

Abstract
Contractile myocytes provide a test of the hypothesis that cells sense their mechanical as well as molecular microenvironment, altering expression, organization, and/or morphology accordingly. Here, myoblasts were cultured on collagen strips attached to glass or polymer gels of varied elasticity. Subsequent fusion into myotubes occurs independent of substrate flexibility. However, myosin/actin striations emerge later only on gels with stiffness typical of normal muscle (passive Young's modulus, E ∼12 kPa). On glass and much softer or stiffer gels, including gels emulating stiff dystrophic muscle, cells do not striate. In addition, myotubes grown on top of a compliant bottom layer of glass-attached myotubes (but not softer fibroblasts) will striate, whereas the bottom cells will only assemble stress fibers and vinculin-rich adhesions. Unlike sarcomere formation, adhesion strength increases monotonically versus substrate stiffness with strongest adhesion on glass. These findings have major implications for in vivo introduction of stem cells into diseased or damaged striated muscle of altered mechanical composition.
Keywords: myofibrillogenesis; patterning; adhesion; differentiation; muscular dystrophy
Introduction
Injection of mesenchymal stem cells into an ever-stiffening fibrotic infarct zone in the heart leads to cellular expression of myocyte markers, but differentiation clearly stops short of myofibril assembly (Shake et al., 2002). The reason is unclear, but physical aspects of the microenvironment, especially mechanical compliance of matrix (Pelham and Wang, 1997) and perhaps adjacent cells, are becoming increasingly linked to gene expression and protein organization. Sufficient substrate stiffness appears particularly important to anchorage-dependent cells, which often rely on a finite resistance to cell-generated forces in order to induce outside-in mechanical signals. Such signals feed back into cell tension (Wang et al., 2002), cell adhesion (Choquest et al., 1997; Beningo et al., 2001), posttranslational modification (Pelham and Wang, 1997; Beningo et al., 2001), protein expression (Cukierman et al., 2001), cytoskeletal organization (Cukierman et al., 2001; Engler et al., 2004a), and even cell viability (Wang et al., 2000). It seems fair to hypothesize that substrate stiffness imparts new meaning to “anchorage dependence.”
With contractile cells such as muscle cells, one might hypothesize that coupling to a soft or stiff matrix is extremely important to cell function. Myotubes ultimately transmit actomyosin contractions through their attachments to matrix. If a matrix or substrate is fully rigid, as in conventional cultures on polystyrene or coverslips, contractile efforts will clearly be isometric. If a matrix is more compliant, such as with a collagen gel, myocytes will be able to contract, but in a manner that depends on the effective stiffness of the underlying substrate (Engler et al., 2004a). Analogous to the wrinkling of films by cells (Harris et al., 1980; Oliver et al., 1995), cells generate traction forces on collagen-coated polyacrylamide (PA) gel substrates. Cells also spread in area to a greater extent on stiff, yet compliant substrates compared with softer substrates (Lo et al., 2000; Engler et al., 2004a).
A number of muscle and related pathologies involve changes in matrix properties. Beyond myocardial infarctions mentioned above, atherosclerosis generally involves smooth muscle cells (SMCs) in a matrix that is certainly altered and perhaps softened (Campbell and Chamley-Campbell, 1981; Glukhova and Koteliansky, 1995; Stenmark and Mecham, 1997). Muscular dystrophies are also associated with changes in matrix: the diaphragm of the dystrophin-deficient mdx mouse is more fibrotic and thus much stiffer than a normal mouse diaphragm (Stedman et al., 1991). In the same studies, muscle precursor satellite cells were found to be as abundant in dystrophic tissue as in normal tissue, but mdx did not seem successful in regenerating differentiated muscle.
The work here attempts to elucidate a role for matrix or tissue stiffness in striated muscle differentiation. Although the conventional in vivo matrix for myotubes is an ∼1-μm-thick (or less) basement membrane of laminin and collagen, tissue compliance convolves compliance of this matrix with cellular compliance. Our ex vivo studies here of normal versus pathological tissue stiffness first set the substrate stiffness scale for subsequent in vitro studies. To controllably culture skeletal muscle on suitably compliant substrates, the strong tendency of fusing myocytes to branch and overlay each other must be limited. Micropatterned, compliant substrates solve this problem and allow for isolation of mechanisms underlying cell–matrix adhesion, extending to its role in differentiation. The process typically begins with myoblast anchorage to the substrate, cell spreading, withdrawal from cell cycle, and cell fusion into nascent myotubes. Increasing expression and assembly of cytoskeleton and adhesions ultimately concludes with characteristic striations along the contractile myotube. This last step proves to be especially sensitive to substrate compliance. The results thus reveal an optimal, tissue-like matrix that is stiff, yet compliant enough to balance cell adhesion, contractility, and ultimately differentiation. Micropatterned, compliant substrates solve this problem (Fig. 1) and allow for isolation of mechanisms underlying cell–matrix adhesion, extending to its role in differentiation.
Figure 1.

Micropatterned collagen on PA gels. (A) Cross-linked PA was first bound to an aminosilanized glass slide or coverslip. Sulfo-SANPAH (x), a heterobifunctional cross-linker, was then applied to the free surface and attached to the gel by exposure to 365 nm light. (B and B′) A micropatterned glass stamp with 20-μm-wide channels was dipped in collagen-I to fill its channels. The “loaded” microstamp was overlaid on the PA gel bearing exposed succinimidyl ester groups and the reaction was allowed to proceed for 12 h at 37°C. (C) This gave a striped pattern of cross-linked collagen as visualized by immunofluorescence (C′). Addition of C2C12 myoblasts showed strong alignment on the patterns after two d (D) on both soft (1 kPa) and stiff (8 kPa) PA gels. Bars, 100 μm.
Results
Skeletal muscle stiffness ex vivo: normal and dystophic
Skeletal muscle is obviously a “soft tissue” rather than hard as bone. To quantitate the transverse, passive elasticity of a representative muscle, extensor digitorum longus (EDL) muscles were excised from both normal (C57) mice and dystrophin-deficient (mdx) mice and probed locally along their lengths by atomic force microscopy (AFM; Fig. 2). The transverse direction is, of course, the direction relevant to lateral adhesion between cells and matrix. Hertz cone model fits of the indenting force yields a transverse Young's modulus, E (Domke and Radmacher, 1998; Rotsch et al., 1999). Data was averaged over multiple locations within each sample (n = 3) from normal mice of different ages, where the predominant modulus was E Muscle = 12 ± 4 kPa (Table I; 85% of data). The result is in agreement with the AFM-determined elasticity of C2C12 cells (up to 1 wk): E C2C12 = 12–15 kPa (Collinsworth et al., 2002).
Figure 2.

Passive stiffness of normal and dystrophic muscle tissue. EDL muscle was dissected from normal (C57) and dystrophic (mdx) mice and probed in buffer by AFM. The inset schematic depicts the AFM probe indenting a thick tissue section on a glass support. Indentations of 1–2 μm were randomly made on ∼100-μm-thick samples at 10 or more locations per left and right EDL samples (n = 387). Force versus indentation curves were fit with a Hertz cone model (Engler et al., 2004a) in order to determine the passive elasticity as a Young's modulus, E. Table I lists the predominant stiffness for both normal C57 muscle and mdx-derived muscle.
Table I.
Passive elasticity of EDL muscle (mean ± SD) dissected from normal (C57) and dystrophic (mdx) mice, and probed by AFM indentation (Fig. 2)
| Mouse | Mean muscle stiffness, E (kPa) |
|---|---|
| C57 | 12 ± 4 |
| mdx | 18 ± 6 |
From mdx mice, EDL appeared significantly stiffer with a predominant modulus E mdx = 18 ± 6 kPa (55% of data). As with the C57 control mice, the average muscle elasticity was statistically similar for all mdx mice regardless of age (P = 0.42). However, the micro-scale measurements of transverse elasticity for mdx muscle also proved more broadly distributed than in normal muscle: mdx probing gave a significant number of indentations with E mdx > 35 kPa (∼40%) and also a few with E mdx < 5 kPa (∼5%).
