

Abstract
Current models of mitotic chromosome structure are based largely on the examination of maximally condensed metaphase chromosomes. Here, we test these models by correlating the distribution of two scaffold components with the appearance of prophase chromosome folding intermediates. We confirm an axial distribution of topoisomerase IIα and the condensin subunit, structural maintenance of chromosomes 2 (SMC2), in unextracted metaphase chromosomes, with SMC2 localizing to a 150–200-nm-diameter central core. In contrast to predictions of radial loop/scaffold models, this axial distribution does not appear until late prophase, after formation of uniformly condensed middle prophase chromosomes. Instead, SMC2 associates throughout early and middle prophase chromatids, frequently forming foci over the chromosome exterior. Early prophase condensation occurs through folding of large-scale chromatin fibers into condensed masses. These resolve into linear, 200–300-nm-diameter middle prophase chromatids that double in diameter by late prophase. We propose a unified model of chromosome structure in which hierarchical levels of chromatin folding are stabilized late in mitosis by an axial “glue.”
Keywords: chromosome structure; condensins; topoisomerase II; mitosis; SMC
Introduction
Understanding the structural and molecular basis of mitotic chromosome condensation remains a basic challenge in cell biology. Historically, three general experimental approaches have been pursued to circumvent technical difficulties associated with direct structural analysis of fully compact, native metaphase chromosomes. These three approaches have led to three alternative conceptual classes of models for metaphase chromosome architecture—radial loop, hierarchical folding, and network models—which are quite different in terms of the structural motifs postulated as giving rise to chromosome condensation (Swedlow and Hirano, 2003). Current microscopy methods cannot distinguish between these different predicted structural motifs in metaphase chromosomes (Belmont, 1998). However, these models make very different predictions in terms of the functional mechanisms underlying chromosome condensation and the types of folding intermediates that would be observed in prophase during early stages of chromosome condensation. Unfortunately, a major deficit in our understanding of mitotic chromosome condensation is the lack of good structural data concerning these early prophase stages and correlation of these condensation stages with the dynamics of chromosomal proteins implicated in the process of chromosome condensation.
A series of experiments, combining removal of all histone and most nonhistone proteins with more gentle extraction methods involving selective extraction of histone H1 and/or chromatin decondensation by lowering ionic conditions, has led to radial loop models of chromosome organization (Paulson and Laemmli, 1977; Laemmli et al., 1978). In these models, loops of radially organized 30-nm chromatin fibers are anchored to an axial chromosome structure, or chromosome “scaffold” (Marsden and Laemmli, 1979), formed by a special class of nonhistone proteins, including topoisomerase IIα (Earnshaw and Heck, 1985; Gasser et al., 1986) and structural maintenance of chromosomes 2 (SMC2) (Saitoh et al., 1994). Anchoring of these loops to the chromosome scaffold is proposed to involve special scaffold/matrix associated region (SAR/MAR) DNA sequences (Razin, 1996). A variation of the basic radial loop model is the radial loop/helical coil model in which a prophase chromatid, itself organized according to the original radial loop model, is then helically folded to form the final metaphase chromosome (Rattner and Lin, 1985; Boy de la Tour and Laemmli, 1988). However, more recent experiments suggest that metaphase chromosomes displaying this apparent helical folding are seen only in hypercondensed metaphase chromosomes isolated from mitotically arrested cells (Maeshima and Laemmli, 2003).
From a functional perspective, radial loop models postulate that the chromatin folding above the level of the 30-nm chromatin fiber is guided by nonhistone scaffold protein interactions with specific DNA sequences. The original model postulated an interconnected protein structure, but an alternative possibility of distributed islands of scaffold proteins anchoring local clusters of radial loops was introduced based on electron microscopy localization of topoisomerase II in swollen chromosomes (Earnshaw and Heck, 1985). The prediction from these models is not only that mitotic chromosome condensation will be dependent on these scaffold proteins, but also that the temporal pattern of scaffold assembly will coincide with or precede the appearance of chromosome condensation above the level of the 30-nm chromatin fiber.
Immunostaining of unextracted chromosomes and in vivo observations have confirmed the existence of an axial core distribution in native metaphase chromosomes for scaffold proteins topoisomerase IIα and condensins (Earnshaw and Heck, 1985; Tavormina et al., 2002; Maeshima and Laemmli, 2003; Ono et al., 2003). A requirement for condensins for chromosome assembly was first indicated in a cell-free extract prepared from Xenopus eggs (Hirano et al., 1997). Genetic analyses have demonstrated an in vivo role for condensin subunits in chromosome organization and segregation (for review see Swedlow and Hirano, 2003); however, the exact defects in mitotic chromosome structure have not been clearly delineated, and significant chromosome compaction has been observed after condensin subunit knockdowns. Knockdown of SMC2 condensin subunit expression by either RNA interference or a conditional knockout reveals defects in metaphase chromosome structure when chromosome spreads are prepared after hypotonic treatment (Hudson et al., 2003; Ono et al., 2003), but chromosome compaction appears normal within intact cells not exposed to hypotonic treatment (Gassmann et al., 2004).
The localization of scaffold proteins in early stages of chromosome condensation remains unclear. In one report, the axial core distribution was observed only for topoisomerase IIα and not condensins in prophase chromosomes (Maeshima and Laemmli, 2003). This led to suggestion of a two-step model of chromosome condensation in which topoisomerase IIα is more central to early stages of chromosome condensation and organization of radial loops, with condensins functioning later. However, functional analyses have indicated that prophase chromosome condensation is delayed in chicken DT40 cells in which the SMC2 gene is knocked out conditionally (Hudson et al., 2003) or in Caenorhabditis elegans embryos depleted of SMC4 (Hagstrom et al., 2002), suggesting a role for condensins early in chromosome condensation.
In hierarchical models of chromosome folding, 10- and 30-nm chromatin fibers are postulated to fold progressively into larger fibers that coil to form the final metaphase chromosomes (Sedat and Manuelidis, 1978; Zatsepina et al., 1983; Belmont et al., 1987; Belmont and Bruce, 1994). In contrast to radial loop models, chromosome condensation in hierarchical models is not dependent on formation of a core protein scaffold, and therefore the temporal pattern of chromosome condensation will not necessarily coincide with scaffold assembly. Successive helical coiling and folded chromonema models are examples of this group of models (Sedat and Manuelidis, 1978; Belmont and Bruce, 1994). Ultrastructural analysis of chromosome decondensation between telophase and early G1 indicated one or more levels of compaction between the 30-nm chromatin fiber and an ∼100–130-nm chromonema fiber, which itself folded into telophase chromosomes (Belmont and Bruce, 1994). However, no comparable work was done to analyze intermediates of prophase chromosome condensation. Recently, a novel imaging method was used to selectively label two transgene chromosome regions approximately half a chromosome band in size (Strukov et al., 2003). Ultrastructural analysis of immunogold staining of this region within intact metaphase chromosomes suggested the existence of a subunit of native metaphase chromosomes ∼250 nm in size. These results suggest a hierarchical folding model, but do not distinguish between a helical coil/radial loop model versus nonradial loop hierarchical models. Still missing is a description of folding levels lying between the 30-nm fiber and the postulated 250-nm coiling subunit.
Meanwhile, chromosome micromanipulation experiments have challenged predictions from both the radial loop and hierarchical models. Brief nuclease treatment leads to a loss of metaphase chromosome elasticity, arguing against a core protein scaffold dominating chromosome mechanical properties (Poirier and Marko, 2002). Furthermore, mechanical stretching experiments show elastic extension of metaphase chromosomes to several times their normal length without obvious changes in diameter or the sequential uncoiling of different folding levels as predicted in hierarchical models (Poirier et al., 2000). These results have led to the proposal of a chromatin network model for chromosome organization (Poirier and Marko, 2002) in which chromosomes are stabilized by protein cross-links between adjacent chromatin fibers present on average every 15 kb. However, no attention to possible intermediates of mitotic condensation intermediates was described for this model.
