

Abstract
Proper regulation of the phosphoinositide 3-kinase–Akt pathway is critical for the prevention of both insulin resistance and tumorigenesis. Many recent studies have characterized a negative feedback loop in which components of one downstream branch of this pathway, composed of the mammalian target of rapamycin and ribosomal S6 kinase, block further activation of the pathway through inhibition of insulin receptor substrate function. These findings form a novel basis for improved understanding of the pathophysiology of metabolic diseases (e.g., diabetes and obesity), tumor syndromes (e.g., tuberous sclerosis complex and Peutz-Jegher's syndrome), and human cancers.
Growth factors of the insulin family, including insulin, insulin-like growth factor (IGF) I, and IGF-II, are critical regulators of cell growth, survival, and metabolism. These molecules exert their effects on cell physiology through activation of insulin/IGF-I receptor tyrosine kinases (RTKs) on the surface of responsive cells. Once activated, these RTKs autophosphorylate, creating phosphotyrosine binding sites for the insulin receptor substrate (IRS) family of scaffolding proteins, among others. IRS proteins are subsequently phosphorylated by the insulin/IGF-I RTKs on several tyrosine residues. The resulting phosphotyrosines initiate signaling cascades by acting as binding sites for proteins containing src homology 2 domains, including the p85 regulatory subunit of class I phosphoinositide 3-kinase (PI3K). IRS-mediated activation of PI3K at the plasma membrane leads to its generation of the lipid second messenger phosphatidylinositol-3,4,5-trisphosphate (PIP3), which recruits a subset of proteins with pleckstrin homology domains. Among these is the Ser/Thr kinase Akt (also known as protein kinase B), which is activated upon recruitment to the plasma membrane. Activated Akt, through subsequent phosphorylation of several downstream targets, is primarily responsible for the ability of the insulin family of growth factors to stimulate cell growth, survival, and glucose uptake (for recent reviews on insulin signaling see Saltiel and Kahn, 2001; White, 2002).
Resistance, or unresponsiveness, to insulin is associated with a wide variety of human disorders, including type 2 diabetes, obesity, and cardiovascular disease. This fact has lead many laboratories to focus their efforts on characterizing molecular events that contribute to the desensitization of cells to insulin. A primary mechanism that has surfaced from these studies is the inhibition of IRS protein function through the phosphorylation of serine residues, degradation, and/or decrease in expression of serine that disconnect the insulin receptor from PI3K–Akt activation (for reviews see Saltiel and Kahn, 2001; White, 2002). Many distinct pathways have been implicated in the down-regulation of IRS-1 and IRS-2. For instance, adipose-derived molecules, such as tumor necrosis factor α (TNF-α) and free fatty acids (FFAs), stimulate an inhibitory phosphorylation of mouse IRS-1 by c-Jun NH2-terminal kinase (JNK; Hotamisligil et al., 1996; Aguirre et al., 2000; Hirosumi et al., 2002). Interestingly, another mechanism of IRS-1 inhibition stems from prolonged exposure to insulin itself and requires activation of the PI3K–Akt pathway (Smith et al., 1995; Rui et al., 2001). Recently, activation of the mammalian target of rapamycin (mTOR) branch downstream of the PI3K–Akt pathway has emerged as the critical event in rendering IRS-1 and IRS-2 unresponsive to insulin/IGF-I. This mTOR-mediated negative feedback loop and its physiological relevance to metabolic disorders and, perhaps, tumorigenesis are the focus of this review.
mTOR and insulin resistance
TOR proteins are Ser/Thr kinases with highly conserved homologues that are found in all eukaryotes (Schmelzle and Hall, 2000). In mammals, the two best-characterized targets of mTOR are the ribosomal S6 kinases (S6K1 and S6K2) and the eukaryotic initiation factor 4E (eIF4E)–binding protein 1 (4E-BP1). mTOR activity leads to S6K1/2 phosphorylation and activation and to 4E-BP1 phosphorylation and release from the cap-dependent translation initiation factor eIF4E. These two events, likely combined with other mTOR targets, lead to an increase in ribosomal biogenesis and the selective translation of specific mRNA populations. This ability to increase the protein synthesis capacity of the cell is responsible, at least in part, for the ability of TOR proteins to drive cell growth and proliferation (for review see Fingar and Blenis, 2004).
