

Abstract
Amember of the family of ATPases associated with diverse cellular activities, called p97 in mammals and Cdc48 in yeast, associates with the cofactor Ufd1–Npl4 to move polyubiquitinated polypeptides from the endoplasmic reticulum (ER) membrane into the cytosol for their subsequent degradation by the proteasome. Here, we have studied the mechanism by which the p97–Ufd1–Npl4 complex functions in this retrotranslocation pathway. Substrate binding occurs when the first ATPase domain of p97 (D1 domain) is in its nucleotide-bound state, an interaction that also requires an association of p97 with the membrane through its NH2-terminal domain. The two ATPase domains (D1 and D2) of p97 appear to alternate in ATP hydrolysis, which is essential for the movement of polypeptides from the ER membrane into the cytosol. The ATPase itself can interact with nonmodified polypeptide substrates as they emerge from the ER membrane. Polyubiquitin chains linked by lysine 48 are recognized in a synergistic manner by both p97 and an evolutionarily conserved ubiquitin-binding site at the NH2 terminus of Ufd1. We propose a dual recognition model in which the ATPase complex binds both a nonmodified segment of the substrate and the attached polyubiquitin chain; polyubiquitin binding may activate the ATPase p97 to pull the polypeptide substrate out of the membrane.
Keywords: p97/Cdc48; ubiquitin; AAA ATPase; protein degradation; ER quality control
Introduction
The ER is the major site of protein folding and assembly in the secretory pathway. Eukaryotic cells have evolved a highly conserved ER quality control system that catalyzes protein folding and prevents unfolded polypeptides or unassembled protein complexes from reaching their final destinations (for review see Ellgaard and Helenius, 2001). Misfolded or unassembled polypeptides that cannot reach their native conformation in the ER are retrotranslocated into the cytosol, where they are degraded by the proteasome, a pathway termed ER-associated protein degradation, or retrotranslocation (for reviews see Brodsky and McCracken, 1999; Tsai et al., 2002). The mechanism of retrotranslocation is largely unknown. However, recent works have shed some light on the decisive step during which polypeptides are released from the ER membrane into the cytosol before they are degraded by the proteasome. The release step seems to require an ATPase, named Cdc48 in yeast and p97 in mammals (Bays et al., 2001; Ye et al., 2001; Braun et al., 2002; Jarosch et al., 2002; Rabinovich et al., 2002).
p97/Cdc48 belongs to a family of ATPases associated with diverse cellular activities (AAA;* for review see Ogura and Wilkinson, 2001). The characteristic feature of this family is the presence of one or two conserved ATPase domains, referred to as AAA cassettes, which contain Walker A and Walker B motifs that mediate ATP binding and hydrolysis, respectively. p97/Cdc48 is similar to the NSF protein involved in vesicle fusion in the secretory pathway. Like NSF, p97/Cdc48 consists of two ATPase domains (D1 and D2) and an additional NH2-terminal domain (N). Structural studies reveal that the ATPase domains form two hexameric rings stacked on top of each other with a central pore of ∼15 Å (Rouiller et al., 2000; Zhang et al., 2000). The N domains are located at the side of the D1 ring. Although both p97 and Cdc48 are abundant cytosolic proteins, a significant fraction of them is tightly associated with the ER membrane (Rabouille et al., 1998; Hitchcock et al., 2001).
The ATPase p97/Cdc48 appears to perform different cellular functions depending on its association with cofactors (Meyer et al., 2000). Mammalian p97 can interact in a mutually exclusive manner with either p47 or a dimer consisting of Ufd1 and Npl4. In mammals, the complex of p97–p47 appears to play a role in the homotypic fusion of ER and Golgi membranes (Kondo et al., 1997; Rabouille et al., 1998; Uchiyama et al., 2002). The p97–p47 complex binds ubiquitinated proteins via a UBA domain in p47, and this domain is essential for its activity in Golgi membrane fusion (Meyer et al., 2002). In Saccharomyces cerevisiae, the complex of Cdc48–Ufd1–Npl4 has been implicated in the release of polypeptides from the ER membrane (for review see Tsai et al., 2002). This release step is essential for both retrotranslocation of misfolded proteins from the ER into the cytosol, and for the activation of the transcription factor Spt23 from its ER-anchored precursor (Hitchcock et al., 2001; Rape et al., 2001). Mammalian p97 has also been shown to function in retrotranslocation (Ye et al., 2001), but a requirement for the cofactor Ufd1–Npl4 has not yet been demonstrated.
Exactly how p97/Cdc48 and its cofactor Ufd1–Npl4 extract polypeptides from the ER membrane during retrotranslocation is unclear. It is not known how the ATPase binds to the ER membrane, nor how the two ATPase domains collaborate to “pull” polypeptides out of the membrane. The mechanism of substrate recognition has been particularly difficult to address because its elucidation requires complex systems in which p97/Cdc48 acts on physiological substrates emerging from the ER membrane. Two possibilities of substrate recognition have been proposed. In one model, p97/Cdc48 would interact directly with an unfolded polypeptide segment as it emerges from the ER membrane. Alternatively, the recognition signal could be a polyubiquitin chain attached to a substrate. This model is based on the fact that all polypeptides emerging from the ER undergo polyubiquitination, and that this modification is required for the release of substrates from the ER membrane into the cytosol (for review see Tsai et al., 2002).
Mammalian Ufd1–Npl4 has been shown to bind polyubiquitin and has been proposed to mediate substrate binding by p97 (Meyer et al., 2002). A zinc-binding motif in Npl4, the Npl4 zinc finger (NZF) domain, has been identified as the major binding site for ubiquitin (Meyer et al., 2002; Wang et al., 2003). In addition, p97/Cdc48 itself can interact with ubiquitin (Dai and Li, 2001; Rape et al., 2001). The relevance of the reported ubiquitin interactions for retrotranslocation is unclear, particularly because the ubiquitin binding to p97/Cdc48 is weak and the yeast homologue of Npl4 lacks an NZF domain.
In this paper, we have addressed the mechanism by which the p97 ATPase and its cofactor Ufd1–Npl4 function in retrotranslocation. Our results provide insight into the ATPase cycle, the mode of interaction of the complex with the membrane, and particularly the mechanism of substrate recognition. We propose a dual recognition mechanism in which a nonubiquitinated segment of a substrate is initially recognized by p97 itself, and subsequently, the polyubiquitin chain is bound by both p97 and the cofactor Ufd1–Npl4.
Results
Coupling between the two ATPase domains of p97
p97 contains an NH2-terminal domain (N) and two AAA cassettes (D1 and D2; Fig. 1 A). To understand how the two ATPase domains of p97 influence one another, we generated a set of point mutants that are defective in the ATPase cycle of either one or both of the two domains. Mutants defective in ATP binding were obtained by altering the lysine residue (K) in the Walker A motifs to an alanine (A), resulting in mutants in the D1 domain (AK mutant), D2 domain (KA mutant), or in both domains (AA mutant). Mutants that allow ATP binding but are defective in hydrolysis were obtained by mutating in the D1 and D2 domains the conserved glutamate residue of the Walker B motifs to glutamine, resulting in the QE, EQ, and QQ mutants. We also generated deletion mutants that lack either the N domain (ΔN) or the second ATPase domain (ΔD2). All these mutants were expressed as His-tagged recombinant proteins in Escherichia coli and purified (Fig. 1 B). The oligomeric state of these proteins was determined by gel filtration (unpublished data). All proteins formed stable hexamers, similar to the wild-type protein. ATP-binding experiments confirmed that the AA mutant had a low affinity for the nucleotide, whereas the KA mutant bound about half the amount of nucleotide as the wild-type protein (Fig. S1, available at http://www.jcb.org/cgi/content/full/jcb.200302169/DC1). Interestingly, the AK mutant behaved similarly to the AA mutant, indicating that nucleotide binding to the D1 ring is required for ATP binding to D2. As expected, the QQ protein bound ATP, but did not hydrolyze it (Fig. S1).
Figure 1.
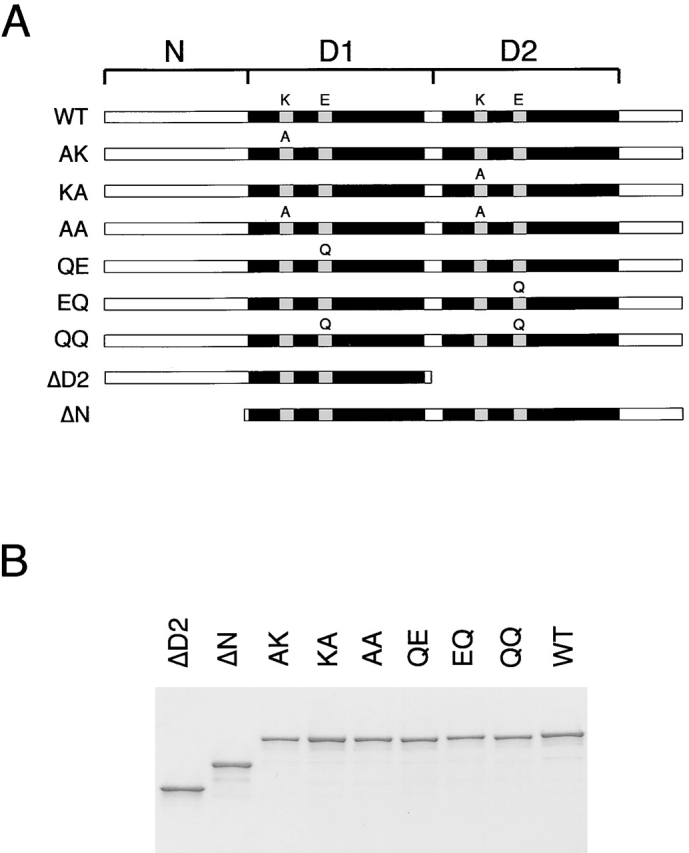

ATPase activity of p97 mutants. (A) Scheme of p97 constructs used in this work (N, NH2-terminal domain; D1, D2, first and second ATPase domain, respectively). The name of each construct is shown on the left. For the wild-type protein (WT), the conserved lysine (K) in the Walker A motif and glutamate (E) in the Walker B motif are indicated. For the mutants, the alanine (A) or glutamine (Q) altered residue is shown. (B) The p97 proteins were expressed in E. coli, purified, and subjected to SDS-PAGE. Shown is the gel after staining with Coomassie blue. (C) The ATPase activity of the different p97 proteins was measured. Shown is the mean of three experiments. (D) Wild-type Cdc48, the yeast homologue of p97, or ATP hydrolysis mutants (QE or EQ) were expressed under the gal promoter in the presence of galactose in the temperature-sensitive cdc48–3 yeast strain at the permissive (30°C) or nonpermissive (37°C) temperature (top). As control, the cells were grown on glucose to repress the genes (bottom).
