

Abstract
14-3-3 proteins are phosphoserine/threonine-binding proteins that play important roles in many regulatory processes, including intracellular protein targeting. 14-3-3 proteins can anchor target proteins in the cytoplasm and in the nucleus or can mediate their nuclear export. So far, no role for 14-3-3 in mediating nuclear import has been described. There is also mounting evidence that nuclear import is regulated by the phosphorylation of cargo proteins, but the underlying mechanism remains elusive. Myopodin is a dual-compartment, actin-bundling protein that functions as a tumor suppressor in human bladder cancer. In muscle cells, myopodin redistributes between the nucleus and the cytoplasm in a differentiation-dependent and stress-induced fashion. We show that importin α binding and the subsequent nuclear import of myopodin are regulated by the serine/threonine phosphorylation-dependent binding of myopodin to 14-3-3. These results establish a novel paradigm for the promotion of nuclear import by 14-3-3 binding. They provide a molecular explanation for the phosphorylation-dependent nuclear import of nuclear localization signal-containing cargo proteins.
Introduction
14-3-3 proteins are abundant, chaperone-like adaptor molecules that act as homo- and heterodimers (Fu et al., 2000). Monomeric 14-3-3 consists of nine α helices, which create a COOH-terminal binding cleft for target binding and an NH2-terminal dimerization domain (Liu et al., 1995; Xiao et al., 1995). 14-3-3 proteins are predominately found in the cytoplasm (Dalal et al., 1999; Kumagai and Dunphy, 1999; Muslin and Xing, 2000), but are also found in the nucleus (Bihn et al., 1997; Todd et al., 1998; Pan et al., 1999), in the Golgi apparatus (Celis et al., 1990), and in chloroplasts (Sehnke et al., 2000). More than 100 14-3-3 interacting proteins have been identified. In most cases, binding occurs through a consensus motif within the target protein, which contains a phosphorylated serine or threonine residue (sequence RSxpS/TxP; Muslin et al., 1996). 14-3-3 proteins have multiple effects on their targets, and the functional variability is a feature of the binding partner rather than an intrinsic 14-3-3 trait (Tzivion et al., 2001). 14-3-3 can regulate the subcellular localization of a binding partner. It may cause the nuclear exclusion (Dalal et al., 1999; Kumagai and Dunphy, 1999; Zhang et al., 1999; Grozinger and Schreiber, 2000; McKinsey et al., 2000; Wang et al., 2000; Cahill et al., 2001; Brunet et al., 2002) or the nuclear (Seimiya et al., 2000) or cytoplasmic retention (Dalal et al., 1999) of an interacting protein. So far, no role for 14-3-3 in promoting the nuclear import of a binding partner has been described.
Nuclear import is regulated at numerous levels, including the posttranslational modification of cargo proteins. Phosphorylation of some cargoes can mask their nuclear localization signal (NLS), thereby abrogating their interaction with the cytoplasmic import receptor, importin α, and blocking nuclear import. Other cargo proteins show enhanced binding to importin α after phosphorylation, resulting in an increased nuclear import rate (Jans et al., 2000). The mechanism underlying the phosphorylation-dependent nuclear import of some NLS-containing cargo proteins remains elusive. Phosphorylation may directly modulate the affinity of the NLS for importin α. Alternatively, it may cause conformational changes within the cargo or alter its binding to another protein, both of which may reveal or mask the NLS (Harreman et al., 2004).
Myopodin is a dual-compartment, actin-bundling protein that is found in the nucleus of undifferentiated myoblasts and at the Z-disc of differentiated myotubes, but it shuttles back to the nucleus during thermal stress (Weins et al., 2001). In normal bladder epithelium, myopodin is localized in the cytoplasm and in the nucleus (Sanchez-Carbayo et al., 2003). Of note, the loss of nuclear myopodin expression can predict the clinical outcome of human progressive bladder cancer, implying that myopodin functions as a tumor suppressor (Sanchez-Carbayo et al., 2003). The mechanism that regulates the subcellular localization of myopodin is unknown. We describe a novel mechanism for regulating the nuclear import of an NLS-containing protein. We show that the phosphorylation-dependent binding of 14-3-3 to myopodin is required for the interaction of myopodin with importin α and the subsequent nuclear import of myopodin. We discuss 14-3-3 binding as a novel, regulatory step that promotes the nuclear import of cargo proteins.
Results
Myopodin is a novel 14-3-3–binding protein
Myopodin contains two classic nuclear localization signals, a PPXY motif, an actin-binding site (Weins et al., 2001), and two consensus 14-3-3–binding domains (Fig. 1 A). To identify myopodin-interacting proteins, we screened a mouse embryonic cDNA library using the yeast two-hybrid system. Because full-length myopodin was autoactive, we used the myopodin fragment Myo-2 (aa 187–420) as bait (Fig. 1 A). Among several others, we found a cDNA clone containing the COOH-terminal part of 14-3-3β (aa 143–246), which creates the binding cleft of 14-3-3 (Fu et al., 2000).
Figure 1.
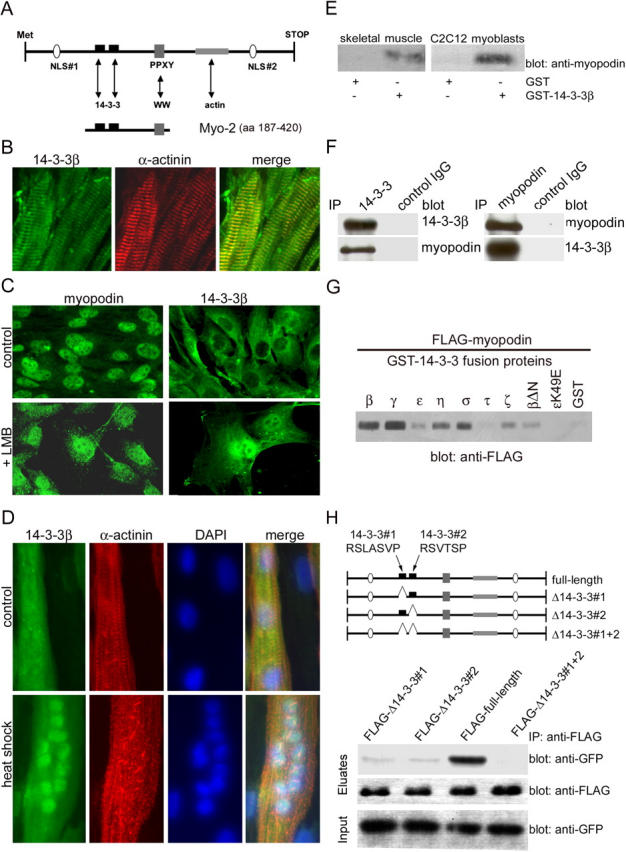
Myopodin interacts with 14-3-3 via two consensus 14-3-3–binding motifs. (A) Myopodin contains two NLSs, a PPXY motif, two 14-3-3–binding motifs, and an actin-binding site. Arrows indicate potential interactions with other proteins or with protein domains (WW). The Myo-2 fragment (aa 187–420) was used as bait in a yeast two-hybrid screen. (B) 14-3-3β colocalizes with the Z-disc marker α-actinin in mouse skeletal muscle. (C) In undifferentiated C2C12 myoblasts, myopodin is predominantly found in the nucleus (top left), whereas 14-3-3β is preferentially found in the cytoplasm (top right). Blocking nuclear export with LMB does not affect the nuclear localization of myopodin (bottom left), but leads to the nuclear accumulation of 14-3-3β (bottom right). (D) Heat shock causes nuclear accumulation of 14-3-3β in differentiated myotubes, as visualized by double labeling with DAPI. (E) Myopodin from mouse skeletal muscle (left) and C2C12 myoblast extracts (right) specifically binds to GST–14-3-3β, but not to GST alone. (F) Coimmunoprecipitation experiments show that endogenous myopodin interacts with 14-3-3β in adult mouse heart. (left) IP with anti–14-3-3β; (right) IP with antimyopodin. No binding was found with a control IgG. (G) Myopodin binds to all 14-3-3 isoforms except 14-3-3τ. 14-3-3β lacking the NH2-terminal dimerization domain (βΔN) can bind to myopodin, albeit to a lesser extent. In contrast, 14-3-3ɛ carrying a point mutation (ɛK49E) that is known to abrogate target binding does not interact with myopodin. (H) The two consensus 14-3-3–binding motifs, 14-3-3#1 (sequence RSLASVP) and 14-3-3#2 (sequence RSVTSP), were deleted separately (Δ14-3-3#1 and Δ14-3-3#2) and together (Δ14-3-3#1 + 2). Both FLAG–Δ14-3-3#1 and FLAG–Δ14-3-3#2 display dramatically reduced binding to 14-3-3β as compared with full-length myopodin (full-length FLAG). Deletion of both 14-3-3–binding motifs (FLAG–Δ14-3-3#1 + 2) abrogates binding to 14-3-3β.