Myoblasts in vitro sense substrate stiffness
Spreading, elongation, and cooperative alignment of myoblasts depend on substrate stiffness. C2C12 skeletal myoblasts were plated at low densities on homogeneous collagen I–coated glass as well as PA gels of varied elastic modulus E. Fig. 3 (A–C) show that myoblasts spread and elongate more on increasingly stiff substrates after just 4 h. By 24 h, cells are spindle-shaped regardless of elastic modulus (Fig. 3 C), although mean cell area still tends to increase with substrate stiffness E (Fig. 3 B). The stiffest gels and glass give statistically similar results for cell area (P = 0.4). Interestingly, this asymptotic behavior occurs at an elastic modulus below E Muscle.
Figure 3.

Initial myoblast responses to collagen-coated PA gels. Myoblasts were plated (5 × 103 cells/cm2) on unpatterned gels to observe initial responses to elasticity. (A) Images of initial cell spreading and elongation on gels. Bars, 20 μm. (B) Spread cell area increases as a function of the substrate elastic modulus as well as time. A hyperbolic fit (Eq. 1) captures the trends at each time point. (C) Cells at 4 h spontaneously elongate (minor axis < major axis) only on stiff gels and glass, but by 24 h the elongated morphology predominates on all substrates. (D) An orientational correlation function (OCF, Eq. 2) evaluated between any two cells on unpatterned PA gels decays exponentially over ∼5–10 cell widths after 24 h For a fully aligned culture, OCF = 1; for a random orientation OCF = 0.5 (indicated by a dashed line); and for anticorrelated interactions OCF = 0. The fit, OCF = B e− Ax + (1 − B), gives a decay constant A and the long range orientation B (B = 0.5 is random). Cells in close proximity thus prefer to align with their nearest neighbors, facilitating formation of myotubes. Error bars are SEM.
The strong dependence of cell spreading at low elastic modulus, E < E Muscle, and the asymptotic behavior of cells spreading on stiff gels and glass substrates fits a simple hyperbola (Engler et al., 2004a) that suggests a specific and saturable mechanosensitive response:
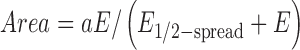 |
(1) |
where E 1/2-spread is a half-max modulus for ECM analogous to dissociation constants for biochemical interactions. Best-fit parameters of E 1/2-spread ∼2.5–5 kPa are listed in Table II. Stiff or rigid substrates, defined by E > E 1/2-spread (e.g., glass), foster cell spreading and include the tissue-like stiffness, E Muscle. Soft substrates with E < E 1/2-spread (e.g., 1 kPa gel) elicit very little cellular response. A decrease in E 1/2-spread with time indicates that spreading (Fig. 3 B) becomes less dependent on substrate elasticity, which is consistent with the cell elongation results (Fig. 3 C). As shown below, subsequent fusion of spindle-shaped myoblasts into nascent myotubes occurs on all substrates regardless of elastic modulus.
Table II.
Hyperbolic fit of myoblast area versus substrate elasticity ( E in Eq. 1)
| Hyperbolic fit | 4 h | 24 h |
|---|---|---|
| a (μm2) | 2,000 | 2,100 |
| E 1/2-spread (kPa) | 5.1 | 2.4 |
For the spindle-shaped myoblasts, Fig. 3 D demonstrates “mechanosensitive” coalignment of adjacent cells. Effects of cell tractions transmitted through gels to neighboring cells were quantified here by an orientational correlation function (OCF; Eq. 2). Over distances of 5–10 cell widths, cells prefer to align (OCF ∼1). Beyond such distances, the correlation of the cell–cell angle decreases toward a random orientation (OCF ∼0.5) with no significant evidence of anticorrelated interactions (OCF ∼0). Both soft (1 kPa) and stiff (8 kPa) gels show statistically overlapping trends, with soft gels suggesting slightly longer-range order. The mechanism of coalignment is understood, from theory (Bischofs and Schwartz, 2003), to involve a propagation of cell-induced substrate contractions (Pelham and Wang, 1997; Wang et al., 2001; Tan et al., 2003). Coalignment thus occurs here to a varying degree on all substrates and appears to be a first step toward fusion.
Nascent myotubes on any substrate
Variability in cell–cell orientation necessitates micropatterned substrates that foster linear fusion and, for later studies of adhesion, inhibit branching. This is achieved by adhesively confining cells. After 2 d on collagen-micropatterned PA gels, C2C12 skeletal myoblasts recognize and align on the collagen pattern, and begin to fuse into myotubes. This occurs regardless of elastic modulus (Fig. 1 D; Fig. 4 A, top). Primary mouse myoblasts (C57 strain) exhibit similar pattern recognition and also clearly show membrane fusion. Up to 2–4 wk in culture (Fig. 4 A, bottom), fully fused C2C12 or C57 myotubes remain nonmotile and nonproliferative, tending to maintain the original patterns. Adhesion proves adequate because, over 4 wk of the culture, overall patterning density calculated as myotube length divided by the total pattern size always remained high (>70%) and decreased little (<10–15%) even with perturbations during needed media changes.
Figure 4.

Myotube striation is substrate stiffness dependent, whereas myocyte patterning is not. (A) By 2 d in culture, myoblasts align on the collagen patterns and begin to fuse into nascent myotubes. This occurs regardless of substrate elastic modulus and is sustainable in culture for weeks. Unbranched, isolated myotubes are consistently >90% of cells throughout the lifetime of all micropatterned cultures. Mouse primary cells (C57 derived) exhibit a similar behavior. (B and C) Several weeks after plating myoblasts on collagen-coated PA gels of varied stiffness or rigid glass, cells were stained for myosin (green), actin (not depicted), and the nucleus (blue) in order to assess cytoskeletal expression and organization. All substrates supported fusion into multi-nucleated myotubes, but after 2 or 4 wk only myotubes on gels of intermediate stiffness showed significant myosin striation. (D) On rigid glass, nascent myotubes showed robust arrays of stress fibers (F-actin is red) and tight substrate adhesion from vinculin by 1 wk and, as extrapolated from the PA gel results, myotubes showed no tendency to striate, even by 4 wk. Note the precision of the end-patterning on glass (see Materials and methods). Bars: (A) 100 μm; (B–D) 20 μm.
Striated myotubes on tissue-like stiffness
Fusion into nascent myotubes is a first, common path in differentiation of skeletal muscle, and to assess the progression of myotube differentiation, C2C12 cells grown for 2 and 4 wk on substrates of varying stiffness were stained for myosin (Fig. 4, B and C) as well as F-actin (not depicted). The multi-nucleated myotubes on both very soft gels (<5 kPa) and very stiff gels or glass show only diffuse actin and myosin staining after 2 wk in culture (n = 35 cells imaged). However, cells on intermediate stiffness (8 kPa and 11 kPa ∼E Muscle) show actin and myosin colocalized as striations in up to 25% of the cell populations at 2 wk (Fig. 5; n = 27). After 4 wk in culture, striated myotubes double in percentage on intermediate stiffness gels. On gels with elastic moduli of 6.5 and 17 kPa, myotubes consistently show a significant but lower percentage of striated cells. In contrast, very soft or very stiff gels (i.e., 1 and 23 kPa) show no more than a small subpopulation of striated myotubes (∼5%) with poor striations and predominantly diffuse premyofibril myosin. Gaussian fits of the differentiation results clearly identify an optimal modulus of E* ∼12 kPa that maximizes myosin striations in muscle.
Figure 5.

Optimum substrate modulus for myotube differentiation. Myoblasts that have fused to myotubes on collagen-coated PA substrates exhibit a striation tendency that is strongly dependent on the substrate's elastic modulus. The percentage of total cells (n > 15 for each sample) exhibiting actin-myosin striation on the collagen-coated PA gels showed an optimum gel modulus of E* = 12 kPa based on simple Gaussian fits with common means and SDs (∼2.5–3 kPa) for 2 and 4 wk. Circles represent differentiation of cells grown on IPN-patterned glass. Error bars are ± SEM.
