

Abstract
Glutathione is the most abundant low molecular weight thiol in the eukaryotic cytosol. The compartment-specific ratio and absolute concentrations of reduced and oxidized glutathione (GSH and GSSG, respectively) are, however, not easily determined. Here, we present a glutathione-specific green fluorescent protein–based redox probe termed redox sensitive YFP (rxYFP). Using yeast with genetically manipulated GSSG levels, we find that rxYFP equilibrates with the cytosolic glutathione redox buffer. Furthermore, in vivo and in vitro data show the equilibration to be catalyzed by glutaredoxins and that conditions of high intracellular GSSG confer to these a new role as dithiol oxidases. For the first time a genetically encoded probe is used to determine the redox potential specifically of cytosolic glutathione. We find it to be −289 mV, indicating that the glutathione redox status is highly reducing and corresponds to a cytosolic GSSG level in the low micromolar range. Even under these conditions a significant fraction of rxYFP is oxidized.
Keywords: green fluorescent protein; glutathione; glutaredoxin; redox; oxidation
Introduction
The redox state of protein thiols is determined by a complex set of factors. Secretory proteins attain disulfide bonds through the action of an oxidative machinery, whereas cytosolic proteins are generally found in the reduced state unless part of an enzymatic or regulatory mechanism involving the reversible oxidation and reduction of vicinal thiols (for reviews see Linke and Jakob, 2003; Tu and Weissman, 2004). Cytosolic thiols are maintained in a reduced state by principally two enzymatic pathways and the abundant low molecular weight thiol glutathione. Both enzyme pathways derive their reducing equivalents from the oxidation of NADPH to NADP+ but are nevertheless fundamentally different in their biophysical properties. In the thioredoxin system, the reduction of thioredoxin is directly linked to NADPH oxidation through the action of the enzyme thioredoxin reductase. This introduces kinetic control on the system, as each step in the reduction chain is accompanied by a large drop in free energy. The glutaredoxin pathway, on the other hand, uses a redox buffer consisting of reduced and oxidized glutathione (GSH and GSSG, respectively). In Escherichia coli, glutaredoxins have been extensively studied as reductants of glutathione-protein mixed disulfides and disulfide bonds formed during the catalytic cycle of certain enzymes (Aslund et al., 1994; Shi et al., 1999; for review see Fernandes and Holmgren, 2004). Reducing equivalents for glutaredoxins are provided by GSH resulting in the formation of GSSG, which in turn is recycled to the reduced form by the enzyme glutathione reductase and NADPH (Holmgren, 1989). The relative rates of reduction of GSSG by glutathione reductase and its subsequent formation through glutaredoxins and other catalyzed and noncatalyzed oxidation reactions determine the steady-state ratio of GSH/GSSG. In whole-cell extracts, this ratio is heavily shifted toward the reduced state (Hwang et al., 1992; Muller, 1996). Such extracts have also provided a rough estimate of the intracellular concentration of GSH, which is typically in the low millimolar (0.5–10 mM) range (Kosower and Kosower, 1978). However, the redox state of glutathione as well as the absolute concentration is expected to vary from one compartment to another, and to our knowledge there is no reliable method that enables targeted measurement of both parameters within a single cellular compartment.
Glutathione homeostasis is maintained through a poorly understood balance between synthesis, uptake, usage, breakdown, and excretion. The biosynthetic pathway has been studied in some detail in the yeast Saccharomyces cerevisiae. Here, synthesis takes place in the cytosol, in two ATP-dependent condensation reactions catalyzed by γ-glutamylcysteine synthetase, encoded by the GSH1 gene, and glutathione synthetase (GSH2), respectively (Ohtake and Yabuuchi, 1991; Grant et al., 1997). Although deletion of the GSH1 gene confers an auxotrophic requirement for glutathione, GSH2 can be disrupted without fatal consequences, suggesting that γ-glutamylcysteine can functionally replace GSH (Grant et al., 1996, 1997; Spector et al., 2001). Uptake of GSH and GSSG from the culture medium is mediated by the high affinity glutathione transporter Hgt1p. As a gsh1 hgt1 double mutant is unable to grow on glutathione as the sole sulfur source, it is likely that Hgt1p is the only glutathione-transporting activity in the yeast plasma membrane (Bourbouloux et al., 2000; Hauser et al., 2000). Glutathione catabolism is thought to be initiated by the hydrolytic cleavage of the unusual γ-peptide bond between glutamate and cysteine, whereby cysteinylglycine and glutamate are released. This reaction is catalyzed by γ-glutamyl transpeptidase (for review see Taniguchi and Ikeda, 1998). A single γ-glutamyl transpeptidase homologue, which is the product of the ECM38 gene, has been identified in yeast (Jaspers and Penninckx, 1984). However, additional pathways must be capable of degrading GSH, as an ECM38 knockout mutant is able to survive on glutathione as the only source of sulfur (Kumar et al., 2003).
A major pool of glutathione appears to be sequestered in the yeast vacuole, where it is actively imported as glutathione and glutathione S-conjugates by the yeast cadmium factor protein (Ycf1p), and to a lesser extent by its paralogue Bpt1p (Li et al., 1996; Rebbeor et al., 1998; Klein et al., 2002). Whether GSH, and perhaps in particular GSSG, is also delivered to the vacuole by vesicular transport as a “spillover” from the secretory pathway is intriguing, but has not yet been explored.
In the present paper, we apply a previously designed GFP-based sensor for disulfide bond formation (Ostergaard et al., 2001) to study the cytosolic thiol-disulfide redox status. The fluorescence of the sensor is modulated by the reversible formation/reduction of a solvent-exposed disulfide bond. Using yeast as a model organism, we show that this sensor is uniquely sensitive to the glutathione redox pair and not targeted by the thioredoxin pathway. Moreover, it possesses a dynamic range that is perfectly suited for measuring the glutathione redox status under normal as well as redox-compromised conditions.