Here, we have attempted to reconcile the different results and predictions of these three classes of models for chromosome organization by describing structural transitions accompanying chromosome condensation during prophase and determining the temporal and spatial patterns of SMC2 and topoisomerase IIα recruitment to chromosomes relative to these transitions. Our results demonstrate folding of large-scale chromatin fibers during early prophase with formation of condensed, linear chromosomes of uniform width in middle prophase preceding formation of a well-defined axial core of either SMC2 or topoisomerase IIα. The measured 150–200-nm diameter for the SMC2 core axial distribution in metaphase chromosomes rules out a radial loop/helical coil model for chromosome organization or any model in which helical gyres are stabilized by a scaffold core lying within these gyres. Integrating key features of previous models, we propose a hierarchical folding, axial glue model of chromosome structure in which hierarchical folding drives chromatin compaction in early mitosis, whereas cross-linking by condensins and other proteins stabilizes chromosome shape and compaction later in mitosis.
Results
Three structural transitions during prophase chromosome condensation are associated with the progressive folding of large-scale chromatin fibers
CHO cells were used for the majority of our structural analysis. The availability of well-characterized cell synchronization procedures, in addition to earlier work from our laboratory on chromosome decondensation from mitosis through S phase (Belmont and Bruce, 1994), made them the cell line of choice. Due to limited cross-reactivity of our anti-condensin and topoisomerase IIα antibodies that were raised against synthetic peptides corresponding to human sequences, these analyses were complemented by selected experiments on human HeLa and HT-1080 cells.
Inspection of large numbers of nuclei, both by light and electron microscopy, suggested that although prophase chromosome condensation was relatively continuous, it could be divided into four distinguishable stages separated by three specific structural transitions.
We defined the first transition as separating a typical G2 interphase DNA distribution from an early prophase stage in which the first clear evidence of chromosome condensation appears. G2 nuclei showed DNA distributed relatively uniformly throughout the nuclear interior (Fig. 1 A). The substructure is highly suggestive of an underlying fibrillar pattern, below the resolution limit of the light microscope. Fiberlike staining can be traced in selected regions for 1–2 μm in length (Fig. 1 A, arrowhead). Transmission electron microscopy (TEM) shows more clearly large-scale chromatin organization discerned as ∼80-nm-diameter fibers (Fig. 2 A, arrows), as described previously during G1 (Belmont and Bruce, 1994). Serial section reconstructions, using 30–40-nm-thick sections, confirmed that these features corresponded to spatially distinct fiber segments that could often be traced as distinct fibers for 0.5–1.0-μm distances (unpublished data). Computational semi-thick sections, formed by projecting multiple individual thin sections, revealed multiple regions in which these large-scale fibers were loosely folded into ∼0.2–0.4-μm-diameter linear segments and larger aggregates (Fig. 3 A).
Figure 1.

Light microscope overview of prophase chromosome condensation. (A) G2 nucleus. Dispersed, apparently fibrillar substructure is distributed throughout the nuclear interior. Arrowhead points to isolated fiber segment. (B and C) Grazing (B) and mid-section (C) of very early prophase nucleus. Chromatin concentration in shell underlying nuclear periphery is becoming apparent. Condensation is highly heterogeneous, with dispersed fibrillar substructure coexisting with localized condensed chromosome regions (arrow). (D and E) Grazing (D) and mid-section (E) of early prophase nucleus. Distinct, individual chromosomes, concentrated near the periphery (E) cannot be visualized as uniform, extended, linear structures; however, condensed chromosomal regions can be followed as linear structures over short segments. (F and G) Grazing (F) and mid-section (G) of middle prophase nucleus. Chromosomes are ∼0.4–0.5 μm in diameter and can be traced (see short arrows) over extended distances. Sister chromatids can be distinguished as parallel structures (long arrow). Chromosomes are closely associated with nuclear periphery, with large regions of the interior free of chromosomes. (H) Late prophase/early prometaphase nucleus. Chromosomes are ∼0.8 μm in diameter; discrimination between sister chromatids is apparent at the tip of some chromosomes (arrowheads). Bottom right panel (a, c, d, f, g, and h) shows selected subregions, marked by long arrows, in A, C, D, F, G, and H. Bars: 2 μm (A–H), 1 μm (bottom right panel).
Figure 2.
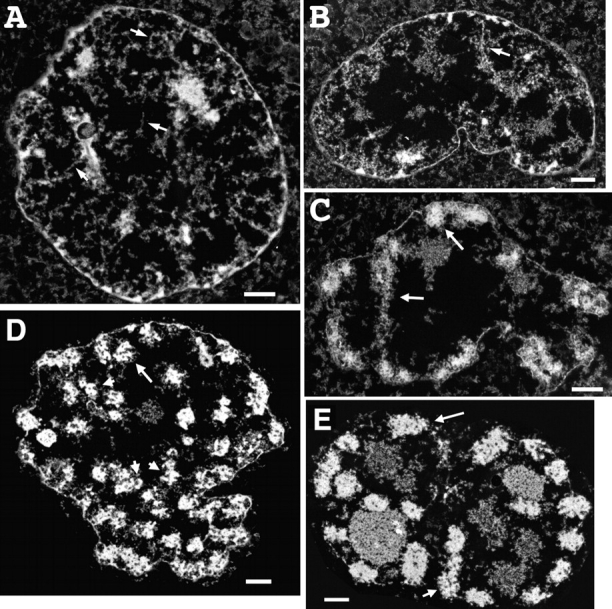
TEM survey of prophase chromosome condensation stages. (A–C) 200-nm-thick sections. (A) Nucleus with condensation typical of late S/G2 population. Chromatin is distributed throughout nuclear interior. Large-scale chromatin fibers ∼60–80 nm in diameter are present as extended fibers (arrows) or loosely folded and kinked within larger, often linear regions, typically 0.2–0.4 μm in diameter. (B) Very early prophase nucleus. Chromatin is now predominately peripherally located in an ∼1-μm shell underlying the nuclear envelope. Individual large-scale chromatin fibers are easily recognizable, but predominately folded within chromatin aggregates ∼1 μm in diameter (arrow points to region enlarged in Fig. 3 B). Distinct chromatids are not apparent. (C) Early prophase nucleus in which the chromosomal peripheral location and extensive interaction with nuclear envelope is obvious. Chromosome substructure consistent with folded large-scale chromatin fibers is evident in most chromosomal regions (arrows point to regions enlarged in Fig. 3, C and D). (D) Middle prophase nucleus. 80-nm-thick section shows condensed chromosomes, ∼0.4–0.5 μm diameter, containing ∼100–130-nm-diameter large-scale chromatin domains (arrowheads). Arrow points to chromosome region showing clear separation of the 0.2–0.25-μm-diameter sister chromatids. (E) Late prophase nucleus. 80-nm section shows nearly uniformly condensed chromosome cross sections in which substructure is difficult to discern, except at chromosome periphery and grazing sections; arrows point to chromosome regions showing substructure consistent with folded chromonema fibers. Bars, 1 μm.
Figure 3.

