

Abstract
Bax and Bak play a redundant but essential role in apoptosis initiated by the mitochondrial release of apoptogenic factors. In addition to their presence at the mitochondrial outer membrane, Bax and Bak can also localize to the ER. Agents that initiate ER stress responses can induce conformational changes and oligomerization of Bax on the ER as well as on mitochondria. In wild-type cells, this is associated with caspase 12 cleavage that is abolished in bax − / − bak − / − cells. In bax − / − bak − / − cells, introduction of Bak mutants selectively targeted to either mitochondria or the ER can induce apoptosis. However, ER-targeted, but not mitochondria-targeted, Bak leads to progressive depletion of ER Ca2+ and induces caspase 12 cleavage. In contrast, mitochondria-targeted Bak leads to enhanced caspase 7 and PARP cleavage in comparison with the ER-targeted Bak. These findings demonstrate that in addition to their functions at mitochondria, Bax and Bak also localize to the ER and function to initiate a parallel pathway of caspase activation and apoptosis.
Keywords: Bak; Bax; Ca2+; caspase 12; ER
Introduction
A eukaryotic cell is a well-integrated unit that consists of a number of subcellular organelles, such as nucleus, mitochondria, the ER, the Golgi apparatus, and lysosomes. These organelles function in an organized fashion to maintain intracellular homeostasis. Each organelle possesses sensors that detect specific cell death–inducing signals and activate different signal transduction pathways, which in turn lead to the ultimate apoptotic events involving the mitochondrial membrane permeabilization and/or the caspase activation cascades (Ferri and Kroemer, 2001).
The ER is the cytoplasmic compartment where proteins and lipids are synthesized and modified. Due to the presence of high concentrations of protein, numerous protein chaperones exist in the ER. These chaperones facilitate the proper folding of proteins, or maintain proteins in a folding-competent state and prevent protein folding intermediates from aggregating. In response to a number of stimuli that disrupt ER function, such as Ca2+ depletion from the ER lumen, inhibition of asparagine (N)-linked glycosylation, reduction of disulfide bonds, or overexpression of some proteins, protein misfolding occurs and unfolded proteins accumulate and aggregate in the ER. The misfolded and aggregated proteins then trigger the unfolded protein response (UPR).* Under less severe situations, UPR results in the reduction of general protein synthesis and selectively activates the expression of proteins facilitating the chaperone function (Kaufman, 1999). Under conditions of severe ER stress that the cells are not able to adapt to, UPR triggers apoptosis. UPR-induced apoptosis is associated with the up-regulation of the transcriptional factor CHOP/GADD153 and with eIF-2α phosphorylation (Kaufman, 1999), which may lead to the activation of a caspase cascade that involves the ER-specific caspase 12 (Nakagawa et al., 2000).
Bcl-2 family proteins play essential roles in regulating apoptosis. Whereas some members of the Bcl-2 family antagonize cell death, others exhibit proapoptotic activities. Bax and Bak are the proapoptotic members of the Bcl-2 family that are ubiquitously expressed in different tissues. They reside in the cytosol or on mitochondria and are kept in an inactive form in healthy cells. Overexpression of the multidomain death-promoting Bax-like proteins results in cell death in both mammalian and yeast cells (Oltvai et al., 1993; Chittenden et al., 1995; Hanada et al., 1995; Greenhalf et al., 1996; Jurgensmeier et al., 1997). When a cell is committed to apoptosis, Bax and Bak undergo conformational change, leading to their oligomerization and insertion in the mitochondria outer membrane (Hsu et al., 1997; Gross et al., 1998; Desagher et al., 1999; Griffiths et al., 1999; Wei et al., 2000; Makin et al., 2001). This process is followed by the release of cytochrome c from mitochondria to the cytosol. Cytochrome c binds to Apaf-1 and caspase 9, resulting in the activation of caspase 9, the subsequent activation of caspases 3 and 7, and ultimately cell death. Studies of the Bax/Bak doubly deficient mice indicate that the multidomain proapoptotic Bax and Bak proteins are required for mitochondria-mediated cell death (Lindsten et al., 2000; Wei et al., 2001; Zong et al., 2001).
Although intensive studies have been focusing on how Bcl-2 family proteins regulate apoptosis at the mitochondria level, evidence exists that Bcl-2 proteins may also function at the ER level. In addition to mitochondria, Bcl-2 is also found in the ER membranes (Cleary and Sklar, 1985; Krajewski et al., 1993; Akao et al., 1994; Lithgow et al., 1994; Nguyen et al., 1994). Overexpression of Bcl-2 and Bcl-xL blocks thapsigargin-induced apoptosis (Srivastava et al., 1999). A Bcl-2–cytochrome b5 (cb5) mutant selectively targeted to the ER blocks apoptosis induced by ER stress resulting from tunicamycin and brefeldin A treatment (Hacki et al., 2000; Annis et al., 2001). This ER-targeted Bcl-2 mutant also protects cells from Bax-induced cell death (Wang et al., 2001). Fibroblasts lacking both Bax and Bak are resistant to ER stress–induced apoptosis, and ectopic expression of Bax in these doubly deficient cells restores their sensitivity (Wei et al., 2001; Zong et al., 2001). These findings suggest that Bax and Bak can either sense the ER damage and function on mitochondria to induce mitochondrial membrane permeabilization–mediated apoptosis, or that there exists an unknown function of Bax and Bak at the ER level. In the present study, we report that the multidomain proapoptotic Bax and Bak proteins also reside at the ER in addition to their mitochondrial localization. ER stress can induce conformational changes and oligomerization of Bax and Bak on the ER membrane. ER stress–induced cleavage of the ER-specific caspase 12 is dependent on Bax and Bak. Targeted expression of Bak in the ER results in ER Ca2+ depletion, caspase 12 processing, and ultimately cell death. These findings indicate that in addition to their roles on mitochondria, Bax/Bak proteins may also be involved in initiating apoptosis from the ER.
Results
Bax and Bak are localized to the ER in addition to mitochondria
Antiapoptotic Bcl-2 proteins have been found in the ER/nuclear envelope in addition to their localization in mitochondria (Krajewski et al., 1993; Akao et al., 1994; Nguyen et al., 1994). It remains unclear whether the proapoptotic proteins Bax and Bak also reside and function at the ER. Subcellular fractionation was performed in wild-type murine embryonic fibroblasts (MEFs), bax − / − bak − / − MEFs, and HeLa cells. In agreement with previous reports, Bax was found in the cytosolic S-100 fraction, as well as in the mitochondria fraction (heavy membrane [HM]). Bak was found in the mitochondrial fraction but not in the cytosol. Despite their differential appearance in the cytosol, both Bax and Bak were found in the ER/microsome fraction (light membrane [LM]) (Fig. 1 A). To further demonstrate the subcellular localizations of Bax and Bak, crude mitochondria from mouse liver were fractionated in a 1–2 M continuous sucrose gradient. The postmitochondria supernatant was fractionated using step sucrose gradient, and the interface between the 1.5 and 1.8 M sucrose was collected as heavy microsomes. Similar to the data shown in Fig. 1 A, there is an enrichment of Bax and Bak in the ER/microsome fraction (Fig. 1 B). The purity of different fractions was determined using markers specifically localized to mitochondria or the ER. The mitochondria fraction was contaminated by the ER membrane during the fractionation procedure, indicated by the presence of the ER membrane protein calnexin in HM. However, the S-100 and LM fractions were not contaminated by mitochondria, as indicated by the absence of the mitochondrial inner membrane protein COX IV or mitochondrial outer membrane Tom40 (Fig. 1 C) in these fractions. An electron micrograph showed that Bax-specific gold labels were associated with the ER and cytosol, and that those of Bak were associated with the ER and mitochondria (Fig. 1 D). These results were specific, as no immunogold labeling was observed in samples of bax − / − bak − / − MEFs processed at the same time (unpublished data). Together, these findings demonstrate that in addition to their location on mitochondria, Bax and Bak also reside in the ER/microsome subcellular compartments.
Figure 1.



