieck K, Dhume ST, Partin JS, Lennarz WJ. The sea urchin egg receptor for sperm: isolation and characterization of the intact, biologically active receptor. J Cell Biol. 1993;122:887–895. doi: 10.1083/jcb.122.4.887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bradd SJ, Dunn MJ. Analysis of membrane proteins by Western blotting and enhanced chemiluminescence. Meth Mol Biol. 1993;19:211–218. doi: 10.1385/0-89603-236-1:211. [DOI] [PubMed] [Google Scholar]