

Abstract
Cystic fibrosis (CF) is a childhood hereditary disease in which the most common mutant form of the CF transmembrane conductance regulator (CFTR) ΔF508 fails to exit the endoplasmic reticulum (ER). Export of wild-type CFTR from the ER requires the coat complex II (COPII) machinery, as it is sensitive to Sar1 mutants that disrupt normal coat assembly and disassembly. In contrast, COPII is not used to deliver CFTR to ER-associated degradation. We find that exit of wild-type CFTR from the ER is blocked by mutation of a consensus di-acidic ER exit motif present in the first nucleotide binding domain. Mutation of the code disrupts interaction with the COPII coat selection complex Sec23/Sec24. We propose that the di-acidic exit code plays a key role in linking CFTR to the COPII coat machinery and is the primary defect responsible for CF in ΔF508-expressing patients.
Introduction
The cystic fibrosis (CF) transmembrane conductance regulator (CFTR) is a cAMP-regulated chloride channel found in the apical membrane of polarized epithelia lining many tissues including the intestine, pancreas, and lung (for reviews see Kopito, 1999; Riordan, 1999; Bertrand and Frizzell, 2003). Although more than 1,000 mutations have been identified in the CFTR gene, ΔF508 accounts for nearly 70% of CF alleles. In homozygous individuals, the ΔF508-CFTR mutation leads to severe forms of CF. CFTR contains 12 transmembrane domains and three cytosolic oriented domains (nucleotide binding domain 1 [NBD1], R, and NBD2) involved in channel gating. Current evidence suggests that deletion of Phe508 in NBD1 prevents proper folding and trafficking of CFTR from the ER to the plasma membrane (Kopito, 1999; Riordan, 1999; Bertrand and Frizzell, 2003). ΔF508-CFTR is a temperature-sensitive mutant, as transfer to the permissive temperature (27–30°C) results in partial export from the ER (Denning et al., 1992; French et al., 1996). Misfolded ΔF508-CFTR that fails to exit the ER is eliminated by the ER-associated degradation (ERAD) pathway (Xiong et al., 1999; Gelman et al., 2002; Lenk et al., 2002).
Export from the ER now appears to involve specific exit codes that couple cargo to a common cytosolic budding machinery (Barlowe, 2003). A conserved di-acidic exit code found in the cytosolic tail of many type I transmembrane proteins was first identified in mammalian cells (Nishimura and Balch, 1997; Nishimura et al., 1999; Sevier et al., 2000) and subsequently in yeast (Votsmeier and Gallwitz, 2001; Malkus et al., 2002). The di-acidic code directs selective (Balch et al., 1994) and efficient (Nishimura and Balch, 1997; Nishimura et al., 1999) ER export by promoting interaction of cargo with the coat complex II (COPII) budding machinery. COPII (for review see Antonny and Schekman, 2001) consists of the Sar1 GTPase, the cargo selection protein complex Sec23/24 that is a Sar1-specific guanine nucleotide activating protein (GAP; Aridor et al., 1998; Miller et al., 2002), and a coat polymer assembly factor, Sec13/31 (Pryer et al., 1993; Antonny et al., 2001, 2003). The Sar1 GTPase is recruited and activated by the ER-localized transmembrane protein Sec12, a guanine nucleotide exchange factor specific for Sar1 (Barlowe and Schekman, 1993; Weissman et al., 2001). Sar1 activation is essential for recruitment of the Sec23/24 complex to the ER membrane to select cargo and to initiate COPII coat assembly.
Assembly and disassembly of the COPII coat can be rigorously controlled in vivo and in vitro by use of biochemically characterized Sar1 mutants. Sar1[T39N] (referred to as Sar1-GDP) is restricted to the GDP-bound state and biochemically functions as a competitive inhibitor of wild-type Sar1 recruitment, thereby preventing Sec23/24 attachment to the ER membrane and coat assembly (Kuge et al., 1994; Weissman et al., 2001). Sar1[H79G] (referred to as Sar1-GTP) has a reduced rate of GAP-stimulated hydrolysis, remaining in the GTP-bound state after activation. Although efficiently recruited to the ER membrane where it promotes cargo recruitment and the assembly of coat polymers, its inability to undergo hydrolysis interferes with subsequent COPII coat disassembly, resulting in inhibition of ER to Golgi transport (Kuge et al., 1994; Aridor et al., 1998, 2001; Huang et al., 2001). In addition, we have demonstrated that the protein kinase inhibitor H89 blocks recruitment of COPII coat components and prevents vesicle formation from the ER (Aridor and Balch, 2000). These probes provide powerful approaches to dissect the role of COPII function in transport of cargo through the secretory pathway.
To develop a therapeutic means to stimulate ΔF508-CFTR trafficking from the ER to the cell surface, it is necessary to understand the mechanisms and pathways that regulate its export from the ER. We have previously shown that CFTR exits the ER in a COPII-dependent fashion (Yoo et al., 2002). A recent paper (Fu and Sztul, 2003) proposed that COPII also regulates the delivery of wild-type CFTR to the ERAD pathway in the yeast Saccharomyces cerevisiae (Xiong et al., 1999; Gelman et al., 2002; Lenk et al., 2002). We have now examined in detail the contribution of COPII to the export of CFTR to the Golgi and its potential contribution to the ERAD pathway in mammalian cells. Our results demonstrate that although COPII is essential for export of wild-type CFTR from the ER, we found no evidence that COPII contributes to the targeting of either wild-type CFTR or the ΔF508-CFTR mutant to ERAD pathway. Intriguingly, we report that export of CFTR is highly sensitive to mutation of a highly conserved, consensus di-acidic exit code (Nishimura and Balch, 1997) found in the NBD1 domain of CFTR. We find that coupling to the Sec23/24 cargo selection complex through the di-acidic code is critical for diverting CFTR from the ERAD pathway. We propose that the di-acidic exit code plays a key role in linking CFTR to the COPII coat machinery and is the primary defect leading to disease in ΔF508-expressing patients.
Results
Degradation of wild-type CFTR is COPII independent
To follow export of CFTR from the ER, baby hamster kidney (BHK) cells were transfected with CFTR using a vaccinia-transient expression system, pulse-labeled with [35S]methionine, and CFTR transport to the Golgi detected by processing of the band B N-linked core-oligosaccharide form to the band C complex form by Golgi-associated glycosyltransferases that has reduced migration on SDS-PAGE (Yoo et al., 2002; Fig. 1 A, inset, CFTR mock). In most tissue culture cell lines engineered to express CFTR, wild-type CFTR is inefficiently exported, resulting in processing of only 20–30% of the ER-associated band B to the Golgi-and post-Golgi mature band C form (Fig. 1 A, inset and bottom left panel; Riordan, 1999). CFTR failing to exit the ER is degraded by ERAD indicated by the loss of band B (Fig. 1 A, inset and top panel; Xiong et al., 1999; Gelman et al., 2002; Lenk et al., 2002).
Figure 1.

Export of CFTR from the ER is COPII dependent. BHK cells were transfected with pcDNA3.1 plasmids containing either wild-type CFTR or ΔF508-CFTR or cotransfected with pcDNA3.1 containing the indicated Sar1 mutants as described previously (Yoo et al., 2002). After a 30-min pulse with [35S]Met, CFTR was immunoprecipitated at the indicated chase time and quantitated using a Molecular Dynamics PhosphoImager as described previously (Yoo et al., 2002). Expression levels of Sar1 mutants (∼5–10-fold endogenous) were monitored by immunoblotting with specific antibody as described previously (Yoo et al., 2002; not depicted). (A and B, top panels) Quantitation of CFTR in band B (ER form) reported as a percentage of total B plus C at the 0 time point. (A and B, bottom panels) Quantitation of the CFTR band C glycosylated form reported as a percentage of total B plus C at the 0 time point. Insets in A and B show representative autoradiographs containing band B and C (arrows). In A (top right panel), a significant increase in amount of band B remaining in six independent experiments for cells transfected with Sar1-GTP compared with Sar1-GDP at 3 h is shown (P < 0.05). The error bars represent SEM. The dashed horizontal line indicates the level of band B remaining in the wild-type (mock) control. In A (bottom right panel), the C/B ratios are shown for the 3-h time point. Results in A and B are typical of at least three independent experiments.
