

Abstract
Activation of the Ras–MAPK signal transduction pathway is necessary for biological responses both to growth factors and ECM. Here, we provide evidence that phosphorylation of S298 of MAPK kinase 1 (MEK1) by p21-activated kinase (PAK) is a site of convergence for integrin and growth factor signaling. We find that adhesion to fibronectin induces PAK1-dependent phosphorylation of MEK1 on S298 and that this phosphorylation is necessary for efficient activation of MEK1 and subsequent MAPK activation. The rapid and efficient activation of MEK and phosphorylation on S298 induced by cell adhesion to fibronectin is influenced by FAK and Src signaling and is paralleled by localization of phospho-S298 MEK1 and phospho-MAPK staining in peripheral membrane–proximal adhesion structures. We propose that FAK/Src-dependent, PAK1-mediated phosphorylation of MEK1 on S298 is central to the organization and localization of active Raf–MEK1–MAPK signaling complexes, and that formation of such complexes contributes to the adhesion dependence of growth factor signaling to MAPK.
Keywords: integrin; adhesion; focal adhesion kinase; Src; extracellular matrix
Introduction
Serum growth factor stimulation of adherent fibroblasts activates MAPK through the sequential activation of Ras, Raf, and MAPK kinase (MEK)* 1/2. Similarly, adhesion of cells to ECM proteins in the absence of growth factors has been reported to stimulate MAPK activity through a Ras-dependent pathway (Schlaepfer et al., 1994; Moro et al., 1998; Wary et al., 1998). Although Ras activation proceeds normally in suspended cells treated with growth factors, MAPK activation is uncoupled at the level of Raf or MEK (Lin et al., 1997; Renshaw et al., 1997). These data are consistent with a model in which adhesion and growth factor signals are integrated downstream of Ras to regulate the functional interactions between Ras and Raf or Raf and MEK. Thus, MEK appears to be well positioned to serve as an anchorage sensor for MAPK signaling.
Adhesion of cells to the ECM stimulates a number of signaling pathways including FAK, Src, and those initiated by the small GTPase Rac (Parsons et al., 2000). Integrin binding to ECM proteins elicits the formation of focal complexes and the recruitment and activation of two protein tyrosine kinases, FAK and Src (Parsons et al., 2000). Integrin-induced activation of FAK results in autophosphorylation of FAK on tyrosine 397 (Y397), which correlates with increased catalytic activity of FAK (Lipfert et al., 1992; Calalb et al., 1995) and serves as a high affinity binding site for the SH2 domain of Src family kinases.
The small GTPase Rac mediates lamellipodia extension and focal complex formation initiated by integrin binding to ECM proteins (Nobes and Hall, 1995). Activation of Rac by cell adhesion stimulates membrane ruffling and the formation of peripheral focal complexes through signaling cascades involving the Rac–Cdc42 effector p21-activated kinase (PAK; Bagrodia and Cerione, 1999). Localization of activated PAK to focal adhesions and membrane ruffles influences cytoskeletal dynamics through phosphorylation of LIM (Edwards et al., 1999) and myosin light chain kinases (Sanders et al., 1999). Signals initiated by Rac activation also influence the Ras–MAPK pathway by synergizing with Raf to activate MAPK (Frost et al., 1997), possibly by sensitizing MEK1 to activation by Raf (Coles and Shaw, 2002). Recently, we demonstrated that Rac–PAK signaling can enhance the association of MEK1 and MAPK and that this pathway is required for the formation of MEK1–MAPK complexes and MAPK activation upon cellular adhesion (Eblen et al., 2002). Moreover, synergy between Rac and Raf promotes anchorage-independent growth of fibroblasts (Qiu et al., 1995).
Because the Ras–MEK–MAPK signal transduction pathway serves as a point of convergence for the regulation of proliferation and migration by growth factors and ECM proteins, we examined the adhesion-dependent activation of both MEK and MAPK. Here, we provide evidence that phosphorylation of MEK1 on S298 by PAK is one point at which these two signaling pathways converge. We show that adhesion to fibronectin (FN) induces PAK1 phosphorylation of MEK1 on S298 and that MEK1 S298 phosphorylation is necessary for efficient activation-specific phosphorylation of MEK1 and subsequent MAPK activation. Adhesion-dependent phosphorylation of MEK1 on S298 is dependent in part upon FAK/Src signaling, consistent with the localization of phospho-S298 MEK1 and phospho-MAPK staining in peripheral membrane–proximal adhesion structures. Moreover, synergistic activation of MEK1 by growth factors and cell adhesion is diminished in cells expressing an MEK1 S298A mutant. We propose that FAK/Src-dependent PAK phosphorylation of MEK1 on S298 is central to the organization and localization of active Raf–MEK1–MAPK signaling complexes and that formation of such complexes underlies the observed adhesion dependence of growth factor signaling to MAPK.
Results
Adhesion-dependent activation of MAPK
Cell adhesion to FN in the absence of growth factors activates both PAK1 and MAPK (Figs. 1 A and 8 B; Aplin et al., 1999; Bagrodia and Cerione, 1999). Suspension of REF52 cells for 1 h in serum-free media greatly reduced phosphorylation of MAPK on the activating sites (i.e., T183/Y185 of ERK2) (Fig. 1, A and B). Replating suspended cells on FN stimulated both MEK activation site phosphorylation (S218/S222) and MAPK phosphorylation within 10 min, with maximal MEK phosphorylation on the activating sites occurring between 5 and 10 min (Fig. 1, A and B). To determine the localization of this active pool of MAPK, REF52 cells suspended for 1 h in serum-free media were replated on FN for 1 h in the absence of serum. Immunostaining with an antibody to the phosphorylated form of MAPK revealed phospho-MAPK colocalized with paxillin in well-defined adhesions (Fig. 1 C, arrowhead). In addition, prominent MAPK staining in peripheral structures resembling Rac-induced focal complexes was observed. These structures were distinguished from focal adhesions in that they did not contain significant levels of paxillin staining (Fig. 1 C, arrow). Cells spreading for shorter periods of time showed poorly organized phospho-MAPK staining (unpublished data).
Figure 1.

Adhesion stimulates MAPK and MEK phosphorylation. REF52 cells were either continuously adherent (A) or suspended (S) and plated on FN for 5, 10, 20, or 40 min. Whole cell lysates were blotted with antiserum specific for (A) phosphorylated MAPK (p-MAPK; top) or ERK2 (bottom), or (B) MEK1 phosphorylated on S218/S222 (p-S218/222MEK1; top) or MEK1 (bottom). (C) REF52 cells were suspended for 1 h and plated on FN for 1 h before co-staining for p-MAPK (red) and paxillin (green). The arrows indicate focal complex-like structures containing p-MAPK; arrowheads indicate paxillin-containing focal adhesions. Bar, 10 μm.
The localization of phosphorylated MAPK in structures resembling Rac-induced focal complexes prompted the examination of the role of Rac and its effector PAK in adhesion-dependent MAPK regulation. As previously reported, Rac signaling to PAK synergizes with Raf to regulate MAPK activity (Frost et al., 1997). In addition, PAK has been reported to phosphorylate MEK1 on S298 in vitro (Coles and Shaw, 2002). To examine PAK phosphorylation of MEK1, an antiphosphopeptide antiserum specific for phosphorylated S298 in MEK1 (p-S298 MEK1) was generated (see Materials and methods). Recombinant group I PAK proteins (PAK1, 2, and 3) stimulated phosphorylation of kinase-defective MEK1 on a site specifically recognized by anti–p-S298 MEK1 (Fig. 2 A, not depicted). Recognition of phosphorylated MEK1 was abolished by preincubation of anti–p-S298 MEK1 with the immunizing phosphopeptide, but not with the corresponding nonphosphopeptide (unpublished data). In addition, anti–p-S298 MEK1 failed to recognize MEK1 containing an alanine substitution at position 298 (S298A; Fig. 2 B). Immunoblotting with anti–p-S298 MEK1 revealed the loss of MEK1 S298 phosphorylation in cells suspended in serum-free media (Fig. 3 A). Replating on FN (Fig. 3 A), laminin, or vitronectin (not depicted) led to a rapid increase in MEK1 S298 phosphorylation (within 5 min), which was maintained throughout the course of the assay and equivalent to levels observed in continuously adherent cells. As shown in Fig. 3 B, anti–p-S298 MEK1 selectively identified endogenous phosphorylated MEK1, but not MEK2, immunoprecipitated from cells. Although MEK2 contains a site equivalent to S298, it does not appear to be a substrate for phosphorylation by PAK (Frost et al., 1997). Finally, adhesion-dependent MEK1 S298 phosphorylation was observed in many cell lines including WI38, mouse embryo fibroblasts, CCL39, COS-1, LNCaP, and MCF7 (unpublished data), indicating the generality of MEK1 S298 phosphorylation.
