

Abstract
Although human immunodeficiency virus type 1 (HIV-1) is generally thought to assemble at the plasma membrane of infected cells, virions have been observed in intracellular compartments in macrophages. Here, we investigated virus assembly in HIV-1–infected primary human monocyte-derived macrophages (MDM). Electron microscopy of cryosections showed virus particles, identified by their morphology and positive labeling with antibodies to the viral p17, p24, and envelope proteins, in intracellular vacuoles. Immunolabeling demonstrated that these compartments contained the late endosomal marker CD63, which was enriched on vesicles within these structures and incorporated into the envelope of budding virions. The virus-containing vacuoles were also labeled with antibodies against LAMP-1, CD81, and CD82, which were also incorporated into the viral envelope. To assess the cellular source of infectious viruses derived from MDM, virus-containing media from infected cells were precipitated with specific antibodies. Only antibodies against antigens found in late endosomes precipitated infectious virus, whereas antibodies against proteins located primarily on the cell surface did not. Our data indicate that most of the infectious HIV produced by primary macrophages is assembled on late endocytic membranes and acquires antigens characteristic of this compartment. This notion has significant implications for understanding the biology of HIV and its cell–cell transmission.
Keywords: HIV; endosome; virus assembly; late endosome; multivesicular body
Introduction
Cell-free transfer and infection of cells by primate lentiviruses (HIV-1, -2, and SIV) requires the assembly and maturation of infectious virus particles. Within an infected cell, virions are formed in a temporally and spatially coordinated manner wherein the components that make up the virus (e.g., the virally encoded envelope, Gag, protease, integrase, reverse transcriptase, and Vpr proteins, together with the RNA genome) assemble in association with a specific cellular membrane from which the viral envelope is derived. Previous EM studies of infected lymphocytes or T cell lines in culture have indicated that lentiviruses can assemble at, and bud through, the plasma membrane of infected cells (Barre-Sinoussi et al., 1983; Levy et al., 1984; Gelderblom et al., 1987). In addition, analysis of purified human immunodeficiency virus type 1 (HIV-1) indicates that, as well as the HIV-1 envelope glycoprotein (Env), glycoproteins found in the plasma membrane of infected cells are also included in the viral membrane (Tremblay et al., 1998; Esser et al., 2001; Ott, 2002).
Despite the view that HIV assembly occurs at the plasma membrane, a number of observations have suggested intracellular organelles may also play a role in HIV-1 production. First, virions and immature or budding HIV particles can occasionally be observed in intracellular vacuoles, even in infected T cell lines (unpublished data), though the nature of these compartments and their role in the pathology of HIV has not been established. Second, the cytoplasmic domain of the HIV Env transmembrane component (TM or gp41) contains a highly conserved tyrosine-based motif that can mediate endocytosis of Env through clathrin-coated pits (Rowell et al., 1995; Egan et al., 1996; Sauter et al., 1996; Bowers et al., 2000). The presence of this signal results in the majority of Env being located in intracellular membranes. Experiments in polarized epithelial cells indicate the distribution of Env can have a dominant effect in determining the site of virus assembly (Lodge et al., 1997). Significantly, disruption of the endocytosis signal in an infectious SIV model leads to attenuation of viral pathogenesis (Fultz et al., 2001). Third, the budding of HIV and some other enveloped viruses uses host cell machineries that normally function in the topologically similar formation of the small internal vesicles of multivesicular bodies (MVBs; for review see Pornillos et al., 2002). HIV particles budding at the cell surface would need to recruit these so-called ESCRT complexes to the plasma membrane, whereas the presence of the ESCRT complexes on endosomes may favor intracellular virus assembly.
Here, we have investigated HIV-1 assembly in infected monocyte-derived macrophages (MDM), a primary cell preparation in which virus has previously been demonstrated to accumulate in intracellular compartments (Gendelman et al., 1988; Orenstein et al., 1988). Although originally suggested to be derived from the Golgi apparatus, recent studies have indicated that the virus-containing structures in these cells are equivalent to the major histocompatibility antigen type II compartment (MIIC) in which maturing major histocompatibility antigen type II (MHCII) molecules are loaded with peptides (Raposo et al., 2002). Here, we confirm these observations and show that in human primary MDM, at least two strains of HIV-1 assemble in intracellular vacuoles at all time points examined, whereas little (if any) virus assembly is seen at the plasma membrane. Using immunolabeling of cryosections, we demonstrate that these intracellular virus-containing vacuoles have characteristics similar to those described for multivesicular late endosomes; they can contain small internal vesicles, and they can be immunolabeled with antibodies against markers for late endosomes. HIV-1 virions were observed to bud directly into this compartment and thereby acquire late endosome membrane proteins, notably CD63 and LAMP-1. MDM were also observed to secrete virions into the culture medium when the virus-containing intracellular vacuoles fused with the plasma membrane. Immunoprecipitation of virus-containing media from infected MDM with antibodies directed against intracellular and cell surface markers demonstrated that most of the infectious HIV released from these cells is produced in an intracellular late endosome compartment, and not at the cell surface.
Results
Infection of MDM with HIV-1
Monocytes were derived from human peripheral blood by adherence and were differentiated in culture for 6 d. At this time, the cultures consisted primarily of macrophages, with ∼90% of the cells positive for the macrophage marker CD14. These MDM could be infected with CCR5 (R5) tropic HIV-1 strains and could be kept in culture for up to 30 d.
To assess the time course of the infections, supernatants were collected from MDM cultures infected with HIV-1 Ba-L, and the amount of released virus was determined by measuring levels of viral capsid, p24, or reverse transcriptase activity (Fig. 1 A). The infectivity of the released viruses was tested in a focus-forming assay after infection of susceptible NP-2 target cells (Fig. 1 B). Virus could be detected in the MDM medium as early as 3 d after infection and accumulated rapidly over the following days. The amount of infectious virus released reached a maximum at 8 d after infection and was maintained thereafter. Measurement of p24 concentration and reverse transcriptase activity demonstrated that the cells continued to release virus for a further two weeks.
Figure 1.
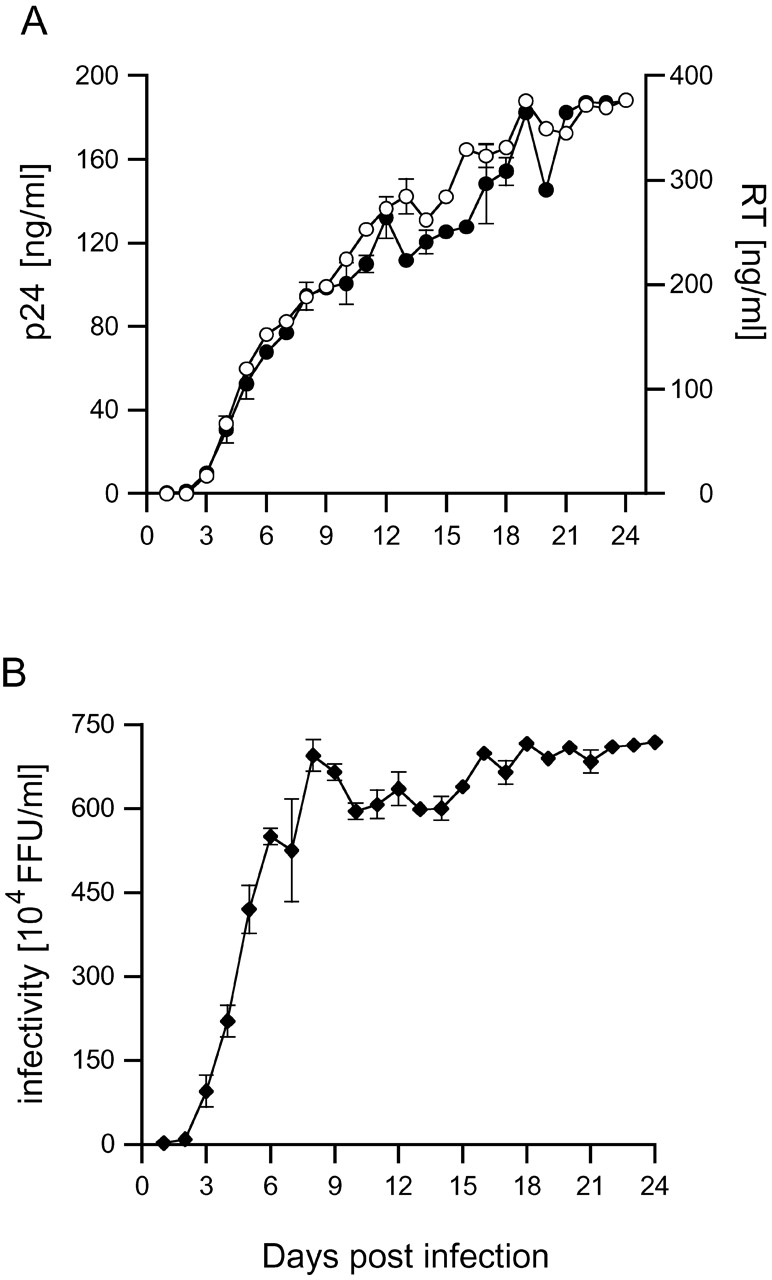
Time course of HIV-1 Ba-L production in MDM. Human MDM were infected with 9.3 × 105 focus-forming units (FFU) of HIV-1 Ba-L. Supernatants were collected daily and analyzed for (A) p24 content (○) or reverse transcriptase (•) and (B) infectivity on NP-2 CD4/CCR5 indicator cells. Activities are corrected for the dilution effect of feeding.
Localization of HIV antigens in infected MDM
To study virus assembly, MDM infected with HIV-1 Ba-L for various times were prepared for cryosection immunolabeling, and semi-thin sections (0.5 μm) were analyzed for expression of viral antigens by fluorescence microscopy. Viral capsid (p24) and matrix (p17) antigens were detected in a large proportion of the cells at all times. At 7 to 20 d, staining with the anti-p24 antibody, which also detects the Gag polyprotein precursor p55, was seen associated with intracellular granules (Fig. 2, A–D), and only rarely were small spots or patches of fluorescence detected at or close to the cell surface. Similar results were obtained with MDM infected with another macrophage-tropic HIV-1 strain (SF162) for 14, 21, or 30 d. Labeling with an anti-matrix (p17) mAb identified this viral antigen in intracellular spots and clusters (Fig. 2, E–G), but not at the cell surface. Thus, HIV-1 appears to accumulate predominantly intracellularly in MDM, in agreement with previous reports (Orenstein et al., 1988; Raposo et al., 2002). Similar results were obtained with MDM infected with the SL-2 and JR-FL strains of HIV-1, though the proportion of infected cells in these cultures was lower.