We have previously reported (Yoo et al., 2002) that Golgi processing of band B to band C is inhibited by cotransfection with the Sar1-GTP mutant when overexpressed ∼5–10-fold, demonstrating that CFTR exits the ER by a COPII-dependent pathway (Fig. 1 A, inset and bottom panels). Inhibition is clearly evident in the ratio of band C to band B (C/B ratio). When compared with the mock control C/B value of ∼1.8 at the 3-h time point, the C/B ratio in the presence of Sar1-GTP was <0.2 (Fig. 1 A, bottom right panel). To pursue the possibility that COPII is required for ER export but not ERAD, we examined the effect of the Sar1-GDP mutant that is restricted to the GDP-bound form on CFTR glycosylation or proteolysis. As Sar1-GDP is a competitive inhibitor of recruitment of wild-type Sar1 to the Sec12 guanine nucleotide exchange factor (Weissman et al., 2001) and Sar1 activation is essential for the recruitment of the cargo selection Sec23/24 complex to the ER membrane (Kuge et al., 1994; Weissman et al., 2001), Sar1-GDP competes with wild-type Sar1 to competitively reduce the efficiency of both COPII recruitment and coat assembly. Although cotransfection of wild-type CFTR with Sar1-GDP (∼5–10-fold overexpression; Kuge et al., 1994; Yoo et al., 2002) strongly inhibited export as measured by processing of band B to band C (Fig. 1 A, inset and bottom panels, C/B ratio is <0.7), wild-type CFTR continued to be degraded efficiently in the presence of the mutant (Fig. 1 A, inset and top panels). Identical results were observed at both early and late time points of transfection (unpublished data), indicating that the potential accumulation of other cargo in the ER in the presence of Sar1-GDP is unlikely to influence the effect of the Sar1-GDP on CFTR trafficking and degradation. In contrast to Sar1-GDP, Sar1-GTP is a dominant-negative mutant that promotes cargo collection into stable COPII-coated intermediates. As a consequence, targeting and fusion of these intermediates to the Golgi is blocked (Kuge et al., 1994; Aridor et al., 1998, 2001; Huang et al., 2001). Interestingly, cotransfection of wild-type CFTR with Sar1-GTP significantly (P < 0.05) protected immature (band B) wild-type CFTR from degradation by the ERAD pathway when compared with degradation rate observed in the presence of the Sar1-GDP mutant (Fig. 1 A, inset and top panels). This result raises the possibility that recruitment of CFTR to the assembling COPII polymer at ER exit sites prevents access to ERAD.
We also examined the effects of both Sar1-GDP– and Sar1-GTP–restricted mutants on ΔF508-CFTR stability given that this protein is not exported from the ER and therefore completely degraded by ERAD (Fig. 1 B). Cotransfection of ΔF508-CFTR with the Sar1-GTP–restricted mutant did not promote processing of band B to C (Fig. 1 B, inset and bottom panel), nor did it affect the kinetics of degradation by ERAD (Fig. 1 B, inset and top panel). An identical result was observed with the Sar1-GDP–restricted mutants (Fig. 1 B). These results are consistent with the idea that failure to export from the ER reflects the inability of ΔF508 to link to COPII.
To provide additional evidence for the role of Sar1 in selection of wild-type CFTR for export, we took advantage of the protein kinase inhibitor H89 that we have previously shown to inhibit Sar1 recruitment and COPII coat assembly (Aridor and Balch, 2000). Addition of H89 to cells stably expressing CFTR strongly reduced export (90%; Fig. 2). ERAD continues efficiently, suggesting that COPII recruitment is not required for targeting for degradation as H89, like Sar1-GDP, uncouples cargo from the COPII coat machinery (Aridor and Balch, 2000).
Figure 2.
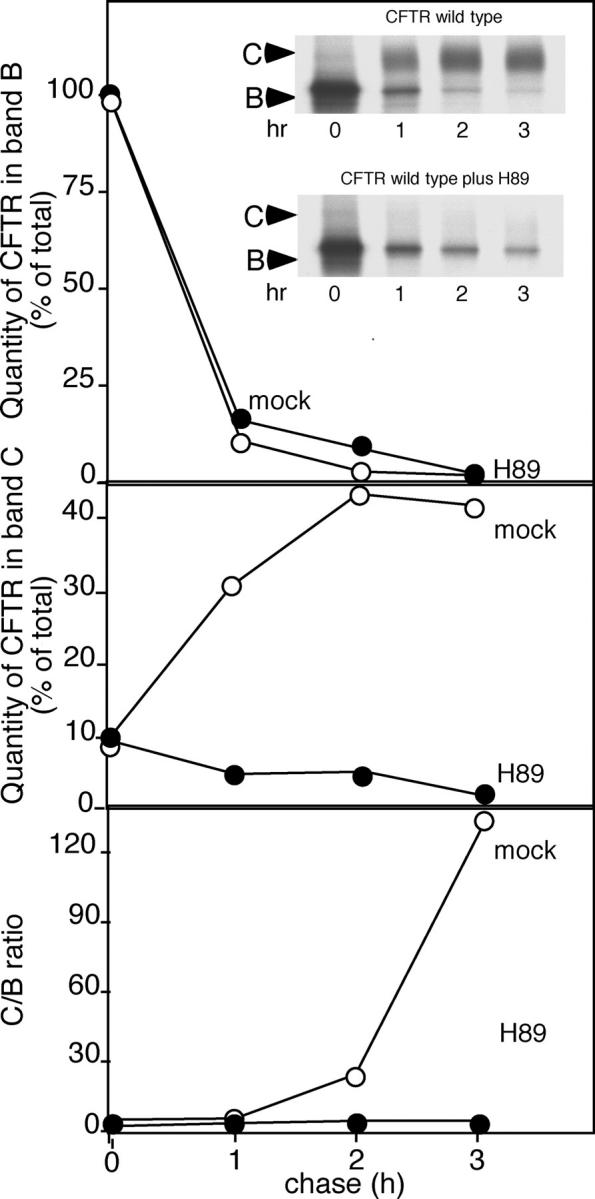
Export of CFTR from the ER but not degradation is inhibited by the kinase inhibitor H89. BHK cells stably expressing either wild-type CFTR or ΔF508-CFTR were labeled with [35S]Met for 30 min as described previously (Yoo et al., 2002). After labeling, cells were chased for the indicated time in the absence or presence of 50 μM H89, and processing of CFTR from the band B ER form to mature band C was quantitated as described previously (Yoo et al., 2002). Results are typical of two independent experiments.
We conclude that conditions that enhance COPII assembly on the ER membrane stabilize wild-type CFTR from ERAD. In contrast, conditions that interfere with COPII recruitment prevent the export of wild-type CFTR, but have little effect on delivery of either wild-type CFTR or ΔF508-CFTR to the ERAD pathway. Thus, the ERAD pathway functions largely independently of the COPII machinery.
Export, but not ERAD, is reversibly sensitive to a novel Sar1 temperature-sensitive (Sar1ts) mutant
To ensure that the aforementioned results with the Sar1 mutants were not a consequence of targeting of either wild-type or mutant CFTR to a nonphysiological pathway, or reflect indirect effects of other cargo in the ER on CFTR export or stability, we developed a temperature-sensitive variant of Sar1. This mutant is dominant-negative at the permissive temperature (32°C), where it is folded properly. It loses this dominant-negative activity when transferred to the restrictive temperature (40°C), where it misfolds. When cotransfected with the type 1 transmembrane protein vesicular stomatitis virus glycoprotein (VSV-G) at 32°C, Sar1ts shows strong inhibition of processing of VSV-G from the endoglycosidase H (endo H)–sensitive ER form to the endo H–resistant Golgi form (Fig. 3 A), indicative of an ER to Golgi transport block. In contrast, at 40°C, a condition that promotes destabilization of the mutant protein, inhibition of export is lost (Fig. 3 A). Thus, Sar1ts shows a thermoreversible temperature-sensitive ER export block.
