

Abstract
Before S phase, cells license replication origins for initiation by loading them with Mcm2-7 heterohexamers. This process is dependent on Cdc6, which is recruited to unlicensed origins. Using Xenopus egg extracts we show that although each origin can load many Mcm2-7 hexamers, the affinity of Cdc6 for each origins drops once it has been licensed by loading the first hexamers. This encourages the distribution of at least one Mcm2-7 hexamer to each origin, and thereby helps to ensure that all origins are licensed. Although Cdc6 is not essential for DNA replication once licensing is complete, Cdc6 regains a high affinity for origins once replication forks are initiated and Mcm2-7 has been displaced from the origin DNA. We show that the presence of Cdc6 during S phase is essential for the checkpoint kinase Chk1 to become activated in response to replication inhibition. These results show that Cdc6 plays multiple roles in ensuring precise chromosome duplication.
Keywords: DNA replication; Cdc6; replication licensing; Xenopus; Chk1
Introduction
To ensure precise chromosome duplication, eukaryotic replication origins are licensed in late mitosis and early G1 to undergo a single initiation event (Blow and Hodgson, 2002; Nishitani and Lygerou, 2002). The origin recognition complex (ORC) first binds to origin DNA and then recruits two other proteins, Cdc6 and Cdt1. The binding of Cdc6 to origins may stabilize the interaction of ORC with chromatin (Mizushima et al., 2000). ORC, Cdc6, and Cdt1 then cooperate to load multiple copies of the Mcm2-7 hexamer onto each origin, forming the pre-replicative complex (pre-RC) and thereby licensing the origin for use in the ensuing S phase. As each origin initiates replication, Mcm2-7 are displaced from it, possibly moving along with or ahead of the replication fork as a DNA helicase (Labib and Diffley, 2001). Once licensing has occurred and Mcm2-7 are loaded onto DNA, ORC, Cdc6, and Cdt1 are no longer required for the continued association of Mcm2-7 with DNA (Donovan et al., 1997; Hua and Newport, 1998; Rowles et al., 1999). Indeed, in Xenopus, the binding of ORC and Cdc6 to chromatin is weakened after licensing has occurred (Rowles et al., 1999; Jares and Blow, 2000; Sun et al., 2002). These observations are consistent with the idea that the major function of the ORC–Cdc6–Cdt1 complex is to clamp the circular Mcm2-7 hexamer around DNA (Fletcher et al., 2003).
The fate of Cdc6 during later cell cycle stages varies in different organisms. In yeasts, phosphorylation of Cdc6 (or its homologue Cdc18 in Schizosaccharomyces pombe) by Cdks targets it for ubiquitin-dependent proteolysis (Drury et al., 1997, 2000; Jallepalli et al., 1997; Kominami and Toda, 1997; Baum et al., 1998; Elsasser et al., 1999). In metazoans, chromatin-bound Cdc6 persists throughout S phase and G2 (Coleman et al., 1996; Coverley et al., 2000; Mendez and Stillman, 2000) and is degraded during G1 by the anaphase promoting complex (Petersen et al., 2000). During S phase and G2, however, the majority of the soluble (nonchromatin bound) Cdc6 appears to be exported out of the nucleus in a Cdk-dependent manner (Saha et al., 1998; Petersen et al., 1999; Pelizon et al., 2000). The majority of the data showing Cdc6 nuclear export in S phase were obtained by overexpression of Cdc6. Surprisingly, a recent study focusing entirely on endogenous Cdc6 showed that even nonchromatin-bound Cdc6 may remain nuclear throughout S phase (Alexandrow and Hamlin, 2004). Despite this persistence of Cdc6 on chromatin or in nuclei later in the cell cycle, it has been shown in Xenopus that once DNA has been licensed, efficient DNA replication no longer requires the presence of Cdc6 (Rowles et al., 1999).
Several lines of evidence suggest that Cdc6 may play another role later in the cell cycle to generate appropriate checkpoint signals for regulated cell cycle progression. During mitotic exit in Saccharomyces cerevisiae, Cdc6 cooperates with Sic1 to directly inactivate Cdks (Calzada et al., 2001). In human cells, overexpression of Cdc6 in G2 phase inhibits activation of Cdk1–cyclin B and blocks entry into mitosis (Clay-Farrace et al., 2003). This latter effect seems to be mediated by activation of the Chk1 checkpoint kinase, because Cdc6 overexpression induced Chk1 phosphorylation, which is indicative of kinase activation. Moreover, addition of a Chk1 inhibitor overcame the Cdc6-induced inhibition of mitotic entry. Finally, results in S. pombe suggest that the Cdc6 homologue Cdc18 is required for checkpoint activation in response to S phase arrest (Murakami et al., 2002). When cdc18 was inactivated after S phase progression had been blocked with hydroxyurea, activation of the Cds1 (Chk2) checkpoint kinase was abolished. Further, the stalled replication forks became destabilized in the absence of Cdc18. These results suggest that Cdc6/Cdc18 has important roles in regulating cell cycle progression in addition to its well-documented role in origin licensing.
We have performed experiments to examine in detail the function and regulation of Xenopus Cdc6. We show that in Xenopus egg extracts, Cdc6 is displaced from chromatin as a direct consequence of origins first becoming licensed. Rebinding of Cdc6 to chromatin occurs in S phase as a consequence of replication forks progressing away from the origin. Finally, we show that the activation of Chk1 that normally occurs as a consequence of replication fork inhibition is dependent on the continued presence of Cdc6.
Results
Cdc6 is displaced from chromatin as a direct consequence of licensing
We investigated the binding of proteins to Xenopus sperm chromatin shortly after sperm nuclei were added to egg extract (Fig. 1 A). Consistent with previous papers (Coleman et al., 1996; Rowles et al., 1996, 1999; Jares and Blow, 2000; Maiorano et al., 2000), Orc2, Cdc6, and Cdt1 associated rapidly with chromatin, reaching approximately maximal levels within 2–3 min. The loading of Mcm5 was more prolonged, taking 20 min to reach peak levels. After 20–30 min, the template DNA became assembled into nuclei, which promotes the conversion of geminin into an active form (Hodgson et al., 2002). Active geminin binds and inhibits Cdt1 (Wohlschlegel et al., 2000; Tada et al., 2001) and can be recruited to chromatin by Cdt1 (Gillespie et al., 2001). Consistent with this, geminin was observed to bind to chromatin 20–30 min after addition of DNA to the extract. The amount of Cdc6 bound to chromatin reached a maximum at 2–3 min and then declined. We have shown previously that this displacement of Cdc6 from chromatin depends on Mcm2-7 binding (Rowles et al., 1999; Jares and Blow, 2000). Consistent with this, Fig. 1 A shows that when Mcm2-7 binding was inhibited by the addition of geminin to the extract, Cdc6 remained at high levels on the chromatin.
Figure 1.

Stable association of Cdc6 with chromatin is abolished after licensing. (A) Sperm nuclei were incubated at 10 ng DNA/μl in interphase extract. At the indicated times chromatin was isolated and immunoblotted for Orc2, Cdc6, Mcm5, Cdt1, and geminin. As controls, extract was incubated for 30 min without sperm, or was supplemented with gemininDEL before incubation for 30 min with sperm. (B) Coomassie-stained gel of recombinant Cdt1 and purified Mcm2-7. (C and D) Unlicensed chromatin was isolated from extract supplemented with recombinant gemininDEL, and was incubated for 30 min with recombinant Cdt1 and/or purified Mcm2-7 or in untreated extract. Chromatin was (C) transferred to extract supplemented with recombinant gemininDEL and α-[32P]dATP, and subsequent DNA synthesis was assayed; or (D) isolated and immunoblotted for Orc2, Cdc6, Cdt1, Mcm2, Mcm4, Mcm5, Mcm6, and histone H3.
To determine whether the displacement of Cdc6 is dependent directly on Mcm2-7 binding or whether it requires additional soluble components, we reconstituted the licensing of chromatin using a semi-fractionated reaction (Chong et al., 1995; Gillespie et al., 2001; Tada et al., 2001). Protocols we have used previously for purifying Mcm2-7 have given material with a markedly low specific activity (Chong et al., 1995; Thömmes et al., 1997; Gillespie and Blow, 2000; Prokhorova and Blow, 2000). Therefore, we developed a new immunoaffinity protocol for Mcm2-7, which gave purified protein with a high level of activity (Fig. 1 B and not depicted). Unlicensed chromatin containing ORC and Cdc6 was isolated from extracts treated with geminin. This chromatin was incubated with fractions containing recombinant Cdt1 and/or purified Mcm2-7 (Fig. 1 B). Incubation of the chromatin with a combination of Cdt1 and Mcm2-7 resulted in efficient origin licensing (Fig. 1 C) and the loading of Mcm2-7 onto chromatin (Fig. 1 D). The combination of Cdt1 and Mcm2-7 also displaced Cdc6 from the chromatin, thus suggesting that Mcm2-7 loading directly displaces Cdc6 without the need for any additional soluble factors.