Results
Monitoring redox sensitive YFP (rxYFP) redox status in living yeast
The redox switch in rxYFP was previously constructed by engineering a solvent-exposed vicinal dithiol (Cys149-Cys202) between adjoining β-strands in the barrel structure of the yellow fluorescent variant of GFP. Reversible formation of a disulfide bond between the two redox-active cysteines resulted in an approximately twofold decrease in the intrinsic fluorescence, sufficient to enable in vivo real-time monitoring of redox changes within the protein (Ostergaard et al., 2001). Yeast was chosen as host for in vivo characterization of rxYFP due to the relative ease with which it can be genetically manipulated, while at the same time maintaining basic eukaryotic redox characteristics. Expression of rxYFP as a cytosolic protein was directed by the constitutive PGK1 promoter. To minimize fluctuations in the expression level between individual cells the expression construct was furthermore integrated into the yeast chromosome. As shown in Fig. 1 A, fluorescence microscopy of transformants revealed a homogenous distribution of the sensor throughout the cytosol and nucleus with little variation in the fluorescence intensity from one cell to another (Fig. 1 A). To determine the redox state of rxYFP from the measured fluorescence intensity, a method had previously been developed whereby the fluorescence of a culture was measured before and after successive addition of the thiol-oxidant 4,4′-dithiodipyridine (4-DPS) and the disulfide-reductant DTT. By relating the initial fluorescence to that of the fully oxidized and reduced reporter, the redox state of rxYFP could be estimated accurately (Ostergaard et al., 2001). However, when applying this methodology to yeast, a prepermeabilization step using 0.1% digitonin was required to get a fast and reproducible fluorescence change. Addition of digitonin accompanied by strong buffer to neutralize the pH of the growth medium resulted in complete permeabilization within minutes. The pH adjustment was required to prevent acid quenching of the fluorescence and to speed up the subsequent oxidation-reduction reactions. Due to the intrinsic pH sensitivity of rxYFP, equilibration of pH across the plasma membrane after digitonin treatment was seen as a transient drop in fluorescence, with subsequent return to a stable baseline (a typical progress curve is given in Fig. 1 B). Given the often rapid kinetics of thiol-disulfide exchange reactions, we were concerned that undesired oxidation/reduction of the sensor might take place during permeabilization. However, at least within the time frame of this step it did not appear to present a problem because essentially identical redox distributions were obtained by quantitative Western blotting on aliquots of the cultures taken immediately before addition of permeant (exemplified in Fig. 1 B, inset). As described previously, the oxidized and reduced state of rxYFP can be separated by nonreducing SDS-PAGE due to differences in their electrophoretic mobilities (Ostergaard et al., 2001). To prevent artificial thiol oxidation before and during electrophoresis, the cultures were first quenched by addition of trichloroacetic acid (TCA) and then treated with the thiol-specific alkylating reagent N-ethyl-malemide (NEM) to block free thiols. In further support of these results, we found the fluorometric redox titration method to give highly reproducible results despite slight variations in the duration of the permeabilization step, which would not be expected if undesired oxidation was taking place.
Figure 1.

Determining intracellular redox properties of cytosolic rxYFP. (A) Fluorescence distribution in wild-type yeast cells (M4745) expressing cytosolic rxYFP from the PGK1 promoter (fluorescence micrograph recorded using a Leitz DMIRBE microscope). (B) A representative fluorometric redox-determination of rxYFP expressed in a glutaredoxin reductase-deficient yeast mutant (M4860). The culture was pretreated with 0.1% digitonin in strong buffer to permeabilize the cells at neutral pH (see Materials and methods). From the fluorescence of the permeabilized cells and the fluorescence after sequential addition of 4-DPS and DTT to final concentrations of 0.3 mM and 93 mM, respectively, the proportion of oxidized rxYFP was calculated to 56%. Due to dilution of the sample during the procedure, the final fluorescence appears to be lower than would be expected from the reported fraction of oxidized reporter. (Inset) Western blot detection of oxidized and reduced rxYFP in the same culture. Cells were quenched by addition of 5% TCA. Subsequently, free thiols were blocked by treatment with 100 mM NEM, and the oxidized and reduced states of rxYFP were separated by nonreducing SDS-PAGE. Quantification of the immunostained bands yielded 53% oxidized rxYFP. (C) Overview of part of the sulfur metabolism in yeast relevant to the present work. The MET15 and GLR1 gene products catalyze the incorporation of sulfide into homocysteine and the NADPH-dependent reduction of GSSG into GSH, respectively. (D) Relationship between the GSSG/GSH ratios determined on whole-cell extracts and the measured proportion of oxidized rxYFP determined by fluorometric redox titration (values were taken from Table I).
rxYFP senses the cytosolic glutathione redox status
Disruption of the yeast GLR1 gene, encoding glutathione reductase, caused a significant increase in the fraction of oxidized reporter, from 10% in the wild type to 56% in the glr1 strain (Table I; Fig. 1 B). Measurement of the total intracellular pools of oxidized and GSH in whole-cell yeast extracts showed a corresponding change in GSSG/GSH ratio from 4% in the wild-type to 19% in the glr1 mutant (Table I). This initially suggested that rxYFP was sensitive to changes in the intracellular glutathione redox status. The existence of oxidized reporter in both strains, despite the presence of an intact thioredoxin pathway, suggested that the redox switch in rxYFP was not a substrate for this reductive system. This was confirmed in vitro using recombinantly expressed and purified His-tagged versions of the two cytosolic thioredoxins Trx1p and Trx2p (Gan, 1991). Although both were active in the insulin reduction assay (Holmgren, 1979), neither was capable of either directly reducing or oxidizing rxYFP (Fig. S1, available at http://www.jcb.org/cgi/content/full/jcb.200402120/DC1).
Table I. Glutathione content and redox state of cytosolic rxYFP in various yeast strains.
| Glutathione content (pmol/106 cells)b |
|||||||
|---|---|---|---|---|---|---|---|
| Strain | Relevant genotype | Suppl.a | GSH | GSSG | GSSG/GSH | Oxidized rxYFPc | E′cytosol d |
| % | % | ||||||
| M4745 | wild type | 206 ± 4 | 9.2 ± 0.4 | 4 | 10 ± 1 | −289 mV | |
| M4860 | glr1 | 172 ± 3 | 33 ± 3 | 19 | 56 ± 3 | −252 mV | |
| M4860 | glr1 | GSSG | 265 ± 9 | 302 ± 5 | 114 | 84 ± 1 | ND |
| M4859 | glr1 met15 | GSH | 267 ± 6 | 161 ± 3 | 60 | 75 ± 2 | ND |
| M4859 | glr1 met15 | GSSG | 186 ± 6 | 417 ± 9 | 225 | 92 ± 1 | ≥ −227 mV |
| M5059 | glrl grx1 grx2 | 225 ± 3 | 67 ± 1 | 30 | ND | −245 mV | |
Strains were grown in SC medium at 30°C. GSH and GSSG indicate supplementation of SC-Met medium with either 1 mM GSH or GSSG.
In order to calculate the amount of GSH or GSSG per 106 cells, one OD600 U was determined for M4745 to be 1.5·107 cells/ml. Values are given as the mean ± SD of measurements on three independent cultures.
The proportion of oxidized cytosolic rxYFP (mean ± SD; n = 3) was determined by spectrofluorometric redox titration as described in Materials and methods.
To further establish a link between the redox state of rxYFP and intracellular glutathione, mutations and growth conditions were identified that resulted in more drastically perturbed intracellular GSSG/GSH ratios. As shown in Fig. 1 C, sulfate assimilation in yeast occurs through a series of reactions whereby sulfate is converted into sulfide and then to homocysteine, the precursor of methionine and cysteine. Owing to the presence of two transsulfuration pathways allowing the interconversion of cysteine and homocysteine, a yeast mutant (Fig. 1 C, met15) disrupted in the sulfate assimilation pathway can survive on cysteine as the sole sulfur source (Hansen and Johannesen, 2000). Oxidized or GSH provide convenient sources of sulfur due to high affinity uptake from the culture medium by the plasma-membrane transporter Hgt1p (Bourbouloux et al., 2000). By combining met15 and glr1 disruptions and growing the resulting double mutant on 1 mM GSSG as the sole sulfur source, we observed a 45-fold increase in intracellular GSSG relative to the wild type (Table I). Under these conditions, the GSSG/GSH ratio was 225%. Intermediate ratios of 60 and 114%, respectively, were obtained by supplementing the growth medium with 1 mM GSH instead of GSSG and by growing the glr1 single disruption mutant in the presence of 1 mM GSSG. Under all these conditions the GSH levels remained essentially unaffected (Table I). Consistent with rxYFP being sensitive to the glutathione redox status, fluorometric redox state assessment in these strains revealed a direct relationship between the fraction of disulfide-bonded rxYFP and the intracellular GSSG/GSH ratio (Fig. 1 D).