Chromonema fibers can be visualized during all stages of prophase chromosome condensation. (A) Late S/G2 nucleus. 175-nm computational projection from five sections, each 35 nm thick. Arrows with curved arrowheads point to large condensed regions, curved arrowheads point to condensed linear segment. Arrow with straight arrowhead points to loose coiling of chromonema fiber into an ∼0.2-μm-wide segment. (B–D) Early prophase chromosomes. Enlarged regions from sections shown in Fig. 2, B and C (arrows). (B) Large-scale chromatin fibers (arrowheads) appear to be coalescing into nascent chromatid. (Fig. 2 B). (C and D) More condensed regions of early prophase chromosomes (Fig. 2 C) appear to contain folded large-scale chromatin fibers (arrowheads). (E–H) Middle prophase chromosomes. Computational projection (E) of 11 80-nm thick serial sections and individual section (F). Arrows (F) point to isolated chromonema fibers in chromosome constriction likely to be a centromere (E, arrow). (G) 0.24-μm projection through three sections and individual section (H) with arrowheads pointing to features suggestive of folded 100–130-nm chromonema fibers. Separation between sister chromatids, each ∼200–250 nm in diameter, are seen over right side of chromosome. (I and J) Late prophase nucleus. (I) computational projection (1.2 μm) through 15 serial sections and an individual section (J). Particularly in grazing sections, ∼100–130-nm-diameter features suggestive of chromonema fibers are present (arrows). Bars: 0.5 μm (long), 0.2 μm (short).
Early prophase nuclei are distinguished from G2 interphase nuclei by the resolution and further compaction of these local chromatin aggregates into more clearly defined, linear chromatids and the collapse of these chromatids toward the nuclear envelope (Fig. 1, B and C). By electron microscopy, the degree of chromatin condensation appears highly heterogeneous throughout the nucleus, but the underlying fibrillar substructure is still apparent (Fig. 2 B, Fig. 3 B). Further condensation during early prophase leads to more easily recognizable, compacted chromosome regions, which in some cases can be traced as linear segments for several μm (Fig. 1, D and E; Fig. 2 C). The fibrillar substructure of these more condensed chromosome regions remains quite apparent (Fig. 2 C; Fig. 3, C and D). However, chromatin packing is still heterogeneous, such that neither linear chromosomes nor distinct sister chromatids are recognizable over extended distances. At the same time, the shell of peripheral chromatin appears to condense more compactly toward the nuclear envelope into these distinct chromosome segments. The percentage of these early prophase stages rises from ∼10 to 35% between 7 and 13 h after release from the late G1/S phase block (unpublished data), with cells with the typical late S/G2 nuclear appearance dropping from ∼85 to 50%.
The second transition corresponds to the change from this early prophase stage to a middle prophase stage defined by the appearance of clearly defined chromosomes that can be easily traced as linear, uniformly condensed chromosomes over distances exceeding several μm in length (Fig. 1, F and G). Chromosomes associate closely with the nuclear envelope, generating large, DNA-free spaces within the interior. Chromosome diameter is uniform and ∼0.4–0.5 μm at this stage—a reduction from the less compact, early prophase chromosomes. Parallel sister chromatids can be recognized at many chromosomal locations (Fig. 1, F and G; long arrows). Discrimination of individual sister chromatids from individual thin TEM sections is more difficult at this stage, although examples are present in which identification of sister chromatids is possible (Fig. 2 D, arrow; Fig. 3 H). Condensation is relatively uniform along the length of the chromosome, allowing chromosomes to be traced over extensive distances within the three-dimensional electron microscopy reconstruction (unpublished data). Within these condensed chromatids, distinct, ∼100-nm-diameter large-scale chromatin fibers can still be visualized (Fig. 2 D, arrows and arrowhead; Fig. 3, G and H). This impression is reinforced by a selected example from a serial section reconstruction (67 sections, each 80 nm thick) showing what appears to be a centromere constriction. The individual serial section (Fig. 3 F) suggests that this constriction is formed by two extended large-scale chromatin fibers, presumably reflecting sister chromatids, which join distally to more condensed chromatids. A chromosome that extended through the center of a nucleolus showed a similar constriction over the chromosome segment within the nucleolus, formed by two parallel large-scale chromatin fibers (Fig. S1, available at http://www.jcb.org/cgi/content/full/jcb.200406049/DC1). We presume this constriction contains a ribosomal gene cluster significantly delayed in condensation.
The third transition during prophase chromosome condensation was considered as the transition from this middle prophase stage to a late prophase stage in which chromosome diameters roughly double to ∼0.8–1.0 μm (Fig. 1 H). Although chromatids were tightly opposed along their length, in favorable orientations individual chromatids could be recognized as lying adjacent and parallel (Fig. 2 E, arrowhead), with suggestions of a slight separation particularly at the chromosome ends. Chromatid arms are straight, uniform in diameter, and show little substructure at light microscopy resolution. At higher resolution, individual electron microscopy sections show uniformly condensed chromosome cross sections in which substructure is also difficult to discern. However, particularly at the chromosome periphery and in grazing sections, there is a strong suggestion again of large-scale chromatin domains, ∼100–130 nm in size (Fig. 3 J, arrows). Typical chromosome arm lengths are 1–3 μm as traced in optical sections, several fold shorter than observed in middle prophase. The overall level of chromosome condensation is intermediate between middle prophase and metaphase, with an average chromatid diameter of ∼0.4 μm versus metaphase diameters of 0.6–0.8 μm and middle prophase diameters of ∼0.2–0.3 μm (Fig. 2 E; Fig. 3, I and J).
Dynamic recruitment and chromosomal distribution of SMC2 and topoisomerase IIα during prophase chromosome condensation
Using the descriptions of distinct prophase stages defined in the previous section, we next determined the timing of chromosome association and spatial distribution of topoisomerase IIα and SMC2 in human HT1080 cells. Our results do not support the establishment of an early axial staining pattern for either topoisomerase IIα or SMC2, but instead indicate a more complicated, dynamic redistribution of both proteins during prophase progression.
In G2 nuclei (Fig. 4, A–C), topoisomerase IIα staining appears relatively diffuse throughout the nucleus, with the exception of bright foci appearing as paired spots (Fig. 4 C, arrowheads). These likely represent replicated centromeres known to concentrate topoisomerase IIα during mitosis, beginning in late S/G2 (Rattner et al., 1996). In contrast, SMC2 is concentrated in small foci, frequently clustered near the periphery of condensed chromatin masses; in some cases these foci appear to overlap less condensed chromosomal regions (Fig. 4 A, arrowheads). At this stage, topoisomerase IIα and SMC2 staining show no apparent correlation (Fig. 4 C).
Figure 4.
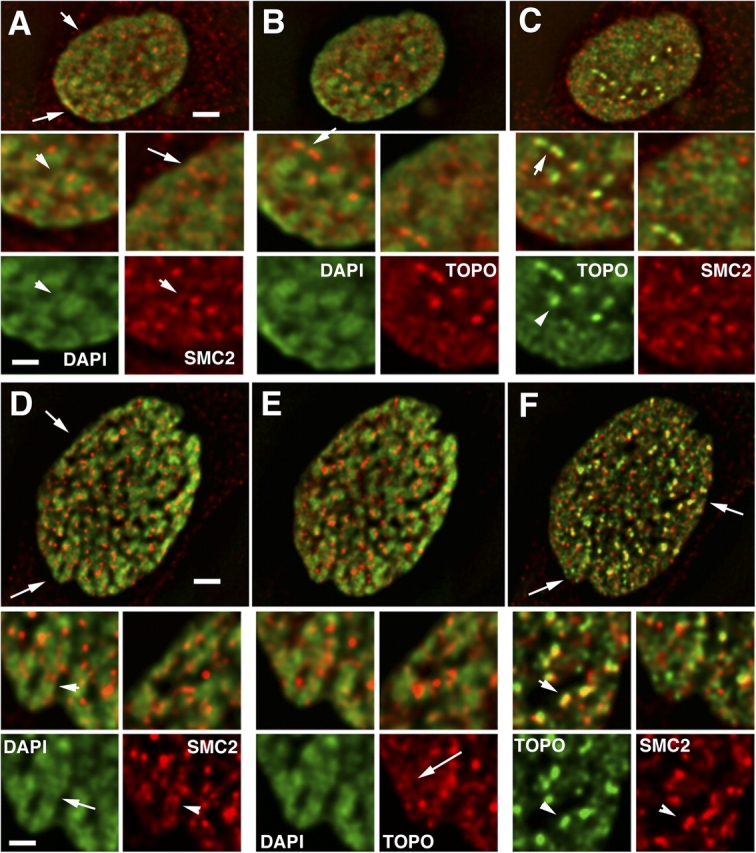
SMC2 and topoisomerase IIα staining in G2 and early prophase nuclei. (A and D) DAPI (green) vs. SMC2 (red); (B and E) DAPI (green) vs. topoisomerase IIα (red); (C and F) topoisomerase IIα (green) vs. SMC2 (red). Arrows (A–C, D–F) point to regions enlarged in insets. G2 nucleus (A–C). SMC2 appears in bright foci, largely between and on periphery of condensed DNA (A, inset, arrow). In some cases, SMC2 staining overlaps less condensed DNA regions (A, insets, arrowheads). Topoisomerase is distributed more diffusely over chromosomal regions, but is concentrated in bright doublets (C, arrowheads), likely representing centromeric regions (see text). Early prophase nucleus (D–F). SMC2 staining is now more obviously concentrated in small foci associated with chromosomes, but largely on the periphery. Short, parallel linear segments are beginning to appear (C, inset, arrowheads), but on the chromosome exterior. Where SMC2 staining is interior, it frequently appears to lie in areas of lower DAPI intensity (D, inset, arrow). Topoisomerase IIα is still located diffusely through chromosome regions (E, inset, arrow). However, now bright doublets of topoisomerase IIα staining frequently overlap bright doublet foci of SMC2 staining (F, inset, arrowheads). Bars: 2 μm (top panels), 1 μm (insets).