Spatial and temporal co-distribution of myopodin and 14-3-3β
In adult heart and skeletal muscles, myopodin is localized at the Z-disc (Weins et al., 2001). By double labeling immunofluorescence with the Z-disc marker α-actinin, 14-3-3β was also found at the Z-disc of the heart (Fig. 1 B) and skeletal muscles (unpublished data). Hence, both myopodin and 14-3-3β are components of the Z-disc. In undifferentiated C2C12 myoblasts, myopodin shows a predominantly nuclear localization (Fig. 1 C, top; Weins et al., 2001), whereas 14-3-3β is mainly found in the cytoplasm (Fig. 1 C, top). This difference in the subcellular localization of myopodin and 14-3-3β seemed counterintuitive to the two-hybrid result and to the observed biochemical interaction between both proteins (Fig. 1, E–H). However, as shown for the U2OS osteosarcoma cell line, 14-3-3σ is also localized in the cytoplasm but accumulates in the nucleus after the blockage of nuclear export by leptomycin B (LMB), implying that 14-3-3σ can shuttle between the cytoplasm and the nucleus (Brunet et al., 2002). Similarly, the inhibition of nuclear export with LMB for 90 min induced the nuclear accumulation of 14-3-3β (Fig. 1 C, bottom). We have previously reported that after heat shock and LMB treatment, myopodin relocates to the nucleus of differentiated myotubes, showing that the nuclear export of myopodin is sensitive to LMB (Weins et al., 2001). Here, we found that 14-3-3β relocates from the cytoplasm to the nucleus after heat shock (Fig. 1 D), suggesting that, similar to myopodin, the subcellular localization of 14-3-3 is altered by cellular stress.
Endogenous myopodin specifically interacts with 14-3-3β in myocytes
Myopodin specifically interacts with 14-3-3β in GST pull-down assays. GST–14-3-3β, but not GST alone, specifically bound to myopodin from adult mouse skeletal muscle and C2C12 myoblast extracts (Fig. 1 E). To further confirm the interaction, endogenous proteins were immunoprecipitated from mouse heart extracts using an anti–14-3-3β antibody and the myopodin-specific antibody SRIB2. Anti–14-3-3 precipitated 14-3-3β from the extract and also coprecipitated myopodin (Fig. 1 F, left). Conversely, antimyopodin coimmunoprecipitated 14-3-3β (Fig. 1 F, right). No interaction was found with a control IgG.
Myopodin interacts with all 14-3-3 isoforms except 14-3-3τ
Mammals express seven different 14-3-3 isoforms (β, γ, ɛ, η, σ, τ, and ζ; Fu et al., 2000), but the yeast two-hybrid screen had identified only the β isoform as a myopodin-interacting protein. The binding of myopodin to other 14-3-3 isoforms was tested in pull-down studies of GST–14-3-3 fusion proteins and purified FLAG-myopodin (Fig. 1 G). Myopodin strongly bound 14-3-3β, γ, η, and σ (Fig. 1 G). The interaction with 14-3-3ɛ and ζ was weaker, and no binding was found for 14-3-3τ (Fig. 1 G). The K49E substitution in the binding groove of 14-3-3 causes the loss of target binding (Zhang et al., 1997), and a 14-3-3ɛ K49E mutant also failed to bind to myopodin (Fig. 1 G), confirming the specificity of the interaction. 14-3-3 dimerization is crucial for target protein binding and 14-3-3 functionality (Tzivion et al., 2001). Therefore, the requirement of 14-3-3 dimerization for myopodin binding was tested with a 14-3-3β construct (14-3-3βΔN) that lacked the NH2-terminal dimerization domain (Seimiya et al., 2000). 14-3-3βΔN also bound to FLAG-myopodin, but to a lesser extent than wild-type 14-3-3β (Fig. 1 G).
Two consensus 14-3-3 motifs mediate the binding of myopodin to 14-3-3β
Myopodin contains two consensus 14-3-3–binding motifs (Fig. 1 H, top) that can mediate phosphorylation-dependent 14-3-3 binding (Yaffe et al., 1997). To map the 14-3-3β–binding sites in myopodin, both motifs were deleted separately (Δ14-3-3#1 and Δ14-3-3#2) or together (Δ14-3-3#1 + 2; Fig. 1 H). Full-length myopodin and deletion constructs were coexpressed with GFP–14-3-3β in HEK-293 cells and were analyzed by coimmunoprecipitation. FLAG full-length myopodin showed a strong interaction with GFP–14-3-3β, whereas a weak binding was found for FLAG–Δ14-3-3#1 and FLAG–Δ14-3-3#2. No binding to 14-3-3β was found after the deletion of both motifs (FLAG–Δ14-3-3#1 + 2). Hence, myopodin contains two functional 14-3-3–binding sites that mediate the interaction between myopodin and 14-3-3β.
14-3-3 function is required for the nuclear import of myopodin
14-3-3 proteins can regulate the subcellular localization of binding partners (Muslin and Xing, 2000), and myopodin is a dual-compartment protein (Weins et al., 2001). This raises the possibility that 14-3-3β participates in regulating the subcellular localization of myopodin. A dominant negative mutant of 14-3-3θ that lacks the NH2-terminal dimerization domain (14-3-3θΔN) redistributes the catalytic subunit of human telomerase from the nucleus to the cytoplasm (Seimiya et al., 2000). Therefore, we tested the effect of 14-3-3βΔN (Fig. 1 G) on the subcellular localization of myopodin in myoblasts. In cells expressing GFP–14-3-3βΔN, myopodin showed a cytoplasmic localization (Fig. 2 A, bottom left), whereas in cells expressing full-length GFP–14-3-3β (Fig. 2 A, top left) or GFP alone (not depicted), myopodin was localized in the nucleus.
Figure 2.
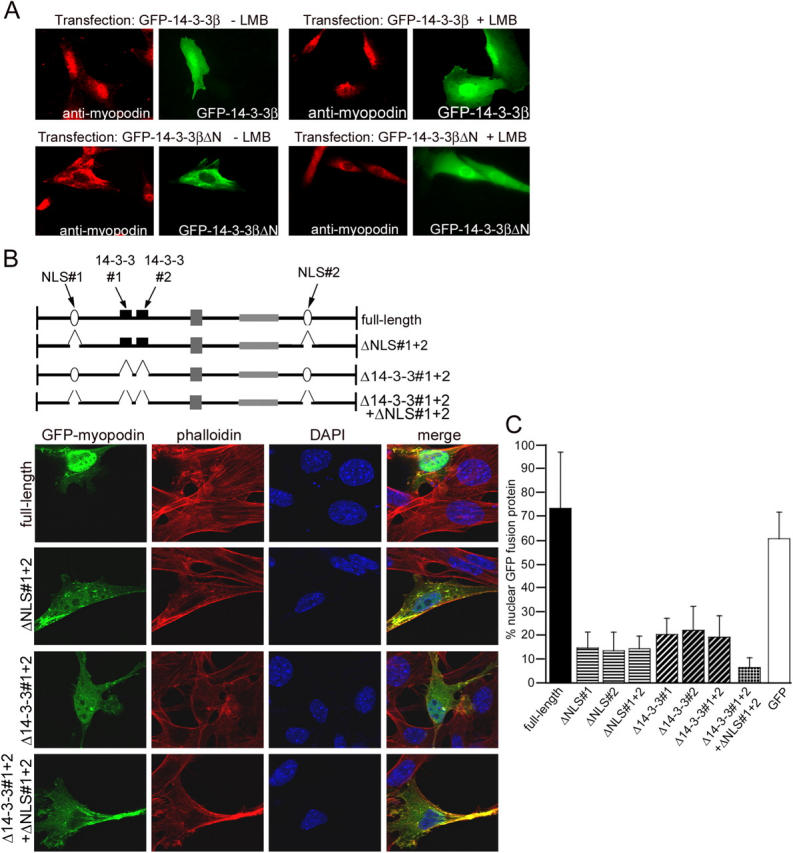
14-3-3β is required for nuclear import of myopodin. (A) Overexpression of full-length GFP–14-3-3β does not change the nuclear localization of endogenous myopodin (top left), whereas the dominant negative form GFP–14-3-3βΔN causes the nuclear exclusion of myopodin (bottom left). LMB does not reverse the cytoplasmic localization of myopodin that is caused by GFP–14-3-3βΔN (bottom right). (B) Confocal microscopy reveals a predominantly nuclear localization of GFP-tagged, full-length myopodin (top). Deletion of both NLSs (ΔNLS#1 + 2) or both 14-3-3–binding sites (Δ14-3-3#1 + 2) dramatically decreases the nuclear localization of GFP–myopodin. The combined deletion of all four binding motifs (Δ14-3-3#1 + 2 + ΔNLS#1 + 2; bottom) virtually abrogates the nuclear localization of myopodin (bottom). (C) Quantitative analysis is presented as a percentage of nuclear GFP–myopodin. 73.4% of GFP full-length myopodin is found in the nucleus. Deletion of both NLSs separately (ΔNLS#1 and ΔNLS#2) or together (ΔNLS#1 + 2) decreases the amount of nuclear GFP–myopodin to 14.8%, 13.4%, and 14.2%, respectively. Removal of 14-3-3–binding sites (Δ14-3-3#1, Δ14-3-3#2, and Δ14-3-3#1 + 2) reduces nuclear myopodin to 20.0%, 21.8%, and 19.1%, respectively. Combined deletion of all four motifs (Δ14-3-3#1 + 2 + ΔNLS#1 + 2) causes a further reduction of nuclear myopodin to 6.4%. 60.5% of GFP (control) is found in the nucleus. Statistical significance was confirmed by analysis of variance between groups (ANOVA; P < 0.001). Error bars indicate standard deviation.