Rapid equilibration of rxYFP in the cytosol
The fluorometric redox titration method is a steady-state technique. As such it provides a snapshot of the intracellular distribution of oxidized and reduced sensor, governed by the balancing action of synthesis of nascent reduced rxYFP and its subsequent oxidation. To evaluate the extent to which the measured steady-state distributions correlated with the equilibrium distribution, the in vivo kinetics of rxYFP oxidation was assessed by pulse-chase immunoprecipitation analysis in the wild-type, glr1, and glr1 met15 mutants, with intracellular GSSG/GSH ratios ranging from low to highly elevated. Cells were labeled with [35S]Met/Cys for 10 min and then chased for 0, 15, 30, and 60 min at which times thiol reactions were quenched by addition of TCA. Subsequently, free thiols were blocked by treatment with the alkylating reagent NEM. SDS-PAGE and phosphorimaging of immunoprecipitated rxYFP revealed that equilibrium was rapidly attained, with a half-time around 10 min irrespective of the intracellular GSSG/GSH ratio (Fig. 2). The fundamental difference between steady state and equilibrium became most apparent in the glr1 mutant, with an intermediate GSSG/GSH ratio. Here, the proportion of oxidized rxYFP was 73% according to the pulse-chase end point distribution, as opposed to 56% by the fluorometric method (compare Fig. 1 B with Fig. 2). This finding showed that whereas the fluorometric method constituted a highly efficient means of monitoring changes in the intracellular GSSG/GSH ratio, pulse-chase end point redox distribution analysis was required to obtain an accurate measure of the glutathione redox status.
Figure 2.

Monitoring the oxidation kinetics of cytosolic rxYFP by pulse labeling and immunoprecipitation. Kinetics of rxYFP oxidation in M4745 (wild type), M4860 (glr1), and M4859 (glr1 met15; grown on 1 mM GSSG as sole sulfur source). Cells were pulse labeled with [35S]Met/Cys for 10 min and chased for the indicated time periods. rxYFP was immunoprecipitated from cell lysates and resolved by nonreducing SDS-PAGE. The number below each lane indicates the proportion of oxidized rxYFP as determined by image analysis (ImageQuant software; Molecular Dynamics). Ox and Red denote oxidized and reduced rxYFP, respectively.
Glutaredoxins catalyze the equilibration of cytosolic rxYFP
As seen above, nascent reduced rxYFP reaches its equilibrium redox distribution fairly rapidly. In vitro, however, oxidation of rxYFP by GSSG proceeds relatively slowly (apparent second order rate constants, k ox, are given in Table II). This suggested that the reaction in vivo was catalyzed by one or more cytosolic components. Being glutathione-dependent thiol/disulfide oxidoreductases, the two cytosolic dithiol-type glutaredoxins, Grx1p and Grx2p, seemed as good candidates for this activity (Luikenhuis et al., 1998). Indeed, when a GRX1 GRX2 double knockout was constructed in the glr1 background, rxYFP oxidation was considerably slowed down and barely reached completion within the extended 180 min chase period (Fig. 3 A). The somewhat higher proportion of oxidized reporter at the last chase point (82%) compared with the glr1 background (73%) agreed well with a higher intracellular content of GSSG in the triple mutant (Table I).
Table II. Kinetic and thermodynamic parameters for the in vitro reaction between rxYFP and glutathione.
| pH | K ox(M)a | k ox(M− 1min− 1)a | k red(M− 2min− 1)b |
|---|---|---|---|
| 6.1 | 11.8 ± 0.9 | 25.5 ± 0.6 | 2.2 ± 0.2 |
| 6.4 | 9.9 ± 0.3 | 32.6 ± 0.3 | 3.3 ± 0.1 |
| 6.7 | 8.2 ± 0.5 | 41.6 ± 0.7 | 5.0 ± 0.3 |
| 7.0c | 6.8 ± 0.2 | 71.7 ± 0.9 | 10.6 ± 0.4 |
| 7.3 | 7.2 ± 0.4 | 123.9 ± 0.5 | 17.2 ± 1.0 |
| 7.6 | 7.6 ± 0.2 | 205.9 ± 1.0 | 27.0 ± 0.8 |
| 7.9 | 6.8 ± 0.2 | 400.4 ± 4.4 | 59.0 ± 0.2 |
Values are given as the mean ± SD (see Materials and methods).
Calculated as k ox/K ox.
An E°′ value of −265 mV for rxYFP was calculated from the measured K ox at pH 7 and an E°′ value of −240 mV for the GSH/GSSG redox couple (Rost and Rapoport, 1964). This redox potential is in good agreement with that determined previously (Ostergaard et al., 2001).
Figure 3.

Estimating the cytosolic concentrations of GSH and GSSG in a glr1 grx1 grx2 strain. (A) Kinetics of rxYFP equilibration in strain M5059 (glr1 grx1 grx2). The pulse chase was performed as described in Fig. 2. (B) A nonlinear least squares fit of Eq. 2 (see Materials and methods) to the progress curve obtained from the pulse chase experiment. Using K ox and k ox values of 6.8 M and 71.7 M−1min−1, determined in vitro at pH 7.0 (see Table III), the cytosolic concentrations of GSH and GSSG were estimated to be 13 and 0.17 mM, respectively, in this strain.
To independently confirm that the glutaredoxins were acting directly on rxYFP, recombinant His-tagged yeast Grx1p (rGrx1p) was expressed and purified from E. coli and tested for its ability to interact with rxYFP in vitro. The catalytic potential of glutaredoxin 1 was evaluated by addition of rGrx1p to a redox buffer containing either oxidized or reduced rxYFP in addition to GSH and GSSG in concentrations that would give ∼50% oxidized redox sensor at equilibrium. As shown in Fig. 4, rGrx1p was found to catalyze the equilibration of rxYFP in both the forward and reverse direction in a concentration-dependent manner. Together, with the in vivo data presented above, this demonstrated that whereas glutaredoxins are normally attributed to disulfide bond reduction they are in fact also capable of catalyzing disulfide bond formation under appropriate redox conditions.
Figure 4.

Grx1p-catalyzed equilibration of rxYFP. Reduced (open symbols) or oxidized (closed symbols) rxYFP was added to a buffer containing 100 mM potassium phosphate, pH 7.0, 1 mM EDTA as well as GSH and GSSG in concentrations chosen to give ∼50% oxidized rxYFP at equilibrium. The different traces correspond to the absence (circles) or the presence of rGrx1p at 1 μM (squares) or 3 μM (diamonds), respectively. The redox state of rxYFP was monitored by the change in fluorescence at 523 nm.
Estimating cytosolic concentrations of GSH and GSSG in the glr1 grx1 grx2 mutant
The slow oxidation of rxYFP in the glr1 grx1 grx2 triple knockout argued against the presence of additional redox catalyst, suggesting that the observed pulse-chase kinetics represented the uncatalyzed equilibration of rxYFP with the intracellular glutathione redox buffer. As detailed in Materials and methods the kinetics of this reaction (Scheme 1) could, formally, be accounted for by an integrated rate equation (Eq. 2; see Materials and methods) incorporating the rate constant for the reaction in the forward direction toward oxidized rxYFP (k ox) as well as the equilibrium constant K ox and the concentrations of GSH and GSSG in the redox buffer.