Figure 3.
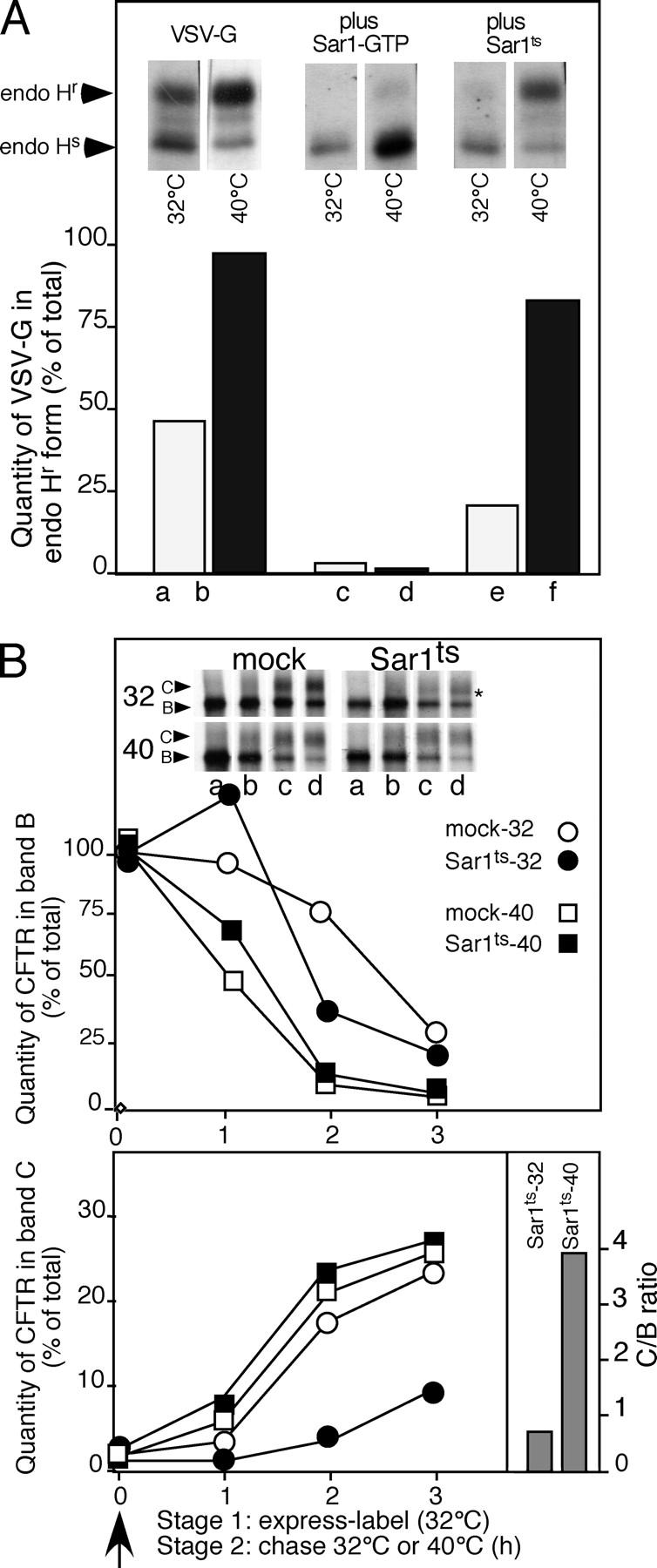
ER export is reversibly sensitive to a novel Sar1 temperature-sensitive (Sar1 ts ) mutant. (A) BHK cells were cotransfected with pcDNA3.1 containing either VSV-G or VSV-G and the indicated Sar1 mutant. The effect of Sar1-GTP (control) or Sar1ts on processing of VSV-G from the endoglycosidase H–sensitive (endo Hs) ER form to the endo H–resistant (endo Hr) Golgi form after a 5-min pulse with [35S]Met at 32°C followed by a 30 min chase at the indicated temperature was determined as described previously (Yoo et al., 2002). (B) BHK cells were cotransfected with pcDNA3.1 containing wild-type CFTR either with vector only (mock) or, where indicated, cotransfected with pcDNA3.1 containing the Sar1ts mutant. The transport and processing of CFTR from band B to C was followed at the indicated temperature as described in Fig. 1. The C/B ratios for the Sar1ts mutant at 32 and 40°C are shown for the 3-h time point. The asterisk in the top panel of B indicates that CFTR is exported very inefficiently from the ER as band C is only weakly processed to early Golgi oligosaccharide forms. White lines indicate that intervening lanes have been spliced out. Results are typical of three independent experiments.
Transient coexpression of Sar1ts and wild-type CFTR at 32°C significantly inhibited the export of wild-type CFTR when compared with the mock control as measured by processing from the band B to the band C forms (Fig. 3 B, bottom right panel, C/B ratio is ∼0.5 at 3 h). Sar1ts had no apparent effect on stabilizing wild-type CFTR from degradation at 32°C (Fig. 3 B, top panel), which is consistent with the interpretation that Sar1ts interferes with COPII coat assembly but does not affect ERAD pathways. Moreover, cotransfection of ΔF508-CFTR with Sar1ts had no effect on either ΔF508-CFTR export or degradation when compared with the controls expressing ΔF508 alone at either 32 or 40°C temperatures (unpublished data). Importantly, we used a two-stage incubation protocol to examine the ability of CFTR to be exported from the ER after accumulation in the presence of the Sar1ts mutant at 32°C. Cells transfected for 14 h at 32°C to prevent CFTR export were labeled for 30 min at 32°C (stage 1). Subsequently, cells were transferred to 40°C (stage 2) to inactivate Sar1ts. In contrast to incubation at 32°C (Fig. 3 B, bottom right panel, C/B ratio is ∼0.5 at 3 h), upon transfer to 40°C we observed normal processing of wild-type CFTR from band B to C (Fig. 3 B, bottom right panel, C/B ratio is >4 at 3 h). Thus, reversible inhibition of processing of wild-type CFTR to band C imposed by incubation at 32°C in the presence of Sar1ts interferes with the normal physiological pathway required for COPII-dependent export but not degradation.
CFTR uses a di-acidic exit code for export from the ER
To understand the requirement for COPII in the export of CFTR from the ER, we examined the CFTR sequence for the presence of the evolutionarily conserved tyrosine-based di-acidic code required for export of VSV-G (Nishimura and Balch, 1997; Nishimura et al., 1999; Sevier et al., 2000) and other transmembrane proteins from the ER in mammalian cells (Bannykh and Balch, 1998; Bannykh et al., 1998) and yeast (Votsmeier and Gallwitz, 2001; Malkus et al., 2002). Notably, CFTR contained a YKDAD motif (residues 563 to 567) in the first nucleotide binding motif (NBD1) that is evolutionarily conserved and similar to that found in VSV-G (YxDxE; Nishimura and Balch, 1997; Sevier et al., 2000). The structure of the NBD1 domain of CFTR (1ROZ.pdb) reveals that the YKDAD motif is located in a surface-exposed loop linking the NH2-terminal helical domain containing Phe508 to the COOH-terminal sheet domain (Lewis et al., 2004; Fig. 4 A). Strikingly, transient expression of CFTR mutants in which either one (DAA-CFTR; Fig. 4 B, inset and bottom panel) or both (AAA-CFTR; unpublished data) conserved Asp residues were mutated to Ala resulted in complete inhibition of export from the ER as measured by the inability of band B to be processed to band C and a morphological distribution consistent with localization to the ER (Bannykh et al., 2000; unpublished data). Like ΔF508-CFTR, cotransfection with Sar1-GTP failed to promote export or stabilize DAA-CFTR from ERAD, suggesting that mutation of the di-acidic motif uncoupled CFTR from COPII (Fig. 4 B). We also examined whether or not, like ΔF508-CFTR, the DAA-CFTR phenotype is temperature-sensitive by examining its rate of export at the lowered temperature (30°C). Whereas transfer of ΔF508-CFTR to 30°C resulted in significant enhancement of transport to the cell surface reflecting its effect on conformational stability of NBD1 as observed previously (Denning et al., 1992; French et al., 1996), we did not observe a similar increase in DAA-CFTR export at 30°C (unpublished data). These results suggest that the DAA mutation does not affect the fold of NBD1 in a similar fashion to the ΔF508 mutation that can be stabilized by incubation at reduced temperature.