Figure 2.
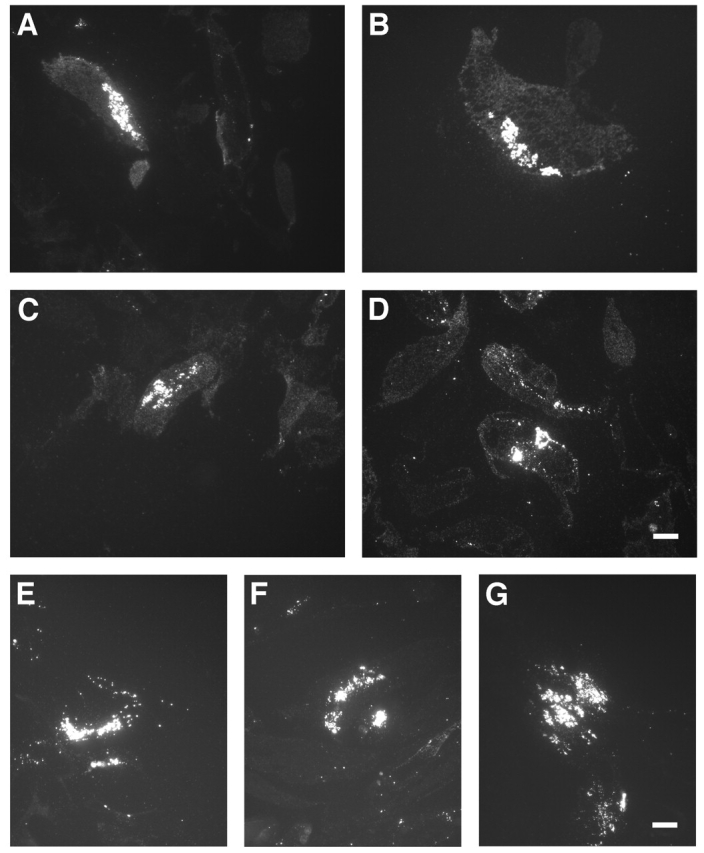
Overview of anti-p24 and -p17 staining on HIV-infected MDM. Semi-thin cryosections of MDM infected with HIV-1 Ba-L and labeled with anti-p24 (A–D), or infected with HIV-1 SF162 and labeled with anti-p17 (E–G), were stained with Alexa® Fluor 488 goat anti–mouse IgG. Cells had been infected for 7 (A), 12 (B), 15 (C), and 20 d (D); or 14 (E), 21 (F), and 30 d (G). Bars, 10 μm.
EM analysis of the virus-containing compartments in MDM
To study the intracellular viral antigens in more detail, ultrathin cryosections (∼50 nm) of infected MDM were labeled with antibodies against p17 or p24 and protein A gold (PAG) and observed by EM. These antibodies stained electron-dense 100–110-nm membrane-containing particles, often with truncated cone-shaped cores typical of mature HIV-1 (Fig. 3). Anti-p17 antibody labeling was often seen close to the membrane of these particles (Fig. 3 A), but was absent over budding figures or particles with the appearance of immature virions, consistent with this antibody recognizing only the cleaved Gag protein (Fig. 3 A′; Ferns et al., 1987). Anti-p24 showed strong labeling over particles with the appearance of both immature and mature virions, with up to 20–30 gold particles per virus particle (Fig. 3, B and B′). The different distributions of the p17 and p24 antigen were confirmed on sections double labeled with these antibodies (Fig. 3, C and C′).
Figure 3.
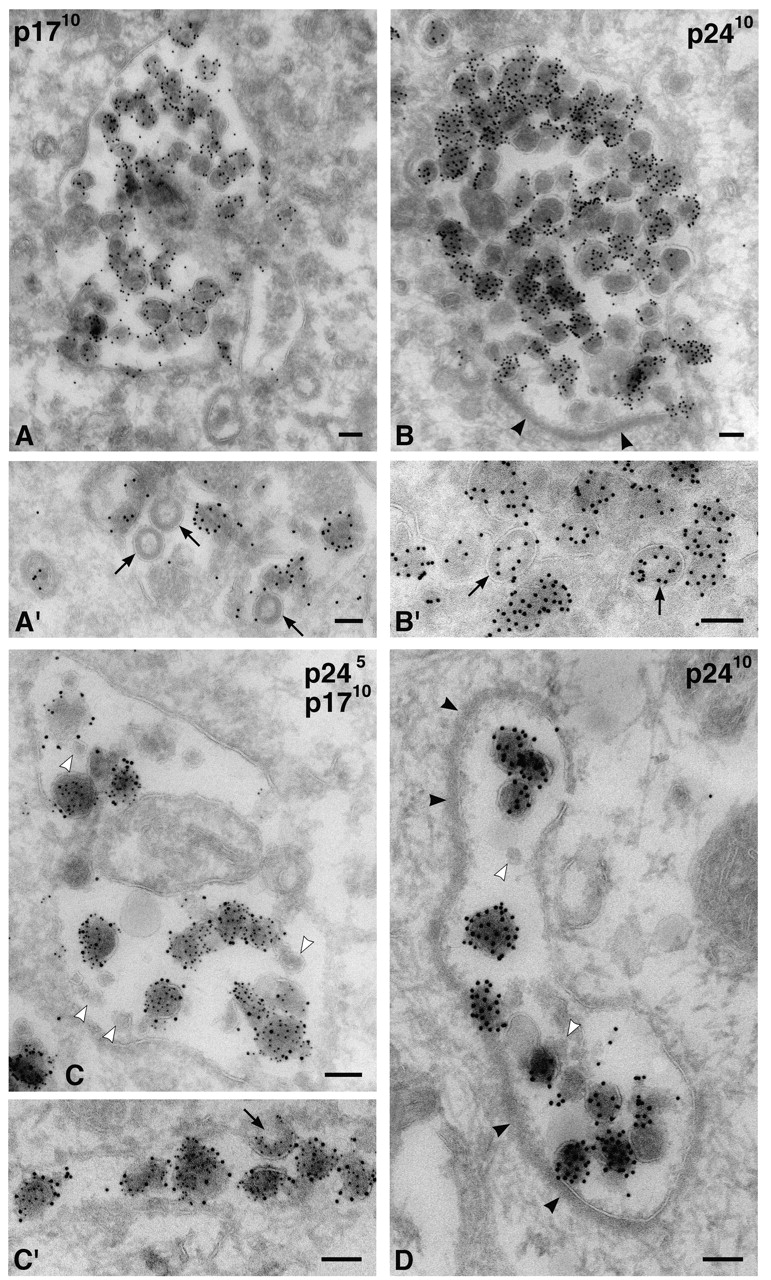
Intracellular HIV-1 in MDM. Cryosections from MDM infected with HIV-1 Ba-L were stained with anti-p17 (A and A′) or anti-p24 antibodies (B and B′) and 10 nm PAG. Alternatively, sections were double labeled for p24 (PAG 5 nm) and p17 (PAG 10 nm) (C and C′). Virus particles were found primarily in intracellular vacuoles at all times, i.e., in these cells infected for 7 (B′), 12 (A′), 14 (A and B), or 20 d (C and C′). Arrows identify budding or immature virions in A′, B′, or C′. D shows a cell infected with HIV-1 SF162 for 14 d, stained with anti-p24 and PAG10. Note the small unlabeled internal vesicles (C and D, white arrowheads) and the flat coat on some of these vacuoles (B and D, black arrowheads). Bars, 100 nm.
Labeled virus particles were seen in intracellular vacuoles, often quite deep in the cell. The presence of occasional budding figures and what appeared to be immature virions (Fig. 3, A′, B′, and C′) indicated that particles containing the p24/p55 Gag protein assembled at the limiting membranes of these intracellular structures. Vacuoles containing the p17- and p24-labeled particles displayed a variety of sizes, from large organelles packed with these particles (Fig. 3 B) to more loosely filled structures (Fig. 3 C), narrow tubules (Fig. 3 C′), or complex intracellular membrane systems. Although some vacuoles contained mainly the labeled particles, others also had internal membrane structures, including small vesicles similar to those found in MVBs (Fig. 3 C). Frequently, the virus vacuoles contained a prominent electron-dense flat coat (Fig. 3, B and D) which, at ∼28 nm (range 25–30 nm), was significantly thicker than the coats lining clathrin-coated pits at the plasma membrane (∼17–23 nm on these preparations). Only rarely were p24/p17-labeled particles found at or close to the plasma membrane. The distribution of these virus-like particles mainly in intracellular vacuoles was not a property unique to the Ba-L virus, as another HIV-1 strain (the macrophage-tropic SF162) also accumulated in similar intracellular compartments (Fig. 3 D).
Envelope expression on intracellular HIV-1 virions
When cryosections of HIV-1 Ba-L–infected MDM were stained with an antibody against the gp120 component of HIV-1 Env (2G12), strong labeling was observed over intracellular particles morphologically identical to those labeled for p24 and/or p17 (Fig. 4 A). These particles were labeled with, on average, five gold particles per virion, suggesting that they had incorporated significant levels of Env. Similarly, viruses could be labeled with another anti-Env antibody (b12), though the labeling was not as strong (unpublished data). Double staining confirmed that Env was enriched on p24-containing particles (Fig. 4 B), consistent with the possibility that these particles are infectious viruses. On the 2G12-stained cryosections, the intracellular virus particles were by far the most strongly labeled structures. However, some labeling was seen on other structures including various tubules, vacuoles, or membrane cisternae, often found in the vicinity of the Golgi apparatus/TGN. Only low levels of labeling were observed at the plasma membrane.
Figure 4.