Next, we analyzed the ATPase activity of the p97 mutants (Fig. 1 C). Deletion of the N domain had no effect on ATPase activity. The QE mutant had ∼50% ATPase activity compared with the wild-type protein, indicating that both the D1 and D2 domains contribute to the overall ATPase activity of p97. This is in contrast to the situation with the homologous NSF protein in which the D2 domain is inactive (Steel and Morgan, 1998). In agreement with the nucleotide-binding experiments, the two ATPase domains appear to influence one another. ATPase activity in D2 required nucleotide binding (but not hydrolysis) by the D1 domain because the mutant AK, in contrast to QE, was inactive. Thus, ATP binding to D1 is required to induce hydrolysis in D2. On the other hand, ATPase activity in D1 required nucleotide binding and hydrolysis in D2 because both the KA and EQ mutants, as well as the deletion mutant ΔD2, were inactive. This result suggests that ADP binding to D2 is required for ATP hydrolysis in D1. Together, these data suggest that the D1 and D2 rings alternate in ATP hydrolysis (see Discussion).
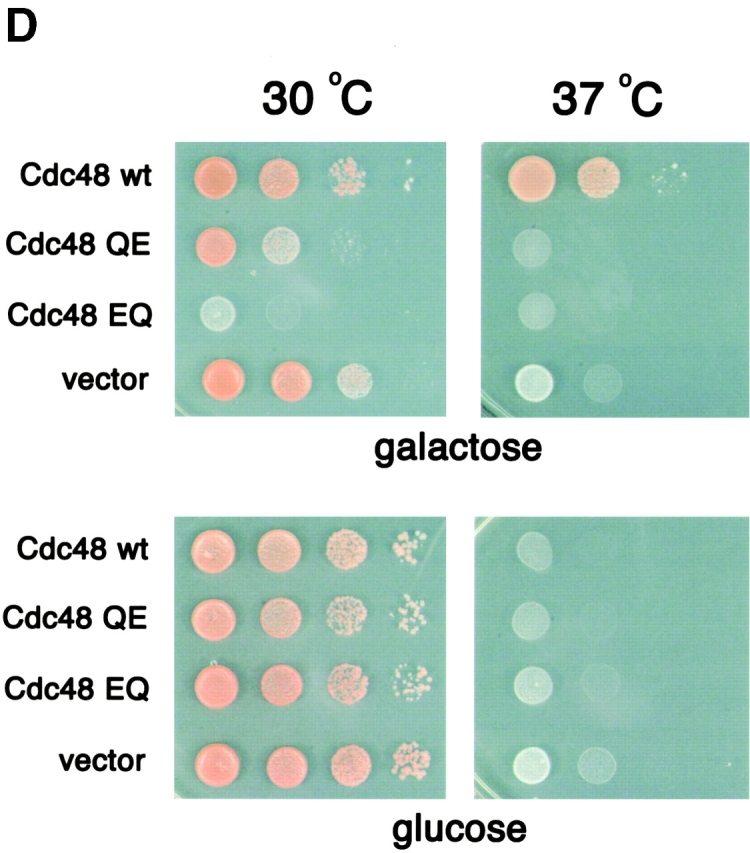
To test the role of the ATPase domains in D1 and D2 in vivo, we made the QE and EQ mutations in Cdc48, the yeast homologue of p97. The mutants were tested for complementation of a cdc48 temperature-sensitive strain at the nonpermissive temperature (Fig. 1 D). When the mutants were expressed under a gal promoter in the presence of galactose, no growth was seen at 37°C, in contrast to the results with the wild-type protein (Fig. 1 D, top right), although the transgenes were expressed at similar levels (Fig. S2). At permissive temperature (30°C), the EQ mutant exerted a strong dominant-negative effect on cell growth, whereas the effect of the QE mutant was much weaker (Fig. 1 D, top left). As expected, when the proteins were not expressed in the presence of glucose, the cells grew at 30°C, but not 37°C (Fig. 1 D, bottom). These data show that ATP hydrolysis in both ATPase domains is required for the function of the ATPase in vivo.
Cofactor and membrane interaction domains of p97
To test the interaction of p97 with its cofactors, purified p97 variants were incubated with rat liver cytosol at high salt to break endogenous complexes. After lowering the salt concentration, complexes were immunoprecipitated with antibodies directed against the NH2-terminal His tag of p97. The precipitates were analyzed by immunoblotting with antibodies to Ufd1 and p47. With the exception of the ΔN protein, all p97 variants were able to interact with both Ufd1–Npl4 and p47 (Fig. 2 A). The results show that the binding of both cofactors requires the N domain of p97.
Figure 2.


p97 binding to cofactors and membrane requires its N domain. (A) p97 binding to cofactors. Purified His-tagged p97 proteins (His-p97) were added to rat liver cytosol in the presence of 1 M NaCl. After dilution of the salt to 150 mM, p97 was immunoprecipitated with His antibodies. Bound proteins were detected by immunoblotting with antibodies to the indicated proteins. (B) Association of p97 with the membrane. Purified His-p97 proteins were added to permeabilized astrocytoma cells. After incubation, the cells were fractionated into membrane (P) and cytosol (S) fractions. The amount of His-p97 in each fraction was determined by immunoblotting with His antibodies (top). Immunoblotting for calnexin served as a loading control for the membrane fraction (bottom).
To determine which domain of p97 is required for its interaction with the ER membrane, we added various p97 proteins to permeabilized astrocytoma cells. After incubation, the membranes were sedimented and washed. Bound p97 protein was detected by immunoblotting with antibodies to the His tag in p97. As a loading control, the samples were also probed with antibodies to the ER membrane protein calnexin. Membrane binding was observed for all the p97 mutants except for the ΔN protein (Fig. 2 B). Binding was abolished when the membranes were pretreated with protease (unpublished data). These results show that the N domain mediates not only the interaction with the cofactors, but also the binding of p97 to the membrane.
Substrate interaction of p97
Next, we investigated the association of the p97 mutants with a retrotranslocation substrate. The substrate chosen was MHC class I heavy chain, which is targeted for retrotranslocation and subsequent proteasomal degradation in cells expressing the human cytomegalovirus protein US11 (Wiertz et al., 1996). The process can be recapitulated in permeabilized cells (Shamu et al., 1999). To determine binding of p97 to MHC class I heavy chains, US11-expressing cells were treated with proteasome inhibitors, labeled with [35S]methionine, and permeabilized with the detergent digitonin. His-tagged p97 variants were added, the cells were incubated for a chase period, and then they were fractionated into membrane (P) and cytosol (S) fractions. A portion of the samples was subjected to immunoprecipitation with heavy chain (HC) antibodies to monitor the amount of substrate in each sample (Fig. 3 , A–C, top panels). The remainder of the samples was subjected to sequential immunoprecipitation with His and HC antibodies to detect p97-associated heavy chains (Fig. 3, A–C, bottom panels). Previous experiments provided evidence that association of the exogenous p97 with heavy chains occurs during retrotranslocation because (1) binding to p97 was seen preferentially with heavy chains on the membrane; (2) the interaction was reduced when the membranes were solubilized before addition of p97; and (3) no binding was seen with heavy chains of cells that did not express US11 (Ye et al., 2001).
Figure 3.
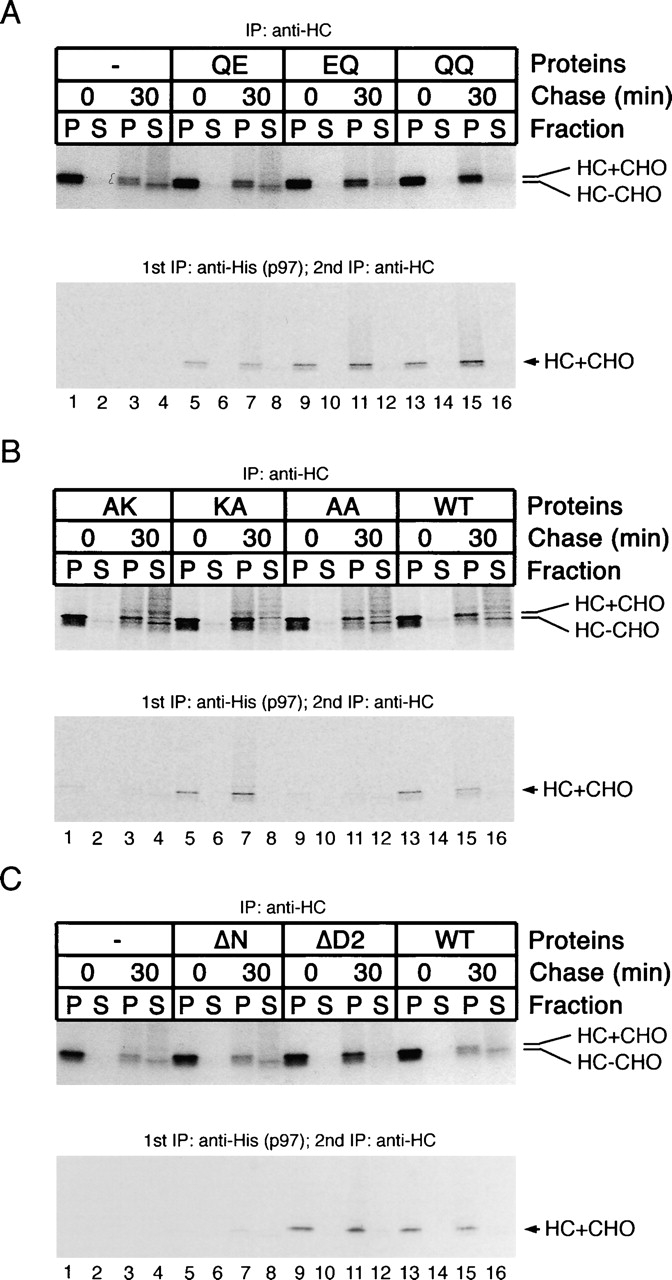
Association of p97 with MHC class I heavy chains. Cells expressing the cytomegalovirus protein US11 were incubated with proteasome inhibitors and [35S]methionine, and were then permeabilized in the presence of various purified His-p97 proteins. After incubation, samples were taken at different time points of the chase period and fractionated into membrane (P) and cytosol (S) fractions. A portion of the samples was subjected to immunoprecipitation with heavy chain (HC) antibodies to monitor the amount of substrate in each sample (A–C, top). The remainder of the samples was subjected to sequential immunoprecipitation with His and HC antibodies to detect heavy chains bound to p97 (A–C, bottom). HC+CHO and HC−CHO indicate the glycosylated and deglycosylated forms of the heavy chains, respectively.