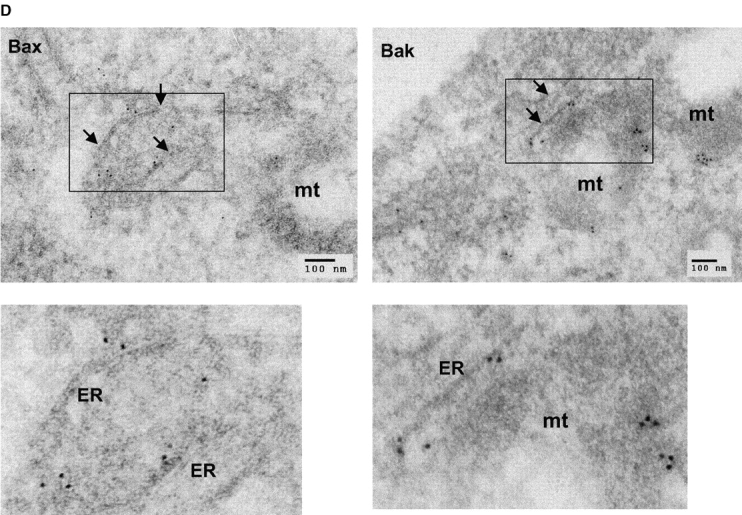
Bax and Bak are localized to the ER in addition to their mitochondrial location. (A) Wild-type and bax − / − bak − / − MEFs as well as HeLa cells were resuspended in hypotonic buffer A and disrupted. Subcellular fractionation was performed to obtain the fractions for cytosol (S-100), HM (mitochondria enriched), and LM (the ER enriched). 20 μg of total protein from each fraction was separated on a 4–12% gradient NuPAGE gel. Antibodies against Bax, Bak, calnexin, and Cox IV were used for immunoblotting. (B) Mouse liver was homogenized in buffer A and fractionated in sucrose gradient as described in the Materials and methods. Fractions from crude mitochondria and the 1.5/1.8 M sucrose interface were probed with indicated antibodies. (C) The LM fraction was not contaminated by mitochondrial outer membrane. Fractions of wild-type MEFs were probed with an anti-Tom40 antibody. (D) Immunoelectron microscopy of Bax (left) and Bak (right) in wild-type MEFs using 10-nm gold particles. The bottom panels are the enlargements of the boxed areas of the top images. The arrowheads point to the ER. mt, mitochondria.
ER stress induces Bax and Bak conformational changes
It has been established that during apoptosis, Bax and Bak undergo conformational change. This process seems to be required for the proapoptotic activity of Bax and Bak (Hsu et al., 1997; Gross et al., 1998; Desagher et al., 1999; Griffiths et al., 1999; Wei et al., 2000; Makin et al., 2001). To determine whether these proapoptotic proteins also undergo conformational changes during ER stress–induced apoptosis, HeLa, MCF7, and 293T cells were treated with thapsigargin and tunicamycin. These drugs trigger ER-mediated apoptosis by perturbing the normal function of the ER. Thapsigargin inhibits the ER Ca2+ uptake from the cytosol. Tunicamycin is an inhibitor of protein N-glycosylation. Antibodies against Bax and Bak that recognize their proapoptotic-competent forms were used. These antibodies were previously used to demonstrate the activation of Bax and Bak in response to other apoptotic stimuli (Griffiths et al., 1999; Mandic et al., 2001). In all three cell lines tested, ER stress treatment resulted in an increase of the active forms of Bax and Bak, indicating that Bax and Bak undergo conformational changes from the inactive forms to the active forms upon ER stress treatment (Fig. 2 A).
Figure 2.


ER stress induces Bax and Bak conformational changes and oligomerization at the ER. (A) ER stresses induce the conformational changes of Bax and Bak. HeLa, MCF7, and 293T cells were treated with thapsigargin (Thap; 2 μM) or tunicamycin (Tuni; 5 μg/ml) for 36 h. Cells were fixed in 0.25% paraformaldehyde in PBS for 5 min. Cells were incubated with a control antibody (mouse IgG1) and conformation-sensitive antibodies against Bax or Bak, followed by incubation with FITC-conjugated secondary antibody. (B) ER stress induces Bax oligomerization at the ER. Wild-type MEFs were treated with brefeldin A (BFA; 10 μg/ml), Thap (2 μM), or Tuni (10 μg/ml) for 24 h. Cells were resuspended in hypotonic buffer A and disrupted. 5 mM BMH cross-linking reagent was added to cross-link the oligomerized proteins. Cells were subjected to subcellular fractionation to obtain the HM and LM fractions. 20 μg of total protein was separated on a 4–12% gradient NuPAGE gel. A polyclonal anti-Bax antibody was used to detect Bax. COX IV and calnexin are shown as indicators of the purity of the fractionation and as loading controls.
ER stress induces Bax oligomerization on the ER
Accompanying the conformational changes, Bax and Bak undergo oligomerization on the mitochondrial outer membrane. As Bax and Bak proteins also exist on the ER membrane (Fig. 1), and they undergo conformational changes upon ER stress treatment (Fig. 2 A), we examined whether these proteins oligomerize on the ER membrane. Wild-type MEF cells were treated with thapsigargin, tunicamycin, or brefeldin A, which is another ER stress agent that blocks the ER–Golgi transportation. After the drug treatment, a sulfhydryl-reactive cross-linker bismaleimidohexane (BMH) was used to cross-link the oligomerized proteins. Cells were fractionated and examined for Bax localization and oligomerization. Consistent with Fig. 1, Bax was found in both mitochondria and ER fractions. ER stress treatment resulted in increased accumulation of Bax in both mitochondria and the ER. Importantly, the amount of oligomerized Bax was also increased in both mitochondria and the ER, indicated by increased amounts of slow mobility bands recognized by Bax antibody after treatments (Fig. 2 B). Similar results were obtained using the NIH3T3 murine fibroblast line (unpublished data). These findings indicate that ER stress can induce the oligomerization of Bax on both mitochondria and the ER, suggesting that Bax may also function at the ER to induce apoptosis in response to apoptotic stimuli.
Caspase 12 activation is Bax/Bak dependent
Caspase 12 is specifically localized to the ER and has been reported to be cleaved during ER stress–induced apoptosis (Nakagawa et al., 2000). To investigate whether ER stress–induced caspase 12 cleavage is dependent on Bax and Bak, wild-type, bax − / − bak − / −, and NIH3T3 cells were treated with brefeldin A, thapsigargin, and tunicamycin. No consistent differences in total caspase 12 expression were observed between wild-type MEFs, NIH3T3 cells, and bax − / − bak − / − MEFs. However, in both wild-type MEFs and NIH3T3 cells, an ∼47-kD fragment was revealed in untreated cells, indicating that there exists a basal processing of caspase 12 in these cells, which does not lead to apoptosis (Fig. 3 A). The ER stress treatment resulted in further caspase 12 cleavage, shown as the appearance of an ∼42-kD fragment in both wild-type MEFs and NIH3T3 cells. In contrast, the caspase 12 processing was not observed in bax − / − bak − / − cells (Fig. 3 A). Interestingly, the UPR-responsive transcriptional factor CHOP was induced by ER stress treatment in all three cell lines, including bax − / − bak − / − cells, indicating that cells deficient in Bax and Bak are still responding to ER stress–induced UPR (Fig. 3 A). If caspase 12 activation is dependent on Bax and Bak, it can be postulated that the killing activity of caspase 12 lies downstream in the apoptotic pathway initiated by these multidomain proapoptotic Bcl-2 proteins. We examined whether enforced activation of caspase 12 can kill cells deficient in Bax and Bak. As expected, ectopically expressed caspase 3 and caspase 9 resulted in apoptosis in both wild-type and bax − / − bak − / − MEFs (Fig. 3 B). Similarly, overexpression of full-length and truncated caspase 12 (t-caspase 12) induced cell death in both wild-type and bax − / − bak − / − cells, and t-caspase 12 had a higher killing activity than the full-length caspase 12, as previously described (Fig. 3 B; Nakagawa and Yuan, 2000).
Figure 3.