Figure 2.

PAK directs MEK1 S298 phosphorylation in vitro. (A) In vitro kinase assays were performed using recombinant MEK1 and recombinant PAK3 as described in Materials and methods. (B) REF52 cells were transfected with HA-tagged wild-type MEK1 or HA-MEK1 S298A, suspended for 1 h and allowed to adhere to FN for 10 min. Anti-HA immunoprecipitates were analyzed and blotted with p-S298MEK1 (top) or MEK1 (bottom).
Figure 3.

MEK1 S298 phosphorylation is regulated by cell adhesion. (A) REF52 cells were treated as described in Fig. 1. Whole cell lysates were blotted with antiserum specific for phospho-S298 MEK1 (p-S298MEK1; top) or MEK1 (bottom). (B) REF52 cells were suspended (S) and replated on FN for 10, 20, or 40 min. Anti-MEK1 or anti-MEK2 antiserum was used to immunoprecipitate endogenous proteins, which were subsequently blotted with anti– p-S298MEK1 or anti-MEK1/2. (C) REF52 cells were suspended and plated on FN for 10 min or 1 h before co-staining for p-S298MEK1 (red) and paxillin (green). The intense staining in the center of the cell represents perinuclear staining that was over-exposed to visualize peripheral structures. Bars, 10 μm.
Because activated PAK is located in peripheral membrane ruffles (Sells et al., 2000), we examined the localization of p-S298 MEK1 in REF52 cells spreading on FN for 10 min. Immunostaining revealed localization of p-S298 MEK1 in peripheral complexes coincident with paxillin staining (Fig. 3 C) as well as in a poorly defined perinuclear compartment (not depicted). After the cells spread for 60 min, paxillin staining became more organized and localized to focal adhesions, whereas anti–p-S298 MEK1 staining appeared diffused with little detectable staining of paxillin-containing focal adhesions. Together, these observations indicate that MEK1 S298 phosphorylation is induced early upon FN adhesion and p-S298 MEK1 is localized in peripheral focal complexes.
PAK regulates adhesion-dependent phosphorylation of MEK1 on S298 and subsequent MEK1 activation
To examine whether PAK regulates adhesion-dependent MEK1 S298 phosphorylation in vivo, COS-1 or REF52 cells expressing kinase-defective PAK1 (or vector control) together with HA-tagged wild-type MEK1 were placed in suspension and replated on FN for the indicated times and S298 phosphorylation was assessed by Western blotting. Expression of kinase-defective PAK1 significantly reduced FN-stimulated S298 phosphorylation of exogenously expressed MEK1 (Fig. 4 A, not depicted), indicating that PAK activation is necessary for adhesion-dependent MEK1 S298 phosphorylation. To determine if PAK activity is sufficient to mediate MEK1 S298 phosphorylation in vivo, MEK1 S298 phosphorylation was examined in cells overexpressing activated PAK (T423E). REF52 cells expressing activated PAK together with wild-type MEK1 exhibited elevated MEK1 S298 phosphorylation whether in suspension or plated on FN (Fig. 4 B). Lastly, endogenous PAK1 immunoprecipitated from REF52 cells was activated by adhesion to FN (see Fig. 8 B), whereas endogenous PAK2 and 4 were not (not depicted). Together, these data indicate that PAK1 activity is both necessary and sufficient to stimulate MEK1 S298 phosphorylation and that PAK1-mediated phosphorylation of MEK1 on S298 is regulated by cell adhesion.
Figure 4.

PAK regulates adhesion-dependent MEK1 S298 phosphorylation. (A) COS-1 cells were transfected with myc-tagged kinase-defective PAK1 (Myc-PAK1-KR) or empty vector control together with HA-tagged MEK1. Cells were suspended (S) or plated on FN for 10, 20, or 30 min. Immunoprecipitates were formed using HA antiserum and blotted for p-S298MEK1 or MEK1. PAK1 expression was determined from whole cell lysates of transfected cells by immunoblotting with Myc antiserum. (B) REF52 cells were transiently cotransfected with HA-MEK1 together with myc-PAK1 T423E or the appropriate empty vector. Cells were suspended for 90 min (S) and allowed to adhere to FN-coated dishes for 20 min (FN). Anti-HA immunoprecipitates were formed and blotted with HA antiserum (middle), and subsequently with anti-pS298MEK1 (top). Western blotting of lysates with anti-myc antiserum confirmed expression of activated PAK1 (bottom).
Figure 8.

Adhesion-dependent c-Raf and PAK activity are affected by inhibiting Src. (A) c-Raf activity was determined from REF52 cells incubated in suspension with 50 μM PP2 or DMSO solvent control (S) and plated on FN for 5 or 10 min. Immunoprecipitates were formed using either c-Raf antiserum, or matched control antiserum raised against the GAL4 transactivation domain, and kinase assays were performed with kinase-defective GST-MEK1 as described in Materials and methods (top). A reaction blank (lane B) confirms the absence of MEK kinase activity in the substrate and c-Raf antiserum, and a reaction lacking GST-MEK1 (lane 8) confirms that MEK1 is the phosphorylated substrate in these assays. Loading controls (bottom) demonstrate similar levels of c-Raf protein in the appropriate assays. (B) PAK1 activity was determined as described above for c-Raf activity (A) except cells were also plated for 5, 10, or 20 min and immunoprecipitates were formed using PAK1 antiserum. PAK1 activity was assessed by blotting the recombinant MEK1 substrate with the p-S298 antiserum (top). The PAK1 loading control is shown in the bottom panel. Lane B denotes a reaction blank in which PAK1 antibody was incubated with lysis buffer instead of lysate before the kinase assay.
MEK1 containing an S298A mutation was reported to bind less efficiently to Raf than wild-type MEK1 indicating that MEK1 S298 phosphorylation regulates the interaction of MEK1 with its upstream activator (Frost et al., 1997). To examine the requirement for S298 phosphorylation on FN-stimulated MEK1 activation, cells expressing HA-tagged MEK1 variants containing T292A, S298A, or T292A/S298A mutations were placed in suspension or plated on FN for 20 min. The MEK1 variants were immunoprecipitated and blotted for p-S218/S222 MEK as an indicator of MEK activation. Phosphorylation of S218/S222 was stimulated by FN in cells expressing both wild-type MEK1 and MEK1 T292A, but not in cells expressing MEK1 S298A or MEK1 T292A/S298A (Fig. 5). Thus, MEK1 S298 is necessary for efficient activation of MEK1 during adhesion.
Figure 5.

Phosphorylation of MEK1 on S298 regulates MEK1 activation. REF52 cells were transiently transfected with HA-MEK1, HA-MEK1 T292A, HA-MEK1 S298A, or HA-MEK1 T292A/S298A. Cells were suspended for 90 min (S) and plated on FN for 20 min (FN). Anti-HA immunoprecipitates were formed and blotted with anti-p-S218/S222MEK1 (top), anti-pS298MEK1 (middle), or anti-HA antiserum (bottom).