Full activation of the mTOR pathway requires signals from both nutrients (e.g., amino acids and glucose) and growth factors. Although the mechanism by which amino acids are sensed by TOR proteins is unknown, their presence is essential for basal activation of mTOR signaling, as well as for further stimulation by growth factors (Hara et al., 1998). Interestingly, amino acids have been shown to modulate insulin signaling through mTOR-dependent effects on IRS-1 (Takano et al., 2001; Tremblay and Marette, 2001). In both adipocytes and myocytes it has been demonstrated that, via mTOR regulation, excess amino acids attenuate and amino acid starvation stimulates Akt activation and subsequent glucose uptake in response to insulin. These effects are concomitant with changes in the state of IRS-1 phosphorylation, localization, and/or degradation. Therefore, the levels of circulating amino acids and other nutrients may have a profound effect on the insulin sensitivity of peripheral tissues, such as fat and skeletal muscle, through mTOR-mediated regulation of IRS-1 function.
The primary pathway by which most growth factors and cytokines activate mTOR and its downstream targets appears to be the PI3K–Akt pathway (for review see Fingar and Blenis, 2004). As with amino acids, several research groups have found that PI3K–Akt-mediated activation of mTOR also leads to attenuation of insulin signaling. Their studies have demonstrated that activation of the PI3K–Akt–mTOR pathway in various cell lines by PDGF (Li et al., 1999), TNF-α (Ozes et al., 2001), or insulin itself (Haruta et al., 2000; Greene et al., 2003; Carlson et al., 2004) leads to IRS-1 and/or IRS-2 serine phosphorylation and down-regulation. Although all of these studies found that IRS protein phosphorylation is sensitive to the mTOR-specific inhibitor rapamycin, the serine residues that are proposed to be involved vary. PDGF-mediated inhibition of IRS-1 required the proline-directed sites S632, S662, and S731 (Li et al., 1999), TNF-α treatment correlated with S632 and S635 phosphorylation (Ozes et al., 2001), and both amino acids and insulin stimulated S307 phosphorylation (Rui et al., 2001; Greene et al., 2003; Carlson et al., 2004; all numbering is for mouse IRS-1). However, S307 (S312 in humans) is the only site demonstrated to be required for insulin-stimulated IRS-1 down-regulation (Greene et al., 2003). Although this site was independently found to be phosphorylated by JNK in response to TNF-α (Aguirre et al., 2000; Hirosumi et al., 2002), JNK does not appear to be responsible for S307 phosphorylation in response to insulin (Rui et al., 2001; Greene et al., 2003), and the identity of the mTOR-dependent kinase that directly phosphorylates this site remains unknown.
The TSC genes and mTOR-dependent IRS-1/2 down-regulation
Tuberous sclerosis complex (TSC) is a syndrome characterized by a pleiotropic array of benign tumors, collectively referred to as hamartomas, often containing abnormally large cells (for a recent review see Kwiatkowski, 2003). Although these tumors rarely become malignant, their presence in various tissues gives rise to severe clinical manifestations, including neurological disorders, skin lesions, cardiac dysfunction, and kidney and lung failure. The disease has been mapped to mutations in either of the two tumor suppressor genes TSC1, which encodes the hamartin protein, and TSC2, which encodes the tuberin protein.
The hamartin and tuberin proteins form a complex that has recently been found to act within the pathway leading from PI3K–Akt activation to mTOR signaling (for review see Manning and Cantley, 2003). The tuberin–hamartin complex potently inhibits TOR-dependent signaling in both flies and mammals (Gao et al., 2002; Goncharova et al., 2002; Kwiatkowski et al., 2002; Tee et al., 2002), and this inhibition is relieved by Akt-directed phosphorylation of tuberin (Inoki et al., 2002; Manning et al., 2002; Potter et al., 2002). Therefore, in response to growth factors such as insulin, the PI3K–Akt pathway activates mTOR signaling through phosphorylation and inhibition of tuberin (Fig. 1 A). Interestingly, the responsiveness of TOR proteins to other signals, such as nutrient and energy availability, also appears to be dependent on the TSC genes (Gao et al., 2002; Inoki et al., 2003, Shaw et al., 2004). Therefore, homozygous loss of TSC1 or TSC2 leads to high constitutive activation of mTOR signaling, as detected in mouse embryonic fibroblasts (MEFs; Jaeschke et al., 2002; Kwiatkowski et al., 2002; Zhang et al., 2003), in the tumors of rodent models of TSC (Kenerson et al., 2002; El-Hashemite et al., 2003a), and in human TSC cells and tumors (Goncharova et al., 2002; El-Hashemite et al., 2003b).
Figure 1.