It has been shown previously that in Xenopus egg extract, ORC saturates sperm chromatin at approximately one copy per 10 kb (Rowles et al., 1996, 1999). This approximately matches the density of replication origins, which are roughly spaced every 5–15 kb (Blow et al., 2001). To understand the physical relationship between Cdc6 and ORC, we examined the quantity of Cdc6 bound to chromatin. Because Cdc6 is displaced from chromatin when licensing occurs, we performed the analysis using extracts supplemented with geminin, which prevents licensing by inhibiting Cdt1. Equal aliquots of Xenopus sperm nuclei were incubated in increasing volumes of geminin-treated extract; after 15 min, the chromatin was isolated and immunoblotted for Cdc6. Fig. 2 A shows that, as reported previously for ORC (Rowles et al., 1996, 1999), the quantity of Cdc6 on chromatin increased in proportion to the volume of extract used, until a plateau was reached and the chromatin became saturated with Cdc6. Quantification by comparison with recombinant standards showed that at saturation there was ∼10 ng Cdc6 bound to each 400 ng of aliquot of DNA (Fig. 2 B), corresponding to ∼2.4 molecules of Cdc6 per 10 kb. This suggests that before licensing occurs, each molecule of ORC can recruit a dimer of Cdc6 onto chromatin.
Figure 2.
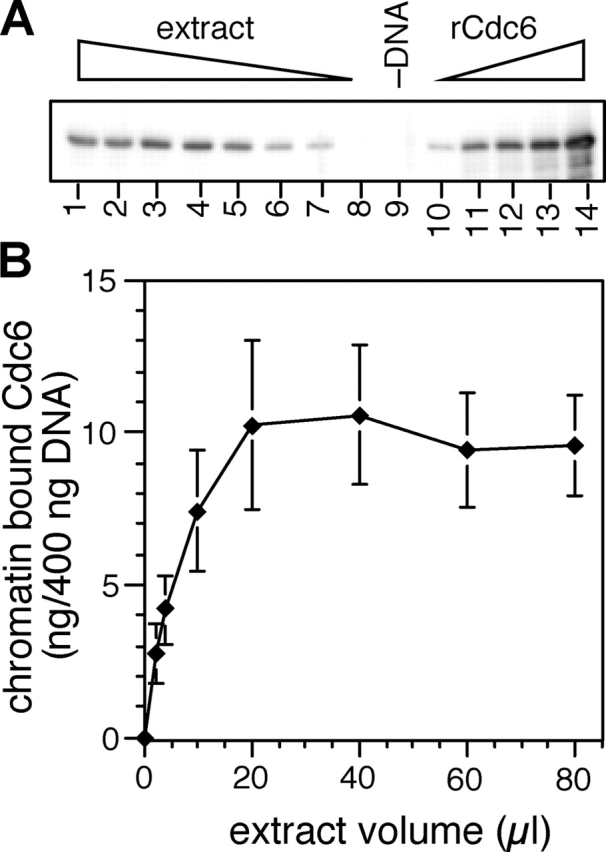
Unlicensed chromatin becomes saturated with approximately two copies of Cdc6 every 10 kb. 400 ng of aliquots of sperm nuclei were incubated for 30 min in increasing volumes of interphase extract supplemented with gemininDEL. (A) Chromatin was isolated and immunoblotted for Cdc6. Lanes 1–8 represent chromatin incubated in 80, 60, 40, 20, 10, 4, 2, and 1 μl extract. Lane 9 shows a mock chromatin isolation from 20 μl extract containing no DNA. Lanes 10–14 show loading standards, containing 5, 10, 15, 20, and 25 ng recombinant Cdc6. (B) The quantity of chromatin-bound Cdc6 was estimated from gels as shown in A. The mean and SD of three separate experiments is shown.
Analogous experiments investigating the binding of Mcm2-7 to chromatin have indicated that 10–20 copies of Mcm2-7 can bind each 10 kb of DNA (Mahbubani et al., 1997; Edwards et al., 2002). However, these levels can be reduced to approximately two copies per 10 kb without significantly reducing the subsequent rate of DNA synthesis. Fig. 1 A shows that Cdc6 is displaced from chromatin when Mcm5 levels are ∼10% of maximal suggesting that one to two Mcm2-7 molecules loaded onto each origin are sufficient to displace Cdc6. To test this idea, we performed an experiment where geminin was added to extracts at different times after addition of sperm nuclei, and the quantity of chromatin-bound Cdc6 and Mcm2 was subsequently analyzed by immunoblotting. When geminin was added at the start of the incubation, Cdc6 was stabilized on the chromatin (Fig. 3 A) and DNA synthesis was inhibited (Fig. 3 B). Addition of geminin to extract after 3 min, when Mcm2 levels were ∼10% of the maximum, resulted in only low levels of Cdc6 remaining on chromatin (Fig. 3 A). Correspondingly, addition of geminin three or more minutes after the start of the incubation had no major effect on the subsequent rate of replication (Fig. 3 B and not depicted). These results are consistent with the idea that the first Mcm2-7 complexes loaded onto each origin are sufficient to both displace Cdc6 and support DNA replication in the subsequent S phase.
Figure 3.
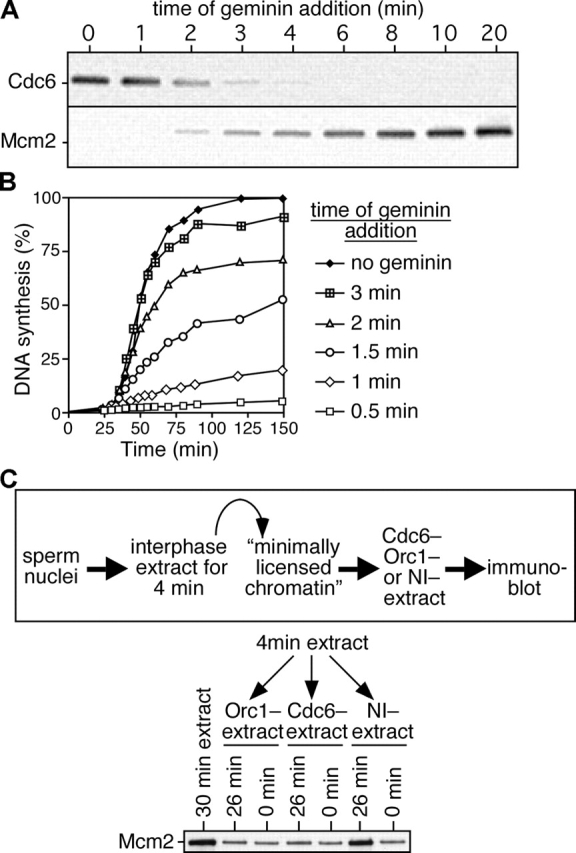
Cdc6 is destabilized on chromatin after the minimal effective quantity of Mcm2-7 is loaded. (A and B) Sperm nuclei were incubated at 10 ng DNA/μl in interphase egg extract, and gemininDEL was added at the indicated times after sperm addition. (A) Chromatin was isolated 5 min after gemininDEL addition and immunoblotted for Cdc6 and Mcm2. (B) DNA synthesis was assayed by α-[32P]dATP incorporation. (C) Sperm nuclei were incubated in interphase egg extract for 4 min; chromatin was isolated and incubated for an additional 0 or 26 min in extract previously immunodepleted of ORC or Cdc6 or mock depleted with nonimmune antibodies (NI−). Chromatin was isolated and immunoblotted for Mcm2. Chromatin isolated after a 30-min incubation in untreated extract is shown as control. The top shows a schematic diagram of the experimental protocol.