The p97 mutants defective in ATP hydrolysis (QQ, EQ, and QE) interacted with heavy chains on the membrane, although the QE mutant had lower affinity (Fig. 3 A, bottom, lanes 5–16). The nucleotide-binding KA mutant, but not the AK and AA proteins, also bound heavy chains (Fig. 3 B, bottom, lanes 5–8 vs. lanes 1–4 and 9–12), suggesting that ATP binding in D1 is required for substrate binding. As might be expected from the lack of membrane interaction, the ΔN protein did not interact with substrate (Fig. 3 C, bottom, lanes 5–8). Because the AK and AA proteins cannot interact with substrate but still bind to membranes (Fig. 2 B), it appears that p97 binds to membranes not solely through an interaction with substrate.
Next, we tested whether the mutant p97 proteins have a dominant-negative effect on the retrotranslocation of MHC class I heavy chains (Fig. 3 A–C, top panels). In the absence of added p97 or in the presence of exogenous wild-type p97, a significant fraction of the heavy chains was retrotranslocated into the cytosol during the chase period (Fig. 3 A, top, lane 4; Fig. 3 B, top, lane 16). The cytosolic heavy chains have a slightly faster mobility in the SDS gel because they are deglycosylated by a cytosolic N-glycanase. Likewise, p97 mutants that were defective in substrate interaction (AK, AA, and ΔN) also did not affect retrotranslocation. In contrast, the p97 variants that allowed substrate binding but did not permit ATP hydrolysis (EQ, QQ, KA, and ΔD2; Fig. 1 C) strongly inhibited heavy chain dislocation; the amount of deglycosylated heavy chains in the cytosol fraction (S) was reduced, and the amount of glycosylated heavy chains in the membrane fraction (P) was increased (Fig. 3, top). The D1 hydrolysis mutant QE, which had lower affinity to substrate and had ∼50% residual ATPase activity (Fig. 1 C), had a weak effect on heavy chain retrotranslocation (Fig. 3 A, top, lane 8 vs. lane 4 and lane 7 vs. lane 3; see also Fig. 4). These data indicate that the dominant-negative effect of the p97 mutants is a consequence of sequestering heavy chains in a defective ATPase complex.
Figure 4.

Effect of p97 mutants on MHC class I heavy chain degradation. US11-expressing cells were labeled and permeabilized in the presence of various purified His-p97 proteins. The samples were chase-incubated for different time periods and subjected to immunoprecipitation with heavy chain (HC) antibodies. The graphs show the quantification of the experiments. Error bars in the bottom graph are derived from three experiments.
These conclusions were confirmed when we directly tested the dominant-negative effect of the p97 mutants on MHC class I heavy chain degradation in the absence of proteasome inhibitors (Fig. 4). Again, the mutants that allowed substrate binding but had a low ATPase activity (EQ, QQ, KA, and ΔD2 mutants) inhibited retrotranslocation and stabilized the substrate. In agreement with the in vivo results in yeast (Fig. 1 D), the QE mutant had a much smaller dominant-negative effect than the EQ mutant. All mutants that failed to bind heavy chains (AK, AA, and ΔN) had no effect. Together, these results show that membrane binding of p97 requires its N domain, whereas substrate binding additionally requires the D1 domain in its nucleotide-bound state; ATP hydrolysis by the D2 domain, and probably also the D1 domain, are needed to move the polypeptide chain into the cytosol.
Ubiquitination is not required for substrate recognition by p97
Although the majority of the heavy chains bound to p97 were unmodified (Fig. 3, bottom), the results did not exclude that they were originally polyubiquitinated when they bound to p97 and were subsequently de-ubiquitinated by an isopeptidase. De-ubiquitination must, in fact, be a very efficient process because the heavy chains accumulating in the cytosol in the presence of proteasome inhibitor are mostly unmodified (Fig. 3 A, top, lane 4), despite the fact that they must have been polyubiquitinated on the membrane (Shamu et al., 2001). To test whether p97 can interact with heavy chains that were never modified, the cytosol in permeabilized US11 cells was replaced with ubiquitin-depleted cow liver cytosol (Shamu et al., 2001). Pulse-chase experiments showed that in the absence of ubiquitin, the heavy chains were stable and remained in the ER membrane (Fig. 5 A, lanes 6 and 7 vs. lanes 2 and 3). To test substrate binding of p97, we added His-tagged p97 to permeabilized cells incubated for different time periods in cytosol containing or lacking ubiquitin, and performed sequential immunoprecipitations with antibodies to the His tag and to the heavy chains. In the absence of ubiquitin, a significant fraction of the heavy chains was bound to p97 (Fig. 5 B, bottom, lanes 7–12). The fraction increased during the chase incubation, indicating that the movement of heavy chains from the ER lumen to the cytoplasmic side of the ER membrane is independent of ubiquitination. When purified ubiquitin was added back to ubiquitin-depleted cytosol, an interaction of substrate with p97 was only seen at early time points during the chase incubation (Fig. 5 B, bottom, lanes 13–18); because ubiquitin restored degradation of the heavy chains (Fig. 5 B, top), there was little material left for interaction with p97 at later time points. As expected, no material was immunoprecipitated when His-tagged p97 was omitted from the incubation (Fig. 5 B, bottom, lanes 1–6). As expected from the previous results (Fig. 3), the QQ, but not the ΔN and AA mutants of p97, interacted with nonubiquitinated substrate (Fig. 5 C). These data suggest that p97 can specifically interact with substrate that has not been modified by a polyubiquitin chain, and that binding of p97 is an early step after emergence of the polypeptide from the ER.
Figure 5.
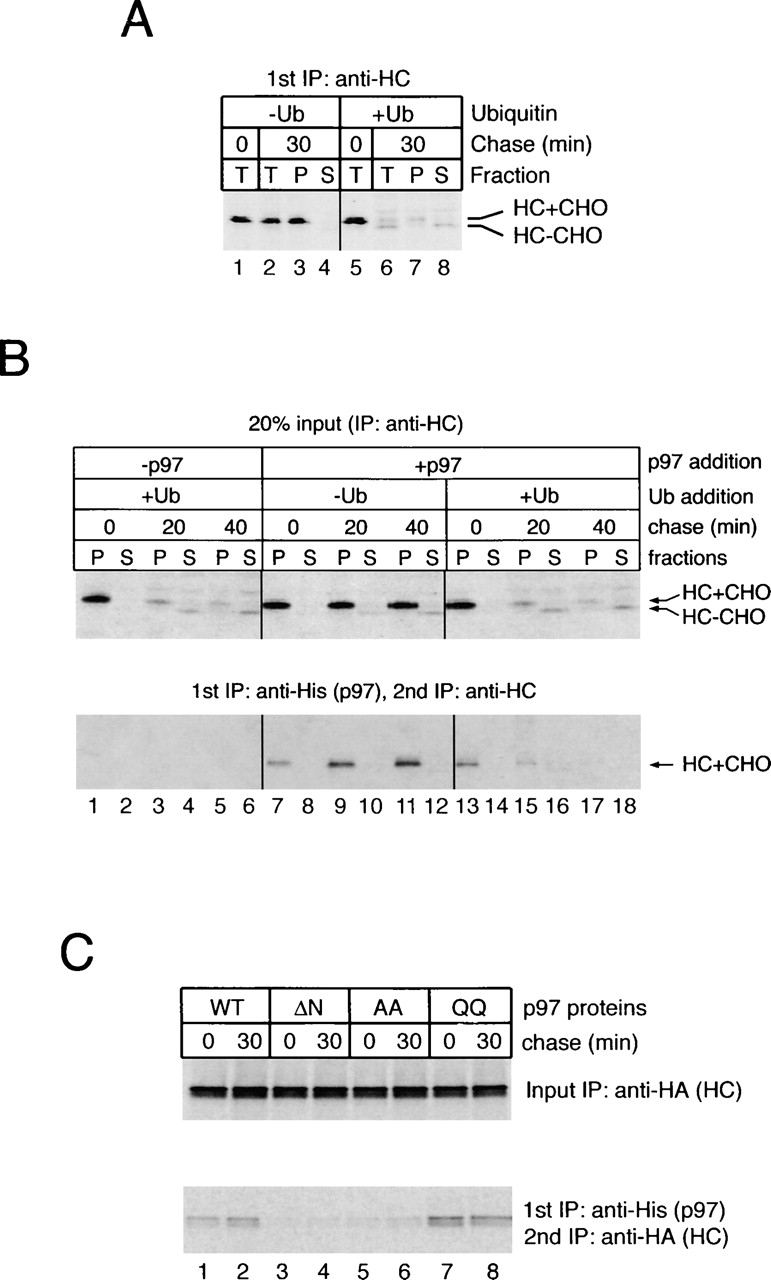
p97 binds to heavy chains that were not ubiquitinated. (A) US11-expressing cells were labeled and permeabilized. The cytosol was removed by high speed centrifugation and replaced with cow liver cytosol that was ubiquitin depleted (−Ub). Where indicated, purified ubiquitin was added back to the depleted cytosol (+Ub). After chase incubation, the samples were either analyzed directly by immunoprecipitation with heavy chain (HC) antibodies (T), or first fractionated into membrane (P) and cytosol (S) fractions before immunoprecipitation with HC antibodies. (B) Purified His-p97 was added during the permeabilization of labeled cells. A portion of the membrane and cytosol fractions was subjected to immunoprecipitation with HC antibodies (top), and the rest was analyzed by sequential immunoprecipitation with His and HC antibodies (bottom). (C) Astrocytoma cells stably expressing hemagglutinin (HA)-tagged MHC class I heavy chains were permeabilized and incubated with ubiquitin-depleted cytosol in the presence of purified wild-type His-p97 or the indicated His-tagged mutant proteins. The membrane fraction was subjected to immunoprecipitation with HA antibodies (top) or His and HA antibodies (bottom). Note that the increase in the amount of p97-associated, HA-tagged heavy chains is less apparent than that in B, which may result from heavy chain overexpression.