Caspase 12 functions downstream of the multidomain proapoptotic Bcl-2 family proteins. (A) ER stress–induced caspase 12 cleavage is dependent on Bax and Bak. Immortalized wild-type and bax − / − bak − / − MEFs and NIH3T3 cells were treated with brefeldin A (BFA; 10 μg/ml), thapsigargin (Thap; 2 μM), or tunicamycin (Tuni; 10 μg/ml) for 30 h. Caspase 12 level and processing were examined by immunoblotting of 20 μg of total cellular protein from samples as indicated. An ∼42-kD caspase 12 fragment is indicated by the arrow. Induction of CHOP expression is shown as an indicator of the ER stress response. A nonspecific band (NS) is shown as a loading control. (B) Caspase 12 kills bax − / − bak − / − cells. Wild-type and bax − / − bak − / − MEFs were cotransfected with pEGFP and constructs expressing caspase 3, caspase 9, caspase 12, and t-caspase 12. 24 h after transfection, cells were stained with DAPI, and cell death percentage was determined by the ratio of DAPI-positive to GFP-positive cells.
Targeting Bak mutants specifically to the ER or mitochondria
To study the function of proapoptotic Bcl-2 proteins selectively expressed on different intracellular organelles, mutants containing either mitochondrial- or ER-targeting sequences were constructed. In these mutants, sequences of the listerial protein ActA, which specifically directs proteins to mitochondria, and that of cb5, which specifically targets proteins to the ER membrane, were used as previously described (Zhu et al., 1996). Although the carboxy-terminal hydrophobic transmembrane domain of Bak is well defined functionally, the membrane-targeting domain of Bax remains controversial. Therefore, we generated Bak mutants specifically targeted to mitochondria and the ER by replacing the carboxy-terminal transmembrane domain of Bak with the cb5 and ActA sequences (Fig. 4 A). To confirm the respective subcellular localization of these mutants, the mutant proteins were tagged with EGFP, and their subcellular localization was examined by microscopy. The Bak mutants were expressed in MCF7 breast carcinoma cells, which lack caspase 3 and thus exhibit delayed apoptotic kinetics in response to Bak transfection, enabling us to look at the subcellular localization of the Bak mutants. GFP-tagged Bak-ActA resulted in superimposition of Bak's localization with that derived from an antibody against COX IV, which is selective for mitochondria, and areas of clear separation in the staining pattern for Bak-ActA and the ER protein calnexin. In contrast, GFP-tagged Bak-cb5 resulted in superimposition of Bak's localization and that of calnexin, whereas there were clear areas of separation of the GFP–Bak-cb5 signals and those of COX IV (Fig. 4 B).
Figure 4.

Generation of mitochondrial- and the ER- targeted Bak mutants. (A) Schemes of the Bak mutants. The carboxy-terminal transmembrane domain of Bak was deleted (ΔC) or replaced with the mitochondria-targeting sequence of ActA (ActA) or the ER-targeting sequence of cb5. (B) Targeting the Bak mutants to mitochondria and the ER. Bak mutants were expressed in MCF7 cells. Mitochondria marker COX IV and the ER marker calnexin were visualized by immunofluorescence. Note that the Bak-ActA mutant colocalized with COX IV but not calnexin, and the Bak-cb5 mutant colocalized with calnexin but not COX IV.
The ER-targeted Bak depletes ER Ca2+
To determine whether the ER-targeted Bak or any of the other Bak mutants can perturb the integrity of the ER membrane, we determined the effects of these mutants on the ER Ca2+ store by assessing thapsigargin-induced ER Ca2+ release. bax − / − bak − / − cells were infected with retroviruses encoding GFP alone, or in combination with wild-type Bak, Bak-ActA, or Bak-cb5, and analyzed for changes in intracellular [Ca2+] after thapsigargin treatment. The thapsigargin-releasable pool of ER Ca2+ was depleted in cells expressing Bak-cb5 (Fig. 5 A). In contrast, cells infected with GFP alone or GFP plus wild-type Bak or Bak-ActA showed a significant thapsigargin-induced release of ER Ca2+ stores, demonstrated by an increase in the cytosolic Ca2+ concentration in calcium-free medium, i.e., in the absence of capacitative Ca2+ entry through the plasma membrane (Fig. 5 A). To demonstrate that the abolishment of the thapsigargin-inducible [Ca2+]i spike is due to the depletion of the ER Ca2+ store in response to Bak-cb5, cells expressing vector, wild-type Bak, Bak-ActA, or Bak-cb5 were incubated in Ca2+-free buffer. Addition of extracellular Ca2+ resulted in higher Ca2+ entry through the plasma membrane in Bak-cb5–expressing cells, indicating that these cells had lower [Ca2+]ER, thus higher store-operated Ca2+ entry (Fig. 5 B).
Figure 5.

The ER-targeted Bak-cb5 induces depletion of the ER Ca2 + pool. (A) Cells with ER-targeted Bak lack thapsigargin-releasable intracellular Ca2+. bax − / − bak − / − cells were infected with retroviruses encoding GFP, Bak-IRES-GFP, Bak-ActA-IRES-GFP, or Bak-cb5-IRES-GFP. 48 h after infection, cells were loaded with Indo-1, and the ER Ca2+ release was measured by flow cytometry. Propidium iodide was added to determine the cell viability. The Ca2+ flux traces were derived from gated live GFP-positive cells. The [Ca2+]i was calibrated from the measured Indo-1 fluorescence ratio as described in the Materials and methods. The arrowhead indicates the time of thapsigargin addition. (B) Expression of ER-targeted Bak depleted intracellular Ca2+ storage in the ER. At the start of measurement, extracellular free Ca2+ concentration was reduced to zero. The arrowhead indicates the time when the extracellular free Ca2+ concentration was brought back to 2 mM. [Ca2+]i was analyzed as in A. (C and D) bax − / − bak − / − cells were infected with the Bak-cb5-IRES-GFP. (C) The measured Indo-1 fluorescence ratio between λ405 and λ475 is presented as an index of change in cytosolic [Ca2+] over time and presented separately for gated live GFP-positive (expressing Bak-cb5) or GFP-negative (not expressing Bak-cb5) cells. The absolute [Ca2+]i in the linear range is indicated on the right. The arrowhead indicates the time of thapsigargin addition. The arrow indicates the time of the addition of 8 mM CaCl2 to the medium. (D) Mitochondrial potential in GFP-positive (expressing Bak-cb5, green) and in GFP-negative (not expressing Bak-cb5, black) cells was measured by tetramethyl rhodamine ethyl ester (TMRE) staining.
The effect of Bak-cb5 on ER Ca2+ is specific because it is only the Bak-cb5–positive population that loses the ability to further release ER Ca2+ in response to thapsigargin. The GFP-negative (untransduced) population within the same sample had a normal thapsigargin-induced response (Fig. 5 C). Upon addition of extracellular Ca2+ to the medium after thapsigargin treatment, rapid Ca2+ influx occurred in both Bak-cb5–expressing and nonexpressing cells within the same sample. This demonstrates that the Bak-cb5–positive cells retain the ability to regulate their intracellular Ca2+ homeostasis to compensate for their unfilled ER Ca2+ stores through capacitative Ca2+ entry. In addition, the Bak-cb5–specific effect on ER Ca2+ is independent of mitochondrial damage because Bak-cb5–expressing cells retained the same mitochondrial potential as that of the uninfected cells in the same culture (Fig. 5 D).
The ER-targeted Bak can induce apoptosis
Expression of wild-type Bak, Bak-ActA, and Bak-cb5 induced apoptosis in wild-type MEFs (Fig. 6), as well as in HeLa, MCF7, and 293T cells (unpublished data), whereas the carboxy terminus–deleted Bak-ΔC mutant did not cause cell death. It is possible that the mitochondria- and ER-targeted Bak mutants kill cells by simply breaking the balance between the endogenous anti- and proapoptotic Bcl-2 family proteins. Previous studies have demonstrated that cells deficient in both Bax and Bak are defective in apoptosis. Therefore, we expressed the mitochondria- and ER-targeted Bak mutants in bax − / − bak − / − cells to examine whether the multidomain proapoptotic Bak-like proteins can directly initiate apoptosis at the ER. As shown in Fig. 6 A, wild type and mitochondria- and ER-targeted Bak mutants all killed the bax − / − bak − / − cells (Fig. 6 A). Quantitation of cell death by DAPI exclusion showed that the ER-targeted Bak has more potent killing activity than the wild type and the mitochondria-targeted Bak in wild-type and bax − / − bak − / − cells (Fig. 6 B). This higher ability of Bak-cb5 to induce apoptosis when compared with wild-type Bax could not be accounted for by a difference in the level of expression, as wild-type Bak was consistently expressed at higher levels than Bak-cb5 after retroviral infection (unpublished data). These data indicate that in the absence of endogenous proapoptotic proteins Bax and Bak, the multidomain proapoptotic Bcl-2 proteins can initiate apoptosis directly from the ER. Overexpression of Bcl-xL blocked the proapoptotic activities of all Bak mutants (Fig. 6 B), indicating that these mutants, including Bak-cb5, induce apoptosis through a physiological manner that is blockable by the antiapoptotic molecule Bcl-xL. Interestingly, the killing activities of the Bak mutants were also blocked by the pan-caspase inhibitor z-VAD-fmk, suggesting that caspase activation may be involved in the death-inducing activities of these mutants (Fig. 6 B).