14-3-3β participates in the nuclear import of myopodin
At this point, two alternative mechanisms for the 14-3-3β–mediated nuclear localization of myopodin were possible: (1) 14-3-3β binding facilitates nuclear import or (2) blocks the LMB-sensitive (Weins et al., 2001) nuclear export of myopodin. To address this matter, we repeated the transfection studies with the GFP–14-3-3βΔN construct in the presence of LMB (Fig. 2 A, top). If 14-3-3β inhibits the nuclear export of myopodin, LMB should rescue GFP–14-3-3βΔN-transfected cells and result in the nuclear relocalization of myopodin. In contrast, if 14-3-3β binding mediates the nuclear import of myopodin, LMB should not affect the cytoplasmic localization of myopodin in cells expressing GFP–14-3-3βΔN. As shown in Fig. 2 A (bottom right), myoblasts expressing GFP–14-3-3βΔN did not show the nuclear localization of endogenous myopodin in the presence of LMB, whereas in untransfected cells or in cells expressing GFP full-length 14-3-3β (Fig. 2 A, top right), myopodin was found in the nucleus. To test the activity of LMB, we performed control experiments with transfected C2C12 myoblasts that expressed GFP–C19, the NH2-terminal fragment (aa 1–276) of integrase interactor 1, which contain an LMB-sensitive nuclear export signal (Craig et al., 2002). In untreated cells, C19 was predominantly located in the cytoplasm and accumulated in the nucleus after the inhibition of CRM1-mediated export by LMB, thereby proving the activity of LMB under these conditions (unpublished data). Together, these results suggest that 14-3-3β is involved in the nuclear import of myopodin.
Two NLSs and two 14-3-3–binding motifs are required for the nuclear import of myopodin
Myopodin contains two putative nuclear localization sequences: NLS#1 and NLS#2 (Fig. 1 A). To assess the functionality of these NLSs, we deleted them separately (ΔNLS#1 and ΔNLS#2) or in combination (ΔNLS#1 + 2), and the subcellular distribution was analyzed by confocal microscopy (Fig. 2 B). In addition, myopodin-induced F-actin filaments (Weins et al., 2001) were visualized with rhodamine-labeled phalloidin, and nuclei were visualized with DAPI. GFP full-length myopodin was mainly detected in the nucleus (Fig. 2 B). The quantitative analysis showed that 73.4% of GFP full-length myopodin was found in the nucleus, compared with 14.8% for ΔNLS#1 and 13.4% for ΔNLS#2 (Fig. 2 C). The simultaneous deletion of both NLSs (ΔNLS#1 + 2) did not further decrease the amount of nuclear myopodin (14.2%; Fig. 2 C). The differences between full-length myopodin and ΔNLS#1, ΔNLS #2, and ΔNLS #1 + 2 were significant (P < 0.01; t test). Hence, both NLSs of myopodin are functional and necessary for an efficient nuclear import of myopodin. However, even in the absence of both NLSs, ∼14% of total GFP fusion protein is still found in the nucleus, suggesting that other domains are also involved in the nuclear import of myopodin. To determine whether the observed nuclear exclusion of myopodin in the presence of 14-3-3βΔN (Fig. 2 A) resulted from a functional loss of the 14-3-3β–myopodin interaction, we next expressed myopodin deletion constructs that lacked the identified 14-3-3–binding motifs as GFP fusion proteins in C2C12 myoblasts (Fig. 2 B). The deletion of either one (Δ14-3-3#1 and Δ14-3-3#2) or both (Δ14-3-3#1 + 2) 14-3-3–binding motifs resulted in a dramatic decrease of nuclear myopodin. 20% of Δ14-3-3#1, 21.8% of Δ14-3-3#2, and 19.1% of Δ14-3-3#1 + 2 were localized in the nucleus (Fig. 2 C), which is a significant difference from the 73.4% for full-length myopodin (P < 0.01; t test). Next, we examined whether 14-3-3β can act synergistically with NLSs to maximize the efficacy of nuclear import. Therefore, we created a GFP–myopodin construct that lacked both 14-3-3–binding motifs and both NLSs (Fig. 2 B, Δ14-3-3#1 + 2 + ΔNLS#1 + 2). GFP–myopodinΔ14-3-3#1 + 2 + ΔNLS#1 + 2 showed 6.4% nuclear localization (Fig. 2 C), which was significantly less when compared with ΔNLS#1 + 2 or Δ14-3-3#1 + 2 (P < 0.01; t test). Together, both NLSs and both 14-3-3–binding sites are required for the efficient nuclear import of myopodin.
The two 14-3-3 motifs are necessary for the binding of recombinant myopodin to endogenous 14-3-3β in myoblasts
To determine whether the various myopodin mutant forms (Fig. 2 C) can bind to endogenous 14-3-3β, FLAG-tagged proteins were expressed in C2C12 myoblasts and were immunoprecipitated with anti-FLAG antibody (Fig. 3 A, bottom). Similar to full-length myopodin (FLAG-full-length), FLAG–Δ14-3-3#1 and FLAG-Δ14-3-3#2 coprecipitated endogenous 14-3-3β, albeit to a lesser extent (Fig. 3 A, top). In contrast, FLAG-Δ14-3-3#1 + 2 did not bind to myopodin. These results corroborate the interaction studies in transfected HEK-293 cells (Fig. 1 H) and suggest that binding to 14-3-3β is required for the nuclear import of myopodin. The deletion of the two NLSs separately (FLAG-ΔNLS#1 and FLAG-ΔNLS#2) or together (FLAG-ΔNLS#1 + 2) did not abrogate the myopodin–14-3-3β interaction. Therefore, the two NLSs in myopodin are not necessary for its binding to 14-3-3β. As expected, FLAG–Δ14-3-3#1 + 2 + ΔNLS#1 + 2 did not interact with endogenous 14-3-3β (Fig. 3 A).
Figure 3.

14-3-3 binding to myopodin is required for the interaction between myopodin and importin α. (A) Endogenous 14-3-3β from C2C12 myoblasts coimmmunoprecipitates with myopodin (FLAG-full-length). FLAG–Δ14-3-3#1 and FLAG–Δ14-3-3#2 show impaired binding to 14-3-3β. No binding to 14-3-3β is found for FLAG–Δ14-3-3#1 + 2. In contrast, deletion of one or both NLSs (FLAG-ΔNLS#1, FLAG-ΔNLS#2, and FLAG-ΔNLS#1 + 2) does not interfere with the binding of myopodin to 14-3-3β. Combined deletion of all four motifs (FLAG–Δ14-3-3#1 + 2 + ΔNLS#1 + 2) abrogates the binding of myopodin to 14-3-3β. (B) Binding of 14-3-3β (top) and importin α (middle) to FLAG-tagged myopodin (bottom). Single NLS deletions (FLAG-ΔNLS#1 and FLAG-ΔNLS#2) do not impair the expression of myopodin or the interaction of myopodin with 14-3-3β or importin α. In contrast, the absence of both NLSs (FLAG-ΔNLS#1 + 2) causes loss of importin α binding, whereas 14-3-3β binding is preserved. Deletion of 14-3-3–binding motifs in myopodin (FLAG–Δ14-3-3#1, FLAG–Δ14-3-3#2, and FLAG–Δ14-3-3#1 + 2) abrogates the interaction of myopodin with 14-3-3β and with importin α. (C) The 14-3-3 inhibitory peptide R18 abrogates the binding of myopodin to 14-3-3β (top) and to importin α (bottom). (D) Immobilized GST–importin α, but not GST alone, binds purified FLAG-myopodin in the presence of purified FLAG–14-3-3β. In contrast, no interaction is found between GST–importin α and FLAG–14-3-3β or FLAG-myopodin alone.