The displacement of Cdc6 from origins does not, however, block continued Mcm2-7 loading (Fig. 1 A and Fig. 3 A). We considered two possible explanations for this. One possibility is that Cdc6 is only required for the loading of the very first Mcm2-7 complex at each origin, after which further Mcm2-7 complexes could be loaded independently of Cdc6 function. Alternatively, the continued presence of Cdc6 may be required for the continued loading of Mcm2-7 onto origins, but this might not require a high affinity interaction between Cdc6 and chromatin. To distinguish between these possibilities, we performed the experiment outlined schematically in Fig. 3 C. Sperm nuclei were incubated for 4 min in an interphase extract to allow minimal licensing and the displacement of Cdc6. This “minimally licensed chromatin” was isolated under high salt conditions to remove ORC and any residual Cdc6 and then incubated for a further 26 min in extract immunodepleted of either Cdc6 or ORC, or in extract mock depleted with nonimmune antibodies. Fig. 3 C shows that only in the presence of Cdc6 and ORC could minimally licensed chromatin load further Mcm2. This suggests that Cdc6 and ORC are required to load all Mcm2-7 complexes and that Cdc6 does not need to be tightly bound to chromatin in order to do so.
Cdc6 is reloaded shortly after origins initiate
Previous studies in Xenopus egg extracts have shown that Cdc6 rebinds to chromatin during the later stages of an incubation (Coleman et al., 1996; Jares and Blow, 2000). Therefore, we performed a detailed time course to determine the relative timing of Cdc6 reloading compared with other known S phase events (Fig. 4). In these extracts, DNA synthesis starts 30–35 min after DNA addition and continues at a maximal rate for a further 20–30 min (Fig. 4 A). Fig. 4 B shows immunoblots of various proteins present on chromatin over this period, and Fig. 4 C shows a quantitation of the blots, each normalized to the maximal level. PCNA, an essential replication fork protein, is present at high levels on chromatin 35–45 min after DNA addition when DNA synthesis is maximal. Mcm2 is displaced from DNA during S phase with levels falling from 40–55 min. Cdt1 and geminin are also displaced from chromatin during S phase. In contrast, Cdc6 is reloaded onto the DNA as it replicates. The loading of Cdc6 mirrors that of PCNA at the start of S phase, though unlike PCNA, Cdc6 levels do not subsequently decline. Fig. 4 D shows that at 120 min, when most of the DNA has replicated, the quantity of Cdc6 on the chromatin is similar to that observed when licensing is blocked by geminin. This behavior is consistent with Cdc6 being reloaded onto origins as they replicate.
Figure 4.

Cdc6 reloading onto chromatin starts at the beginning of S phase. Sperm nuclei were incubated at 7 ng DNA/μl in interphase egg extract. (A) Time course of DNA synthesis. (B and C) Chromatin was isolated at the indicated times and immunoblotted for PCNA, Cdc6, Mcm2, Cdt1, and geminin. The immunoblots are shown in B and quantitation of the intensity of Mcm2, PCNA, and Cdc6 is shown in C. (D) Chromatin from either untreated extract or extract supplemented with geminin was isolated at the indicated times and immunoblotted for Mcm2 and Cdc6.
To test this idea, we supplemented extracts with roscovitine, which inhibits Cdks and therefore prevents the initiation of DNA replication (Meijer et al., 1997). Fig. 5 (A and B) shows that roscovitine efficiently inhibits both DNA synthesis and the reloading of Cdc6 onto chromatin. This inhibition is unlikely to be mediated by the ATM–ATR checkpoint pathway, as it is insensitive to caffeine. To determine whether Cdc6 reloading is dependent on DNA synthesis we supplemented extracts with various concentrations of aphidicolin, a competitive inhibitor of replicative DNA polymerases. With 7.5 μM aphidicolin, only ∼20% DNA synthesis occurred (Fig. 5 C) and very little Cdc6 was reloaded onto chromatin after 90 min (Fig. 5 D).
Figure 5.

Cdc6 reloading onto chromatin is dependent on the initiation of replication. Sperm nuclei were incubated at 10 ng DNA/μl in interphase egg extract supplemented with combinations of 1 mM of roscovitine, 5 mM of caffeine, DMSO, or different concentrations of aphidicolin. After 90 min, samples were assayed for DNA synthesis (A and C) or chromatin was isolated and immunoblotted for Cdc6 and Mcm2 or Mcm7 (B and D).
Aphidicolin affects DNA replication in two ways. By inhibiting the progression of active replication forks, it decreases the size of nascent strands. The inhibition of replication forks in turn activates an ATM–ATR checkpoint response that inhibits further origin firing (Santocanale and Diffley, 1998; Shirahige et al., 1998; Luciani, M.G., personal communication; unpublished data). To prevent this checkpoint response and allow all origins to fire in the presence of aphidicolin, we repeated the aphidicolin titration in extracts treated with caffeine to inhibit ATM and ATR (Fig. 6 A). In the presence of caffeine, 80 μM aphidicolin was required to block Cdc6 reloading, significantly more than is required in the absence of caffeine (compare Fig. 5 D with Fig. 6 A). Examination of the nascent strand length on alkaline gels showed that with 80 μM aphidicolin, nascent strands were <125 nt in size (Fig. 6 B). With 40 μM aphidicolin, where significant Cdc6 reloading was still observed, nascent strands were ∼500 nt long. These results suggest that in order for Cdc6 to be reloaded onto a replication origin, the replication forks must progress a few hundred base pairs away from it.
Figure 6.
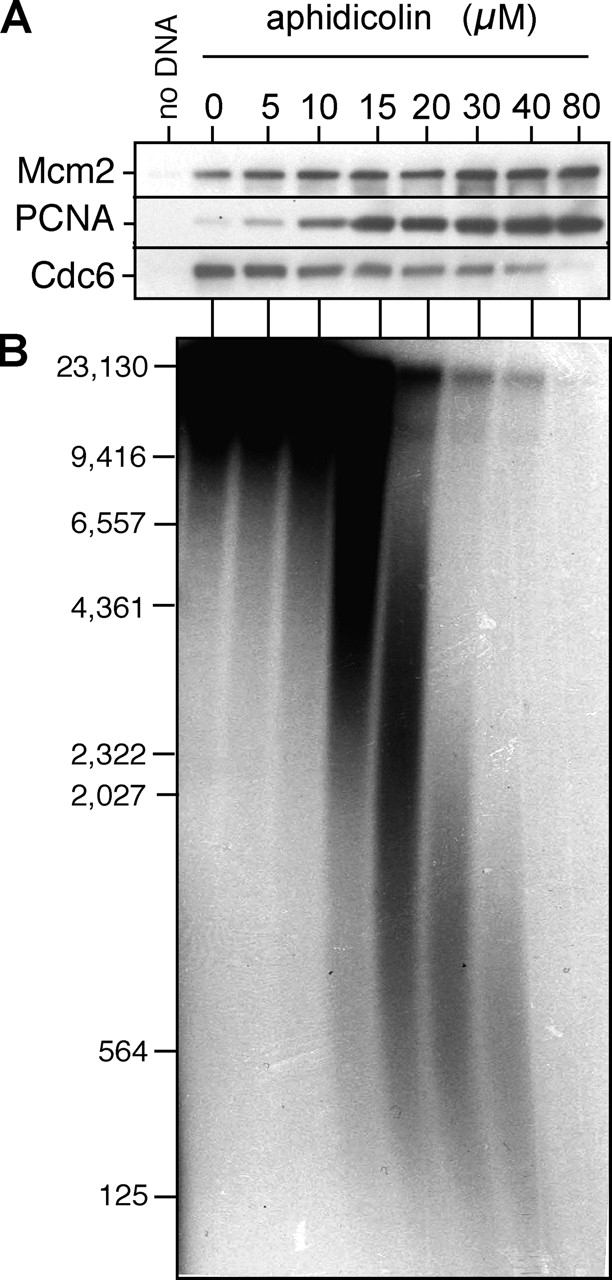
Cdc6 reloading requires fork elongation. Sperm nuclei were incubated at 10 ng DNA/μl in interphase egg extract treated with 0–80 μM aphidicolin in the presence of 5 mM of caffeine and α-[32P]dATP. (A) At 90 min, chromatin was isolated and immunoblotted for Mcm2, PCNA, and Cdc6. (B) DNA was separated on an alkaline agarose gel and autoradiographed. The migration of molecular weight markers (in bp) is shown on the left.