E* is both well-defined and nearly constant from 2 to 4 wk. The optimum curves are also relatively narrow with a SD of 2–3 kPa. This implies of course that a majority of striated cells (63% for the Gaussian fits) are found on substrates with stiffness within just 25% of E*. The fact that E* = E Muscle is discussed below.
Cells grown on patterned glass substrates were stained after 1 and 4 wk for myosin and F-actin (Fig. 4 D), showing little to no striation (Fig. 5, circles). Stress fibers and vinculin-enriched focal adhesions are instead very prominent in these cells by 1 wk on glass. This is consistent with stress fiber assembly in cells on very stiff gels and in cell types ranging from fibroblasts (Beningo et al., 2001) to SMCs (Gaudet et al., 2003; Engler et al., 2004a). Stress fiber disassembly was seen later, but even by 4 wk on glass striations still did not appear.
Cell-on-cell differentiation
The initial apparent elasticity of C2C12 myotubes has been reported to be E C2C12 = 12–15 kPa (Collinsworth et al., 2002), which closely corresponds to the optimal gel stiffness E* for striation. It also closely corresponds to normal skeletal muscle tissue stiffness, E Muscle. An initial layer of myoblasts was therefore plated on patterned glass surfaces to mimic E Muscle with an underlayer of C2C12 cells. After 1–2 d, a second plating of myoblasts was made in order to generate a cell-on-cell arrangement of myotubes (Fig. 6 A). The initial, bottom cell (in red) has well-defined ends and appears locked by stress fibers and vinculin-enriched focal adhesions onto the interpenetrating polymer network (IPN) pattern (per Fig. 4 D). The second, top cell layer (with green vertical stripes) spreads over the exposed apical surface of the bottom, patterned cells. With time the top myotube striates, whereas the bottom myotube does not (Fig. 6, B and C).
Figure 6.

Myotube underlayers provide optimal stiffness for myotube striation. Myoblasts were plated in two sequential layers on patterned glass. (A) An initial, bottom layer of cells was added and is schematically represented as nascent myotubes with square ends “pinned” by stress fibers. Subsequent addition of cells generates an top myotube that progressively differentiates. Within 1 wk after plating (B) and even more prominent at 4 wk (C), the top layer of cells striate both F-actin and myosin although clear striation of F-actin seems slower. Merged images of the top myotube at 4 wk demonstrate actin-myosin colocalization. Bars, 10 μm. (D) Striated myotubes in the top layer, after 1 and 4 wk, respectively constituted 68% (n = 34) and 85% of the total myotube population (repeated in duplicate). Immunostaining of vinculin in cells on top of cells shows diffuse labeling and no profound focal adhesions (not depicted).
Compared with cells of comparable age growing on gels of similar stiffness to that of the bottom layer of undifferentiated cells (i.e., gels ∼E Muscle), the top cells in these patterned cell-on-cell arrangements exhibited an approximate twofold higher percentage of myosin striation. After 4 wk, the percentage of myosin-striated myotubes in the top layer approached 100% (Fig. 6 D). In contrast, cells in the bottom layer showed little indication of striation, despite their intimate contact with a soft and stably differentiated cell above.
Fibroblasts (FC7) are much softer than C2C12 cells with an AFM-determined elasticity of E Fibroblast = 2–5 kPa (Wu et al., 1998; Bushell et al., 1999; Rotsch and Radmacher, 2000; Mahaffy et al., 2004). FC7's were also used to create a first, patterned underlayer of cells (Fig. 7 A). Consistent with the softness of fibroblasts, myotubes grown on FC7's appear unable to striate myosin (Fig. 7 B).
Figure 7.
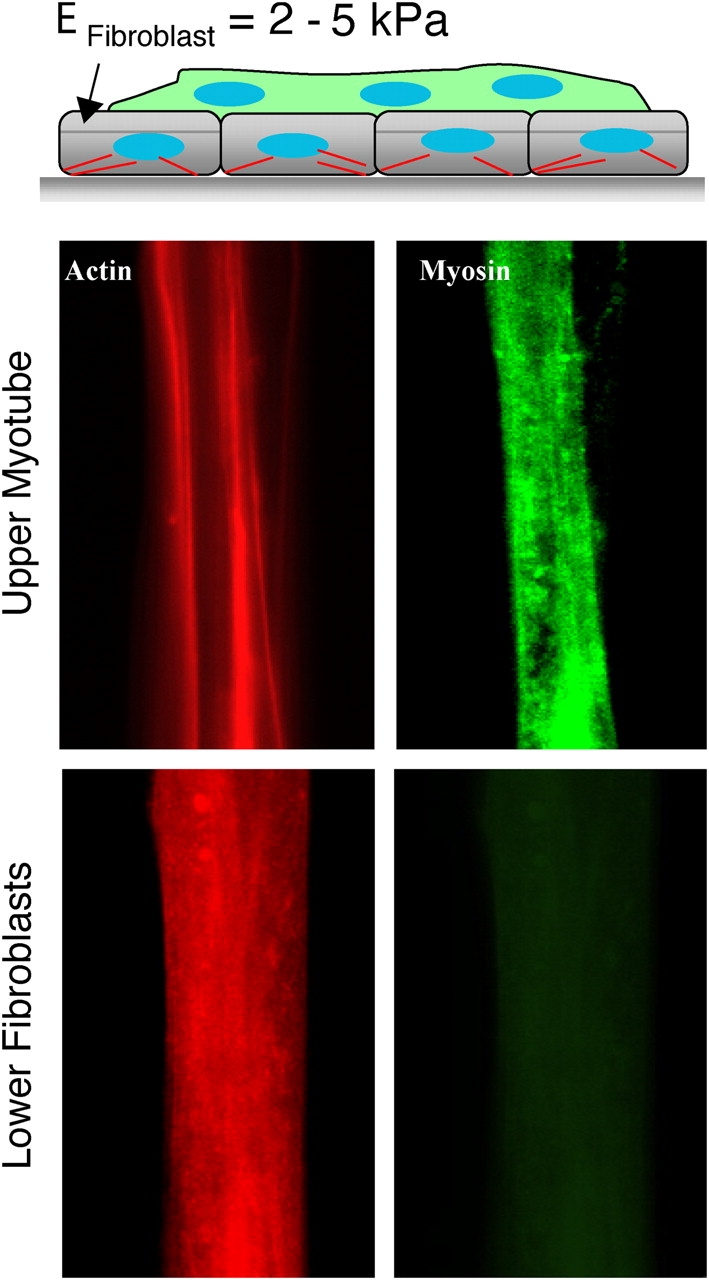
Fibroblast underlayers are too soft for myotube striation. (A) Fibroblasts were plated first on micropatterned glass with subsequent addition of myoblasts. The latter generates an top myotube (B), where differentiation is limited to fusion after 1–2 wk as there is no indication of striation. Myosin expression is nonetheless more prominent in the nascent myotubes than in the bottom fibroblasts that appear to contain similar amounts of F-actin.
Adhesion increases with substrate stiffness
Cell adhesion after 4 wk in culture was studied on various substrates. Isolated myotubes were controllably peeled by a method of micropipette aspiration (Fig. 8, A and B) that is well-suited to elucidating adhesion along the length of single cells (Griffin et al., 2004). As individual myotubes are peeled, molecular adhesions break and limit the rate of peeling along the length of the myotubes (Gallant et al., 2002; Griffin et al., 2004).
Figure 8.