FAK signaling regulates phosphorylation of MEK1
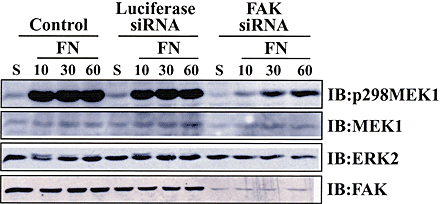
FAK and Src regulate a well-defined pathway that becomes activated after integrin engagement (Parsons et al., 2000). In addition, FAK/Src signaling has been shown to play a role in FN-stimulated MAPK activation (Schlaepfer and Hunter, 1996; Schlaepfer et al., 1994). We examined the role of this pathway in regulating MEK1 S298 phosphorylation using FAK-deficient cells or PP2, an inhibitor of Src family kinases. FAK-null fibroblasts or controls from wild-type littermates were placed in suspension for 1 h and stimulated to spread on FN for the indicated times (Fig. 6 A). Western blot analysis of whole cell lysates using anti–p-S298 MEK1 demonstrated decreased levels (∼70% wild-type levels determined by densitometry) and delayed time course of MEK1 S298 phosphorylation in FAK-null cells compared with FAK-expressing wild-type fibroblasts (Fig. 6 A). Similarly, decreasing FAK protein levels in REF52 cells using siRNAs also resulted in decreased and delayed MEK1 S298 phosphorylation upon adhesion of these cells to FN (unpublished observations; Fig. S1). In both FAK-null and -expressing cells, MAPK phosphorylation paralleled MEK1 S298 phosphorylation (Fig. 6 A).
Figure 6.

FAK signaling regulates MEK1 S298 phosphorylation. (A) FAK-null fibroblasts or cells from wild-type littermate controls were suspended (S), plated on FN for 5, 15, or 45 min, or remained adherent (A). Whole cell lysates were blotted with p-S298MEK1, MEK1, p-MAPK, or ERK2 antisera. (B) FAK-null fibroblasts transfected with myc-tagged wild-type FAK, FAK Y397F, or empty vector control together with HA-tagged wild-type MEK1 were suspended (S) and replated on FN for 5, 10, or 20 min. Immunoprecipitates were formed using HA antiserum and blotted for p-S298MEK1 or MEK1. FAK expression was determined from whole cell lysates of transfected cells using Myc antiserum. FAK autophosphorylation was determined by immunoblotting with Y397 FAK phospho-specific antibodies.
The role of FAK in MEK1 phosphorylation was assessed by reconstituting FAK-null cells with wild-type FAK or the FAK autophosphorylation mutant Y397F. Phosphorylation of cotransfected MEK1 on S298 was measured in suspended cells or cells allowed to adhere to FN for the indicated times. Expression of wild-type FAK, but not the FAK autophosphorylation mutant, Y397F, restored MEK1 S298 phosphorylation in FN-stimulated cells when compared with the empty vector control (Fig. 6 B). Transient overexpression of wild-type FAK under these conditions resulted in adhesion-independent FAK activation, as indicated by constitutive FAK Y397 phosphorylation in suspended cells (Fig. 6 B). Concomitantly, overexpression of wild-type FAK also stimulated MEK1 S298 phosphorylation in suspended cells (Fig. 6 B), which paralleled the increased phosphorylation of S298 observed in suspended cells overexpressing activated PAK1 (Fig. 4 B). The FAK-directed increase in MEK1 S298 phosphorylation was inhibited by coexpression of kinase-defective PAK1, whereas activated PAK1 stimulated MEK1 S298 phosphorylation in cells expressing Y397F FAK (unpublished data). Together, these observations indicate a direct role for FAK activity in stimulating PAK1-directed MEK1 S298 phosphorylation.
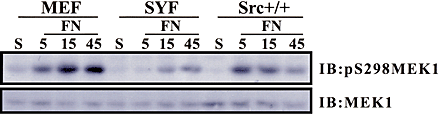
Adhesion-mediated activation of FAK results in the stimulation of Src after binding to FAK phosphorylated on Y397 (Cobb et al., 1994; Schaller et al., 1994, 1999; Reiske et al., 1999). Because Y397F FAK was unable to restore MEK1 S298 phosphorylation, we assessed the role of Src in regulating adhesion-dependent MEK1 S298 phosphorylation using the Src family kinase inhibitor, PP2. REF52 cells were suspended in serum-free media in the presence or absence of PP2, and replated on FN in the continued presence or absence of PP2 (Fig. 7 A). PP2 treatment decreased the extent and delayed the time course of S298 phosphorylation after FN stimulation (Fig. 7 A), similar to the observations for FAK-null cells (Fig. 6 A). MEK1 S298 phosphorylation in cells stimulated to adhere to FN for 5 min in the presence of PP2 was ∼28% the level of untreated cells when normalized to MEK1 loading control. Overexpression of activated PAK (PAK1 T423E) rescued the decrease in MEK1 S298 phosphorylation induced by PP2 treatment (Fig. 7 B). In addition, active PAK-stimulated MEK1 S218/S222 phosphorylation on wild-type MEK1 and to a lesser degree on MEK1 S298A. Finally, cells deficient for Src, Yes, and Fyn also showed decreased and delayed MEK1 S298 phosphorylation upon plating on FN (Fig. S2). These results indicate that Src or a Src family kinase is involved in regulating the pathway(s) leading to MEK1 S298 phosphorylation.
Figure 7.

Src inhibition alters PAK-mediated MEK phosphorylation. (A) REF52 cells were incubated in suspension with 50 μM PP2 or DMSO control (S) and plated on 10 μg/ml FN for 5, 10, 20, or 40 min in the continued presence or absence of PP2. Whole cell lysates were blotted with p-S298MEK1, p-S218/S222MEK1, MEK1, p-MAPK, or ERK2 antisera. Densitometric analysis demonstrated that the intensity of p-S298MEK1 after a 5-min plating on FN in the presence of 50 μM PP2 was ∼28% the level of the DMSO-treated control when normalized to an MEK1 loading control. The observed decrease in p-MAPK levels in lane 9 was not reproducible. (B) REF52 cells were cotransfected with HA-tagged MEK1 constructs and PAK1 T423E or vector control, incubated in suspension with 50 μM PP2 or DMSO control (S) and plated on FN for 20 min. Anti-HA immunoprecipitates were formed and blotted with pS218/S222 antiserum (top), HA antiserum (second [from top] panel), and subsequently with anti-pS298MEK1 (third [from top] panel). Western blotting of lysates with anti-myc antiserum confirmed expression of activated PAK1 (bottom).
In addition to regulating MEK1 S298 phosphorylation, PP2 treatment virtually eliminated MEK activation measured by S218/S222 phosphorylation and significantly decreased MAPK phosphorylation in response to FN stimulation (Fig. 7 A). At least two mechanisms exist by which MEK1 activation could be blocked by the Src inhibitor. Because S298 phosphorylation is necessary for MEK1 activation upon adhesion and is blocked by PP2, Src might regulate activation through its effects on S298 phosphorylation. Alternatively, because Src is reported to phosphorylate and activate Raf (Fabian et al., 1993; Marais et al., 1995), PP2 might function to decrease Raf activation upon adhesion in addition to regulating S298 phosphorylation. To distinguish between these possibilities, we examined the effect of inhibiting Src on Raf and PAK activity. REF52 cells were suspended for 1 h in the presence or absence of PP2 and stimulated with FN for 10 min in the continued presence or absence of PP2. C-Raf was immunoprecipitated and kinase activity was assessed in vitro by measuring 32P incorporation into recombinant kinase–defective MEK1. In the absence of PP2, FN induced little change in c-Raf activity (Fig. 8 A). On the other hand, PP2 treatment resulted in increased c-Raf activity in cells adhering to FN (Fig. 8 A). Despite the observed increase in c-Raf activity after PP2 treatment, phosphorylation of endogenous MEK1 on the activating sites was not observed (Fig. 7 A; not depicted).
The simplest interpretation of these observations is that the block in MEK1 activation in response to PP2 treatment results from inhibiting PAK1-mediated phosphorylation of MEK1 on S298. Therefore, we examined the effect of PP2 treatment on PAK1 activity in response to FN stimulation. Endogenous PAK1 was immunoprecipitated from REF52 cells suspended in the presence or absence of PP2 or replated on FN in the continued presence or absence of PP2. Activity was assessed by measuring the ability of immunoprecipitated PAK1 to phosphorylate recombinant kinase–defective MEK1 on S298 in vitro (Fig. 8 B). FN stimulated immune complex PAK1 activity within 5 min of replating, whereas PP2 treatment delayed the activation of PAK1 from 5 to 20 min (Fig. 8 B). Thus, the effects of PP2 on activation of PAK by FN parallel the effects of PP2 on endogenous MEK1 S298 phosphorylation. Together, these observations indicate that FAK and Src signal transduction cascades contribute to the FN-stimulated MEK1 and MAPK activation primarily by stimulating PAK-mediated phosphorylation of MEK1 on S298.