Recognition of the polyubiquitin chain by the p97–Ufd1–Npl4 complex
Because the p97-dependent release of substrate from the membrane requires polyubiquitination, we tested how the polyubiquitin chain is recognized by p97 and its cofactor Ufd1–Npl4. To address interactions between polyubiquitin and the ATPase complex, we used polyubiquitin chains synthesized in vitro using purified recombinant proteins. The polyubiquitination reaction contained ubiquitin, the ubiquitin-activating enzyme (E1), the ubiquitin-conjugating enzyme Ubc7 (E2), and a GST fusion to a cytosolic fragment of the ubiquitin ligase gp78 (E3; the purity of the recombinant E2 and E3 proteins is shown in Fig. 6 A, lanes 1 and 2). We used Ubc7 and gp78 because both are known to be involved in the degradation of at least some misfolded ER proteins (Fang et al., 2001; Tiwari and Weissman, 2001). When all components were incubated together in the presence of ATP, ubiquitin conjugates consisting of 2 to ∼40 ubiquitin molecules were synthesized (Fig. 6 B, lane 4 vs. lanes 1–3). The majority of chains are not associated with the E3 enzyme because they did not bind to glutathione beads with the GST-gp78 protein (unpublished data). No polyubiquitin chains were formed when wild-type ubiquitin was replaced by methylated ubiquitin, in which all lysines are blocked, or by the ubiquitin K48R mutant, which carries an arginine instead of the lysine at position 48 (Fig. 6 C). Moreover, when ubiquitin derivatives with single lysines at different positions were tested, only the mutant with a lysine at position 48 (K48) gave efficient polyubiquitination (Fig. 6 D, lane 7). These data show that polyubiquitin chains synthesized in this system are linked through lysine 48, the same linkage required for protein degradation by the proteasome.
Figure 6.
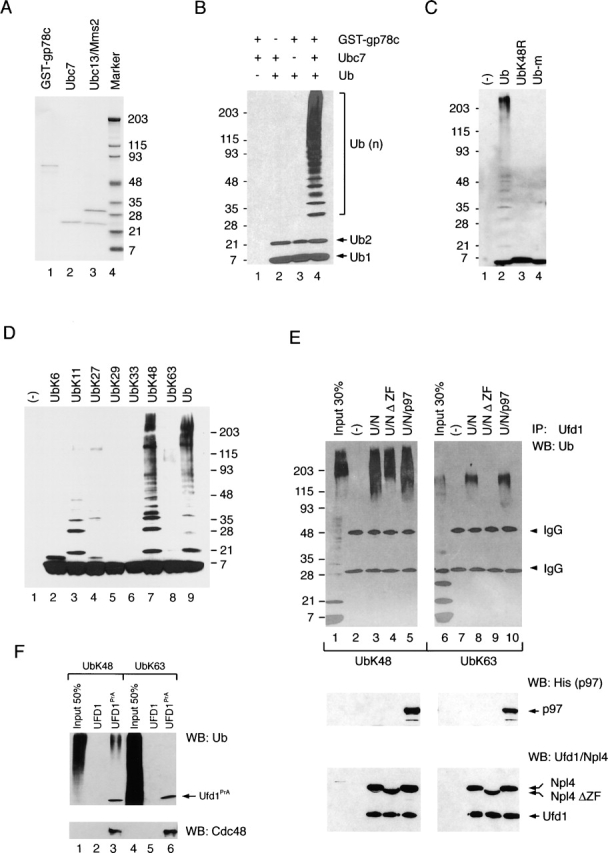
Ubiquitin recognition by the p97–Ufd1–Npl4 complex. (A) A GST fusion to the cytosolic fragment of the ubiquitin ligase gp78, the ubiquitin-conjugating enzyme Ubc7, and the ubiquitin-conjugating complex consisting of the Ubc13 and Mms2 were all expressed in E. coli and purified. Shown is a Coomassie-stained gel after SDS-PAGE with mol wt markers on the right side. (B) In vitro ubiquitination reactions were performed with the indicated components in the presence of ubiquitin activation enzyme (E1) and ATP. The samples were analyzed by immunoblotting with ubiquitin antibodies. Ub, Ub2, and Ub(n) indicate the positions of ubiquitin, di-ubiquitin, and polyubiquitin chains, respectively. (C) Shown is a comparison of wild-type ubiquitin (Ub) with methylated ubiquitin (Ub-m) and a mutant ubiquitin carrying a substitution of lysine 48 by arginine (UbK48R). The reactions were performed as in B. (D) Ubiquitin mutants with single lysines at the indicated position were tested in reactions similar to those in B. (E) Polyubiquitin chains were synthesized with either Ubc7/GST-gp78c (lanes 1–5) or Ubc13/Mms2 (lanes 6–10), generating chains with lysine 48 and lysine 63 linkages, respectively. These chains were incubated with the indicated proteins, and the samples were subjected to immunoprecipitation with Ufd1 antibodies (lanes 2–5 and lanes 7–10). Bound ubiquitin chains were detected by immunoblotting with ubiquitin antibodies (Ub; top). A portion of the immunoprecipitates was analyzed by immunoblotting with His (middle) or Ufd1 and Npl4 antibodies (bottom). UbK48 and UbK63 indicate chains with lysine 48 and lysine 63 linkages, respectively. (F) Yeast cytosol from a control strain (UFD1) or a strain expressing protein A–tagged UFD1 (UFD1PrA) was incubated with IgG beads. The beads were incubated with in vitro–synthesized polyubiquitin chains, and the bound material was analyzed by immunoblotting with ubiquitin antibodies (top) and Cdc48 antibodies (bottom).
Then, we tested whether the cofactor Ufd1–Npl4 interacts with these polyubiquitin chains. In vitro–synthesized polyubiquitin chains were incubated with either the cofactor complex Ufd1–Npl4 (U/N) or with the ternary complex Ufd1–Npl4–His-p97 (U/N/p97). Antibodies to Ufd1 were used to precipitate the complexes, and indeed brought down all components, as demonstrated by immunoblotting with different antibodies (Fig. 6 E, bottom). Bound ubiquitin chains were detected by immunoblotting with anti-ubiquitin antibodies (Fig. 6 E, top). Both the U/N and U/N/p97 complexes efficiently bound polyubiquitin chains (Fig. 6 E, top, lane 3 and lane 5). These data confirm the recent finding that the cofactors provide a binding site for polyubiquitin chains (Meyer et al., 2002). Interestingly, however, deletion of the NZF domain of Npl4, the only known binding site for polyubiquitin chains in the cofactor complex, reduced (but did not abolish) the interaction (lane 4 vs. lane 3). Thus, a hitherto unidentified polyubiquitin-binding domain must exist in other portions of the Ufd1–Npl4 proteins.
To test the specificity of the ubiquitin interaction with the cofactor Ufd1–Npl4, we used in vitro–synthesized polyubiquitin chains with linkages through lysine 63 (K63). These chains were synthesized with the yeast ubiquitin–conjugating enzyme (E2) consisting of Ubc13 and Mms2 (Hofmann and Pickart, 1999; the purity of the dimer is shown in Fig. 6 A, lane 3). These polyubiquitin chains were also bound to both the U/N and U/N/p97 complexes (Fig. 6 E, top, lane 8 and lane 10). However, the lysine 63 chains were not bound when the NZF domain was missing in Npl4 (Fig. 6 E, top, lane 10; <5% of the input material was bound). Thus, in the absence of the NZF domain, the interaction of the Ufd1–Npl4 complex with polyubiquitin chains is specific for the lysine 48 linkage. The NZF domain, on the other hand, appears to be promiscuous, and interacts with both lysine 48– and lysine 63–linked ubiquitin chains.
Because the yeast homologue of Npl4 lacks the NZF domain, one might expect that the yeast Cdc48–Ufd1–Npl4 complex interacts with lysine 48– but not lysine 63–linked polyubiquitin chains. To test this possibility, we replaced the wild-type Ufd1 protein in S. cerevisiae with a fusion of Ufd1 to an IgG-binding domain (Ufd1–PrA). The cells were homogenized and the Cdc48–Ufd1–Npl4 complex isolated by binding to IgG beads. As expected, Cdc48 was bound to the beads together with the tagged Ufd1 protein (Fig. 6 F, bottom, lane 3 and lane 6). When incubated with in vitro–synthesized polyubiquitin chains, the complex indeed bound chains with lysine 48 linkage (Fig. 6 F, top, lane 3), but not those with lysine 63 linkage (Fig. 6 F, lane 6). Because both the yeast Cdc48–Ufd1–Npl4 complex and the mammalian Ufd1–Npl4ΔZF lacking the NZF domain discriminate between lysine 48 and lysine 63 linkages, these data suggest that the unidentified ubiquitin-binding site in the Ufd1–Npl4 complex may be the one relevant for the conserved function in ER protein degradation.