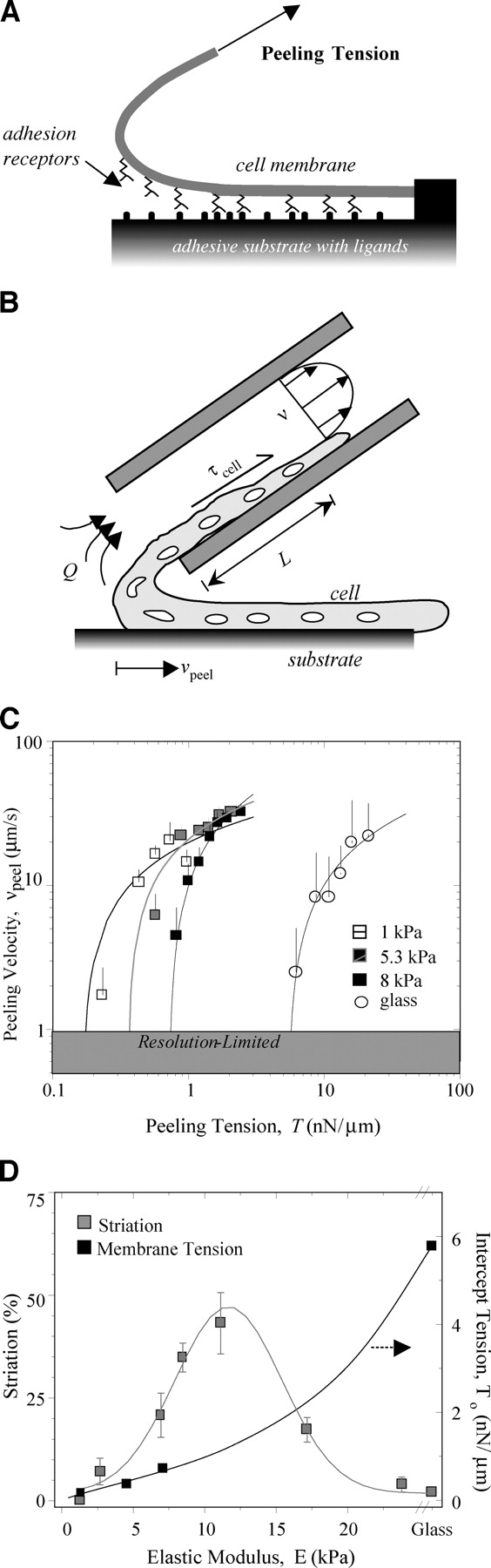
Substrate compliance influences myotube adhesion. Myotubes were mechanically peeled from their matrix after 4 wk in culture. (A) By applying a tension to a single myotube, the receptor-ligand bonds mediating adhesion were forcibly disrupted, and the myotube was peeled from the surface. (B) Cell peeling was accomplished by aspirating myotubes into micropipettes. The wall shear stress imposed by the flowing fluid along the length of the myotube pulled the cell into the micropipette. (C) Mean peeling velocity is plotted as a function of the imposed tension, as calculated across the width of a cell and for aspiration of at least three cells at each substrate modulus (Griffin et al., 2004). Although each myotube peels at a different rate due to adhesive differences, a common resting tension could be extrapolated by simple log fits for results at each substrate stiffness. (D) Comparing the zero-velocity tension for myotubes cultured for 4 wk on various substrates to their state of differentiation, i.e., striation per Fig. 4, demonstrates a disconnect. Adhesive strength simply increases with substrate modulus, but striation is minimal on very stiff and rigid substrates, showing instead an optimum at intermediate cell-like compliances. All error bars are SEM.
Although cell peeling velocity, v peel, fluctuates with the imposed peeling tension, T, it is possible to separate out the effects of focal adhesion clusters for which v peel ∼0 (Griffin et al., 2004). Thresholding v peel measurements to include only those points where cells are most rapidly detaching from the surface gives relatively smooth peeling curves (Fig. 8 C). Clearly, sufficient tension on the cell, regardless of the substrate, will increase the disruption rate of receptor-matrix bonds. Fig. 8 C shows, nonetheless, that v peel versus T curves are increasingly shifted to the right as substrate stiffness increases.
Cell peeling curves from each stiffness are consistently of a mathematical form (Griffin et al., 2004) v peel = a log T + b. Evaluating each curve fit at v peel = 0 gives a zero-velocity tension T o = exp(-b/a) that is the minimum tension required to detach a cell from its substrate. T o also represents the maximum contractile prestress sustainable by the cells on a given substrate. Fig. 8 D shows that T o increases monotonically as a function of substrate E, mirroring the rightward shifts to tighter adhesion with increasing substrate stiffness.
Discussion
Muscle differentiation in cell culture has been described for many years. However, the results here may motivate a careful reexamination of what matrix or microenvironment compliance has been used in the past and/or whether differentiated myotubes were growing on top of less differentiated myotubes in a more three-dimensional arrangement (Figs. 6 and 7). Recent efforts with muscle tissue constructs for implantation probably fit the latter description (Borschel et al., 2004). Micropatterning used here keeps cells well separated and well defined, leading to this first set of observations of how cell differentiation, specifically myofibrillogenesis, proceeds only within a small range of microenvironmental stiffness (Fig. 5). The results also appear the first to show that membrane adhesion strength increases exponentially with substrate stiffness (Fig. 8 D).
Tissue, matrix, and cell stiffness
Muscle tissue is compliant as well as contractile, and many types of muscle passively resist extension with effective or average moduli of E ∼5–15 kPa at strains up to 10–30% (Fung, 1993). The transverse elasticity E Muscle = 12 kPa here of C57 EDL muscle from AFM indentation fits within the published range of similar measurements on cultured myotubes, E C2C12 (Collinsworth et al., 2002). Elson and coworkers (Pasternak et al., 1995) used a glass microprobe (R p = 1 μm) to “poke” myotubes grown from C57-derived satellite cells and reported an average indentation force f = 12.3 ± 0.04 nN per δ = 1 μm; assuming a Hertz indentation model for an incompressible solid, this early method of myotube poking yields E poke ∼f /(δ3/2 R p 1/2) = 12.3 kPa. Although this is exactly as reported here for E Muscle, the same method for partially differentiated (fused but not striated) mdx-isolated satellite cells gave E poke-mdx ∼3.4 kPa. This is significantly softer than the E mdx found here by AFM, and does not represent the large heterogeneity found in vivo. The difference may reflect compensatory mechanisms in vivo such as an ∼10-fold overexpression of filamin C at the sarcolemma in fully differentiated dystrophic muscle compared with normal sarcolemma (Thompson et al., 2000).
The passive elasticity of adherent muscle cells appears set by their contractility (Wang et al., 2002) because inhibition of myosin activity (with BDM) can soften myotubes up to 10-fold in 30 min (Collinsworth et al., 2002). Basal contractility differences might also explain the difference between mdx tissue and cell culture. Consistent with an important role for myosin, soft fibroblasts with E fibroblast < 5 kPa, show much less myosin compared with myotubes (Fig. 7 B). Isolated adherent myotubes grown on top of fibroblasts seem to sense this softness and thus lack striations.
The important finding that E Muscle, E C2C12, and E poke are all close to the optimum E* for normal myotube differentiation (i.e., myofibrillogenesis) on compliant substrates reinforces the conclusion that substrate stiffness is a common critical factor in differentiation to striated muscle. Moreover, our finding that E mdx from excised EDL muscle is significantly greater than E Muscle is consistent with past findings on diaphragm (Stedman et al., 1991) that suggested fibrotic stiffening mechanisms. An increased stiffness is further suggestive of a pathological impact of microenvironmental elasticity (be it a synthetic or natural ECM) on muscle differentiation.
Well before striation, even myoblast spreading is found to be responsive to substrate compliance, with a characteristic for greatest responsiveness of E 1/2-spread = 2–5 kPa. This is generally less than E Muscle and is suggestive of a genuine myoblast-myotube distinction in substrate sensitivity. Like myoblasts, SMCs have been found to exhibit similar stiffness-dependent spreading, E 1/2-spread = 8–10 kPa (Engler et al., 2004a), near the elasticity of the SMC-rich arterial media, E media = 5–8 kPa (Engler et al., 2004b). Stem cells exhibit similar sensitivity to substrate elasticity in spreading and morphology (Fig. S1, available at http://www.jcb.org/cgi/content/full/jcb.200405004/DC1). Paxillin phosphorylation, for example, occurs in fibroblasts plated on stiff substrates for which spreading has been shown to be myosin dependent (Pelham and Wang, 1997). Recent work with gadolinium, a calcium channel inhibitor, has suggested that calcium entry through the plasma membrane strongly influences cell tractions, making calcium a key second messenger in substrate sensing (Munevar et al., 2004). The results collectively suggest that, by whatever molecular mechanism, the more an attached cell pulls on its substrate without being able to deform the substrate, the more the cell and its molecules deform rather than the substrate.