Because Src-dependent MEK1 S298 phosphorylation is required for adhesion-mediated activation of MEK (Fig. 5) and subsequent MAPK activation (Eblen et al., 2002), we examined the effect of Src inhibition on phospho-MAPK localization in REF52 cells. Cells were suspended for 1 h and allowed to spread on FN for 1 h in the continued presence or absence of PP2 and immunostained for phospho-MAPK (red) and paxillin (green). Consistent with a previous report (Fincham et al., 2000), inhibition of Src activity had no effect on the localization of phospho-MAPK to focal adhesions (Fig. 9, arrowheads). However, PP2 decreased the extent of phospho-MAPK staining in peripheral structures reminiscent of Rac-induced adhesion complexes. Indeed, only 22% of the cells treated with PP2 exhibited peripheral adhesion complexes containing phospho-MAPK, whereas almost all (98%) of the untreated REF52 cells displayed peripheral adhesion complexes rich in phospho-MAPK staining (Fig. 9, arrows). These observations indicate that, in addition to decreasing MAPK activation, inhibition of Src activity reduces the pool of activated MAPK recruited to newly formed adhesion complexes.
Figure 9.

Src inhibition alters the location of phospho-MAPK. REF52 cells were suspended in the presence or absence of 50 μM PP2 and plated on FN for 1 h in the continued presence or absence of PP2 before co-staining for p-MAPK (red) and paxillin (green). The arrows indicate peripheral adhesion complexes containing p-MAPK; arrowheads indicate paxillin-containing focal adhesions. Bar, 10 μm.
MEK1 S298 phosphorylation modulates growth factor–stimulated MEK1 activation
ECM adhesion has been reported to influence serum growth factor–stimulated MAPK activation at the level of Raf or MEK (Lin et al., 1997; Renshaw et al., 1997). Indeed, REF52 cells suspended for 90 min and stimulated with growth factors or replated on FN for 30 min resulted in a modest induction of MAPK phosphorylation (Fig. 10 A). Stimulating fibroblasts with growth factors while they adhere to FN for 30 min resulted in an enhanced activation of MAPK phosphorylation (Fig. 10, A and B) as reported previously (Roovers et al., 1999). The ability of FN to amplify growth factor–stimulated MAPK activity was most evident at lower, more physiologically relevant concentrations of EGF (0.125 ng/ml; Fig. 10 B, lanes 9 and 10), whereas higher concentrations of EGF (10 ng/ml) overcame the adhesion requirement to efficiently activate MAPK (Fig. 10 B, lanes 1 and 2). In addition, MEK1 activation was maximally activated by stimulation with both growth factors and adhesion to FN (Fig. 10 C). To determine whether MEK1 S298 phosphorylation plays a role in MEK1 activation in response to growth factor stimulation, HA-tagged wild-type MEK1 or MEK1 S298A was immunoprecipitated from REF52 cells that were serum-starved, suspended, and either stimulated with EGF in suspension, replated onto FN, or stimulated and replated. In the absence of adhesion, growth factor stimulation of REF52 cells resulted in equivalent S218/S222 phosphorylation of exogenously expressed wild-type MEK1 or MEK1 S298A (Fig. 10 D, lanes 2 and 8). However, growth factor stimulation of adherent cells expressing MEK1 S298A resulted in ∼50% less activation of MEK S218/S222 compared with cells expressing wild-type MEK1 (Fig. 10 D, lanes 4–6 and 10–12). Together, these results indicate that adhesion-dependent MEK1 S298 phosphorylation regulates, at least in part, the maximal activation of MEK1 by serum growth factors.
Figure 10.

Adhesion-dependent MEK1 S298 phosphorylation promotes maximal MEK1 activation in response to growth factor stimulation. (A) REF52 cells were suspended for 90 min and either stimulated for 30 min with EGF and IGF-1 in suspension, replated on FN for 30 min, or stimulated with EGF and IGF-1 while they attached to FN for 30 min. Whole cell lysates were blotted with p-MAPK or ERK2 antisera. (B) REF52 cells were placed in suspension for 90 min and either stimulated in suspension for 30 min with the indicated concentrations of EGF or replated on FN in the presence of the indicated concentrations of EGF for 30 min. Whole cell lysates were blotted with p-MAPK or ERK2 antisera. (C) Cells were treated as in A, and whole cell lysates were blotted with p-S218/222 MEK or MEK1 antisera. (D) REF52 cells were transiently transfected with HA-MEK1 (lanes 1–6) or HA-MEK1 S298A (lanes 7–12). Cells were suspended in serum-free media for 90 min and either kept unstimulated in suspension (lanes 1 and 7), stimulated in suspension with EGF for 20 min (lanes 2 and 8), allowed to adhere to FN (lanes 3 and 9) or allowed to attach to FN while stimulated with EGF for the indicated times (lanes 4–6 and 10–12). Anti-HA immunoprecipitates were formed and blotted with p-S218/222 MEK1, p-S298MEK1 or HA antisera. Densitometry and normalization to the loading controls revealed that S218/222 phosphorylation of MEK1 S298A was ∼50% that seen in the wild-type protein.
Discussion
This paper provides evidence that phosphorylation of MEK1 on S298 is a convergence point for integrating growth factor signaling and adhesion signaling via the MAPK pathway. PAK1 efficiently phosphorylated MEK1 on S298 in vitro and in vivo. MEK1 S298 phosphorylation was necessary for efficient activation of MEK1 and subsequent MAPK activation. The rapid phosphorylation of MEK on S298, and its efficient activation in response to adhesion to FN required FAK and Src and were paralleled by localization of phospho-S298 MEK1 and phospho-MAPK staining in peripheral membrane–proximal adhesion structures. Thus, these experiments identify MEK1 as a target of combinatorial signaling through growth factor and integrin receptors.
The adhesion-dependent PAK1 phosphorylation of MEK1 on S298 indicates that MEK1, through its functional coupling of Raf and MAPK, might serve as a sensor for the adhesive status of cells or cellular compartments (e.g., protrusions at the leading edge). In addition, our work provides a molecular framework for the observation that stimulation of cells in suspension with physiologic concentrations of growth factor fails to activate MEK and MAPK despite activating Ras and Raf. These observations suggest that anchorage-independent activation of PAK and consequent phosphorylation of MEK1 might contribute to increased migration and anchorage-independent proliferation during malignancy.
Role of Rac, PAK, and S298 phosphorylation in MAPK activation
Previous studies have shown that Rac signaling to PAK synergizes with Raf to regulate MAPK activity (Frost et al., 1997). The PAK effects on MAPK signal transduction appear to be mediated by phosphorylation of Raf on S338 (King et al., 1998; Chaudhary et al., 2000). PAK-mediated phosphorylation of c-Raf has been reported to stimulate its activity (King et al., 1998). Furthermore, adhesion of COS7 cells overexpressing c-Raf mutants has been reported to stimulate c-Raf S338 phosphorylation through a phosphoinositide 3-kinase (PI3K)–dependent mechanism (Chaudhary et al., 2000). We did not observe stimulation of endogenous c-Raf S338 phosphorylation (not depicted) or endogenous c-Raf activity (Fig. 8 A) when cells were replated on FN. Moreover, inhibiting PI3K activity with LY294002 had no effect on MEK1 S298 phosphorylation (unpublished data). These discrepancies could reflect differences in the pathways used by different cell types or an alteration in pathway usage when proteins are overexpressed. Indeed, overexpression of active PAK1 led to a modest stimulation of the activation of MEK1 S298A as judged by increased phosphorylation of S218/S222. This observation suggests the possibility that overexpression of activated PAK1 may obviate normal signaling constraints by phosphorylating Raf on S338, thus enhancing MEK activation in a S298-independent manner. This is consistent with observations showing adhesion-dependent Raf activation after PAK phosphorylation of Raf-S338 in COS7 cells overexpressing c-Raf mutants (Chaudhary et al., 2000). Based on these observations, we suggest that the Raf–MEK–MAPK pathway may be exquisitely sensitive to the level of PAK1 activation and in the case of adhesion of REF52 cells to FN, PAK1 is primarily a modulator of MEK1 activation.