Scheme I.

Applying this equation on the equilibrium data obtained from the pulse-chase analysis allowed us to estimate the absolute cytosolic concentrations of GSH and GSSG (Fig. 3 A). Because the precise pH of the cytosol was not known, values for the two predefined parameters K ox and k ox were measured in vitro in the pH range from 6.1 to 7.9 (Table II). Using a nonlinear least squares procedure and K ox and k ox parameters determined at pH 7, a close fit to the pulse-chase data was obtained which yielded GSH and GSSG concentrations of 13 and 0.17 mM, respectively. Due to the robustness of K ox to changes in pH around 7 (Table II) the estimated concentrations only displayed a minor pH dependency. Accordingly, if the fit was extended to cover pH values ranging from 6.7 to 7.3, which should include most of the cytosolic pH values reported in the literature (Melvin and Shanks, 1996; Breeuwer and Abee, 2000), GSH and GSSG concentrations were found to lie within a range of 10–19 mM and 0.1–0.3 mM, respectively.
The wild-type cytosolic glutathione redox potential is surprisingly reducing
As mentioned above, steady-state measurements will underestimate the fraction of oxidized rxYFP compared with pulse-labeling end points. As the latter reflect the equilibrium of rxYFP with the glutathione redox pair, the intracellular glutathione redox potential, in turn, can be derived from the end point redox distribution. Using the Nernst equation and −265 mV as the standard redox potential (E°′) of rxYFP calculated from K ox at pH 7.0 (Table II), we found the redox potential of the cytosolic glutathione pool in wild-type yeast cells to be −289 mV (Table I). In our whole-cell measurements the amount of GSH relative to cell density only displayed very little variation (<10% from wild type to glr1 grx1 grx2) in spite of large differences in GSSG/GSH ratios (Table I). This strongly argued that the GSH concentration (13 mM) that could be derived from the glr1 grx1 grx2 mutant was also valid for the wild type. Thus, using this concentration and a redox potential of −289 mV as calculated above, the concentration of GSSG in the cytosol of wild-type yeast was estimated to be ∼4 μM; a value considerably lower that would be anticipated on basis of glutathione measurements on whole-cell extracts.
Targeting of rxYFP to other cellular compartments
One of the obvious virtues of using a GFP-based sensor is its genetic nature, which, in principle, allows it to be targeted to noncytosolic compartments like the ER. We would like to note that we have found that several signal peptides commonly used for secretion of heterologous proteins (α-factor, carboxypeptidase Y, invertase) did not promote efficient translocation of rxYFP (unpublished data). We have nevertheless constructed a variant that by several criteria appears to be translocated efficiently. However, the redox potential of rxYFP is, in its current version, not optimally suited for reliable redox measurements in this or other secretory compartments.
Discussion
With a GFP-based redox sensor, like rxYFP, at hand it would be tempting to ask what “the redox potential” of the cytoplasm is. However, measuring this hypothetical property of given compartment is not like measuring pH and indeed makes no sense. The proton is involved in many different reactions, and buffered through the interaction with a vast number of acids it has a high mobility in an aqueous biological solution like the cytosol of a cell. On the other hand, although thiol-disulfide reactions display a degree of promiscuity and the activation energy is generally lower than many other cellular reactions, they are sluggish compared with acid-base reactions. Thus, the conceptual difference between measuring pH and general thiol-disulfide redox status can be explained by the very low activation energy for the proton's binding and release compared with the thiol redox reaction. Indeed, in the E. coli periplasm thiol-disulfide oxidative and reductive pathways run side-by-side in opposite directions (Bader et al., 2001). Therefore, it was imperative to determine which cellular redox components interacted with rxYFP. To gain insight into what redox system was actually measured by rxYFP, we expressed the redox sensor in the yeast cytosol, as this would allow us to rapidly analyze the effects of mutations in a number of well-characterized genes involved in cytosolic thiol-disulfide redox homeostasis (Grant, 2001).
We found that at steady state ∼10% of rxYFP was oxidized in the cytosol of wild-type yeast. The degree of oxidation was consistent from determinations on a growing culture of yeast cells by fluorescence, from quantification of Western blots (unpublished data), and from pulse-labeling experiments. By introduction of various mutations and changing the growth medium we were able to manipulate the ratio between total GSSG and GSH from 4% in the wild type to 225%. The most extreme situation was attained by combining the glr1 mutation with a mutation in the sulfur assimilation pathway (met15) and growing the cells on GSSG as the sole sulfur source (Fig. 1 C). To our knowledge, such a GSSG/GSH ratio has not previously been measured in any living cells. Interestingly, the cellular glutathione homeostasis maintains essentially the same total pool of GSH whereas the GSSG varies (see Table I). Moreover, there is a consistent correlation between the GSSG/GSH ratio in the various mutants and the measured fraction of oxidized rxYFP, which increases to >90% in the most extreme case, suggesting a direct effect of the glutathione redox status on the redox status of rxYFP (Fig. 1 D).
As any protein is initially produced in the reduced state, the steady-state fraction of oxidized rxYFP might reflect a complete, but very slow, oxidation. To determine the oxidation kinetics and equilibrium redox distribution of rxYFP, pulse-labeling experiments were performed, followed by immunoprecipitation and separation of oxidized and reduced forms by nonreducing SDS-PAGE. In these experiments it could be shown that equilibrium was reached fairly rapidly in the wild type and in the glr1 and glr1 met15 mutants (Fig. 2), much more rapidly than in vitro kinetics would predict for an uncatalyzed reaction (Table II). We find that glutaredoxins catalyze the rapid equilibration of the sensor with the intracellular glutathione buffer, a conclusion we base on two observations. In vivo the rate of oxidation is significantly slowed down in glr1 grx1 grx2 mutant cells, deleted for the two cytosolic dithiol-type glutaredoxins (Fig. 2 and Fig. 3 A). In support of this, we show that glutaredoxin can catalyze the oxidation of reduced rxYFP as well as the reverse reaction in vitro (Fig. 4). Although this makes perfect sense from a biochemical point of view, it exposes a rather unconventional role for a glutaredoxin in vivo, where it is normally attributed to reduction of disulfide bonds (Grant, 2001). Similar in vitro experiments using yeast cytosolic thioredoxins showed catalysis of neither oxidation nor reduction (Fig. S1). This is an important observation as reduction of rxYFP by thioredoxin could in principle have formed a rapid kinetic link to NADPH oxidation through thioredoxin reductase. Instead, however, we find that the properties of rxYFP make it uniquely suited to the glutathione redox pair in the cytosol and that equilibration with this redox buffer is catalyzed by glutaredoxins.