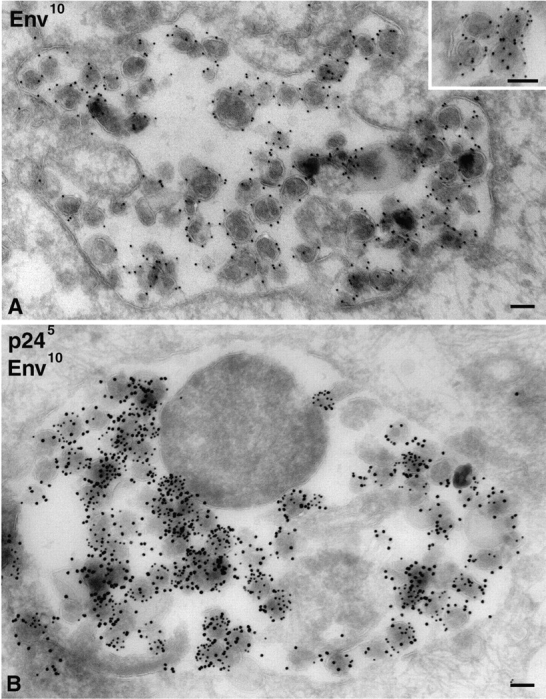
Detection of HIV-1 envelope on intracellular HIV-1 virions in MDM. Cryosections from MDM infected with HIV-1 Ba-L for 14 d were stained with anti-Env 2G12 and PAG10 (A) or double labeled for p24 with PAG5 and Env with PAG10 (B). The inset in A shows a detailed view with gold particles over the viral membrane on equatorially sectioned virions, or all over virions cut more tangentially. Bars, 100 nm.
Identification of the virus-containing vacuoles as late endosomes
To identify the intracellular vacuoles into which the Env/p24-labeled virus particles were budding, we colabeled ultrathin cryosections with antibodies against various intracellular markers. Recent analyses of HIV-1–infected macrophages indicated that the MHCII protein HLA-DR is incorporated into virions budding into MIICs (Raposo et al., 2002). Because MIICs are a specialized late endosome compartment, we stained infected MDM with antibodies against CD63, a tetraspanin found in late endosomes and MVBs, particularly in the membranes of the internal vesicles. In macrophages, a small amount of CD63 is also found at the plasma membrane. On MDM cryosections, virus-containing vacuoles were strongly labeled with anti-CD63 (Fig. 5). Gold particles identified CD63 within the envelope of individual virions, as well as over small membrane vesicles similar to the internal vesicles of MVBs (Fig. 5 A). This suggests that the compartment into which HIV-1 is budding in MDM is equivalent to the late endosome/MVB compartment. The MDM used in these analyses did not contain many classical MVBs, with numerous internal vesicles, similar to those found in tissue culture cell lines. Instead, anti-CD63 strongly labeled various small membrane vacuoles, vesicles, and tubular profiles that contained few internal vesicles. Virions were seen to bud into these compartments, incorporating CD63 into their membrane envelope as they assembled at the limiting membrane (Fig. 5 C).
Figure 5.
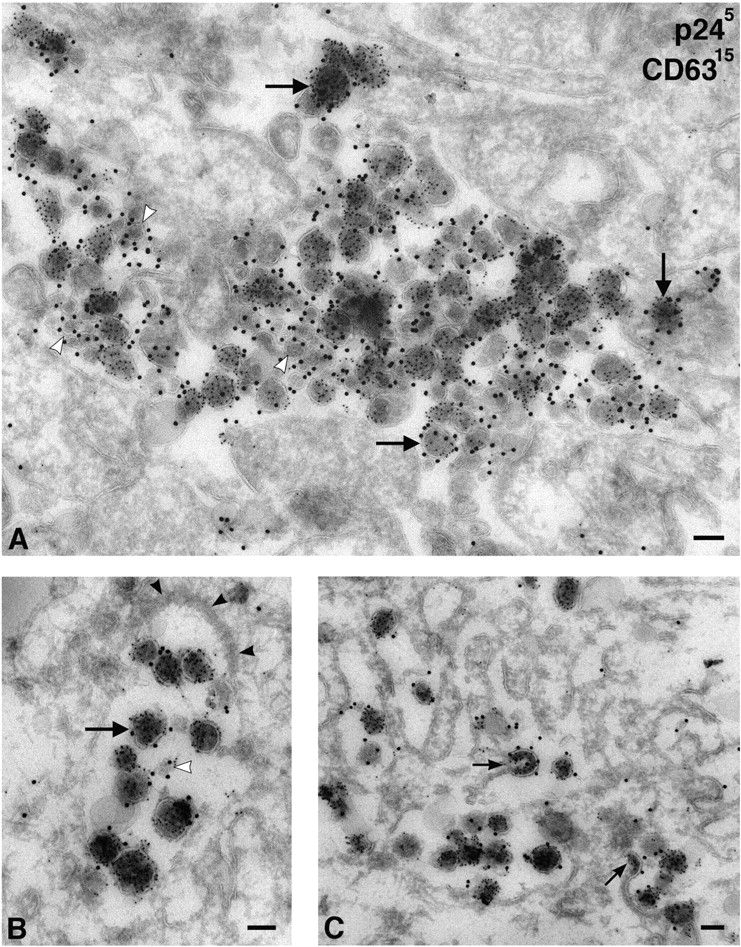
HIV-1 virions in MDM accumulate in CD63-containing compartments. Cryosections from MDM infected with HIV-1 Ba-L for 14 d were double labeled for p24 with PAG5 and for CD63 with PAG15. (A) Overview of a large complex intracellular vacuole filled with virions and clusters of small vesicles containing CD63 (white arrowheads). Virions are identified by PAG5 and are frequently labeled with the larger gold particles marking CD63 (e.g., at the large arrows), whereas the small vesicles are labeled only with CD63/PAG15. (B) A smaller vacuole containing virus particles also labeled with CD63/PAG15. Note the electron-dense coat (black arrowheads). (C) Virions budding into a complex vacuole labeled with anti-CD63. Small arrows indicate budding virus particles incorporating CD63 into their membranes. Bars, 100 nm.
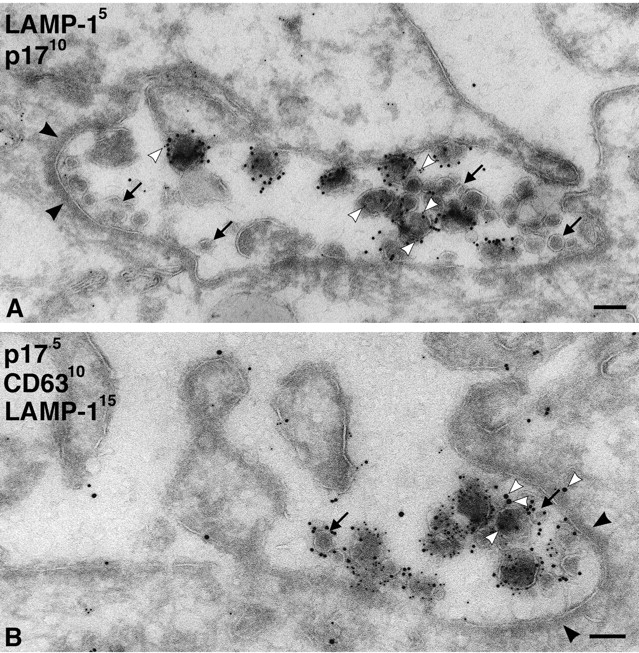
We also stained MDM with antibodies against lysosomal markers. Although antibodies against LAMP-1, LAMP-2, or the macrophage-specific lysosomal glycoprotein CD68/macrosialin occasionally identified electron-dense lysosomes, the majority of the labeling was found over small vacuoles and membrane tubules, some of which contained internal vesicles or other structures similar to those labeled with anti-CD63. The anti-LAMP-1 antibody showed strong labeling on cryosections and was seen over the virus-containing vacuoles (Fig. 6), occasionally even over viral envelopes (Fig. 6, A and C). However, labeling of the virus-containing vacuoles was weak compared with virus-negative, LAMP-1–labeled membrane tubules and vesicles that were frequently found nearby (Fig. 6, A and B). This was clear in triple labeling, where sections were costained for viral p17, CD63, and LAMP-1 (Fig. 6 C). Although high levels of CD63 were associated with virus vacuoles and found over the virions, the level of LAMP-1 labeling over this compartment was low. This suggests that the CD63-containing virus vacuoles represent a late endosome compartment, which differs from the LAMP-1–enriched membranes nearby. Other lysosomal markers such as LAMP-2 or CD68 were expressed at much lower levels than LAMP-1 in these MDM. Nonetheless, these markers were also occasionally found associated with virus-containing vacuoles (Fig. 7 A; unpublished data).
Figure 6.