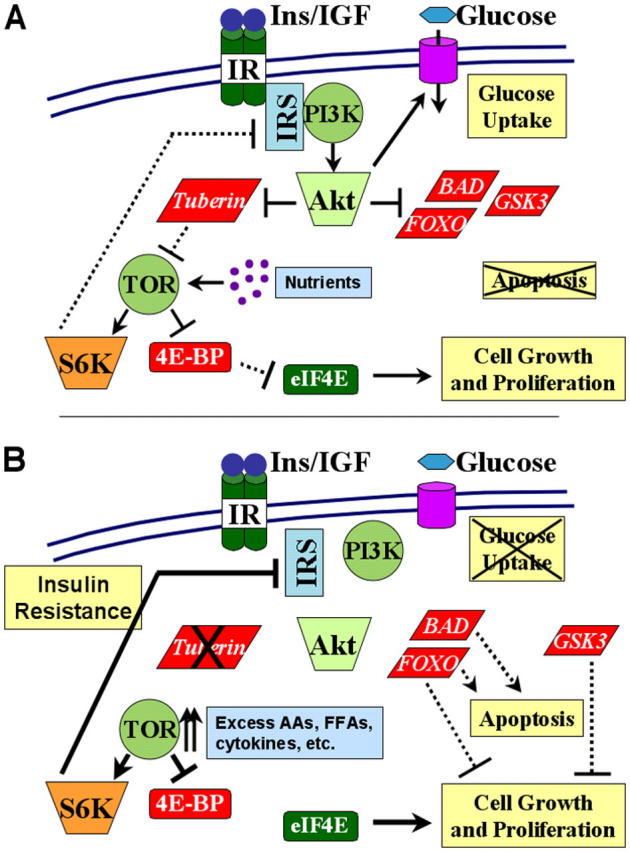
Model of the PI3K–Akt–TSC–TOR pathway and feedback regulation of insulin/IGF-I signaling. (A) Under normal conditions, insulin/IGF-I engagement of the insulin or IGF-I receptor (IR) leads to Tyr phosphorylation of IRS proteins and subsequent recruitment and activation of PI3K. PI3K activity leads to activation of Akt, which phosphorylates and inhibits many downstream substrates, including BAD, FOXO transcription factors, GSK3, and tuberin (TSC2). Through these and other targets, Akt activity stimulates glucose uptake and cell growth and proliferation, and inhibits apoptosis. Akt-directed phosphorylation of tuberin relieves its inhibition of TOR, via activation of the small G protein Rheb (not depicted). TOR activation by this pathway requires the presence of nutrients and results in activation of S6K and inhibition of 4E-BP1. S6K phosphorylates the ribosomal S6 protein (not depicted) and can feedback and inhibit IRS proteins (see S6K1: closing the loop section for details). Inhibition of 4E-BP1 relieves its inhibition of the translation initiation factor eIF4E. (B) Under atypical conditions of constitutive TOR activation, arising from chronic insulin/IGF-I exposure, excess nutrients (e.g., amino acids [AAs] and free fatty acids [FFAs]), inflammatory cytokines, or genetic loss of specific tumor suppressor genes, such as TSC2 (encoding tuberin), aberrantly high S6K activity shuts down insulin/IGF-I signaling. Therefore, although this unregulated TOR activity leads to constitutive eIF4E activation, it leads to insulin/IGF-I resistance through down-regulation of IRS protein function. This feedback inhibition prevents Akt-mediated glucose uptake and regulation of its downstream substrates. Dashed versus solid lines represent hypothetical signal strength.
The study of TSC1 − / − and TSC2 − / − MEFs has uncovered a striking inability of serum, PDGF, and, especially, insulin and IGF-I to activate the PI3K–Akt pathway in these cells (Jaeschke et al., 2002; Kwiatkowski et al., 2002; Zhang et al., 2003). This strong attenuation of Akt activation is also seen in Drosophila melanogaster TSC1 − / − cells and larvae (Radimerski et al., 2002). The insensitivity of TSC-deficient MEFs to PDGF is due to a loss of expression of both PDGF receptor α and PDGF receptor β (Zhang et al., 2003). However, two groups have now characterized the mechanism of insulin/IGF-I resistance in these MEFs (Harrington et al., 2004; Shah et al., 2004). They found that the resistance is a consequence of constitutive down-regulation of IRS-1 and IRS-2 that is due to sustained and unregulated mTOR signaling. Harrington et al. (2004) found that in the absence of TSC2, both IRS-1 and IRS-2 shift to a slower mobility during SDS-PAGE, which is generally indicative of an increase in Ser/Thr phosphorylation, and this shift is blocked by short (1 h) treatments with rapamycin. This group found that the mRNA and protein levels of IRS-1 alone are decreased in TSC-deficient MEFs, whereas Shah et al. (2004) found that the levels and stability of both the IRS-1 and IRS-2 proteins are decreased in these cells. Importantly, both studies found that long-term (24 h) treatment of TSC-deficient MEFs with rapamycin completely restores IRS-1 protein levels and the insulin/IGF-I responsiveness of the PI3K–Akt pathway. Together, these studies demonstrate that high levels of constitutive mTOR activity strongly down-regulate insulin/IGF-I signaling through combined effects on IRS-1 expression and IRS-1 and IRS-2 serine phosphorylation and protein stability.