Figure 6.
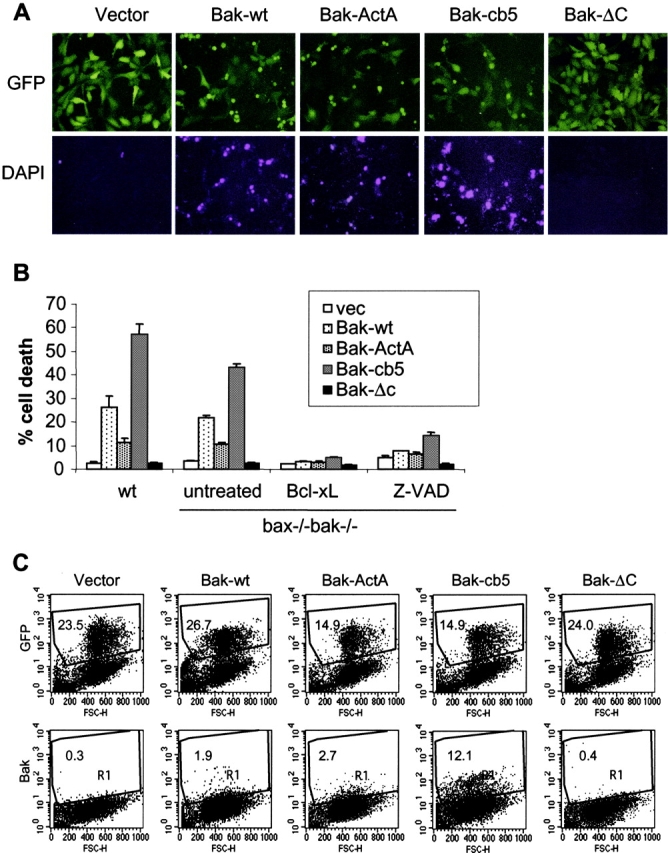
The ER-targeted Bak can induce apoptosis in the absence of endogenous Bax and Bak. Wild-type and bax − / − bak − / − MEFs were infected with retroviruses expressing GFP or GFP together with wild-type murine Bak, Bak-ActA, Bak-cb5, or Bak-ΔC. 48 h after infection, 1 μg/ml DAPI was added to stain the apoptotic cells. (A) bax − / − bak − / − cells were photographed using a FITC or DAPI filter. (B) The percentage of dead cells was determined by the ratio of DAPI-positive cells to GFP-positive cells. In addition to the retroviral infection of the Bak mutants, bax − / − bak − / − cells were also coinfected with retrovirus expressing Bcl-xL, or infected in the presence of 20 μg/ml Z-VAD-fmk. Data shown are the average of three independent experiments. (C) 24 h after infection, bax − / − bak − / − cells were trypsinized. A portion of each sample was subjected to FACS® to determine the expression efficiency indicated by GFP-positive cells. The rest of the cells were fixed in 0.25% paraformaldehyde and stained with an anti-Bak antibody that recognizes the active form of murine Bak. The number in each dot plot represents the percentage of gated events (R1).
It is interesting to note that the Bak-cb5 mutant induced apoptosis in a more potent fashion than wild-type Bak and Bak-ActA (Fig. 6 A). A possible reason is that the replacement of the Bak carboxy-terminal transmembrane domain with the cb5 sequence may change the protein to its active death-inducing conformation. To address this possibility, Bak mutants were expressed in bax − / − bak − / − cells, with GFP as a marker expressed from the internal ribosomal entry site (IRES) site. At 24 h after infection, when only marginal cell death could be observed, all mutants were expressed at comparable levels, indicated by GFP-positive cells. Interestingly, an anti-Bak antibody that recognizes specifically the active proapoptotic form of Bak revealed that most of the Bak-cb5–expressing cells had undergone a conformational change indicative of Bak activation (Fig. 6 C). Thus, the higher death rate in Bak-cb5–expressing cells may be due to the activated conformation of Bak caused by alteration of its carboxy terminus in Bak-cb5 and membrane insertion in the ER.
The ER-targeted Bak mutant specifically induces caspase 12, but not caspase 7, cleavage
As caspase 12 cleavage is dependent on Bax and Bak, and the ER-targeted Bak can lead to apoptosis, we determined whether expression of the ER-targeted Bak in bax − / − bak − / − cells results in caspase 12 processing. Wild-type Bak and the ER- and mitochondria-targeted Bak mutants were retrovirally expressed in bax − / − bak − / − MEFs. Expression of Bak-cb5 resulted in cleavage of caspase 12, whereas little caspase 12 cleavage was observed in wild-type Bak–expressing cells, and virtually no caspase 12 cleavage was observed in Bak-ActA–expressing cells (Fig. 7). In contrast, expression of either wild-type Bak or Bak-ActA mutant resulted in cleavage of caspase 7 and PARP, whereas little to no caspase 7 and PARP cleavage was observed in Bak-cb5–infected cells (Fig. 7). Thus, despite the fact that Bak-cb5 induces apoptosis, it does not appear to activate the mitochondrial apoptosis machinery in the bax − / − bak − / −cells that lack the mitochondrial Bax and Bak. Moreover, these findings indicate that increased cleavage of caspase 12 in Bak-cb5–expressing cells is not merely due to higher levels of cell death, but rather that distinct caspases are selectively activated depending on whether Bak is localized primarily to the ER or mitochondria.
Figure 7.
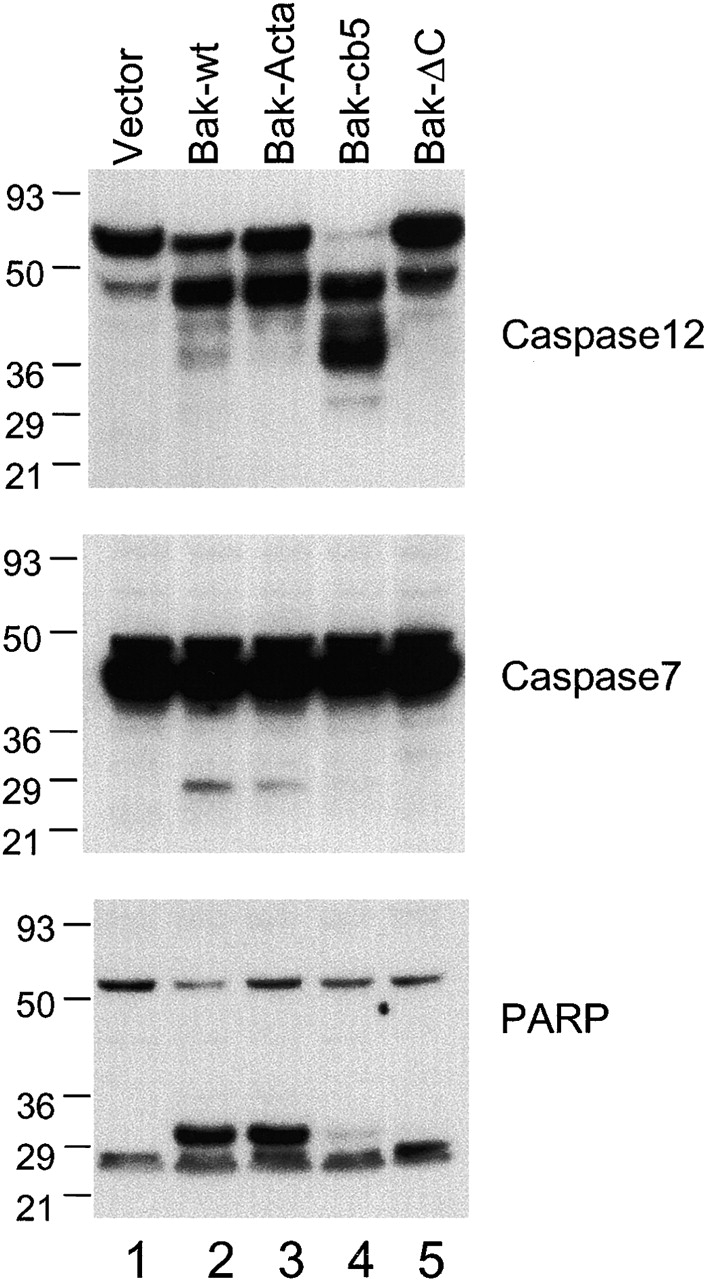
ER-targeted Bak can induce selective cleavage of caspase 12, but not of caspase 7. bax − / − bak − / − cells were infected with GFP vector control, Bak-IRES-GFP, Bak-ActA-IRES-GFP, Bak-cb5-IRES-GFP, and Bak-ΔC-IRES-GFP at a high multiplicity. After infection, cells were lysed. Caspase 12, caspase 7, or PARP were detected by immunoblotting using respective antibodies.