Figure 4.
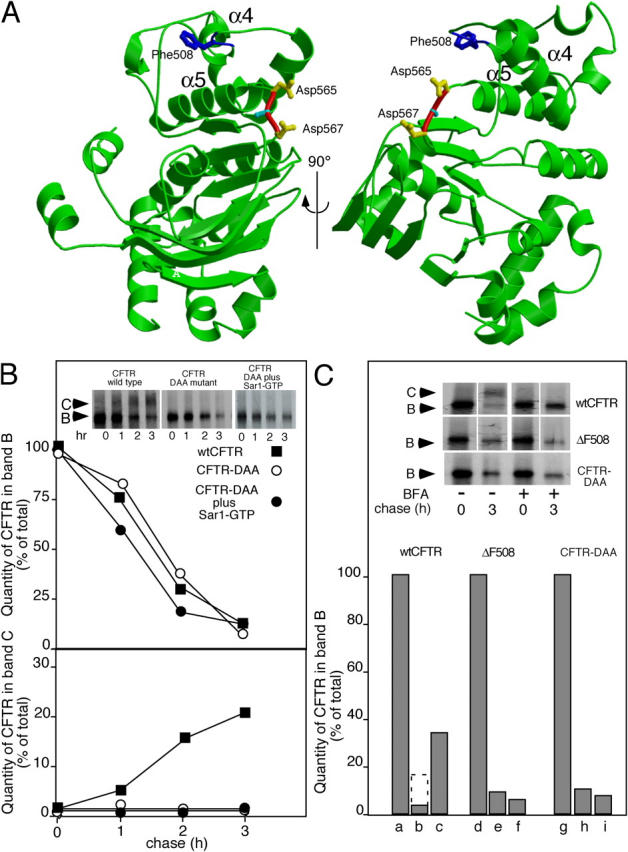
CFTR uses a di-acidic code for export from the ER. (A) Location of Phe508 and the di-acidic code in the NBD1 domain of CFTR (Lewis et al., 2004; 1ROX.pdb). Shown in blue is Phe508; shown in red (carbon backbone) and yellow (side-chains) is the orientation of residues Asp565 and Asp567 (di-acidic code). (B) Cells were either transfected or cotransfected with the indicated wild-type or mutant CFTR and the Sar1-GTP mutant as indicated. Processing from band B to band C was quantitated as described in Fig. 1. (C) Effect of BFA on stability of wild-type CFTR, ΔF508-CFTR, or the DAA-CFTR mutant in the ER. White lines indicate that intervening lanes have been spliced out. Cells were pulse-labeled for 30 min with [35S]Met at 37°C. 10 μg/ml BFA was added and incubation continued for 3 h at 37°C. (lanes a, d, and g) Total CFTR found in band B after pulse (expressed as a value of 100%). (lanes b, e, and h) Fraction of CFTR found in band B after 3-h incubation in the absence of BFA. (lanes c, f, and i) Fraction of CFTR found in band B after 3-h incubation in the presence of BFA. The dashed bar in lane b indicates the total B + C at the 3-h time point in the absence of BFA. Results are typical of at least two independent experiments for each condition shown.
To further characterize the biochemical properties of the DAA-CFTR mutant, we took advantage of the previous observation that treatment of CFTR-expressing cells with brefeldin A (BFA) reduces degradation of wild-type CFTR, but not ΔF508-CFTR (Ostedgaard et al., 1999). BFA triggers the collapse of the Golgi to the ER by preventing the recruitment of coat complex I to Golgi membranes (for review see Chardin and McCormick, 1999). In contrast to its potent effect on coat complex I function in the Golgi, BFA has no effect on COPII function during ER export. As a consequence, in BFA-treated cells ER-associated cargo is collected in export intermediates that accumulate in the absence of functional Golgi compartments (Lippincott-Schwartz et al., 1989). As expected, addition of BFA prevented processing of wild-type CFTR from band B to C and partially stabilized wild-type CFTR from ERAD, likely reflecting recruitment to COPII intermediates (Fig. 4 C; Lukacs et al., 1994; Ward and Kopito, 1994). In contrast, BFA did not prevent degradation of either the ΔF508- or the DAA-CFTR mutant (Fig. 4 C). These results are consistent with the interpretation that both mutants fail to couple to the COPII machinery (Nishimura and Balch, 1997; Nishimura et al., 1999), resulting in their delivery to the ERAD pathway.
The biochemical interaction of CFTR with COPII is disrupted by the ΔF508 and DAA mutations
Although the results described in the previous section raise the possibility that CFTR may exit the ER through the di-acidic code, it remained to be shown that wild-type CFTR can engage the COPII machinery and whether interaction with COPII can be disrupted by the ΔF508- or DAA-CFTR mutations. For this purpose, we first examined the ability of CFTR to bind the Sec23/Sec24 cargo selection complex in immunoprecipitates recovered from detergent lysates of HEK293 cell lines stably expressing either wild-type CFTR or ΔF508-CFTR. As shown in Fig. 5 A, in cells expressing wild-type CFTR, the protein is largely in the band C mature form with <5% in the band B ER form, an expected result given that wild-type CFTR accumulates in post-Golgi compartments. In contrast, in ΔF508-CFTR–expressing HEK293 cells, the only form is band B reflecting its ER localization, its level of expression reflecting continuous degradation by ERAD (Fig. 5 A). Despite the large difference in the steady-state level of CFTR in wild-type and ΔF508-expressing cells, when we quantitated the recovery of Sec24 with respect to the total amount of wild-type CFTR or ΔF508-CFTR in band B using immunoblotting, we observed that ΔF508-CFTR bound <25% of that observed for wild-type CFTR (Fig. 5 A). Sec24 was not recovered from HEK293 cells that do not express CFTR (unpublished data). Because immunoprecipitations were performed on ice, it is likely that the temperature-sensitive phenotype of ΔF508-CFTR resulted in partial folding after transfer to ice, accounting for the observed level of recovery of Sec24. Although the band B ER form would be expected to be the only form of CFTR that would bind the ER-specific COPII coat machinery, we examined whether or not immunoprecipitation of Sec24 would recover band C, the cell surface form. Despite the large excess of band C in the cell (Fig. 5 A), immunoprecipitation of Sec24 followed by immunoblotting for CFTR only recovered the band B forms of either wild type or ΔF508 (Fig. 5 B).
Figure 5.

Wild-type CFTR but not DAA-CFTR binds COPII. (A) HEK293 stably expressing wild type or ΔF508 were lysed in the presence of 1% Triton X-100, and Sec23/Sec24 complex recovered by immunoprecipitation with a CFTR-specific mAb was quantitated as described in Materials and methods. CFTR (bands B and C) and Sec24 were detected by immunoblotting with specific antibody as described in Materials and methods. (B) As in A except the amount CFTR recovered by immunoprecipitation with a Sec24-specific polyclonal antibody was quantitated as described in Materials and methods using immunoblotting. HEK293 cells not expressing CFTR were used as a control. CFTR (bands B and C) were detected by immunoblotting with specific antibody. (C) HEK293 cells were transfected with pcDNA3.1 plasmids containing either wild-type CFTR or DAA-CFTR as described previously (Yoo et al., 2002). Cells were lysed in the presence of 1% Triton X-100, and the amount of Sec23/Sec24 complex recovered bound to band B of CFTR (expressed as a Sec24/band B ratio) was quantitated as described in Materials and methods. CFTR (bands B and C) and Sec24 were detected by immunoblotting with specific antibodies. (D) HEK293 stably expressing wild type or ΔF508 incubated at 37°C (wild type) or for the indicated time at 30°C (ΔF508) were lysed in the presence of 1% Triton X-100, and the amount of Sec23/Sec24 complex recovered bound to CFTR by immunoprecipitation with a CFTR-specific antibody was quantitated as described in Materials and methods. Error bars indicates the SD for samples generated for each time point in triplicate.