Myofibrillogenesis, membrane adhesion, and mechanosensing
Myofibrillogenesis is a protracted process that begins at the membrane. In a nascent myotube, α-actinin rich “Z-bodies” assemble with F-actin and nonmuscle myosin II in cross-linked premyofibrils (McKeena et al., 1986; Sanger et al., 2002). Premyofibrils occur along the sides and at the ends of the myotube, spanning vinculin-rich adhesive contacts with the substrate (Terai et al., 1989). Recruitment of muscle myosin II filaments and titin converts the premyofibrils into “nascent” myofibrils that lose nonmuscle myosin II and generate contractile tension, prestress, or force, f. With sufficient tension, mature myofibrils assemble from nascent myofibrils as the small Z-bodies laterally fuse and align into Z-bands. Inhibitors of muscle myosin II, including BDM (and BTS, a skeletal muscle myosin inhibitor), clearly block late stage myofibril bundling and so f clearly stimulates myofibrillogenesis (Ramachandran et al., 2003). Although it is not difficult to understand that a nonlinear sensitivity of myofibrillogenesis to f implies a nonlinear sensitivity to substrate elasticity, what molecules or complexes are especially sensitive to force or substrate displacements are presently unclear.
One candidate mechanoreceptor is N-RAP, which appears at the appropriate place and time for myofibrillogenesis to be sensitive to substrate elasticity. N-RAP is a multidomain, nebulin-related, actin-binding protein with a LIM domain critical for myofibrillogenesis (Carroll et al., 2001). N-RAP also binds α-actinin and filamin (Lu et al., 2003) and is thought to form a membrane-bound scaffold for nucleating actin filament growth (Carroll et al., 2004). Due to N-RAP's large size (>100 kD), it is believed to orient parallel to the membrane in order to mediate force transmission (Mohiddin et al., 2003). Force, f, might drive important conformational changes as part of N-RAP's scaffolding function and cause the N-RAP LIM domain to act as a transcriptional regulator of cellular differentiation (Sadler et al., 1992). Another LIM domain protein, skeletal muscle LIM protein 1 (or SLIM1), is known to induce myoblast elongation by a α5β1 integrin-dependent mechanism (McGrath et al., 2003). The dependence shown here of myoblast elongation on substrate stiffness at initial times (Fig. 3) might thus reflect similar LIM domain-dependent mechanisms as the later striation in myotubes. N-RAP and LIM domain proteins are, however, only illustrative of the types of proteins or complexes that might facilitate myofibrillogenesis on only those matrices with a suitable tissue-like stiffness suited to fostering force generation (Fig. 9).
Figure 9.

Differentiation is controlled by substrate compliance and adhesive tension. Myofibrillogenesis begins with membrane-nucleated premyofibrils and generates mature myofibrils in a contractile force-dependent manner. On substrates of tissue-like stiffness, the adhesion is moderate (based on peeling tension), which allows premyofibrils to couple optimally to contractile forces, f*, fostering striation and myofibril maturation. On rigid or soft substrates, differentiation is delayed, if not stopped. Cells on rigid substrates (e.g., glass) produce large isometric forces from well-formed stress fibers and numerous vinculin-enriched focal adhesions, all of which limit actin reorganization into striations. Cell adhesion and contractile forces on soft substrates are extremely weak leading to poorly spread cells that lack striations or even stress fibers.
However, an important question still remains: what might antagonize myofibrillogenesis on rigid substrates (e.g., glass) and thus push the system beyond its optimum of Fig. 5 while, at the same time, not disrupt focal adhesions (Fig. 4 D)? Multiple mechanisms and/or factors probably contribute to this phenomenon. N-RAP might become phosphorylated, dissociating it from actin (Chew et al., 2002) and thus limiting premyofibril nucleation. High stresses generated on the stiffest substrates by myosin II would also tend to forcibly dissociate key interactions such as those between N-RAP and its binding partners; these interactions would otherwise be critical to extending N-RAP as a conformationally active mechanoreceptor. Such a pathway might be stimulated by phorbol esters, which have long been known to cause sarcomere disassembly without loss of vinculin- and talin-rich focal adhesions (Lin et al., 1989). Integrin ligation activates PKC in muscle cells as do phorbol esters, with FAK activation downstream of these contributing to cell spreading and survival (Disatnik and Rando, 1999). FAK regulation could also occur by calcium-dependent mechanisms (Lo et al., 2000) or stretch-activation (Nakamura et al., 2001; Munevar et al., 2004) that should also be sensitive to substrate stiffness (Wang et al., 2002). Strong and stable focal adhesions that are found for many cell types only on the most rigid matrices (Pelham and Wang, 1997; Engler et al., 2004a) and also found here (Fig. 4 D) might activate the PKC pathway(s) far more strongly than is conducive for myofibrillogenesis (Fig. 9, rigid matrix).
In the absence of stiff ECM resistance, however, assembly and phosphorylation of adhesion proteins are inhibited (Pelham and Wang, 1997), and thus F-actin assembly is reduced (Engler et al., 2004a). Thus, on extremely soft substrates (1–5 kPa), it can only be observed from the studies here that cell attachment is extremely weak (Fig. 8) and, of course, striation is absent. Poor adhesion, generating only a small adhesion force, is consistent with reports of reduced mechanical loading inducing decreased myogenic expression (Torgan et al., 2000) and with other cell types grown on soft gels (Pelham and Wang, 1997; Beningo et al., 2001; Engler et al., 2004a) that prefer moderately stiff mechanical environments in vivo. Given the mechanosensitive down-regulation of focal and cytoskeletal proteins observed on soft gels, we speculate that nucleation of premyobrils requires more mature and suitably stable adhesion structures and cytoskeletal filaments that are signaled by normal muscle stiffness (Fig. 9, soft matrix).
A first illustrative estimate of the force needed for myofibrillogenesis might be made from the minimum tension, T o, needed to peel myotubes off of the gels of tissue-like stiffness (Fig. 8). The maturation force per premyofibril, f*, when suitably calculated across the cell width w should not exceed T o otherwise the cell will self-peel. If N is the average number of premyofibrils across w, then f* < T o × w/N. Using the focal adhesion staining in Fig. 4 D to estimate N = 10, w = 20 μm, and from Fig. 8 D T o = 1–2 nN/μm, the optimal force for differentiation on an E* substrate is f* ≤ 1 nN. This is much less than past estimates of cell forces on glass (∼5–10 nN), and suggests a need for lability in premyobril formation and myogenic progression (Fig. 9).
Conclusion
Muscle has well-recognized active and passive components, and so it is not surprising that striation requires more than cell anchorage. The studies here show that differentiation in culture depends intimately on optimal outside-in signaling of matrix elasticity. Most surprising, however, is the narrowness of the optimum substrate compliance, which has important implications for muscle disease and stiffened as well as softened tissue. This is particularly relevant to attempts to introduce stem cells into dystrophic skeletal muscle or infarcted regions of myocardium (Mueller et al., 2002; De Bari et al., 2003; Kobinger et al., 2003). Our results suggest that as both dystrophic and infarct processes induce tissue stiffening, implanted stem cells are unlikely to fully differentiate into contractile muscle. Indeed, stem cells injected into fibrotic, dystrophic skeletal muscle have been shown to differentiate into connective tissue rather than muscle (Huard et al., 2003), which could be a function of mechanical signaling as well as well-accepted cytokine signaling to the cells. On the other hand, injection of stem cells into an ever-stiffening infarct zone of the heart resulted in the cells beginning to express myocyte markers, but stopping differentiation short of myofibrillogenesis, highly analogous to our cells on a stiff matrix (Shake et al., 2002). Instruction of stem cells for tissue repair will undoubtedly require an understanding of the signaling pathways involved. Our results suggest that it may also require modification of tissue compliance if tissue repair is ultimately going to be successful.