Cdc6 is required to activate Chk1 when replication forks are stalled
Cdc6 is required for loading of Mcm2-7 onto chromatin and the licensing of replication origins. Once licensing has occurred, Cdc6 is no longer required for subsequent DNA replication (Rowles et al., 1999; Jares and Blow, 2000). Therefore, it is unclear what function, if any, may be performed by the Cdc6 reloaded onto chromatin. Before S phase, the recruitment of Xenopus Cdc6 to chromatin depends on ORC (Coleman et al., 1996), so we first set out to determine whether Cdc6 reloading is also ORC dependent. To achieve this, we exploited the observation that when licensed chromatin is isolated in the presence of high salt, ORC and Cdc6 are removed but functional Mcm2-7 remains bound to the chromatin (Rowles et al., 1999). We then incubated this high salt-washed chromatin for 90 min in a second extract, which had either been immunodepleted of ORC or mock depleted with nonimmune antibodies (Fig. 7 A). Consistent with previous results (Rowles et al., 1999), the high salt-washed chromatin replicated equally well in the Orc1-depleted and mock-depleted extracts (Fig. 7 B). Cdc6 was efficiently reloaded onto chromatin in the mock-depleted extract, but not in the Orc1-depleted extract (Fig. 7 C). This suggests that, as in pre-RC formation, Cdc6 reloading is dependent on the presence of ORC.
Figure 7.
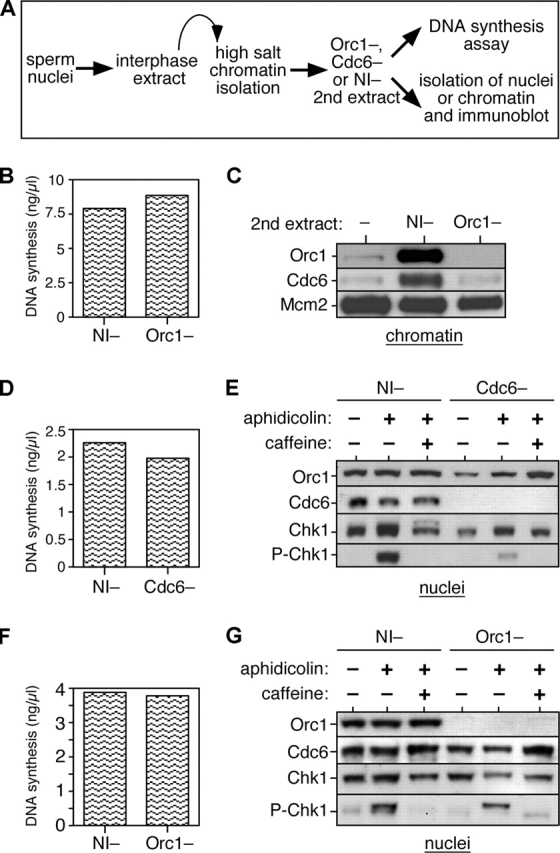
Cdc6 reloading is ORC dependent and Cdc6 is required in S phase for Chk1 activation. (A) Schematic of the experimental procedure. Sperm nuclei were incubated for 15 min at 15 ng DNA/μl in interphase egg extract. Chromatin was isolated under high salt conditions to remove ORC. Chromatin was incubated in a second extract depleted of either ORC or Cdc6 or mock depleted with nonimmune antibodies (NI−). DNA synthesis in the second extract (without aphidicolin or caffeine) was measured by incorporation of α-[32P]dATP (B, D, and F). The second extract was optionally supplemented with combinations of 40 μM aphidicolin or 5 mM of caffeine, and after 90 min in the second extract, chromatin, or intact nuclei were isolated and immunoblotted with the indicated antibodies (C, E, and G). (B and C) Licensed high salt-washed chromatin was incubated in Orc1- or nonimmune-depleted extracts. Chromatin was isolated and immunoblotted for Orc1, Cdc6, and Mcm2. (D and E) Licensed high salt-washed chromatin was incubated in Cdc6- or nonimmune-depleted extracts. Nuclei were isolated and immunoblotted for Orc1, Cdc6, Chk1, and phospho-Chk1. (F and G) Licensed high salt-washed chromatin was incubated in Orc1- or nonimmune-depleted extracts. Nuclei were isolated and immunoblotted for Orc1, Cdc6, Chk1, and phospho-Chk1.
Recent work in fission yeast and human cells has suggested that Cdc6 plays a role in activating a Chk1-dependent checkpoint during S phase or G2 (Murakami et al., 2002; Clay-Farrace et al., 2003). Therefore, we adapted the experiment described in Fig. 7 (B and C) to address whether the reloading of Cdc6 is required for Chk1 activation in the Xenopus system. Chk1 is predominantly nuclear, though not chromatin bound, and is activated by phosphorylation (Guo et al., 2000; Liu et al., 2000). Licensed, high salt-washed chromatin (lacking Cdc6) was therefore prepared and transferred to extract which had either been immunodepleted of Cdc6 or mock depleted with nonimmune antibodies. Aliquots of these extracts were supplemented with either aphidicolin or aphidicolin plus caffeine, and after a further incubation, nuclei were isolated and blotted for the presence of Orc1 (as loading control), Cdc6, Chk1, or phosphorylated Chk1 (indicative of Chk1 activation). Fig. 7 (D and E) shows that in the mock-depleted extracts, addition of aphidicolin induced Chk1 phosphorylation. This phosphorylation of Chk1 was blocked by caffeine, consistent with it being a consequence of activation of ATM–ATR checkpoint pathways. In the absence of Cdc6, however, the ability of aphidicolin to induce Chk1 was severely impaired. This suggests that Cdc6 is necessary for activating Chk1 in response to aphidicolin. This experiment was repeated, but transferring the salt-washed chromatin into extract depleted of ORC (Fig. 7, F and G). In the absence of ORC, Chk1 phosphorylation was still observed when replication was blocked by aphidicolin. Therefore, although Cdc6 is required for Chk1 phosphorylation, Cdc6 does not need to be recruited to chromatin to perform this function. Interestingly, although there is no chromatin-bound Cdc6 in the absence of ORC (Fig. 7 C), the nuclei contained high levels of Cdc6 even in the absence of ORC (Fig. 7 G), suggesting that endogenous Cdc6 is retained inside the nuclei by another mechanism.
Next, we wanted to confirm that the role of Cdc6 in checkpoint activation is distinct from its role in Mcm2-7 chromatin loading. Although Mcm2-7 loading does not normally occur once S phase has started, it is formally possible that in the presence of aphidicolin Cdc6 is required to reload or stabilize Mcm2-7 complexes at stalled forks. If forks became destabilized in the absence of Cdc6 they may not be able to maintain the checkpoint signal that leads to Chk1 phosphorylation. Thus, Cdc6 might be required for Chk1 activation in response to aphidicolin simply by performing its normal function in Mcm2-7 loading. To address this point, we allowed licensing to occur in Xenopus extracts, and then supplemented aliquots with an excess of a mutant form of geminin, gemininDEL, which is constitutively active (McGarry and Kirschner, 1998; Li and Blow, 2004), to prevent any further Mcm2-7 loading. Fig. 8 A shows that the mutant geminin had no effect on the ability of extracts to maintain the phosphorylation of Chk1 in response to aphidicolin. This indicates that Chk1 phosphorylation is not dependent of ongoing Mcm2-7 loading. To directly investigate the stability of fork proteins in the absence of Cdc6, we added high salt-washed chromatin to nonimmune- and Cdc6-depleted extracts treated with aphidicolin, as in Fig. 7 E. Chromatin was then isolated at different times over a 4-h period and immunoblotted for the presence of the replication fork proteins Mcm2, RPA, Cdc45, and PCNA (Fig. 8 B). There was no evidence for the destabilization of any of these proteins in the absence of Cdc6. Since RPA binds to single-stranded DNA this suggests that there are similar quantities of single-stranded DNA (which in turn is thought to be a major signal for the activation of the ATM–ATR checkpoint pathway) in the presence or absence of Cdc6. Together, these results suggest that the requirement of Cdc6 for Chk1 phosphorylation represents a function of Cdc6 distinct from its known role in the loading of Mcm2-7 onto chromatin.
Figure 8.

The checkpoint function of Cdc6 does not depend on Mcm2-7 loading. (A) Sperm nuclei were incubated in extract supplemented with 40 μM aphidicolin. At 15 min, after licensing had occurred, recombinant gemininDEL was added to an aliquot of the reaction. At 30, 60, 90, 120, and 150 min from the start of the incubation, nuclei were isolated and immunoblotted for Chk1 and phospho-Chk1. Nuclei isolated at 60 min from extract lacking aphidicolin is also shown as control. (B) Sperm nuclei were incubated for 15 min at 15 ng DNA/μl in interphase egg extract. Chromatin was isolated under high salt conditions to remove ORC. Chromatin was then incubated in a second extract depleted of Cdc6 or mock depleted with nonimmune antibodies (NI−). Both extracts were supplemented with 40 μM aphidicolin. At 0, 1, 2, 3, or 4 h afterwards, chromatin was isolated and immunoblotted for Mcm2, Cdc45, RPA-70, or PCNA.