An alternative possibility to explain PAK-Raf synergy is that PAK-dependent phosphorylation of MEK1 on S298 leads to enhancement of MAPK by modulating both the physical and functional interactions between Raf, MEK, and MAPK. Frost and co-workers showed that MEK1 with mutations of T292 and S298 to alanine bound c-Raf less efficiently than did wild-type MEK1 (Frost et al., 1997). Indeed, recent evidence has shown that PAK phosphorylation of S298 sensitizes MEK1 to activation by Raf (Coles and Shaw, 2002). Here, we show that MEK1 S298 phosphorylation is dependent on cell adhesion (Fig. 3 A) and that adhesion-dependent phosphorylation of MEK1 on S298 was required for phosphorylation and activation of MEK1 in response to FN stimulation (Fig. 5). However, MEK1 activation occurred in the absence of a measurable increase in c-Raf activity. One possible explanation is that adhesion-dependent activation of MEK1 is regulated by phosphorylation from MEK kinases other than Raf, including MEK kinase (MEKK) 1, 2, and 3, which have been implicated in stimulating the MAPK pathway (Ellinger-Ziegelbauer et al., 1997; Xia et al., 1998). We cannot rule out the possibility that other MEK kinases are activated by adhesion to FN, however, PAK1 phosphorylation of MEK1 would be required for MEKK to activate MEK1 because MEK1 S298A was not phosphorylated on the activating sites upon plating on FN (Fig. 5). An alternative possibility is that adhesion-dependent PAK1 activation and subsequent phosphorylation of MEK1 on S298 is sufficient to stimulate MEK1 phosphorylation and activation by low levels of c-Raf activity merely by enhancing the interaction between c-Raf and MEK1. In both scenarios, adhesion-dependent PAK1 phosphorylation sensitizes MEK1 for activation from the upstream activator. Previous observations demonstrated that MEK1 was constitutively phosphorylated on S298 in the presence or absence of serum (Catling et al., 1995), signifying that MEK1 S298 phosphorylation is regulated uniquely by cell adhesion. Furthermore, in a separate study we show that adhesion-dependent phosphorylation of MEK1 on S298 is necessary for the formation of MEK1–MAPK complexes and subsequent MAPK activation (Eblen et al., 2002). Therefore, we suggest a unique activation mechanism by which cell adhesion stimulates MAPK activity by sensitizing MEK1 to activation by basal levels of c-Raf activity or a different MEKK and by promoting the assembly of MEK1–MAPK signaling complexes.
In addition to regulating the amplitude of MAPK activity, PAK1 likely influences the spatial regulation of MAPK by stimulating localized assembly of MEK1-signaling complexes in focal adhesions and peripheral focal complexes. Activated PAK1 localizes to focal adhesions and membrane ruffles (Sells et al., 2000). Indeed, PAK1 activity, reflected by phospho-S298 MEK1 staining, was observed in peripheral membrane structures 10 min after replating onto FN (Fig. 3 C). We propose that MEK1 phosphorylated on S298 stimulates MAPK activity in peripheral membrane structures by promoting activation complexes containing Raf and MAPK. MAPK, once activated, might down-regulate components of its activation pathway (Brunet et al., 1994; Dong et al., 1996), which could result in the dissociation of MEK from MAPK, allowing MEK to translocate to additional cellular compartments as was observed at later time points after replating (Fig. 3 C, 60 min FN).
FAK and Src as regulators of MAPK
FAK and Src positively regulate phosphorylation of MEK1 on S298 by PAK1. Cells deficient in FAK expression displayed decreased MEK1 S298 phosphorylation and a delayed time course of phosphorylation after FN stimulation (Fig. 6 A), which could be rescued by activated PAK (not depicted). The observation that wild-type FAK, but not FAK Y397F, which is incapable of interacting with Src, stimulated MEK1 S298 phosphorylation in FAK-deficient cells strongly indicates that these kinases function coordinately to regulate downstream signals to Rac and PAK. This is supported by similar observations in Src/Yes/Fyn-deficient cells and the observation that PP2 was an effective inhibitor of MEK1 298 phosphorylation. No change in MEK1 S298 phosphorylation was observed in cells treated with LY294002 (unpublished data), suggesting that PI3K, which also binds FAK phosphorylated on Y397 (Reiske et al., 1999), is not required for MEK1 S298 phosphorylation.
Inhibition of Src family kinases with PP2 not only inhibited MEK1 S298 phosphorylation, but also decreased phosphorylation of both MEK and MAPK on their respective activating sites in response to adhesion. Furthermore, PP2 inhibited the appearance of activated MAPK at the cell periphery. PP2 might inhibit Raf-mediated activation of MEK1 by reducing MEK1 S298 phosphorylation, or alternatively might reduce Raf activity directly, because Src phosphorylation of Raf has been shown to increase its activity (Fabian et al., 1993; Marais et al., 1995). However, adhesion to FN failed to stimulate Raf activity. Interestingly, Raf activity was actually increased after PP2 treatment even though phosphorylation of its downstream substrate MEK1 was suppressed. In contrast, adhesion-dependent PAK1 activation was decreased by PP2 treatment consistent with the hypothesis that FN-stimulated MEK1 activation is regulated primarily by PAK1 phosphorylation of MEK1 on S298. These observations, together with data showing that phospho-MAPK staining was decreased in peripheral focal complexes after PP2 treatment, indicate that the activation and localization of active MAPK to Rac-like focal complexes is regulated by Src-mediated activation of PAK1 and subsequent phosphorylation of MEK1.
Integrin activation of PAK
Integrin engagement leads to PAK activation and the targeting of PAK to adhesion complexes (Manser et al., 1998; del Pozo et al., 2000; West et al., 2001). The small GTPases Rac and Cdc42 are potent activators of PAK and are activated in response to integrin ligation. The molecular pathways leading to Rac–Cdc42 and PAK activation are likely numerous, however, the data presented in this paper indicate that in the setting of cell adhesion to FN, FAK, and Src play a significant (but not exclusive) role in the activation of PAK1. Previous studies have implicated PI3K in the integrin-dependent activation of PAK1, Raf, and MAPK (Chaudhary et al., 2000). In our work, the PI3K inhibitor LY294002 had little effect on the adhesion-induced phosphorylation of MEK1 on S298, consistent with activation via the integrin FAK/Src-directed pathway. Of interest is the recent observation that DOCK180, when combined with its binding partner ELMO, is an efficient activator of Rac (Brugnera et al., 2002). This suggests the possibility that FAK/Src-dependent activation of p130 Crk–associated substrate and Crk may lead to the activation of DOCK180/ELMO and the subsequent activation of Rac and PAK.
Biological importance of adhesion signaling to MEK
Active MAPK and pS298 MEK1 were found proximal to immature focal complexes and focal adhesions after FN stimulation. As pS298 MEK1 is a surrogate for active PAK, these findings support a role for these kinases in the regulation of focal adhesion dynamics and cell migration. MAPK and PAK activities have both been found to be important for cell migration (Klemke et al., 1997; Kiosses et al., 1999; Sells et al., 1999). Moreover, Rac overexpression enhances MAPK-dependent migration of cells stimulated with low levels of EGF while having little effect on migration in response to high concentrations of growth factor (Leng et al., 1999). PAK phosphorylation of MEK1 on S298, which is required for MEK1 activation-specific phosphorylation in the absence of inducible Raf activity, provides a mechanism whereby suboptimal Raf activation likely achieved with shallow gradients of growth factor stimulation could promote maximal MAPK activity in a localized manner.
In addition to stimulating MAPK activity, cell adhesion also regulates growth factor–induced activation of MAPK (Renshaw et al., 1997). Without an appropriate ECM, serum growth factors stimulate Ras activity, however, cell adhesion is required to activate MAPK at the level of Raf or MEK (Lin et al., 1997; Renshaw et al., 1997). Furthermore, the adhesion requirement for growth factor–induced MAPK activation is lost in cells overexpressing activated FAK (Renshaw et al., 1999) or PAK (Howe and Juliano, 2000). Our findings that adhesion-dependent PAK1 activation is required for MEK1 activation by Raf and for MEK1–MAPK coupling (Eblen et al., 2002) provide a mechanism to account for the disconnect between Raf and MEK in serum-stimulated suspended cells. In the absence of cell adhesion and MEK1 S298 phosphorylation, growth factor–stimulated Raf activity resulted in reduced phosphorylation and activation of MEK, thereby rendering MAPK activation dependent on cell adhesion to ECM. Because sustained MAPK activity regulates adhesion-dependent growth (Assoian and Schwartz, 2001), anchorage-independent growth may result from continuous PAK phosphorylation of MEK1 on S298. Indeed, PAK has been implicated in regulating anchorage-independent growth (Tang et al., 1997, 1999; Vadlamudi et al., 2000). Furthermore, we find adhesion-independent MEK1 S298 phosphorylation in highly tumorigenic human prostate cell lines (unpublished observations). Therefore, the loss of adhesion-dependent regulation of PAK activity would have a profound impact on tumorigenesis and metastatic progression by affecting cellular proliferation and migration.