Next, we determined that the novel binding site is contained in Ufd1. A purified mammalian Npl4 protein lacking the NZF motif (Npl4ΔZF) did not bind ubiquitin chains in the absence of Ufd1 (unpublished data), whereas a GST fusion to full-length Ufd1 interacted with polyubiquitin chains containing lysine 48 linkages (Fig. 7 B, lane 3). A GST fusion to the NH2-terminal 215 residues of Ufd1 (UT3 domain; Fig. 7 A), which was predicted to contain a double Ψ barrel fold (Coles et al., 1999; Golbik et al., 1999), also bound ubiquitin chains (Fig. 7 B, lane 4). On the other hand, a GST fusion to the COOH-terminal UT6 domain (Fig. 7 A), containing binding sites for both p97 and Npl4 (Hetzer et al., 2001), was inactive (Fig. 7 B, lane 5).
Figure 7.
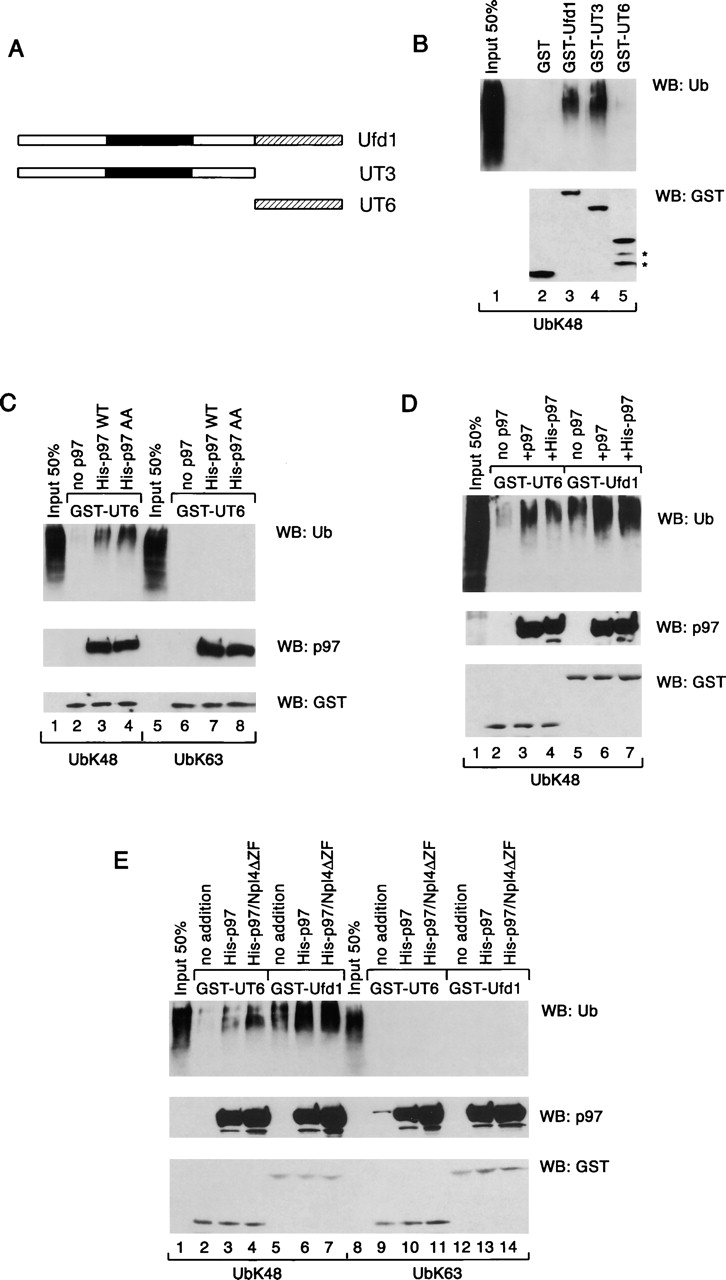
Polyubiquitin binding to p97 and Ufd1. (A) Scheme of Ufd1 and mutant constructs used in the binding assays. The black box indicates the double Ψ barrel domain. The shaded box shows the p97- and Npl4-binding domain UT6. (B) Polyubiquitin chains synthesized with Ubc7/GST-gp78c were added to glutathione beads containing bound GST fusions to full-length Ufd1 or fragments of it. The bound fraction was analyzed by immunoblotting with ubiquitin and GST antibodies (top and bottom panels, respectively). The stars in the bottom panel indicate degradation products of GST-UT6. (C) A GST fusion to the UT6 domain was immobilized on glutathione beads in the absence (no p97) or presence of His-tagged wild-type p97 or AA mutant. Binding of polyubiquitin chains with lysine 48 linkages was tested as in B. Bound proteins were also analyzed by immunoblotting with p97 (middle) or GST (bottom) antibodies. As a control, binding of polyubiquitin chains with lysine 63 linkages was tested (lanes 5–8). (D) GST fusions to the UT6 domain or Ufd1 were immobilized on glutathione beads in the absence (no p97) or presence of excess untagged or His-tagged p97. Polyubiquitin binding was analyzed as in C. (E) GST fusions to the UT6 fragment and Ufd1 were immobilized in the presence of p97 or p97 plus Npl4 lacking the NZF domain (Npl4ΔZF). Polyubiquitin binding was tested as in D. Lanes 8–14 show controls with lysine 63–linked polyubiquitin chains.
To test whether p97 can also bind polyubiquitin chains, the His-tagged p97 was immobilized via a GST-UT6 fragment before incubation with polyubiquitin chains. With chains containing lysine 48 linkages, binding was indeed observed (Fig. 7 C, lane 3 vs. lane 2). As with Ufd1, the interaction was not seen with lysine 63 linkages (Fig. 7 C, lanes 6–8). The AA mutant of p97, which is unable to recognize substrate (Fig. 3), also bound polyubiquitin chains (Fig. 7 C, lane 4), indicating that substrate and ubiquitin binding can be separated. Together, these data suggest that both p97 and Ufd1 can bind polyubiquitin chains.
The interactions of p97 and Ufd1 with polyubiquitin appear to be synergistic. When both components were immobilized together, polyubiquitin binding was significantly stronger than with either component alone (Fig. 7 D, lane 6 and 7 vs. lanes 3–5). A similarly strong binding was seen when all three components (p97, Ufd1, and Npl4ΔZF) were together in a ternary complex (Fig. 7 E, lane 7). These interactions are again specific for lysine 48–linked polyubiquitin chains (Fig. 7 E, lanes 1–7 vs. lanes 8–14). Together, our data suggest that the Ufd1–Npl4 complex has an evolutionarily conserved function in mediating ubiquitin binding of the ATPase complex. The Ufd1–Npl4–p97 complex contains two conserved and cooperating polyubiquitin-binding sites, one in p97 itself, and one in the UT3 domain of Ufd1; the mammalian complex also contains the NZF domain that may further increase the binding of polyubiquitin.
The ubiquitin-binding domain of Ufd1 is required for retrotranslocation
Next, we tested whether the ubiquitin-binding domain in Ufd1 is important for the dislocation and degradation of MHC class I heavy chains. We reasoned that a mutant Ufd1–Npl4 complex lacking the UT3 ubiquitin-binding site would act as a dominant-negative mutant by sequestering endogenous p97 in a defective complex that could no longer efficiently interact with ubiquitin chains on the substrate. Indeed, although addition of either wild-type Ufd1–Npl4 (U/N) or Ufd1–Npl4ΔZF (U/NΔZF) had no effect on heavy chain degradation (Fig. 8 A, top; for quantification, see top right panel), the corresponding complexes lacking the UT3 domain (UΔUT3/N and UΔUT3/NΔZF) were inhibitory (Fig. 8 A, bottom). Because all complexes tested bind p97 with similar affinity (unpublished data), the results suggest that lack of ubiquitin interaction may cause the dominant-negative effect. With the mutant lacking UT3 (Ufd1ΔUT3/Npl4), polyubiquitinated heavy chains accumulated on the membrane (Fig. 8 B, bottom, lane 11 vs. lane 3), as they did when the ATPase mutant KA was added (Fig. 8 B, lane 7; quantification given above the lanes). In controls, the ubiquitinated heavy chains were released into the cytosol (Fig. 8 B, lane 4). Thus, both ubiquitin binding and ATP hydrolysis by p97–Ufd1–Npl4 complex are required for the release of polyubiquitinated substrates into the cytosol. Ubiquitin binding to UT3, and not to the NZF domain of Npl4, appears to be important for retrotranslocation.
Figure 8.