To test the ability of the DAA-CFTR mutant to bind the Sec23/Sec24, we transfected HEK293 cells with wild-type CFTR or DAA-CFTR using the vaccinia expression system. Strikingly, we observed that band B of the DAA-CFTR mutant bound <5% of that recovered in immunoprecipitates with wild-type CFTR (Fig. 5 B). These results support the conclusion that the marked inefficiency of DAA-CFTR to exit the ER is a consequence of its inability to couple to the cargo selection complex Sec23/Sec24.
To correlate the effects of the di-acidic code DAA-CFTR mutant on COPII binding with the effects of Phe508 on the ability of the ΔF508-CFTR to exit the ER after transfer to 30°C, we performed a temperature-shift experiment. As shown in Fig. 5 D, with increasing incubation time at 30°C, ΔF508 showed a strong increase in its interaction with Sec24, approaching the value observed for wild-type CFTR at 37°C at steady-state. These results suggest that reduced temperature promotes maturation of CFTR and presentation of the exit code for interaction with the COPII coat machinery for export from the ER.
Discussion
COPII-dependent export of CFTR
Using Sar1 mutants with well-characterized biochemical properties, we found that export of wild-type CFTR is dependent on COPII coat assembly in mammalian cells. This would be the expected result if wild-type CFTR, but not ΔF508-CFTR, acquires the normal protein fold necessary for cargo selection by COPII. Several lines of evidence support this conclusion. First, the Sar1-GTP mutant that stabilizes coat assembly markedly reduces the rate of ERAD through capture into COPII transport intermediates that accumulate in the presence of activated Sar1 (Aridor et al., 1995, 1998, 2001). Second, the Sar1-GDP mutant (that prevents COPII coat recruitment) inhibited export of wild-type CFTR, but did not stabilize the ER band B form. It is unlikely that the effects of the GDP mutant on CFTR export were influenced by build-up of other cargo in the ER during the transient expression protocol as we observed inhibition of transport at the earliest time points of transfection when the inhibitory properties of the Sar1-GDP mutant begin to take effect. This conclusion is supported by the ability of H89 to rapidly and efficiently uncouple CFTR from export in cells stably expressing wild-type CFTR. Furthermore, in the two-stage assay using the Sar1ts mutant, we found that accumulation of CFTR in the ER at 32°C had no effect on export after inactivation of Sar1ts at 40°C. Because we did not detect significant effects of Sar1-GDP (which uncouples CFTR from COPII) on degradation of CFTR, we suggest that the export and ERAD pathways operate independently.
COPII does not participate in ERAD
A recent paper (Fu and Sztul, 2003) suggested that targeting of CFTR to the ERAD pathway in S. cerevisiae requires COPII because in the absence of COPII function CFTR accumulates in an ER-like subcompartment rather than being degraded (Kiser et al., 2001; Fu and Sztul, 2003; Sullivan et al., 2003). In contrast, we found that COPII does not participate in ERAD of either wild-type CFTR or ΔF508-CFTR in mammalian cells. The reasons for this discrepancy are now apparent. First, CFTR will not exit the ER in yeast under any condition, limiting interpretation of the results (Kiser et al., 2001; Fu and Sztul, 2003; Sullivan et al., 2003). Second, all experiments used overexpression of a GFP-tagged variant of CFTR (GFP-CFTR). A similar GFP-CFTR construct, when expressed in mammalian cells, has a markedly reduced rate of export and accumulates in ER tubular elements at 30°C (comparable to experiments in yeast where cells are incubated at 24°C) that significantly overlaps with calnexin (Fig. S1, available at http://www.jcb.org/cgi/content/full/jcb.200401035/DC1). This overlap is consistent with the fact that folding of nascent CFTR in the ER is likely to involve calnexin (Pind et al., 1994). However, upon transfer to 37°C, GFP-CFTR is efficiently exported to the cell surface of mammalian cells (Fig. S1). Thus, in mammalian cells, GFP-CFTR can accumulate transiently in a bona fide ER-like tubular network at reduced temperature that is unlikely to be a specialized COPII-regulated degradative subcompartment as suggested for yeast where export is blocked at all temperatures (Fu and Sztul, 2003; Sullivan et al., 2003).
A third concern is that all experiments relied on temperature-sensitive COPII mutants to prevent degradation, leading to the interpretation that COPII is required for ERAD. These experiments required prolonged incubation of yeast at the restrictive temperature (40°C) to assess the effect of the mutant COPII component on ERAD. It is well-established that elevated temperature (40°) in mammalian cells markedly stabilizes (approximately twofold) both wild-type CFTR and ΔF508-CFTR from ERAD in response to induction of heat shock folding chaperones (Strickland et al., 1997; Loo et al., 1998; Meacham et al., 1999; Choo-Kang and Zeitlin, 2001; Farinha et al., 2002; unpublished data). Thus, the twofold effect of the temperature-sensitive Sec12 variant on preventing CFTR degradation in yeast can be largely explained by the twofold difference in stability observed upon shift from the permissive to the restrictive temperature in control incubations in the absence of the Sec12ts (Fu and Sztul, 2003).
Fourth, unlike the Sar1-GDP and -GTP mutants, the biochemical properties of the Sec13 and Sec23 temperature-sensitive mutants have not been characterized. For example, both Sec13 and Sec23 temperature-sensitive variants may not be inactivated by shifting to the restrictive temperature as suggested (Fu and Sztul, 2003). Rather they may promote stable coat assembly leading to partial segregation of CFTR from ERAD as we have observed during COPII coat polymer assembly promoted by the Sar1-GTP mutant. We conclude that role of COPII is to direct CFTR selection into COPII vesicles, removing the protein from the ERAD COPII-independent targeted pool.
A di-acidic code mediates CFTR export
An important finding that provides new insight into the role of COPII in export of CFTR from the ER was that processing of band B to C was sensitive to mutation of the conserved di-acidic code exit motif originally detected in the cytoplasmic tail of VSV-G (Nishimura and Balch, 1997; Nishimura et al., 1999). Consistent with this finding, a natural variant in the cystic fibrosis mutation database (http://www.genet.sickkids.on.ca/cftr/) has a substitution of G for D at position 565 (565DAD to 565GAD) that results in a severe deficiency phenotype, although this allele is complex (Tzetis et al., 2001). The requirement for a di-acidic code in CFTR export is consistent with a similar requirement for a di-acidic motif for export of the multi-membrane-spanning K+ channel (Ma et al., 2001; Ma and Jan, 2002) and yeast GAP permease (Malkus et al., 2002) from the ER. Given that transfer of the di-acidic code to an ER-retained protein is sufficient to promote efficient export (Nishimura and Balch, 1997; Nishimura et al., 1999) and the fact that the di-acidic code recognizes the Sec24 subunit of the Sec23/24 complex (Miller et al., 2002), it is now evident that the di-acidic motif may be used by CFTR to engage the Sec24. Our CFTR coimmunoprecipitation results provide direct evidence that the DAD motif in NBD1 of CFTR serves as a functional ER exit code as loss of the code results in impaired binding to Sec23/24. In the case of the GAP permease, it has been suggested that the di-acidic code, although necessary for export, is not sufficient ((Miller et al., 2002). A similar constraint may apply to CFTR as the protein has a complicated cytosolic folding pathway involving numerous chaperones and cochaperones (Pind et al., 1994; Loo et al., 1998; Brodsky, 2001; Zhang et al., 2002) that may facilitate recognition of the Sec23/Sec24 complex (unpublished data).