Materials and methods
AFM on skeletal muscle tissue samples
Left and right whole EDL muscles were excised from C57 normal (3, 13, and 21 mo old) and mdx dystrophic mice (3, 9, and 15 mo old). Muscle was carefully divided into thinner bundles with a razor and wet mounted onto coverglass. Tissue section thickness was confirmed by light microscopy to be ∼100 μm, and samples were kept in DME plus antibiotics. Samples were probed within 8 h of dissection to minimize rigor. Samples were placed on an Asylum 1-D AFM (Asylum Research) and indented perpendicular to the myotube axis by a pyramid-tipped probe (Veeco) with a thermally determined spring constant, k sp ∼60 pN/nm. Force-indentation profiles were obtained at >10 locations of each muscle section, and each profile was fit up to a 2-μm tip deflection with a Hertz cone model (Domke and Radmacher, 1998; Rotsch et al., 1999) to determine the elastic modulus. Histograms of all elastic moduli were plotted, and peak moduli were reported.
Cell culture
C2C12 cells (<10 passage; American Type Culture Collection) were maintained in polystyrene flasks in a 37°C, 5% CO2 incubator, and cultured in growth media: DME, supplemented with 20% of FBS, 0.5% chicken embryo extract, plus antibiotics. 1–2 d after plating on various substrates detailed below, cell media was switched to differentiation media, containing DME with 10% horse serum, 1% rat brain extract (Portier et al., 1999), and antibiotics. FC7 primary human dermal fibroblasts were derived from a normal 21-yr-old female donor (anonymous) and used between passage numbers 6 and 10. FC7 human fibroblasts were cultured in identical media conditions to C2C12 cells. Mesenchymal stem cells used in the supplemental information section were obtained from Osiris Therapeutics and cultured in low glucose DME with 10% FBS.
Cell multilayers were created by plating an initial layer of cells on patterned glass substrates detailed below. A subsequent layer of C2C12 cells was plated on top of the initial layer 1–2 d after the initial plating and the media was switched to differentiation media 1–2 d later. The bottom cell layer consisted of either C2C12 cells or FC7 fibroblasts cultured in identical conditions to C2C12 cells.
Cell culture products were purchased from Invitrogen Life Technologies, whereas all other products were purchased from Sigma-Aldrich unless noted. Statistical image analyses were preformed with Scion Image and displayed as cell averages (±SD) with paired t tests to determine significance. Live cell experiments were preformed for no more than 1 h at RT, with frequent media renewal.
Gel substrates
Cells were plated on PA gel substrates of variable stiffness and observed for up to 4 wk. PA gels were prepared on glass slides as previously established by Pelham and Wang (1997) and illustrated in Fig. 1 A. In brief, N,N′ methylene-bis-acrylamide (ranging from 0.03% to 0.3%) was added to solutions of 5 or 10% acrylamide (C3H5NO) in distilled water and cross-linked by the addition of 10% ammonium persulfate (1/200 vol) and N,N,N′,N′-tetramethylethylenediamine (1/2,000 vol). 25 μl of the polymerizing solution was placed on an aminosilanized glass slide or coverslip, which covalently bound the gel samples to a rigid support; coverslips coated with dichlorodimethylsilane were placed on top to ensure a smooth gel surface and easy detachment for the final PA gel bound the aminosilinated coverslip. When polymerized, PA gels were ∼70 μm thick as confirmed by microscopy. Their elastic modulus was set by the acrylamide and bis-acrylamide concentrations (Engler et al., 2004a).
Due to the antiadhesive properties of PA gel, chemical cross-linking of rat-tail collagen I (BD Biosciences) to the gel surface was done with heterobifunctional sulfosuccinimidyl-6-(4′-azido-2′-nitrophenylamino) hexanoate (sulfo-SANPAH; Pierce Chemical Co.). The phenylazide group of sulfo-SANPAH, upon photoactivation at 365 nm, covalently attaches to the gel, leaving the succinimidyl ester to react with lysine ɛ-NH2 of collagen.
Cell patterning on gels
To create isolated myotube strips, strips of collagen on PA gels were made with a micropatterned glass stamp (Fig. 1 B). A mask with 100-μm-wide reflective lines spaced 20 μm apart was designed and applied to a photoresist-coated glass slide. The photoresist (Shipley) was excited by 365 nm light, binding it to the glass, and then baked to ensure attachment. When placed in an HF glass-etch for 10 min, glass in the 20-μm spaces without photoresist was removed, whereas photoresist-protected glass was not etched, thus giving 20-μm-wide grooves spaced 100 μm apart along the length of the glass (Fig. 1 B′). Residual photoresist was then removed by acetone. Collagen solution (1 mg/ml) was drawn into the grooves in this glass “stamp”, and the stamp was inverted onto the PA gel in the presence of the sulfo-SANPAH cross-linker. The sample was kept at pH 8.5 in a 37°C incubator overnight to allow the collagen to react with the gel. Due to the confinement of collagen, collagen bound solely to PA in the grooved areas of the stamp (Fig. 1, B, C, and C′), thus promoting cell attachment only within these 20-μm strips (Fig. 1, C′ and D).
Mechanics of gels
The elastic modulus of various gels was determined by AFM as well as macroscopic methods (Dimitriadis et al., 2002; Engler et al., 2004a). AFM measurements were averaged over several trials and multiple locations per gel; collagen attachment had no detectable effect.
Glass micropatterns
Cells were also grown on glass, patterned by an IPN to produce isolated myotubes on glass (Griffin et al., 2004). In brief, clean glass was patterned with photoresist and the exposed glass was bonded to allyltrichlorosilane. The photoresist was then stripped by acetone and the allyltrichlorosilane-covered glass was bound to the IPN. The remaining clean glass was bonded to 3-aminopropyltriethoxysilane and a layer of collagen. The final pattern was a series of adhesive collagen-adsorbed glass strips, 20-μm wide and 200–1,000-μm long, surrounded by the antiadhesive IPN.
Myoblast spreading and alignment
Myoblasts were first observed on unpatterned PA gel of various stiffnesses for initial spreading and alignment tendencies (as a ratio of the cell's major axis to its minor axis equals “elongation ratio”) after 4 and 24 h in culture. An orientational correlation function (OCF, Eq. 2) between the reference and incident cell (Bates and Frenkel, 2000), was used to assess the spontaneous alignment of unpatterned cells.
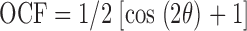 |
(2) |
Immunofluorescence
Patterned cells after 1, 2, and/or 4 wk were labeled with rhodamine-phalloidin, Hoechst 33342 nuclear stain (Molecular Probes), and immunofluorescent antibodies to myosin (Zymed Laboratories) or vinculin. Cells were fixed with formaldehyde and blocked in 5% BSA for 1 h at 37°C. Patterned cells were permeabilized with 0.5% Triton X-100 (Fisher Scientific) and incubated at 4°C for 8–10 h in antimyosin (Zymed Laboratories) or vinculin, diluted 1:100 in PBS. Cells were permeabilized again and incubated in 1:100 FITC-conjugated anti–mouse IgG, and 60 μg/ml TRITC-phalloidin at 37°C. Finally, cells were incubated in 1:100 Hoechst 33342 (Molecular Probes) at RT for 10 min. The samples were mounted onto coverslips with Gel/Mount (Biomedia) and viewed under epifluorescence at 60× (1.4 NA oil objective) or total internal reflectance fluorescence microscopy. Images were obtained using a liquid nitrogen cooled CCD camera (Photometrics).
Micropipette aspiration
Isolated myotubes were controllably peeled from gel and IPN-coated glass substrates to determine the dependence of minimal peeling tension on substrate stiffness. In brief, a fluid shear stress with Poiseuille pipe flow was created in a 75-μm aspirating pipette by a syringe pump with known flow rates (Griffin et al., 2004). When positioned over a detached cell end, the local shear force induced cell peeling at the leading edge of a patterned myotube, aspirating the cell into the pipette. Peeling tension was calculated from the shear stress integrated along the length of the cell in the pipette, given corrections for pipette entrance and gel displacement effects. The resultant peeling velocities from these experiments were very heterogeneous due to randomly distributed focal adhesion within numerous cells tested; thus velocity-tension plots were only made using binned averages of active peeling (and not focal adhesion), defined as a velocity at least 50% of the previous point.