In early prophase, SMC2 staining still appears in bright foci, but is more obviously associated with the condensing chromosomes (Fig. 4 D; Fig. S2, available at http://www.jcb.org/cgi/content/full/jcb.200406049/DC1). Interestingly, small linear staining segments are appearing at this stage, in some cases as paired structures (Fig. 4 D, insets, arrowheads; Fig. S2, arrows), but these map to the chromosome edges rather than to a centrally located chromatid axis. Where present within the chromosomal masses, the bright foci of SMC2 staining frequently overlay regions of lower DAPI density (Fig. 4 D, bottom left inset, arrow). Again, topoisomerase IIα staining is concentrated over bright foci likely representing centromere regions (Fig. 4 F). Less intense staining elsewhere is more diffuse than SMC2 and is distributed throughout chromosomal regions (Fig. 4 E, bottom right inset, arrow).
In contrast to the G2 nuclei, at this stage bright, paired foci of SMC2 staining now colocalizes with a significant fraction of the topoisomerase IIα doublet foci (Fig. 4 F, bottom insets, arrowheads). This colocalization persists into middle prophase (Fig. 5 C). Double staining with antibodies to both the centromeric protein, CENP-B, and SMC2 revealed a significant colocalization of SMC2 over a subset of centromeric foci during early and middle prophase (unpublished data). However, outside of these bright foci, SMC2 and topoisomerase staining remain largely uncorrelated (Fig. 4 F). The intensity of the SMC2 staining foci observed within these late G2 and early prophase nuclei is several fold lower than the intensity of SMC2 axial staining observed during metaphase.
Figure 5.
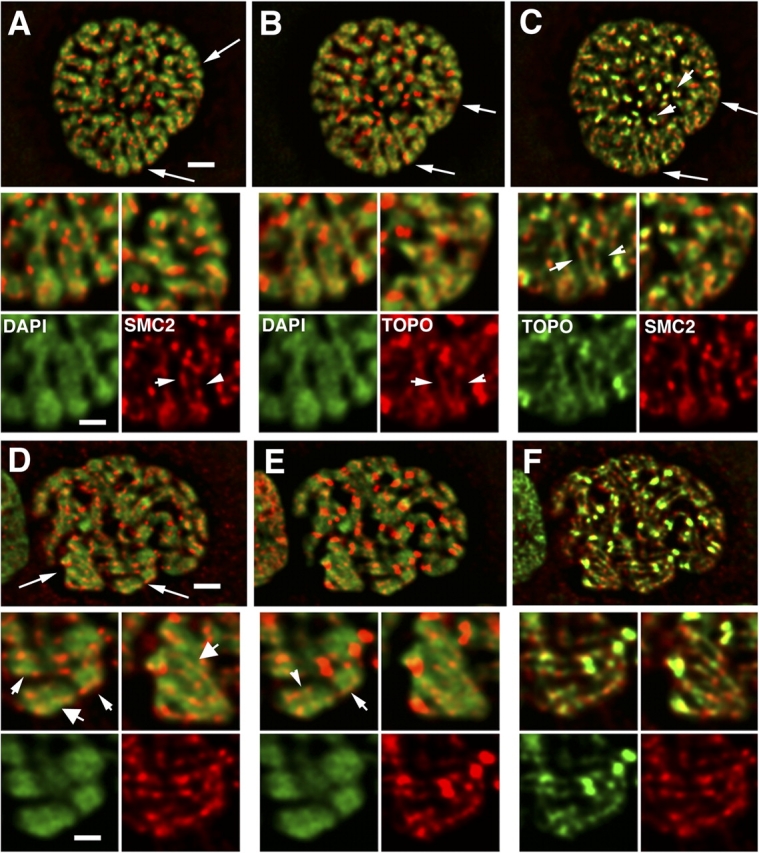
SMC2 and topoisomerase IIα staining in middle and late prophase nuclei. (A and D) DAPI (green) vs. SMC2 (red); (B and E) DAPI (green) vs. topoisomerase IIα (red); (C and F) topoisomerase IIα (green) vs. SMC2 (red). Arrows (A–C, D–F) point to regions enlarged in insets. Middle prophase (A–C). SMC2 and topoisomerase IIα staining is now becoming correlated, forming colinear segments, but largely on the chromosome exterior (A–C, insets, arrowheads). Arrowheads (C) point to colocalization of both in doublets, likely to be paired centromere regions. Late prophase (D–F). Correlation between SMC2 and topoisomerase IIα now very obvious, again forming colinear segments localizing sometimes on chromosome exterior (small arrowheads, insets), but in other locations toward the chromatid axis (large arrowheads, insets). Bars: 2 μm (A–F), 1 μm (insets).
In middle prophase, SMC2 and topoisomerase IIα staining now begins to become correlated, with the degree of this correlation varying in different chromosome regions (Fig. 5 C; Fig. S3, available at http://www.jcb.org/cgi/content/full/jcb.200406049/DC1). Again, SMC2 staining remains nonaxial, with staining foci frequently appearing in these optical sections at the edges of condensed chromosome regions rather than centered over the regions of brightest DAPI staining. Topoisomerase IIα staining also appears in foci which, like the SMC2 staining, often appears at edges of condensed regions. However, there appears to be more diffuse staining throughout the interior of the DAPI regions than observed for SMC2. However, many regions of staining appear in which SMC2 and topoisomerase IIα are highly correlated. These regions of correlated SMC2 and topoisomerase IIα staining include 1–2-μm-long linear segments that fall largely on the periphery of the chromosomes, and not in a central axis (Fig. 5, A–C, insets, arrowheads; Fig. S3, arrows). Interestingly, these linear segments of SMC2 and topoisomerase IIα staining in some cases appear colinear, but do not exactly colocalize. One exception is over centromeric regions, in which bright foci of SMC2 and topoisomerase IIα staining overlap (Fig. 5 C, top, arrowheads).
Strikingly, in late prophase, linear SMC2 and topoisomerase IIα staining as well as close correlation between the SMC2 and topoisomerase IIα staining now becomes quite pronounced throughout the nucleus (Fig. 5, D–F; Fig. S4). Topoisomerase IIα is strongly recruited to centromere regions. However, rescaling the topoisomerase IIα image to saturate the centromeric staining now reveals very well-defined, linear staining for topoisomerase IIα, as well as SMC2, along chromosome arms. Again, the staining does not exactly colocalize. The location of the SMC2 and topoisomerase IIα staining though is ambiguous, with some regions showing staining at the outside chromatid edges (Fig. 5, D and E, top insets, small arrowheads; Fig. S4, arrows, available at http://www.jcb.org/cgi/content/full/jcb.200406049/DC1) and others showing what appears to be internal staining along the chromatid center axis (Fig. 5 D, top insets, larger arrowheads).