NBD1 domains of a large variety of ABC transporters have a similar structural organization (Schmitt and Tampe, 2002). Structural alignment of the NBD1 domain of CFTR (Lewis et al., 2004) with the NBD1 domain of the ABC transporter BtuCD reveals that the di-acidic code is found in a solvent-exposed loop connecting the NH2-terminal helical domain containing Phe508 with the more COOH-terminal sheet domain (Fig. 4 A and Fig. 6). Given that the structure of Sar1 and the Sec23/24 complex containing a bound di-acidic peptide are available (Huang et al., 2001; Bi et al., 2002; Mossessova et al., 2003), we built a homology model with the NBD1 of CFTR replacing the similar structural fold of NBD1 of BtuCD (Fig. 6). This new structure (BtuCD-NBD1CFTR) was then combined with the structure of the Sec23/24 -Sar1 complex to illustrate the ability of the di-acidic code loop in NBD1CFTR to insert directly into the Sec23/24 di-acidic code binding pocket (Fig. 6, yellow residues; and Fig. S2 for a three-dimensional view, available at http://www.jcb.org/cgi/content/full/jcb.200401035/DC1). Docking reveals little steric interference between residues within the binding pocket, or between residues defining the tertiary structure of BtuCD-NBD1CFTR and the Sar1/Sec24/23 complex. Moreover, the orientation of the NH2 terminus of Sar1 (Fig. 6, red β-strand; residues 25–34), which is linked to the flexible NH2-terminal tail involved in anchoring Sar1 to the membrane (Fig. 6, black dashed line; not observed in the crystal structure; Huang et al., 2001; Bi et al., 2002), is oriented correctly to facilitate recruitment of Sec23/24 by activated Sar1. It is apparent that Sar1 and BtuCD-NBD1CFTR interact with Sec23/Sec24 independently as shown for the holo cargo-selection complex. Thus, the general structural features of the CFTR-NBD1 domain presented within the structure of BtuCD are consistent with the role of the di-acidic code in directing interaction with COPII for exit from the ER.
Figure 6.

CFTR-COPII cargo selection complex. (bottom) The structure of CFTR-NBD1 (1ROX.pdb) is oriented to show its structural homology with the NBD1 structural domain of the bacterial ABC transporter BtuCD (1L7V.pdb). In CFTR-NBD1, Phe508 is shown in blue and residues Asp565 and Asp567 (di-acidic code) are shown in yellow. In BtuCD, the cytosolic domains are shown in green and putative transmembrane spanning domains are shown in red. The short loop shown in red in BtuCD corresponds structurally to the di-acidic code-containing loop in CFTR-NBD1. (middle) Homology structure containing CFTR-NBD1 and BtuCD (Btu-NBD1CFTR) docked to the Sar1/Sec23/24 complex (Bi et al., 2002; Mossessova et al., 2003; see Materials and methods). Gray rectangle illustrates the putative location of the membrane bilayer. (top) The interaction of the CFTR-NBD1 di-acidic code with the binding pocket of Sec23/24 (Bi et al., 2002; Mossessova et al., 2003). Shown in blue are Arg230, 235, 559, and 561 basic residues defining the di-acidic code pocket of Sec24 (Bi et al., 2002; Mossessova et al., 2003). Only a minor steric interference is observed between Asp565 (CFTR-NBD1) and Leu616 (Sec24) in the homology model. See Fig. S2 for a three-dimensional rendering of the interface between CFTR-NBD1 and the Sec23/24 cargo complex. Fig. S2 is available at http://www.jcb.org/cgi/content/full/jcb.200401035/DC1.
Given the likely importance of the aromatic side-chain of Phe508 in stabilizing the conserved structural fold of the NH2-terminal helical domain of NBD1 (Fig. 6, blue residue), deletion of Phe508 in the ΔF508-CFTR mutant may alter the orientation of the di-acidic motif-containing loop, thereby compromising its interaction with Sec24. This notion was directly tested by the CFTR coimmunoprecipitation experiments where we found that binding of band B of ΔF508 to Sec24 was much lower than that of wild-type CFTR. Consistent with this interpretation, ΔF508-CFTR-Sec24 interaction and transport to cell surface can be rescued by promoting folding at 30°C. Moreover, export of ΔF508-CFTR from the ER can also be rescued in cis by additional point mutations in NBD1 that stabilize the structural fold (DeCarvalho et al., 2002). Given that over 1,000 mutants have been reported to affect CFTR export and function leading to varying degrees of disease, it is not surprising that residues flanking the di-acidic code have also been reported in the mutation database. Structural considerations strongly suggest that many of these flanking mutations, unlike the di-acidic code that is found in the solvent-exposed loop, are likely to affect the fold of the α-helical and β-strands that contribute to the structural organization of NBD1 and indirectly compromise presentation of the code (unpublished data). We propose that the inability of the ΔF508-CFTR mutant to engage the COPII machinery through Sec24 is the primary basis for the most common form of CF.
Materials and methods
Materials
Reagents for vaccinia transient expression were prepared as described previously (Yoo et al., 2002). HEK293 stably expressing wild-type CFTR and ΔF508-CFTR were obtained from N. Bradbury (University of Pittsburgh School of Medicine, Pittsburgh, PA). The M3A7 antibody was provided by J. Riordan (Mayo Clinic, Scottsdale, AZ).
Vaccinia transient expression
Vaccinia transient expression and quantification was performed exactly as described previously (Yoo et al., 2002). SEM were determined using the software program Prism.
Immunoprecipitation of the CFTR-Sec24 complex
HEK293 cells stably expressing wild-type CFTR or ΔF508-CFTR, or vaccinia-transfected HEK293 cells transiently expressing wild-type CFTR and DAA-CFTR were lysed on ice in Triton X-100 lysis buffer (50 mM Tris-HCl, pH 7.4, 150 mM NaCl, and 1% Triton X-100) supplemented with complete protease inhibitor cocktail tablets (Roche Diagnostics GmbH). Soluble fraction of the lysates were incubated with protein G beads coupled with anti-CFTR antibody M3A7 or anti-Sec24 polyclonal antibodies. The immunocomplexes were washed and eluted with 50 mM Tris-HCl, pH 6.8, and 1% SDS. The protein components of the complexes were separated by SDS-PAGE. CFTR and Sec24 were detected by immunoblot. CFTR and Sec24 were quantitated by densitometry using a Molecular Dynamics Imaging System. In brief, for CFTR detected by immunoblotting, immunoblots exposed in the linear range were quantitated by measuring the densities of band B or C. A similar area immediately adjacent to bands B or C in each lane (background) were subtracted from the B or C value to obtain the indicated values shown in the Results.
Structural alignment of CFTR-NBD1, BtuCD, and the Sec23/24 complex
Fitting of CFTR-NBD1 to BtuCD and Sec23/24 was performed as follows. The Sec24/NBD1 complex model was built from the Sec24/DXE peptide complex (1PD1.pdb) and NBD1 (1R0X.pdb) coordinates by superimposing DXE peptide onto the corresponding part of NBD1. The Sec24/NBD1/Sec23 complex model was built from the Sec24/NBD1 model and Sec24/Sec23 complex (1M2V.pdb) by superimposing Sec24 in both coordinates. The Sec24/NBD1/Sec23/Sar1 complex model was built from Sec24/NBD1/Sec23 model and Sec23/Sar1 complex (1M2O.pdb) by superimposing Sec23 in both coordinates. The Sec24/NBD1/Sec23/Sar1/BtuCD complex model was built from the Sec24/NBD1/Sec23/Sar1 model and BtuCD (1L7V.pdb) by superimposing CFTR-NBD1 and BtuD in both coordinates. Fitting was performed using the McLachlan algorithm (McLachlan, 1982) as implemented in the program ProFit (http://www.bioinf.org.uk/software/).
Online supplemental material
Fig. S1 shows GFP-CFTR accumulates in the ER at 28°C. Fig. S2 is a three-dimensional rendering of CFTR-NBD1 di-acidic code docked to the Sar1/Sec23/24 cargo selection complex. Online supplemental material is available at http://www.jcb.org/cgi/content/full/jcb.200401035/DC1.