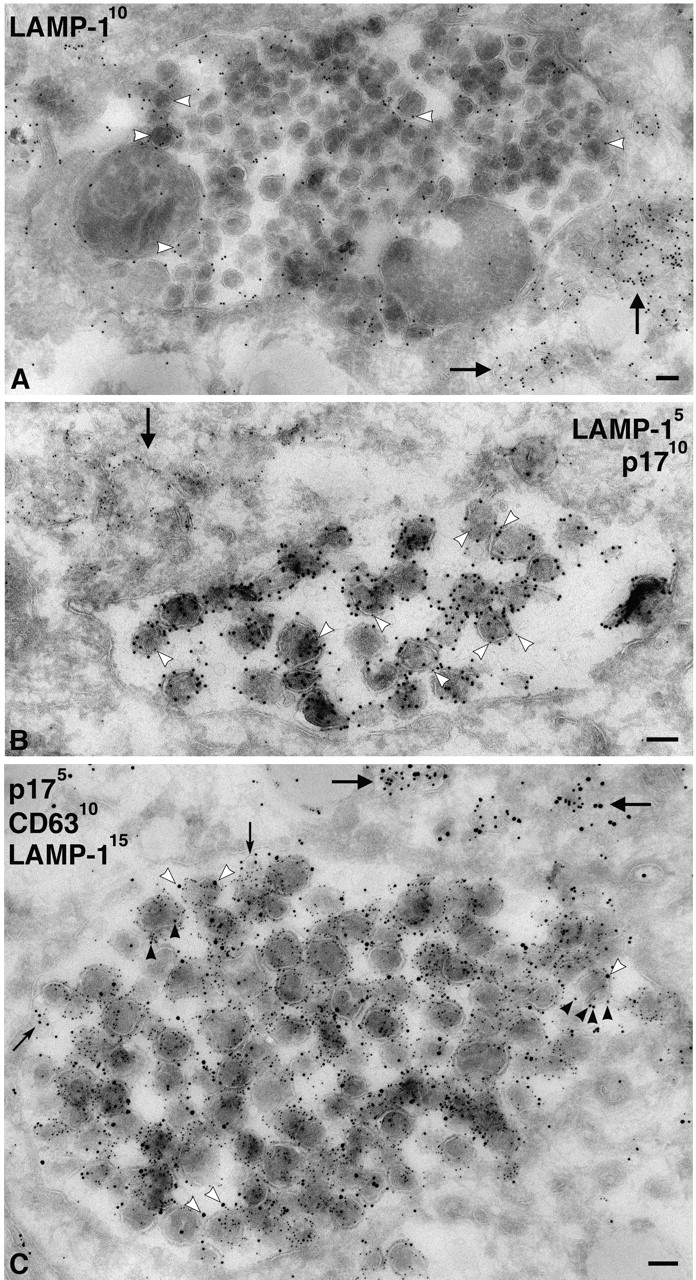
Virus-containing vacuoles in MDM labeled with antibodies to LAMP-1. (A) Cryosections from MDM infected with HIV-1 Ba-L for 7 d were labeled with a rabbit antiserum to LAMP-1 and PAG10. Note gold labeling associated with the large virus-containing vacuole, including gold particles on individual virions (white arrowheads). However, lysosomal membranes nearby contain significantly more gold particles (arrows). (B) Cryosection double labeled with rabbit anti-LAMP-1/PAG5 and anti-p17/PAG10. The large virus- containing vacuole contains many p17-labeled virions, whereas a few 5 nm gold particles identify associated LAMP-1 (e.g., at the white arrowheads). More strongly LAMP-1–labeled membranes are observed nearby (arrow). (C) A large virus vacuole triple labeled with anti-p17/PAG5, anti-CD63/PAG10, and rabbit anti-LAMP-1/PAG15. Many virions are labeled with the anti-CD63/PAG10 (black arrowheads), and some also contain a few LAMP-1/PAG15 particles, some of which are identified by white arrowheads. The rare, small internal vesicles contain just CD63 (small arrows). CD63 and LAMP-1–labeled membranes are also observed nearby (large arrows). Bars, 100 nm.
Figure 7.
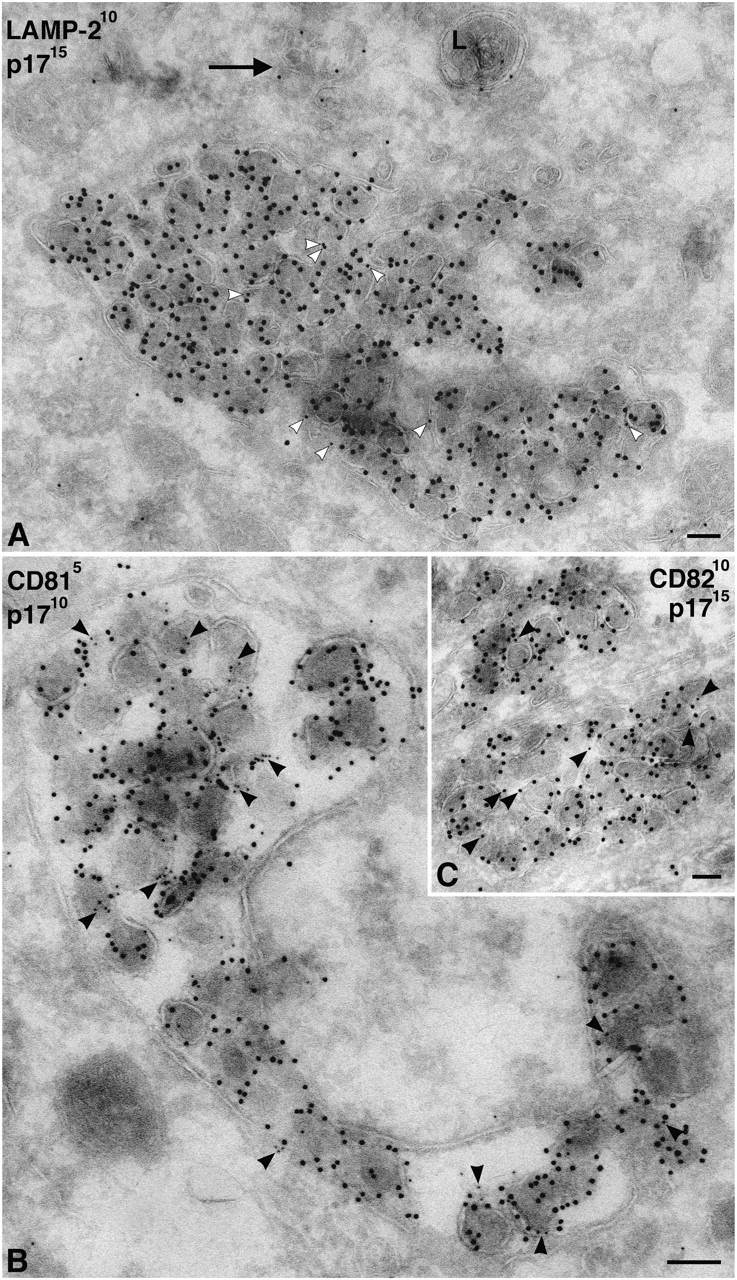
Cellular markers associated with virus-containing vacuoles in MDM. (A) Cryosections of MDM infected with HIV-1 Ba-L were double labeled with rabbit anti-LAMP-2 and PAG10, and virions were identified with anti-p17/PAG15. LAMP-2 was not highly expressed on these MDM, and hence only few gold particles could be observed, but there was some labeling of virus-containing vacuoles (white arrowheads). In addition, a lysosome (L) and membrane tubules are labeled nearby (arrow). (B and C) Cryosections of MDM infected with HIV-1 Ba-L were double labeled with antibodies against CD81 (B) or CD82 (C), whereas virions were identified with anti-p17. Both CD81 and CD82 are associated with the virus-containing vacuoles and can sometimes be found on the virions (black arrowheads). Labeling with anti-CD81 is significantly stronger than with anti-CD82. Bars, 100 nm.
Because CD63 was enriched on viruses, and because other tetraspanins have been found in the internal vesicles of MVBs (Escola et al., 1998), we stained MDM with antibodies against two other tetraspanins, CD81 (Fig. 7 B) and CD82 (Fig. 7 C). These proteins can be found on the plasma membrane of MDM, but were also seen to be associated with the virus-containing vacuoles. In particular, CD81 labeled the virus vacuoles strongly and could be seen associated with viral envelopes (Fig. 7 B). CD82 had a similar distribution, though the staining was weaker (Fig. 7 C).
These analyses suggest that in MDM, HIV assembles in intracellular vacuoles containing markers for late endosomes and MVBs. As the virus buds into this compartment, it acquires various cellular proteins characteristic of late endosomes/MVBs into its envelope, including the tetraspanins CD63, CD81 and CD82, and some LAMP-1.
Virus precipitation with antibodies against cellular proteins
HIV-infected MDM cultures release infectious virus into the medium (Fig. 1 B). Our EM observations suggest that the majority of HIV particles produced by MDM are assembled in late endosomes. Whether these are the source of the infectious virus in the cells' media is not clear. To determine the cellular origin of the infectious HIV derived from MDM, we precipitated viruses collected from MDM culture media with antibodies directed against ectodomain epitopes of a number of transmembrane and GPI-linked proteins associated with intracellular and/or plasma membranes (Table I). The selection of these proteins was based on their reported expression in specific membrane systems and/or their presence in HIV-1 envelopes (Tremblay et al., 1998; Ott, 2002). Expression of the antigens and their cellular distribution was determined on (or in) MDM by FACS® analysis and by immunofluorescent staining of fixed infected and uninfected MDM cultures or of semi-thin cryosections (Table I).
Table I. Immunoprecipitation of HIV-1 Ba-L–containing supernatant media from infected MDM cultures.
| Virus precipitation
|
Expression in MDM
|
||||
|---|---|---|---|---|---|
| Percentage of infectivity in supernatanta | Percentage of p24 precipitated | n b | Cell surfacec | Intracellulard | |
| Viral antigens | |||||
| HIV-1 Env (b12) | 0.3 ± 0.7 | 92.2 ± 8.1 | 5 | ND | ND |
| Control | 100 | 14.2 ± 2.7 | 5 | − | − |
| MHCII | |||||
| HLA-DR | 29.4 ± 8.4 | 79.6 ± 4.3 | 5 | +++ | ++ |
| Tetraspanin | |||||
| CD63 (1B5) | 5.6 ± 2.1 | 95.5 ± 3.4 | 4 | + | ++++ |
| CD63 (FC5.01) | 6.6 | 100 | 1 | ND | ++++ |
| CD81 | 84.5 ± 9.5 | 39.6 ± 16.5 | 3 | +++ | ++ |
| CD82 | 75.1 ± 18.8 | 51.2 ± 7.0 | 3 | + | + |
| CD53 | 58.7 ± 8.0 | 58.6 ± 16.3 | 3 | ++ | ND |
| CD151 | 85.0 ± 21.3 | 12.2 ± 5.5 | 3 | ++ | +/− |
| Lysosome associated | |||||
| LAMP-1 | 54.5 | 40.2 | 1 | − | +++ |
| LAMP-2 | 115.8 | 21.2 | 1 | − | ++ |
| GPI anchored | |||||
| CD14 | 87.8 ± 16.8 | 11.9 ± 7.3 | 3 | ++ | +/− |
| CD52 | 94.6 ± 11.6 | 18.4 ± 8.0 | 3 | ++ | +/− |
| CD55 | 88.3 ± 16.7 | 9.5 ± 5.2 | 3 | + | +/− |
| CD59 | 45.4 ± 17.6 | 67.0 ± 9.0 | 3 | +/+ | + |
| Adhesion | |||||
| CD11a | 110.3 ± 28.4 | 18.4 ± 5.1 | 3 | ++ | +/− |
| CD54 | 56.3 | 20.0 | 1 | +++ | ND |
| CD169 | 96.8 ± 17.5 | 17.5 ± 1.7 | 3 | ++ | +/− |
| Scavenger receptor | |||||
| CD163 | 103.2 ± 20.3 | 17.1 ± 4.4 | 3 | + | +/− |
ND, not determined.