Immunogold staining reveals a broad axial core of SMC2 staining in metaphase chromosomes
By metaphase, the centralized axial staining of both topoisomerase IIα and SMC2 is clear with extensive colocalization of topoisomerase IIα and SMC2 (Fig. 6, A–C). The axial staining observed for both SMC2 and topoisomerase IIα corresponds to a staining diameter roughly half the chromatid diameter and comparable to the diffraction limit of light microscopy. Therefore, it is not possible to determine the true diameter of the axial condensin core by conventional light microscopy. Previous immunogold localization of topoisomerase II was done on metaphase chromosomes greatly swollen in low salt buffers, precluding measurement of core staining diameter (Earnshaw and Heck, 1985). We used immunogold staining and TEM for higher resolution visualization of the axial SMC2 staining in native metaphase chromosomes. This revealed SMC2 staining distributed over an ∼0.15–0.2-μm width in HeLa metaphase chromatids, roughly one third of the ∼0.5-μm chromatid diameter (Fig. 6 D). Gold staining is relatively diffuse, although this may reflect limitations in antibody staining as opposed to the true protein distribution. Stereopairs reveal hints of a possible underlying organization to the staining (unpublished data), possibly helical, but this remains speculative at this time and awaits detailed structural analysis from three-dimensional reconstructions beyond the scope of this work.
Figure 6.
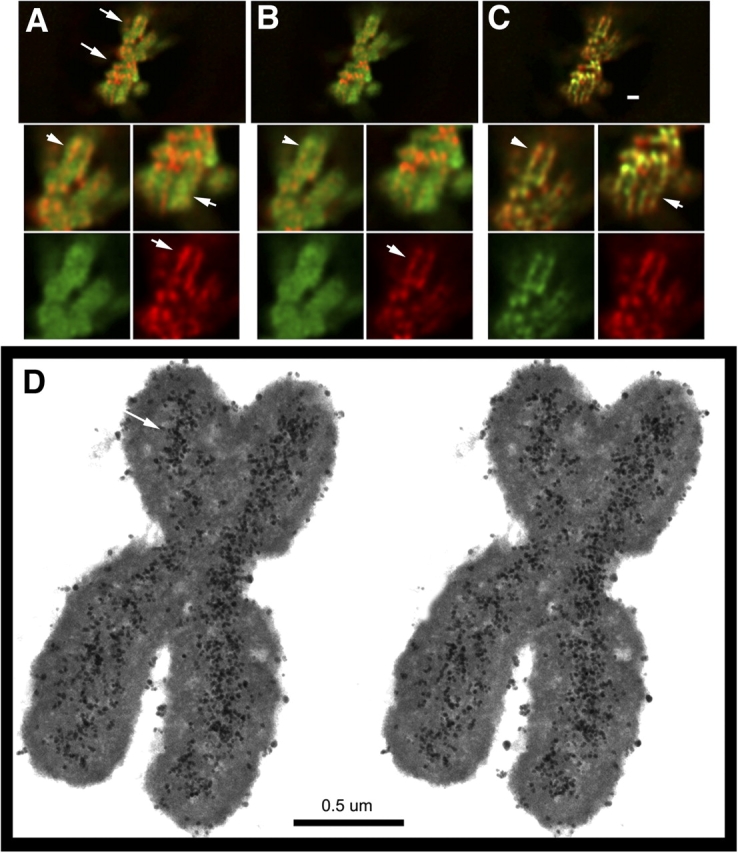
Axial staining of SMC2 and topoisomerase IIα in metaphase chromosomes. (A–C) SMC2 and topoisomerase IIα staining. (A) DAPI (green), SMC (red); (B) DAPI (green), topoisomerase IIa (red); (C) topoisomerase IIα (green), SMC2 (red). Arrows point to enlarged regions in insets. Arrowheads in insets point to axial staining. Bar, 1 μm. (D) Immunogold SMC2 staining. Stereopair of semi-thick sections through metaphase chromosome. Silver-enhanced gold staining (arrows) reveals axial staining of SMC2 of ∼0.15–0.2 μm in width.
Discussion
A major stumbling block in understanding the biochemical mechanisms underlying chromosome condensation is our very incomplete knowledge of the structural motifs underlying mitotic chromosome condensation. Here, we describe three structural transitions underlying prophase mitotic chromosome condensation and correlate these transitions with the dynamic redistribution of topoisomerase IIα and the condensin SMC2 subunit (Fig. 7 A). We show that folding of large-scale chromatin fibers is a prominent feature of these early stages of condensation. SMC2 and topoisomerase IIα staining first appears in foci distributed throughout the chromosome width or even at the chromosome exterior. A further doubling of the chromatid diameter and the appearance of a well-defined, central axis of topoisomerase IIα and condensin SMC2 staining occurs well after formation of uniformly condensed chromosomes and a defined chromosome axis in middle prophase. The temporally coordinated appearance of axial staining for both of these proteins down the chromatid center appears as a relatively late event in prophase chromosome condensation. Electron microscopy immunogold staining shows that this axial SMC2 staining spans roughly one third of the metaphase chromosome diameter.
Figure 7.

Models for chromosome condensation. (A) Stages of condensation. Changes in large-scale chromatin folding (blue) versus SMC2 distribution (red) from early S (a), G2 (b), early prophase (c), middle prophase (d), late prophase (e), and metaphase (f). See text for details. (B) “Hierarchical folding, axial glue” model of metaphase chromosome structure. (a) 30-nm fiber folds into 100–130-nm chromonema fiber, which folds into 200–250-nm middle prophase chromatid, which folds into 500–750-nm metaphase chromatid. Only one chromatid is shown here. (b) Axial condensin distribution (green) occupies approximately one third of the chromatid diameter, acting as cross-linking “glue” to stabilize metaphase chromosome.
Implications for structural and functional models of mitotic chromosome condensation
Our findings have strong implications for models of mitotic chromosome structure. In radial loop models, chromosome condensation and formation of a defined chromosome axis was conceptualized as occurring through the organization of chromatin loops by the formation and compaction of a chromosome scaffold. Recently, linear axial topoisomerase IIα staining was reported as appearing in prophase before the appearance of condensin axial staining (Maeshima and Laemmli, 2003). These observations led to a two-step chromosome assembly model in which early chromosome condensation was driven through organization of radial loops by axial topoisomerase IIα, with recruitment of condensins to the center chromatid axis occurring later.
Assuming fixed radial loop sizes, one would predict chromosome condensation as beginning by the coalescence of loops into early prophase chromosomes with condensed foci of the same diameter as metaphase chromosomes. These condensed foci might be separated by less dense, elongated early prophase chromatids with lower numbers of loops per unit length, but again the diameter of these less dense regions should be comparable to metaphase chromosomes. These predictions are in contradiction to our observed data that show early prophase condensation accompanied by progressive folding and compaction of large-scale chromatin fibers and the doubling of prophase chromatid diameters between middle and late prophase. Moreover, by systematically analyzing changes in chromosome condensation throughout the different stages of prophase, we were able to better time the recruitment of topoisomerase IIα and condensin subunit SMC2. We did not observe axial chromosome staining for topoisomerase IIα and SMC2 until late prophase, and even at this stage the localization to the chromatid center axis was ambiguous over some chromosome regions where the linear staining appeared toward the chromosome exterior. More importantly, the timing of axial staining for these two proteins appeared very close temporally.