Discussion
Destabilization of Cdc6 loading when minimal licensing has occurred
We show here that before licensing, approximately two copies of Cdc6 are recruited to each replication origin. Previous work has suggested that Cdc6 can form multimers (Saha et al., 1998; Frolova et al., 2002). Frolova et al. (2002) used two different Cdc6 mutants each with an inactivating mutation in either the Walker A or Walker B ATPase motifs. When both mutants were added together to Xenopus extracts, Cdc6 activity was restored. This result is consistent with active Cdc6 functioning as a dimer at replication origins, and would fit the stoichiometry that we report here. When origins become licensed, however, the quantity of Cdc6 associated with chromatin drops markedly (Rowles et al., 1999; Jares and Blow, 2000). We show here that this reduction in Cdc6 association with chromatin occurs as a direct consequence of the Mcm2-7 loading onto the DNA, because the displacement of Cdc6 occurs when unlicensed chromatin is incubated with purified Cdt1 and Mcm2-7. Consistent with other researchers (Mahaffey et al., 2003), we see no evidence for ubiquitin-dependent proteolysis of Cdc6 in this process as had been reported previously (Hua and Newport, 1998).
In normal Xenopus egg extract, an average of 10–20 copies of Mcm2-7 are loaded onto chromatin for each replicon (Mahbubani et al., 1997; Edwards et al., 2002). Maximum replication rates can be achieved, however, with an average of only one to two copies of Mcm2-7 per 10-kb replicon. If, as seems likely, the Mcm2-7 hexamer functions as a helicase ahead of each replication fork, then two copies would be needed at each origin to drive its two replication forks. We show here that the affinity of Cdc6 for chromatin declines once the chromatin has obtained the minimal quantity of Mcm2-7 required for efficient DNA replication. However, loading of further Mcm2-7 onto each origin is still dependent on the presence of Cdc6 and ORC. This behavior has some interesting implications for how replication origins are established. Once the first one to two Mcm2-7 hexamers are loaded onto each origin, the origin becomes licensed to support efficient DNA replication in the subsequent S phase (Fig. 9, A and B). The affinity of Cdc6 for the origin then drops, so that Cdc6 will then be targeted toward ORC at other currently unlicensed origins. Therefore, under circumstances where Cdc6 or Mcm2-7 are limiting, this mechanism ensures that most ORC binding sites become licensed and bind at least one to two Mcm2-7 hexamers. This reduces the danger of chromosomes being incompletely replicated as a consequence of too few origins being licensed (Blow, 2001; Blow et al., 2001; Hyrien et al., 2003).
Figure 9.

A model for Cdc6 function. (A) Dimers of Cdc6 bind tightly to unlicensed chromatin. (B) Although 10–20 copies of Mcm2-7 are normally loaded onto each origin, loading of the first one to two copies of Mcm2-7 (M) is sufficient to minimally license the origin. Once this has happened, the affinity of Cdc6 for the origin declines. This potentially directs Cdc6 and Mcm2-7 to other unlicensed origins. (C) Once replication forks have initiated and moved a small distance from the origin, Cdc6 can rebind to ORC at the origin with high affinity. This permits recruitment of cyclin E to the origin. Cdc6 is also required during S phase to allow Chk1 activation in response to replication fork inhibition, though ORC-dependent chromatin binding is not required for this function.
This explanation is consistent with the curiously linear relationship observed between the replication rate and the total amount of Mcm2-7 loaded onto DNA under conditions when licensing is limited (Fig. 3 B; Mahbubani et al., 1997). If Mcm2-7 hexamers were loaded onto different origins at random, not all origins would be loaded with the same number of Mcm2-7 hexamers and some origins might not have any Mcm2-7 at all. With a completely random distribution, the probability that an origin would have no Mcm2-7 loaded onto it is given by e–r, where r is the ratio between the total number of Mcm2-7 hexamers loaded and the total number of origins (see Materials and methods for the derivation). Therefore, when the number of origins is equal to the number of Mcm2-7 hexamers loaded (i.e., when r = 1), ∼37% of the origins should have no Mcm2-7 loaded. However, this is approximately the Mcm/origin ratio of minimally licensed chromatin, which replicates at near-maximal rates and so presumably has almost all of its origins licensed (Rowles et al., 1999; Blow et al., 2001). It would take at least a threefold excess of Mcm2-7 over origins (sixfold if two hexamers are minimally required at each origin) to give a probability of >95% that all origins are licensed. When Mcm2-7 loading is limited, the observed replication is more efficient than this random distribution would predict. The displacement of Cdc6 from origins once they have been loaded with the minimal essential complement of Mcm2-7 generates a nonrandom distribution of Mcm2-7 at origins, skewing the distribution so that each origin has a high probability of being loaded with the minimal essential complement of Mcm2-7.
Reloading of Cdc6 after initiation
Later in the cell cycle, Cdc6 regains its ability to bind chromatin with high affinity (Coleman et al., 1996; Jares and Blow, 2000). We show here that this reassociation with chromatin is dependent on the continued presence of ORC and results in similar levels of Cdc6 on chromatin as is seen on unlicensed DNA, and so probably represents a simple reestablishment of the ORC-Cdc6 interaction that is observed before licensing. The reloading of Cdc6 starts just at the beginning of S phase and is dependent on Cdk activity, suggesting that it is dependent on the initiation of DNA replication. Consistent with this, Cdc6 reloading was also inhibited in extracts treated with low levels of aphidicolin, where an intra–S phase checkpoint has been activated which inhibits initiation from late-firing replication origins (Luciani, M.G., personal communication; unpublished data). When the intra–S phase checkpoint was abolished by caffeine, only high levels of aphidicolin (>40 μM) could block Cdc6 reloading. This suggests that some DNA synthesis and movement of the replication fork away from origins is required for Cdc6 to rebind with high affinity to ORC (Fig. 9 C).
Previous work has shown that Cdc6 is required to recruit cyclin E–Cdk2 to chromatin in Xenopus egg extracts (Furstenthal et al., 2001a). Recruitment of cyclin E–Cdk2 to chromatin is thought to be required for the ubiquitination of proteins such as the Cdk inhibitor Xic1, which are recognized by the SCF (Skp1–Cullin–F-box) ubiquitin ligases (Furstenthal et al., 2001b). Xic1, which binds to cyclin E–Cdk2, is degraded during S phase after the initiation of replication has taken place (You et al., 2002). Since we show here that Cdc6 reloading is dependent on initiation, this provides a potential explanation for why Xic1 degradation is also dependent on initiation. This system may represent part of a positive feedback loop to ensure complete genome replication, whereby low Cdk levels lead to limited initiation and Cdc6 reloading, causing Xic1 degradation and further Cdk activation (Li and Blow, 2001).
Role of Cdc6 in checkpoint activation
We also show that Cdc6 is required during S phase to activate the Chk1 checkpoint kinase in response to inhibition of replication elongation by the DNA polymerase inhibitor aphidicolin (Fig. 9 C). This is reminiscent of results in S. pombe, which showed that the Cdc6 homologue Cdc18 is required to activate the Cds1 checkpoint kinase in response to the replication inhibitor hydroxyurea (Murakami et al., 2002). It has also been shown in mammalian cells that overexpression of Cdc6 in G2 cells can block progression into mitosis by activation of Chk1 (Clay-Farrace et al., 2003). Therefore, our results add weight to the idea that Cdc6 plays an important regulatory role that is distinct from its contribution to the pre-RC and licensing. The ability of Cdc6 to bind Cdks (Elsasser et al., 1996; Brown et al., 1997; Furstenthal et al., 2001a) raises the possibility that Cdc6 functions in some way to integrate cell cycle checkpoint activity with the activity of Cdks.