Acknowledgments
This work was supported by National Institutes of Health (NIH) grant GM42336 to W.E. Balch and DK 51870 to J. Riordan and W. Balch, an individual National Research Service Award NIH post-doctoral fellowship to B.D. Moyer, and a Cystic Fibrosis Foundation postdoctoral fellowship to X. Wang. The Scripps Research Institute manuscript number 1234-CB.
X. Wang and J. Matteson contributed equally to this paper.
Abbreviations used in this paper: BFA, brefeldin A; BHK, baby hamster kidney; CF, cystic fibrosis; CFTR, CF transmembrane conductance regulator; COPII, coat complex II; endo H, endoglycosidase H; ERAD, ER-associated degradation; GAP, guanine nucleotide activating protein; NBD1, nucleotide binding domain 1; VSV-G, vesicular stomatitis virus glycoprotein.
References
- Antonny, B., P. Gounon, R. Schekman, and L. Orci. 2003. Self-assembly of minimal COPII cages. EMBO Rep. 4:419–424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antonny, B., D. Madden, S. Hamamoto, L. Orci, and R. Schekman. 2001. Dynamics of the COPII coat with GTP and stable analogues. Nat. Cell Biol. 3:531–537. [DOI] [PubMed] [Google Scholar]
- Antonny, B., and R. Schekman. 2001. ER export: public transportation by the COPII coach. Curr. Opin. Cell Biol. 13:438–443. [DOI] [PubMed] [Google Scholar]
- Aridor, M., and W.E. Balch. 2000. Kinase signaling initiates COPII recruitment and export from the mammalian endoplasmic reticulum. J. Biol. Chem. 275:35673–35676. [DOI] [PubMed] [Google Scholar]
- Aridor, M., S.I. Bannykh, T. Rowe, and W.E. Balch. 1995. Sequential coupling between COPII and COPI vesicle coats in endoplasmic reticulum to Golgi transport. J. Cell Biol. 131:875–893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aridor, M., K.N. Fish, S. Bannykh, J. Weissman, T.H. Roberts, J. Lippincott-Schwartz, and W.E. Balch. 2001. The Sar1 GTPase coordinates biosynthetic cargo selection with endoplasmic reticulum export site assembly. J. Cell Biol. 152:213–229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aridor, M., J. Weissman, S. Bannykh, C. Nouffer, and W.E. Balch. 1998. Cargo selection by the COPII budding machinery during export from the endoplasmic reticulum. J. Cell Biol. 141:61–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balch, W.E., J.M. McCaffery, H. Plutner, and M.G. Farquhar. 1994. Vesicular stomatitis virus glycoprotein is sorted and concentrated during export from the endoplasmic reticulum. Cell. 76:841–852. [DOI] [PubMed] [Google Scholar]
- Bannykh, S.I., and W.E. Balch. 1998. Selective transport of cargo between the endoplasmic reticulum and Golgi compartments. Histochem. Cell Biol. 109:463–475. [DOI] [PubMed] [Google Scholar]
- Bannykh, S.I., G.I. Bannykh, K.N. Fish, B.D. Moyer, J.R. Riordan, and W.E. Balch. 2000. Traffic pattern of cystic fibrosis transmembrane regulator through the early exocytic pathway. Traffic. 1:852–870. [DOI] [PubMed] [Google Scholar]
- Bannykh, S.I., N. Nishimura, and W.E. Balch. 1998. Getting into the Golgi. Trends Cell Biol. 8:21–25. [DOI] [PubMed] [Google Scholar]
- Barlowe, C. 2003. Signals for COPII-dependent export from the ER: what's the ticket out? Trends Cell Biol. 13:295–300. [DOI] [PubMed] [Google Scholar]
- Barlowe, C., and R. Schekman. 1993. SEC12 encodes a guanine-nucleotide-exchange factor essential for transport vesicle budding from the ER. Nature. 365:347–349. [DOI] [PubMed] [Google Scholar]
- Bertrand, C.A., and R.A. Frizzell. 2003. The role of regulated CFTR trafficking in epithelial secretion. Am. J. Physiol. Cell Physiol. 285:C1–C18. [DOI] [PubMed] [Google Scholar]
- Bi, X., R.A. Corpina, and J. Goldberg. 2002. Structure of the Sec23/24-Sar1 pre-budding complex of the COPII vesicle coat. Nature. 419:271–277. [DOI] [PubMed] [Google Scholar]
- Brodsky, J.L. 2001. Chaperoning the maturation of the cystic fibrosis transmembrane conductance regulator. Am. J. Physiol. Lung Cell. Mol. Physiol. 281:L39–L42. [DOI] [PubMed] [Google Scholar]
- Chardin, P., and F. McCormick. 1999. Brefeldin A: the advantage of being uncompetitive. Cell. 97:153–155. [DOI] [PubMed] [Google Scholar]
- Choo-Kang, L.R., and P.L. Zeitlin. 2001. Induction of HSP70 promotes ΔF508 CFTR trafficking. Am. J. Physiol. Lung Cell. Mol. Physiol. 281:L58–L68. [DOI] [PubMed] [Google Scholar]
- DeCarvalho, A.C., L.J. Gansheroff, and J.L. Teem. 2002. Mutations in the nucleotide binding domain 1 signature motif region rescue processing and functional defects of cystic fibrosis transmembrane conductance regulator ΔF508. J. Biol. Chem. 277:35896–35905. [DOI] [PubMed] [Google Scholar]
- Denning, G.M., M.P. Anderson, J.F. Amara, J. Marshall, A.E. Smith, and M.J. Welsh. 1992. Processing of mutant cystic fibrosis transmembrane conductance regulator is temperature-sensitive. Nature. 358:761–764. [DOI] [PubMed] [Google Scholar]
- Farinha, C.M., P. Nogueira, F. Mendes, D. Penque, and M.D. Amaral. 2002. The human DnaJ homologue (Hdj)-1/heat-shock protein (Hsp) 40 co-chaperone is required for the in vivo stabilization of the cystic fibrosis transmembrane conductance regulator by Hsp70. Biochem. J. 366:797–806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- French, P.J., J.H. van Doorninck, R.H. Peters, E. Verbeek, N.A. Ameen, C.R. Marino, H.R. de Jonge, J. Bijman, and B.J. Scholte. 1996. A ΔF508 mutation in mouse cystic fibrosis transmembrane conductance regulator results in a temperature-sensitive processing defect in vivo. J. Clin. Invest. 98:1304–1312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu, L., and E. Sztul. 2003. Traffic-independent function of the Sar1p/COPII machinery in proteasomal sorting of the cystic fibrosis transmembrane conductance regulator. J. Cell Biol. 160:157–163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gelman, M.S., E.S. Kannegaard, and R.R. Kopito. 2002. A principal role for the proteasome in endoplasmic reticulum-associated degradation of misfolded intracellular cystic fibrosis transmembrane conductance regulator. J. Biol. Chem. 277:11709–11714. [DOI] [PubMed] [Google Scholar]
- Huang, M., J.T. Weissman, S. Beraud-Dufour, P. Luan, C. Wang, W. Chen, M. Aridor, I.A. Wilson, and W.E. Balch. 2001. Crystal structure of Sar1-GDP at 1.7 Å resolution and the role of the NH2 terminus in ER export. J. Cell Biol. 155:937–948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiser, G.L., M. Gentzsch, A.K. Kloser, E. Balzi, D.H. Wolf, A. Goffeau, and J.R. Riordan. 2001. Expression and degradation of the cystic fibrosis transmembrane conductance regulator in Saccharomyces cerevisiae. Arch. Biochem. Biophys. 390:195–205. [DOI] [PubMed] [Google Scholar]
- Kopito, R.R. 1999. Biosynthesis and degradation of CFTR. Physiol. Rev. 79:S167–S173. [DOI] [PubMed] [Google Scholar]
- Kuge, O., C. Dascher, L. Orci, T. Rowe, M. Amherdt, H. Plutner, M. Ravazzola, G. Tanigawa, J.E. Rothman, and W.E. Balch. 1994. Sar1 promotes vesicle budding from the endoplasmic reticulum but not Golgi compartments. J. Cell Biol. 125:51–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lenk, U., H. Yu, J. Walter, M.S. Gelman, E. Hartmann, R.R. Kopito, and T. Sommer. 2002. A role for mammalian Ubc6 homologues in ER-associated protein degradation. J. Cell Sci. 115:3007–3014. [DOI] [PubMed] [Google Scholar]