Materials and methods
Reagents
REF52 and COS-1 (American Type Culture Collection) cells were grown in DME supplemented with 10% FBS (Life Technologies). FAK-null fibroblasts, provided by D. Ilic (Ilic et al., 1995) were grown in DME supplemented with 10% FBS (Life Technologies), 1% sodium pyruvate, 1% nonessential aa, and 0.5 μM β-mercaptoethanol (Bio-Rad Laboratories). PP2 (Calbiochem) and Src family kinase inhibitor were reconstituted in DMSO and used at a final concentration of 50 μM. The following antibodies were used: phospho-MAPK (New England Biolabs, Inc.; Zecevic et al., 1998); MEK1/2 (New England Biolabs, Inc.); phospho-MEK1 S218/S222 (Sigma-Aldrich); ERK2 (clone B3B9; Upstate Biotechnology); MEK1, MEK2, and paxillin (Transduction Labs); HA (clone 12CA5); myc (clone 9E10; Transduction Labs); p-FAK Y397 (Biosource International); PAK (anti-N20 for immunoprecipitation; anti-C19 for Western analysis); c-Raf (anti–C-12); and Gal4 transactivation domain (Santa Cruz Biotechnology, Inc.).
Generation and characterization of phospho-specific MEK1 antibody
The anti-MEK (pS298) antibody was prepared by Research Genetics. Rabbits were immunized with the KLH-conjugated phosphopeptide, R-T-P-G-R-P-L-pS-S-Y-G-M-D-S. Antibodies from pooled bleeds were affinity purified over a phosphopeptide column and passed over a nonphosphorylated peptide column to remove antibodies specific for the nonphosphorylated peptide. The purified antibody was tested for specificity both in ELISA assays, using bound phosphopeptide versus nonphosphopeptide, and on immunoblots of purified GST-MEK. Recombinant MEK1 was purified as described previously (Catling et al., 2001). Recombinant His-tagged PAK3 was a gift from S. Bagrodia and R.A. Cerione (Cornell University, Ithaca, NY). Purified MEK1 was incubated with or without recombinant PAK3 in 0.5 mM γ-[32P]ATP (∼8,400 cpm/pmol), 10 mM MgCl2, 1 mM DTT, and 25 mM Hepes-NaOH, pH 7.4, for 20 min at 30°C. Reactions were terminated with SDS-PAGE sample buffer and resolved on 10% gels. Incorporation was analyzed by autoradiography and immunoblotting. Control reactions lacking MEK1 showed no incorporation or p-S298 MEK1 reactivity, and Cerenkov counting indicated that PAK3 rapidly phosphorylates MEK1 to stoichiometry under these conditions.
Plasmids and transfection assays
All constructs in this work used a cytomegalovirus promoter to drive expression of peptide epitope–tagged protein. Wild-type MEK1, MEK1 T292A, MEK1 S298A, MEK1 T292A/S298A, and kinase-defective MEK1 (K97A) subcloned into pCMVHA (Catling et al., 1995), wild-type FAK, and the autophosphorylation mutant (Y397F) of FAK subcloned into pCMV-myc (Xiong and Parsons, 1997), and myc-tagged PAK1-K299R and PAK T423E subcloned into pRK5-myc (Weed et al., 1998).
To test the phosphorylation of MEK1 mutants (T292A, S298A, or T292A/S298A), two 150-mm dishes containing 2 × 106 REF52 were each transfected with 0.25 μg each MEK1 construct and 12.25 μg pCMVHA using Superfect. The effect of kinase-defective PAK on MEK1 S298 phosphorylation was tested by transfecting two 150-mm dishes containing 1.5 × 106 COS-1 cells each with 3 μg PAK1 K299R or empty vector together with 1.25 μg wild-type MEK1 using Lipofectamine. The effect of activated PAK on MEK1 phosphorylation was tested by transfecting two 150-mm dishes containing 1.5 × 106 REF52 cells each with 4 μg PAK1 T423E or empty vector together with 1 μg wild-type MEK1 using Superfect (QIAGEN). To assess the role of wild-type FAK or FAK Y397F on MEK1 phosphorylation, 4 × 106 FAK-deficient cells were plated on each of two 150-mm plates per construct and transfected each with 10.75 μg FAK, FAK Y397F, or empty vector together with 1.25 μg wild-type MEK1 using Polyfect (QIAGEN) according to the manufacturer's instructions. 24 h later, the cells were harvested, pooled from two dishes, suspended for 60–90 min in serum-free media, and replated on dishes coated with 10 μg/ml FN (Sigma-Aldrich) for 5–30 min. To test the effects of growth factors and cell adhesion on MEK and MAPK activation, REF52 cells were serum-starved for 16 h in DME containing 0.2% FBS, suspended in serum-free DME for 90 min, and either kept in suspension, stimulated in suspension with the indicated concentrations of EGF or with 2 ng/ml EGF and 100 ng/ml IGF-1, or allowed to adhere to 10 μg/ml FN in the presence or absence of growth factors for 30 min. The role of MEK1 S298 phosphorylation in MEK1 activation was determined by transfecting REF52 cells with HA-tagged wild-type MEK1 or HA-tagged MEK1 S298A. After 32 h, the cells were serum-starved in DME containing 0.2% FBS for 16 h. Cells were suspended in serum-free DME for 90 min and either treated with 3 ng/ml EGF in suspension, allowed to adhere to 10 μg/ml FN, or allowed to adhere to FN in the presence of EGF for the indicated times. For all transfection experiments, MEK1 phosphorylation was determined after immunoprecipitation using HA antibodies as described in the next section.
Immunoprecipitation and Western analysis
Suspended cells or those that were allowed to reattach to 10 μg/ml FN were lysed in supplemented RIPA buffer (50 mM Hepes, 0.15 M NaCl, 2 mM EDTA, 0.1% Nonidet P-40, and 0.05% sodium deoxycholate, pH 7.2) containing 1 mM PMSF, 100 mM leupeptin, and 0.05 TIU/ml aprotinin (1 mM Na3VO4, 40 mM NaF, and 10 mM Na4P2O7). Attached cells were lysed directly on the plate in supplemented RIPA buffer. Lysates were incubated for 1 h at 4°C with protein A–Sepharose beads (Amersham Biosciences) preconjugated with specific IgG. The immunoprecipitated proteins were resolved on 10% SDS-PAGE gels, transferred to nitrocellulose, and blotted with p-S218/S222 MEK1, 12CA5 (mAb to the HA tag), MEK1, or MEK2 followed by HRP-conjugated secondary antibody. The immunoblots were stripped (100 mM β-mercaptoethanol, 2% SDS, and 62.5 mM Tris-HCl, pH 6.7) and blotted with p-S298 MEK1 followed by incubation with HRP-conjugated goat anti–rabbit secondary antibodies and visualization by ECL (Amersham Biosciences). Western analysis of whole cell lysates was performed using 25–50 μg protein.
Kinase assays
REF52 cells were suspended in the presence of PP2 (50 μM) or DMSO for 90 min and allowed to adhere to 10 μg/ml FN in the continued presence of PP2 for the indicated times. Endogenous PAK1 or matched antibody-specific GAL4 control immunoprecipitates were formed. Kinase reactions were initiated by adding a mix containing 1 μg of recombinant, kinase-defective MEK1 (MEK1 K97A; a gift from T. Vomastek (University of Virginia, Charlottesville, VA), 100 μM ATP, 10 mM MgCl2, 1 mM DTT, and 25 mM Hepes-NaOH, pH 7.4, for 20 min at 30°C. Reactions were terminated with SDS-PAGE sample buffer and resolved on 10% gels. C-Raf kinase assays were performed as described previously (Slack et al., 1999).