Discussion
Bcl-2 family proteins play a central role in regulating apoptosis. It has been well established that these proteins function at mitochondria to prevent or promote the release of apoptogenic factors such as cytochrome c, AIF, Smac/Diablo, endonuclease G, and Omi (Wang, 2001). In addition to their mitochondrial localization, some antiapoptotic members of the Bcl-2 family have also been found to be present at the ER and perinuclear membrane regions, suggesting a role of the Bcl-2 family proteins at sites other than mitochondria (Cleary and Sklar, 1985; Krajewski et al., 1993; Akao et al., 1994; Lithgow et al., 1994; Nguyen et al., 1994). Here we demonstrate that the multidomain proapoptotic Bcl-2 family members Bax and Bak also reside at the ER in addition to their mitochondrial localization. ER stress induces conformational changes and oligomerization of these proteins on the ER membrane, which may lead to a lesion of the ER membrane integrity. The existence of a Bax/Bak-dependent apoptotic pathway initiated from the ER may explain the observations that cells lacking Apaf-1, caspase 9, or cytochrome c showed only attenuated, but not complete, resistance to certain death stimuli (Cecconi et al., 1998; Kuida et al., 1998; Li et al., 2000), whereas the bax − / − bak − / − cells are resistant to virtually all death stimuli in a prolonged manner (Lindsten et al., 2000; Cheng et al., 2001; Wei et al., 2001; Zong et al., 2001). Consistent with this theory is the finding that Bcl-2 overexpression inhibits broader death pathways than deficiency of Apaf-1 and caspase 9 (Marsden et al., 2002).
The ER is the main intracellular storage compartment for Ca2+, which is an important secondary messenger that is required for numerous cellular functions. Apoptosis occurs upon the perturbation of cellular Ca2+ homeostasis, such as cytosolic Ca2+ overload, ER Ca2+ depletion, and mitochondrial Ca2+ increase. The close physical contact of mitochondria and the ER results in a higher exposure to Ca2+ of mitochondria than the rest of the cytosol, when Ca2+ is released from the ER (Rizzuto et al., 1998). Bcl-2 family proteins have been previously implicated in controlling apoptosis by affecting cellular Ca2+ homeostasis. Overexpression of Bcl-2 leads to reduced mitochondrial Ca2+ stores and enhanced ER sequestration, whereas overexpression of either wild-type Bax or Bak has the opposite effect (Pinton et al., 2000; Pan et al., 2001; Abeele et al., 2002; Li et al., 2002; Nutt et al., 2002a,b). This has been interpreted to mean that Bax and Bak interfere with mitochondrial Ca2+ homeostasis or initiate apoptosis by affecting the function of the mitochondrial permeability transition pore. The present data suggest that Bax and Bak localized to the ER can directly affect ER Ca2+ stores. In bax − / − bak − / − cells, targeting the Bak-cb5 mutant to the ER resulted in acute ER Ca2+ depletion, indicating a direct effect of the multidomain proapoptotic proteins on ER Ca2+ stores (Fig. 5). The effect on ER Ca2+ appears to result from the fact that the ER-targeted Bak-cb5 protein undergoes activation directly upon insertion in the ER membrane, as demonstrated by its adopting an active Bak configuration when introduced into cells by retroviral infection. In contrast, secondary factors, such as endogenous BH3-only proteins, must activate wild-type Bak, as it retains an inactivated configuration and does not induce significant Ca2+ release. When apoptosis is initiated in wild-type Bak–expressing cells, it induces a mixture of events associated with apoptosis from the two targeted Bak constructs.
In mitochondria-initiated apoptosis, the activation of Bax and Bak involves BH3-only proteins. The presence of several BH3-only proteins, such as tBid, Bim, Noxa, and Puma, at mitochondria when apoptosis is triggered has suggested that the activation of Bax/Bak by BH3-only proteins may take place at this site (Li et al., 1998; Luo et al., 1998; Puthalakath et al., 1999; Oda et al., 2000; Nakano and Vousden, 2001; Yu et al., 2001). It is possible that some of the BH3-only proteins may also localize to the ER and trigger Bax and Bak activation at the ER, as suggested by recent findings that the ER localization of the BH3-only protein BIK/NBK was induced by apoptotic stimuli, and that a BIK/NBK mutant specifically targeted to the ER retained the capability of inducing cytochrome c release and apoptosis (Germain et al., 2002; Mathai et al., 2002). It is of interest to determine whether other BH3-only proteins can also function at the ER and whether novel ER-specific BH3-only proteins may exist.
Caspase 12 has been demonstrated to be specifically localized to the ER (Nakagawa et al., 2000). Although a required role for caspase 12 in the induction of apoptosis has not been established, the finding that caspase 12 cleavage is dependent on Bax and Bak suggests that caspase 12 processing lies downstream of Bax and Bak. Interestingly, expression of the ER-targeted Bak-cb5 resulted in a significant increase in caspase 12 processing, whereas expression of wild-type or the mitochondrial Bak-ActA resulted in preferential cleavage of caspase 7 and PARP. These data suggest that in addition to their role in mitochondrial apoptosis, Bax and Bak localized to the ER can initiate an alternative pathway of caspase activation that results in selective caspase 12 activation (Fig. 8).
Figure 8.

Bax and Bak localized to the ER can initiate apoptosis. In addition to their mitochondrial localization and activity, Bax and Bak also reside at the ER. Upon ER stress treatment, Bax and Bak can initiate apoptosis from the ER.
As in mitochondria, Bax/Bak may initiate apoptosis from the ER by inducing the release of apoptogenic factors from the ER lumen into the cytosol. Although we have observed selective ER Ca2+ depletion by the ER-targeted Bak, a number of additional molecules sequestered in the ER may also be released and contribute to the initiation of apoptosis. Based on recent identification of a number of mitochondrial intermembrane proteins, which act as proapoptotic factors when released into the cytosol, it will be important in the future to pursue the possibility that other ER-sequestered factors may contribute to the apoptotic pathway initiated by the activation of Bax and Bak localized to the ER.
Materials and methods
Cell culture
Wild-type and bax − / − bak − / − murine fibroblasts were immortalized by spontaneous immortalization. MEF, MCF7, and Phoenix cells were cultured in DME supplemented with 10% FBS, 100 U/ml penicillin, and 100 μg/ml streptomycin.
DNA constructs
Murine Bak cDNA was cloned into pBabeMN-IRES-GFP to generate plasmid pBabe-Bak-GFP (Bak-IRES-GFP). A stop codon was introduced after amino acid 183 by PCR to generate plasmid pBabe-Bak-ΔC-IRES-GFP (Bak-ΔC-IRES-GFP). Cb5 and ActA DNA fragments (Zhu et al., 1996) were generated by PCR and subsequently subcloned after amino acid 183 to make pBabe-Bak-cb5-IRES-GFP (Bak-cb5-IRES-GFP) and pBabe-Bak-ActA-IRES-GFP (Bak-ActA-IRES-GFP), respectively. Plasmids used for localization experiments were made by subcloning appropriate DNA fragments from pBabeMN-IRES-GFP into pEGFP-C1 (CLONTECH Laboratories, Inc.). Expression constructs for caspase 3 (pBK5-caspase 3) and caspase 9 (pBK5-caspase 9) are a gift from X. Yang (University of Pennsylvania, Philadelphia, PA). Expression constructs for caspase 12 (pcDNA3.1-caspase 12) and t-caspase 12 (pcDNA3.1-t-caspase 12) were previously described (Nakagawa and Yuan, 2000).