Removal of 14-3-3 motifs results in the loss of myopodin binding to importin α
Lysine-rich, classical NLSs (sequence K-K/R-X-K/R) that were originally identified in the SV40 T antigen (Kalderon et al., 1984; Lanford and Butel, 1984) are recognized by the importin α/β dimer. Importin α directly binds to the basic NLSs of cargo proteins but needs importin β to cross the nuclear pore complex (Weis, 1998). To study the interaction between the NLSs of myopodin and importin α, recombinant FLAG-myopodin proteins were analyzed for their ability to bind importin α (Fig. 3 B). FLAG-myopodin proteins were expressed in HEK-293 cells, were immunoprecipitated with anti-FLAG antibody, and were incubated with total protein extract from Xenopus laevis embryos. Eluates were analyzed with anti–14-3-3β (Fig. 3 B, top) or anti–importin α (Hachet et al., 2004; Fig. 3 B, middle) antibodies. Immunolabeling with anti-FLAG antibody demonstrated that each binding reaction included equal amounts of the various FLAG-tagged myopodin proteins (Fig. 3 B, bottom). Using full-length FLAG-myopodin, 14-3-3β and importin α were coprecipitated, showing an interaction between myopodin and both proteins (Fig. 3 B). As shown for the binding of endogenous 14-3-3β in myoblasts (Fig. 3 A), the deletion of the NLSs (FLAG-ΔNLS#1, FLAG-ΔNLS#2, and FLAG-ΔNLS#1 + 2) did not impair the myopodin–14-3-3β interaction (Fig. 3 B). In contrast, FLAG-ΔNLS#1 + 2 did not bind importin α. The deletion of either NLS (FLAG-ΔNLS#1 or FLAG-ΔNLS#2) did not abrogate the interaction between myopodin and importin α (Fig. 3 B). The removal of the two 14-3-3–binding domains, separately (FLAG-Δ14-3-3#1 and FLAG-Δ14-3-3#2) or together (FLAG-Δ14-3-3#1 + 2), resulted in the loss of the 14-3-3 and importin α binding (Fig. 3 B). Hence, the presence of 14-3-3 motifs is necessary for the interaction of myopodin with importin α, whereas the two NLSs are dispensable for the binding of myopodin to 14-3-3.
Inhibition of 14-3-3 binding with a high-affinity peptide antagonist abrogates the interaction between myopodin and importin α
To further confirm this finding with another approach, the experiment was repeated with full-length myopodin in the absence or presence of the R18 peptide, a well-established, high-affinity competitive inhibitor of 14-3-3 binding (Wang et al., 1999). R18 abolished the interaction between FLAG-myopodin and endogenous 14-3-3β and between the binding of myopodin and importin α (Fig. 3 C). This result lends further support to our hypothesis that the binding of myopodin to importin α critically depends on the interaction of myopodin with 14-3-3.
The direct binding of myopodin to importin α requires the presence of 14-3-3
To determine whether myopodin can directly bind to importin α, we conducted in vitro reconstitution studies with purified importin α, myopodin, and 14-3-3β. To do so, purified GST-tagged importin α was incubated with purified, FLAG-tagged myopodin, FLAG-tagged 14-3-3β, or both together and analyzed by GST pull-down studies (Fig. 3 D). When importin α was incubated with myopodin (Fig. 3 D, left) or 14-3-3β (Fig. 3 D, middle) alone, no interaction was found. In contrast, when all three proteins were present, both myopodin and 14-3-3β could be eluted together with importin α, but not with GST alone (Fig. 3 D, right). These results confirm that 14-3-3 cannot directly bind to importin α, as expected from the absence of a functional NLS (Brunet et al., 2002). Furthermore, the direct binding of purified myopodin to importin α only occurs in the presence of 14-3-3. Finally, these results demonstrate that the interaction between 14-3-3β and importin α is indirect and is mediated by myopodin.
Myopodin is phosphorylated in vivo
14-3-3 proteins are phosphoserine/threonine-binding proteins (Muslin et al., 1996), and many 14-3-3 interactions depend on the phosphorylation of the target protein (Yaffe, 2002). Therefore, we examined whether the myopodin–14-3-3 interaction is regulated by phosphorylation. To this end, purified FLAG-myopodin was dephosphorylated with λ protein phosphatase (λ-PPase), which reduced the molecular size of myopodin (Fig. 4 A). Moreover, λ-PPase treatment converted the fuzzier signal of the phosphorylated protein into a more compact and sharper band (Fig. 4 A, top). These findings show that myopodin is phosphorylated in vivo.
Figure 4.

Two phosphorylated residues in myopodin mediate 14-3-3 binding. (A) SDS-PAGE analysis showing reduced molecular weight of purified FLAG-myopodin after dephosphorylation with λ-PPase. (B) Dephosphorylation of myopodin abrogates binding to GST–14-3-3β. The interaction is also prevented by the 14-3-3–blocking peptide R18. (C) Binding of purified FLAG-myopodin to endogenous 14-3-3β from C2C12 myoblasts is abrogated by dephosphorylation of myopodin (top). In contrast, binding of endogenous myopodin to purified FLAG–14-3-3β is not affected by the dephosphorylation of 14-3-3β (bottom). (D) Dephosphorylation with λ-PPase abrogates the interaction of FLAG-myopodin (bottom) with endogenous 14-3-3β (top) and importin α (middle) from X. laevis extracts. (E) Putative phosphoacceptor sites within the 14-3-3–binding motifs of myopodin. S225 in motif#1, as well as T272 and S273 in motif#2, were substituted with alanine to remove putative phosphorylation sites. Replacement with aspartic acid or glutamic acid was done to mimic phosphorylation. (F) Purified FLAG-tagged, wild-type myopodin interacts with GST–14-3-3β. Substitution of S225 or T272 with alanine (S225A, T272A, and S225AT272A) abrogates 14-3-3β binding. Replacement of S273 with alanine (S273A) does not interfere with the binding of myopodin to 14-3-3β. (G) Substitutions of S225 or T272 with aspartic or glutamic acid, respectively, does not alter the binding of myopodin to 14-3-3 (top). However, after dephosphorylation, only S225DT272E retains strong binding to GST–14-3-3β. Single mutations (S225D and T272E) bind significantly less, and wild-type binding is abrogated.
The binding to 14-3-3β requires the phosphorylation of myopodin
Next, we determined whether the phosphorylation of myopodin is required for binding to 14-3-3β (Fig. 4 B). In the phosphorylated state (Fig. 4 B, left), myopodin bound to 14-3-3β, but not after dephosphorylation with λ-PPase (Fig. 4 B, right). This binding was also abrogated by the 14-3-3 inhibitory peptide R18 (Fig. 4 B). Therefore, we concluded that the interaction between myopodin and 14-3-3β is dependent on the phosphorylation of myopodin. To confirm this finding, we analyzed the ability of dephosphorylated, recombinant myopodin to bind endogenous 14-3-3β from C2C12 myoblasts (Fig. 4 C). FLAG-tagged myopodin was purified from HEK-293 cells and was incubated with C2C12 protein extract. As shown above for recombinant proteins (Fig. 2 A), myopodin binds endogenous 14-3-3β, whereas the dephosphorylation with λ-PPase abolished the interaction (Fig. 4 C, top). 14-3-3 proteins, including 14-3-3β, can also be phosphorylated (Fu et al., 2000). In a converse experiment, to determine whether the phosphorylation of 14-3-3β is required for myopodin binding, purified FLAG–14-3-3β was incubated with C2C12 protein extract. 14-3-3β could bind endogenous myopodin independent of λ-PPase treatment (Fig. 4 C, bottom). This finding is consistent with the ability of GST–14-3-3β that is purified from bacteria to bind myopodin (Fig. 1 E). Together, the phosphorylation of myopodin is required for 14-3-3 binding, whereas the phosphorylation of 14-3-3 is not required for myopodin binding.