Immunofluorescence
REF52 cells were suspended in serum-free DME for 1 h at 37°C and plated on coverslips coated with 10 μg/ml FN for 10 min or 1 h. The cells were rinsed three times with PBS before fixing and staining. P-MAPK staining was performed as described previously (Zecevic et al., 1998). P-S298 MEK1 staining was performed after fixing the cells with 4% PFA in PBS, permeabilizing with 0.5% Triton X-100, and blocking in 20% NGS and 2% BSA. Cells were incubated with anti–p-S298 MEK1 and antipaxillin antibodies diluted in 10% NGS; 1% BSA followed by FITC-conjugated goat anti–mouse IgG, and Texas red–conjugated goat anti–rabbit IgG in 0.5% BSA at a final concentration of 1.5–2 μg/ml each.
Online supplemental material
Online supplemental figures are available at http://www.jcb.org/cgi/content/full/jcb.200212141/DC1. The figure legends associated with each figure provide descriptions as to how the experiments were performed.
Supplemental Material
Acknowledgments
We are grateful to D. Ilic, T. Vomastek, S. Bagrodia, and R.A. Cerione for providing reagents and R. Tilghman for providing FAK knockout cells. We thank our colleagues for helpful discussions.
This work was supported by the National Institutes of Health grants CA 40042 (to M.J. Weber and J.T. Parsons), CA 29243 (to J.T. Parsons), and CA 80606 (to J.T. Parsons).
The online version of this article includes supplemental material.
M. Zecevic's present address is Cancer Prevention Fellowship Program, Division of Cancer Prevention, National Cancer Institute, 6130 Executive Blvd., Suite 309, MSC 7361, Rockville, MD 20852.
Footnotes
Abbreviations used in this paper: FN, fibronectin; MEK, MAPK kinase; MEKK, MEK kinase; PAK, p21-activated kinase; PI3K, phosphoinositide 3-kinase.
References
- Aplin, A.E., A.K. Howe, and R.L. Juliano. 1999. Cell adhesion molecules, signal transduction and cell growth. Curr. Opin. Cell Biol. 11:737–744. [DOI] [PubMed] [Google Scholar]
- Assoian, R.K., and M.A. Schwartz. 2001. Coordinate signaling by integrins and receptor tyrosine kinases in the regulation of G1 phase cell-cycle progression. Curr. Opin. Genet. Dev. 11:48–53. [DOI] [PubMed] [Google Scholar]
- Bagrodia, S., and R.A. Cerione. 1999. Pak to the future. Trends Cell Biol. 9:350–355. [DOI] [PubMed] [Google Scholar]
- Brugnera, E., L. Haney, C. Grimsley, M. Lu, S.F. Walk, A.C. Tosello-Trampont, I.G. Macara, H. Madhani, G.R. Fink, and K.S. Ravichandran. 2002. Unconventional Rac-GEF activity is mediated through the Dock180-ELMO complex. Nat. Cell Biol. 4:574–582. [DOI] [PubMed] [Google Scholar]
- Brunet, A., G. Pages, and J. Pouyssegur. 1994. Growth factor-stimulated MAP kinase induces rapid retrophosphorylation and inhibition of MAP kinase kinase (MEK1). FEBS Lett. 346:299–303. [DOI] [PubMed] [Google Scholar]
- Calalb, M.B., T.R. Polte, and S.K. Hanks. 1995. Tyrosine phosphorylation of focal adhesion kinase at sites in the catalytic domain regulates kinase activity: a role for Src family kinases. Mol. Cell. Biol. 15:954–963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Catling, A.D., H.J. Schaeffer, C.W. Reuter, G.R. Reddy, and M.J. Weber. 1995. A proline-rich sequence unique to MEK1 and MEK2 is required for raf binding and regulates MEK function. Mol. Cell. Biol. 15:5214–5225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Catling, A.D., S.T. Eblen, H.J. Schaeffer, and M.J. Weber. 2001. Scaffold protein regulation of mitogen-activated protein kinase cascade. Methods Enzymol. 332:368–387. [DOI] [PubMed] [Google Scholar]
- Chaudhary, A., W.G. King, M.D. Mattaliano, J.A. Frost, B. Diaz, D.K. Morrison, M.H. Cobb, M.S. Marshall, and J.S. Brugge. 2000. Phosphatidylinositol 3-kinase regulates Raf1 through Pak phosphorylation of serine 338. Curr. Biol. 10:551–554. [DOI] [PubMed] [Google Scholar]
- Cobb, B.S., M.D. Schaller, T.H. Leu, and J.T. Parsons. 1994. Stable association of pp60src and pp59fyn with the focal adhesion-associated protein tyrosine kinase, pp125FAK. Mol. Cell. Biol. 14:147–155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coles, L.C., and P.E. Shaw. 2002. PAK1 primes MEK1 for phosphorylation by Raf-1 kinase during cross-cascade activation of the ERK pathway. Oncogene. 21:2236–2244. [DOI] [PubMed] [Google Scholar]
- del Pozo, M.A., L.S. Price, N.B. Alderson, X.D. Ren, and M.A. Schwartz. 2000. Adhesion to the extracellular matrix regulates the coupling of the small GTPase Rac to its effector PAK. EMBO J. 19:2008–2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong, C., S.B. Waters, K.H. Holt, and J.E. Pessin. 1996. SOS phosphorylation and disassociation of the Grb2-SOS complex by the ERK and JNK signaling pathways. J. Biol. Chem. 271:6328–6332. [DOI] [PubMed] [Google Scholar]
- Eblen, S.T., J.K. Slack, M.J. Weber, and A.D. Catling. 2002. Rac-PAK signaling stimulates extracellular signal-regulated kinase (ERK) activation by regulating formation of MEK1-ERK complexes. Mol. Cell. Biol. 22:6023–6033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edwards, D.C., L.C. Sanders, G.M. Bokoch, and G.N. Gill. 1999. Activation of LIM-kinase by Pak1 couples Rac/Cdc42 GTPase signalling to actin cytoskeletal dynamics. Nat. Cell Biol. 1:253–259. [DOI] [PubMed] [Google Scholar]
- Ellinger-Ziegelbauer, H., K. Brown, K. Kelly, and U.J. Siebenlist. 1997. Direct activation of the stress-activated protein kinase (SAPK) and extracellular signal-regulated protein kinase (ERK) pathways by an inducible mitogen-activated protein kinase/ERK kinase kinase 3 (MEKK) derivative. J. Biol. Chem. 272:2668–2674. [DOI] [PubMed] [Google Scholar]
- Fabian, J.R., I.O. Daar, and D.K. Morrison. 1993. Critical tyrosine residues regulate the enzymatic and biological activity of Raf-1 kinase. Mol. Cell. Biol. 13:7170–7179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fincham, V.J., M. James, M.C. Frame, and S.J. Winder. 2000. Active ERK/MAP kinase is targeted to newly forming cell-matrix adhesions by integrin engagement and v-Src. EMBO J. 19:2911–2923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frost, J.A., H. Steen, P. Shapiro, T. Lewis, N. Ahn, P.E. Shaw, and M.H. Cobb. 1997. Cross-cascade activation of ERKs and ternary complex factors by Rho family proteins. EMBO J. 16:6426–6438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howe, A.K., and R.L. Juliano. 2000. Regulation of anchorage-dependent signal transduction by protein kinase A and p21-activated kinase. Nat. Cell Biol. 2:593–600. [DOI] [PubMed] [Google Scholar]
- Ilic, D., Y. Furuta, S. Kanazawa, N. Takeda, K. Sobue, N. Nakatsuji, S. Nomura, J. Fujimoto, M. Okada, and T. Yamamoto. 1995. Reduced cell motility and enhanced focal adhesion contact formation in cells from FAK-deficient mice. Nature. 377:539–544. [DOI] [PubMed] [Google Scholar]
- King, A.J., H. Sun, B. Diaz, D. Barnard, W. Miao, S. Bagrodia, and M.S. Marshall. 1998. The protein kinase Pak3 positively regulates Raf-1 activity through phosphorylation of serine 338. Nature. 396:180–183. [DOI] [PubMed] [Google Scholar]