The reasons for the difference between our results and those reported by Maeshima and Laemmli (2003) are unclear. The topoisomerase IIα axial staining they observed in the absence of condensin axial staining was for an example that we would have classified as late prophase, based on the measured chromosome diameter. Our observed nonaxial SMC2 staining intensity in early prophase is significantly weaker than the axial staining observed in metaphase, but appears specific, as it was competed with excess antigenic peptide (unpublished data). Typical exposure times for early prophase nuclei were roughly 8–10-fold longer than that used for metaphase examples, and roughly fivefold longer for late prophase nuclei than that used for metaphase examples. In Maeshima and Laemmli (2003), detergent extraction before fixation was used in a subset of experiments. It is possible that condensin is more loosely associated with prophase chromosomes than with metaphase chromosomes and is removed by this treatment, resulting in a further and artificial enhancement of the contrast between prophase and metaphase staining.
Our results also contradict predictions of a modified radial loop model in which loops of 30-nm chromatin fibers are organized by an axial distribution of chromosome scaffold proteins in prophase chromatids, with this prophase chromatid itself folding into a helix to give rise to the metaphase chromosome (Rattner and Lin, 1985; Boy de la Tour and Laemmli, 1988). At our middle prophase stage, marked by uniformly high chromatin condensation, a uniform chromosome diameter, and clearly defined chromosome arms, topoisomerase IIα and SMC2 staining is still not axial. Moreover, in metaphase chromosomes, our electron microscopy immunogold localization reveals SMC2 core staining covering approximately the middle third of the chromatid diameter, with an ∼0.15–0.2-μm staining diameter. There is no evidence for an unlabeled metaphase chromatid core formed by radial loops extending in all directions from a helically coiled scaffold as expected in a radial loop/helical coil model.
Instead, the early events in chromosome condensation described in this paper are more consistent with a hierarchical folding model of chromosome organization involving the folding of 30-nm fibers into large-scale chromatin fibers which themselves are folded further to form the mitotic chromatid. Previous work, examining chromosome decondensation during early G1, demonstrated the existence of large-scale chromatin fibers, or “chromonema fibers,” as discrete, spatially distinct entities (Belmont and Bruce, 1994). The observed chromosomal decondensation during late telophase through late G1 and early S phase was consistent with a progressive uncoiling of large-scale chromatin fibers from telophase through G1. Use of the lac operator/lac repressor system to tag gene-amplified chromosome regions allowed tracing of distinct large-scale fibers in live interphase cells for over 5 μm in length (Robinett et al., 1996). Conceptually, one might still imagine that the large-scale chromatin fibers visualized beginning in telophase and through G1 represent a dramatic chromatin reorganization from a different chromosome organization present throughout most of mitosis. However, we now complement our previous work, using the same experimental cell system, by showing that all stages of chromosome condensation during prophase are in fact consistent with the progressive folding and coiling of large-scale chromatin fibers.
Regions showing loose coiling are already evident in G2 nuclei and a coalescence of these large-scale chromatin fibers into nascent chromatids, together with a collapse of chromatin toward the nuclear envelope, were used by us to define early prophase morphologically. During later stages of chromosome condensation, although large-scale chromatin fibers are no longer traced, unambiguously, within the condensed, tightly packed chromosomes, it is clear that they can still be recognized in favorable views, particularly within grazing chromosome sections.
However, two important aspects of our results do not fit easily into hierarchical models. One is the apparent lack of structural order observed for the folding of large-scale chromatin fibers in the nascent, condensing chromatids. Implicit in hierarchical models is the idea of self-assembly driven by successive and regular coiling of chromatin fibers. The absence of a high degree of structural order at these early stages of chromosome condensation therefore suggests the possibility of underlying, inherently asymmetric molecular mechanisms, provided by nonhistone chromosomal proteins, driving condensation. Second, hierarchical models would predict a folding of the 200–250-nm-diameter middle prophase chromatid to form the late prophase and metaphase chromatid, consistent with our previous observation of a ∼250-nm subunit within metaphase chromosomes (Strukov et al., 2003). However, the observed width of the axial SMC2 core seems too large to fit within the middle of a 500-nm metaphase chromatid formed by helical folding of a 200–300-nm middle prophase chromatid, and there is no evidence for a chromatin-free hole of this dimension. This suggests that the SMC2 distribution in metaphase chromatids spans across different hierarchical subunits. The alternative is that there is a fundamental local reorganization of chromatin from late prophase into metaphase associated with the observed changes in topoisomerase IIα and SMC2 distribution. Because large-scale chromatin fibers can still be recognized in sections through the chromatid exterior, presumably this chromatin reorganization would primarily effect the center chromatid core.
As described in the Introduction, network models of chromosome compaction focus on the concept of fiber–fiber cross-linking within a chromatin gel and the absence of a distinct protein scaffold chromosome core. Our observations of distinct chromosome folding intermediates contradict simple network models in which chromosome condensation occurs through a progressive increase in chromosome diameter and contraction of chromosome length. However, the recent estimate of an ∼15-kb distance between cross-links (Poirier and Marko, 2002) corresponds to a distance of ∼125 nm for a 30-nm chromatin fiber with a compaction ratio of 40. This would more closely match the size scale of the ∼100-nm-diameter large-scale chromatin fibers we have observed as compared with the 50–200-kb loops predicted in radial loop models. This frequency of cross-linking could occur through SMC2 interactions between chromatin fibers, both through contact between the large-scale chromatin fibers on the chromosome periphery with the axial SMC2 core and/or through SMC2 fiber interactions within the metaphase chromosome core.
Unified working model for mitotic chromosome condensation
The previous discussion highlights discrepancies between previous models of chromosome organization and our observations of early stages of chromosome condensation. Therefore, we propose a “hierarchical folding, axial glue” working model for chromosome condensation (Fig. 7 B). Key features from previous hierarchical folding, radial loop, and chromatin network models are incorporated into this model. Initial folding of large-scale chromatin fibers into early prophase chromosomes, with release of chromatin attachments from interior nuclear structures, is followed by a condensation into the uniform, tight middle prophase chromatids, ∼200–300 nm in diameter. A second coiling of this middle prophase chromatid during late prophase through prometaphase yields the final metaphase chromosome.
During the transition from middle prophase to the fully condensed metaphase chromosome, an axial distribution of condensins and topoisomerase IIα forms. We envision a dynamic chromosome core, which may resemble a network of cross-linked chromatin more than the unitary protein scaffold envisioned in the original radial loop models, but which may still serve to provide chromosome stability. This model would explain the observations of large-scale chromatin folding in late G2 through middle prophase, but also the failure to see a progressive unfolding of these levels of organization in mechanical stretching experiments of metaphase chromosomes or a dependence of chromosome mechanical properties on a protein core.
This model would also be consistent with a recent report indicating that the condensin subunit SMC2 plays a functional role in the stabilization of metaphase chromosome structure as opposed to chromosome compaction during mitotic progression (Hudson et al., 2003). Subsequent work from the Earnshaw laboratory further confirms no significant defects in the degree of compaction after near total knockdown of SMC2 if intact mitotic cells are examined with no hyptonic pretreatment (Gassmann et al., 2004). These observations are in agreement with our hierarchical folding/axial glue model in which chromosome condensation would occur largely through molecular mechanisms driving hierarchical folding, whereas axial concentration of condensins and topoisomerase IIα would serve to lock these folding subunits together within the condensed metaphase chromosome.
The value in proposing this working model is that it leads to a different way of conceptualizing the functional process of chromosome condensation. For instance, our model predicts that the in vitro appearance of radial loops in isolated chromosomes after exposure to low salt buffers is simply the manifestation of the cross-linking that occurs late in the process of chromosome condensation, and does not reflect the actual structural motifs responsible for chromosome condensation (i.e., condensation driven by formation of radial loops). At the same time this model makes clear predictions that can be tested experimentally. For example, our model predicts that chromosome stretching experiments should reveal the progressive unfolding of hierarchical folding patterns if the cross-linking provided by condensins are eliminated by condensin depletion. Proposing this working model also serves to focus attention on key questions for future work. What are the specific roles of condensins and topoisomerase IIα during early chromosome condensation (Swedlow and Hirano, 2003), and what molecular mechanisms independent of condensins may drive chromosome condensation? Is there a fundamental local reorganization of chromatin between middle and late prophase, or are the changes simply a result of global folding of the middle prophase chromatid? Functional dissection of the biochemical mechanisms underlying chromosome condensation will require a detailed understanding of the different structural stages of chromosome condensation, as we have begun to outline in this paper.