Our estimates of absolute concentrations of GSH and GSSG are based on only two assumptions: (1) a cytosolic pH between 6.7 and 7.3; and (2) the absence of other catalysts than glutaredoxin 1 and 2. Although we have no direct evidence for the latter, it seems, however, highly reasonable given the slow equilibration kinetics observed in the glr1 grx1 grx2 triple knockout. It should also be emphasized that the presence of additional redox catalysts would imply an even lower cytosolic GSSG concentration than the one estimated here. If the GSH concentration of this mutant is close to that of the wild type, as supported by whole cell measurements, we find the cytosolic GSSG concentration of the wild type to be surprisingly low, ∼4 μM. Although it may be intuitively pleasing to know the absolute concentrations of oxidized and GSH in the cytosol, these figures may be difficult to interpret in a cytosolic environment where conditions are probably far from ideal due to high concentrations of proteins and low molecular weight solutes. In terms of disulfide bond stability the glutathione redox potential is, however, the central redox parameter on which to gauge the cytosolic redox environment. We find a value of −289 mV, which can be derived from the redox status of rxYFP using the Nernst equation. Although there is no clear consensus in the literature, this figure is significantly lower than previous estimates (e.g., −232 mV determined in a work by Hwang et al., 1992). Indeed, GSSG/GSH ratios measured on whole-cell extracts is considerably higher (4% in the wild type) than what we find in the cytosol (0.03%). Even under the most extreme conditions in the grl1 met15 strain grown on 1 mM GSSG the cytosol remains fairly reducing (Table I, ∼ −227 mV, which is equivalent to ∼4% GSSG), suggesting that the vast majority of the more than twofold excess of GSSG measured in cell extracts of this strain is sequestered (Table I). It is not unlikely that there is a considerable excess of GSSG in the secretory pathway including the vacuole and ER (Hwang et al., 1992; Li et al., 1996; Cuozzo and Kaiser, 1999). As redox potentials and GSH/GSSG ratios for the cytosolic compartment have generally been derived from whole-cell extracts, similar, albeit less extreme, sequestration may explain the difference between the redox potential we find in vivo and published data (Hwang et al., 1992; for review see Schafer and Buettner, 2001). Our results, which are very robust as discussed above, predict a significantly more reducing environment in the cytosol than previously estimated. In recent work, a GFP-based redox sensor has been applied to mitochondria and cytosol of mammalian cells in culture (Dooley et al., 2004; Hanson et al., 2004). However, it has not been determined what redox pair is measured in these cells nor has the kinetics for reaching equilibrium been investigated.
The engineered disulfide bond in rxYFP is of moderate stability as compared with many structural disulfide bonds in secretory proteins (Gilbert, 1995). Therefore, it is quite remarkable that it is found to be 10 and 53% oxidized under steady-state conditions in yeast and E. coli, respectively (Ostergaard et al., 2001). This suggests that the conspicuous absence of disulfide bonds in cytosolic proteins is a consequence of an evolutionary selection and not simply the result of reducing conditions.
Materials and methods
Strain and plasmid constructions
Saccharomyces cerevisiae was grown in YPD medium or SC medium (adjusted to pH 5.5) and manipulated as described elsewhere (Sherman, 2002). The genotypes of strains used in this work are given in Table III.
Table III. Yeast strains.
| Strain | Genotype | Source |
|---|---|---|
| M4745 | MATα his3-Δ1 leu2-Δ0 lys2-Δ0 ura3-Δ0 | EUROSCARF (BY4742) |
| M4860 | MATα his3-Δ1 leu2-Δ0 lys2-Δ0 ura3-Δ0 glr1::kanMX4 | EUROSCARF |
| M4859 | MATa his3-Δ1 leu2-Δ0 ura3-Δ0 met15-Δ0 glr1::kanMX4 | EUROSCARF |
| M4975 | MATα his3-Δ1 leu2-Δ0 lys2-Δ0 ura3-Δ0 grx2::kanMX4 | EUROSCARF |
| M5059 | MATα his3-Δ1 leu2-Δ0 lys2-Δ0 ura3-Δ0 glr1::URA3 grx1::HIS3 grx2::kanMX4 | This work |
A LEU2/TRP1-marked integration plasmid expressing rxYFP under PGK1 control was assembled as follows. A 1.6-kbp fragment containing LEU2 from YDp-L (Berben et al., 1991) was cloned into the BamHI site of pRS304 (Sikorski and Hieter, 1989). The 770-bp 5′ untranslated region of PGK1 was amplified from genomic DNA by PCR with primers introducing 5′ and 3′ XbaI and SpeI sites, respectively. The XbaI–SpeI cut amplicon was then ligated into the SpeI site of the pRS304-LEU2 plasmid destroying the XbaI site. Behind the PGK1 promoter was inserted a SpeI–SacI pHOJ104-derived fragment encoding rxYFP (Ostergaard et al., 2001). The 518-bp 3′ untranslated region of TDH3 was PCR-amplified from genomic DNA with primers introducing flanking SacI sites. The PCR fragment was cut and ligated into the SacI site, to generate pHOJ150. Chromosomal integration of the construct into the TRP1 locus of yeast was achieved by standard transformation of the PmeI-cut plasmid.
Chromosomal deletions of GLR1 and GRX1 genes in M4975 (Table III) were generated by standard transformation with linear DNA fragments containing >300 bp of sequence derived from the regions immediately 5′ and 3′ to the start and stop codons, respectively, separated by an appropriate selection marker (Table III) from the YDp-series of plasmids (Sikorski and Hieter, 1989). The DNA fragments were constructed by PCR splicing by overlap extension (Horton et al., 1989).
For heterologous overexpression of Grx1p, the yeast GRX1 gene was amplified from genomic DNA using primers (5′-GGCGGCGCATATGGTATCTCAAGAAACTATC and 5′-GCCGCCCTCGAGATTTGCAAGAATAGGTTCTAAC) incorporating a 5′ NdeI site overlapping the initiating Met codon and a 3′ XhoI site. The cut amplicon was subsequently ligated into the corresponding sites of pET-24a(+) (Novagen) to give pHOJ167, expressing Grx1p with a COOH-terminal hexa-histidine tag.
Analytical techniques
The concentration of GSH (Sigma-Aldrich) in stock solutions was determined using 5,5′-dithiobis(2-nitrobenzoic acid) and 13,600 M−1cm−1 as the molar extinction coefficient (ɛ) of nitrothiobenzoate at 412 nm (Ellman, 1959). Stock solutions of GSSG (Sigma-Aldrich) were quantified from its absorbance at 248 nm (ɛ = 382 M−1cm−1; Chau and Nelson, 1991). Reduction of rxYFP and rGrx1p was performed by a 1–2-h incubation in the presence of 5 mM DTT at pH 7.0. Reductant was subsequently removed by gel filtration on a prepacked NAP-5 column (Sephadex G-25; Amersham Biosciences) equilibrated with an appropriate buffer. Before all in vitro experiments, buffers were thoroughly purged with argon to prevent interference from dissolved molecular oxygen.
Expression and purification of Grx1p
A 1-liter culture of Rosetta(DE3) (Novagen) cells harboring pHOJ167 was grown to the mid-log phase in terrific broth at 37°C (Sambrook et al., 1989), at which time IPTG was added to 1 mM. After 3 h of induction, cells were harvested, lysed, and rGrx1p purified on a His-Bind resin (Novagen) according to the manufacturer's protocol. To remove trace amounts of contaminating proteins, the elutate, dialyzed overnight against 20 mM potassium phosphate, pH 7.5, was subjected to anion exchange chromatography on a Resource Q column (Amersham Biosciences). rGrx1p, eluted with a linear gradient from 0–0.5 M NaCl in dialysis buffer, was >95% pure as judged by SDS-PAGE and Coomassie staining. The protein was stored at −80°C in 100 mM potassium phosphate, pH 7.0, 1 mM EDTA. Treatment with the thiol-specific alkylating reagent 4-acetamido-4′-maleimidylstilbene-2,2′-disulfonic acid (Molecular Probes) before and after incubation with DTT revealed that the purified rGrx1p was completely oxidized.