Dephosphorylation of myopodin abrogates importin α binding
To further determine whether the phosphorylation of myopodin is not only required for 14-3-3 binding but also for importin α binding, FLAG-tagged myopodin was treated with λ-PPase and was incubated with X. laevis protein extract (Fig. 4 D). In the absence of λ-PPase treatment, FLAG-myopodin bound to 14-3-3β and importin α (Fig. 4 D, left). In contrast, the dephosphorylation of myopodin with λ-PPase abrogated the interaction with 14-3-3β and importin α (Fig. 4 D, right). These findings further support the hypothesis that the interaction of myopodin with importin α requires the phosphorylation-dependent binding of myopodin to 14-3-3.
S225 and T272 mediate the phosphorylation-dependent binding of myopodin to 14-3-3β
The removal of the 14-3-3–binding sites (Fig. 1 H and Fig. 3, A and B) and the dephosphorylation of myopodin (Fig. 4, B–D) abrogate 14-3-3 binding, suggesting that the amino acids mediating the phosphorylation-dependent interaction are localized within the two 14-3-3–binding motifs. According to the consensus 14-3-3–binding motifs RSxpS/TxP (mode 1) and RxxxpS/TxP (mode 2; Yaffe et al., 1997), we predicted that S225 and T272 were phosphoacceptor sites (Fig. 4 E). To test this hypothesis, we mutated these amino acids to alanine (S225A and T272A), which cannot be phosphorylated by protein kinases. As a control, S273, which was not expected to be crucial for 14-3-3 binding, was also mutated (S273A; Fig. 4 E). Myopodin and its point mutation variants were expressed as FLAG fusion proteins in HEK-293 cells and were purified and incubated with immobilized GST–14-3-3β. Wild-type myopodin could bind GST–14-3-3β, whereas no interaction was found for S225A, T272A, or S225AT272A (Fig. 4 F). In contrast, S273A showed normal 14-3-3 binding. As expected from the single mutation S225A, the combined removal of serine 225 and serine 273 (S225AS273A) also resulted in the loss of 14-3-3 binding (Fig. 4 F). Together, S225 and T272, but not S273, are crucial for the interaction of myopodin with 14-3-3β. It also confirms the finding that the two 14-3-3–binding motifs in myopodin are necessary and sufficient for the efficient interaction with 14-3-3β (Fig. 1 H). To determine whether the phosphorylation of S225 and T272 is required for 14-3-3 binding, the phosphorylation of both residues was mimicked by the substitution of negatively charged amino acids. S225 was replaced by aspartic acid (S225D) and T272 by glutamic acid (T272E; Fig. 4 G). The single (S225D and T272E) or combined substitutions (S225DT272E) did not alter binding to GST–14-3-3β (Fig. 4 G, top). In contrast, after dephosphorylation with λ-PPase and before GST–14-3-3β binding (Fig. 4 G, bottom), the binding of wild-type myopodin was abrogated, whereas the binding of S225D and T272E was dramatically reduced. In contrast, S225DT272E retained strong binding to 14-3-3β, demonstrating that the phosphorylation of S225 and S272 is necessary for the efficient binding of myopodin to 14-3-3β.
S225 and T272 mediate the phosphorylation-dependent nuclear import of myopodin
To determine whether the aa S225 and T272 of myopodin are not only required for 14-3-3 binding but are also required for the nuclear import of myopodin, the subcellular distribution of GFP-tagged wild-type and mutant myopodin was analyzed by confocal microscopy (Fig. 5 A) and was quantified as described above (Fig. 2 C). As shown previously (Fig. 2 B; Weins et al., 2001), wild-type myopodin was mainly localized in the nucleus (Fig. 5 A). The quantitative analysis showed 77.4% nuclear localization (Fig. 5 B). The removal of single phosphorylation sites (S225A and T272A) resulted in a significantly decreased nuclear localization (20.1% for S225A and 20.5% for T272A; Fig. 5 B) versus 77.4% for wild-type myopodin (P < 0.01; t test). The combined replacement of S225 and T272 (Fig. 5 A, S225AT272A) further decreased nuclear myopodin to 10.5% (Fig. 5 B), a significant difference from the single mutations S225A and T272A (P < 0.01; t test). In contrast, S273A showed normal nuclear localization (78.8% nuclear; Fig. 5 B). The active mutants S225D, T272E, or S273D showed 75.6%, 80.3%, and 75.7% nuclear localization, respectively. The highest percentage of nuclear myopodin was found for S225DT272E (Fig. 5 A). However, the difference to wild type (83.5% for S225DT272E versus 77.4% for wild-type myopodin) was not significant. Altogether, these experiments show that the phosphorylation of S225 and T272 is required for the efficient nuclear import of myopodin.
Figure 5.

Two phosphorylated residues regulate nuclear localization of myopodin. (A) Confocal imaging of GFP-tagged myopodin in C2C12 myoblasts. Wild-type myopodin shows a predominantly nuclear localization (top), and S225AT272A displays a dramatically decreased nuclear localization (middle). In contrast, S225DT272E shows a primarily nuclear localization (bottom). Rhodamine-labeled phalloidin identifies myopodin-induced actin bundles, and DAPI visualizes nuclei. (B) Quantitative analysis is presented as a percentage of nuclear GFP–myopodin. 77.4% of wild-type myopodin is detected in the nucleus. S225A and T272A show 20.1% and 20.5% nuclear myopodin, respectively. S225AT272A shows a further decrease to 10.5%. In contrast, S273A does not alter nuclear localization (78.8%). S225D, S273D, and T272E show 75.6%, 75.7%, and 80.3% nuclear localization, respectively. S225DT272E displays 83.5% nuclear localization. Statistical significance was confirmed by ANOVA (P < 0.001). Error bars indicate standard deviation.
Discussion
This study uncovered a novel mechanism for the phosphorylation-dependent nuclear import of NLS-containing proteins. To the best of our knowledge, this is the first study to demonstrate that 14-3-3 promotes the nuclear import of a cargo protein. In particular, we established that phosphorylated myopodin directly interacts with 14-3-3 proteins, and that this interaction is functionally significant because it promotes the binding of myopodin to importin α and, thereby, promotes the nuclear import of myopodin.
Nuclear import is regulated at several levels. The binding of NLS-containing proteins to cytosolic receptors of the importin/karyopherin superfamily is critical because the affinity of this interaction determines transport efficiency (Weis, 2003). Therefore, targeting sequence recognition is a key control point in the regulation of nuclear import (Jans et al., 2000). One way to regulate nuclear transport is by changing the phosphorylation status of the cargo protein. Phosphorylation of the SV40 large tumor antigen and the Drosophila melanogaster morphogen dorsal upstream of the NLSs directly enhances the affinity between NLSs and importin α/β1 (Hubner et al., 1997; Briggs et al., 1998). The deletion of the phosphorylation sites in these nuclear cargoes causes a decreased nuclear import rate, which reveals the importance of serine/threonine phosphorylation for nuclear import efficiency. Phosphorylation of a cargo protein may modulate the affinity between NLS and importin α or may cause a conformational change in the cargo, thereby exposing an NLS (Harreman et al., 2004). In addition, phosphorylation may cause the release or binding of a heterologous, NLS-masking protein.
Recent proteomic analyses of 14-3-3–binding proteins revealed that importins were putative 14-3-3 targets (Meek et al., 2004). However, 14-3-3 proteins do not have an intrinsic NLS, and, therefore, the direct binding of importins to 14-3-3 is unlikely (Brunet et al., 2002). Instead, these interactions are indirect and are mediated by proteins like myopodin that bind both 14-3-3 proteins and importin α (Fig. 3 D).
14-3-3 proteins are highly acidic, chaperone-like molecules that can induce conformational changes in their binding partners. Myopodin, on the other hand, is a very basic protein with an isolectric point of 9.34 (Weins et al., 2001). Hence, it seems conceivable that 14-3-3 binding neutralizes the negative charge of myopodin. This, in turn, may change the structure of myopodin, thereby unmasking and exposing NLSs. In this study, we have shown that myopodin contains two functional 14-3-3–binding sites that are required for efficient 14-3-3 binding (Fig. 1 H). Because each monomer of a 14-3-3 dimer binds its target in opposite directions (Yaffe et al., 1997), the binding of a 14-3-3 dimer could change the predicted linear structure of myopodin (Weins et al., 2001) to a U-shaped conformation. This is a potential regulatory conformational change in myopodin that could alter its biochemical features, such as the capability to interact with other proteins (e.g., importin α). Clearly, structural analysis of the myopodin–14-3-3 interaction will be required to confirm or refute this hypothesis.
14-3-3 proteins can participate in protein translocation into mitochondria (Alam et al., 1994) and chloroplasts (May and Soll, 2000) by serving as cytoplasmic chaperones. In this study, we describe a novel role of 14-3-3 as a regulator of nuclear import by showing that 14-3-3 binding enables myopodin to interact with importin α. Protein import into mitochondria and chloroplasts is mechanistically and evolutionary very different from nuclear import. Nevertheless, the results of this study suggest that cargoes with different subcellular destinations may share 14-3-3 as a cytoplasmic chaperone in order to achieve an import-compatible structure.