The mechanism by which Cdc6 activates checkpoint kinases is currently unclear. We present data suggesting that the requirement of Cdc6 for Chk1 phosphorylation does not depend on its ability to load Mcm2-7 onto chromatin. We also show that activation of Chk1 in response to aphidicolin in Xenopus does not depend on the continued presence of ORC, and so does not require the Cdc6 to be chromatin bound. However, we also show that much endogenous Cdc6 remains associated with nuclei in the absence of ORC, where it is in an ideal position to mediate the phosphorylation of nuclear Chk1 in response to stalled replication forks. This result is in apparent contrast with results reported in a number of different metazoans, including Xenopus, that the majority of the soluble Cdc6 is exported out of the nucleus during S and G2 in a Cdk-dependent manner (Saha et al., 1998; Petersen et al., 1999; Pelizon et al., 2000). However, many of these experiments reported the export of ectopically expressed or recombinant protein, and it is possible that endogenous protein behaves differently (Alexandrow and Hamlin, 2004). Perhaps in the absence of ORC, Cdc6 can either shuttle between nucleus and cytoplasm, or can be retained within the nucleus associated with some nonchromatin structure (Coleman et al., 1996; Fujita et al., 2002) where it can still interact with replication forks and Chk1.
Materials and methods
Preparation and use of extracts
Activated and metaphase-arrested Xenopus egg extracts were prepared as described previously (Chong et al., 1997). For replication assays, the extracts were supplemented with 100 μg/ml cycloheximide, 25 mM phosphocreatine, 15 μg/ml creatine phosphokinase, 0.3 mM CaCl2, and α-[32P]dATP. Geminin-treated extracts were prepared in a similar way, except 3 μg/ml gemininDEL (McGarry and Kirschner, 1998) was added 5 min before use. Immunodepletion of interphase extracts with specific antibodies or with antibodies from nonimmune rabbit serum was performed as described previously (Chong et al., 1997). Immediately after, immunodepletion extracts were snap frozen in liquid nitrogen in 10 μl of aliquots for future use. DNA synthesis was assessed by measuring incorporation of α-[32P]dATP into acid insoluble material as described previously (Chong et al., 1997). Final DNA concentrations in the assays were kept at 1–15 ng of DNA/μl extract. All incubations were performed at 23°C.
Recombinant proteins and antibodies
Recombinant proteins were produced as described previously: Xenopus Cdc6 (Gillespie et al., 2001), Xenopus Cdt1 (Tada et al., 2001), and Xenopus gemininDEL (McGarry and Kirschner, 1998; Tada et al., 2001). The antibodies used in this work were as follows: Xenopus Orc1 (Rowles et al., 1996), Xenopus Orc2 (raised against full-length 6X His-tagged Xenopus Orc2), Xenopus Cdc6 (Tada et al., 1999), human Mcm2 (BM28; BD Transduction Labs), Xenopus Mcm3 (Mahbubani et al., 1997), Xenopus Mcm5 (raised against 6X His-tagged amino acids 289–721 of Xenopus Mcm5), Xenopus Mcm4, 6, and 7 (Prokhorova and Blow, 2000), Xenopus Cdt1 (Tada et al., 2001), Xenopus geminin (Tada et al., 2001), histones H3 and H4 (rabbit polyclonal; Upstate Biotechnology), PCNA (PC10; Sigma-Aldrich), phospho-Chk1 (Ser345; Cell Signaling Technology), and Chk1 (sc-7898; Santa Cruz Biotechnology, Inc.).
Chromatin and nuclear templates
Demembranated Xenopus sperm nuclei were prepared as described previously (Chong et al., 1997). They were assembled into chromatin by incubation at 5–20 ng DNA/μl for appropriate times in treated extracts. In the case of immunodepleted extracts, sperm nuclei were incubated at 2–7 ng DNA/μl, to compensate for the threefold dilution that occurs during depletion. Extract was diluted 10–20-fold in nuclear isolation buffer (Chong et al., 1997; NIB: 50 mM KCl, 50 mM Hepes-KOH, pH 7.6, 5 mM MgCl2, 2 mM DTT, 0.5 mM spermidine-3HCl, 0.15 mM spermine-4HCl, 1 μg/ml aprotinin, 1 μg/ml leupeptin, 1 μg/ml pepstatin), supplemented with 0.1% Triton X-100, 2.5 mM Mg-ATP, and underlayered with the same buffer containing 15% sucrose (wt/vol). The chromatin was pelleted at 2,100 g in a swinging bucket centrifuge for 5 min at 4°C. The diluted extract and the top part of the cushion were carefully removed, and the cushion was carefully washed with 150 μl NIB supplemented with 0.1% Triton X-100, 2.5 mM Mg-ATP. The majority of the overlying cushion was then removed, and the chromatin was recentrifuged at 10,000 g in a fixed-angle rotor. The chromatin pellet was resuspended in loading buffer and subjected to immunoblotting by standard techniques using 4–12% bis-Tris gradient SDS-PAGE (Invitrogen) and ECL detection (SuperSignal® West Pico Chemiluminescent). For the Cdc6 quantification experiments, the chemiluminescence signal was measured using a cooled CCD camera (model LAS-1000; Fuji).
When chromatin was isolated that had to be reincubated in another extract, the isolation was performed in a similar way except that Triton X-100 was omitted from the buffers and only the first centrifugation was performed, after which the chromatin was resuspended to ∼100 ng DNA/μl and snap frozen in liquid nitrogen in 5 μl of aliquots for later use. For high salt chromatin isolation the extract was diluted in NIB plus 2.5 mM Mg-ATP and 200 mM KCl, and was underlayered with two cushions: the first containing NIB plus 2.5 mM Mg-ATP, 200 mM KCl, and 5% sucrose (wt/vol); and the second containing NIB plus 2.5 mM Mg-ATP and 10% sucrose (wt/vol).
For isolation of intact nuclei (Kumagai et al., 1998), extract containing nuclei was underlayered with 10 vol of a buffer containing 40% sucrose, 50 mM Hepes-KOH, pH 7.5, 100 mM KCl, and 2.5 mM Mg-MgCl2, and pelleted at 5,000 g in a fixed-angle rotor for 3 min at 4°C. The pellets were resuspended in 1 ml of the same buffer and recentrifuged under the same conditions. For extracts that had been treated with caffeine, 5 mM of caffeine was included in the isolation buffer.
Purification of Xenopus Mcm2-7
Mcm2-7 was purified from interphase Xenopus egg extract by immunoaffinity purification. All purification steps were performed at 4°C. 400 μl recombinant protein A beads (Amersham Biosciences) were mixed with 800 μl of antiserum against Xenopus Mcm3 or Mcm7. 2 ml of interphase egg extract was diluted fourfold in LFB3/10 (20 mM Hepes-KOH, pH 8, 10% (wt/vol) sucrose, 2 mM DTT, 10 mM KCl, 1 μg/ml aprotinin, 1 μg/ml leupeptin, 1 μg/ml pepstatin) before addition to the beads. After incubation for 45 min on a rotary mixer, the beads were washed three times (5 min, 5 min, and 30 min) in 15 ml LFB1/50 (40 mM Hepes-KOH, pH 8, 10% sucrose (wt/vol), 2 mM DTT, 50 mM KCl, 20 mM K2HPO4/KH2PO4, 2 mM MgCl2, 1 mM EGTA, 1 μg/ml aprotinin, 1 μg/ml leupeptin, 1 μg/ml pepstatin) for 5 min, 5 min, and 30 min on the mixer. Elution was performed in three successive one-bead volumes of LFB1/600 (LFB1 containing 600 mM KCl). Mcm3/5 from the anti-Mcm7 antibody beads and Mcm2/4/6/7 from the anti-Mcm3 beads were recovered by centrifugation for 2 min at 700 g in a swinging bucket centrifuge through a 25-μm CellMicroSievesTM mesh (BioDesign Inc. of New York) and dialyzed in LFB3/10. The Mcm2-7 heterohexamer was recovered via stoichiometric addition of Mcm3/5 to Mcm2/4/6/7 after analysis by SDS PAGE, immunoblotting, or gel filtration.
Alkaline agarose gels
For alkaline agarose gel analysis, Xenopus sperm nuclei were incubated at 15 ng/μl for 90 min in 20 μl of aliquots of treated extracts containing α-[32P]dATP before addition of 300 μl StopN (20 mM Tris-HCl, pH 8, 200 mM NaCl, 5 mM EDTA, 0.5% SDS) containing 2 μg/ml RNase, and digestion for 10 min at 37°C. Proteinase K was added to 200 μg/ml and incubated for a further 30 min at 37°C. Protein was extracted with 300 μl phenol/chloroform/isoamyl alcohol (25:24:1) using Phase Lock GelTM tubes (Eppendorf). 2.5 vol of ethanol were added and the DNA was precipitated for 20 min in dry ice before centrifugation for 10 min at 16,000 g in a fixed-angle rotor. The pellet was washed in 90% ethanol and resuspended in 20 μl 2× alkaline loading buffer: 50 mM NaOH, 6 mM EDTA, 2.5% Ficoll, 0.025% Bromocresol green. 15 cm 0.9% agarose gels were equilibrated for >1 h at RT in alkaline gel running buffer (50 mM NaCl, 1 mM EDTA). Gels were run at 2 V/cm for 10–12 h at 4°C and fixed in 7% TCA (wt/vol) and 1.4% (wt/vol) sodium pyrophosphate for 20 min. Gels were then dried between paper and exposed to X-ray film.