Rate and equilibrium constants for the reaction rxYFPred + GSSG ⇔ rxYFPox + 2 GSH
Reactions were performed at 30°C in buffers containing 1 mM EDTA and either 100 mM K-MES (pH 6.1–6.7) or 100 mM K-MOPS (pH 7.0–7.9). The apparent second order rate constant (k ox) for oxidation of rxYFP by GSSG was determined by diluting reduced rxYFP ∼300-fold into 2 ml of prewarmed buffer to a final concentration of ∼0.3 μM. When a stable base line was attained, the reaction was initiated by the addition of a high molar excess of GSSG to ensure pseudo first-order conditions, and the reaction was followed by the change in fluorescence at 523 nm. At each pH, a minimum of four independent measurements were performed at varying concentrations of GSSG. The rate constant k ox was then obtained by a linear fit to the estimated pseudo first-order rate constants.
Equilibrium constants (K ox) at varying pH were determined directly in the fluorometer by incubating ∼0.2 μM reduced rxYFP in 2.5 ml MES or MOPS buffer (as above) containing 30–150 μM GSSG and 19 mM GSH, adjusted to give 20–70% oxidized rxYFP at equilibrium. To reduce the time required to reach equilibrium, reactions were performed in the presence of 5 μM rGrx1p. As shown in Fig. 4, rGrx1p functions as a simple redox-catalyst by speeding up the forward and reverse reactions to exactly the same extent. At equilibrium, the proportion of reduced redox sensor (f red) was determined by relating the fluorescence signal to that of the fully oxidized and reduced sensor obtained by addition of 20 μl 1.0 M GSSG to the reaction mixture followed by 20 μl 2.5 M DTT. To determine the exact amount of GSSG in the redox buffer at equilibrium, 180 μl of the reaction mixture were removed before addition of GSSG and DTT, quenched by addition of formic acid to 20%, and then analyzed by HPLC essentially as described previously (Takahashi and Creighton, 1996). At each pH, K ox was estimated from the relationship f red = 1/(1+ K ox·[GSSG]/[GSH]2) and reported as the mean of at least three independent reactions at varying [GSSG].
Equilibration of reduced rxYFP in a glutathione redox buffer
Progression of rxYFP toward equilibrium in a glutathione redox buffer can be described by the following differential equation
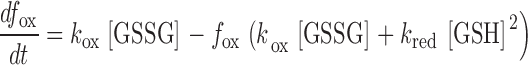 |
(1) |
in which f ox symbolizes the proportion of oxidized rxYFP at time t, and k ox and k red are the rate constants for the forward oxidation and reverse reduction reaction, respectively. Applying the boundary condition f ox = 0 (t = 0) and the thermodynamic linkage K ox = k ox/k red, integration of Eq. 1 yields the following expression (Takahashi and Creighton, 1996)
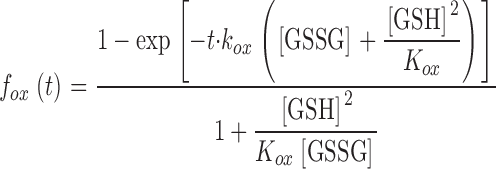 |
(2) |
where GSH and GSSG concentrations are assumed to remain constant during the reaction.
Measurement of low molecular weight thiols
The intracellular content of GSH and GSSG was quantified by HPLC after derivatization with N-(1-pyrenyl)maleimide (NPM) using a protocol (Winters et al., 1995) optimized for yeast. Cells were grown in SC medium (when supplemented with either 1 mM GSH or GSSG, methionine was omitted from the medium) for more than five generations in the exponential phase before analysis. At an OD600 of 0.8–1.0, between 15 and 30 OD600 U were harvested by filtration onto a 25-mm glass filter membrane (Advantec) and immediately washed with 5 ml water. The filter was then rapidly transferred to 1 ml prewarmed 1% (wt/vol) 5-sulfosalicylic acid (Sigma; Anderson, 1985) and boiled for 4 min, interrupted by vigorous shaking every minute. The supernatant containing the extracted low molecular-weight thiols was separated from filter and cell debris by centrifugation at 15,000 g for 15 min (4°C) and kept on ice until further analysis. For determination of reduced thiols, 20 μl of the supernatant (appropriately diluted) were combined with 50 μl 100 mM Tris-HCl, pH 8.0, and 20 μl 0.1 M NaOH, raising the pH to ∼8, and immediately derivatized by addition of 750 μl 2.67 mM NPM (Sigma-Aldrich) dissolved in acetonitrile. After 10 min of incubation at room temperature, the reaction was quenched by addition of 2 μl 2 M HCl. To determine cellular disulfides, free thiols were blocked by adding 20 μl of the cell extract to a tube containing 50 μl 100 mM Tris-HCl, pH 8.0, 20 μl 0.1 M NaOH, 20 μl 10 mM NEM (Sigma-Aldrich), and 110 μM water. The mixture was incubated 10 min at 70°C. Excess NEM was subsequently quenched by addition of 20 μl 14.3 mM β-mercaptoethanol (Sigma-Aldrich) followed by 10-min incubation at 70°C. Finally, disulfides were reduced by addition of 10 μl 20 mM tris-(2-carboxyethyl)phosphine (Sigma-Aldrich). After 10 min incubation at 70°C, the released thiols were labeled with NPM as described above. Quantification was performed by HPLC using a ReliaSil C18-AQ column (5 mm, 250 × 4.6 mm; Column Engineering Inc.). A 20-μl aliquot was injected onto the column and eluted isocractically in 65% acetonitrile, 0.1% acetic acid, 0.1% o-phosphoric acid (all vol/vol in water) at a flow rate of 0.5 ml/min. The NPM-derivatized thiols were detected by fluorescence with excitation and emission at 330 and 375 nm, respectively. Thiol concentrations were determined by relating the integrated peak areas to a standard curve based on known amounts of glutathione. Linearity was observed in the range from 300 fmol to 16 pmol thiol. The slope of the GSSG standard curve was found to be exactly twice that of the GSH standard curve, implying quantitative reduction of disulfides by tris-(2-carboxyethyl)phosphine. The reliability of the method was ascertained by the full recovery of exogenously added GSH or GSSG to cell extracts.
Determining the redox state of rxYFP in yeast by fluorescence
Strains were grown in SC-Leu medium (when supplemented with 1 mM GSH or GSSG, methionine was also omitted from the medium) for more than five generations before measurement. At an OD600 of 0.25–0.45, 840 μl of the culture was transferred to a prewarmed cuvette (30°C) and fluorescence monitored continuously using a Perkin-Elmer Luminescence Spectrometer LS50B equipped with an XF3074 emission filter (Omega Optical Inc.). Excitation wavelength was set at 512 nm (4-nm slit width). The pH of the medium was raised to ∼7 by addition of 60 μl 1.5 M K-MOPS, pH 7.9, 15 mM EDTA, immediately followed by 20 μl 4.6% (wt/vol) digitonin (Merck). After complete permeabilization of the cells (<3 min), observed as a slight drop in fluorescence intensity before return to a stable baseline (denoted Finit), the oxidation state of rxYFP was determined by reading the fluorescence after successive addition of 50 μl 6.3 mM 4-DPS (Sigma-Aldrich; Fox) and 100 μl 1 M DTT (Fred) to respectively fully oxidize and reduce the protein. The fraction of oxidized rxYFP was then calculated from the expression 1 − (Finit − Fox)/(Fred − Fox), taking into account dilution by the added reagents.