S6K1: closing the loop
It is now well established that mTOR signaling is intimately involved in many pathways that lead to insulin resistance through IRS-1/2 down-regulation. The finding in Drosophila that larvae doubly mutant for dTSC1 and dS6K have normal levels of Akt activity, relative to the attenuated state of dTSC1 − / − larvae, was the first suggestion that this feedback mechanism downstream of mTOR is likely to be mediated by S6K (Radimerski et al., 2002). Harrington et al. (2004) have now found that small interfering RNA knockdown of S6K1 or S6K2 can restore the expression of IRS-1 mRNA in TSC2 − / − MEFs. Furthermore, in overexpression experiments, Shah et al. (2004) found that a kinase-dead version of S6K1 can block this negative feedback loop and activate Akt.
Although the mechanism of transcriptional regulation of IRS-1 by S6K1/2 remains unknown, it appears that the effect of S6K1 on IRS-1 serine phosphorylation is direct. In in vitro kinase assays, S6K1 directly phosphorylates S302 of mouse IRS-1 (Harrington et al., 2004), a site matching its preferred substrate recognition motif (Alessi et al., 1996). Furthermore, S302 is constitutively phosphorylated in TSC2 − / − MEFs, and this phosphorylation is inhibited by treatment with either rapamycin or S6K1 small interfering RNAs. The results from in vitro binding assays suggest that phosphorylation of this site by S6K hinders the ability of IRS-1 to associate with the insulin receptor (Harrington et al., 2004). A recent independent study also found that insulin and IGF-I stimulate mTOR-dependent S302 phosphorylation in various cell lines, as well as in skeletal muscle (Giraud et al., 2004). However, the results of overexpression experiments led this group to conclude that S302 phosphorylation enhances, rather than inhibits, insulin signaling. Although additional experiments are necessary to resolve this discrepancy, two other research groups have recently provided further evidence that S302 phosphorylation inhibits signaling from IRS-1 (Greene et al., 2004; Werner et al., 2004).
A critical in vivo role for S6K1 in desensitizing tissues to insulin was demonstrated in a recent study on S6K1-deficient mice (Um et al., 2004). The researchers found that these mice are hypersensitive to insulin and impervious to the obesity-induced insulin resistance detected in wild-type mice. In the adipose tissue of wild-type mice, a dramatic increase in S6K1 activation is observed in response to a high fat diet or in genetic models of obesity, and this is accompanied by an increase in IRS-1 phosphorylation on S307, S632, and S635 and by attenuation of insulin-induced Akt activation. However, under these same conditions in S6K1 − / − mice, these sites on IRS-1 are not phosphorylated, and Akt remains responsive to insulin. Although this study did not examine S302, an increase in phosphorylation of this site has recently been demonstrated in mouse models of obesity and insulin resistance (Werner et al., 2004).
These results demonstrate that S6K1 is essential to multiple pathways that render cells unresponsive to insulin, including those originating from chronic exposure to insulin, FFAs, and perhaps TNF-α. Interestingly, mice lacking either S6K1 or JNK1 are similarly protected from obesity-induced insulin resistance and display loss of IRS-1 phosphorylation on S307 (Hirosumi et al., 2002; Um et al., 2004), a site found to be a direct target of JNK (Aguirre et al., 2000). It appears that phosphorylation of S307 and other serines on IRS-1 might, therefore, exhibit cooperation with one another in response to some stimuli. Because mTOR signaling appears to be required for the effects of several pathways on IRS-1, it seems likely that there is an initial S6K1-mediated phosphorylation event, perhaps on S302, that is required for other serine kinases to subsequently phosphorylate IRS-1. If this model is correct, this mechanism would prevent any pathway from blocking insulin signaling when mTOR activity is low, such as during glucose or amino acid starvation. This would allow cells, and mTOR, to remain maximally sensitive to the insulin–PI3K–Akt pathway, which is involved in nutrient uptake.
Impact on our understanding of human disease states
As a downstream target, S6K is poised to sense the degree of activation of mTOR, which is stimulated by both nutrients and growth factors (Fig. 1 A). As a critical component of the negative feedback loop, S6K then has the ability to modify accordingly the level of input from some growth factors, such as insulin and IGF-I. However, under nutritional conditions in which mTOR is chronically activated, such as in the presence of excess amino acids or during high fat diets, S6K will constitutively shut down the responsiveness of certain cells to insulin (Fig. 1 B). This suggests that rapamycin (or, more specifically, an inhibitor of S6K) might be an effective treatment for certain metabolic disorders, such as type 2 diabetes, characterized by insulin resistance (Um et al., 2004).