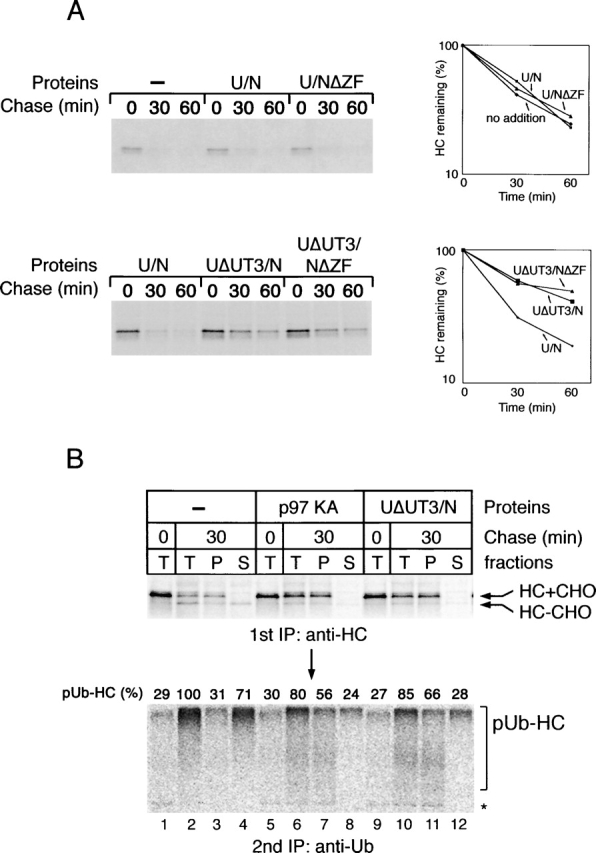
The UT3 domain of Ufd1 is required for retrotranslocation. (A) US11-expressing cells were labeled and permeabilized in the presence of different Ufd1–Npl4 (U/N) variants. The added U/N complexes included wild-type U/N or U/N lacking either the NZF domain in Npl4 (U/NΔZF), the UT3 domain in Ufd1 (UΔUT3/N), or both the NZF domain and the UT3 domains (UΔUT3/NΔZF). After incubation for the indicated time periods, the samples were analyzed by immunoprecipitation with heavy chain (HC) antibodies. The graphs show the quantification of the experiments. (B) Comparison of the dominant-negative effects of the p97 ATPase mutant KA and of the cofactor complex UΔUT3/N. The experiment was performed as in A, but proteasome inhibitors were included during incubation. The samples were analyzed both directly (T) and after fractionation into membrane (P) or cytosol (S) fractions. A portion of the precipitated HC was subjected to a second round of immunoprecipitation with ubiquitin (Ub) antibodies (bottom). Quantification of the polyubiquitin chains is given above the lanes, with the results in lane 2 set to 100%.
Discussion
Our results shed light on how the AAA ATPase p97 functions together with its cofactor Ufd1–Npl4 in protein retrotranslocation. One conclusion from the study of different mutants is that the two ATPase domains of p97 are functionally coupled. The activity of the second ATPase domain (D2) requires nucleotide binding, but not hydrolysis by the first ATPase domain (D1), indicating that the D2 domain may hydrolyze nucleotides when D1 is in its ATP-bound state. It appears that D2 can only bind ATP when D1 is in its nucleotide-bound state. In contrast, the ATPase activity of D1 depends on both ATP binding and hydrolysis by the D2 domain, suggesting that the D1 domain likely hydrolyzes ATP when D2 is in its ADP-bound form. Therefore, we propose an ATPase cycle in which there is alternate ATP hydrolysis in the two ATPase rings (Fig. 9 A). In this model, ATP hydrolysis in D2 precedes that in D1. Conformational changes consistent with this proposal have been observed (Rouiller et al., 2002). Previous results had suggested that the D1 domain does not hydrolyze ATP (Song et al., 2003), but our results both in vitro and in yeast cells indicate that activity of D1 is required for the function of the protein. The previous results may perhaps be explained by the use of a different mutation in the active site of D1 (K to T mutation).
Figure 9.


ATPase cycle and substrate recognition of p97. (A) Proposed communication between the two ATPase rings (D1 and D2) of p97. The D2 domain hydrolyzes ATP (T) and releases an orthophosphate (Pi), first converting the ring into its ADP-bound state (D). ATP hydrolysis in D1 follows. (B) A dual recognition model for p97–Ufd1–Npl4 function in retrotranslocation. Protein retrotranslocation is proposed to proceed in four steps. (I) A polypeptide chain emerging from the ER may be able to slide back and forth through the membrane (arrows), probably through a protein-conducting channel. The p97–Ufd1–Npl4 complex is recruited to the membrane by an unknown receptor (green). (II) A nonubiquitinated substrate is initially recognized by p97 (the ATPase domains D1 and D2 are shown in yellow, the N domain in blue). This interaction may prevent the substrate from backsliding into the ER lumen. The substrate also undergoes ubiquitination (red) at the membrane. (III) Once the ubiquitin chain (red) reaches a certain length, it interacts with p97 and the cofactor complex Ufd1–Npl4 (black). (IV) p97 uses ATP hydrolysis to move the polypeptide into the cytosol.
Also, we have shown that substrate binding by p97 requires D1, but not D2, to be in a nucleotide-bound state. Other ATPases (such as GroEL/S, Hsp70, or ClpA) also require ATP binding, but not hydrolysis, for substrate binding. In fact, the proposed alternate hydrolysis in the two ATPase rings of p97 is reminiscent of the mechanism of GroEL/S (for review see Bukau and Horwich, 1998). Another AAA ATPase, Hsp104, also displays a similar communication between the two ATPase rings (Cashikar et al., 2002).
We propose that the polypeptide substrate is transported through the central pore in the double barrel structure of p97 in the direction of the D1 to the D2 ring. Our data indeed suggest that the D1 ring is proximal to the membrane because the N domain, located at its side, is required for membrane interaction of p97. However, because the N domain is also involved in cofactor binding, it is unclear which component makes direct contact with the membrane. Because a mutant of p97 defective in substrate binding could still bind to the membrane, substrate binding cannot be the only linkage of the ATPase complex with the membrane. An interaction with an unidentified membrane receptor may explain why a large fraction of p97 and of its yeast homologue Cdc48 is firmly bound to the ER.
Our results provide insight into how the p97–Ufd1–Npl4 complex recognizes retrotranslocation substrates. p97 itself recognizes both polyubiquitinated and nonubiquitinated polypeptides. The strongest evidence that p97 can interact with nonmodified substrate comes from our experiments in which polyubiquitination was prevented. Because p97 binding to nonmodified substrate was similarly observed in a crude permeabilized cell system in the presence of ubiquitin, the interaction is not caused by ubiquitin depletion and is of physiological relevance. Although direct contact of p97 with substrate remains to be demonstrated, our data show that the interaction is nucleotide dependent, as observed for other ATPases (for review see Bukau and Horwich, 1998; Hoskins et al., 2000). An appealing possibility is that p97 recognizes unfolded hydrophobic polypeptide segments as demonstrated with model substrates (Thoms, 2002). The VAT protein, a homologue of p97 in archaebacteria, must also recognize nonubiquitinated substrates, because no ubiquitination exists in these organisms.
In a pulse-chase experiment, the amount of p97-associated substrate increased with time when ubiquitination was abolished, suggesting that p97 may interact with nonmodified polypeptides as soon as they emerge from the ER membrane, preventing their backsliding into the ER lumen (Fig. 9 B). Although it has been proposed that ubiquitin mediates the initial interaction of the transcription factor Spt23 with the Cdc48–Ufd1–Npl4 complex (Rape et al., 2001), binding to an unfolded segment of the polypeptide has not been excluded. At least for retrotranslocation substrates, it is likely that polyubiquitination succeeds substrate binding to p97.
Our data suggest that the polyubiquitin chain is recognized by both p97 and the cofactor Ufd1–Npl4. The finding that p97 can weakly interact with polyubiquitin chains is in agreement with previous results (Dai and Li, 2001; Rape et al., 2001). The interaction of p97 with ubiquitin appears to be distinct from that with nonmodified substrate because it can be observed with the AA mutant of p97 that is defective in substrate binding. Polyubiquitin binding to the Ufd1–Npl4 complex was previously ascribed to the NZF domain of Npl4 (Meyer et al., 2002). With a more sensitive binding assay, we now demonstrate that the UT3 domain of Ufd1 contains an additional ubiquitin-binding site that has a lower affinity than the NZF domain, but appears to be more important for retrotranslocation. This domain is similar in sequence among all eukaryotic Ufd1 proteins, consistent with a conserved function in retrotranslocation, and a cofactor complex lacking this domain acts as a dominant-negative mutant in retrotranslocation. These data also demonstrate that the mammalian cofactor Ufd1–Npl4, like its homologue in yeast, plays a role in ER protein degradation. The UT3 domain recognizes polyubiquitin chains in a lysine 48 (but not lysine 63) linkage, as might be expected from the similar preference by the proteasome acting downstream of p97 in the ER degradation pathway. Longer polyubiquitin chains are the preferred substrate for Ufd1, in agreement with our previous finding that a certain chain length is required for substrate release from the membrane (Shamu et al., 2001). Whether the ubiquitin-binding NZF domain of Npl4 plays a role in retrotranslocation is unclear because it is missing in the yeast protein, does not show specificity for the ubiquitin linkage, and because a mammalian cofactor complex lacking this domain is not dominant negative. It could play a supporting role in ER protein degradation in mammalian cells, or could be involved in p97–Ufd1–Npl4 dependent processes other than retrotranslocation.
Our data suggest that p97 and Ufd1 act synergistically in polyubiquitin binding. One possibility is that the proteins together form a binding site with higher affinity. Specifically, double Ψ barrel folds are present in both the UT3 domain of Ufd1 and the N domain of p97 (Coles et al., 1999; Golbik et al., 1999), and these could both bind weakly to polyubiquitin chains containing lysine 48 linkages. When present together in a complex, the simultaneous interaction of the double Ψ barrel folds with polyubiquitin chains could increase the affinity.
On the basis of our results, we propose a model of dual substrate recognition by the p97–Ufd1–Npl4 complex; a nonubiquitinated, unfolded segment of a retrotranslocation substrate would initially be recognized by p97 itself (Fig. 9 B). Once a polyubiquitin chain has been attached to the substrate, it would be recognized by both p97 and the cofactor Ufd1 in the complex. This interaction may activate the ATPase to pull the polypeptide chains out of the membrane. Purified p97 has a relatively high intrinsic ATPase activity, and therefore stimulation of the ATPase activity may require that the basal activity in the cell be inhibited, perhaps by the membrane receptor of p97. In the cytosol, the basal ATPase activity of p97 is inhibited by the cofactor p47 (Meyer et al., 1998). Consistent with our model, mutants in the p97–Ufd1–Npl4 complex that can bind the substrate, but either lack ubiquitin interaction or are defective in the ATPase cycle, are dominant negative for retrotranslocation. The dual recognition model explains why p97 interacts with nonubiquitinated substrates and why polyubiquitination is nevertheless required for its function. The unmodified segments of the polypeptide chain may be inserted as a loop into the central cavity of the p97 double barrel, whereas the bulky polyubiquitin chains may stay bound at the side of the barrel. Loop insertion of a substrate polypeptide is also discussed for the proteasome (Liu et al., 2003).