Online supplemental material
Fig. S1 shows that mesenchymal stem cells exhibit myoblast morphology on muscle-like stiffness. Online supplemental material is available at http://www.jcb.org/cgi/content/full/jcb.200405004/DC1.
Acknowledgments
The authors would like to thank Professors Jean and Joe Sanger and Professor Yu Li Wang for very insightful discussions on premyobril formation, Dr. Huisheng Feng and Masataka Kawana for technical assistance and helpful discussions of methods, and the SEAS Microfabrication Center.
Support for this work was provided by grants from the National Institutes of Health (to H.L. Sweeney and D. Discher), NSF-PECASE (to D. Discher), and the MDA (to D. Discher).
Abbreviations used in this paper: AFM, atomic force microscopy; EDL, extensor digitorum longus; IPN, interpenetrating polymer network; PA, polyacrylamide; SMC, smooth muscle cell.
References
- Bates, M.A., and D. Frenkel. 2000. Phase behavior of two-dimensional hard rod fluids. J. Chem. Phys. 112:10034–10041. [Google Scholar]
- Beningo, K.A., M. Dembo, I. Kaverina, J.V. Small, and Y.L. Wang. 2001. Nascent focal adhesions are responsible for the generation of strong propulsive forces in migrating fibroblasts. J. Cell Biol. 153:881–888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bischofs, I.B., and U. Schwartz. 2003. Cell organization in soft media due to active mechanosensing. Proc. Natl. Acad. Sci. USA. 100:9274–9279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borschel, G.H., R.G. Dennis, and W.M. Kuzon Jr. 2004. Contractile skeletal muscle tissue-engineered on an acellular scaffold. Plast. Reconstr. Surg. 113:595–602, discussion 603-604. [DOI] [PubMed] [Google Scholar]
- Bushell, G.R., C. Cahill, F.M. Clarke, C.T. Gibson, S. Myhra, and G.S. Watson. 1999. Imaging and force-distance analysis of human fibroblasts in vitro by atomic force microscopy. Cytometry. 36:254–264. [DOI] [PubMed] [Google Scholar]
- Campbell, G.R., and J.H. Chamley-Campbell. 1981. Smooth muscle phenotypic modulation: role in atherogenesis. Med. Hypotheses. 7:729–735. [DOI] [PubMed] [Google Scholar]
- Carroll, S.L., A.H. Herrera, and R. Horowits. 2001. Targeting and functional role of N-RAP, a nebulin-related LIM protein, during myofibril assembly in cultured chick cardiomyocytes. J. Cell Sci. 114:4229–4238. [DOI] [PubMed] [Google Scholar]
- Carroll, S.L., S. Lu, A.H. Herrera, and R. Horowits. 2004. N-RAP scaffolds I-Z-I assembly during myofibrillogenesis in cultured chick cardiomyocytes. J. Cell Sci. 117:105–114. [DOI] [PubMed] [Google Scholar]
- Chew, C.S., X. Chen, J.A. Parente, S. Tarrer, C. Okamoto, and H.Y. Qin. 2002. Lasp-1 binds to non-muscle F-actin in vitro and is localized within multiple sites of dynamic actin assembly in vivo. J. Cell Sci. 115:4787–4799. [DOI] [PubMed] [Google Scholar]
- Choquest, D., D. Felsenfeld, and M. Sheetz. 1997. Extracellular matrix rigidity causes strengthening of integrin-cytoskeleton linkages. Cell. 88:39–48. [DOI] [PubMed] [Google Scholar]
- Collinsworth, A.M., S. Zhang, W.E. Kraus, and G.A. Truskey. 2002. Apparent elastic modulus and hysteresis of skeletal muscle cells throughout differentiation. Am. J. Physiol. Cell Physiol. 283:C1219–C1227. [DOI] [PubMed] [Google Scholar]
- Cukierman, E., R. Pankov, D.R. Stevens, and K.M. Yamada. 2001. Taking cell-matrix adhesions to the third dimension. Science. 294:1708–1712. [DOI] [PubMed] [Google Scholar]
- De Bari, C., F. Dell'Accio, F. Vandenabeele, J.R. Vermeesch, J.M. Raymackers, and F.P. Luyten. 2003. Skeletal muscle repair by adult human mesenchymal stem cells from synovial membrane. J. Cell Biol. 160:909–918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dimitriadis, E.K., F. Horkay, J. Maresca, B. Kachar, and R.S. Chadwick. 2002. Determination of elastic moduli of thin layers of soft material using the atomic force microscope. Biophys. J. 82:2798–2810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Disatnik, M.H., and T.A. Rando. 1999. Integrin-mediated muscle cell spreading. The role of protein kinase c in outside-in and inside-out signaling and evidence of integrin cross-talk. J. Biol. Chem. 274:32486–32492. [DOI] [PubMed] [Google Scholar]
- Domke, J., and M. Radmacher. 1998. Measuring the elastic properties of thin polymer films with the atomic force microscope. Langmuir. 14:3320–3325. [Google Scholar]
- Engler, A.J., L. Bacakova, C. Newman, A. Hategan, M. Griffin, and D. Discher. 2004. a. Substrate compliance versus ligand density in cell on gel responses. Biophys. J. 86:617–628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engler, A.J., L. Richert, J. Wong, C. Picart, and D. Discher. 2004. b. Surface probe measurements of the elasticity of sectioned tissue, thin gels and polyelectrolyte multilayer films: correlations between substrate stiffness and cell adhesion. Surf. Sci. In press. [Google Scholar]
- Fung, Y.-C. 1993. Biomechanics: mechanical properties of living tissues. Springer-Verlag New York Inc., New York. 568 pp.
- Gallant, N.D., J.R. Capadona, A.B. Frazier, D.M. Collard, and A.J. Garcia. 2002. Micropatterned surfaces to engineer focal adhesions for analysis of cell adhesion strengthening. Langmuir. 18:5579–5584. [Google Scholar]
- Gaudet, C., W.A. Marganski, S. Kim, C.T. Brown, V. Gunderia, M. Dembo, and J.Y. Wong. 2003. Influence of type I collagen surface density on fibroblast spreading, motility, and contractility. Biophys. J. 85:3329–3335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glukhova, M.A., and V.E. Koteliansky. 1995. Integrins, cytoskeletal and extracellular matrix proteins in developing smooth muscle cells of human aorta. In The Vascular Smooth Muscle Cell. Molecular and Biological Responses to the Extracellular Matrix. S.M. Schwartz and R.P. Mecham, editors. Academic Press, New York. 37–79.