Pulse labeling and immunoprecipitation
Before metabolic labeling, strains were grown overnight in SC-Leu-Met medium (when necessary, supplemented with 1 mM GSH or GSSG) to a final OD600 of ∼0.5 and then resuspended in the same medium at an OD600 of 0.5. After 30 min of preincubation at 30°C, cells were radiolabeled with 120 μCi [35S]Met/Cys (Pro-Mix; Amersham Biosciences) per ml of culture for 10 min and then chased by addition of excess methionine and cysteine. At indicated time points, 950 μl of cells were withdrawn and quenched by addition of 5% TCA (Sigma-Aldrich). Cells were spun down and washed twice with 0.5 ml ice-cold acetone. The pellet was allowed to dry and then redissolved in 100 μl of lysis buffer (100 mM K-MOPS, pH 7.0, 2% SDS, 1× complete protease inhibitor cocktail; Roche Diagnostics) containing 40 mM NEM. Cell lysis and immunoprecipitation were performed as described previously (Norgaard et al., 2001). rxYFP was precipitated using 10 μl polyclonal rabbit anti-GFP antibody (Molecular Probes) per time point.
Online supplemental material
Fig. S1 shows that yeast thioredoxins do not react with rxYFP. Online supplemental material is available at http://www.jcb.org/cgi/content/full/jcb.200402120/DC1.
Acknowledgments
The technical assistance of Anette W. Bruun and Anders Barfod is gratefully acknowledged. We thank Morten Kielland-Brandt, Kresten Lindorff-Larsen, and Olof Björnberg for critically reading the manuscript, and Flemming G. Hansen for stimulating discussions.
H. Østergaard's present address is Novo Nordisk A/S, Novo Nordisk Park, DK-2760 Måløv, Denmark
Abbreviations used in this paper: 4-DPS, 4,4′-dithiodipyridine; GSH, reduced glutathione; GSSG, oxidized glutathione; NEM, N-ethyl-maleimide; NPM, N-(1-pyrenyl)maleimide; rGrx1p, recombinant His-tagged yeast Grx1p; rxYFP, redox sensitive YFP; TCA, trichloroacetic.
References
- Anderson, M.E. 1985. Determination of glutathione and glutathione disulfide in biological samples. Methods Enzymol. 113:548–555. [DOI] [PubMed] [Google Scholar]
- Aslund, F., B. Ehn, A. Miranda-Vizuete, C. Pueyo, and A. Holmgren. 1994. Two additional glutaredoxins exist in Escherichia coli: glutaredoxin 3 is a hydrogen donor for ribonucleotide reductase in a thioredoxin/glutaredoxin 1 double mutant. Proc. Natl. Acad. Sci. USA. 91:9813–9817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bader, M.W., A. Hiniker, J. Regeimbal, D. Goldstone, P.W. Haebel, J. Riemer, P. Metcalf, and J.C. Bardwell. 2001. Turning a disulfide isomerase into an oxidase: DsbC mutants that imitate DsbA. EMBO J. 20:1555–1562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berben, G., J. Dumont, V. Gilliquet, P.A. Bolle, and F. Hilger. 1991. The YDp plasmids: a uniform set of vectors bearing versatile gene disruption cassettes for Saccharomyces cerevisiae. Yeast. 7:475–477. [DOI] [PubMed] [Google Scholar]
- Bourbouloux, A., P. Shahi, A. Chakladar, S. Delrot, and A.K. Bachhawat. 2000. Hgt1p, a high affinity glutathione transporter from the yeast Saccharomyces cerevisiae. J. Biol. Chem. 275:13259–13265. [DOI] [PubMed] [Google Scholar]
- Breeuwer, P., and T. Abee. 2000. Assessment of the intracellular pH of immobilized and continuously perfused yeast cells employing fluorescence ratio imaging analysis. J. Microbiol. Methods. 39:253–264. [DOI] [PubMed] [Google Scholar]
- Chau, M.H., and J.W. Nelson. 1991. Direct measurement of the equilibrium between glutathione and dithiothreitol by high performance liquid chromatography. FEBS Lett. 291:296–298. [DOI] [PubMed] [Google Scholar]
- Cuozzo, J.W., and C.A. Kaiser. 1999. Competition between glutathione and protein thiols for disulphide-bond formation. Nat. Cell Biol. 1:130–135. [DOI] [PubMed] [Google Scholar]
- Dooley, C.T., T.M. Dore, G.T. Hanson, W.C. Jackson, S.J. Remington, and R.Y. Tsien. 2004. Imaging dynamic redox changes in mammalian cells with green fluorescent protein indicators. J. Biol. Chem. 279:22284–22293. [DOI] [PubMed] [Google Scholar]
- Ellman, G.L. 1959. Tissue sulfhydryl groups. Arch. Biochem. Biophys. 82:70–77. [DOI] [PubMed] [Google Scholar]
- Fernandes, A.P., and A. Holmgren. 2004. Glutaredoxins: glutathione-dependent redox enzymes with functions far beyond a simple thioredoxin backup system. Antioxid. Redox Signal. 6:63–74. [DOI] [PubMed] [Google Scholar]
- Gan, Z.R. 1991. Yeast thioredoxin genes. J. Biol. Chem. 266:1692–1696. [PubMed] [Google Scholar]
- Gilbert, H.F. 1995. Thiol/disulfide exchange equilibria and disulfide bond stability. Methods Enzymol. 251:8–28. [DOI] [PubMed] [Google Scholar]
- Grant, C.M. 2001. MicroReview: role of the glutathione/glutaredoxin and thioredoxin systems in yeast growth and response to stress conditions. Mol. Microbiol. 39:533–541. [DOI] [PubMed] [Google Scholar]
- Grant, C.M., F.H. MacIver, and I.W. Dawes. 1996. Glutathione is an essential metabolite required for resistance to oxidative stress in the yeast Saccharomyces cerevisiae. Curr. Genet. 29:511–515. [DOI] [PubMed] [Google Scholar]
- Grant, C.M., F.H. MacIver, and I.W. Dawes. 1997. Glutathione synthetase is dispensable for growth under both normal and oxidative stress conditions in the yeast Saccharomyces cerevisiae due to an accumulation of the dipeptide gamma-glutamylcysteine. Mol. Biol. Cell. 8:1699–1707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansen, J., and P.F. Johannesen. 2000. Cysteine is essential for transcriptional regulation of the sulfur assimilation genes in Saccharomyces cerevisiae. Mol. Gen. Genet. 263:535–542. [DOI] [PubMed] [Google Scholar]
- Hanson, G.T., R. Aggeler, D. Oglesbee, M. Cannon, R.A. Capaldi, R.Y. Tsien, and S.J. Remington. 2004. Investigating mitochondrial redox potential with redox-sensitive green fluorescent protein indicators. J. Biol. Chem. 279:13044–13053. [DOI] [PubMed] [Google Scholar]
- Hauser, M., A.M. Donhardt, D. Barnes, F. Naider, and J.M. Becker. 2000. Enkephalins are transported by a novel eukaryotic peptide uptake system. J. Biol. Chem. 275:3037–3041. [DOI] [PubMed] [Google Scholar]