14-3-3 proteins are involved in the regulation of cell proliferation, differentiation, and cell death (van Hemert et al., 2001). For example, the epithelial 14-3-3 isoform σ serves as a tumor suppressor (Hermeking, 2003), and the loss of 14-3-3σ expression can cause cell transformation (Dellambra et al., 2000). Several studies have shown a decrease or loss of 14-3-3σ expression in transformed cells (Simooka et al., 2004; Urano et al., 2004), including transitional urinary bladder carcinomas (Ostergaard et al., 1997; Moreira et al., 2004). Myopodin not only binds to 14-3-3σ but also acts as a tumor suppressor in bladder carcinomas (Sanchez-Carbayo et al., 2003). In fact, the relocalization of myopodin from the nucleus to the cytoplasm predicts the clinical outcome of these tumors (Sanchez-Carbayo et al., 2003). Based on the results of this study, it is tempting to speculate that the loss of nuclear myopodin in invasive bladder tumors results from the loss of 14-3-3σ expression or from the loss of myopodin phosphorylation, which, in turn, abrogates 14-3-3 and importin α binding. Future studies will explore the phosphorylation stage of myopodin in normal urothelium and in bladder tumors.
To summarize, this study has helped to identify a functional role for 14-3-3 in promoting nuclear import. In particular, we have shown that importin α binding and the subsequent nuclear import of the tumor suppressor myopodin are regulated by the serine/threonine phosphorylation-dependent binding of myopodin to 14-3-3 (Fig. 6). Altogether, these data provide a novel paradigm for the regulation of nuclear import by 14-3-3 in that 14-3-3 regulates the binding of a phosphorylated cargo protein to importin α and the nuclear import machinery.
Figure 6.

A model for phosphorylation- and 14-3-3-dependent nuclear import of myopodin. (1) When phosphoacceptor sites in 14-3-3–binding motifs#1 (S225) and #2 (T272) are not phosphorylated, myopodin cannot interact with 14-3-3. Therefore, the NLSs in myopodin are not accessible for importin α binding, and myopodin cannot enter the nucleus. (2) After phosphorylation by serine/threonine protein kinases, myopodin binds to 14-3-3, rendering the NLSs accessible for importin α binding. (3) Importin α binds to the NLSs and mediates the nuclear import of myopodin (4).
Materials and methods
Yeast two-hybrid screen
The mouse myopodin fragment Myo-2 (nt 559–1260) that was fused to the GAL4 DNA-binding domain was used to screen a pretransformed mouse embryonic (E17.5) cDNA library according to the manufacturer's protocol (MATCHMAKER Two-Hybrid System 3; CLONTECH Laboratories, Inc.).
Cloning and vectors
All cDNA fragments used in this study were amplified by RT-PCR using Pfu DNA polymerase (Stratagene) and were cloned into pEGFP-N1 (CLONTECH Laboratories, Inc.), pFLAG-cytomegalo virus-5a, b, and c (Sigma-Aldrich), or a modified glutathione S-transferase fusion vector (pGEX; Ron and Dressler, 1992), which was provided by Ben Margolis (University of Michigan Medical School, Ann Arbor, MI). Point mutations were generated with the QuikChange Multi Site-Directed Mutagenesis Kit (Stratagene). All constructs were verified by DNA sequencing. Mouse myopodin cDNA and its mutated variants were cloned into pEGFP-N1 and pFLAG vectors. GST–14-3-3β, ɛ, and η cDNAs were provided by Andrey Shaw (Washington University School of Medicine, St. Louis, MO), and GST–14-3-3ɛ K49E, γ, σ, τ, and ζ were provided by Michael Yaffe (Massachusetts Institute of Technology, Cambridge, MA). The 14-3-3β cDNA and the NH2-terminal deletion mutant βΔN (aa 128–246) were subcloned into pEGFP-N1, pFLAG, and pGEX. The X. laevis importin α-1a cDNA (provided by Ian Mattaj, European Molecular Biology Laboratory, Heidelberg, Germany) was cloned into pGEX. The GFP–C19 construct was provided by Ganjam Kalpana (Albert Einstein College of Medicine, Bronx, NY).
Antibodies
Rabbit antimyopodin (Weins et al., 2001) and rabbit anti–X. laevis importin α (Hachet et al., 2004) have been previously described. Rabbit anti–14-3-3β (C-20) was purchased from Santa Cruz Biotechnology, Inc.; the mouse monoclonal anti–α-actinin antibody (sarcomeric, clone EA-53) was purchased from Sigma-Aldrich; and the goat anti-GST was purchased from Amersham Biosciences. HRP-coupled secondary antibodies were purchased from Promega. For immunofluorescence, Texas red–conjugated goat anti–rabbit (Jackson ImmunoResearch Laboratories) and AlexaFluor488 (Molecular Probes) or Cy3-conjugated goat anti–mouse (Jackson ImmunoResearch Laboratories) were used. For immunoprecipitation of FLAG fusion proteins, monoclonal anti–FLAG-M2 antibody that was covalently attached to agarose (Sigma-Aldrich) was used. GFP fusion proteins were detected with rabbit anti-GFP (Living Colors A.v. peptide antibody; CLONTECH Laboratories, Inc.), and FLAG fusion proteins were detected with anti–FLAG-M2 antibody.
Cell culture and transfection
Cell culture of C2C12 myoblasts and HEK-293 cells as well as heat shock were performed as described previously (Weins et al., 2001). For biochemical analyses, transient transfections were performed using Lipofectamin2000 (Invitrogen).
Immunofluorescence and confocal microscopy
Immunofluorescence microscopy was performed as described previously (Weins et al., 2001). Anti–14-3-3β was used at 1:50; anti–α-actinin was used at 1:1,000; rhodamine-labeled phalloidin (Molecular Probes) was used at 1:500; and DAPI was used at 1:5,000.
Quantification of nuclear myopodin
We used a high resolution Nomarski objective (60×, NA 1.4; Olympus) and a high resolution epifluorescence with a precision stepper motor (model 99S001; Ludl) to measure the depth of individual cells. The adherent cells ranged from 5 to 8 μm in depth. The plasma membrane was stretched tightly over the top and bottom of the nuclei in these cells, and the nucleus appeared to span the entire depth of the cell. For the quantitative analysis of the localization of GFP fusion proteins, phase-contrast images of transfected myoblasts were collected. Using a widefield microscope system (model IX70; Olympus), the axial depth that was detected was deeper than 5 μm, and, therefore, fluorescence was accurately detected from the entire depth of the cells. To analyze transfected cells, whole cell areas, nuclei, and backgrounds of 25 cells per construct were traced using IPLab Image Analysis software (Scanalytics). For further calculations, Excel software (Microsoft) was used. The mean background level was subtracted from the means of the entire cell and the nucleus alone. To determine the content of GFP fusion protein in the whole cell versus that in the nucleus, the mean intensity of the entire cell versus the nucleus was multiplied by the area of each. Total GFP fusion protein in the cytoplasm was calculated by subtracting the mass of the nucleus from the mass of the total cell. The percentage of nuclear myopodin was calculated by dividing the mass of the nucleus by the mass of the cytoplasm.
Tissue and cell lysates
GST pull-down experiments were performed as described previously (Schwarz et al., 2001). For coimmunoprecipitation of endogenous proteins from the mouse heart, the tissue was homogenized in extraction buffer (50 mM Tris, pH 7.5, 150 mM NaCl, 0.1% SDS, 0.5% deoxycholic acid, and 1% NP-40) in a 1:3 wt/vol ratio. To purify FLAG-tagged proteins, HEK-293 cells were cultured in a 10-cm dish to ∼90% confluence and were transfected with FLAG cDNA constructs. After 24 h, cells were washed with PBS and were harvested with a cell scraper in 900 μl of modified radioimmunoprecipitation buffer (40 mM Tris, pH 7.4, 200 mM NaCl, 1% Triton X-100, 0.25% deoxycholic acid, 1 mM EDTA, and 1 mM EGTA). Lysates were incubated on ice for 30 min and were cleared at 14,000 g for 15 min at 4°C. For coimmunoprecipitation of FLAG- or GFP-tagged fusion proteins, HEK-293 cells were grown on a 10-cm dish, were cotransfected at a confluence of ∼90%, and were harvested on ice after 24 h using 5 ml PBS/50 mM EDTA. Cells were pelleted using centrifugation at 1,000 g for 5 min at 4°C and were washed twice with 5 ml PBS. For cell lysis, the pellet was resuspended in 1 ml of immunoprecipitation (IP) buffer (50 mM Tris, pH 8.0, 150 mM NaCl, 50 mM KCl, 10 mM EDTA, 10 mM EGTA, 1.5% Triton X-100, and 0.75% NP-40) and was incubated on ice for 30 min. The cell lysate was cleared by centrifugation for 10 min at 5,000 g. To coimmunoprecipitate endogenous 14-3-3β from myoblasts, C2C12 cells were grown on a 10-cm dish and were transfected with FLAG constructs at a confluence of ∼90%. FLAG fusion proteins were isolated as described for HEK-293 cells. For X. laevis protein extracts, 100 2-d-old embryos were homogenized in 2 ml IP buffer. After centrifuging at 10,000 g for 30 min at 4°C, the supernatant was used for binding studies.