Retroviral infection
Phoenix packaging cells were transfected with retroviral constructs. Viral supernatants were collected during the 48–96-h period after transfection and centrifuged at 2,000 rpm for 10 min to remove contaminating packaging cells. MEFs were infected with the viral supernatant in the presence of 10 μg/ml of polybrene (Sigma-Aldrich).
Cell death assay
Cells were retrovirally infected with GFP or Bak mutant–GFP. 48 h after infection, a total of 1 μg/ml DAPI was added to cell culture. Cells were observed and photographed with the Nikon Eclipse TE300 microscope and subjected to flow cytometry to determine the number of DAPI-positive cells over that of GFP-positive cells. In transient transfection assays, cells were transfected with 0.3 μg pEGFP together with 1.8 μg pBK5-caspase 3, pBK5-caspase 9, pcDNA3.1-caspase 12, or pcDNA3.1-t-caspase 12 using Geneporter2 transfection reagent (Gene Therapy Systems). 24 h later, cells were stained with 1 μg/ml DAPI, and the percentage of cell death was determined by the ratio of DAPI-positive cells to GFP-positive cells.
Immunoelectronic microscopy
Cells were rinsed briefly with prewarmed serum-free medium and then fixed with 2% paraformaldehyde and 0.05% glutaraldehyde for 30 min. Cells were collected to make a pellet by centrifugation, washed with PBS, and dehydrated at −25°C with ethanol. Cells were infiltrated with a mixture of ethanol/Lowicryl K4M medium, embedded in pure Lowicryl K4M, and polymerized at −25°C for 5 d with UV light at 365 nm. Ultrathin sections (80 nm) were cut with a diamond knife, mounted on Formvar-coated thin-bar nickel grids, and labeled with established protocols (Allen et al., 1996) using the antibodies against Bax (6A7; BD Biosciences) and Bak (NT; Upstate Biotechnology).
Subcellular fractionation
Cells were resuspended in hypotonic buffer A (250 mM sucrose, 20 mM Hepes, pH 7.5, 10 mM KCl, 1.5 mM MgCl2, 1 mM EDTA, 1 mM EGTA, 1× protease inhibitor complex [Roche]) on ice for 30 min. Cells were disrupted by passing through 26-gauge needles 30 times, and then 30-gauge needles 20 times. Cell lysates were centrifuged at 750 g for 10 min at 4°C to get rid of unlysed cells and nuclei. The supernatant was centrifuged at 10,000 g for 20 min at 4°C. The pellet was saved as the HM (mitochondria) fraction. The supernatant was centrifuged at 100,000 g for 1 h at 4°C. The supernatant was saved as the S-100 (cytosol) fraction and the pellet as the LM (ER/microsome) fraction. The HM and LM fractions were lysed in RIPA buffer (1% sodium deoxycholine, 0.1% SDS, 1% Triton X-100, 10 mM Tris, pH 8.0, 0.14 M NaCl) for Western blotting. For Fig. 1 B, a mouse liver was homogenized in buffer A with four strokes using Dounce homogenizer. The lysate was centrifuged at 750 g for 10 min at 4°C to get rid of unlysed cells and nuclei. The postnuclei supernatant was centrifuged at 6,000 g for 30 min to obtain the pellet as crude mitochondria. Crude mitochondria were resuspended in 0.5 ml buffer A, top loaded to a 1–2 M continuous sucrose gradient, and centrifuged at 25,000 rpm in an SW28 rotor for 90 min. Fractions were collected using a 16G needle into collection tubes at a rate of 0.75 ml/tube. The postmitochondrial supernatant was adjusted with 2.5 M sucrose to the final concentration of 1.8 M and bottom loaded into an SW41 centrifuge tube. 1.5 ml each of 1.5, 1.3, 1.0, 0.86, and 0.25 M sucrose was added to fill the centrifuge tube. The sample was centrifuged at 41,000 rpm for 6 h at 4°C. The material formed at the interface between the 1.5 and 1.8 M sucrose layers was collected. All fractions were diluted with threefold buffer A and centrifuged at 41,000 rpm for 90 min in an SW55 rotor. Pellets were dissolved in 100 μl RIPA buffer. 10 μl of each fraction was loaded into a 4–12% NuPAGE gradient gel for immunoblotting analysis.
Measurement of [Ca2+]i
Cells were resuspended in RPMI medium 1640 with 1% FBS and loaded with 3 μM Indo-1 (Molecular Probes) in the presence of 4 mM probenecid at 37°C for 30 min. Loaded cells were washed twice and resuspended in HBSS without calcium (Invitrogen). The measurement of cytosolic free Ca2+ concentration ([Ca2+]i) was performed on an LSR flow cytometer (Becton Dickinson). In brief, 2 min before analysis, EGTA was added to each sample to a final concentration of 1.5 mM. After stable basal [Ca2+]i was achieved, cells were treated with thapsigargin (5 μM) to release the ER Ca2+ pool. Data analysis was performed by using FlowJo software (Tree Star), and [Ca2+]i was calculated from the equation [Ca2+]i = K d · (R − R min) · F(λ = 475)min/(R max − R) · F(λ = 475)max, where the values of R min, R max, Fmin, and Fmax were determined by in situ calibration. Indo-1–loaded cells were resuspended in RPMI.
Detection of Bax and Bak conformational changes
Analysis of the conformational changes of Bax and Bak was performed as described previously (Griffiths et al., 1999; Mandic et al., 2001). Cells were treated with thapsigargin (2 μM) or tunicamycin (5 μg/ml) for 48 h. Cells were fixed in 0.25% paraformaldehyde in PBS for 5 min. Cells were washed three times with PBS and incubated with 1:50 anti–mouse IgG1 (BD Biosciences), anti-Bax (clone 6A7; BD Biosciences), anti-Bak (AM03; Oncogene Research Products), or anti-Bak (NT; Upstate Biotechnology) in 100 μg/ml digitonin in PBS for 30 min. After being washed with PBS three times, cells were incubated with 1:100 FITC-conjugated anti–mouse Ig and 1 μg/ml DAPI for 30 min. Cells were subjected to analysis using an LSR cytometer.
Chemical cross-linking
Wild-type MEFs or NIH3T3 cells were treated with brefeldin A (10 μg/ml), thapsigargin (2 μM), or tunicamycin (1 μg/ml) for 24 h. Cells were harvested and disrupted with 26-gauge needles. The cell lysates were incubated with 5 mM BMH (Pierce Chemical Co.) for 30 min at room temperature. Subcellular fractionation was performed to obtain HM and LM pellets. The pellets were lysed in RIPA buffer for immunoblotting.
Immunoblotting and immunofluorescence
20 μg of total protein was resolved in precast 4–12% NuPAGE gradient gels. Immunoblotting was performed by use of antibodies against Bax (N-20; Santa Cruz Biotechnology, Inc.), Bak (Upstate Biotechnology), COX IV (Molecular Probes), Tom40 (Santa Cruz Biotechnology, Inc.), calnexin (C-20; Santa Cruz Biotechnology, Inc.), caspase 12, and CHOP (B-3; Santa Cruz Biotechnology, Inc.). For immunofluorescence, cells were fixed in 4% paraformaldehyde and permeabilized for 10 min in PBS containing 0.2% Triton X-100. Cells were washed with PBS containing 0.02% Triton X-100 and 1.5% FBS, followed by incubation with antibodies against COX IV or calnexin (Santa Cruz Biotechnology, Inc.) for 1 h at room temperature. Cells were incubated with rhodamine red– or FITC-conjugated secondary antibodies (Jackson ImmunoResearch Laboratories). Nuclei were visualized by staining with 1 μg/ml DAPI. Images were captured on a 510 LSM confocal microscope (Carl Zeiss MicroImaging, Inc.).
Acknowledgments
We thank Drs. John Wagner and Sita Awasthi for their assistance on sucrose gradient fractionation. We thank Dr. Xiaolu Yang for the caspase 3 and caspase 9 constructs. We thank Drs. Carl June, James Alwine, and J. Kevin Foskett for their comments and members of the Thompson laboratory for their inspiration and discussion.