- Holmgren, A. 1979. Thioredoxin catalyzes the reduction of insulin disulfides by dithiothreitol and dihydrolipoamide. J. Biol. Chem. 254:9627–9632. [PubMed] [Google Scholar]
- Holmgren, A. 1989. Thioredoxin and glutaredoxin systems. J. Biol. Chem. 264:13963–13966. [PubMed] [Google Scholar]
- Horton, R.M., H.D. Hunt, S.N. Ho, J.K. Pullen, and L.R. Pease. 1989. Engineering hybrid genes without the use of restriction enzymes: gene splicing by overlap extension. Gene. 77:61–68. [DOI] [PubMed] [Google Scholar]
- Hwang, C., A.J. Sinskey, and H.F. Lodish. 1992. Oxidized redox state of glutathione in the endoplasmic reticulum. Science. 257:1496–1502. [DOI] [PubMed] [Google Scholar]
- Jaspers, C.J., and M.J. Penninckx. 1984. Glutathione metabolism in yeast Saccharomyces cerevisiae. Evidence that gamma-glutamyltranspeptidase is a vacuolar enzyme. Biochimie. 66:71–74. [DOI] [PubMed] [Google Scholar]
- Klein, M., Y.M. Mamnun, T. Eggmnn, C. Schuller, H. Wolfger, E. Martinoia, and K. Kuchler. 2002. The ATP-binding cassette (ABC) transporter Bpt1p mediates vacuolar sequestration of glutathione conjugates in yeast. FEBS Lett. 520:63–67. [DOI] [PubMed] [Google Scholar]
- Kosower, N.S., and E.M. Kosower. 1978. The glutathione status of cells. Int. Rev. Cytol. 54:109–160. [DOI] [PubMed] [Google Scholar]
- Kumar, C., R. Sharma, and A.K. Bachhawat. 2003. Investigations into the polymorphisms at the ECM38 locus of two widely used Saccharomyces cerevisiae S288C strains, YPH499 and BY4742. Yeast. 20:857–863. [DOI] [PubMed] [Google Scholar]
- Li, Z.S., M. Szczypka, Y.P. Lu, D.J. Thiele, and P.A. Rea. 1996. The yeast cadmium factor protein (YCF1) is a vacuolar glutathione S-conjugate pump. J. Biol. Chem. 271:6509–6517. [DOI] [PubMed] [Google Scholar]
- Linke, K., and U. Jakob. 2003. Not every disulfide bond lasts forever: disulfide bond formation as a redox switch. Antioxid. Redox Signal. 5:425–434. [DOI] [PubMed] [Google Scholar]
- Luikenhuis, S., G. Perrone, I.W. Dawes, and C.M. Grant. 1998. The yeast Saccharomyces cerevisiae contains two glutaredoxin genes that are required for protection against reactive oxygen species. Mol. Biol. Cell. 9:1081–1091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melvin, B.K., and J.V. Shanks. 1996. Influence of aeration on cytoplasmic pH of yeast in an NMR airlift bioreactor. Biotechnol. Prog. 12:257–265. [DOI] [PubMed] [Google Scholar]
- Muller, E.G. 1996. A glutathione reductase mutant of yeast accumulates high levels of oxidized glutathione and requires thioredoxin for growth. Mol. Biol. Cell. 7:1805–1813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Norgaard, P., V. Westphal, C. Tachibana, L. Alsoe, B. Holst, and J.R. Winther. 2001. Functional differences in yeast protein disulfide isomerases. J. Cell Biol. 152:553–562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohtake, Y., and S. Yabuuchi. 1991. Molecular cloning of the gamma-glutamylcysteine synthetase gene of Saccharomyces cerevisiae. Yeast. 7:953–961. [DOI] [PubMed] [Google Scholar]
- Ostergaard, H., A. Henriksen, F.G. Hansen, and J.R. Winther. 2001. Shedding light on disulfide bond formation: engineering a redox switch in green fluorescent protein. EMBO J. 20:5853–5862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rebbeor, J.F., G.C. Connolly, M.E. Dumont, and N. Ballatori. 1998. ATP-dependent transport of reduced glutathione on YCF1, the yeast orthologue of mammalian multidrug resistance associated proteins. J. Biol. Chem. 273:33449–33454. [DOI] [PubMed] [Google Scholar]
- Rost, J., and S. Rapoport. 1964. Reduction-potential of glutathione. Nature. 201:185. [DOI] [PubMed] [Google Scholar]
- Sambrook, J., E.F. Fritsch, and T. Maniatis. 1989. Molecular Cloning. A Laboratory Manual. Appendix A2. C. Nolan, editor. Cold Spring Harbor Laboratory, Cold Spring Harbor, NY.
- Schafer, F.Q., and G.R. Buettner. 2001. Redox environment of the cell as viewed through the redox state of the glutathione disulfide/glutathione couple. Free Radic. Biol. Med. 30:1191–1212. [DOI] [PubMed] [Google Scholar]
- Sherman, F. 2002. Getting started with yeast. Methods Enzymol. 350:3–41. [DOI] [PubMed] [Google Scholar]
- Shi, J., A. Vlamis-Gardikas, F. Aslund, A. Holmgren, and B.P. Rosen. 1999. Reactivity of glutaredoxins 1, 2, and 3 from Escherichia coli shows that glutaredoxin 2 is the primary hydrogen donor to ArsC-catalyzed arsenate reduction. J. Biol. Chem. 274:36039–36042. [DOI] [PubMed] [Google Scholar]
- Sikorski, R.S., and P. Hieter. 1989. A system of shuttle vectors and yeast host strains designed for efficient manipulation of DNA in Saccharomyces cerevisiae. Genetics. 122:19–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spector, D., J. Labarre, and M.B. Toledano. 2001. A genetic investigation of the essential role of glutathione: mutations in the proline biosynthesis pathway are the only suppressors of glutathione auxotrophy in yeast. J. Biol. Chem. 276:7011–7016. [DOI] [PubMed] [Google Scholar]
- Takahashi, N., and T.E. Creighton. 1996. On the reactivity and ionization of the active site cysteine residues of Escherichia coli thioredoxin. Biochemistry. 35:8342–8353. [DOI] [PubMed] [Google Scholar]
- Taniguchi, N., and Y. Ikeda. 1998. gamma-Glutamyl transpeptidase: catalytic mechanism and gene expression. Adv. Enzymol. Relat. Areas Mol. Biol. 72:239–278. [DOI] [PubMed] [Google Scholar]
- Tu, B.P., and J.S. Weissman. 2004. Oxidative protein folding in eukaryotes: mechanisms and consequences. J. Cell Biol. 164:341–346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winters, R.A., J. Zukowski, N. Ercal, R.H. Matthews, and D.R. Spitz. 1995. Analysis of glutathione, glutathione disulfide, cysteine, homocysteine, and other biological thiols by high-performance liquid chromatography following derivatization by n-(1-pyrenyl)maleimide. Anal. Biochem. 227:14–21. [DOI] [PubMed] [Google Scholar]