To calculate the percentage of infectivity in the supernatant, the infectivity of samples treated with the control antibody against VSV-G was set at 100%.
Number of experiments.
Determined by FACS® analysis or by immunofluorescence.
Determined by immunofluorescence staining of semi-thin cryosections or of permeabilized cells.
Cell-free media from HIV-1 Ba-L–infected MDM were mixed with the respective antibody and subsequently bound to fixed Staphylococcus aureus. Samples were centrifuged to produce a pellet and a supernatant. The supernatants were analyzed for unprecipitated infectious virus by infecting NP-2 cells. Both the pellet and supernatant were assayed for viral p24 by ELISA. As positive controls for precipitation, we used the anti-Env antibody b12 as well as an anti-HLA-DR antibody, as HLA-DR has been reported to be readily incorporated into macrophage-derived HIV-1 Ba-L (Esser et al., 2001). An antibody against the vesicular stomatitis virus glycoprotein (VSV-G) was used to determine nonspecific virus precipitation.
Nonspecific precipitation of infectious virus was low, as the anti-VSV-G antibody produced only a small (<10%) reduction in the number of infectious focus-forming units (FFU; unpublished data). By contrast, precipitation with the anti-Env antibody completely eliminated infectivity in the supernatant. The antibody against the cellular protein HLA-DR also precipitated a significant amount of infectious virus, in agreement with previous reports that this protein is incorporated into the HIV-1 envelope (Esser et al., 2001; Raposo et al., 2002). Of the antibodies against tetraspanin markers, anti-CD63 showed the most efficient removal of infectious virus from the supernatant; this was observed with two different anti-CD63 antibodies (Table I). The other tetraspanin antibodies, anti-CD81, -CD82, -CD53, and -CD151 also precipitated some of the infectious virus, though much less efficiently, suggesting that not all virus particles incorporated sufficient levels of these molecules into their envelopes for immunoadsorption to occur (Fig. 8 A). Precipitation with antibodies against LAMP proteins showed that LAMP-1 was incorporated into some of the infectious virus, but LAMP-2 was not detectable (Table I). Antibodies against well-characterized cell surface proteins, including GPI-anchored proteins (CD14, CD52, and CD55) and various adhesion proteins (CD11a and CD169), did not precipitate significant amounts of infectious virus. However, some HIV infectivity was removed by antibodies to the GPI-anchored protein CD59, and by CD54 (ICAM-1; Fig. 8 A and Table I).
Figure 8.
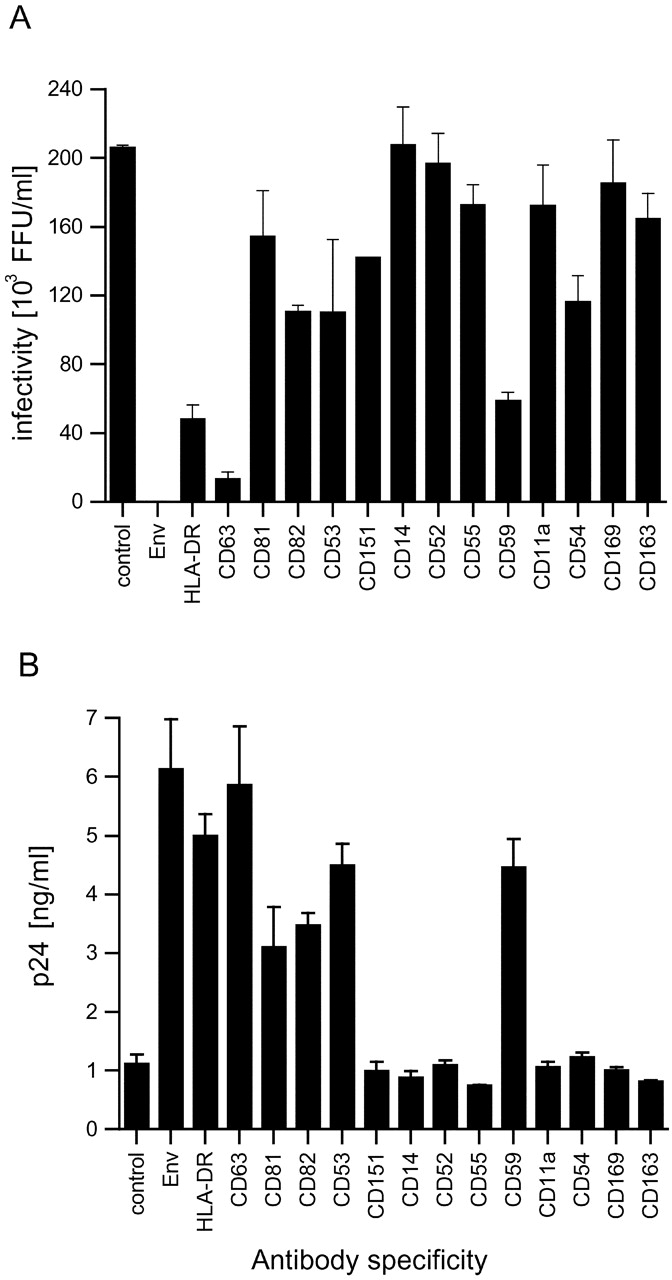
Virus precipitation with antibodies against cellular proteins. Cell-free supernatants of MDM-derived HIV-1 Ba-L were precipitated with the antibodies indicated. Unprecipitated supernatants were analyzed for remaining infectious virus by a focus-forming assay on NP-2 CD4/CCR5 cells (A). Pellets were analyzed for precipitated viral protein by p24 ELISA (B).
Measurement of the amount of viral p24 precipitated by these antibodies showed a corresponding result, as low levels of the viral protein were precipitated with the VSV-G antibody and high amounts with the anti-Env and -HLA-DR antibodies (Fig. 8 B). This demonstrates directly that the anti-Env antibody b12, a neutralizing antibody (which could account for the observed reduction of infectious virus), is able to precipitate the virus (Fig. 8 A). The anti-CD63 antibodies precipitated similar levels of p24 as the Env antibody, confirming that the majority of virus incorporates CD63 in its envelope (Fig. 8 B and Table I). The anti-CD81 antibody precipitated comparable amounts of viral protein to the anti-CD82 and -CD53 antibodies, although it did not remove as many infectious particles from the supernatant (compare Fig. 8 B with Fig. 8 A). The CD151 antibodies did not precipitate detectable levels of p24. Pellets of precipitates with anti-LAMP-1, but not with anti-LAMP-2, contained viral p24 (Table I). Antibodies against various cell surface proteins including CD14, CD11a, and CD54 failed to precipitate a significant amount of viral p24 (Fig. 8 B and Table I). Out of all these cell surface markers, only antibodies to the GPI-linked protein CD59 were able to precipitate some p24.
Together, these results show that virtually all the infectious HIV-1 that accumulated in the media of the MDM had acquired late endosome–associated proteins, in particular CD63, whereas the infectious virions were devoid of a number of cell surface markers. Together with the EM observations, these data suggest that all the infectious HIV and most (if not all) virus particles released from HIV-infected MDM are derived from late endosomes/MVBs.
Virus release from infected MDM
In hematopoietic cells, late endocytic compartments can function as secretory organelles (Blott and Griffiths, 2002). Moreover, MIICs have been proposed to fuse with the plasma membrane, either directly or via membrane tubules (Raposo et al., 1996; Boes et al., 2002; Chow et al., 2002). Our EM observations of infected MDM suggested that such a mechanism might provide a route for virus release. Virions were not generally found on the surface of the infected cells, but occasionally accumulations of virions were seen in invaginations or deep pockets of the plasma membrane (Fig. 9). The virions in these invaginations were often interspersed with small vesicles resembling exosomes, the MVB-derived vesicles released by certain hematopoietic cells when their MVBs fuse with the plasma membrane (Stoorvogel et al., 2002; Thery et al., 2002), which could be labeled with antibodies against LAMP-1 and CD63 (Fig. 9). This suggests that the plasma membrane pockets represent exocytic profiles resulting from the fusion of the virus-containing vacuoles with the cell surface. Thus, in human MDM, infectious HIV-1 is produced in intracellular compartments with features of late endosomes or MVBs, and these viruses can be released to the extracellular medium by secretion.
Figure 9.
Exocytosis of virions on infected MDM. Plasma membrane invaginations probably resulting from the fusion of intracellular virus-containing vacuoles with the plasma membrane. Cryosection in A was double labeled for LAMP-1/PAG5 and p17/PAG10, whereas B was triple labeled for p17/PAG5, CD63/PAG10, and LAMP-1/PAG15. Although these invaginations are continuous with the cell surface, they show labeling for LAMP-1 (white arrowheads) and CD63. Virions are interspersed with many small vesicles (black arrows) that are strongly labeled for CD63 (B, black arrows). Also, note the thick electron-dense coats on some parts of these invaginations (black arrowheads). Bars, 100 nm.
Discussion
Enveloped viruses have to assemble on, and bud through, a cellular membrane to acquire their own membrane. For HIV and related viruses, this budding has been, in the main, considered to occur at the plasma membrane of infected cells, possibly involving particular cell surface microdomains (Ono and Freed, 2001). These views have prevailed despite the fact that soon after the discovery of HIV, ultrastructural analysis of infected monocytes and macrophages showed the presence of virions in intracellular vacuoles in the vicinity of the Golgi apparatus (Gendelman et al., 1988; Orenstein et al., 1988). Now, we have used immunolabeling of ultrathin cryosections with specific antibodies directed against not only the viral Gag and Env proteins, but also a number of well-defined cellular antigens, to examine virus assembly in more detail. Our analyses confirm that in primary human MDM, multiple R5-tropic HIV-1 strains assemble into and accumulate in intracellular vacuoles. The virus-containing compartment is not part of the Golgi complex or TGN, but instead has the properties of late endosomes/MVBs, in agreement with the observations of Raposo et al. (2002), who showed that in MDM, HIV assembly occurs in the late endocytic MIIC compartment. Furthermore, using immunoprecipitation, we show that the majority of the infectious virus derived from cultured MDM assembles in these organelles, and not at the plasma membrane.