W.X. Zong is a recipient of the postdoctoral fellowship from the Cancer Research Institute. C. Li is supported by National Institutes of Health training grant T32 (CA09140). G. Hatzivassiliou is a Damon Runyon Fellow supported by the Damon Runyon Cancer Research Foundation (DRG1714-02).
W.-X. Zong and C. Li contributed equally to this work.
Footnotes
Abbreviations used in this paper: BMH, bismaleimidohexane; cb5, cytochrome b5; HM, heavy membrane; IRES, internal ribosomal entry site; LM, light membrane; MEF, murine embryonic fibroblast; t-caspase, truncated caspase; UPR, unfolded protein response.
References
- Abeele, F.V., R. Skryma, Y. Shuba, F. Van Coppenolle, C. Slomianny, M. Roudbaraki, B. Mauroy, F. Wuytack, and N. Prevarskaya. 2002. Bcl-2-dependent modulation of Ca(2+) homeostasis and store-operated channels in prostate cancer cells. Cancer Cell. 1:169–179. [DOI] [PubMed] [Google Scholar]
- Akao, Y., Y. Otsuki, S. Kataoka, Y. Ito, and Y. Tsujimoto. 1994. Multiple subcellular localization of bcl-2: detection in nuclear outer membrane, endoplasmic reticulum membrane, and mitochondrial membranes. Cancer Res. 54:2468–2471. [PubMed] [Google Scholar]
- Allen, E., Q.C. Yu, and E. Fuchs. 1996. Mice expressing a mutant desmosomal cadherin exhibit abnormalities in desmosomes, proliferation, and epidermal differentiation. J. Cell Biol. 133:1367–1382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Annis, M.G., N. Zamzami, W. Zhu, L.Z. Penn, G. Kroemer, B. Leber, and D.W. Andrews. 2001. Endoplasmic reticulum localized Bcl-2 prevents apoptosis when redistribution of cytochrome c is a late event. Oncogene. 20:1939–1952. [DOI] [PubMed] [Google Scholar]
- Cecconi, F., G. Alvarez-Bolado, B.I. Meyer, K.A. Roth, and P. Gruss. 1998. Apaf1 (CED-4 homolog) regulates programmed cell death in mammalian development. Cell. 94:727–737. [DOI] [PubMed] [Google Scholar]
- Cheng, E.H., M.C. Wei, S. Weiler, R.A. Flavell, T.W. Mak, T. Lindsten, and S.J. Korsmeyer. 2001. BCL-2, BCL-X(L) sequester BH3 domain-only molecules preventing BAX- and BAK-mediated mitochondrial apoptosis. Mol. Cell. 8:705–711. [DOI] [PubMed] [Google Scholar]
- Chittenden, T., E.A. Harrington, R. O'Connor, C. Flemington, R.J. Lutz, G.I. Evan, and B.C. Guild. 1995. Induction of apoptosis by the Bcl-2 homologue Bak. Nature. 374:733–736. [DOI] [PubMed] [Google Scholar]
- Cleary, M.L., and J. Sklar. 1985. Nucleotide sequence of a t(14;18) chromosomal breakpoint in follicular lymphoma and demonstration of a breakpoint-cluster region near a transcriptionally active locus on chromosome 18. Proc. Natl. Acad. Sci. USA. 82:7439–7443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desagher, S., A. Osen-Sand, A. Nichols, R. Eskes, S. Montessuit, S. Lauper, K. Maundrell, B. Antonsson, and J.C. Martinou. 1999. Bid-induced conformational change of Bax is responsible for mitochondrial cytochrome c release during apoptosis. J. Cell Biol. 144:891–901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferri, K.F., and G. Kroemer. 2001. Organelle-specific initiation of cell death pathways. Nat. Cell Biol. 3:E255–E263. [DOI] [PubMed] [Google Scholar]
- Germain, M., J.P. Mathai, and G.C. Shore. 2002. BH-3-only BIK functions at the endoplasmic reticulum to stimulate cytochrome c release from mitochondria. J. Biol. Chem. 277:18053–18060. [DOI] [PubMed] [Google Scholar]
- Greenhalf, W., C. Stephan, and B. Chaudhuri. 1996. Role of mitochondria and C-terminal membrane anchor of Bcl-2 in Bax induced growth arrest and mortality in Saccharomyces cerevisiae. FEBS Lett. 380:169–175. [DOI] [PubMed] [Google Scholar]
- Griffiths, G.J., L. Dubrez, C.P. Morgan, N.A. Jones, J. Whitehouse, B.M. Corfe, C. Dive, and J.A. Hickman. 1999. Cell damage-induced conformational changes of the pro-apoptotic protein Bak in vivo precede the onset of apoptosis. J. Cell Biol. 144:903–914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gross, A., J. Jockel, M.C. Wei, and S.J. Korsmeyer. 1998. Enforced dimerization of BAX results in its translocation, mitochondrial dysfunction and apoptosis. EMBO J. 17:3878–3885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hacki, J., L. Egger, L. Monney, S. Conus, T. Rosse, I. Fellay, and C. Borner. 2000. Apoptotic crosstalk between the endoplasmic reticulum and mitochondria controlled by Bcl-2. Oncogene. 19:2286–2295. [DOI] [PubMed] [Google Scholar]
- Hanada, M., C. Aime-Sempe, T. Sato, and J.C. Reed. 1995. Structure-function analysis of Bcl-2 protein. Identification of conserved domains important for homodimerization with Bcl-2 and heterodimerization with Bax. J. Biol. Chem. 270:11962–11969. [DOI] [PubMed] [Google Scholar]
- Hsu, Y.T., K.G. Wolter, and R.J. Youle. 1997. Cytosol-to-membrane redistribution of Bax and Bcl-X(L) during apoptosis. Proc. Natl. Acad. Sci. USA. 94:3668–3672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jurgensmeier, J.M., S. Krajewski, R.C. Armstrong, G.M. Wilson, T. Oltersdorf, L.C. Fritz, J.C. Reed, and S. Ottilie. 1997. Bax- and Bak-induced cell death in the fission yeast Schizosaccharomyces pombe. Mol. Biol. Cell. 8:325–339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaufman, R.J. 1999. Stress signaling from the lumen of the endoplasmic reticulum: coordination of gene transcriptional and translational controls. Genes Dev. 13:1211–1233. [DOI] [PubMed] [Google Scholar]
- Krajewski, S., S. Tanaka, S. Takayama, M.J. Schibler, W. Fenton, and J.C. Reed. 1993. Investigation of the subcellular distribution of the bcl-2 oncoprotein: residence in the nuclear envelope, endoplasmic reticulum, and outer mitochondrial membranes. Cancer Res. 53:4701–4714. [PubMed] [Google Scholar]
- Kuida, K., T.F. Haydar, C.Y. Kuan, Y. Gu, C. Taya, H. Karasuyama, M.S. Su, P. Rakic, and R.A. Flavell. 1998. Reduced apoptosis and cytochrome c-mediated caspase activation in mice lacking caspase 9. Cell. 94:325–337. [DOI] [PubMed] [Google Scholar]
- Li, C., C.J. Fox, S.R. Master, V.P. Bindokas, L.A. Chodosh, and C.B. Thompson. 2002. Bcl-xL affects Ca2+ homeostasis by altering expression of inositol 1,4,5-trisphosphate receptors. Proc. Natl. Acad. Sci. USA. 99:9830–9835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, H., H. Zhu, C.J. Xu, and J. Yuan. 1998. Cleavage of BID by caspase 8 mediates the mitochondrial damage in the Fas pathway of apoptosis. Cell. 94:491–501. [DOI] [PubMed] [Google Scholar]
- Li, K., Y. Li, J.M. Shelton, J.A. Richardson, E. Spencer, Z.J. Chen, X. Wang, and R.S. Williams. 2000. Cytochrome c deficiency causes embryonic lethality and attenuates stress-induced apoptosis. Cell. 101:389–399. [DOI] [PubMed] [Google Scholar]