- Kiosses, W.B., R.H. Daniels, C. Otey, G.M. Bokoch, and M.A. Schwartz. 1999. A role for p21-activated kinase in endothelial cell migration. J. Cell Biol. 147:831–844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klemke, R.L., S. Cai, A.L. Giannini, P.J. Gallagher, P. de Lanerolle, and D.A. Cheresh. 1997. Regulation of cell motility by MAPK. J. Cell Biol. 137:481–492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leng, J., R.L. Klemke, A.C. Reddy, and D.A. Cheresh. 1999. Potentiation of cell migration by adhesion-dependent cooperative signals from the GTPase Rac and Raf kinase. J. Biol. Chem. 274:37855–37861. [DOI] [PubMed] [Google Scholar]
- Lin, T.H., Q. Chen, A. Howe, and R.L. Juliano. 1997. Cell anchorage permits efficient signal transduction between ras and its downstream kinases. J. Biol. Chem. 272:8849–8852. [PubMed] [Google Scholar]
- Lipfert, L., B. Haimovich, M.D. Schaller, B.S. Cobb, J.T. Parsons, and J.S. Brugge. 1992. Integrin-dependent phosphorylation and activation of the protein tyrosine kinase pp125FAK in platelets. J. Cell Biol. 119:905–912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manser, E., T.H. Loo, C.G. Koh, Z.S. Zhao, X.Q. Chen, L. Tan, I. Tan, T. Leung, and L. Lim. 1998. PAK kinases are directly coupled to the PIX family of nucleotide exchange factors. Mol. Cell. 1:183–192. [DOI] [PubMed] [Google Scholar]
- Marais, R., Y. Light, H.F. Paterson, and C.J. Marshall. 1995. Ras recruits Raf-1 to the plasma membrane for activation by tyrosine phosphorylation. EMBO J. 14:3136–3145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moro, L., M. Venturino, C. Bozzo, L. Silengo, F. Altruda, L. Beguinot, G. Tarone, and P. Defilippi. 1998. Integrins induce activation of EGF receptor: role in MAP kinase induction and adhesion-dependent cell survival. EMBO J. 17:6622–6632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nobes, C.D., and A. Hall. 1995. Rho, rac, and cdc42 GTPases regulate the assembly of multimolecular focal complexes associated with actin stress fibers, lamellipodia, and filopodia. Cell. 81:53–62. [DOI] [PubMed] [Google Scholar]
- Parsons, J.T., K.H. Martin, J.K. Slack, J.M. Taylor, and S.A. Weed. 2000. Focal adhesion kinase: a regulator of focal adhesion dynamics and cell movement. Oncogene. 19:5606–5613. [DOI] [PubMed] [Google Scholar]
- Qiu, R.G., J. Chen, D. Kirn, F. McCormick, and M. Symons. 1995. An essential role for Rac in Ras transformation. Nature. 374:457–459. [DOI] [PubMed] [Google Scholar]
- Reiske, H.R., S.C. Kao, L.A. Cary, J.L. Guan, J.F. Lai, and H.C. Chen. 1999. Requirement of phosphatidylinositol 3-kinase in focal adhesion kinase-promoted cell migration. J. Biol. Chem. 274:12361–12366. [DOI] [PubMed] [Google Scholar]
- Renshaw, M.W., X.D. Ren, and M.A. Schwartz. 1997. Growth factor activation of MAP kinase requires cell adhesion. EMBO J. 16:5592–5599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Renshaw, M.W., L.S. Price, and M.A. Schwartz. 1999. Focal adhesion kinase mediates the integrin signaling requirement for growth factor activation of MAPK. J. Cell Biol. 147:611–618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roovers, K., G. Davey, X. Zhu, M.E. Bottazzi, and R.K. Assoian. 1999. Alpha5beta1 integrin controls cyclin D1 expression by sustaining mitogen-activated protein kinase activity in growth factor-treated cells. Mol. Biol. Cell. 10:3197–3204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanders, L.C., F. Matsumura, G.M. Bokoch, and P. de Lanerolle. 1999. Inhibition of myosin light chain kinase by p21-activated kinase. Science. 283:2083–2085. [DOI] [PubMed] [Google Scholar]
- Schaller, M.D., J.D. Hildebrand, J.D. Shannon, J.W. Fox, R.R. Vines, and J.T. Parsons. 1994. Autophosphorylation of the focal adhesion kinase, pp125FAK, directs SH2-dependent binding of pp60src. Mol. Cell. Biol. 14:1680–1688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaller, M.D., J.D. Hildebrand, and J.T. Parsons. 1999. Complex formation with focal adhesion kinase: a mechanism to regulate activity and subcellular localization of Src kinases. Mol. Biol. Cell. 10:3489–3505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlaepfer, D.D., and T. Hunter. 1996. Evidence for in vivo phosphorylation of the Grb2 SH2-domain binding site on focal adhesion kinase by Src-family protein-tyrosine kinases. Mol. Cell. Biol. 16:5623–5633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlaepfer, D.D., S.K. Hanks, T. Hunter, and P. van der Geer. 1994. Integrin-mediated signal transduction linked to Ras pathway by GRB2 binding to focal adhesion kinase. Nature. 372:786–791. [DOI] [PubMed] [Google Scholar]
- Sells, M.A., J.T. Boyd, and J. Chernoff. 1999. p21-activated kinase 1 (Pak1) regulates cell motility in mammalian fibroblasts. J. Cell Biol. 145:837–849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sells, M.A., A. Pfaff, and J. Chernoff. 2000. Temporal and spatial distribution of activated Pak1 in fibroblasts. J. Cell Biol. 151:1449–1458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slack, J.K., A.D. Catling, S.T. Eblen, M.J. Weber, and J.T. Parsons. 1999. c-Raf-mediated inhibition of epidermal growth factor-stimulated cell migration. J. Biol. Chem. 274:27177–27184. [DOI] [PubMed] [Google Scholar]
- Tang, Y., Z. Chen, D. Ambrose, J. Liu, J.B. Gibbs, J. Chernoff, and J. Field. 1997. Kinase-deficient Pak1 mutants inhibit Ras transformation of Rat-1 fibroblasts. Mol. Cell. Biol. 17:4454–4464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang, Y., J. Yu, and J. Field. 1999. Signals from the Ras, Rac, and Rho GTPases converge on the Pak protein kinase in Rat-1 fibroblasts. Mol. Cell. Biol. 19:1881–1891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vadlamudi, R.K., L. Adam, R.A. Wang, M. Mandal, D. Nguyen, A. Sahin, J. Chernoff, M.C. Hung, and R. Kumar. 2000. Regulatable expression of p21-activated kinase-1 promotes anchorage-independent growth and abnormal organization of mitotic spindles in human epithelial breast cancer cells. J. Biol. Chem. 275:36238–36244. [DOI] [PubMed] [Google Scholar]
- Wary, K.K., A. Mariotti, C. Zurzolo, and F.G. Giancotti. 1998. A requirement for caveolin-1 and associated kinase Fyn in integrin signaling and anchorage-dependent cell growth. Cell. 94:625–634. [DOI] [PubMed] [Google Scholar]
- Weed, S.A., Y. Du, and J.T. Parsons. 1998. Translocation of cortactin to the cell periphery is mediated by the small GTPase Rac1. J. Cell Sci. 111:2433–2443. [DOI] [PubMed] [Google Scholar]
- West, K.A., H. Zhang, M.C. Brown, S.N. Nikolopoulos, M.C. Riedy, A.F. Horwitz, and C.E. Turner. 2001. The LD4 motif of paxillin regulates cell spreading and motility through an interaction with paxillin kinase linker (PKL). J. Cell Biol. 154:161–176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia, Y., Z. Wu, B. Su, B. Murray, and M. Karin. 1998. JNKK1 organizes a MAP kinase module through specific and sequential interactions with upstream and downstream components mediated by its amino-terminal extension. Genes Dev. 12:3369–3381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiong, W., and J.T. Parsons. 1997. Induction of apoptosis after expression of PYK2, a tyrosine kinase structurally related to focal adhesion kinase. J. Cell Biol. 139:529–539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zecevic, M., A.D. Catling, S.T. Eblen, L. Renzi, J.C. Hittle, T.J. Yen, G.J. Gorbsky, and M.J. Weber. 1998. Active MAPK in mitosis: localization at kinetochores and association with the motor protein CENP-E. J. Cell Biol. 142:1547–1558. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}