Materials and methods
Constructs
To generate the ATP hydrolysis mutants, codons E305 and/or E578 in plasmid pQE-p97 (Meyer et al., 2000) were mutated to Q. The ATP-binding mutants were generated by changing codons K251 and/or K524 to A. The ΔN and the ΔD2 constructs carry deletions of the NH2-terminal 185 residues or the COOH-terminal 348 residues, respectively. Plasmids encoding mammalian Npl4, a construct lacking the NZF domain (Npl4ΔZF), COOH-terminally His-tagged Ufd1, and GST fusions to Ufd1, UT3, and UT6 were all described previously (Meyer et al., 2000, 2002). UT3-HIS was derived from pET–Ufd1–His by deleting the UT6 coding segment; His-UT6 was generated by cloning the UT6 coding DNA segment into pTrcHis (Invitrogen). The plasmids pGEX-gp78c and pGEX-MmUBC7 were provided by A. Weissman (National Cancer Institute, Bethesda, MD). Genes coding for yeast Ubc13 and Mms2 were amplified by PCR from yeast genomic DNA and cloned into the pTrcHis-TOPO® vector (Invitrogen). Wild-type Cdc48 was cloned into pYC2NT (Invitrogen), and amino acids E315 and E588 were altered to Q.
Antibodies, chemicals, and proteins
Antibodies to MHC class I heavy chain, ubiquitin, and the His tag have been described previously (Ye et al., 2001). Antibodies to mammalian Ufd1, Npl4, and p47 were described by Meyer et al. (2000). The Cdc48 antibodies were a gift from M. Latterich (McGill University, Montreal, Canada). Calnexin and GST antibodies were purchased from StressGen Biotechnologies and Amersham Biosciences, respectively; proteasome inhibitor MG132 was purchased from Calbiochem; bovine ubiquitin was purchased from Sigma-Aldrich; and ubiquitin K48R, methylated ubiquitin, and ubiquitin activating enzyme (E1) was purchased from Affinity BioReagents, Inc.
Protein purification
His-tagged proteins were all purified using Ni-NTA beads (QIAGEN). With the exception of p97 variants, all proteins eluted from the Ni column were further fractionated on a Mono-Q column using a 0–500-mM potassium chloride gradient. The p97 variants were further purified by size-exclusion chromatography on a Superdex™ 200 HR (10/30) column in 50 mM Tris/HCl, pH 8.0, 150 mM potassium chloride, 5% glycerol, and 2 mM magnesium chloride. The complex of His-Ubc13/His-Mms2 was assembled on the same size-exclusion column using the same conditions. GST-gp78c and GST-Ubc7 were purified with glutathione beads. GST-gp78c was eluted with reduced glutathione, whereas Ubc7 was cleaved off from the beads using thrombin. The eluted proteins were further purified on a Mono-Q column. The purification of GST-Ufd1, GST-UT3, GST-UT6, Npl4, Npl4ΔZF, Ufd1-His, UT3-His, and His-UT6, as well as the assembly of various Ufd1–Npl4 complexes was described previously (Meyer et al., 2002). His-UT6 and Npl4 as well as His-UT6 and Npl4ΔZF were coexpressed in E. coli and purified on Ni-NTA. All purified proteins were stored at −80°C in 50 mM Tris/HCl, pH 8.0, 150 mM potassium chloride, 2 mM magnesium chloride, and 5% glycerol.
ATPase measurements, nucleotide and cofactor binding, and membrane association
The ATPase activity of p97 variants was measured as described previously (Meyer et al., 1998). To test nucleotide binding by the p97 variants, 100 μg proteins were incubated with 0.2 mM ATP plus 2 μCi α[32P]ATP in 50 mM Tris/HCl, pH 8.0, 150 mM KCl, 1 mM magnesium chloride, and 15% glycerol at 4°C for 20 min, and fractionated by a gel filtration column (NAP5; Amersham Biosciences). Nucleotides in each fraction were analyzed by TLC. The cofactor-binding assay was performed as before with slight modification (Meyer et al., 2000). 2-μg purified His-tagged p97 proteins were added to 500 μg rat liver cytosol in 1 M NaCl, 50 mM Hepes, pH 7.6, 250 mM sucrose, 50 mM potassium acetate, 6 mM magnesium chloride, 1 mM EDTA, and 1 mM DTT plus protease inhibitor. The salt concentration was adjusted to 150 mM with 20 mM Hepes, pH 7.6 and 1 mM DTT. p97 proteins were immunoprecipitated with His antibodies, and the bound proteins were analyzed by immunoblotting with p47, Ufd1, and His antibodies. For membrane-binding experiments, US11 cells were permeabilized with 0.028% digitonin in the presence of various p97 proteins at a concentration of 1 μM. After incubation, the membranes were sedimented and washed with permeabilization buffer without detergent (25 mM Hepes, pH 7.3, 115 mM potassium acetate, 5 mM sodium acetate, 2.5 mM magnesium chloride, and 0.5 mM EGTA). Proteins in the membrane and cytosol fractions were solubilized in sample buffer and analyzed by immunoblotting.
Dislocation and degradation of heavy chains and substrate binding
The experiments were performed as described previously (Ye et al., 2001). Recombinant proteins were added at a 1-μM concentration. Immunoprecipitation with heavy chain antibodies and reprecipitation with ubiquitin antibodies were done as described previously (Shamu et al., 1999). The interaction of p97 with MHC heavy chains was analyzed as described previously (Ye et al., 2001). Ubiquitin depletion experiments were performed as described previously by incubating cow liver cytosol with a GST fusion to a mutant of the ubiquitin-conjugating enzyme Ubc2B, which carries a Ser instead of a Cys in the active site (Shamu et al., 2001).
In vitro ubiquitination and ubiquitin binding
Polyubiquitin chains were synthesized at 37°C with either 4 μM Ubc7, 1μM GST-gp78c, or 2 μM His-Ubc13/His-Mms2 in buffers containing 25 mM Tris/HCl, pH 7.4, 2 mM magnesium/ATP, 0.1 mM DTT, 110 nM E1 enzyme, and 20 μM ubiquitin. The binding experiments were performed at 4°C in 0.3 ml of 50 mM Hepes, pH 7.3, 150 mM potassium chloride, 2.5 mM magnesium chloride, 5% glycerol, 2 mM β-mercaptoethanol, 0.1% Triton X-100, and 1 mg/ml BSA. 20-μl polyubiquitin chains were incubated with 1 μg of the various purified recombinant proteins. The proteins were then precipitated with specific antibodies, and bound ubiquitin chains were detected by immunoblotting with ubiquitin antibodies. To test ubiquitin chain binding to GST fusion proteins, the latter were first immobilized on glutathione beads. The ubiquitin chains were preabsorbed with glutathione beads to remove the E3 and ubiquitin chains associated with it, before addition to the immobilized proteins. To test ubiquitin binding of the yeast Cdc48–Ufd1–Npl4 complex, the endogenous gene for Ufd1 was replaced by a gene coding for a fusion of Ufd1 with the IgG-binding domain of protein A. Cytosol from the mutant yeast cells was incubated with IgG beads to isolate the Cdc48–Ufd1–Npl4 complex, and was then incubated with polyubiquitin chains.
Experiments with yeast
The yeast cdc48–3 strain was transformed with vector or plasmids coding for either wild-type Cdc48 or mutant Cdc48 proteins. The cells were serially diluted and plated on galactose- or glucose-containing agar. They were incubated at 30 or 37°C for 3 d.
Online supplemental material
Fig. S1 shows nucleotide binding to various His-tagged p97 proteins, and Fig. S2 shows that mutant Cdc48 defective in ATP hydrolysis is expressed at similar levels as the wild-type protein in S. cerevisiae. Online supplemental material available at http://www.jcb.org/cgi/content/full/jcb.200302169/DC1.
Supplemental Material
Acknowledgments
We thank Alexej Kisselev (Harvard Medical School, Boston, MA) for ubiquitin mutants containing single lysines; Alan Weissman for Ubc7 and gp78 constructs; Dennis Flierman (Harvard Medical School) for ubiquitin-depleted cytosol; Byron DeLaBarre (Stanfod University, Stanford, CA) for untagged p97 protein; and Caroline Shamu and Andrew Osborne for critical reading of the manuscript. We thank Graham Warren for support and helpful discussions.
Y. Ye is supported by the Helen Hay Whitney postdoctoral fellowship. H.H. Meyer is supported by grants to Graham Warren from the National Institutes of Health (NIH) and the Human Frontier Science Program, and T.A. Rapoport by NIH grants (R01GM52586 and R01GM54238). T.A. Rapoport is a Howard Hughes Medical Institute Investigator.
The online version of this article includes supplemental material.
Footnotes
Abbreviations used in this paper: AAA, ATPase associated with diverse cellular activities; NZF, Npl4 zinc finger.