- Lewis, H.A., S.G. Buchanan, S.K. Burley, K. Conners, M. Dickey, M. Dorwart, R. Fowler, X. Gao, W.B. Guggino, W.A. Hendrickson, et al. 2004. Structure of nucleotide-binding domain 1 of the cystic fibrosis transmembrane conductance regulator. EMBO J. 23:282–293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lippincott-Schwartz, J., L.C. Yuan, J.S. Bonifacino, and R.D. Klausner. 1989. Rapid redistribution of Golgi proteins into the ER in cells treated with brefeldin A: evidence for membrane cycling from Golgi to ER. Cell. 56:801–813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loo, M.A., T.J. Jensen, L. Cui, Y. Hou, X.B. Chang, and J.R. Riordan. 1998. Perturbation of Hsp90 interaction with nascent CFTR prevents its maturation and accelerates its degradation by the proteasome. EMBO J. 17:6879–6887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lukacs, G.L., A. Mohamed, N. Kartner, X.B. Chang, J.R. Riordan, and S. Grinstein. 1994. Conformational maturation of CFTR but not its mutant counterpart (ΔF508) occurs in the endoplasmic reticulum and requires ATP. EMBO J. 13:6076–6086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma, D., and L.Y. Jan. 2002. ER transport signals and trafficking of potassium channels and receptors. Curr. Opin. Neurobiol. 12:287–292. [DOI] [PubMed] [Google Scholar]
- Ma, D., N. Zerangue, Y.F. Lin, A. Collins, M. Yu, Y.N. Jan, and L.Y. Jan. 2001. Role of ER export signals in controlling surface potassium channel numbers. Science. 291:316–319. [DOI] [PubMed] [Google Scholar]
- Malkus, P., F. Jiang, and R. Schekman. 2002. Concentrative sorting of secretory cargo proteins into COPII-coated vesicles. J. Cell Biol. 159:915–921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLachlan, A.D. 1982. Rapid comparison of protein structures. Acta Crystallogr. 38:871–873. [Google Scholar]
- Meacham, G.C., Z. Lu, S. King, E. Sorscher, A. Tousson, and D.M. Cyr. 1999. The Hdj-2/Hsc70 chaperone pair facilitates early steps in CFTR biogenesis. EMBO J. 18:1492–1505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller, E., B. Antonny, S. Hamamoto, and R. Schekman. 2002. Cargo selection into COPII vesicles is driven by the Sec24p subunit. EMBO J. 21:6105–6113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mossessova, E., L.C. Bickford, and J. Goldberg. 2003. SNARE selectivity of the COPII coat. Cell. 114:483–495. [DOI] [PubMed] [Google Scholar]
- Nishimura, N., and W.E. Balch. 1997. A di-acidic signal required for selective export from the endoplasmic reticulum. Science. 277:556–558. [DOI] [PubMed] [Google Scholar]
- Nishimura, N., S. Bannykh, S. Slabough, J. Matteson, Y. Altschuler, K. Hahn, and W.E. Balch. 1999. A di-acidic (DXE) code directs concentration of cargo during export from the endoplasmic reticulum. J. Biol. Chem. 274:15937–15946. [DOI] [PubMed] [Google Scholar]
- Ostedgaard, L.S., B. Zeiher, and M.J. Welsh. 1999. Processing of CFTR bearing the P574H mutation differs from wild-type and ΔF508-CFTR. J. Cell Sci. 112:2091–2098. [DOI] [PubMed] [Google Scholar]
- Pind, S., J.R. Riordan, and D.B. Williams. 1994. Participation of the endoplasmic reticulum chaperone calnexin (p88, IP90) in the biogenesis of the cystic fibrosis transmembrane conductance regulator. J. Biol. Chem. 269:12784–12788. [PubMed] [Google Scholar]
- Pryer, N.K., N.R. Salama, R. Schekman, and C.A. Kaiser. 1993. Cytosolic Sec13p complex is required for vesicle formation from the endoplasmic reticulum in vitro. J. Cell Biol. 120:865–875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riordan, J.R. 1999. Cystic fibrosis as a disease of misprocessing of the cystic fibrosis transmembrane conductance regulator glycoprotein. Am. J. Hum. Genet. 64:1499–1504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sevier, C.S., O.A. Weisz, M. Davis, and C.E. Machamer. 2000. Efficient export of the vesicular stomatitis virus G protein from the endoplasmic reticulum requires a signal in the cytoplasmic tail that includes both tyrosine-based and di-acidic motifs. Mol. Biol. Cell. 11:13–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmitt, K., and R. Tampe. 2002. Structure and mechanism of ABC transporters. Curr. Opin. Struct. Biol. 12:754–760. [DOI] [PubMed] [Google Scholar]
- Strickland, E., B.H. Qu, L. Millen, and P.J. Thomas. 1997. The molecular chaperone Hsc70 assists the in vitro folding of the N-terminal nucleotide-binding domain of the cystic fibrosis transmembrane conductance regulator. J. Biol. Chem. 272:25421–25424. [DOI] [PubMed] [Google Scholar]
- Sullivan, M.L., R.T. Youker, S.C. Watkins, and J.L. Brodsky. 2003. Localization of the BiP molecular chaperone with respect to endoplasmic reticulum foci containing the cystic fibrosis transmembrane conductance regulator in yeast. J. Histochem. Cytochem. 51:545–548. [DOI] [PubMed] [Google Scholar]
- Tzetis, M., A. Efthymiadou, S. Strofalis, P. Psychou, A. Dimakou, E. Pouliou, S. Doudounakis, and E. Kanavakis. 2001. CFTR gene mutations–including three novel nucleotide substitutions–and haplotype background in patients with asthma, disseminated bronchiectasis and chronic obstructive pulmonary disease. Hum. Genet. 108:216–221. [DOI] [PubMed] [Google Scholar]
- Votsmeier, C., and D. Gallwitz. 2001. An acidic sequence of a putative yeast Golgi membrane protein binds COPII and facilitates ER export. EMBO J. 20:6742–6750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ward, C.L., and R.R. Kopito. 1994. Intracellular turnover of cystic fibrosis transmembrane conductance regulator. Inefficient processing and rapid degradation of wild-type and mutant proteins. J. Biol. Chem. 269:25710–25718. [PubMed] [Google Scholar]
- Weissman, J.T., H. Plutner, and W.E. Balch. 2001. The mammalian guanine nucleotide exchange factor mSec12 is essential for activation of the Sar1 GTPase directing endoplasmic reticulum export. Traffic. 2:465–475. [DOI] [PubMed] [Google Scholar]
- Xiong, X., E. Chong, and W.R. Skach. 1999. Evidence that endoplasmic reticulum (ER)-associated degradation of cystic fibrosis transmembrane conductance regulator is linked to retrograde translocation from the ER membrane. J. Biol. Chem. 274:2616–2624. [DOI] [PubMed] [Google Scholar]
- Yoo, J.S., B.D. Moyer, S. Bannykh, H.M. Yoo, J.R. Riordan, and W.E. Balch. 2002. Non-conventional trafficking of the cystic fibrosis transmembrane conductance regulator through the early secretory pathway. J. Biol. Chem. 277:11401–11409. [DOI] [PubMed] [Google Scholar]
- Zhang, H., K.W. Peters, F. Sun, C.R. Marino, J. Lang, R.D. Burgoyne, and R.A. Frizzell. 2002. Cysteine string protein interacts with and modulates the maturation of the cystic fibrosis transmembrane conductance regulator. J. Biol. Chem. 277:28948–28958. [DOI] [PubMed] [Google Scholar]