Chronic activation of mTOR signaling can also result from genetic loss of certain tumor suppressor genes, including PTEN, TSC1/2, and LKB1 (Neshat et al., 2001; Kwiatkowski, 2003; Shaw et al., 2004). Interestingly, mutations in these genes are associated with the specific tumor syndromes Cowden's disease (PTEN), TSC (TSC1/2), and Peutz-Jegher's syndrome (LKB1), which are all characterized by the development of benign tumors classified as hamartomas. Although aberrant mTOR activation appears to be a common biochemical link between cells lacking these tumor suppressors, the activation state of the PI3K–Akt pathway varies. Loss of PTEN (phosphatase and tensin homologue deleted on chromosome 10), which counters the activity of PI3K, leads to constitutive activation of Akt (for review see Cantley and Neel, 1999). However, as discussed above for TSC1/2, MEFs lacking LKB1 also exhibit some attenuation of Akt activation (Shaw et al., 2004). This discrepancy might have profound effects on the nature of the tumors that arise in patients with these different syndromes. The overall development of a hamartoma might be the result of constitutive mTOR activity, but the severity and malignancy potential of the tumor might depend on the ability to overcome the feedback inhibition of Akt. The attenuation of Akt is strongest in the absence of the TSC genes, and the inability of Akt to phosphorylate and inhibit its downstream substrates other than tuberin, such as specific members of the FOXO family of transcription factors, GSK3, and Bad, could limit the proliferation and survival capacity of TSC hamartomas (Fig. 1 B). Consistent with this idea, malignant tumors are very rare in TSC patients (Kwiatkowski, 2003), whereas in Cowden's disease, where loss of PTEN activates Akt independently of growth factor signaling, the development of malignant tumors is much more common (Cantley and Neel, 1999).
Because rapamycin relieves this feedback inhibition of Akt (Harrington et al., 2004; Shah et al., 2004), a difficult question in the field is whether this mTOR inhibitor would be beneficial or detrimental to patients with hamartomas exhibiting aberrant mTOR activation. Consistent with mTOR activation driving initial tumor development in these related syndromes, rapamycin has been proven to be effective at blocking the formation of tumors in rodent models of TSC (Kenerson et al., 2002). However, it is less clear whether rapamycin would be a useful treatment once the tumor has already been established, as is the case in patients diagnosed with TSC. In fact, studies of TSC1/2- and LKB1-deficient MEFs have demonstrated that under some growth conditions rapamycin actually enhances survival of cells devoid of these tumor suppressors (Inoki et al., 2003; Harrington et al., 2004; Shah et al., 2004; Shaw et al., 2004), and this is likely due to the restoration of critical survival pathways controlled by Akt. However, these studies are done on a short-term basis and under very specific conditions. It seems likely that, although rapamycin's inhibition of S6K activation would eliminate the negative feedback mechanism, it would also shut down the aberrant activation of eIF4E and cap-dependent translation. Based on many studies suggesting that eIF4E is a potent oncogene (e.g., for review see Mamane et al., 2004; Ruggero et al., 2004; Wendel et al., 2004), it would appear that the mTOR-dependent activation of eIF4E, through 4E-BP1 phosphorylation, may be at the heart of tumorigenesis in syndromes exhibiting constitutive mTOR signaling. If this is true, then long-term treatment with rapamycin would block the primary cellular process driving tumor growth, irrespective of Akt activity. Furthermore, it is currently unknown whether the down-regulation of PDGF receptors observed in TSC-deficient MEFs is sensitive to rapamycin (Zhang et al., 2003).
It remains to be established whether the state of this mTOR-dependent feedback mechanism is what delineates the severity of some tumors and metabolic diseases. However, studies on this pathway, and the diseases to which it contributes, underscore the importance of elucidating the complex wiring of seemingly basic signal transduction pathways. The days of the linear genetic or biochemical pathway are long past.
Acknowledgments
I thank Drs. Reuben Shaw, Lewis Cantley, and Gokhan Hotamisligil for insightful discussions and critical comments on this manuscript.
This work was supported by a Rothberg Courage Award from the Tuberous Sclerosis Alliance and a Career Development Grant from the Leukemia and Lymphoma Society.
Abbreviations used in this paper: 4E-BP1, eIF4E-binding protein 1; eIF4E, eukaryotic initiation factor 4E; FFA, free fatty acid; IGF, insulin-like growth factor; IRS, insulin receptor substrate; JNK, c-Jun NH2-terminal kinase; MEF, mouse embryonic fibroblast; PI3K, phosphoinositide 3-kinase; RTK, receptor tyrosine kinase; S6K, ribosomal S6 kinase; TNF-α, tumor necrosis factor α; TOR, target of rapamycin; TSC, tuberous sclerosis complex.