Modeling a random distribution of Mcm2-7
Suppose there are x replication origins and m Mcm2-7 hexamers, such that the ratio r between them is given by m = rx. If the Mcm2-7 hexamers are distributed at random amongst all the origins, the probability P that any randomly selected origin gets no Mcm2-7 hexamers is given by
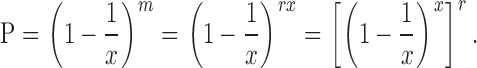 |
For a very large number of origins, consider the case where x → ∞. Since
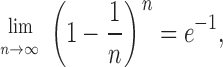 |
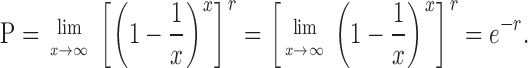 |
Acknowledgments
We thank Neil Perkins, Anatoliy Li, and Anna Woodward for comments on the manuscript, and Dyce Davidson for help with the mathematical modeling.
This work was funded by a CR-UK studentship to M. Oehlmann (STU063/001) and CR-UK program grants SP2385/0101 and C303/A3135.
Abbreviations used in this paper: NIB, nuclear isolation buffer; ORC, origin recognition complex; pre-RC, pre-replicative complex.
References
- Alexandrow, M.G., and J.L. Hamlin. 2004. Cdc6 chromatin affinity is unaffected by serine-54 phosphorylation, S-phase progression, and overexpression of cyclin A. Mol. Cell. Biol. 24:1614–1627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baum, B., H. Nishitani, S. Yanow, and P. Nurse. 1998. Cdc18 transcription and proteolysis couple S phase to passage through mitosis. EMBO J. 17:5689–5698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blow, J.J. 2001. Control of chromosomal DNA replication in the early Xenopus embryo. EMBO J. 20:3293–3297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blow, J.J., P.J. Gillespie, D. Francis, and D.A. Jackson. 2001. Replication origins in Xenopus egg extract are 5–15 kilobases apart and are activated in clusters that fire at different times. J. Cell Biol. 152:15–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blow, J.J., and B. Hodgson. 2002. Replication licensing—defining the proliferative state? Trends Cell Biol. 12:72–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown, G.W., P.V. Jallepalli, B.J. Huneycutt, and T.J. Kelly. 1997. Interaction of the S phase regulator cdc18 with cyclin-dependent kinase in fission yeast. Proc. Natl. Acad. Sci. USA. 94:6142–6147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calzada, A., M. Sacristan, E. Sanchez, and A. Bueno. 2001. Cdc6 cooperates with Sic1 and Hct1 to inactivate mitotic cyclin-dependent kinases. Nature. 412:355–358. [DOI] [PubMed] [Google Scholar]
- Chong, J.P., H.M. Mahbubani, C.Y. Khoo, and J.J. Blow. 1995. Purification of an MCM-containing complex as a component of the DNA replication licensing system. Nature. 375:418–421. [DOI] [PubMed] [Google Scholar]
- Chong, J.P., P. Thömmes, A. Rowles, H.M. Mahbubani, and J.J. Blow. 1997. Characterization of the Xenopus replication licensing system. Methods Enzymol. 283:549–564. [DOI] [PubMed] [Google Scholar]
- Clay-Farrace, L., C. Pelizon, D. Santamaria, J. Pines, and R.A. Laskey. 2003. Human replication protein Cdc6 prevents mitosis through a checkpoint mechanism that implicates Chk1. EMBO J. 22:704–712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coleman, T.R., P.B. Carpenter, and W.G. Dunphy. 1996. The Xenopus Cdc6 protein is essential for the initiation of a single round of DNA replication in cell-free extracts. Cell. 87:53–63. [DOI] [PubMed] [Google Scholar]
- Coverley, D., C. Pelizon, S. Trewick, and R.A. Laskey. 2000. Chromatin-bound Cdc6 persists in S and G(2) phases in human cells, while soluble Cdc6 is destroyed in a cyclin A-cdk2 dependent process. J. Cell Sci. 113:1929–1938. [DOI] [PubMed] [Google Scholar]
- Donovan, S., J. Harwood, L.S. Drury, and J.F. Diffley. 1997. Cdc6p-dependent loading of Mcm proteins onto pre-replicative chromatin in budding yeast. Proc. Natl. Acad. Sci. USA. 94:5611–5616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drury, L.S., G. Perkins, and J.F.X. Diffley. 1997. The Cdc4/34/53 pathway targets Cdc6p for proteolysis in budding yeast. EMBO J. 16:5966–5976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drury, L.S., G. Perkins, and J.F.X. Diffley. 2000. The cyclin-dependent kinase Cdc28p regulates distinct modes of Cdc6p proteolysis during the budding yeast cell cycle. Curr. Biol. 10:231–240. [DOI] [PubMed] [Google Scholar]
- Edwards, M.C., A.V. Tutter, C. Cvetic, C.H. Gilbert, T.A. Prokhorova, and J.C. Walter. 2002. MCM2-7 complexes bind chromatin in a distributed pattern surrounding the origin recognition complex in Xenopus egg extracts. J. Biol. Chem. 277:33049–33057. [DOI] [PubMed] [Google Scholar]
- Elsasser, S., F. Lou, B. Wang, J.L. Campbell, and A. Jong. 1996. Interaction between yeast Cdc6 protein and B-type cyclin/Cdc28 kinases. Mol. Biol. Cell. 7:1723–1735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elsasser, S., Y. Chi, P. Yang, and J.L. Campbell. 1999. Phosphorylation controls timing of Cdc6p destruction: a biochemical analysis. Mol. Biol. Cell. 10:3263–3277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fletcher, R.J., B.E. Bishop, R.P. Leon, R.A. Sclafani, C.M. Ogata, and X.S. Chen. 2003. The structure and function of MCM from archaeal M. Thermoautotrophicum. Nat. Struct. Biol. 10:160–167. [DOI] [PubMed] [Google Scholar]
- Frolova, N.S., N. Schek, N. Tikhmyanova, and T.R. Coleman. 2002. Xenopus Cdc6 performs separate functions in initiating DNA replication. Mol. Biol. Cell. 13:1298–1312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujita, M., Y. Ishimi, H. Nakamura, T. Kiyono, and T. Tsurumi. 2002. Nuclear organization of DNA replication initiation proteins in mammalian cells. J. Biol. Chem. 277:10354–10361. [DOI] [PubMed] [Google Scholar]
- Furstenthal, L., B.K. Kaiser, C. Swanson, and P.K. Jackson. 2001. a. Cyclin E uses Cdc6 as a chromatin-associated receptor required for DNA replication. J. Cell Biol. 152:1267–1278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furstenthal, L., C. Swanson, B.K. Kaiser, A.G. Eldridge, and P.K. Jackson. 2001. b. Triggering ubiquitination of a CDK inhibitor at origins of DNA replication. Nat. Cell Biol. 3:715–722. [DOI] [PubMed] [Google Scholar]
- Gillespie, P.J., and J.J. Blow. 2000. Nucleoplasmin-mediated chromatin remodelling is required for Xenopus sperm nuclei to become licensed for DNA replication. Nucleic Acids Res. 28:472–480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gillespie, P.J., A. Li, and J.J. Blow. 2001. Reconstitution of licensed replication origins on Xenopus sperm nuclei using purified proteins. BMC Biochem. 2:15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo, Z., A. Kumagai, S.X. Wang, and W.G. Dunphy. 2000. Requirement for Atr in phosphorylation of Chk1 and cell cycle regulation in response to DNA replication blocks and UV-damaged DNA in Xenopus egg extracts. Genes Dev. 14:2745–2756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hodgson, B., A. Li, S. Tada, and J.J. Blow. 2002. Geminin becomes activated as an inhibitor of Cdt1/RLF-B following nuclear import. Curr. Biol. 12:678–683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hua, X.H., and J. Newport. 1998. Identification of a preinitiation step in DNA replication that is independent of origin recognition complex and cdc6, but dependent on cdk2. J. Cell Biol. 140:271–281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hyrien, O., K. Marheineke, and A. Goldar. 2003. Paradoxes of eukaryotic DNA replication: MCM proteins and the random completion problem. Bioessays. 25:116–125. [DOI] [PubMed] [Google Scholar]