In macrophages and other antigen-presenting cells, the endocytic pathway, in particular late endosomes and lysosomes, is morphologically and functionally complex, with roles requiring the degradative properties of conventional lysosomes as well as the secretory functions of exocytic organelles. Classical MVBs were rare in the MDM that we studied, but various tubular and cisternal membranes were labeled with markers frequently associated with late endosomes and lysosomes, including CD63 and LAMP-1. The morphology of these organelles resembles the tubular lysosomes previously described in macrophages (for review see Rabinowitz et al., 1992). The virus-containing vacuoles had a complex morphology and frequently contained a few small vesicles resembling the internal vesicles of MVBs. Virus-containing vacuoles were characterized by high levels of CD63. In addition, they contained LAMP-1 and, to a lesser extent, LAMP-2 and the macrophage-specific lysosomal membrane glycoprotein CD68. However, they did not represent the main LAMP-1–labeled compartment in the MDM cells. Electron-dense lysosomes were only rarely observed, and most of the cellular LAMP-1 labeling was seen over tubular and vesicular membrane elements, which were frequently seen close to virus-containing vacuoles. Thus, the CD63high/LAMP-1low virus-containing compartment may represent an earlier endocytic compartment to the LAMP-enriched tubular lysosomes, i.e., a compartment equivalent to late endosomes or MVBs. The late endosomal lipid lysobisphosphatidic acid (LBPA) could not be detected in our MDM preparations (unpublished data); whether MDM lack LBPA or express an antigenic variant of LBPA that does not react with the 6C4 antibody (Kobayashi et al., 1998) is unclear.
Another characteristic of the intracellular virus-containing vacuoles were thick, electron-dense coats that frequently extended over a large proportion of the cytoplasmic aspect of the vacuole membrane (Fig. 3 D). The nature of these coats is not clear. They were thicker than the clathrin coats observed at the cell surface, did not show the “bilayered” appearance of endosomal clathrin coats containing Hrs (for review see Sachse et al., 2002), and were not labeled with anti-clathrin antibodies (unpublished data). Given that the intracellular budding of HIV and the formation of the internal vesicles of MVBs are topologically similar and require the same cellular ESCRT machinery (Garrus et al., 2001; Pornillos et al., 2002; Freed, 2003), it is possible that these coats contain components of ESCRT complexes. In T cells, where the majority of virus assembly occurs at the plasma membrane, these cellular components would have to be recruited to the cell surface, but why virus is generated at different sites in these different cell types is at present unclear.
In contrast to studies with macrophage-derived HIV-1 Ada, which contained only very low levels of Env (Orenstein et al., 1988; Meltzer et al., 1990), our immunolabeling experiments suggested that the intracellular virions observed in MDM acquired significant amounts of Env. Indeed, on many anti-Env–stained sections, the intracellular virions represented the most strongly labeled structures, suggesting that Env is enriched on the budding virions. How these Env levels compare with those in viruses budding from the cell surface in other cell types is under investigation. The Env proteins of primate immunodeficiency viruses contain at least one conserved endocytic sorting signal in their cytoplasmic domains (Rowell et al., 1995; Egan et al., 1996; Sauter et al., 1996; Bowers et al., 2000; Wyss et al., 2001), which accounts for the low levels of surface Env seen in our labeling experiments, but how Env is sorted into the endosomal budding profiles is unknown. The fact that high levels of Env are apparent on virions assembling into late endosomes suggests these particles are likely to be infectious.
In addition to the viral Env protein, HIV virions also accumulate a number of cellular glycoproteins into their membranes, most notably CD63, as well as CD81 and CD82, and some LAMP-1. Furthermore, HIV-1 has been shown to acquire MHCII (HLA-DR) in this compartment (Raposo et al., 2002). It has been known for some time that HIV incorporates a number of other cellular proteins into its envelope (Tremblay et al., 1998; Esser et al., 2001; Ott, 2002). These have been proposed to facilitate infection, and viruses containing HLA-DR have been reported to be more infectious (Cantin et al., 1997a,b). Whether the inclusion of late endosomal proteins such as CD63 has any role in virus transmission is not known, though CD63 in the viral membrane may contribute to the ability of anti-CD63 antibodies to inhibit HIV infection of macrophages (von Lindern et al., 2003).
The inclusion of late endosomal proteins in the viral membrane offers a useful experimental marker to facilitate the identification of the cellular source of infectious virus released from MDM. When infected MDM are homogenized, the number of infectious particles in the supernatant is increased significantly, suggesting that a large intracellular pool of infectious virus does exist in these cells (unpublished data); however, infectious virus can also be collected from the media of infected MDM. Because it was possible that this might include virus that assembled at the cell surface, but was not detected in our EM analysis, we tried to identify the source of infectious particles in the tissue culture media by analyzing the composition of the virions using immunoprecipitation analysis. These experiments demonstrated that the virions were derived from an intracellular compartment because markers for internal proteins, which have been shown by EM to be present on the intracellular virus particles, can precipitate virus. Most notable among these was CD63, which consistently removed >95% of the infectious particles from the medium. Although the MDM express low levels of CD63 at the PM, the majority of this protein was found in the intracellular late endosome compartment, where we have seen it to be incorporated into budding virions. Similarly, virions could be precipitated with an antibody to HLA-DR, another marker incorporated into virions as they bud into this intracellular compartment (Raposo et al., 2002). The late endosome/lysosome protein LAMP-1 also precipitated some infectious HIV-1, as did antibodies against CD81 and CD82, markers that were also shown by EM to be associated with intracellular virions. By contrast, antibodies against cell surface markers including CD11a, CD52, CD55, CD163, CD169, and the macrophage-specific CD14 all failed to precipitate significant amounts of infectious virus (Fig. 8 and Table I). Interestingly, antibodies against CD59 and CD54 precipitated infectious virus. Although usually considered to be plasma membrane markers, recent work indicated that both of these antigens can be incorporated into exosomes, and thus must be present in late endosomes/MVBs (Thery et al., 2002; Clayton et al., 2003). Previous reports comparing HIV derived from different cell types noted that macrophage-derived viruses may exclude some cell surface markers, compared with T cell–derived virus, consistent with the former undergoing intracellular assembly (Esser et al., 2001).
Our experiments show that in infected MDM, HIV is assembled in late endosomes and is subsequently released to the medium, most likely by fusion of the virus-containing vacuoles with the plasma membrane (Raposo et al., 2002). Indeed, virion-filled deep invaginations of the plasma membrane could sometimes be observed (Fig. 9). Although these might represent phagocytic structures or specialized plasma membrane virus assembly zones, several observations argue against this. First, if the invaginations were associated with phagocytosis of virions from the media, they might be expected to be seen on all cells, but they were mainly found on cells with intracellular virus vacuoles. Second, few virions were found near the surface of MDM, so it is unclear how virus particles might be concentrated into these structures. Third, the virions were frequently interspersed with small vesicles resembling exosomes that labeled strongly with antibodies to CD63 (Fig. 9 B). Hence, these pockets are more likely to be exocytic profiles resulting from the fusion of virus-containing vacuoles with the cell surface. Apart from these profiles, very few virions were seen at the plasma membrane.
In many cells, compartments of the late endocytic pathway have some capacity to undergo regulated exocytosis (Andrews, 2000), but in hematopoietic cells, these organelles have specialized secretory functions (Blott and Griffiths, 2002). In antigen-presenting cells, MIICs or MIIC-derived tubules can fuse with the plasma membrane to increase MHCII expression at the cell surface (Chow et al., 2002). The virus-containing vacuoles in MDM may also be secretory organelles, and virus release might be triggered by specific stimuli to the cells. Indeed, it has been shown that macrophages can release virions onto epithelial cell monolayers (Bourinbaiar and Phillips, 1991). Directed secretion of virions could be particularly important during antigen presentation, when macrophages present loaded MHCII to T cells. As a macrophage stimulates the T cell, reverse signaling from the T cell to the macrophage may induce directed release of virions onto the T cell target. Thus, macrophage transmission of HIV may be mechanistically similar to the proposed sequestration and release of virus from dendritic cells. These cells can sequester virus, possibly in an endosome compartment, without themselves becoming infected, and subsequently release virus during their interactions with T cells (Kwon et al., 2002; McDonald et al., 2003).
Macrophages play a crucial role in HIV infection and AIDS. They are among the first cells infected by HIV, and have been found to be less susceptible to the cytopathic effects of HIV and more resistant to HIV-induced apoptosis, allowing them to survive for a long time (weeks to months) after infection (Herbein et al., 2002; Kedzierska et al., 2003). Infected macrophages, laden with infectious virions, can potentially pass virus onto T cells during antigen presentation. In addition, virions assembled intracellularly in macrophages may differ from T cell–released virus in their spectrum of host-derived molecules, as well as, perhaps, in the amount of Env expressed, and therefore, in their infectivity. Because macrophages are a major reservoir for HIV-1 in infected individuals, the intracellular assembly of virus in these cells may be an important component of pathogenesis in vivo. Our analyses suggest a key role for late endosomes in the biogenesis of HIV. The full functional significance of this is a focus for ongoing investigation.
Materials and methods
Reagents
Tissue culture reagents and Nunc tissue culture plastic were from Invitrogen. EM chemicals were from Agar Scientific and TAAB Laboratories. Other chemicals were from Sigma-Aldrich, unless otherwise indicated. The antibodies used in this work, and their sources, are listed in Table SI (available at http://www.jcb.org/cgi/content/full/jcb.200304008/DC1).
Cells and infections
Peripheral blood mononuclear cells were isolated from buffy coats of healthy, HIV-1 seronegative donors (National Blood Service, Essex, UK) by density gradient centrifugation (Axis-Shield PoC) and plated onto gelatin-coated tissue culture dishes in RPMI 1640 containing 5% pooled human serum (HS; National Blood Service) at 37°C, 5% CO2. After 2 h, unattached cells were washed off and the remaining adherent monocytes were incubated overnight in RPMI 1640, 10% HS. The next day, cells were detached with PBS/5 mM EDTA, washed twice in PBS, and replated in 25-cm2 flasks at 5–6 × 106 cells/flask. After 2 h, excess cells were removed and the adherent cells were differentiated to macrophages in RPMI 1640, 10% HS until required. Cultures were fed every 2–3 d by replacing half of the medium with fresh RPMI 1640, 10% HS.