References
- Aguirre, V., T. Uchida, L. Yenush, R. Davis, and M.F. White. 2000. The c-Jun NH(2)-terminal kinase promotes insulin resistance during association with insulin receptor substrate-1 and phosphorylation of Ser(307). J. Biol. Chem. 275:9047–9054. [DOI] [PubMed] [Google Scholar]
- Alessi, D.R., F.B. Caudwell, M. Andejelkovic, B.A. Hemmings, and P. Cohen. 1996. Molecular basis for the substrate specificity of protein kinase B; comparison with MAPKAP kinase-1 and p70 S6 kinase. FEBS Lett. 399:333–338. [DOI] [PubMed] [Google Scholar]
- Cantley, L.C., and B.G. Neel. 1999. New insights into tumor suppression: PTEN suppresses tumor formation by restraining the phosphoinositide 3-kinase/AKT pathway. Proc. Natl. Acad. Sci. USA. 96:4240–4245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carlson, C.J., M.F. White, and C.M. Rondinone. 2004. Mammalian target of rapamycin regulates IRS-1 serine 307 phosphorylation. Biochem. Biophys. Res. Commun. 316:533–539. [DOI] [PubMed] [Google Scholar]
- El-Hashemite, N., V. Walker, H. Zhang, and D.J. Kwiatkowski. 2003. a. Loss of Tsc1 or Tsc2 induces vascular endothelial growth factor production through mammalian target of rapamycin. Cancer Res. 63:5173–5177. [PubMed] [Google Scholar]
- El-Hashemite, N., H. Zhang, E.P. Henske, and D.J. Kwiatkowski. 2003. b. Mutation in TSC2 and activation of mammalian target of rapamycin signalling pathway in renal angiomyolipoma. Lancet. 361:1348–1349. [DOI] [PubMed] [Google Scholar]
- Fingar, D.C., and J. Blenis. 2004. Target of rapamycin (TOR): an integrator of nutrient and growth factor signals and coordinator of cell growth and cell cycle progression. Oncogene. 23:3151–3171. [DOI] [PubMed] [Google Scholar]
- Gao, X., Y. Zhang, P. Arrazola, O. Hino, T. Kobayashi, R.S. Yeung, B. Ru, and D. Pan. 2002. Tsc tumour suppressor proteins antagonize amino acid-TOR signalling. Nat. Cell Biol. 4:699–704. [DOI] [PubMed] [Google Scholar]
- Giraud, J., R. Leshan, Y.H. Lee, and M.F. White. 2004. Nutrient-dependent and insulin-stimulated phosphorylation of insulin receptor substrate-1 on serine 302 correlates with increased insulin signaling. J. Biol. Chem. 279:3447–3454. [DOI] [PubMed] [Google Scholar]
- Goncharova, E.A., D.A. Goncharov, A. Eszterhas, D.S. Hunter, M.K. Glassberg, R.S. Yeung, C.L. Walker, D. Noonan, D.J. Kwiatkowski, M.M. Chou, et al. 2002. Tuberin regulates p70 S6 kinase activation and ribosomal protein S6 phosphorylation: a role for the TSC2 tumor suppressor gene in pulmonary lymphangioleiomyomatosis (LAM). J. Biol. Chem. 277:30958–30967. [DOI] [PubMed] [Google Scholar]
- Greene, M.W., H. Sakaue, L. Wang, D.R. Alessi, and R.A. Roth. 2003. Modulation of insulin-stimulated degradation of human insulin receptor substrate-1 by Serine 312 phosphorylation. J. Biol. Chem. 278:8199–8211. [DOI] [PubMed] [Google Scholar]
- Greene, M.W., N. Morrice, R.S. Garofalo, and R.A. Roth. 2004. Modulation of human insulin receptor substrate-1 tyrosine phosphorylation by protein kinase Cdelta. Biochem. J. 378:105–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hara, K., K. Yonezawa, Q.P. Weng, M.T. Kozlowski, C. Belham, and J. Avruch. 1998. Amino acid sufficiency and mTOR regulate p70 S6 kinase and eIF-4E BP1 through a common effector mechanism. J. Biol. Chem. 273:14484–14494. [DOI] [PubMed] [Google Scholar]
- Harrington, L.S., G.M. Findlay, A. Gray, T. Tolkacheva, S. Wigfield, H. Rebholz, J. Barnett, N.R. Leslie, S. Cheng, P.R. Shepherd, et al. 2004. The TSC1-2 tumor suppressor controls insulin–PI3K signaling via regulation of IRS proteins. J. Cell Biol. 166:213–223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haruta, T., T. Uno, J. Kawahara, A. Takano, K. Egawa, P.M. Sharma, J.M. Olefsky, and M. Kobayashi. 2000. A rapamycin-sensitive pathway down-regulates insulin signaling via phosphorylation and proteasomal degradation of insulin receptor substrate-1. Mol. Endocrinol. 14:783–794. [DOI] [PubMed] [Google Scholar]