- Griffin, M., A.J. Engler, T. Barber, K. Healy, H.L. Sweeney, and D. Discher. 2004. Patterning, prestress and peeling dynamics of myocytes. Biophys. J. 86:1209–1222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris, A.K., P. Wild, and D. Stopak. 1980. Silicone rubber substrata: a new wrinkle in the study of cell locomotion. Science. 208:177–179. [DOI] [PubMed] [Google Scholar]
- Huard, J., B. Cao, and Z. Qu-Petersen. 2003. Muscle-derived stem cells: potential for muscle regeneration. Birth Defects Res Part C Embryo Today. 69:230–237. [DOI] [PubMed] [Google Scholar]
- Kobinger, G.P., J.P. Louboutin, E.R. Barton, H.L. Sweeney, and J.M. Wilson. 2003. Correction of the dystrophic phenotype by in vivo targeting of muscle progenitor cells. Hum. Gene Ther. 14:1441–1449. [DOI] [PubMed] [Google Scholar]
- Lin, Z.X., J. Eshleman, C. Grund, D.A. Fischman, T. Masaki, W.W. Franke, and H. Holtzer. 1989. Differential response of myofibrillar and cytoskeletal proteins in cells treated with phorbol myristate acetate. J. Cell Biol. 108:1079–1091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lo, C.M., H.B. Wang, M. Dembo, and Y.L. Wang. 2000. Cell movement is guided by the rigidity of the substrate. Biophys. J. 79:144–152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu, S., S.L. Carroll, A.H. Herrera, B. Ozanne, and R. Horowits. 2003. New N-RAP-binding partners alpha-actinin, filamin and Krp1 detected by yeast two-hybrid screening: implications for myofibril assembly. J. Cell Sci. 116:2169–2178. [DOI] [PubMed] [Google Scholar]
- Mahaffy, R.E., S. Park, E. Gerde, J. Kas, and C.K. Shih. 2004. Quantitative analysis of the viscoelastic properties of thin regions of fibroblasts using atomic force microscopy. Biophys. J. 86:1777–1793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGrath, M.J., C.A. Mitchell, I.D. Coghill, P.A. Robinson, and S. Brown. 2003. Skeletal muscle LIM protein 1 (SLIM1/FHL1) induces alpha 5 beta 1-integrin-dependent myocyte elongation. Am. J. Physiol. Cell Physiol. 285:C1513–C1526. [DOI] [PubMed] [Google Scholar]
- McKeena, N.M., C.S. Johnson, and Y.-L. Wang. 1986. Formation and alignment of Z lines in living chick myotubes microinjected with rhodamine-labeled α-actinin. J. Cell Biol. 103:2163–2171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohiddin, S.A., S. Lu, J.P. Cardoso, S.L. Carroll, S. Jha, R. Horowits, and L. Fananapazir. 2003. Genomic organization, alternative splicing, and expression of human and mouse N-RAP, a nebulin-related LIM protein of striated muscle. Cell Motil. Cytoskeleton. 55:200–212. [DOI] [PubMed] [Google Scholar]
- Mueller, G.M., T. O'Day, J.F. Watchko, and M. Ontell. 2002. Effect of injecting primary myoblasts versus putative muscle-derived stem cells on mass and force generation in mdx mice. Hum. Gene Ther. 13:1081–1090. [DOI] [PubMed] [Google Scholar]
- Munevar, S., Y.L. Wang, and M. Dembo. 2004. Regulation of mechanical interactions between fibroblasts and the substratum by stretch-activated Ca2+ entry. J. Cell Sci. 117:85–92. [DOI] [PubMed] [Google Scholar]
- Nakamura, T.Y., Y. Iwata, M. Sampaolesi, H. Hanada, N. Saito, M. Artman, W.A. Coetzee, and M. Shigekawa. 2001. Stretch-activated cation channels in skeletal muscle myotubes from sarcoglycan-deficient hamsters. Am. J. Physiol. Cell Physiol. 281:C690–C699. [DOI] [PubMed] [Google Scholar]
- Oliver, T., M. Dembo, and K. Jacobson. 1995. Traction forces in locomoting cells. Cell Motil. Cytoskeleton. 31:225–240. [DOI] [PubMed] [Google Scholar]
- Pasternak, C., S. Wong, and E.L. Elson. 1995. Mechanical function of dystrophin in muscle cells. J. Cell Biol. 128:355–361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pelham, R.J., and Y.-L. Wang. 1997. Cell locomotion and focal adhesions are regulated by substrate flexibility. Proc. Natl. Acad. Sci. USA. 94:13661–13665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Portier, G., A. Benders, A. Oosterhof, J. Veerkamp, and T. Kuppevelt. 1999. Differentiation markers of mouse C2C12 and rat L6 myogenic cell lines and the effect of differentiation medium. In Vitro Cell. Dev. Biol. Anim. 35:219–227. [DOI] [PubMed] [Google Scholar]
- Ramachandran, I., M. Terry, and M.B. Ferrari. 2003. Skeletal muscle myosin cross-bridge cycling is necessary for myofibrillogenesis. Cell Motil. Cytoskeleton. 55:61–72. [DOI] [PubMed] [Google Scholar]
- Rotsch, C., K. Jacobson, and M. Radmacher. 1999. Dimensional and mechanical dynamics of active and stable edges in motile fibroblasts investigated by using atomic force microscopy. Proc. Natl. Acad. Sci. USA. 96:921–926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rotsch, C., and M. Radmacher. 2000. Drug-induced changes of cytoskeletal structure and mechanics in fibroblasts: an atomic force microscopy study. Biophys. J. 78:520–535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sadler, I., A.W. Crawford, J.W. Michelsen, and M.C. Beckerle. 1992. Zyxin and cCRP: two interactive LIM domain proteins associated with the cytoskeleton. J. Cell Biol. 119:1573–1587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanger, J.W., P. Chowrashi, N.C. Shaner, S. Spalthoff, J. Wang, N.L. Freeman, and J.M. Sanger. 2002. Myofibrillogenesis in skeletal muscle cells. Clin. Orthop. 403:S153–S162. [DOI] [PubMed] [Google Scholar]
- Shake, J.G., P.J. Gruber, W.A. Baumgartner, G. Senechal, J. Meyers, J.M. Redmond, M.F. Pittenger, and B.J. Martin. 2002. Mesenchymal stem cell implantation in a swine myocardial infarct model: engraftment and functional effects. Ann. Thorac. Surg. 73:1919–1925, discussion 1926. [DOI] [PubMed] [Google Scholar]
- Stedman, H.H., H.L. Sweeney, J.B. Shrager, H.C. Maguire, R.A. Panettieri, B. Petrof, M. Narusawa, J.M. Leferovich, J.T. Sladky, and A.M. Kelly. 1991. The mdx mouse diaphragm reproduces the degenerative changes of Duchenne muscular dystrophy. Nature. 352:536–539. [DOI] [PubMed] [Google Scholar]
- Stenmark, K.R., and R.P. Mecham. 1997. Cellular and molecular mechanisms of pulmonary vascular remodeling. Annu. Rev. Physiol. 59:89–144. [DOI] [PubMed] [Google Scholar]
- Tan, J.L., J. Tien, D.M. Pirone, D.S. Gray, K. Bhadriraju, and C.S. Chen. 2003. Cells lying on a bed of microneedles: an approach to isolate mechanical force. Proc. Natl. Acad. Sci. USA. 100:1484–1489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terai, M., M. Komiyama, and Y. Shimada. 1989. Myofibril assembly is linked with vinculin, alpha-actinin, and cell-substrate contacts in embryonic cardiac myocytes in vitro. Cell Motil. Cytoskeleton. 12:185–194. [DOI] [PubMed] [Google Scholar]
- Thompson, T.G., Y.M. Chan, A.A. Hack, M. Brosius, M. Rajala, H.G. Lidov, E.M. McNally, S. Watkins, and L.M. Kunkel. 2000. Filamin 2 (FLN2): A muscle-specific sarcoglycan interacting protein. J. Cell Biol. 148:115–126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torgan, C.E., S.S. Burge, A.M. Collinsworth, G.A. Truskey, and W.E. Kraus. 2000. Differentiation of mammalian skeletal muscle cells cultured on microcarrier beads in a rotating cell culture system. Med. Biol. Eng. Comput. 38:583–590. [DOI] [PubMed] [Google Scholar]
- Wang, H.B., M. Dembo, and Y.L. Wang. 2000. Substrate flexibility regulates growth and apoptosis of normal but not transformed cells. Am. J. Physiol. Cell Physiol. 279:C1345–C1350. [DOI] [PubMed] [Google Scholar]
- Wang, N., K. Naruse, D. Stamenovic, J.J. Fredberg, S.M. Mijailovich, I.M. Tolic-Norrelykke, T. Polte, R. Mannix, and D.E. Ingber. 2001. Mechanical behavior in living cells consistent with the tensegrity model. Proc. Natl. Acad. Sci. USA. 98:7765–7770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, N., I.M. Tolic-Norrelykke, J. Chen, S.M. Mijailovich, J.P. Butler, J.J. Fredberg, and D. Stamenovic. 2002. Cell prestress. I. Stiffness and prestress are closely associated in adherent contractile cells. Am. J. Physiol. Cell Physiol. 282:C606–C616. [DOI] [PubMed] [Google Scholar]
- Wu, H.W., T. Kuhn, and V.T. Moy. 1998. Mechanical properties of L929 cells measured by atomic force microscopy: effects of anticytoskeletal drugs and membrane crosslinking. Scanning. 20:389–397. [DOI] [PubMed] [Google Scholar]