Western blotting and endogenous coimmunoprecipitation
SDS-PAGE and Western blotting were performed as described previously (Mundel et al., 1997; Weins et al., 2001). Antibodies were used at the following ratios: myopodin-specific antibody SRIB2 at 1:300; anti–14-3-3β antibody at 1:2,000; anti–importin α antibody at 1:1,000; antibody against FLAG at 1:10,000; antibody against GFP at 1:300; anti-GST at 1:10,000; and HRP-conjugated secondary antibodies at 1:20,000. The immunoreaction was visualized by ECL (Amersham Biosciences). To immunoprecipitate endogenous protein complexes, 1 ml of mouse heart extract was incubated overnight with 2 μg of antibodies at 4°C under rotation. Immune complexes were precipitated with 50 μl of protein A/G–Sepharose (Sigma-Aldrich) and were eluted in 100 μl SDS sample buffer. Anti-GFP IgG served as a negative control.
GST-binding assays
GST pull-down studies from tissue and cell extracts were performed as reported previously (Schwarz et al., 2001). To study the interaction between GST fusion proteins and FLAG-tagged proteins, GST proteins were expressed in bacteria and were purified using glutathione-coupled agarose as described previously (Schwarz et al., 2001). 1 μg of purified FLAG fusion protein in 500 μl PBS was added to the glutathione beads, and the reaction was incubated at 4°C under rotation for 2 h. For 14-3-3 inhibition studies, 1 mM R18 peptide (provided by Thomas McDonald, Albert Einstein College of Medicine; Wang et al., 1999) was added to the FLAG fusion protein in PBS. For the triple binding assay with purified importin α, myopodin, and 14-3-3β, the immobilized GST fusion protein was incubated with 1 μg each of two purified FLAG proteins. Silver staining and Western blot analysis confirmed the purity of FLAG-tagged myopodin and FLAG-tagged 14-3-3β. The beads were collected by centrifugation and were washed five times in 1 ml PBS, and bound proteins were eluted by boiling in 150 μl Laemmli buffer. Eluates were analyzed by SDS-PAGE and by immunoblotting.
Acknowledgments
We thank Svetlana Ratner and Michael Cammer for help with confocal and quantitative microscopy and Matthew Schmerer for technical help.
Jun Oh was supported by the Deutsche Forschungsgemeinschaft and the Kidney and Urology Foundation of America. This work was supported by the National Institutes of Health grants DA18886, DK57683, and DK062472 to P. Mundel and grant AR41480 to R.H. Singer.
Abbreviations used in this paper: λ-PPase, λ protein phosphatase; IP, immunoprecipitation; LMB, leptomycin B; NLS, nuclear localization signal; pGEX, glutathione S-transferase fusion vector.
References
- Alam, R., N. Hachiya, M. Sakaguchi, S. Kawabata, S. Iwanaga, M. Kitajima, K. Mihara, and T. Omura. 1994. cDNA cloning and characterization of mitochondrial import stimulation factor (MSF) purified from rat liver cytosol. J. Biochem. (Tokyo). 116:416–425. [DOI] [PubMed] [Google Scholar]
- Bihn, E.A., A.L. Paul, S.W. Wang, G.W. Erdos, and R.J. Ferl. 1997. Localization of 14-3-3 proteins in the nuclei of arabidopsis and maize. Plant J. 12:1439–1445. [DOI] [PubMed] [Google Scholar]
- Briggs, L.J., D. Stein, J. Goltz, V.C. Corrigan, A. Efthymiadis, S. Hubner, and D.A. Jans. 1998. The cAMP-dependent protein kinase site (Ser312) enhances dorsal nuclear import through facilitating nuclear localization sequence/importin interaction. J. Biol. Chem. 273:22745–22752. [DOI] [PubMed] [Google Scholar]
- Brunet, A., F. Kanai, J. Stehn, J. Xu, D. Sarbassova, J.V. Frangioni, S.N. Dalal, J.A. DeCaprio, M.E. Greenberg, and M.B. Yaffe. 2002. 14-3-3 transits to the nucleus and participates in dynamic nucleocytoplasmic transport. J. Cell Biol. 156:817–828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cahill, C.M., G. Tzivion, N. Nasrin, S. Ogg, J. Dore, G. Ruvkun, and M. Alexander-Bridges. 2001. Phosphatidylinositol 3-kinase signaling inhibits DAF-16 DNA binding and function via 14-3-3-dependent and 14-3-3-independent pathways. J. Biol. Chem. 276:13402–13410. [DOI] [PubMed] [Google Scholar]
- Celis, J.E., B. Gesser, H.H. Rasmussen, P. Madsen, H. Leffers, K. Dejgaard, B. Honore, E. Olsen, G. Ratz, J.B. Lauridsen, et al. 1990. Comprehensive two-dimensional gel protein databases offer a global approach to the analysis of human cells: the transformed amnion cells (AMA) master database and its link to genome DNA sequence data. Electrophoresis. 11:989–1071. [DOI] [PubMed] [Google Scholar]
- Craig, E., Z.K. Zhang, K.P. Davies, and G.V. Kalpana. 2002. A masked NES in INI1/hSNF5 mediates hCRM1-dependent nuclear export: implications for tumorigenesis. EMBO J. 21:31–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dalal, S.N., C.M. Schweitzer, J. Gan, and J.A. DeCaprio. 1999. Cytoplasmic localization of human cdc25C during interphase requires an intact 14-3-3 binding site. Mol. Cell. Biol. 19:4465–4479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dellambra, E., O. Golisano, S. Bondanza, E. Siviero, P. Lacal, M. Molinari, S. D'Atri, and M. De Luca. 2000. Downregulation of 14-3-3σ prevents clonal evolution and leads to immortalization of primary human keratinocytes. J. Cell Biol. 149:1117–1130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu, H., R.R. Subramanian, and S.C. Masters. 2000. 14-3-3 proteins: structure, function, and regulation. Annu. Rev. Pharmacol. Toxicol. 40:617–647. [DOI] [PubMed] [Google Scholar]
- Grozinger, C.M., and S.L. Schreiber. 2000. Regulation of histone deacetylase 4 and 5 and transcriptional activity by 14-3-3-dependent cellular localization. Proc. Natl. Acad. Sci. USA. 97:7835–7840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hachet, V., T. Kocher, M. Wilm, and I.W. Mattaj. 2004. Importin alpha associates with membranes and participates in nuclear envelope assembly in vitro. EMBO J. 23:1526–1535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harreman, M.T., T.M. Kline, H.G. Milford, M.B. Harben, A.E. Hodel, and A.H. Corbett. 2004. Regulation of nuclear import by phosphorylation adjacent to nuclear localization signals. J. Biol. Chem. 279:20613–20621. [DOI] [PubMed] [Google Scholar]
- Hermeking, H. 2003. The 14-3-3 cancer connection. Nat. Rev. Cancer. 3:931–943. [DOI] [PubMed] [Google Scholar]
- Hubner, S., C.Y. Xiao, and D.A. Jans. 1997. The protein kinase CK2 site (Ser111/112) enhances recognition of the simian virus 40 large T-antigen nuclear localization sequence by importin. J. Biol. Chem. 272:17191–17195. [DOI] [PubMed] [Google Scholar]
- Jans, D.A., C.Y. Xiao, and M.H. Lam. 2000. Nuclear targeting signal recognition: a key control point in nuclear transport? Bioessays. 22:532–544. [DOI] [PubMed] [Google Scholar]
- Kalderon, D., B.L. Roberts, W.D. Richardson, and A.E. Smith. 1984. A short amino acid sequence able to specify nuclear location. Cell. 39:499–509. [DOI] [PubMed] [Google Scholar]
- Kumagai, A., and W.G. Dunphy. 1999. Binding of 14-3-3 proteins and nuclear export control the intracellular localization of the mitotic inducer Cdc25. Genes Dev. 13:1067–1072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lanford, R.E., and J.S. Butel. 1984. Construction and characterization of an SV40 mutant defective in nuclear transport of T antigen. Cell. 37:801–813. [DOI] [PubMed] [Google Scholar]