- Hirosumi, J., G. Tuncman, L. Chang, C.Z. Gorgun, K.T. Uysal, K. Maeda, M. Karin, and G.S. Hotamisligil. 2002. A central role for JNK in obesity and insulin resistance. Nature. 420:333–336. [DOI] [PubMed] [Google Scholar]
- Hotamisligil, G.S., P. Peraldi, A. Budavari, R. Ellis, M.F. White, and B.M. Spiegelman. 1996. IRS-1-mediated inhibition of insulin receptor tyrosine kinase activity in TNF-alpha- and obesity-induced insulin resistance. Science. 271:665–668. [DOI] [PubMed] [Google Scholar]
- Inoki, K., Y. Li, T. Zhu, J. Wu, and K.L. Guan. 2002. TSC2 is phosphorylated and inhibited by Akt and suppresses mTOR signalling. Nat. Cell Biol. 4:648–657. [DOI] [PubMed] [Google Scholar]
- Inoki, K., T. Zhu, and K.L. Guan. 2003. TSC2 mediates cellular energy response to control cell growth and survival. Cell. 115:577–590. [DOI] [PubMed] [Google Scholar]
- Jaeschke, A., J. Hartkamp, M. Saitoh, W. Roworth, T. Nobukuni, A. Hodges, J. Sampson, G. Thomas, and R. Lamb. 2002. Tuberous sclerosis complex tumor suppressor–mediated S6 kinase inhibition by phosphatidylinositide-3-OH kinase is mTOR independent. J. Cell Biol. 159:217–224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kenerson, H.L., L.D. Aicher, L.D. True, and R.S. Yeung. 2002. Activated mammalian target of rapamycin pathway in the pathogenesis of tuberous sclerosis complex renal tumors. Cancer Res. 62:5645–5650. [PubMed] [Google Scholar]
- Kwiatkowski, D.J. 2003. Tuberous sclerosis: from tubers to mTOR. Ann. Hum. Genet. 67:87–96. [DOI] [PubMed] [Google Scholar]
- Kwiatkowski, D.J., H. Zhang, J.L. Bandura, K.M. Heiberger, M. Glogauer, N. el-Hashemite, and H. Onda. 2002. A mouse model of TSC1 reveals sex-dependent lethality from liver hemangiomas, and up-regulation of p70S6 kinase activity in TSC1 null cells. Hum. Mol. Genet. 11:525–534. [DOI] [PubMed] [Google Scholar]
- Li, J., K. DeFea, and R.A. Roth. 1999. Modulation of insulin receptor substrate-1 tyrosine phosphorylation by an Akt/phosphatidylinositol 3-kinase pathway. J. Biol. Chem. 274:9351–9356. [DOI] [PubMed] [Google Scholar]
- Mamane, Y., E. Petroulakis, L. Rong, K. Yoshida, L.W. Ler, and N. Sonenberg. 2004. eIF4E–from translation to transformation. Oncogene. 23:3172–3179. [DOI] [PubMed] [Google Scholar]
- Manning, B.D., and L.C. Cantley. 2003. Rheb fills a GAP between TSC and TOR. Trends Biochem. Sci. 28:573–576. [DOI] [PubMed] [Google Scholar]
- Manning, B.D., A.R. Tee, M.N. Logsdon, J. Blenis, and L.C. Cantley. 2002. Identification of the tuberous sclerosis complex-2 tumor suppressor gene product tuberin as a target of the phosphoinositide 3-kinase/Akt pathway. Mol. Cell. 10:151–162. [DOI] [PubMed] [Google Scholar]
- Neshat, M.S., I.K. Mellinghoff, C. Tran, B. Stiles, G. Thomas, R. Petersen, P. Frost, J.J. Gibbons, H. Wu, and C.L. Sawyers. 2001. Enhanced sensitivity of PTEN-deficient tumors to inhibition of FRAP/mTOR. Proc. Natl. Acad. Sci. USA. 98:10314–10319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ozes, O.N., H. Akca, L.D. Mayo, J.A. Gustin, T. Maehama, J.E. Dixon, and D.B. Donner. 2001. A phosphatidylinositol 3-kinase/Akt/mTOR pathway mediates and PTEN antagonizes tumor necrosis factor inhibition of insulin signaling through insulin receptor substrate-1. Proc. Natl. Acad. Sci. USA. 98:4640–4645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Potter, C.J., L.G. Pedraza, and T. Xu. 2002. Akt regulates growth by directly phosphorylating Tsc2. Nat. Cell Biol. 4:658–665. [DOI] [PubMed] [Google Scholar]