- Jallepalli, P.V., G.W. Brown, M. Muzi-Falconi, D. Tien, and T.J. Kelly. 1997. Regulation of the replication initiator protein p65(cdc18) by CDK phosphorylation. Genes Dev. 11:2767–2779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jares, P., and J.J. Blow. 2000. Xenopus Cdc7 function is dependent on licensing but not on XORC, XCdc6, or CDK activity and is required for XCdc45 loading. Genes Dev. 14:1528–1540. [PMC free article] [PubMed] [Google Scholar]
- Kominami, K., and T. Toda. 1997. Fission yeast WD-repeat protein pop1 regulates genome ploidy through ubiquitin-proteasome-mediated degradation of the CDK inhibitor Rum1 and the S-phase initiator Cdc18. Genes Dev. 11:1548–1560. [DOI] [PubMed] [Google Scholar]
- Kumagai, A., Z. Guo, K.H. Emami, S.X. Wang, and W.G. Dunphy. 1998. The Xenopus Chk1 protein kinase mediates a caffeine-sensitive pathway of checkpoint control in cell-free extracts. J. Cell Biol. 142:1559–1569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Labib, K., and J.F. Diffley. 2001. Is the MCM2-7 complex the eukaryotic DNA replication fork helicase? Curr. Opin. Genet. Dev. 11:64–70. [DOI] [PubMed] [Google Scholar]
- Li, A., and J.J. Blow. 2001. The origin of CDK regulation. Nat. Cell Biol. 3:E182–E184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, A., and J.J. Blow. 2004. Non-proteolytic inactivation of geminin requires CDK-dependent ubiquitination. Nat. Cell Biol. 6:260–267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu, Q., S. Guntuku, X.S. Cui, S. Matsuoka, D. Cortez, K. Tamai, G. Luo, S. Carattini-Rivera, F. DeMayo, A. Bradley, et al. 2000. Chk1 is an essential kinase that is regulated by Atr and required for the G(2)/M DNA damage checkpoint. Genes Dev. 14:1448–1459. [PMC free article] [PubMed] [Google Scholar]
- Mahaffey, D.T., C. Gorbea, and M. Rechsteiner. 2003. Evidence that DNA replication is not regulated by ubiquitin-dependent proteolysis in Xenopus egg extract. Exp. Cell Res. 288:225–234. [DOI] [PubMed] [Google Scholar]
- Mahbubani, H.M., J.P. Chong, S. Chevalier, P. Thömmes, and J.J. Blow. 1997. Cell cycle regulation of the replication licensing system: involvement of a Cdk-dependent inhibitor. J. Cell Biol. 136:125–135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maiorano, D., J. Moreau, and M. Mechali. 2000. XCDT1 is required for the assembly of pre-replicative complexes in Xenopus laevis. Nature. 404:622–625. [DOI] [PubMed] [Google Scholar]
- McGarry, T.J., and M.W. Kirschner. 1998. Geminin, an inhibitor of DNA replication, is degraded during mitosis. Cell. 93:1043–1053. [DOI] [PubMed] [Google Scholar]
- Meijer, L., A. Borgne, O. Mulner, J.P.J. Chong, J.J. Blow, N. Inagaki, M. Inagaki, J.G. Delcros, and J.P. Moulinoux. 1997. Biochemical and cellular effects of roscovitine, a potent and selective inhibitor of the cyclin-dependent kinases cdc2, cdk2 and cdk5. Eur. J. Biochem. 243:527–536. [DOI] [PubMed] [Google Scholar]
- Mendez, J., and B. Stillman. 2000. Chromatin association of human origin recognition complex, Cdc6, and minichromosome maintenance proteins during the cell cycle: Assembly of prereplication complexes in late mitosis. Mol. Cell. Biol. 20:8602–8612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mizushima, T., N. Takahashi, and B. Stillman. 2000. Cdc6p modulates the structure and DNA binding activity of the origin recognition complex in vitro. Genes Dev. 14:1631–1641. [PMC free article] [PubMed] [Google Scholar]
- Murakami, H., S.K. Yanow, D. Griffiths, M. Nakanishi, and P. Nurse. 2002. Maintenance of replication forks and the S-phase checkpoint by Cdc18p and Orp1p. Nat. Cell Biol. 4:384–388. [DOI] [PubMed] [Google Scholar]
- Nishitani, H., and Z. Lygerou. 2002. Control of DNA replication licensing in a cell cycle. Genes Cells. 7:523–534. [DOI] [PubMed] [Google Scholar]
- Pelizon, C., M.A. Madine, P. Romanowski, and R.A. Laskey. 2000. Unphosphorylatable mutants of Cdc6 disrupt its nuclear export but still support DNA replication once per cell cycle. Genes Dev. 14:2526–2533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petersen, B.O., J. Lukas, C.S. Sorensen, J. Bartek, and K. Helin. 1999. Phosphorylation of mammalian CDC6 by cyclin A/CDK2 regulates its subcellular localization. EMBO J. 18:396–410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petersen, B.O., C. Wagener, F. Marinoni, E.R. Kramer, M. Melixetian, E.L. Denchi, C. Gieffers, C. Matteucci, J.M. Peters, and K. Helin. 2000. Cell cycle- and cell growth-regulated proteolysis of mammalian CDC6 is dependent on APC-CDH1. Genes Dev. 14:2330–2343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prokhorova, T.A., and J.J. Blow. 2000. Sequential MCM/P1 subcomplex assembly is required to form a heterohexamer with replication licensing activity. J. Biol. Chem. 275:2491–2498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rowles, A., J.P. Chong, L. Brown, M. Howell, G.I. Evan, and J.J. Blow. 1996. Interaction between the origin recognition complex and the replication licensing system in Xenopus. Cell. 87:287–296. [DOI] [PubMed] [Google Scholar]
- Rowles, A., S. Tada, and J.J. Blow. 1999. Changes in association of the Xenopus origin recognition complex with chromatin on licensing of replication origins. J. Cell Sci. 112:2011–2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saha, P., J.J. Chen, K.C. Thome, S.J. Lawlis, Z.H. Hou, M. Hendricks, J.D. Parvin, and A. Dutta. 1998. Human CDC6/Cdc18 associates with Orc1 and cyclin-cdk and is selectively eliminated from the nucleus at the onset of S phase. Mol. Cell. Biol. 18:2758–2767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santocanale, C., and J.F. Diffley. 1998. A Mec1- and Rad53-dependent checkpoint controls late-firing origins of DNA replication. Nature. 395:615–618. [DOI] [PubMed] [Google Scholar]
- Shirahige, K., Y. Hori, K. Shiraishi, M. Yamashita, K. Takahashi, C. Obuse, T. Tsurimoto, and H. Yoshikawa. 1998. Regulation of DNA-replication origins during cell-cycle progression. Nature. 395:618–621. [DOI] [PubMed] [Google Scholar]
- Sun, W.H., T.R. Coleman, and M.L. DePamphilis. 2002. Cell cycle-dependent regulation of the association between origin recognition proteins and somatic cell chromatin. EMBO J. 21:1437–1446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tada, S., J.P.J. Chong, H.M. Mahbubani, and J.J. Blow. 1999. The RLF-B component of the replication licensing system is distinct from Cdc6 and functions after Cdc6 binds to chromatin. Curr. Biol. 9:211–214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tada, S., A. Li, D. Maiorano, M. Mechali, and J.J. Blow. 2001. Repression of origin assembly in metaphase depends on inhibition of RLF-B/Cdt1 by geminin. Nat. Cell Biol. 3:107–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thömmes, P., Y. Kubota, H. Takisawa, and J.J. Blow. 1997. The RLF-M component of the replication licensing system forms complexes containing all six MCM/P1 polypeptides. EMBO J. 16:3312–3319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wohlschlegel, J.A., B.T. Dwyer, S.K. Dhar, C. Cvetic, J.C. Walter, and A. Dutta. 2000. Inhibition of eukaryotic replication by geminin binding to Cdt1. Science. 290:2309–2312. [DOI] [PubMed] [Google Scholar]
- You, Z., K. Harvey, L. Kong, and J. Newport. 2002. Xic1 degradation in Xenopus egg extracts is coupled to initiation of DNA replication. Genes Dev. 16:1182–1194. [DOI] [PMC free article] [PubMed] [Google Scholar]