- Lindsten, T., A.J. Ross, A. King, W.X. Zong, J.C. Rathmell, H.A. Shiels, E. Ulrich, K.G. Waymire, P. Mahar, K. Frauwirth, et al. 2000. The combined functions of proapoptotic Bcl-2 family members bak and bax are essential for normal development of multiple tissues. Mol. Cell. 6:1389–1399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lithgow, T., R. van Driel, J.F. Bertram, and A. Strasser. 1994. The protein product of the oncogene bcl-2 is a component of the nuclear envelope, the endoplasmic reticulum, and the outer mitochondrial membrane. Cell Growth Differ. 5:411–417. [PubMed] [Google Scholar]
- Luo, X., I. Budihardjo, H. Zou, C. Slaughter, and X. Wang. 1998. Bid, a Bcl2 interacting protein, mediates cytochrome c release from mitochondria in response to activation of cell surface death receptors. Cell. 94:481–490. [DOI] [PubMed] [Google Scholar]
- Makin, G.W., B.M. Corfe, G.J. Griffiths, A. Thistlethwaite, J.A. Hickman, and C. Dive. 2001. Damage-induced Bax N-terminal change, translocation to mitochondria and formation of Bax dimers/complexes occur regardless of cell fate. EMBO J. 20:6306–6315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mandic, A., K. Viktorsson, M. Molin, G. Akusjarvi, H. Eguchi, S.I. Hayashi, M. Toi, J. Hansson, S. Linder, and M.C. Shoshan. 2001. Cisplatin induces the proapoptotic conformation of Bak in a δMEKK1-dependent manner. Mol. Cell. Biol. 21:3684–3691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marsden, V.S., L. O'Connor, L.A. O'Reilly, J. Silke, D. Metcalf, P.G. Ekert, D.C. Huang, F. Cecconi, K. Kuida, K.J. Tomaselli, et al. 2002. Apoptosis initiated by Bcl-2-regulated caspase activation independently of the cytochrome c/Apaf-1/caspase-9 apoptosome. Nature. 419:634–637. [DOI] [PubMed] [Google Scholar]
- Mathai, J.P., M. Germain, R.C. Marcellus, and G.C. Shore. 2002. Induction and endoplasmic reticulum location of BIK/NBK in response to apoptotic signaling by E1A and p53. Oncogene. 21:2534–2544. [DOI] [PubMed] [Google Scholar]
- Nakagawa, T., and J. Yuan. 2000. Cross-talk between two cysteine protease families. Activation of caspase-12 by calpain in apoptosis. J. Cell Biol. 150:887–894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakagawa, T., H. Zhu, N. Morishima, E. Li, J. Xu, B.A. Yankner, and J. Yuan. 2000. Caspase-12 mediates endoplasmic-reticulum-specific apoptosis and cytotoxicity by amyloid-β. Nature. 403:98–103. [DOI] [PubMed] [Google Scholar]
- Nakano, K., and K.H. Vousden. 2001. PUMA, a novel proapoptotic gene, is induced by p53. Mol. Cell. 7:683–694. [DOI] [PubMed] [Google Scholar]
- Nguyen, M., P.E. Branton, P.A. Walton, Z.N. Oltvai, S.J. Korsmeyer, and G.C. Shore. 1994. Role of membrane anchor domain of Bcl-2 in suppression of apoptosis caused by E1B-defective adenovirus. J. Biol. Chem. 269:16521–16524. [PubMed] [Google Scholar]
- Nutt, L.K., J. Chandra, A. Pataer, B. Fang, J.A. Roth, S.G. Swisher, R.G. O'Neil, and D.J. McConkey. 2002. a. Bax-mediated Ca2+ mobilization promotes cytochrome c release during apoptosis. J. Biol. Chem. 277:20301–20308. [DOI] [PubMed] [Google Scholar]
- Nutt, L.K., A. Pataer, J. Pahler, B. Fang, J. Roth, D.J. McConkey, and S.G. Swisher. 2002. b. Bax and Bak promote apoptosis by modulating endoplasmic reticular and mitochondrial Ca2+ stores. J. Biol. Chem. 277:9219–9225. [DOI] [PubMed] [Google Scholar]
- Oda, E., R. Ohki, H. Murasawa, J. Nemoto, T. Shibue, T. Yamashita, T. Tokino, T. Taniguchi, and N. Tanaka. 2000. Noxa, a BH3-only member of the Bcl-2 family and candidate mediator of p53-induced apoptosis. Science. 288:1053–1058. [DOI] [PubMed] [Google Scholar]
- Oltvai, Z.N., C.L. Milliman, and S.J. Korsmeyer. 1993. Bcl-2 heterodimerizes in vivo with a conserved homolog, Bax, that accelerates programmed cell death. Cell. 74:609–619. [DOI] [PubMed] [Google Scholar]
- Pan, Z., M.B. Bhat, A.L. Nieminen, and J. Ma. 2001. Synergistic movements of Ca(2+) and Bax in cells undergoing apoptosis. J. Biol. Chem. 276:32257–32263. [DOI] [PubMed] [Google Scholar]
- Pinton, P., D. Ferrari, P. Magalhaes, K. Schulze-Osthoff, F. Di Virgilio, T. Pozzan, and R. Rizzuto. 2000. Reduced loading of intracellular Ca(2+) stores and downregulation of capacitative Ca(2+) influx in Bcl-2-overexpressing cells. J. Cell Biol. 148:857–862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puthalakath, H., D.C. Huang, L.A. O'Reilly, S.M. King, and A. Strasser. 1999. The proapoptotic activity of the Bcl-2 family member Bim is regulated by interaction with the dynein motor complex. Mol. Cell. 3:287–296. [DOI] [PubMed] [Google Scholar]
- Rizzuto, R., P. Pinton, W. Carrington, F.S. Fay, K.E. Fogarty, L.M. Lifshitz, R.A. Tuft, and T. Pozzan. 1998. Close contacts with the endoplasmic reticulum as determinants of mitochondrial Ca2+ responses. Science. 280:1763–1766. [DOI] [PubMed] [Google Scholar]
- Srivastava, R.K., S.J. Sollott, L. Khan, R. Hansford, E.G. Lakatta, and D.L. Longo. 1999. Bcl-2 and Bcl-X(L) block thapsigargin-induced nitric oxide generation, c-Jun NH(2)-terminal kinase activity, and apoptosis. Mol. Cell. Biol. 19:5659–5674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, N.S., M.T. Unkila, E.Z. Reineks, and C.W. Distelhorst. 2001. Transient expression of wild-type or mitochondrially targeted Bcl-2 induces apoptosis, whereas transient expression of endoplasmic reticulum-targeted Bcl-2 is protective against Bax-induced cell death. J. Biol. Chem. 276:44117–44128. [DOI] [PubMed] [Google Scholar]
- Wang, X. 2001. The expanding role of mitochondria in apoptosis. Genes Dev. 15:2922–2933. [PubMed] [Google Scholar]
- Wei, M.C., T. Lindsten, V.K. Mootha, S. Weiler, A. Gross, M. Ashiya, C.B. Thompson, and S.J. Korsmeyer. 2000. tBID, a membrane-targeted death ligand, oligomerizes BAK to release cytochrome c. Genes Dev. 14:2060–2071. [PMC free article] [PubMed] [Google Scholar]
- Wei, M.C., W.X. Zong, E.H. Cheng, T. Lindsten, V. Panoutsakopoulou, A.J. Ross, K.A. Roth, G.R. MacGregor, C.B. Thompson, and S.J. Korsmeyer. 2001. Proapoptotic BAX and BAK: a requisite gateway to mitochondrial dysfunction and death. Science. 292:727–730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu, J., L. Zhang, P.M. Hwang, K.W. Kinzler, and B. Vogelstein. 2001. PUMA induces the rapid apoptosis of colorectal cancer cells. Mol. Cell. 7:673–682. [DOI] [PubMed] [Google Scholar]
- Zhu, W., A. Cowie, G.W. Wasfy, L.Z. Penn, B. Leber, and D.W. Andrews. 1996. Bcl-2 mutants with restricted subcellular location reveal spatially distinct pathways for apoptosis in different cell types. EMBO J. 15:4130–4141. [PMC free article] [PubMed] [Google Scholar]
- Zong, W.X., T. Lindsten, A.J. Ross, G.R. MacGregor, and C.B. Thompson. 2001. BH3-only proteins that bind pro-survival Bcl-2 family members fail to induce apoptosis in the absence of Bax and Bak. Genes Dev. 15:1481–1486. [DOI] [PMC free article] [PubMed] [Google Scholar]