- Radimerski, T., J. Montagne, M. Hemmings-Mieszczak, and G. Thomas. 2002. Lethality of Drosophila lacking TSC tumor suppressor function rescued by reducing dS6K signaling. Genes Dev. 16:2627–2632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruggero, D., L. Montanaro, L. Ma, W. Xu, P. Londei, C. Cordon-Cardo, and P.P. Pandolfi. 2004. The translation factor eIF-4E promotes tumor formation and cooperates with c-Myc in lymphomagenesis. Nat. Med. 10:484–486. [DOI] [PubMed] [Google Scholar]
- Rui, L., V. Aguirre, J.K. Kim, G.I. Shulman, A. Lee, A. Corbould, A. Dunaif, and M.F. White. 2001. Insulin/IGF-1 and TNF-alpha stimulate phosphorylation of IRS-1 at inhibitory Ser307 via distinct pathways. J. Clin. Invest. 107:181–189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saltiel, A.R., and C.R. Kahn. 2001. Insulin signalling and the regulation of glucose and lipid metabolism. Nature. 414:799–806. [DOI] [PubMed] [Google Scholar]
- Schmelzle, T., and M.N. Hall. 2000. TOR, a central controller of cell growth. Cell. 103:253–262. [DOI] [PubMed] [Google Scholar]
- Shah, O.J., Z. Wang, and T. Hunter. 2004. Inappropriate activation of the TSC/Rheb/mTOR/S6K cassette induces IRS1/2 depletion, insulin resistance, and cell survival deficiencies. Curr. Biol. 14:1650–1656. [DOI] [PubMed] [Google Scholar]
- Shaw, R.J., N. Bardeesy, B.D. Manning, L. Lopez, M. Kosmatka, R.A. DePinho, and L.C. Cantley. 2004. The LKB1 tumor suppressor negatively regulates mTOR signaling. Cancer Cell. 6:91–99. [DOI] [PubMed] [Google Scholar]
- Smith, L.K., C.J. Vlahos, K.K. Reddy, J.R. Falck, and C.W. Garner. 1995. Wortmannin and LY294002 inhibit the insulin-induced down-regulation of IRS-1 in 3T3-L1 adipocytes. Mol. Cell. Endocrinol. 113:73–81. [DOI] [PubMed] [Google Scholar]
- Takano, A., I. Usui, T. Haruta, J. Kawahara, T. Uno, M. Iwata, and M. Kobayashi. 2001. Mammalian target of rapamycin pathway regulates insulin signaling via subcellular redistribution of insulin receptor substrate 1 and integrates nutritional signals and metabolic signals of insulin. Mol. Cell. Biol. 21:5050–5062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tee, A.R., D.C. Fingar, B.D. Manning, D.J. Kwiatkowski, L.C. Cantley, J. Blenis. 2002. Tuberous sclerosis complex-1 and -2 gene products function together to inhibit mammalian target of rapamycin (mTOR)-mediated downstream signaling. Proc. Natl. Acad. Sci. 99:13571–13576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tremblay, F., and A. Marette. 2001. Amino acid and insulin signaling via the mTOR/p70 S6 kinase pathway. A negative feedback mechanism leading to insulin resistance in skeletal muscle cells. J. Biol. Chem. 276:38052–38060. [DOI] [PubMed] [Google Scholar]
- Um, S.H., F. Frigerio, M. Watanabe, F. Picard, M. Joaquin, M. Sticker, S. Fumagalli, P.R. Allegrini, S.C. Kozma, J. Auwerx, and G. Thomas. 2004. Absence of S6K1 protects against age- and diet-induced obesity while enhancing insulin sensitivity. Nature. 431:200–205. [DOI] [PubMed] [Google Scholar]
- Wendel, H.G., E. De Stanchina, J.S. Fridman, A. Malina, S. Ray, S. Kogan, C. Cordon-Cardo, J. Pelletier, and S.W. Lowe. 2004. Survival signalling by Akt and eIF4E in oncogenesis and cancer therapy. Nature. 428:332–337. [DOI] [PubMed] [Google Scholar]
- Werner, E.D., J. Lee, L. Hansen, M. Yuan, and S.E. Shoelson. 2004. Insulin resistance due to phosphorylation of insulin receptor substrate-1 at serine 302. J. Biol. Chem. 279:35298–35305. [DOI] [PubMed] [Google Scholar]
- White, M.F. 2002. IRS proteins and the common path to diabetes. Am. J. Physiol. Endocrinol. Metab. 283:E413–E422. [DOI] [PubMed] [Google Scholar]
- Zhang, H., G. Cicchetti, H. Onda, H.B. Koon, K. Asrican, N. Bajraszewski, F. Vazquez, C.L. Carpenter, and D.J. Kwiatkowski. 2003. Loss of Tsc1/Tsc2 activates mTOR and disrupts PI3K-Akt signaling through downregulation of PDGFR. J. Clin. Invest. 112:1223–1233. [DOI] [PMC free article] [PubMed] [Google Scholar]