Materials and methods
Buffers
Buffers include CMF-PBS (calcium, magnesium-free Dulbecco's PBS solution, 2.68 mM KCl, 1.47 mM KH2PO4, 137 mM NaCl, and 8.06 mM Na2HPO4-7H2O), PBS* (CMF-PBS plus 5 mM MgCl2 and 0.1 mM EDTA), buffer A (80 mM KCl, 20 mM NaCl, 2 mM EDTA, 0.5 mM EGTA, 15 mM Pipes, 15 mM β-mercaptoethanol, 0.5 mM spermidine, 0.2 mM spermine, and 10 μg/ml turkey egg white inhibitor, pH 7.0), and buffer C (0.25 M sucrose, 10 mM Pipes, 1.5 mM MgCl2, 1.0 mM CaCl2 and 10 μg/ml turkey egg white protease inhibitor, pH 6.8).
Tissue culture and synchronization
Human HeLa and HT1080 cells were grown in Dulbecco's minimum essential medium with 10% FBS and supplemented with nonessential amino acids. CHO cells were grown in Ham's F10 media supplemented with 15% bovine calf serum (Hyclone). Synchronization used a three-step procedure. Cells were first blocked in G1 using a 36-h incubation in isoleucine-deficient Ham's F10 media with 10% dialyzed FCS (Irvine Science), then released from the G1 block and blocked at the G1/S boundary by 10 h in complete Ham's F10 with 1 mM hydroxyurea (HU) (Tobey and Crissman, 1972; Tobey et al., 1990). Cells were washed 2× in Ham's F10 media to remove HU and were allowed to progress through S into G2. Mitotic cells were removed using a nocodazole block and shake-off. Because CHO cells escape a nocodazole block after several hours, multiple shake-offs were used. Specifically, 5 h after HU release, cells were incubated in 600 ng/ml nocodazole (Sigma-Aldrich) for 2 h, followed by shake-off to remove mitotic cells; mitotic shake-off was repeated several times at 2-h intervals with nocodazole present.
Synchronization was monitored by flow cytometry. After the isoleucine minus block, 90–94% of cells had 2c DNA content, with 94% of cells with 2c DNA content after the HU block. The HU block was reversible for >95% of the cells, as assayed by increased DNA content after release. 9 h after the HU block, 78% of cells showed 4c DNA content, increasing to 86% after an additional 4 h. Cell numbers decreased to 57 and 23% the values from the S phase block release, suggesting 43 and 77% of the cells had progressed into mitosis 9 and 13 h after HU release, respectively.
Light microscopy preparation and immunostaining
CHO cells were plated directly on #11/2 coverslips and were grown to 60–70% confluence. Synchronized CHO cells were harvested with trypsin-EDTA, washed 2× in CMF-PBS, and spun onto coverslips. Cells were fixed 30 min in CMF-PBS with 2% glutaraldehyde at RT, followed by12 h at 4°C. After fixation, coverslips were washed in CMF-PBS and stained with 0.5 μg/ml DAPI.
For immunostaining, coverslips were washed in PBS*, fixed with 1.6% freshly prepared PFA in PBS*, and then permeabilized in PBS* with 0.1% Triton X-100. Staining conditions were as described elsewhere (Li et al., 1998). Primary antibodies were a rabbit anti-hCAP-E/SMC2 peptide (Kimura et al., 2001) used at 0.23 μg/ml, and a mouse monoclonal anti-topoisomerase IIα antibody (Topogen) used at 1:500 dilution. PBS* with 0.1% Triton X-100, 5% donkey serum, and 5% goat serum was used throughout primary and secondary antibody staining. Secondaries were a Texas red–labeled, F(ab′)2 donkey anti–rabbit IgG (1:500 dilution) and an FITC labeled goat anti–mouse IgG (1:1,000 dilution; Jackson ImmunoResearch Laboratories). After immunostaining, samples were mounted using ProLong AntiFade (Molecular Probes, Inc.). To confirm the specificity of SMC2 staining we used peptide competition experiments.
Light microscopy
All light microscopy used optical sectioning deconvolution methods as described previously (Belmont et al., 1993). Exposure times were adjusted for cell cycle stage to compensate for differential staining intensity. To reduce chromatic aberration between the DAPI and FITC (topoisomerase IIα) images, the same FITC band-pass emission filter, but different excitation filters, were used.
Electron microscopy
Cells were washed in buffer A, permeabilized in detergent, fixed with glutaraldehyde, dehydrated in EtOH, postfixed in osmium tetroxide, and embedded in Epon 812. Serial sectioning, electron microscopy, and display were as described previously (Belmont and Bruce, 1994).
Immunogold staining of condensin
Buffer A was used without β-mercaptoethanol. Log phase HeLa cells were treated with 600 ng/ml nocodazole for 2 h. Mitotic cells were collected by three shake-offs, separated by 15 min. Mitotic cells were put on ice between shake-offs, and then washed in PBS*, followed by incubation in 75 mM KCl for 10 min. Cells were centrifuged, resuspended in buffer A with 0.1 mg/ml digitonin, and vortexed 2 × 30 s to release chromosomes. Cells and nuclei were pelleted by centrifugation, and the supernatant was applied to glass coverslips. Chromosomes were stuck to the glass coverslips by low speed centrifugation. Chromosomes were placed into either buffer C and fixed in 1.85% freshly prepared PFA for 10 min. Coverslips were washed 3 × 5 min in buffer C, 3 × 5 min in buffer A, 3 × 5 min in buffer A plus 20 mM glycine, then blocked in 6% normal goat serum in buffer A for 1 h. Primary and secondary antibody staining was done in buffer A as described for immunofluorescence, but using a Nanogold goat anti–rabbit secondary antibody at 1:400 dilution for 20 h at 4°C. Coverslips were washed in buffer C and fixed in 2% glutaraldehyde for 2 h. Silver enhancement was done as described elsewhere (Burry, 1995; Gilerovitch et al., 1995), followed by embedding in Epon. Images were taken on a transmission electron microscope (CM200; Philips) using a CCD camera (Tietz).
Online supplemental material
Three additional figures are available online that show examples of SMC2 and topoisomerase IIα staining in early, middle, and late prophase from Fig. 6 and Fig. 7, at higher magnification. These supplemental figures also show the SMC2 and topoisomerase IIα staining for the entire nuclei as separate, grayscale images rather than after merging with DAPI staining in color images as shown in Fig. 6 and 7. Online supplemental material available at http://www.jcb.org/cgi/content/full/jcb.200406049/DC1.
Acknowledgments
We thank Dmitri Novikov for drawing the models in Fig. 7.
This work was supported by NIH grant GM53926 to T. Hirano and NIH grant GM42516 to A. Belmont.
N. Kireeva and I. Kereev's present address is Department of Electron Microscopy, A.N. Belozersky Institute for Physical Chemical Biology, Moscow State University, Moscow 119899, Russia.
Abbreviations used in this paper: SMC2, structural maintenance of chromosomes 2; TEM, transmission electron microscopy.