References
- Bays, N.W., S.K. Wilhovsky, A. Goradia, K. Hodgkiss-Harlow, and R.Y. Hampton. 2001. HRD4/NPL4 is required for the proteasomal processing of ubiquitinated ER proteins. Mol. Biol. Cell. 12:4114–4128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braun, S., K. Matuschewski, M. Rape, S. Thoms, and S. Jentsch. 2002. Role of the ubiquitin-selective CDC48(UFD1/NPL4)chaperone (segregase) in ERAD of OLE1 and other substrates. EMBO J. 21:615–621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brodsky, J.L., and A.A. McCracken. 1999. ER protein quality control and proteasome-mediated protein degradation. Semin. Cell Dev. Biol. 10:507–513. [DOI] [PubMed] [Google Scholar]
- Bukau, B., and A.L. Horwich. 1998. The Hsp70 and Hsp60 chaperone machines. Cell. 92:351–366. [DOI] [PubMed] [Google Scholar]
- Cashikar, A.G., E.C. Schirmer, D.A. Hattendorf, J.R. Glover, M.S. Ramakrishnan, D.M. Ware, and S.L. Lindquist. 2002. Defining a pathway of communication from the C-terminal peptide binding domain to the N-terminal ATPase domain in a AAA protein. Mol. Cell. 9:751–760. [DOI] [PubMed] [Google Scholar]
- Coles, M., T. Diercks, J. Liermann, A. Groger, B. Rockel, W. Baumeister, K.K. Koretke, A. Lupas, J. Peters, and H. Kessler. 1999. The solution structure of VAT-N reveals a ‘missing link’ in the evolution of complex enzymes from a simple βαββ element. Curr. Biol. 9:1158–1168. [DOI] [PubMed] [Google Scholar]
- Dai, R.M., and C.C. Li. 2001. Valosin-containing protein is a multi-ubiquitin chain-targeting factor required in ubiquitin-proteasome degradation. Nat. Cell Biol. 3:740–744. [DOI] [PubMed] [Google Scholar]
- Ellgaard, L., and A. Helenius. 2001. ER quality control: towards an understanding at the molecular level. Curr. Opin. Cell Biol. 13:431–437. [DOI] [PubMed] [Google Scholar]
- Fang, S., M. Ferrone, C. Yang, J.P. Jensen, S. Tiwari, and A.M. Weissman. 2001. The tumor autocrine motility factor receptor, gp78, is a ubiquitin protein ligase implicated in degradation from the endoplasmic reticulum. Proc. Natl. Acad. Sci. USA. 98:14422–14427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Golbik, R., A.N. Lupas, K.K. Koretke, W. Baumeister, and J. Peters. 1999. The Janus face of the archaeal Cdc48/p97 homologue VAT: protein folding versus unfolding. Biol. Chem. 380:1049–1062. [DOI] [PubMed] [Google Scholar]
- Hetzer, M., H.H. Meyer, T.C. Walther, D. Bilbao-Cortes, G. Warren, and I.W. Mattaj. 2001. Distinct AAA-ATPase p97 complexes function in discrete steps of nuclear assembly. Nat. Cell Biol. 3:1086–1091. [DOI] [PubMed] [Google Scholar]
- Hitchcock, A.L., H. Krebber, S. Frietze, A. Lin, M. Latterich, and P.A. Silver. 2001. The conserved npl4 protein complex mediates proteasome-dependent membrane-bound transcription factor activation. Mol. Biol. Cell. 12:3226–3241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hofmann, R.M., and C.M. Pickart. 1999. Noncanonical MMS2-encoded ubiquitin-conjugating enzyme functions in assembly of novel polyubiquitin chains for DNA repair. Cell. 96:645–653. [DOI] [PubMed] [Google Scholar]
- Hoskins, J.R., S.K. Singh, M.R. Maurizi, and S. Wickner. 2000. Protein binding and unfolding by the chaperone ClpA and degradation by the protease ClpAP. Proc. Natl. Acad. Sci. USA. 97:8892–8897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jarosch, E., C. Taxis, C. Volkwein, J. Bordallo, D. Finley, D.H. Wolf, and T. Sommer. 2002. Protein dislocation from the ER requires polyubiquitination and the AAA-ATPase Cdc48. Nat. Cell Biol. 4:134–139. [DOI] [PubMed] [Google Scholar]
- Kondo, H., C. Rabouille, R. Newman, T.P. Levine, D. Pappin, P. Freemont, and G. Warren. 1997. p47 is a cofactor for p97-mediated membrane fusion. Nature. 388:75–78. [DOI] [PubMed] [Google Scholar]
- Liu, C.W., M.J. Corboy, G.N. DeMartino, and P.J. Thomas. 2003. Endoproteolytic activity of the proteasome. Science. 299:408–411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer, H.H., H. Kondo, and G. Warren. 1998. The p47 co-factor regulates the ATPase activity of the membrane fusion protein, p97. FEBS Lett. 437:255–257. [DOI] [PubMed] [Google Scholar]
- Meyer, H.H., J.G. Shorter, J. Seemann, D. Pappin, and G. Warren. 2000. A complex of mammalian ufd1 and npl4 links the AAA-ATPase, p97, to ubiquitin and nuclear transport pathways. EMBO J. 19:2181–2192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer, H.H., Y. Wang, and G. Warren. 2002. Direct binding of ubiquitin conjugates by the mammalian p97 adaptor complexes, p47 and Ufd1-Npl4. EMBO J. 21:5645–5652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogura, T., and A.J. Wilkinson. 2001. AAA+ superfamily ATPases: common structure–diverse function. Genes Cells. 6:575–597. [DOI] [PubMed] [Google Scholar]
- Rabinovich, E., A. Kerem, K.U. Frohlich, N. Diamant, and S. Bar-Nun. 2002. AAA-ATPase p97/Cdc48p, a cytosolic chaperone required for endoplasmic reticulum-associated protein degradation. Mol. Cell. Biol. 22:626–634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rabouille, C., H. Kondo, R. Newman, N. Hui, P. Freemont, and G. Warren. 1998. Syntaxin 5 is a common component of the NSF- and p97-mediated reassembly pathways of Golgi cisternae from mitotic Golgi fragments in vitro. Cell. 92:603–610. [DOI] [PubMed] [Google Scholar]
- Rape, M., T. Hoppe, I. Gorr, M. Kalocay, H. Richly, and S. Jentsch. 2001. Mobilization of processed, membrane-tethered SPT23 transcription factor by CDC48(UFD1/NPL4), a ubiquitin-selective chaperone. Cell. 107:667–677. [DOI] [PubMed] [Google Scholar]
- Rouiller, I., V.M. Butel, M. Latterich, R.A. Milligan, and E.M. Wilson-Kubalek. 2000. A major conformational change in p97 AAA ATPase upon ATP binding. Mol. Cell. 6:1485–1490. [DOI] [PubMed] [Google Scholar]
- Rouiller, I., B. DeLaBarre, A.P. May, W.I. Weis, A.T. Brunger, R.A. Milligan, and E.M. Wilson-Kubalek. 2002. Conformational changes of the multifunction p97 AAA ATPase during its ATPase cycle. Nat. Struct. Biol. 9:950–957. [DOI] [PubMed] [Google Scholar]
- Shamu, C.E., C.M. Story, T.A. Rapoport, and H.L. Ploegh. 1999. The pathway of US11-dependent degradation of MHC class I heavy chains involves a ubiquitin-conjugated intermediate. J. Cell Biol. 147:45–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shamu, C.E., D. Flierman, H.L. Ploegh, T.A. Rapoport, and V. Chau. 2001. Polyubiquitination is required for US11-dependent movement of MHC class I heavy chain from endoplasmic reticulum into cytosol. Mol. Biol. Cell. 12:2546–2555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steel, G.J., and A. Morgan. 1998. Selective stimulation of the D1 ATPase domain of N-ethylmaleimide-sensitive fusion protein (NSF) by soluble NSF attachment proteins. FEBS Lett. 423:113–116. [DOI] [PubMed] [Google Scholar]
- Song, C., Q. Wang, and C.C. Li. 2003. ATPase activity of p97-valosin-containing protein (VCP). D2 mediates the major enzyme activity, and D1 contributes to the heat-induced activity. J. Biol. Chem. 278:3648–3655. [DOI] [PubMed] [Google Scholar]
- Thoms, S. 2002. Cdc48 can distinguish between native and non-native proteins in the absence of cofactors. FEBS Lett. 520:107–110. [DOI] [PubMed] [Google Scholar]
- Tiwari, S., and A.M. Weissman. 2001. Endoplasmic reticulum (ER)-associated degradation of T cell receptor subunits. Involvement of ER-associated ubiquitin-conjugating enzymes (E2s). J. Biol. Chem. 276:16193–16200. [DOI] [PubMed] [Google Scholar]
- Tsai, B., Y. Ye, and T.A. Rapoport. 2002. Retro-translocation of proteins from the endoplasmic reticulum into the cytosol. Nat. Rev. Mol. Cell Biol. 3:246–255. [DOI] [PubMed] [Google Scholar]
- Uchiyama, K., E. Jokitalo, F. Kano, M. Murata, X. Zhang, B. Canas, R. Newman, C. Rabouille, D. Pappin, P. Freemont, and H. Kondo. 2002. VCIP135, a novel essential factor for p97/p47-mediated membrane fusion, is required for Golgi and ER assembly in vivo. J. Cell Biol. 159:855–866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, B., S.L. Alam, H.H. Meyer, M. Payne, T.L. Stemmler, D.R. Davis, and W.I. Sundquist. 2003. Structure and ubiquitin interactions of the conserved zinc finger domain of Npl4. J. Biol. Chem. 278:20225–20234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiertz, E.J., T.R. Jones, L. Sun, M. Bogyo, H.J. Geuze, and H.L. Ploegh. 1996. The human cytomegalovirus US11 gene product dislocates MHC class I heavy chains from the endoplasmic reticulum to the cytosol. Cell. 84:769–779. [DOI] [PubMed] [Google Scholar]
- Ye, Y., H.H. Meyer, and T.A. Rapoport. 2001. The AAA ATPase Cdc48/p97 and its partners transport proteins from the ER into the cytosol. Nature. 414:652–656. [DOI] [PubMed] [Google Scholar]
- Zhang, X., A. Shaw, P.A. Bates, R.H. Newman, B. Gowen, E. Orlova, M.A. Gorman, H. Kondo, P. Dokurno, J. Lally, et al. 2000. Structure of the AAA ATPase p97. Mol. Cell. 6:1473–1484. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}