Virus strains
Stocks of HIV-1 Ba-L (a gift from R. Shattock, St. Georges Hospital, London, UK) were prepared by infecting a single preparation of MDM with 8 × 105 TCID50 for 2 h in 2 ml/25 cm2 flask, after which the volume was increased to 6 ml. Cell-free supernatants were collected from the infected cultures and stored in liquid nitrogen until required. Stocks of SF162 (a gift from A. McKnight and P. Clapham, University College London, London, UK) were prepared likewise with an initial inoculum of 105 FFU/ml. Virus stocks were titrated using the focus-forming assay described later in this section.
Virus precipitation assay
Virus immunoprecipitation was adapted from the method described previously (Esser et al., 2001). Virus samples, diluted to 106 FFU/ml in PBS containing 3% BSA, were mixed with the mAbs at a concentration of 10 μg/ml in a final volume of 100 μl, and binding was performed overnight at 4°C. Pansorbin cells (formalin-fixed S. aureus; Calbiochem) were preblocked for 1 h with PBS/3% BSA, and 25 μl of a 10% solution was added directly to the virus/mAb mixture. After incubation for 30 min at RT, captured viruses were precipitated by centrifugation (4,000 rpm for 30 min). Supernatants were analyzed for unprecipitated infectious virus by a focus-forming assay. Precipitated and unprecipitated material was analyzed for p24 content by ELISA.
p24 ELISA
96-well plates (Maxisorb; Nunc) were coated with 50 μg/ml anti-p24 (D7320) antibody in NaCO3 coating buffer overnight. Plates were washed several times in TBS (144 mM NaCl and 25 mM Tris, pH 7.5), and nonspecific binding sites were blocked with 4% milk powder in TBS for 1 h. Supernatants of infected cells were harvested and cell debris were removed by centrifugation. Samples were mixed with 1% Empigen (Surfachem, Ltd.) and incubated for at least 20 min. Multiple dilutions of the samples in 1% Empigen in TBS were made. Serial dilutions of recombinant p24 (EVA620; NIBSC Centralised Facility for AIDS Reagents, Potters Bar, UK) served as a standard. The samples were bound for 4 h at RT or overnight at 4°C, followed by several washes with TBS. Adsorbed p24 was detected with an alkaline phosphatase–coupled antibody (EH12-AP at 0.5 μg/ml in 4% milk powder, 20% sheep serum, and 0.5% Tween 20 in TBS) for 1 h at RT. After extensive washing with TBS, LumiPhos® Plus substrate (Aureon Biosystems) was added for 1 h in the dark, and the enzymatic activity was measured in a luminometer (Lucy 1; Rosys Anthos) using the Stingray software (DazDaq, Ltd.).
Reverse transcriptase assay
Reverse transcriptase activity in viral supernatants was measured using the Lenti RT activity kit (Cavidi Tech).
Focus-forming assay
NP-2 CD4/CCR5 cells (a gift from A. McKnight and P. Clapham) were plated in 48-well plates at a density of 1.5 × 104 cells/well, 1 d before infection. Cells were infected for 3 h, washed, and cultivated for 60 h in DME containing 5% FCS and 100 U/μm penicillin/100 mg/μl streptomycin. Cells were fixed in methanol/acetone (1:1) and incubated for 1 h with a mixture of two anti-HIV-1 p24 mAbs (38:97K and EF7) diluted 1:40 in PBS/1% FCS, washed in PBS/1% FCS, and incubated for 1 h with a secondary anti–mouse β-galactosidase–coupled antibody (2.5 μg/ml in PBS/1% FCS). After washing with PBS/1% FCS, β-galactosidase substrate solution (0.5 mg/ml 5-bromo-4-chloro-3-indolyl-β-galactoside in PBS containing 3 mM potassium ferricyanide and 1 mM magnesium chloride) was added, and the cells were incubated overnight. Blue-stained infected cell foci were counted microscopically and virus titers were expressed as FFU/ml.
EM immunolabeling
HIV-infected MDM or uninfected control cells were fixed by adding an equal volume of prewarmed double-strength fixative (8% PFA in 0.1 M sodium phosphate buffer, pH 7.4) directly into the culture medium. After 10 min, the medium was replaced with single-strength fixative (4% PFA) for 2 h. Fixed cells were rinsed in PBS/20 mM glycine, embedded in 12% gelatin, infiltrated with 2.3 M sucrose, and frozen in liquid nitrogen as described previously (Raposo et al., 1997).
Cryosections (∼50 nm) were quenched in 50 mM glycine/50 mM NH4Cl and labeled with primary antibodies and PAG (5, 10, or 15 nm; from The EM Lab, Utrecht University, Netherlands). Sections stained with mouse IgG1 mAb were incubated with a rabbit anti–mouse bridging antibody before labeling with PAG. For double-labeling experiments, cells were first stained with mouse IgG1 mAbs, the rabbit anti–mouse bridging antibody, and PAG, and were fixed in 1% glutaraldehyde for 10 min before requenching and staining with a second primary antibody. When the second primary antibody was also a mouse mAb, unoccupied binding sites on the bridging antibody were first blocked with an irrelevant murine IgG1 (the anti-VSV-G antibody P5D4) and glutaraldehyde fixation, before staining with the second primary antibody (1B5 anti-CD63 IgG2b or 4C9 anti-p17 IgG2a) and a second size of PAG. Sections were examined with a transmission electron microscope (model EM420; Philips).
Immunofluorescence staining
Cells fixed in 3% PFA or semi-thin cryosections (0.5 μm) were quenched in 50 mM glycine/50 mM NH4Cl, blocked in PBS containing 0.2% gelatin or 1% BSA, and labeled with various antibodies diluted in the blocking buffer. Cells or sections were washed and stained with fluorescent secondary antibodies, washed extensively, and mounted in moviol. For permeabilized cells, 0.05% saponin was added to the blocking step and each antibody incubation.
FACS®
Cells were incubated in blocking buffer (PBS containing 1% FCS, 0.01% NaN3, and 10 μg/ml human IgG), for 1 h at 4°C and gently scraped off the dish. Primary antibodies were diluted in blocking buffer to a final concentration of 10 μg/ml. The secondary antibodies (Alexa® Fluor 488–conjugated goat anti–mouse, or goat anti–rat rhodamine) were diluted in PBS containing 1% FCS and 0.01% NaN3. Antibody incubations were performed at 4°C for 1 h, and were followed by several washes in PBS, 1% FCS, and 0.01% NaN3. Cells were fixed overnight in PBS, 1% PFA, 2% FCS, and 0.02% NaN3, and were analyzed using a FACSCalibur™ flow cytometer (Becton Dickinson).
Online supplemental material
Table SI lists the antibodies used in this work and their sources. Online supplemental material available at http://www.jcb.org/cgi/content/full/jcb.200304008/DC1.
Supplemental Material
Acknowledgments
We thank the many colleagues who have contributed ideas and reagents to this project, in particular A. McKnight and R. Weiss (University College London, London, UK) for access to their containment laboratory, know-how, and reagents; P. Clapham, G. Simmons, and P.J. Klasse, who contributed to the early experiments; R. Shattock for providing viruses; and those listed in Table SI for providing antibodies. We are grateful to N. Signoret, A. Fraile-Ramos, and M. Malim for critical comments on the manuscript.
The work was supported by the UK Medical Research Council and a European Union Training and Mobility of Researchers Programme grant (FMRX-CT96-0058).
The online version of this article includes supplemental material.
Abbreviations used in this paper: Env, HIV-1 envelope glycoprotein; FFU, focus-forming units; HIV, human immunodeficiency virus; MDM, monocyte-derived macrophages; MHCII, major histocompatibility antigen type II; MIIC, MHCII compartment; MVB; multivesicular body; PAG, protein A gold; VSV-G, vesicular stomatitis virus glycoprotein.