References
- Belmont, A.S. 1998. Nuclear ultrastructure: transmission electron microscopy and image analysis. Methods Cell Biol. 53:99–124. [DOI] [PubMed] [Google Scholar]
- Belmont, A.S., and K. Bruce. 1994. Visualization of G1 chromosomes: a folded, twisted, supercoiled chromonema model of interphase chromatid structure. J. Cell Biol. 127:287–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belmont, A.S., J.W. Sedat, and D.A. Agard. 1987. A three-dimensional approach to mitotic chromosome structure: evidence for a complex hierarchical organization. J. Cell Biol. 105:77–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belmont, A.S., Y. Zhai, and A. Thilenius. 1993. Lamin B distribution and association with peripheral chromatin revealed by optical sectioning and electron microscopy tomography. J. Cell Biol. 123:1671–1685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boy de la Tour, E., and U.K. Laemmli. 1988. The metaphase scaffold is helically folded: sister chromatids have predominately opposite helical handedness. Cell. 55:937–944. [DOI] [PubMed] [Google Scholar]
- Burry, R.W. 1995. Pre-embedding immunocytochemistry with silver-enhanced small gold particles. In Immunogold-Silver Staining: Principles, Methods, and Applications. M.A. Hayat, editor. CRC Press, Boca Raton, FL. 217–230.
- Earnshaw, W.C., and M.M.S. Heck. 1985. Localization of topoisomerase 2 in mitotic chromosomes. J. Cell Biol. 100:1716–1725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gasser, S.M., T. Laroche, J. Falquet, E. Boy de la Tour, and U.K. Laemmli. 1986. Metaphase chromosome structure. Involvement of topoisomerase II. J. Mol. Biol. 188:613–629. [DOI] [PubMed] [Google Scholar]
- Gassmann, R., P. Vagnarelli, D. Hudson, and W.C. Earnshaw. 2004. Mitotic chromosome formation and the condensin paradox. Exp. Cell Res. 296:35–42. [DOI] [PubMed] [Google Scholar]
- Gilerovitch, H.G., G.A. Bishop, J.S. King, and R.W. Burry. 1995. The use of electron microscopic immunocytochemistry with silver-enhanced 1.4-nm gold particles to localize GAD in the cerebellar nuclei. J. Histochem. Cytochem. 43:337–343. [DOI] [PubMed] [Google Scholar]
- Hagstrom, K.A., V.F. Holmes, N.R. Cozzarelli, and B.J. Meyer. 2002. C. elegans condensin promotes mitotic chromosome architecture, centromere organization, and sister chromatid segregation during mitosis and meiosis. Genes Dev. 16:729–742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirano, T., R. Kobayashi, and M. Hirano. 1997. Condensins, chromosome condensation protein complexes containing XCAP-C, XCAP-E and a Xenopus homolog of the Drosophila Barren protein. Cell. 89:511–521. [DOI] [PubMed] [Google Scholar]
- Hudson, D.F., P. Vagnarelli, R. Gassmann, and W.C. Earnshaw. 2003. Condensin is required for nonhistone protein assembly and structural integrity of vertebrate mitotic chromosomes. Dev. Cell. 5:323–336. [DOI] [PubMed] [Google Scholar]
- Kimura, K., O. Cuvier, and T. Hirano. 2001. Chromosome condensation by a human condensin complex in Xenopus egg extracts. J. Biol. Chem. 276:5417–5420. [DOI] [PubMed] [Google Scholar]
- Laemmli, U.K., S.M. Cheng, K.W. Adolph, J.R. Paulson, J.A. Brown, and W.R. Baumbach. 1978. Metaphase chromosome structure: the role of nonhistone proteins. Cold Spring Harb. Symp. Quant. Biol. 42:351–360. [DOI] [PubMed] [Google Scholar]
- Li, G., G. Sudlow, and A.S. Belmont. 1998. Interphase cell cycle dynamics of a late-replicating, heterochromatic homogeneously staining region: precise choreography of condensation/decondensation and nuclear positioning. J. Cell Biol. 140:975–989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maeshima, K., and U.K. Laemmli. 2003. A two-step scaffolding model for mitotic chromosome assembly. Dev. Cell. 4:467–480. [DOI] [PubMed] [Google Scholar]
- Marsden, M.P.F., and U.K. Laemmli. 1979. Metaphase chromosome structure: evidence for a radial loop model. Cell. 17:849–858. [DOI] [PubMed] [Google Scholar]
- Ono, T., A. Losada, M. Hirano, M.P. Myers, A.F. Neuwald, and T. Hirano. 2003. Differential contributions of condensin I and condensin II to mitotic chromosome architecture in vertebrate cells. Cell. 115:109–121. [DOI] [PubMed] [Google Scholar]
- Paulson, J.R., and U.K. Laemmli. 1977. The structure of histone depleted chromosomes. Cell. 12:817–828. [DOI] [PubMed] [Google Scholar]
- Poirier, M.G., and J.F. Marko. 2002. Mitotic chromosomes are chromatin networks without a mechanically contiguous protein scaffold. Proc. Natl. Acad. Sci. USA. 99:15393–15397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poirier, M., S. Eroglu, D. Chatenay, and J.F. Marko. 2000. Reversible and irreversible unfolding of mitotic newt chromosomes by applied force. Mol. Biol. Cell. 11:269–276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rattner, J.B., and C.C. Lin. 1985. Radial loops and helical coils coexist in metaphase chromosomes. Cell. 42:291–296. [DOI] [PubMed] [Google Scholar]
- Rattner, J.B., M.J. Hendzel, C.S. Furbee, M.T. Muller, and D.P. Bazett-Jones. 1996. Topoisomerase II α is associated with the mammalian centromere in a cell cycle– and species-specific manner and is required for proper centromere/kinetochore structure. J. Cell Biol. 134:1097–1107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Razin, S.V. 1996. Functional architecture of chromosomal DNA domains. Crit. Rev. Eukaryot. Gene Expr. 6:247–269. [DOI] [PubMed] [Google Scholar]
- Robinett, C.C., A. Straight, G. Li, C. Willhelm, G. Sudlow, A. Murray, and A.S. Belmont. 1996. In vivo localization of DNA sequences and visualization of large-scale chromatin organization using lac operator/repressor recognition. J. Cell Biol. 135:1685–1700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saitoh, N., I.G. Goldberg, E.R. Wood, and W.C. Earnshaw. 1994. ScII: an abundant chromosome scaffold protein is a member of a family of putative ATPases with an unusual predicted tertiary structure. J. Cell Biol. 127:303–318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sedat, J., and L. Manuelidis. 1978. A direct approach to the structure of eukaryotic chromosomes. Cold Spring Harb. Symp. Quant. Biol. 42:331–350. [DOI] [PubMed] [Google Scholar]
- Strukov, Y.G., Y. Wang, and A.S. Belmont. 2003. Engineered chromosome regions with altered sequence composition demonstrate hierarchical large-scale folding within metaphase chromosomes. J. Cell Biol. 162:23–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swedlow, J.R., and T. Hirano. 2003. The making of the mitotic chromosome: modern insights into classical questions. Mol. Cell. 11:557–569. [DOI] [PubMed] [Google Scholar]
- Tavormina, P.A., M.G. Come, J.R. Hudson, Y.Y. Mo, W.T. Beck, and G.J. Gorbsky. 2002. Rapid exchange of mammalian topoisomerase IIα at kinetochores and chromosome arms in mitosis. J. Cell Biol. 158:23–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tobey, R.A., and H.A. Crissman. 1972. Preparation of large quantities of synchronized mammalian cells in late G1 in the pre-DNA replicative phase of the cell cycle. Exp. Cell Res. 75:460–464. [DOI] [PubMed] [Google Scholar]
- Tobey, R.A., N. Oishi, and H.A. Crissman. 1990. Cell cycle synchronization: reversible induction of G2 synchrony in cultured rodent and human diploid fibroblasts. Proc. Natl. Acad. Sci. USA. 87:5104–5108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zatsepina, O.V., V.Y. Polyakov, and Y.S. Chentsov. 1983. Chromonema and chromomere: structural units of mitotic and interphase chromosomes. Chromosoma. 88:91–97. [Google Scholar]