- Liu, D., J. Bienkowska, C. Petosa, R.J. Collier, H. Fu, and R. Liddington. 1995. Crystal structure of the zeta isoform of the 14-3-3 protein. Nature. 376:191–194. [DOI] [PubMed] [Google Scholar]
- May, T., and J. Soll. 2000. 14-3-3 proteins form a guidance complex with chloroplast precursor proteins in plants. Plant Cell. 12:53–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKinsey, T.A., C.L. Zhang, and E.N. Olson. 2000. Activation of the myocyte enhancer factor-2 transcription factor by calcium/calmodulin-dependent protein kinase-stimulated binding of 14-3-3 to histone deacetylase 5. Proc. Natl. Acad. Sci. USA. 97:14400–14405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meek, S.E., W.S. Lane, and H. Piwnica-Worms. 2004. Comprehensive proteomic analysis of interphase and mitotic 14-3-3-binding proteins. J. Biol. Chem. 279:32046–32054. [DOI] [PubMed] [Google Scholar]
- Moreira, J.M., P. Gromov, and J.E. Celis. 2004. Expression of the tumor suppressor protein 14-3-3 sigma is down-regulated in invasive transitional cell carcinomas of the urinary bladder undergoing epithelial-to-mesenchymal transition. Mol. Cell. Proteomics. 3:410–419. [DOI] [PubMed] [Google Scholar]
- Mundel, P., H.W. Heid, T.M. Mundel, M. Kruger, J. Reiser, and W. Kriz. 1997. Synaptopodin: an actin-associated protein in telencephalic dendrites and renal podocytes. J. Cell Biol. 139:193–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muslin, A.J., and H. Xing. 2000. 14-3-3 proteins: regulation of subcellular localization by molecular interference. Cell. Signal. 12:703–709. [DOI] [PubMed] [Google Scholar]
- Muslin, A.J., J.W. Tanner, P.M. Allen, and A.S. Shaw. 1996. Interaction of 14-3-3 with signaling proteins is mediated by the recognition of phosphoserine. Cell. 84:889–897. [DOI] [PubMed] [Google Scholar]
- Ostergaard, M., H.H. Rasmussen, H.V. Nielsen, H. Vorum, T.F. Orntoft, H. Wolf, and J.E. Celis. 1997. Proteome profiling of bladder squamous cell carcinomas: identification of markers that define their degree of differentiation. Cancer Res. 57:4111–4117. [PubMed] [Google Scholar]
- Pan, S., P.C. Sehnke, R.J. Ferl, and W.B. Gurley. 1999. Specific interactions with TBP and TFIIB in vitro suggest that 14-3-3 proteins may participate in the regulation of transcription when part of a DNA binding complex. Plant Cell. 11:1591–1602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ron, D., and H. Dressler. 1992. pGSTag—a versatile bacterial expression plasmid for enzymatic labeling of recombinant proteins. Biotechniques. 13:866–869. [PubMed] [Google Scholar]
- Sanchez-Carbayo, M., K. Schwarz, E. Charytonowicz, C. Cordon-Cardo, and P. Mundel. 2003. Tumor suppressor role for myopodin in bladder cancer: loss of nuclear expression of myopodin is cell-cycle dependent and predicts clinical outcome. Oncogene. 22:5298–5305. [DOI] [PubMed] [Google Scholar]
- Schwarz, K., M. Simons, J. Reiser, M.A. Saleem, C. Faul, W. Kriz, A.S. Shaw, L.B. Holzman, and P. Mundel. 2001. Podocin, a raft-associated component of the glomerular slit diaphragm, interacts with CD2AP and nephrin. J. Clin. Invest. 108:1621–1629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sehnke, P.C., R. Henry, K. Cline, and R.J. Ferl. 2000. Interaction of a plant 14-3-3 protein with the signal peptide of a thylakoid-targeted chloroplast precursor protein and the presence of 14-3-3 isoforms in the chloroplast stroma. Plant Physiol. 122:235–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seimiya, H., H. Sawada, Y. Muramatsu, M. Shimizu, K. Ohko, K. Yamane, and T. Tsuruo. 2000. Involvement of 14-3-3 proteins in nuclear localization of telomerase. EMBO J. 19:2652–2661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simooka, H., T. Oyama, T. Sano, J. Horiguchi, and T. Nakajima. 2004. Immunohistochemical analysis of 14-3-3 sigma and related proteins in hyperplastic and neoplastic breast lesions, with particular reference to early carcinogenesis. Pathol. Int. 54:595–602. [DOI] [PubMed] [Google Scholar]
- Todd, A., N. Cossons, A. Aitken, G.B. Price, and M. Zannis-Hadjopoulos. 1998. Human cruciform binding protein belongs to the 14-3-3 family. Biochemistry. 37:14317–14325. [DOI] [PubMed] [Google Scholar]
- Tzivion, G., Y.H. Shen, and J. Zhu. 2001. 14-3-3 proteins; bringing new definitions to scaffolding. Oncogene. 20:6331–6338. [DOI] [PubMed] [Google Scholar]
- Urano, T., S. Takahashi, T. Suzuki, T. Fujimura, M. Fujita, J. Kumagai, K. Horie-Inoue, H. Sasano, T. Kitamura, Y. Ouchi, and S. Inoue. 2004. 14-3-3sigma is down-regulated in humar prostate cancer. Biochem. Biophys. Res. Commun. 319:795–800. [DOI] [PubMed] [Google Scholar]
- van Hemert, M.J., H.Y. Steensma, and G.P. van Heusden. 2001. 14-3-3 proteins: key regulators of cell division, signalling and apoptosis. Bioessays. 23:936–946. [DOI] [PubMed] [Google Scholar]
- Wang, B., H. Yang, Y.C. Liu, T. Jelinek, L. Zhang, E. Ruoslahti, and H. Fu. 1999. Isolation of high-affinity peptide antagonists of 14-3-3 proteins by phage display. Biochemistry. 38:12499–12504. [DOI] [PubMed] [Google Scholar]
- Wang, A.H., M.J. Kruhlak, J. Wu, N.R. Bertos, M. Vezmar, B.I. Posner, D.P. Bazett-Jones, and X.J. Yang. 2000. Regulation of histone deacetylase 4 by binding of 14-3-3 proteins. Mol. Cell. Biol. 20:6904–6912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weins, A., K. Schwarz, C. Faul, L. Barisoni, W.A. Linke, and P. Mundel. 2001. Differentiation- and stress-dependent nuclear cytoplasmic redistribution of myopodin, a novel actin-bundling protein. J. Cell Biol. 155:393–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weis, K. 1998. Importins and exportins: how to get in and out of the nucleus. Trends Biochem. Sci. 23:185–189. [DOI] [PubMed] [Google Scholar]
- Weis, K. 2003. Regulating access to the genome: nucleocytoplasmic transport throughout the cell cycle. Cell. 112:441–451. [DOI] [PubMed] [Google Scholar]
- Xiao, B., S.J. Smerdon, D.H. Jones, G.G. Dodson, Y. Soneji, A. Aitken, and S.J. Gamblin. 1995. Structure of a 14-3-3 protein and implications for coordination of multiple signalling pathways. Nature. 376:188–191. [DOI] [PubMed] [Google Scholar]
- Yaffe, M.B. 2002. How do 14-3-3 proteins work?–Gatekeeper phosphorylation and the molecular anvil hypothesis. FEBS Lett. 513:53–57. [DOI] [PubMed] [Google Scholar]
- Yaffe, M.B., K. Rittinger, S. Volinia, P.R. Caron, A. Aitken, H. Leffers, S.J. Gamblin, S.J. Smerdon, and L.C. Cantley. 1997. The structural basis for 14-3-3: phosphopeptide binding specificity. Cell. 91:961–971. [DOI] [PubMed] [Google Scholar]
- Zhang, L., H. Wang, D. Liu, R. Liddington, and H. Fu. 1997. Raf-1 kinase and exoenzyme S interact with 14-3-3zeta through a common site involving lysine 49. J. Biol. Chem. 272:13717–13724. [DOI] [PubMed] [Google Scholar]
- Zhang, S., H. Xing, and A.J. Muslin. 1999. Nuclear localization of protein kinase U-alpha is regulated by 14-3-3. J. Biol. Chem. 274:24865–24872. [DOI] [PubMed] [Google Scholar]