References
- Andrews, N.W. 2000. Regulated secretion of conventional lysosomes. Trends Cell Biol. 10:316–321. [DOI] [PubMed] [Google Scholar]
- Barre-Sinoussi, F., J.C. Chermann, F. Rey, M.T. Nugeyre, S. Chamaret, J. Gruest, C. Dauguet, C. Axler-Blin, F. Vezinet-Brun, C. Rouzioux, et al. 1983. Isolation of a T-lymphotropic retrovirus from a patient at risk for acquired immune deficiency syndrome (AIDS). Science. 220:868–871. [DOI] [PubMed] [Google Scholar]
- Blott, E.J., and G.M. Griffiths. 2002. Secretory lysosomes. Nat. Rev. Mol. Cell Biol. 3:122–131. [DOI] [PubMed] [Google Scholar]
- Boes, M., J. Cerny, R. Massol, M. Op den Brouw, T. Kirchhausen, J. Chen, and H.L. Ploegh. 2002. T-cell engagement of dendritic cells rapidly rearranges MHC class II transport. Nature. 418:983–988. [DOI] [PubMed] [Google Scholar]
- Bourinbaiar, A.S., and D.M. Phillips. 1991. Transmission of human immunodeficiency virus from monocytes to epithelia. J. Acquir. Immune Defic. Syndr. 4:56–63. [PubMed] [Google Scholar]
- Bowers, K., A. Pelchen-Matthews, S. Honing, P.J. Vance, L. Creary, B.S. Haggarty, J. Romano, W. Ballensiefen, J.A. Hoxie, and M. Marsh. 2000. The simian immunodeficiency virus envelope glycoprotein contains multiple signals that regulate its cell surface expression and endocytosis. Traffic. 1:661–674. [DOI] [PubMed] [Google Scholar]
- Cantin, R., J.F. Fortin, G. Lamontagne, and M. Tremblay. 1997. a. The acquisition of host-derived major histocompatibility complex class II glycoproteins by human immunodeficiency virus type 1 accelerates the process of virus entry and infection in human T-lymphoid cells. Blood. 90:1091–1100. [PubMed] [Google Scholar]
- Cantin, R., J.F. Fortin, G. Lamontagne, and M. Tremblay. 1997. b. The presence of host-derived HLA-DR1 on human immunodeficiency virus type 1 increases viral infectivity. J. Virol. 71:1922–1930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chow, A., D. Toomre, W. Garrett, and I. Mellman. 2002. Dendritic cell maturation triggers retrograde MHC class II transport from lysosomes to the plasma membrane. Nature. 418:988–994. [DOI] [PubMed] [Google Scholar]
- Clayton, A., C.L. Harris, J. Court, M.D. Mason, and B.P. Morgan. 2003. Antigen-presenting cell exosomes are protected from complement-mediated lysis by expression of CD55 and CD59. Eur. J. Immunol. 33:522–531. [DOI] [PubMed] [Google Scholar]
- Egan, M.A., L.M. Carruth, J.F. Rowell, X. Yu, and R.F. Siliciano. 1996. Human immunodeficiency virus type 1 envelope protein endocytosis mediated by a highly conserved intrinsic internalization signal in the cytoplasmic domain of gp41 is suppressed in the presence of the Pr55gag precursor protein. J. Virol. 70:6547–6556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Escola, J.M., M.J. Kleijmeer, W. Stoorvogel, J.M. Griffith, O. Yoshie, and H.J. Geuze. 1998. Selective enrichment of tetraspan proteins on the internal vesicles of multivesicular endosomes and on exosomes secreted by human B-lymphocytes. J. Biol. Chem. 273:20121–20127. [DOI] [PubMed] [Google Scholar]
- Esser, M.T., D.R. Graham, L.V. Coren, C.M. Trubey, J.W. Bess, Jr., L.O. Arthur, D.E. Ott, and J.D. Lifson. 2001. Differential incorporation of CD45, CD80 (B7-1), CD86 (B7-2), and major histocompatibility complex class I and II molecules into human immunodeficiency virus type 1 virions and microvesicles: implications for viral pathogenesis and immune regulation. J. Virol. 75:6173–6182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferns, R.B., R.S. Tedder, and R.A. Weiss. 1987. Characterization of monoclonal antibodies against the human immunodeficiency virus (HIV) gag products and their use in monitoring HIV isolate variation. J. Gen. Virol. 68:1543–1551. [DOI] [PubMed] [Google Scholar]
- Freed, E.O. 2003. The HIV-TSG101 interface: recent advances in a budding field. Trends Microbiol. 11:56–59. [DOI] [PubMed] [Google Scholar]
- Fultz, P.N., P.J. Vance, M.J. Endres, B. Tao, J.D. Dvorin, I.C. Davis, J.D. Lifson, D.C. Montefiori, M. Marsh, M.H. Malim, and J.A. Hoxie. 2001. In vivo attenuation of simian immunodeficiency virus by disruption of a tyrosine-dependent sorting signal in the envelope glycoprotein cytoplasmic tail. J. Virol. 75:278–291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garrus, J.E., U.K. von Schwedler, O.W. Pornillos, S.G. Morham, K.H. Zavitz, H.E. Wang, D.A. Wettstein, K.M. Stray, M. Cote, R.L. Rich, et al. 2001. Tsg101 and the vacuolar protein sorting pathway are essential for HIV-1 budding. Cell. 107:55–65. [DOI] [PubMed] [Google Scholar]
- Gelderblom, H.R., E.H. Hausmann, M. Ozel, G. Pauli, and M.A. Koch. 1987. Fine structure of human immunodeficiency virus (HIV) and immunolocalization of structural proteins. Virology. 156:171–176. [DOI] [PubMed] [Google Scholar]
- Gendelman, H.E., J.M. Orenstein, M.A. Martin, C. Ferrua, R. Mitra, T. Phipps, L.A. Wahl, H.C. Lane, A.S. Fauci, D.S. Burke, et al. 1988. Efficient isolation and propagation of human immunodeficiency virus on recombinant colony-stimulating factor 1-treated monocytes. J. Exp. Med. 167:1428–1441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herbein, G., A. Coaquette, D. Perez-Bercoff, and G. Pancino. 2002. Macrophage activation and HIV infection: can the Trojan horse turn into a fortress? Curr. Mol. Med. 2:723–738. [DOI] [PubMed] [Google Scholar]
- Kedzierska, K., S.M. Crowe, S. Turville, and A.L. Cunningham. 2003. The influence of cytokines, chemokines and their receptors on HIV-1 replication in monocytes and macrophages. Rev. Med. Virol. 13:39–56. [DOI] [PubMed] [Google Scholar]
- Kobayashi, T., E. Stang, K.S. Fang, P. de Moerloose, R.G. Parton, and J. Gruenberg. 1998. A lipid associated with the antiphospholipid syndrome regulates endosome structure and function. Nature. 392:193–197. [DOI] [PubMed] [Google Scholar]
- Kwon, D.S., G. Gregorio, N. Bitton, W.A. Hendrickson, and D.R. Littman. 2002. DC-SIGN-mediated internalization of HIV is required for trans-enhancement of T cell infection. Immunity. 16:135–144. [DOI] [PubMed] [Google Scholar]
- Levy, J.A., A.D. Hoffman, S.M. Kramer, J.A. Landis, J.M. Shimabukuro, and L.S. Oshiro. 1984. Isolation of lymphocytopathic retroviruses from San Francisco patients with AIDS. Science. 225:840–842. [DOI] [PubMed] [Google Scholar]
- Lodge, R., J.P. Lalonde, G. Lemay, and E.A. Cohen. 1997. The membrane-proximal intracytoplasmic tyrosine residue of HIV-1 envelope glycoprotein is critical for basolateral targeting of viral budding in MDCK cells. EMBO J. 16:695–705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDonald, D., L. Wu, S.M. Bohks, V.N. KewalRamani, D. Unutmaz, and T.J. Hope. 2003. Recruitment of HIV and its receptors to dendritic cell-T cell junctions. Science. 300:1295–1297. [DOI] [PubMed] [Google Scholar]
- Meltzer, M.S., M. Nakamura, B.D. Hansen, J.A. Turpin, D.C. Kalter, and H.E. Gendelman. 1990. Macrophages as susceptible targets for HIV infection, persistent viral reservoirs in tissue, and key immunoregulatory cells that control levels of virus replication and extent of disease. AIDS Res. Hum. Retroviruses. 6:967–971. [DOI] [PubMed] [Google Scholar]
- Ono, A., and E.O. Freed. 2001. Plasma membrane rafts play a critical role in HIV-1 assembly and release. Proc. Natl. Acad. Sci. USA. 98:13925–13930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orenstein, J.M., M.S. Meltzer, T. Phipps, and H.E. Gendelman. 1988. Cytoplasmic assembly and accumulation of human immunodeficiency virus types 1 and 2 in recombinant human colony-stimulating factor-1-treated human monocytes: an ultrastructural study. J. Virol. 62:2578–2586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ott, D.E. 2002. Potential roles of cellular proteins in HIV-1. Rev. Med. Virol. 12:359–374. [DOI] [PubMed] [Google Scholar]
- Pornillos, O., J.E. Garrus, and W.I. Sundquist. 2002. Mechanisms of enveloped RNA virus budding. Trends Cell Biol. 12:569–579. [DOI] [PubMed] [Google Scholar]
- Rabinowitz, S., H. Horstmann, S. Gordon, and G. Griffiths. 1992. Immunocytochemical characterization of the endocytic and phagolysosomal compartments in peritoneal macrophages. J. Cell Biol. 116:95–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raposo, G., H.W. Nijman, W. Stoorvogel, R. Liejendekker, C.V. Harding, C.J. Melief, and H.J. Geuze. 1996. B lymphocytes secrete antigen-presenting vesicles. J. Exp. Med. 183:1161–1172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raposo, G., M.J. Kleijmeer, G. Posthuma, J.W. Slot, and H.J. Geuze. 1997. Immunogold labeling of ultrathin cryosections: application in immunology. Handbook of Experimental Immunology. L.A. Herzenberg, D.M. Weir, and C. Blackwell, editors. Blackwell Science Inc., Oxford, UK. 1–11.
- Raposo, G., M. Moore, D. Innes, R. Leijendekker, A. Leigh-Brown, P. Benaroch, and H. Geuze. 2002. Human macrophages accumulate HIV-1 particles in MHC II compartments. Traffic. 3:718–729. [DOI] [PubMed] [Google Scholar]
- Rowell, J.F., P.E. Stanhope, and R.F. Siliciano. 1995. Endocytosis of endogenously synthesized HIV-1 envelope protein. Mechanism and role in processing for association with class II MHC. J. Immunol. 155:473–488. [PubMed] [Google Scholar]
- Sachse, M., S. Urbe, V. Oorschot, G.J. Strous, and J. Klumperman. 2002. Bilayered clathrin coats on endosomal vacuoles are involved in protein sorting toward lysosomes. Mol. Biol. Cell. 13:1313–1328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sauter, M.M., A. Pelchen-Matthews, R. Bron, M. Marsh, C.C. LaBranche, P.J. Vance, J. Romano, B.S. Haggarty, T.K. Hart, W.M. Lee, and J.A. Hoxie. 1996. An internalization signal in the simian immunodeficiency virus transmembrane protein cytoplasmic domain modulates expression of envelope glycoproteins on the cell surface. J. Cell Biol. 132:795–811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stoorvogel, W., M.J. Kleijmeer, H.J. Geuze, and G. Raposo. 2002. The biogenesis and functions of exosomes. Traffic. 3:321–330. [DOI] [PubMed] [Google Scholar]
- Thery, C., L. Zitvogel, and S. Amigorena. 2002. Exosomes: composition, biogenesis and function. Nat. Rev. Immunol. 2:569–579. [DOI] [PubMed] [Google Scholar]
- Tremblay, M.J., J.F. Fortin, and R. Cantin. 1998. The acquisition of host-encoded proteins by nascent HIV-1. Immunol. Today. 19:346–351. [DOI] [PubMed] [Google Scholar]
- von Lindern, J.J., D. Rojo, K. Grovit-Ferbas, C. Yeramian, C. Deng, G. Herbein, M.R. Ferguson, T.C. Pappas, J.M. Decker, A. Singh, et al. 2003. Potential role for CD63 in CCR5-mediated human immunodeficiency virus type 1 infection of macrophages. J. Virol. 77:3624–3633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wyss, S., C. Berlioz-Torrent, M. Boge, G. Blot, S. Honing, R. Benarous, and M. Thali. 2001. The highly conserved C-terminal dileucine motif in the cytosolic domain of the human immunodeficiency virus type 1 envelope glycoprotein is critical for its association with the AP-1 clathrin adaptor. J. Virol. 75:2982–2992. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.